

Abstract
Obesity is a risk factor for cancer incidence and cancer mortality. The association of obesity and cancer is attributed to multiple factors, but the tightest linkage is with the chronic, low grade inflammation that accompanies obesity. MDSC are known facilitators of cancer progression that act by suppressing the activation and function of tumor-reactive T cells. Because MDSC quantity and function are driven by chronic inflammation, we hypothesized that MDSC may accumulate in obese individuals and facilitate tumor growth by suppressing antitumor immunity. To test this hypothesis, tumor-bearing mice on a high fat or low fat diet (HFD or LFD) were assessed for tumor progression and the metabolic dysfunction associated with obesity. HFD enhanced the accumulation of MDSC and the resulting MDSC had both beneficial and detrimental effects. HFD-induced MDSC protected mice against diet-induced metabolic dysfunction and reduced HFD-associated inflammation, but also increased the accumulation of fat, enhanced tumor progression and spontaneous metastasis, and reduced survival time. HFD-induced MDSC facilitated tumor growth by limiting the activation of tumor-reactive CD8+ T cells. Leptin, an adipokine that regulates appetite satiety and is over-expressed in obesity, undergoes cross-talk with MDSC in which leptin drives the accumulation of MDSC while MDSC down-regulate the production of leptin. Collectively, these studies demonstrate that although MDSC protect against some metabolic dysfunction associated with HFD, they enhance tumor growth in HFD mice, and that leptin is a key regulator linking HFD, chronic inflammation, immune suppression, and tumor progression.
Keywords: obesity, inflammation, immune suppression, programmed death ligand 1
Summary sentence:
High fat diet drives the accumulation of immune suppressive myeloid-derived suppressor cells which promote cancer growth but provide protection against metabolic dysfunction that accompanies obesity.
Introduction
Epidemiological and laboratory studies strongly indicate that obesity is a risk factor for both cancer incidence and cancer mortality [1–4]. There is a strong correlation for many cancers; however, the association between obesity and breast cancer is particularly evident, especially in post-menopausal women [5, 6]. Cancer risk for multiple cancers has been attributed to a variety of physiological changes that occur during obesity, including changes in growth factors (e.g. insulin, insulin-like growth factor) [7], sex steroid hormones (e.g. estrogen) [8], adipokines (e.g. leptin and adiponectin) [9]), and the intestinal microbiome [10]. Perhaps the tightest linkage between obesity and cancer is that of the chronic, low grade inflammation that accompanies obesity, since chronic inflammation is also an established risk factor for cancer [11–13]. Adipose tissue produces a number of factors that drive and sustain inflammation, and macrophages residing in adipose tissue are a major source. They produce high levels of the pro-inflammatory mediators IL-6, TNFα, and IL-1β which contribute to the chronic low grade inflammation that accompanies obesity [14]. In addition to their pro-obesity activity, IL-1β, TNFα, and IL-6 also drive malignancy. IL-1β and TNFα facilitate the conversion of non-malignant cells to malignant cells by enhancing proliferation and survival [15] and by favoring epithelial to mesenchymal transition [16]. IL-6 activation of STAT3 results in multiple cellular changes that promote malignancy by enhancing cell proliferation, stemness, survival, metastatic spread, and neovascularization [17].
Chronic production of IL-1β, TNFα, and IL-6 also facilitates the progression of cancer by inducing myeloid-derived suppressor cells (MDSC) [18–22]. MDSC are profoundly immune suppressive cells that use a variety of immune and non-immune mechanisms to facilitate tumor growth by inhibiting innate and adaptive immunity, as well as by promoting angiogenesis and metastasis [23–26]. Given earlier studies indicating that obesity increases MDSC levels in mice [27], we hypothesized that the elevated levels of MDSC in obese individuals may be an important mechanism by which obesity facilitates cancer progression.
We have tested our hypothesis using mice fed a high fat diet (HFD) vs. a low fat diet (LFD), conditions that are known to produce obese and lean mice, respectively [28]. Our results demonstrate that MDSC induced by HFD are protective against some of the metabolic dysfunction associated with weight gain, while suppressing T cell function and increasing tumor progression. Mechanistically, the adipokine leptin, which regulates appetite satiety and is up-regulated in HFD mice, is responsible for driving the accumulation of MDSC.
Materials and Methods
Mice, 4T1 tumor inoculation, MDSC
BALB/c, C57BL/6, transgenic BALB/c clone 4 (H-2Kd-restricted, TcR specific for influenza hemagglutinin peptide518–526), transgenic C57BL/6 OT1 (H-2Kb-restricted, TcR specific for ovalbumin peptide SIINFEKL), and BALB/c PDL1−/− mice were bred in the UMBC animal facility from stock obtained from The Jackson Laboratory (Bar Harbor, ME) or breeding pairs provided by Dr. D. Pardoll (PDL1−/−; Johns Hopkins). Mice were fed chow with 60% energy derived from fat (high fat diet HFD), or matched chow containing 10% of energy derived from fat (low fat diet, LFD) (Research Diets, Inc., New Brunswick, NJ; D12492 and D12450J, respectively). Unless noted otherwise “HFD” and “LFD” mice refer to groups that were maintained on a HFD or LFD, respectively, for a minimum of 8 weeks. BALB/c-derived 4T1 tumor cells were maintained as described [29].
Mice were inoculated orthotopically in the abdominal mammary fat pad with 7×103 4T1 cells in 100μl DMEM. Primary tumors were measured with digital calipers as described [29]. Tumor volume = 4/3πr3 (where r= ½ average of two perpendicular diameters). Survival time was recorded when mice became moribund and were euthanized. Metastatic disease was quantified as described using the clonogenic assay [30].
MDSC were obtained from the blood of tumor-free and 4T1-tumor-bearing mice when tumors were ~ 8–10 mm in diameter. MDSC from the blood of tumor-free mice were purified using anti-Gr1 antibody (RB6.8C5, IgG2b; BioXcell, (West Lebanon, NH), magnetic beads, and LS columns (Miltenyi Biotech) as previously described [31]. Leukocyte content of RBC-depleted blood from 4T1-tumor-bearing mice typically contained 85–90% MDSC so further purification was not performed. MDSC from dissociated tumors were purified using a myeloid-derived suppressor cell purification kit (Miltenyi Biotech, Auburn, CA). MDSC used in experiments were >85–90 % Gr1+CD11b+ as assessed by flow cytometry. All animal procedures were approved by the UMBC Institutional Animal Care and Use Committee.
Antibodies, other reagents, and flow cytometry
Fluorescently labeled anti-mouse mAbs CD11b-Pacific Blue (M1/70), Gr1-APC (RB6–8C5), CD8-PE (53–6.7), CD3-APC (145–2C11), CD4-Pacific Blue (GK1.5), Ly6G-APC (1A8), Ly6C-PE (HK1.4), F4/80-APC (BM8), CD69-PE-Cy7 (H1–2F3), anti-PDL1-PE (10F.9G2), anti-Rat IgG2A-FITC, and IgG isotype controls were from BioLegend (San Diego, CA). CD25-FITC (7D4), CD44-FITC (IM7), anti-mouse arginase 1 (clone 19; mIgG1), and rat-anti-mouse IgG1-FITC (A85–1) were from BD Biosciences (San Jose, CA). Uncoupled anti-PDL1 (10F.9G2) was from BioXcell (West Lebanon, NH). Anti-mouse leptin, IFNγ, and CD80-Fc were from R&D Systems (Minneapolis, MN). Novolin-R human insulin was from ADW Diabetes Suppliers (Pompano Beach, FL). Live cells were cell surface stained with Abs and analyzed on a Beckman/Coulter Cyan ADP flow cytometer using Summit v4.3 software as described [32]. Internal staining was performed following fixation with formaldehyde and permeabilization with saponin as described [33]. %CD11b+Ly6G+ or CD11b+Ly6C+ cells = (%CD11b+Ly6C+ or %CD11b+ Ly6G+)/(%CD11b+Ly6G+ + %CD11b+Ly6C+)
Tumor, lung, and liver dissociations
Tumors were dissociated as previously described [31]. Briefly, resected tumors were minced, placed into GentleMACS C tubes (Miltenyi Biotech) with dissociation medium (DMEM with 300 U/ml collagenase IV, 0.1% hyaluronidase, and 2 kU/ml DNase I), processed on the GentleMACS dissociator, rotated (10 rpm; Glas-Col rotator) at 37 °C for 40 min, then processed twice on the GentleMACS dissociator. Remaining debris was removed by passing through a 70-μm mesh. For some samples dead cells were removed by ficoll density gradient centrifugation. Livers were dissociated into a single cell suspension as described by [34] except Gey’s solution was used to lyse RBC instead of ACK solution. Lungs were dissociated as previously described [35].
MDSC and T cell depletions
Mice were MDSC-depleted or control antibody-treated by twice weekly IP injection of 200 μg Gr1 mAb (RB6–8C5; rat IgG2b) or isotype-matched control IgG (LTF-2; rat IgG2b), respectively, both from BioXcell. For CD4+ and CD8+ T cell depletions, mice were treated with GK1.5 plus 2.43 mAbs (100μl of each ascites or 200μg of purified mAb) i.p. on days −6, −3, and −1, followed by twice weekly injections for up to 8 weeks until moribund. Depletion was confirmed by flow cytometry.
Fasting glucose and insulin tolerance
For the fasting glucose test, mice were fasted for 6 hrs and then blood glucose levels were measured using a One-Touch Ultra II glucometer (LifeScan, Inc., Johnson & Johnson). For insulin tolerance tests, mice were injected with insulin (0.75 IU/kg body weight diluted in PBS; 40–60μl/mouse) and blood glucose was measured using the One-Touch Ultra glucometer every 30 min over a two hour period by collecting one drop of blood (~50 μl) from the tail vein. Area under the curve (AUC) for insulin tolerance was calculated using https://www.statstodo.com/AUC_Pgm.php.
MDSC suppression
MDSC suppression of T cell activation was assayed as described [29]. Briefly, splenocytes from TcR transgenic mice were cultured with cognate peptide and tumor-infiltrating MDSC from 4T1-bearing BALB/c mice. Cultures were pulsed with 3H-thymidine on day 3 and harvested on day 4. Supernatants from replicate plates without 3H-thymidine were collected and analyzed for IFNγ by ELISA (R&D Systems). For suppression assays in which PDL1 was blocked, PDL1 antibody (10F.9G2) was added to each well at 5.47 ng/ml. Percent suppression = [1-(T cells+MDSC/T cells)] × 100%.
Leptin
Plasma leptin levels were assayed using a Quantikine anti-mouse and rat Leptin ELISA kit per the manufacturer’s directions (R&D, MOB00).
Reactive oxygen species
ROS production was measured by staining with dichlorodihydrofluorescein diacetate (DCFDA) as described [29]. Briefly, 1×106 MDSC were incubated in serum-free DMEM containing 0.2 μM DCFDA for 20 min at 37°C. Stained cells were washed twice with excess cold PBS and analyzed by flow cytometry.
T cell adoptive transfer and in vivo activation
CD8+ T cells were isolated from the spleens of OT-1 transgenic mice using an EasyStep Mouse CD8+ T Cell Isolation Kit (Stem Cell Technologies), and labeled using the CellTrace Violet Cell Proliferation Kit (Life Technologies, Carlsbad, CA) at a dye concentration of 5mM, according to the manufacturers’ protocol. Female C57BL/6 mice were adoptively transferred via the retro-orbital sinus with 5×106-1×107 violet-labeled CD8+ T cells in 100 μl PBS. Twenty-four hours later mice were injected in the retro-orbital sinus with cognate peptide (SIINFEKL, 50μg/100 μl PBS). Forty-eight hours later, the mice were euthanized and their splenocytes stained and assayed by flow cytometry.
Inhibition of leptin receptor
BALB/c mice that had been maintained on a HFD for a minimum of eight weeks were inoculated daily for three consecutive days in the retro-orbital sinus with soluble mouse Leptin Receptor-Fc (Ob-R; Fc is IgG2a; R&D Systems) (100 μg/200 μl sterile PBS). Control mice were injected with an irrelevant recombinant protein (nonfunctional custom-made CD80-Fc) or IgG2a (R&D Systems). Seventy-two hrs after the last injection mice were bled and the level of circulating MDSC was assessed by flow cytometry.
IFNγ induction of PDL1
Blood MDSC (1–2 × 105/well/200μl medium (HL1, 5×10−5M β-mercaptoethanol, 1% penicillin/streptomycin, 1% glutamax, 1% gentamycin)) were incubated at 37°C for ~24 hrs. with 100 units/ml mouse IFNγ. Cells were harvested, stained with antibodies to Gr1, CD11b, and PDL1, and the gated Gr1+CD11b+ cells analyzed for PDL1 expression by flow cytometry.
qRT-PCR
Tumor or parametrial fat pads were dissected and immediately placed in RNAlater Solution (Ambion, Life Technologies) and then stored at 4°C overnight. Tumor tissue was transferred to TRIzol Reagent (Ambion, Life Technologies), homogenized using the Tissue Tearor homogenizer (Biospec Products, Inc., Bartlesville, OK), and RNA was then isolated following the manufacturer’s protocol. Parametrial fat pad RNA was isolated using the RNeasy Lipid Tissue Mini Kit (Qiagen, Promega, Madison, WI) according to the manufacturer’s protocol. RNA concentrations were determined using a BioTek Synergy 2 microplate reader. cDNA was created from each RNA sample using the Maxima cDNA kit (Thermo Fisher). Transcripts were amplified using the KiCqStart SYBR Green qPCR ReadyMix, iQ (Sigma Life Science, Darmstadt, Germany) following the manufacturer’s protocol, and detected using a CFX96 Real-Time PCR Detection System (Bio-Rad). Each sample was analyzed in triplicate and each primer set was tested by a melting curve. Relative expression of each experimental transcript was normalized to mRNA expression of the ribosomal protein L32 or β-actin.
Primers
Mouse IL-10 forward 5’-GCCGGGAAGACAATAACTGC-3’
Mouse IL-10 reverse 5’-TGTCCAGCTGGTCCTTTGTT-3’
Mouse TNF-α forward 5’-ACGGCATGGATCTCAAAGAC-3’
Mouse TNF-α reverse 5’-CGGACTC CGCAAAGTCTA AG-3’
Mouse IFNγ forward 5’- TTGCCAAGTTTGAGGTCAACAAC-3’
Mouse IFNγ reverse 5’-CGAATCAGCAGCGACTCCTT-3’
Mouse Leptin forward 5’- TCACACACGCAGTCGGTATC-3’
Mouse Leptin reverse 5’- CACATTTTGGGAAGGCAGGC-3’
Mouse L32 forward 5’- CTGCCATCTGTTTTACGGCA-3’
Mouse L32 reverse 5’- ATCAGGATCTGGCCCTTGAAC-3’
Mouse β-actin forward 5’- TGAGCTGCGTTTTACACCCT-3’
Mouse β-actin reverse 5’ - TTTGGGGGATGTTTGCTCCA-3’
Statistical analyses
Differences in tumor volume growth rates were determined by pairwise comparison test (compare groups of growth curves); survival curves were analyzed by log rank test, using the Walter & Eliza Hall Bioinformatics website (http://bioinf.wehi.edu.au/software/compareCurves/ and http://bioinf.wehi.edu.au/software/russell/logrank/, respectively. Data from insulin tolerance tests were calculated by determining the area under the curve (AUC). Other results were analyzed using Student’s unpaired 2-tailed t test (Microsoft Excel 2013). P values <0.05 were considered significant. * indicates p<0.05; ** indicates p<0.01; *** indicates p<0.001. Error bars represent SD from the mean unless noted otherwise.
Results
High fat diet induces weight gain and increases the accumulation of immune suppressive MDSC
Given the well documented effects of nutritional overload on the risk and progression of breast cancer, we have focused our studies on females. Six to eight week old female BALB/c and C57BL/6 mice were started on a HFD or LFD and maintained on their respective diet for twelve weeks. Pre-diet weights for the HFD and LFD groups of each strain were not significantly different (Fig. 1A). Overall, the BALB/c post-diet LFD and HFD groups were 28.5% and 53.1% heavier than their pre-diet weights, respectively, and the C57BL/6 post-diet LFD and HFD groups were 34.8% and 126.2% heavier than their pre-diet weights, respectively, demonstrating that a HFD causes a greater weight gain than a LFD.
Figure 1. HFD increases body weight and the quantity of immune suppressive MDSC in blood.
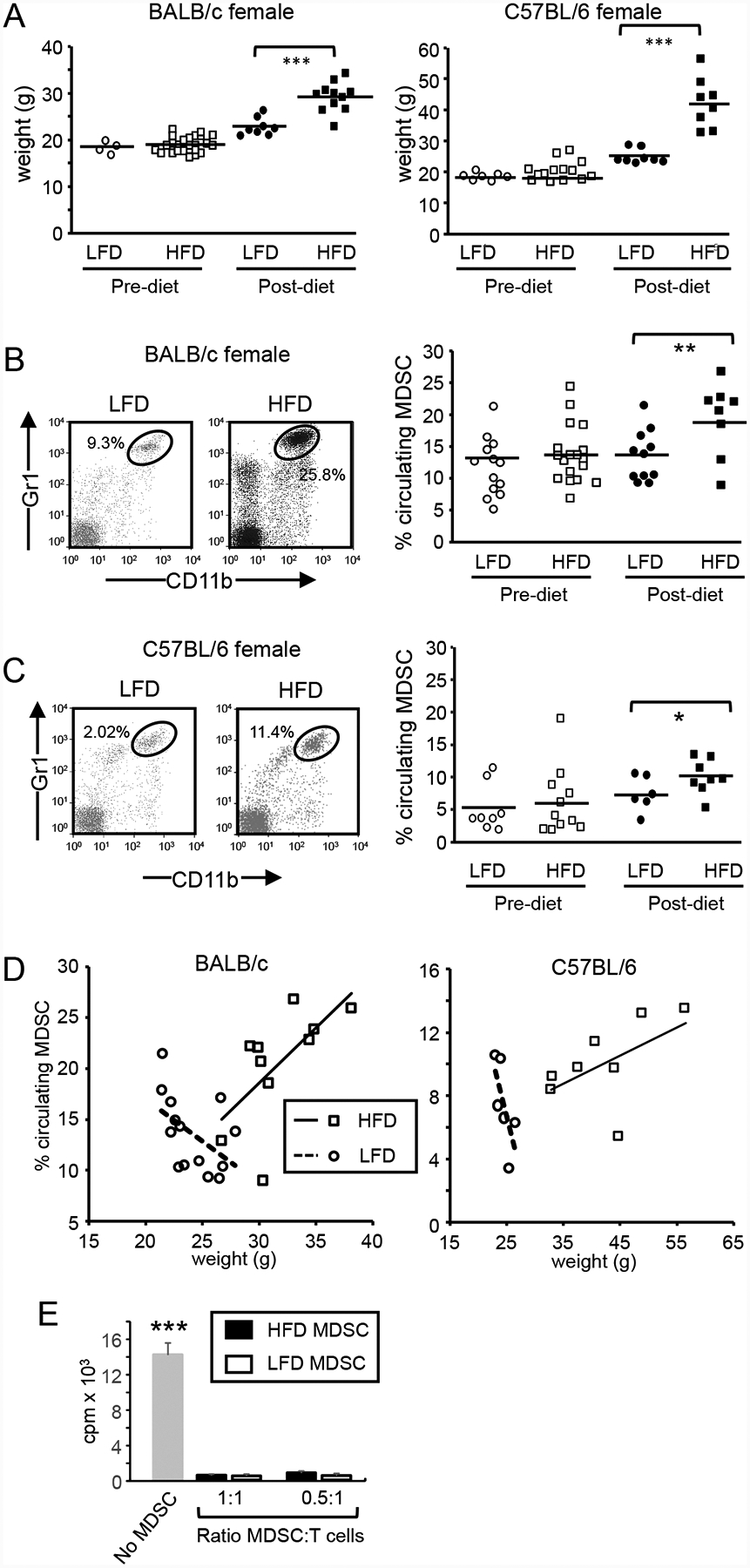
Female 6–8 week old BALB/c and C57BL/6 mice were maintained on a HFD or LFD for 12 weeks and (A) weighed. Six to 8 week old female BALB/c (B) and C57BL/6 (C) mice were bled and then placed on a HFD or LFD for 12 weeks and the percent of circulating total, PMN-MDSC and M-MDSC was determined by flow cytometry pre and post-diet. (D) HFD increases and LFD decreases the number of circulating MDSC in proportion to weight. The % of MDSC in the blood of female BALB/c and C57BL/6 female mice on a HFD or LFD was plotted relative to weight. (E) Gr1+CD11b+ cells from HFD and LFD mice are immune suppressive. Gr1+CD11b+ cells from the spleens of 8 HFD mice were purified by magnetic bead sorting, and cultured with splenocytes from Clone4 TcR transgenic mice plus cognate peptide at ratios of 1:1 and 0.5:1 MDSC:T cells. T cell activation was assessed by incorporation of tritiated thymidine. All values with MDSC are statistically significantly different from no MDSC at p<0.001.
To determine if HFD impacted MDSC levels, pre-diet (6–8 weeks old) and post-diet (18–20 weeks old) female BALB/c (Fig. 1B) and C57BL/6 (Fig. 1C) mice were bled and the levels of circulating total MDSC (Gr1+CD11b+ cells) were assessed by flow cytometry. Pre-diet levels of circulating MDSC Gr1+CD11b+ cells in the BALB/c LFD and HFD groups were not statistically significantly different (average of 14.3% ± 7.8 and 13.8% ± 4.5 Gr1+CD11b+ cells, respectively). After being on their diets for 12 weeks, LFD and HFD BALB/c mice contained 9.2% and 38.8% more total MDSC, respectively, than their pre-diet counterparts. For the C57BL/6 HFD group, diet increased total MDSC by 19.7%, while for the C57BL/7 LFD group, diet decreased total MDSC by 13.6%.
We also assessed levels of polymorphonuclear/granulocytic MDSC (PMN-MDSC; CD11b+Ly6G+Ly6C+/−) and monocytic MDSC (M-MDSC; CD11b+Ly6G-Ly6C+ cells) for both pre and post-diet BALB/c and C57BL/6 mice (Supplemental Fig. 1). The ratio of M-MDSC to PMN-MDSC did not significantly change for either BALB/c or C57BL/6 mice pre and post diet (PMN-MDSC:M-MDSC for LFD pre vs post diet: for BALB/c LFD 41.5:1 vs 39.3:1; for BALB/c HFD pre vs post diet: 45.1:1 vs 37.7:1; for C57BL/6 LFD pre vs post diet: 11.3:1 vs 4.2:1; for C57BL/6 HFD pre vs post diet: 5.6:1 vs 4.1:1, indicating that HFD does not preferentially increase M-MDSC or PMN-MDSC.
To determine if MDSC levels are impacted by weight, we plotted MDSC levels (% blood Gr1+CD11b+ cells) of HFD and LFD mice as a function of weight (Fig. 1D). For both BALB/c and C57BL/6 HFD mice, MDSC levels increased with increasing weight. In contrast, MDSC levels of LFD BALB/c and C57BL/6 mice remained unchanged or decreased with increased weight.
To confirm that the cells of HFD and LFD mice were immune suppressive MDSC, Gr1+CD11b+ cells were magnetic bead purified from whole blood of HFD and LFD mice that had been on their diets for >12 weeks and tested for their ability to suppress the proliferation of antigen-specific transgenic T cells, the hallmark of MDSC (Fig. 1E). Gr1+CD11b+ cells from both HFD and LFD mice were profoundly immune suppressive, identifying the cells as MDSC.
These results indicate that as mice on a HFD gain weight, MDSC levels in the blood also increase. Mice on a LFD also gain weight; however, their levels of circulating MDSC do not significantly increase, suggesting that a LFD protects against the accumulation of MDSC.
High fat diet-induced MDSC protect against metabolic dysfunction and reduce HFD-associated inflammation, but promote the accumulation of fat
Metabolic dysfunction is frequent in obese individuals or individuals consuming a HFD. Common problems include elevated fasting glucose levels and elevated insulin tolerance which are characteristic of type II diabetes [36]. To determine if HFD-induced MDSC contribute to either of these metabolic dysfunctions, 6–8 week old female BALB/c and C57BL/6 mice were placed on a HFD or LFD for eight weeks. Some groups were also concurrently antibody-depleted for MDSC or treated with an isotype matched antibody as a control. At the end of the eight weeks, BALB/c and C57BL/6 HFD mice were significantly heavier than the LFD mice (Fig. 2A). HFD mice of both strains had significantly higher fasting glucose levels (Fig. 2B) and significantly increased insulin tolerance (Fig. 2C) compared to their corresponding LFD mice. Depletion of MDSC further increased fasting glucose levels (Fig. 2B) and insulin tolerance levels (Fig. 2C), demonstrating that MDSC protect against these obesity-related dysfunctions.
Figure 2. HFD-induced MDSC protect against metabolic dysfunction and reduce inflammation, but promote adiposity.
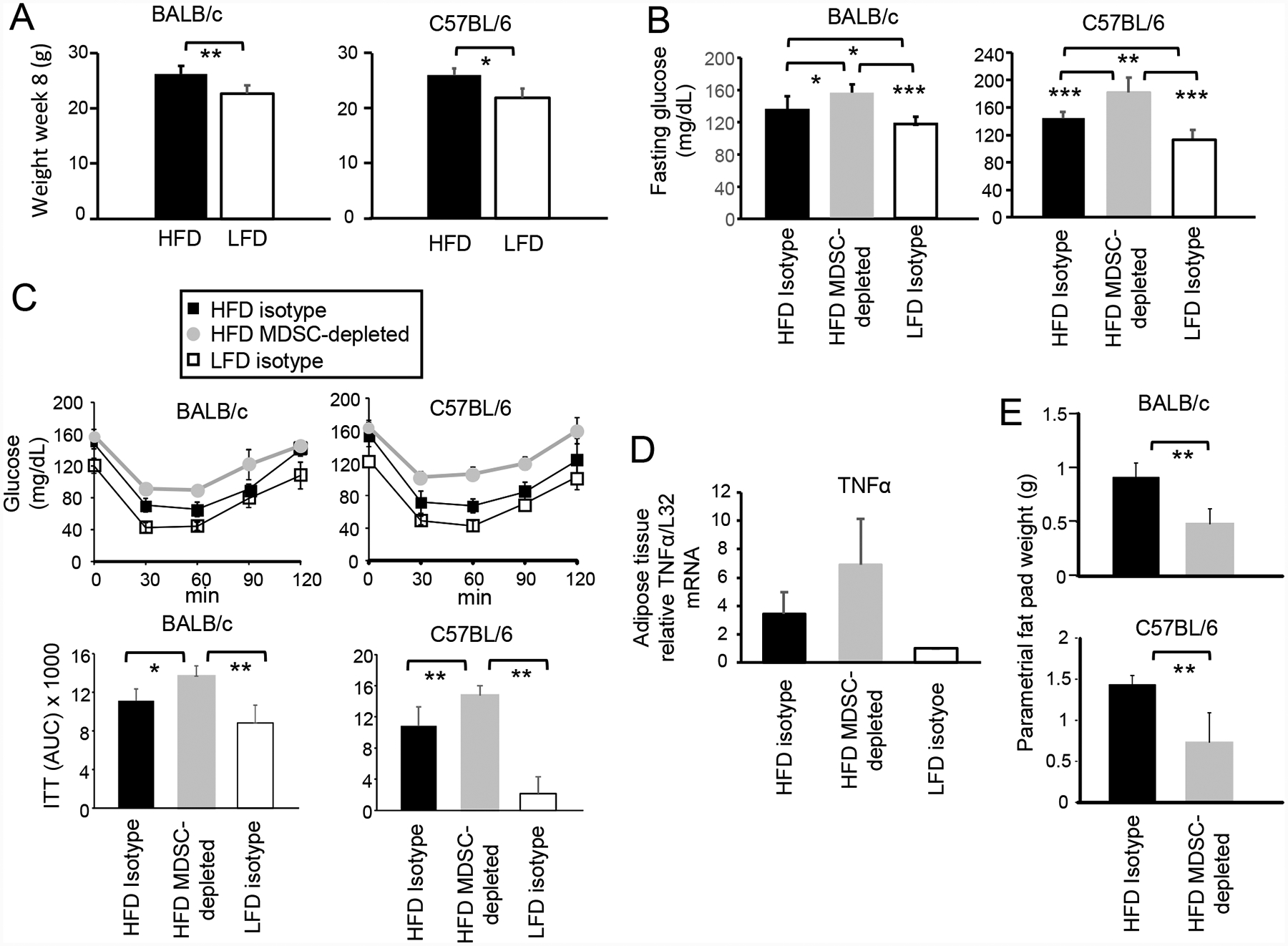
BALB/c and C57BL/6 female mice were maintained on a HFD or LFD diet for eight weeks. Some groups were concurrently depleted for MDSC or treated with isotype control antibodies. (A) HFD increases body weight. Mice on a HFD or LFD for 8 weeks were weighed. BALB/c: HFD n=20, LFD n=10; C57BL/6: HFD n=5, LFD n=5. (B) MDSC protect against HFD-induced elevated serum glucose. BALB/c and C57BL/6 mice on a HFD or LFD for 8 weeks were fasted for 6 hr and then tested for glucose levels in the blood. BALB/c: HFD and LFD n=10 mice/group; C57BL/6: HFD n= 5 mice/group, LFD n = 4 mice/group. (C) MDSC protect against insulin tolerance. Mice were injected with insulin, bled at 30 minute intervals, and the blood tested for glucose (insulin tolerance test). Area under the curve (AUC) values are pooled for 5 mice/group for each strain. (D) MDSC reduce inflammation in adipose tissue. RNA was isolated from the parametrial fat pads of BALB/c mice and assayed by qRT-PCR for expression of TNFα. Data are fold-change in HFD tissue relative to the same tissue from LFD mice. n=5 mice/group for HFD and LFD mice, and n=4 mice/group for HFD MDSC-depleted group. Data are from one of two independent experiments. (E) MDSC promote the accumulation of visceral adipose tissue. BALB/c and C57BL/6 HFD mice were MDSC-depleted or treated with control isotype mAb and their parametrial fat pads were dissected and weighed. n=4–5 mice/group.
To ascertain that the HFD induces a low grade chronic inflammation comparable to obesity, the level of TNFα in adipose tissue of HFD and LFD mice was analyzed by qRT-PCR. Adipose tissue from MDSC-depleted HFD mice was included in the assay to determine if HFD-induced MDSC impact TNFα levels (Fig. 2D). HFD diet adipose tissue contained significantly higher levels of TNFα mRNA as compared to LFD adipose tissue. Depletion of MDSC in HFD mice further increased the TNFα mRNA level in adipose tissue, demonstrating that HFD-induced MDSC down-regulate the inflammatory milieu. These results identify another mechanism by which HFD-induced MDSC protect against a complication of nutrient overload.
Adipose tissue and its associated infiltrating adaptive and innate immune cells are a major source for many of the cytokines and chemokines that drive metabolic dysfunction, so that increased quantities of adipose tissue exacerbate dysfunction. To determine if HFD-induced MDSC impact the quantity of adipose tissue in HFD mice, parametrial fat pads from BALB/c and C57BL/6 HFD and MDSC-depleted HFD mice were dissected and weighed (Fig. 2E). Mice depleted for MDSC had significantly less adipose tissue compared to mice with MDSC, indicating that MDSC facilitate the accumulation of adipose tissue. These results demonstrate that in contrast to the ability of MDSC to diminish metabolic dysfunction and inflammation, MDSC help drive obesity by facilitating the accumulation of adipose tissue.
HFD-induced MDSC facilitate tumor progression
Since MDSC are a known facilitator of tumor progression, we hypothesized that HFD-induced MDSC may contribute to more rapid tumor growth and reduced survival of mice on a HFD. To test this hypothesis we used the mouse 4T1 mammary carcinoma breast cancer model since tumor progression is particularly accelerated in obese women with breast cancer [5]. BALB/c mice were started on a HFD or LFD and eight weeks later were inoculated in the mammary fat pad with 4T1 cells. One group of HFD mice was also treated twice weekly with antibody to Gr1 to deplete MDSC. Three weeks after tumor inoculation the mice were bled and the levels of circulating MDSC determined by flow cytometry (Fig. 3A). MDSC levels in the blood of HFD tumor-bearing mice were higher than MDSC levels in LFD tumor-bearing mice. Depletion significantly reduced the level of circulating MDSC in HFD mice, but consistent with previous findings, depletion did not completely eliminate MDSC due to their homeostatic regulation [31]. MDSC depletion reduced tumor progression in HFD mice; however, these tumors still grew more rapidly than tumors in LFD mice (Fig. 3B). HFD mice had a significantly shorter survival time (MST 35.14 ± 5.42) than LFD mice (MST ± 47.47 ± 6.96; p<0.001) and HFD MDSC-depleted mice (MST 41.8 ± 10.83; p<0.05) (Fig. 3C). Survival times of HFD MDSC-depleted and LFD mice were not significantly different. These results demonstrate that HFD reduces the survival time of tumor-bearing mice, and that MDSC are responsible for the reduced survival time.
Figure 3. HFD-induced MDSC facilitate primary tumor growth and metastatic disease, and reduce survival time.
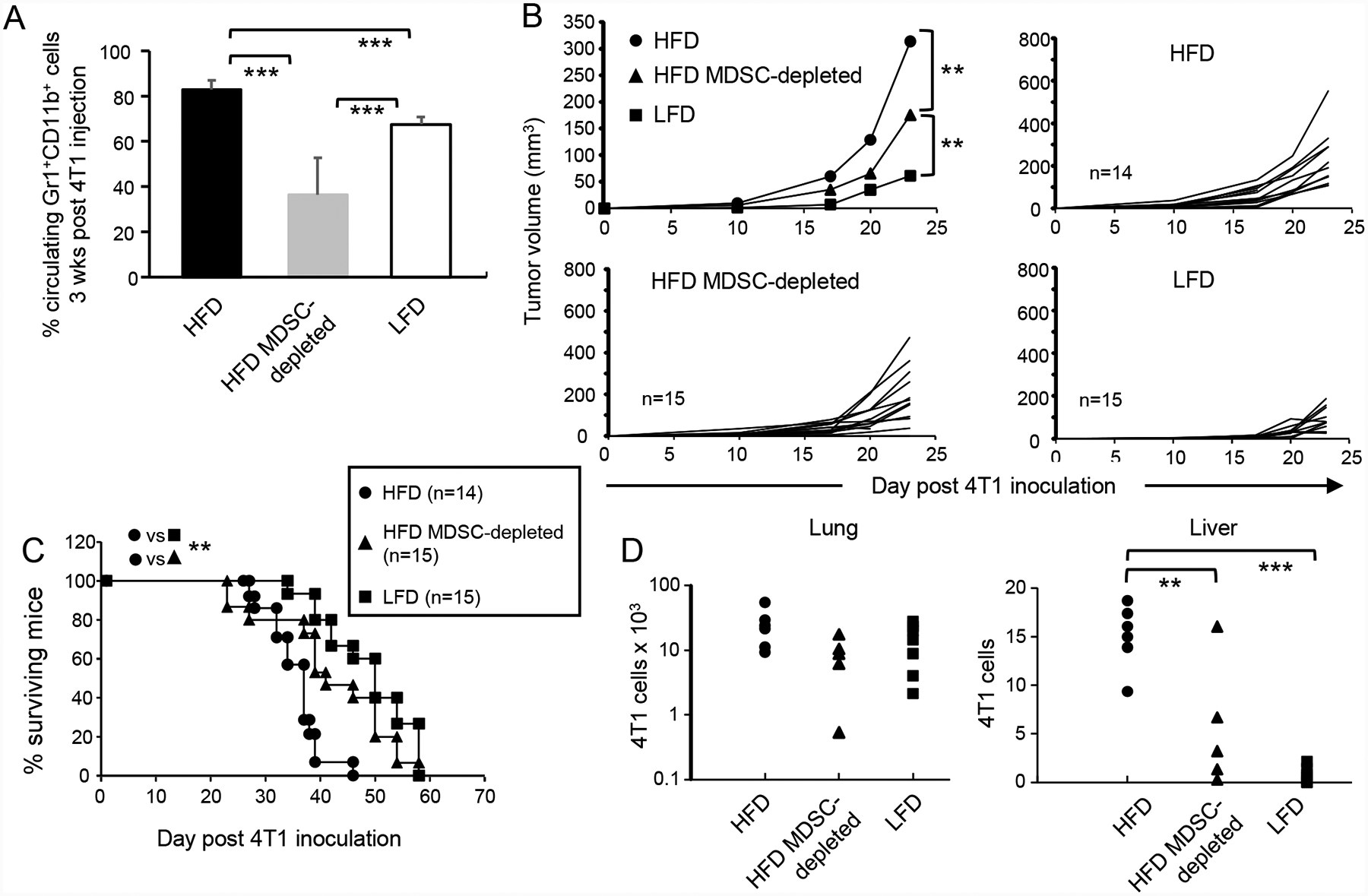
Female BALB/c mice were maintained on a HFD or LFD for eight weeks and then inoculated with 7000 4T1 mammary carcinoma cells. Mice were then either depleted for MDSC or treated with PBS. (A) MDSC levels are elevated in HFD tumor-bearing mice. Three weeks after tumor inoculation, the level of MDSC in the blood was determined. n=9, 6, and 8 mice/group for HFD, HFD-MDSC-depleted, and LFD, respectively. (B) HFD-induced MDSC facilitate primary tumor growth and (C) HFD-induced MDSC decrease survival time. Mice were followed for primary tumor growth and survival time. (D) HFD-induced MDSC enhance spontaneous metastatic disease. Twenty-eight days post tumor inoculation, mice were sacrificed and their lungs and livers assayed by clonogenic assay for metastatic 4T1 cells. Each marker represents an individual mouse. Data for A-C are pooled from two independent experiments.
4T1 is a spontaneously metastatic tumor [30]. To determine if HFD-induced MDSC impact metastasis, female BALB/c mice were maintained on a HFD or LFD for eight weeks and then inoculated in the mammary fat pad with 4T1 cells. One group of HFD mice was also depleted for MDSC. Twenty-eight days later the lungs and livers of tumor-bearing mice were removed and assessed by clonogenic assay for metastatic 4T1 tumor cells (Fig. 3D). Numbers of metastatic cells in the lungs was the same for all groups. However, HFD mice had significantly more metastatic 4T1 cells in their livers as compared to LFD mice, and MDSC depletion significantly reduced the number of metastatic 4T1 cells in the livers of HFD mice. There was no difference in the levels of MDSC in either the lungs or livers of tumor-bearing HFD vs LFD mice (supplemental Fig. 2).
These results demonstrate that HFD-induced MDSC are responsible for the more rapid tumor growth in HFD mice, the shortened survival time of tumor-bearing HFD mice, and the higher level of metastatic cells in the liver of HFD mice.
HFD-induced MDSC facilitate tumor progression by suppressing T cell activation
To determine if tumor progression in HFD mice is regulated by T cells, HFD mice were inoculated with 4T1 tumor cells and depleted for CD4+ plus CD8+ T cells or treated with an irrelevant control mAb (Supplemental Fig. 3). Tumors grew more rapidly in the T cell depleted mice indicating that T cells in HFD can delay tumor progression.
To determine if HFD-induced MDSC facilitate tumor growth by acting on T cells, HFD and LFD mice were inoculated with 4T1 tumor and either depleted for MDSC, concurrently depleted for CD4+ and CD8+ T cells, or untreated (Fig. 4A). As previously observed (Fig. 3B) tumors grew more rapidly in HFD vs. LFD mice and MDSC depletion reduced tumor progression. Tumor progression in HFD mice depleted for MDSC plus T cells was significantly faster than tumor progression in the other groups. HFD mice depleted for MDSC plus T cells had reduced survival times that were equivalent to isotype control-treated HFD mice (Fig. 4B). These results demonstrate that in HFD mice, MDSC reduce survival by acting on T cells.
Figure 4. HFD-induced MDSC facilitate tumor progression by suppressing T cell activation.
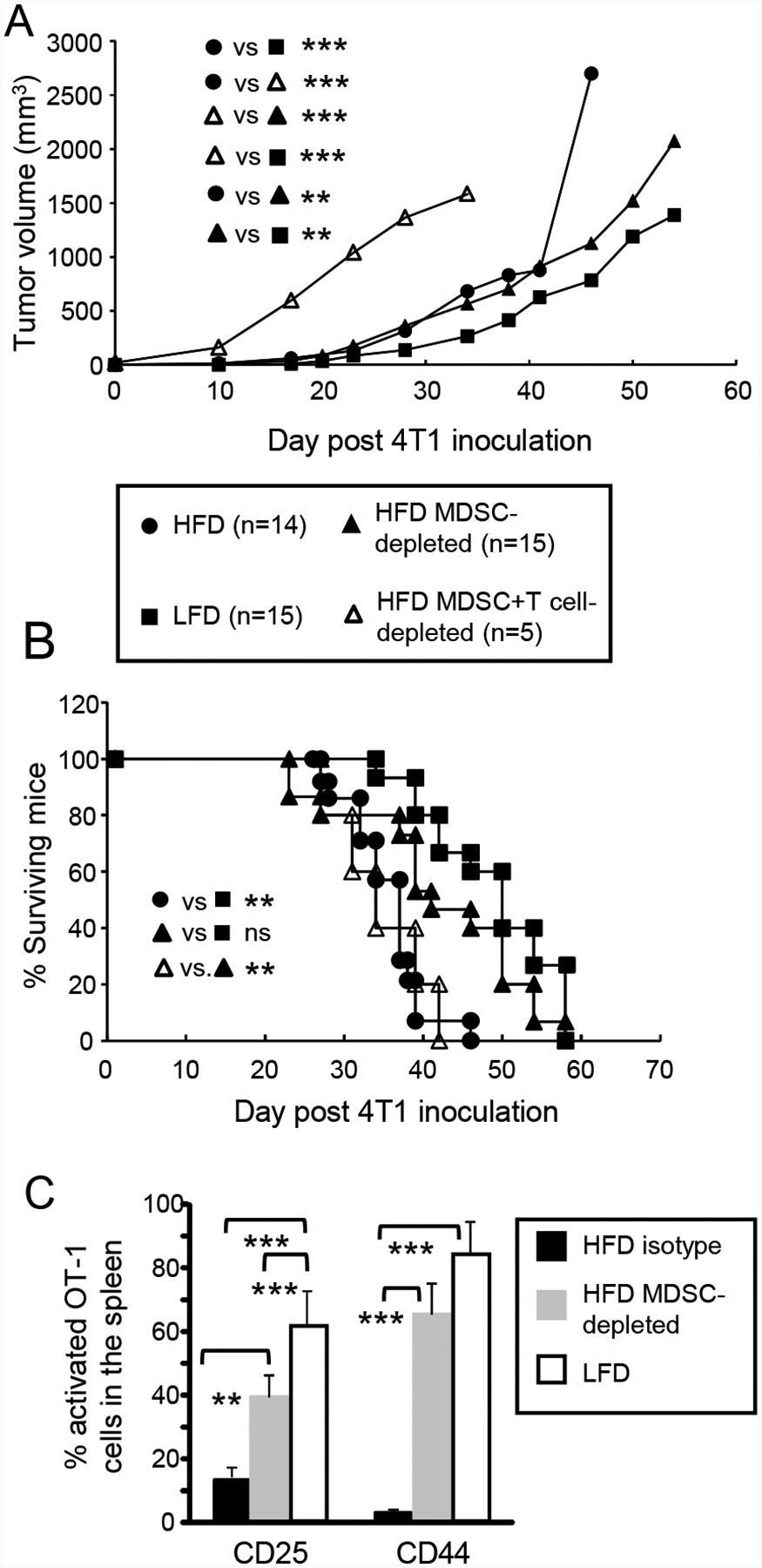
Female BALB/c mice were maintained on a HFD or LFD for nine weeks and then inoculated with 4T1 mammary carcinoma cells. HFD mice were then either depleted for MDSC, depleted for MDSC and CD4+ and CD8+ T cells, or treated with an isotype-matched control mAb. (A, B) MDSC enhance tumor progression and survival by acting on T cells. Mice were monitored for (A) primary tumor progression and (B) survival time. Legend applies to both (A) and (B). (C) HFD-induced MDSC inhibit T cell activation in vivo. Tumor-bearing BALB/c HFD, HFD MDSC-depleted, and LFD mice were adoptively transferred with violet-labeled CD8+ OT1 cells and one day later were injected with cognate peptide. Two days after peptide inoculation, mice were sacrificed and their spleens analyzed for activated CD3+violet+ OT1 T cells as assessed by the activation markers CD25 and CD44. n=3–4 mice/group.
MDSC use a variety of mechanisms to obstruct anti-tumor immunity and promote tumor progression [25], but their canonical function is their ability to suppress T cell activation [26]. To determine if HFD-induced MDSC enhance tumor growth by inhibiting T cell activation, HFD, HFD diet MDSC-depleted, and LFD mice carrying 4T1 tumors were adoptively transferred with violet-labeled CD8+ OT-1 transgenic T cells and challenged with cognate peptide. Two days later the mice were sacrificed and their splenic OT-1+ CD8+ T cells were analyzed by flow cytometry for the activation markers CD25 and CD44 (Fig. 4C). LFD mice contained significantly more activated OT-1 cells compared to HFD mice. MDSC depletion of HFD mice significantly increased the level of activated OT-1 T cells. These results demonstrate that T cell activation in tumor-bearing HFD mice is impaired by MDSC.
HFD increases the suppressive potency of tumor-infiltrating MDSC
Chronic inflammation is characteristic of obesity and HFD mice have elevated levels of the pro-inflammatory mediator TNFα in their adipose tissue (Fig. 2D) and elevated levels of IL-6 in their blood [27]. To determine if heightened inflammation also exists in the tumor microenvironment, 4T1 tumors of HFD and LFD mice were assayed by qRT-PCR for IL-1β, an inflammatory cytokine that is frequently present in inflamed tumors. IL-1β was elevated in the HFD tumors (Fig. 5A). Since chronic inflammation is an established driver of MDSC suppressive potency [18, 37], we tested if tumor-infiltrating MDSC from HFD mice are more suppressive than tumor-infiltrating MDSC from LFD mice. MDSC were magnetic bead purified from 4T1 tumors of HFD and LFD mice and tested for their ability to inhibit the activation of antigen-specific Clone 4 transgenic T cells (Fig. 5B). HFD-induced MDSC were more suppressive on a per cell basis than LFD MDSC, consistent with the concept that HFD elevates both systemic and intratumoral inflammation and increases MDSC suppressive activity.
Figure 5. The tumor microenvironment induces MDSC expression of PDL1 and enhances tumor progression. BALB/c HFD and LFD mice were inoculated with 4T1 tumor cells.
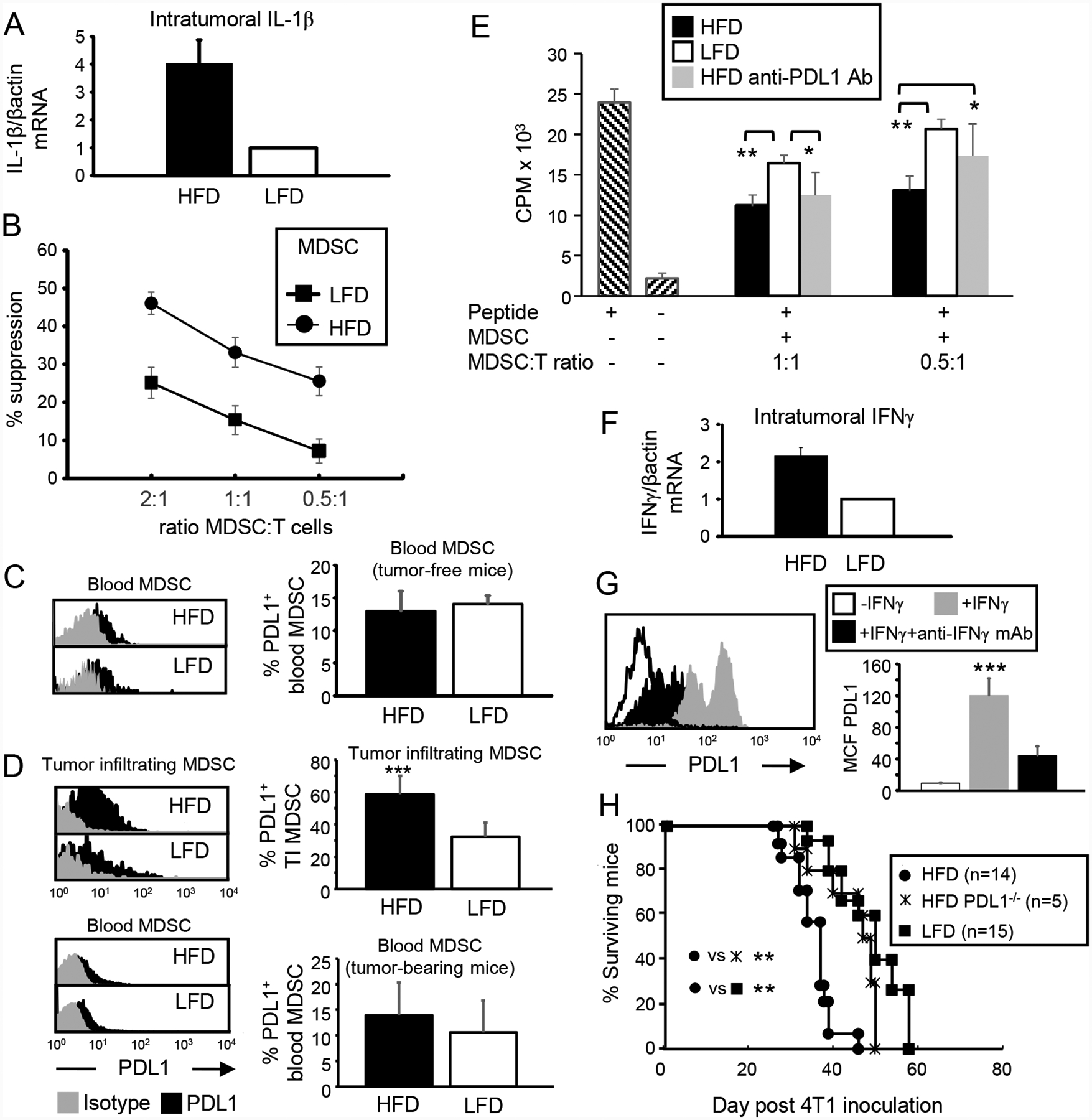
(A) Tumor microenvironment in HFD mice has elevated levels of IL-1β. RNA from 4T1 tumors was assayed by qRT-PCR for IL-1β. Data are the pooled average ± SEM of 3 independent experiments with a total of 9 mice/group. (B) Tumor-infiltrating HFD-induced MDSC have enhanced suppressive activity. Splenocytes from Clone 4 TCR transgenic mice were incubated with cognate peptide in the presence of varying amounts of 4T1-induced HFD or LFD MDSC. Data are pooled from 3 independent experiments with 2 mice/group/experiment. (C) Blood MDSC from non-tumor-bearing mice express similar levels of PDL1. Mice were bled and their leukocytes labeled for Gr1, CD11b, and PDL1. Gated Gr1+CD11b+ cells were analyzed for PDL1. n=4 mice/group. (D) Tumor-infiltrating HFD-induced MDSC express more PDL1. Tumor-bearing mice were sacrificed on days 30–40 after 4T1 inoculation and their blood cells and tumor-infiltrating MDSC analyzed for PDL1. Blood: HFD n= 9, LFD n=8; TIMDSC: HFD n=8, LFD n=3. (E) Antibody blocking of PDL1 reduces the suppressive potency of HFD-induced MDSC. Splenocytes from Clone 4 transgenic mice were incubated with cognate peptide ± varying amounts of 4T1-induced MDSC ± antibody to PDL1. n=3,5,3 mice/group for HFD, HFD PDL1 antibody-treated, and LFD MDSC, respectively. (F) IFNγ is elevated in the tumor microenvironment of HFD mice. RNA from 4T1 tumors was assayed by qRT-PCR for IFNγ. Data are the pooled average ± SEM of 4 independent experiments with a total of 9 mice/group. (G) MDSC expression of PDL1 is up-regulated by IFNγ. Blood MDSC were cultured for 24 hrs ± 100 units/ml IFNγ ± antibody to IFNγ, then stained for Gr1, CD11b, and PDL1. Gated Gr1+CD11b+ cells were analyzed for PDL1. Representative histogram; data are pooled from 3 mice and 3 independent experiments. (H) BALB/c HFD, BALB/c LFD, and BALB/c PDL1−/− mice were inoculated with 4T1 cells and followed for survival time.
The tumor microenvironment induces MDSC expression of PDL1 and thereby enhances tumor progression
MDSC in BALB/c HFD mice are >95% PMN-MDSC (see Supplemental Fig. 1). PMN-MDSC mediate their suppressive effects by their content and release of arginase and reactive oxygen species (ROS). Therefore, the increased suppressive potency of tumor-infiltrating HFD-induced MDSC could be due to higher levels of arginase and/or ROS. However, MDSC from HFD and LFD mice contain the same levels of arginase and ROS as assessed by flow cytometry (Supplemental Fig. 4). Therefore, higher levels of ROS or arginase are not responsible for the increased suppressive activity of tumor-infiltrating HFD-induced MDSC. Since some MDSC express PDL1, we speculated that HFD might induce or increase PDL1 expression on MDSC and thereby increase MDSC suppressive activity. This possibility was tested by PDL1 staining of blood MDSC from tumor-free HFD and LFD mice (Fig. 5C) and blood and tumor-infiltrating MDSC from HFD and LFD mice carrying 4T1 tumors (Fig. 5D). Few blood MDSC from tumor-free HFD and LFD BALB/c mice expressed PDL1 (12.95% ± 3.08 and 14.06% ± 1.3, respectively), and the percent of PDL1+ MDSC did not differ between HFD and LFD mice. In contrast, 58.8% ± 11.4 of tumor-infiltrating MDSC in HFD mice were PDL1+ as compared to 32% ± 8.9 of tumor-infiltrating MDSC in LFD mice (p<0.001), suggesting that tumor-infiltrating MDSC expression of PDL1 in HFD mice might increase MDSC-mediated immune suppression.
If MDSC expression of PDL1 enhances HFD MDSC suppressive activity, then blocking PDL1 on HFD MDSC should reduce suppressive potency. This possibility was tested by comparing the ability of LFD and HFD-induced MDSC to suppress T cell activation in the presence or absence of antibodies that block PDL1 (Fig. 5E). As previously observed (Fig. 5B), tumor-induced HFD MDSC were significantly more suppressive than tumor-induced LFD MDSC. At a 0.5:1 ratio of MDSC to antigen-specific T cells, PDL1-blocked HFD MDSC were equally suppressive to LFD MDSC and significantly less suppressive than HFD MDSC.
IFNγ is an established inducer of PDL1 on immune and some non-immune cells, raising the possibility that increased PDL1 levels of TIMDSC in HFD mice are due to increases in IFNγ in the tumor microenvironment of HFD vs. LFD mice. To examine this possibility, IFNγ expression in 4T1 tumors of HFD and LFD mice was assessed by qRT-PCR (Fig 5F). Tumors from HFD BALB/c mice contained approximately twice as much IFNγ mRNA as tumors from LFD mice. To determine if IFNγ increases PDL1 expression on MDSC, blood MDSC, which express very low levels of PDL1 (Fig. 5C), were incubated with or without IFNγ. Incubation in low levels of IFNγ increased MDSC expression of PDL1, and inclusion of antibodies to IFNγ prevented the increase. (Fig. 5G), demonstrating that IFNγ enhances MDSC expression of PDL1.
To determine if PDL1 affects tumor progression, HFD, LFD and HFD PDL1−/− mice were inoculated with 4T1 cells and followed for survival time (Fig. 5H). LFD and HFD PDL1−/− mice had similar survival times, and survived significantly longer than HFD mice.
These results indicate that HFD increases MDSC expression of PDL1 and that PDL1 expression contributes to more rapid tumor growth.
Leptin-MDSC crosstalk regulates MDSC accumulation and leptin levels
Leptin is a pleiotropic molecule that regulates multiple metabolic, endocrine, and immunological functions, as well as impacting wound healing, angiogenesis, and hematopoiesis. It regulates the balance between food consumption and metabolism and is responsible for decreasing appetite. It is induced by pro-inflammatory mediators and is typically over-expressed in obese individuals who become non-responsive to its effects [38]. To determine if MDSC and leptin impact each other, female BALB/c mice on a HFD or LFD were antibody depleted for MDSC or treated with an isotype control antibody and their levels of blood leptin measured by ELISA (Fig. 6A). As expected, blood levels of leptin were elevated in HFD mice as compared to LFD mice. MDSC-depleted HFD mice had significantly increased blood leptin levels, while the relatively low levels of leptin in LFD mice were not significantly affected by MDSC depletion. These results indicate that MDSC in HFD mice decrease leptin.
Figure 6. Leptin drives the expansion of MDSC.
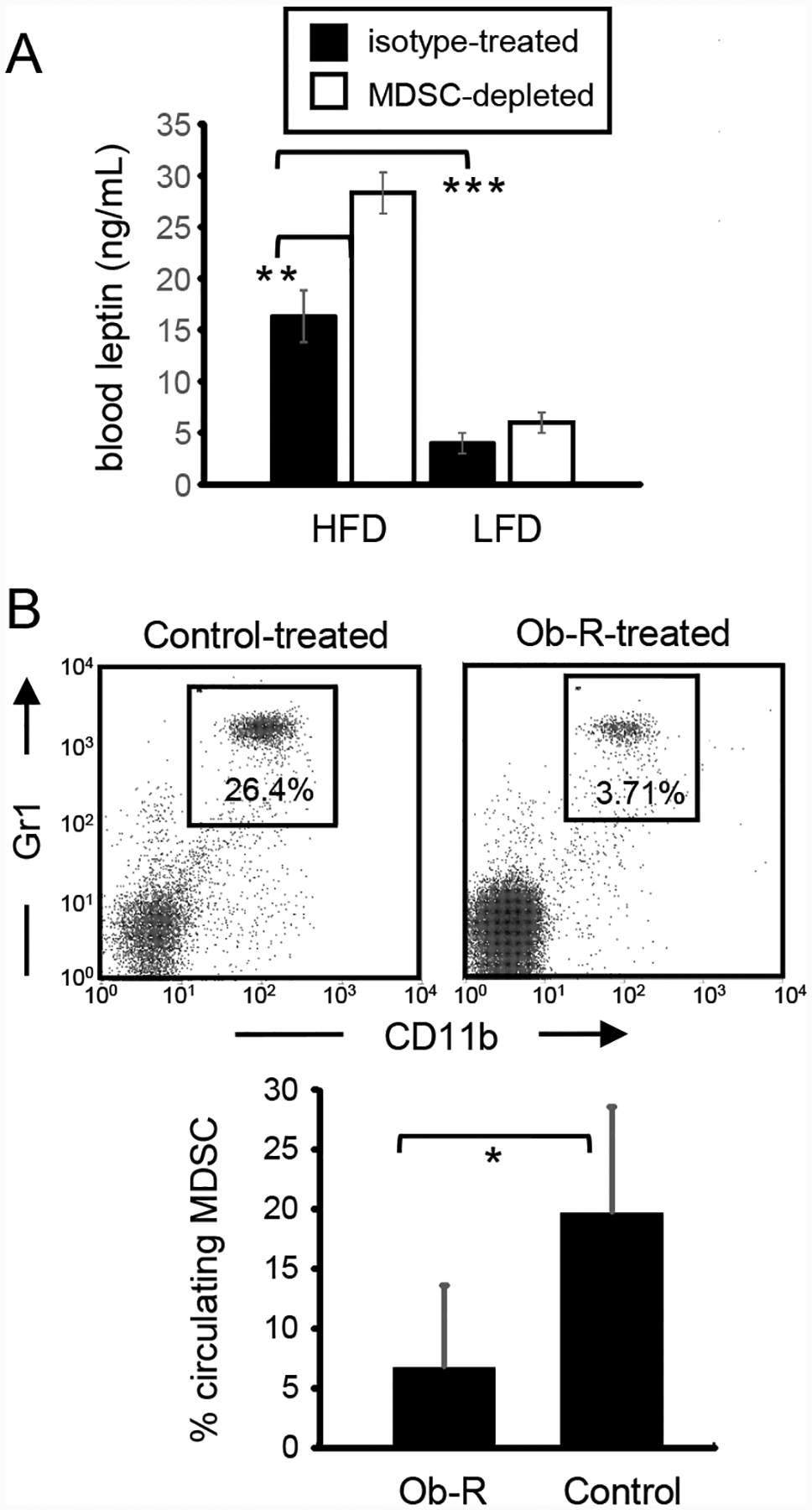
(A) HFD mice have elevated levels of leptin. Leptin levels in the plasma of HFD, LFD, and HFD-MDSC-depleted mice were measured by ELISA (n=3 mice/group). (B) In vivo blocking of the leptin receptor in HFD mice decreases the level of circulating MDSC. Female BALB/c mice on a HFD were administered soluble leptin receptor (Ob-R-Fc) or an irrelevant soluble recombinant protein or mouse IgG1 (control). MDSC levels in the blood were determined three days later. Data are the average of 3 Ob-R-treated and 14 control-treated mice pooled from two independent experiments.
In addition to being induced by inflammation, leptin also contributes to inflammation by increasing T cell production of pro-inflammatory cytokines [39, 40]. Since MDSC are driven by inflammation, we hypothesized that leptin may facilitate the expansion of MDSC in HFD mice. If leptin drives the accumulation of MDSC, then blocking the leptin receptor (Ob-R) should reduce the level of MDSC. To test this possibility, female BALB/c mice on a HFD were administered a soluble form of the Ob-R (Ob-R-Fc) or an irrelevant recombinant protein or mouse IgG daily for three consecutive days. Seventy-two hours after the last dose, the level of circulating MDSC in the blood was determined by flow cytometry (Fig. 6B). Mice receiving the soluble receptor had significantly reduced numbers of circulating MDSC relative to the control group demonstrating that MDSC accumulation in HFD mice is positively regulated by leptin.
Collectively, these results demonstrate that leptin and MDSC engage in cross-talk in which leptin facilitates the accumulation of MDSC, while MDSC down-regulate the level of leptin.
DISCUSSION
Obesity is an established risk factor for developing cancer and is an indicator for poor prognosis for individuals with established disease [1, 4]. Obesity drives multiple metabolic and endocrine abnormalities that contribute to cancer risk and cancer progression. In addition, obesity perturbs the immune system and polarizes macrophages and T cells towards a type 2 phenotype that favors tumor growth [28]. We now report another immune mechanism by which nutritional overload facilitates tumor progression. Our studies demonstrate that a high fat diet and the accompanying weight gain induce the accumulation of excess numbers of MDSC in mice. The resulting MDSC promote more rapid growth of primary tumor and facilitate spontaneous metastasis by preventing the activation of tumor-reactive T cells. The induction of MDSC is regulated by leptin, a molecule that is over-expressed in HFD mice and the accumulation of MDSC can be reduced by blocking the leptin receptor. Although HFD-induced MDSC are detrimental with respect to tumor progression, they protect against HFD-induced elevated glucose levels and insulin tolerance, reduce inflammation in adipose tissue, and down-regulate leptin levels. Therefore, MDSC play a schizophrenic role in nutritional overload in that they are beneficial with respect to the metabolic dysfunction that accompanies HFD, but they are detrimental with respect to cancer progression. Fig. 7 summarizes MDSC-leptin crosstalk and its effects on metabolic dysfunction, adiposity, and tumor progression.
Figure 7. MDSC-leptin crosstalk enhances immune suppression and promotes tumor progression, while increasing adiposity but limiting metabolic dysfunction in mice on a high fat diet.
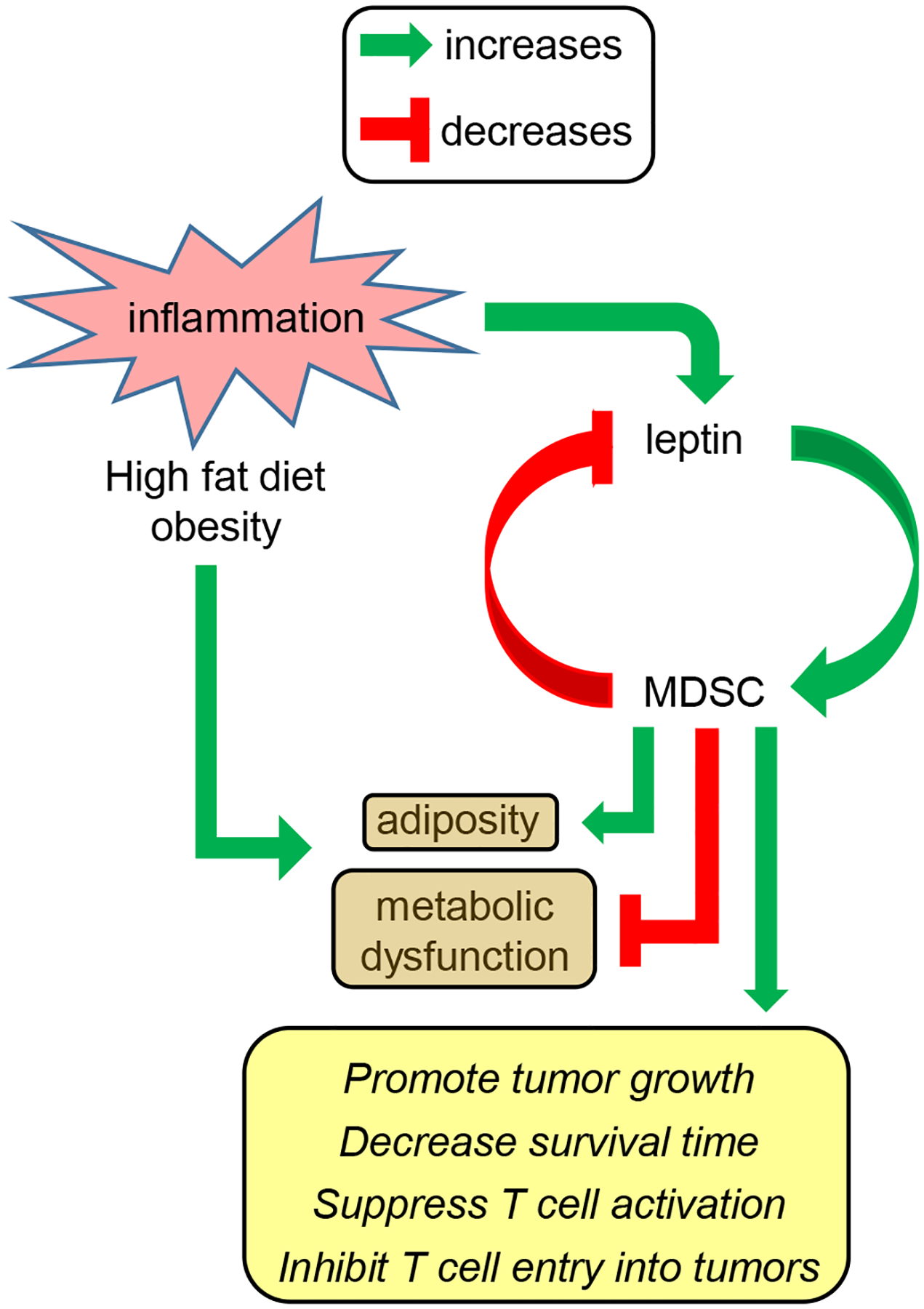
The chronic low grade inflammation that is induced by and accompanies obesity results in the upregulation of the adipokine leptin, the accumulation of adipose tissue, and metabolic dysfunction. Leptin and MDSC undergo cross-talk in which leptin induces the accumulation of MDSC while MDSC down-regulate the production of leptin. The elevated levels of MDSC that exist in mice on a high fat diet promote adiposity and enhance tumor progression while diminishing some of the metabolic dysfunction that accompanies high fat diet and obesity.
MDSC have traditionally been considered as detrimental cells due to their widespread presence in cancer patients where they inhibit anti-tumor immunity. However, in settings such as autoimmunity, MDSC may be beneficial by down-regulating immune responses against self [26]. Recent studies have also demonstrated that MDSC accumulate in pregnant women [41] and are essential for successful pregnancy in mice because they inhibit allo responses against genetically disparate fetuses [42]. These beneficial functions of MDSC are the result of their immune suppressive activity and ability to counteract non-desirable immunity. The studies described here identify additional beneficial effects of MDSC. However, it is unclear whether the protection conferred by MDSC against metabolic dysfunction involves the immune suppressive function of MDSC.
Effects of specific types of fat on various disease states have been investigated. In particular, the polyunsaturated fatty acid (PUFA) α-linolenic acid (omega-3 fatty acid) is associated with a reduction in cardiovascular disease [43] and protection against some inflammatory and autoimmune diseases [44]. A recent study demonstrated that these PUFAs increase the accumulation and suppressive potency of MDSC [45], consistent with the concept that these PUFAs reduce autoimmunity by promoting MDSC-mediated immune suppression. In contrast to diets high in PUFAs, the fat in the HFD used in the current studies consists of 26.5% polyunsaturated fat and 73.5% saturated and monosaturated fats. Therefore, PUFA, saturated, and monosaturated fats all appear to drive the accumulation of MDSC.
The studies reported here identify a new mechanism by which leptin, a product of adipose tissue which is over-expressed in obesity, impacts the immune system and thereby modulates both metabolic dysfunction and tumor growth. Leptin has previously been shown to drive multiple immune functions, such as the proliferation of circulating naïve T cells [39], inhibition of proliferation of T memory cells [40], antagonism of T regulatory cells [46], impairment of NK cytotoxicity [47], maturation of DC and DC function [48], inhibition of neutrophil chemotaxis and infiltration [49, 50], and induction of mast cells producing inflammatory mediators IL-1β, TNFα, and IL-6 which in turn polarize macrophages towards an M1 phenotype [51]. Enhancement of MDSC accumulation and increasing MDSC suppressive potency are now added to the list of functions regulated by leptin.
It has been reported that MDSC levels increase as mice age [33, 52, 53]. Limited data in older cancer patients suggest that MDSC levels also increase in humans as they age [54]. However, in many cases, body mass also increases with ageing, raising the possibility that the observed increases in MDSC with ageing are at least partially due to increased body mass. The studies presented here support this possibility since HFD-induced weight increase directly correlated with MDSC accumulation. Since levels of MDSC in LFD mice were inversely correlated with weight, our studies further suggest that fat intake may also be a variable regulating MDSC levels in older individuals. Studies tracking weight gain and diet along with age are needed to parse out which parameters regulate MDSC accumulation during ageing.
The beneficial effects of MDSC identified here provide an evolutionary rationale for why these cells exist. Given the varied cell populations that tumors have co-opted to promote their progression, it is not surprising that tumors have also co-opted MDSC. Whether the expansion of MDSC that accompanies HFD diet and nutritional overload increases the risk of cancer remains unknown. However, the studies presented here demonstrate that once malignant cells are present, HFD-induced MDSC contribute to the more rapid progression of tumors.
Supplementary Material
Supplemental Fig. 1. HFD and LFD do not significantly affect the ration of PMN-MDSC to M-MDSC.
Supplemental Fig. 2. The lungs and liver of HFD and LFD tumor-bearing mice contain the same percentages of MDSC.
Supplemental Fig. 3. Tumor progression in HFD mice is regulated by CD4+ and CD8+ T cells.
Supplemental Fig. 4. ROS and arginase content are similar for HFD and LFD MDSC.
Acknowledgements
We thank Dr. D. Pardoll (Johns Hopkins) for providing breeding pairs of the BALB/c PDL1−/− mice and Dr. T. Greten for providing the procedure for analyzing liver-infiltrating MDSC. These studies were partially supported by NIH RO1GM021248, RO1CA84232, and RO1CA115880.
ABBREVIATIONS
- AUC
area under the curve
- HFD
high fat diet
- IFNγ
interferon gamma
- LFD
low fat diet
- MDSC
myeloid-derived suppressor cell(s)
- Ob-R
obesity/leptin receptor
- PDL1
programmed death ligand 1
- PUFA
polyunsaturated fatty acid
- STAT3
signal transducer activator of transcription 3
- TcR
T cell receptor for antigen
- TIL
tumor-infiltrating lymphocytes
- TIMDSC
tumor-infiltrating MDSC
- TNFα
tumor necrosis factor alpha
Footnotes
Conflict of Interest Disclosure
The authors declare no conflicts of interest
References
- 1.Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ (2003) Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N. Engl. J. Med 348, 1625–38. [DOI] [PubMed] [Google Scholar]
- 2.Renehan AG, Tyson M, Egger M, Heller RF, Zwahlen M (2008) Body-mass index and incidence of cancer: a systematic review and meta-analysis of prospective observational studies. Lancet 371, 569–78. [DOI] [PubMed] [Google Scholar]
- 3.Font-Burgada J, Sun B, Karin M (2016) Obesity and Cancer: The Oil that Feeds the Flame. Cell. Metab 23, 48–62. [DOI] [PubMed] [Google Scholar]
- 4.van Kruijsdijk RC, van der Wall E, Visseren FL (2009) Obesity and cancer: the role of dysfunctional adipose tissue. Cancer Epidemiol. Biomarkers Prev 18, 2569–78. [DOI] [PubMed] [Google Scholar]
- 5.Feola A, Ricci S, Kouidhi S, Rizzo A, Penon A, Formisano P, Giordano A, Di Carlo A, Di Domenico M (2017) Multifaceted Breast Cancer: The Molecular Connection With Obesity. J. Cell. Physiol 232, 69–77. [DOI] [PubMed] [Google Scholar]
- 6.Matthews SB and Thompson HJ (2016) The Obesity-Breast Cancer Conundrum: An Analysis of the Issues. Int. J. Mol. Sci 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garcia-Jimenez C, Gutierrez-Salmeron M, Chocarro-Calvo A, Garcia-Martinez JM, Castano A, De la Vieja A (2016) From obesity to diabetes and cancer: epidemiological links and role of therapies. Br. J. Cancer 114, 716–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lorincz AM and Sukumar S (2006) Molecular links between obesity and breast cancer. Endocr Relat Cancer 13, 279–92. [DOI] [PubMed] [Google Scholar]
- 9.Berger NA (2014) Obesity and cancer pathogenesis. Ann. N. Y. Acad. Sci 1311, 57–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rogers CJ, Prabhu KS, Vijay-Kumar M (2014) The microbiome and obesity-an established risk for certain types of cancer. Cancer J 20, 176–80. [DOI] [PubMed] [Google Scholar]
- 11.Balkwill F and Mantovani A (2001) Inflammation and cancer: back to Virchow? Lancet 357, 539–45. [DOI] [PubMed] [Google Scholar]
- 12.Coussens LM and Werb Z (2002) Inflammation and cancer. Nature 420, 860–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mantovani A, Allavena P, Sica A, Balkwill F (2008) Cancer-related inflammation. Nature 454, 436–44. [DOI] [PubMed] [Google Scholar]
- 14.Grivennikov SI, Greten FR, Karin M (2010) Immunity, inflammation, and cancer. Cell 140, 883–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karin M and Lin A (2002) NF-kappaB at the crossroads of life and death. Nat. Immunol 3, 221–7. [DOI] [PubMed] [Google Scholar]
- 16.Kim S, Takahashi H, Lin WW, Descargues P, Grivennikov S, Kim Y, Luo JL, Karin M (2009) Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature 457, 102–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taniguchi K and Karin M (2014) IL-6 and related cytokines as the critical lynchpins between inflammation and cancer. Semin Immunol. 26, 54–74. [DOI] [PubMed] [Google Scholar]
- 18.Ostrand-Rosenberg S and Sinha P (2009) Myeloid-derived suppressor cells: linking inflammation and cancer. J. Immunol 182, 4499–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bunt SK, Sinha P, Clements VK, Leips J, Ostrand-Rosenberg S (2006) Inflammation induces myeloid-derived suppressor cells that facilitate tumor progression. J. Immunol 176, 284–90. [DOI] [PubMed] [Google Scholar]
- 20.Bunt SK, Yang L, Sinha P, Clements VK, Leips J, Ostrand-Rosenberg S (2007) Reduced inflammation in the tumor microenvironment delays the accumulation of myeloid-derived suppressor cells and limits tumor progression. Cancer Res. 67, 10019–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sade-Feldman M, Kanterman J, Ish-Shalom E, Elnekave M, Horwitz E, Baniyash M (2013) Tumor necrosis factor-alpha blocks differentiation and enhances suppressive activity of immature myeloid cells during chronic inflammation. Immunity 38, 541–54. [DOI] [PubMed] [Google Scholar]
- 22.Zhao X, Rong L, Li X, Liu X, Deng J, Wu H, Xu X, Erben U, Wu P, Syrbe U, Sieper J, Qin Z (2012) TNF signaling drives myeloid-derived suppressor cell accumulation. J. Clin. Invest 122, 4094–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ostrand-Rosenberg S, Sinha P, Beury DW, Clements VK (2012) Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression. Semin. Cancer Biol 22, 275–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V (2012) Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol 12, 253–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ostrand-Rosenberg S (2010) Myeloid-derived suppressor cells: more mechanisms for inhibiting antitumor immunity. Cancer Immunol. Immunother 59, 1593–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Parker KH, Beury DW, Ostrand-Rosenberg S (2015) Myeloid-Derived Suppressor Cells: Critical Cells Driving Immune Suppression in the Tumor Microenvironment. Adv. Cancer Res 128, 95–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xia S, Sha H, Yang L, Ji Y, Ostrand-Rosenberg S, Qi L (2011) Gr-1+ CD11b+ myeloid-derived suppressor cells suppress inflammation and promote insulin sensitivity in obesity. J. Biol. Chem 286, 23591–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deng T, Lyon CJ, Bergin S, Caligiuri MA, Hsueh WA (2016) Obesity, Inflammation, and Cancer. Annu. Rev. Pathol 11, 421–49. [DOI] [PubMed] [Google Scholar]
- 29.Sinha P, Clements VK, Ostrand-Rosenberg S (2005) Reduction of myeloid-derived suppressor cells and induction of M1 macrophages facilitate the rejection of established metastatic disease. J. Immunol 174, 636–45. [DOI] [PubMed] [Google Scholar]
- 30.Pulaski BA and Ostrand-Rosenberg S (1998) Reduction of established spontaneous mammary carcinoma metastases following immunotherapy with major histocompatibility complex class II and B7.1 cell-based tumor vaccines. Cancer Res. 58, 1486–93. [PubMed] [Google Scholar]
- 31.Beury DW, Carter KA, Nelson C, Sinha P, Hanson E, Nyandjo M, Fitzgerald PJ, Majeed A, Wali N, Ostrand-Rosenberg S (2016) Myeloid-Derived Suppressor Cell Survival and Function Are Regulated by the Transcription Factor Nrf2. J. Immunol 196, 3470–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Srivastava MK, Sinha P, Clements VK, Rodriguez P, Ostrand-Rosenberg S (2010) Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res. 70, 68–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hanson EM, Clements VK, Sinha P, Ilkovitch D, Ostrand-Rosenberg S (2009) Myeloid-derived suppressor cells down-regulate L-selectin expression on CD4+ and CD8+ T cells. J. Immunol 183, 937–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ma C, Kapanadze T, Gamrekelashvili J, Manns MP, Korangy F, Greten TF (2012) Anti-Gr-1 antibody depletion fails to eliminate hepatic myeloid-derived suppressor cells in tumor-bearing mice. J Leukoc Biol 92, 1199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sinha P, Chornoguz O, Clements VK, Artemenko KA, Zubarev RA, Ostrand-Rosenberg S (2011) Myeloid-derived suppressor cells express the death receptor Fas and apoptose in response to T cell-expressed FasL. Blood 117, 5381–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Iyengar NM, Hudis CA, Dannenberg AJ (2015) Obesity and cancer: local and systemic mechanisms. Annu. Rev. Med 66, 297–309. [DOI] [PubMed] [Google Scholar]
- 37.Ostrand-Rosenberg S, Sinha P, Chornoguz O, Ecker C (2012) Regulating the suppressors: apoptosis and inflammation govern the survival of tumor-induced myeloid-derived suppressor cells (MDSC). Cancer Immunol. Immunother 61, 1319–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Perez-Perez A, Vilarino-Garcia T, Fernandez-Riejos P, Martin-Gonzalez J, Segura-Egea JJ, Sanchez-Margalet V (2017) Role of leptin as a link between metabolism and the immune system. Cytokine Growth Factor Rev. [DOI] [PubMed] [Google Scholar]
- 39.Lord GM, Matarese G, Howard JK, Baker RJ, Bloom SR, Lechler RI (1998) Leptin modulates the T-cell immune response and reverses starvation-induced immunosuppression. Nature 394, 897–901. [DOI] [PubMed] [Google Scholar]
- 40.Lord GM, Matarese G, Howard JK, Bloom SR, Lechler RI (2002) Leptin inhibits the anti-CD3-driven proliferation of peripheral blood T cells but enhances the production of proinflammatory cytokines. J. Leukoc. Biol 72, 330–8. [PubMed] [Google Scholar]
- 41.Kostlin N, Hofstadter K, Ostermeir AL, Spring B, Leiber A, Haen S, Abele H, Bauer P, Pollheimer J, Hartl D, Poets CF, Gille C (2016) Granulocytic Myeloid-Derived Suppressor Cells Accumulate in Human Placenta and Polarize toward a Th2 Phenotype. J. Immunol 196, 1132–45. [DOI] [PubMed] [Google Scholar]
- 42.Ostrand-Rosenberg S, Sinha P, Figley C, Long R, Park D, Carter D, Clements VK (2017) Frontline Science: Myeloid-derived suppressor cells (MDSCs) facilitate maternal-fetal tolerance in mice. J. Leukoc. Biol 101, 1091–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ander BP, Dupasquier CM, Prociuk MA, Pierce GN (2003) Polyunsaturated fatty acids and their effects on cardiovascular disease. Exp. Clin. Cardiol 8, 164–72. [PMC free article] [PubMed] [Google Scholar]
- 44.Harbige LS (1998) Dietary n-6 and n-3 fatty acids in immunity and autoimmune disease. Proc. Nutr. Soc 57, 555–62. [DOI] [PubMed] [Google Scholar]
- 45.Yan D, Yang Q, Shi M, Zhong L, Wu C, Meng T, Yin H, Zhou J (2013) Polyunsaturated fatty acids promote the expansion of myeloid-derived suppressor cells by activating the JAK/STAT3 pathway. Eur. J. Immunol 43, 2943–55. [DOI] [PubMed] [Google Scholar]
- 46.Wagner NM, Brandhorst G, Czepluch F, Lankeit M, Eberle C, Herzberg S, Faustin V, Riggert J, Oellerich M, Hasenfuss G, Konstantinides S, Schafer K (2013) Circulating regulatory T cells are reduced in obesity and may identify subjects at increased metabolic and cardiovascular risk. Obesity 21, 461–8. [DOI] [PubMed] [Google Scholar]
- 47.Wrann CD, Laue T, Hubner L, Kuhlmann S, Jacobs R, Goudeva L, Nave H (2012) Short-term and long-term leptin exposure differentially affect human natural killer cell immune functions. Am J Physiol Endocrinol Metab 302, E108–16. [DOI] [PubMed] [Google Scholar]
- 48.Moraes-Vieira PM, Larocca RA, Bassi EJ, Peron JP, Andrade-Oliveira V, Wasinski F, Araujo R, Thornley T, Quintana FJ, Basso AS, Strom TB, Camara NO (2014) Leptin deficiency impairs maturation of dendritic cells and enhances induction of regulatory T and Th17 cells. Eur. J. Immunol 44, 794–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kordonowy LL, Burg E, Lenox CC, Gauthier LM, Petty JM, Antkowiak M, Palvinskaya T, Ubags N, Rincon M, Dixon AE, Vernooy JH, Fessler MB, Poynter ME, Suratt BT (2012) Obesity is associated with neutrophil dysfunction and attenuation of murine acute lung injury. Am J Respir Cell Mol Biol 47, 120–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Naylor C, Burgess S, Madan R, Buonomo E, Razzaq K, Ralston K, Petri WA Jr. (2014) Leptin receptor mutation results in defective neutrophil recruitment to the colon during Entamoeba histolytica infection. mBio 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhou Y, Yu X, Chen H, Sjoberg S, Roux J, Zhang L, Ivoulsou AH, Bensaid F, Liu CL, Liu J, Tordjman J, Clement K, Lee CH, Hotamisligil GS, Libby P, Shi GP (2015) Leptin Deficiency Shifts Mast Cells toward Anti-Inflammatory Actions and Protects Mice from Obesity and Diabetes by Polarizing M2 Macrophages. Cell Metab 22, 1045–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Enioutina EY, Bareyan D, Daynes RA (2011) A role for immature myeloid cells in immune senescence. J. Immunol 186, 697–707. [DOI] [PubMed] [Google Scholar]
- 53.Grizzle WE, Xu X, Zhang S, Stockard CR, Liu C, Yu S, Wang J, Mountz JD, Zhang HG (2007) Age-related increase of tumor susceptibility is associated with myeloid-derived suppressor cell mediated suppression of T cell cytotoxicity in recombinant inbred BXD12 mice. Mech. Ageing Dev 128, 672–80. [DOI] [PubMed] [Google Scholar]
- 54.Verschoor CP, Johnstone J, Millar J, Dorrington MG, Habibagahi M, Lelic A, Loeb M, Bramson JL, Bowdish DM (2013) Blood CD33(+)HLA-DR(−) myeloid-derived suppressor cells are increased with age and a history of cancer. J. Leukoc. Biol 93, 633–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Fig. 1. HFD and LFD do not significantly affect the ration of PMN-MDSC to M-MDSC.
Supplemental Fig. 2. The lungs and liver of HFD and LFD tumor-bearing mice contain the same percentages of MDSC.
Supplemental Fig. 3. Tumor progression in HFD mice is regulated by CD4+ and CD8+ T cells.
Supplemental Fig. 4. ROS and arginase content are similar for HFD and LFD MDSC.