

Abstract
Cancer treatment has been founded traditionally on the three approaches of surgery, radiation and chemotherapy with the latter recognized as the obvious systemic treatment approach applicable to disease that has spread. While significant progress has been made over nearly 100 years of developing systemic treatments, it remains clear that use of the toxic agents involved is a two-edged sword with normal organ toxicities always needing to be balanced with and against administration of relevant therapeutic doses. With the advent of monoclonal antibodies targeted against tumor associated antigens that could be used as carriers of potently toxic chemotherapy drugs, it was thought that such antibody-drug conjugates (ADCs) could engender the answer to the toxicity/therapeutic equation by shifting the equation more towards beneficial therapeutic efficacy. However, over 40 or so years, antibody-drug conjugates have not significantly affected the toxicity/therapy balance paradigm in most cancer indications, especially in solid tumors. Ideally, a further step may be required in that non-tumor targeted antibody-drug conjugate should be essentially non-toxic in its native administered form, with toxic effects unleashed only at the site of targeted tumors. A new approach that employs this principle is the use of an antibody-drug conjugate that is essentially non-toxic to normal tissues by virtue of requiring an extra step of light activation to become potent. We describe the preclinical data and first clinical results gained over the past few years by use of antibody-drug conjugates wherein the drug comprises a near-infrared photoactivatable dye delivered to tumors by a monoclonal antibody and subsequently activated to a toxic entity solely at sites of tumors.
Keywords: antibody-drug conjugate, near infrared photoimmunotherapy, MAb-IR700, cetuximab, cancer
Table of Contents Graphic
Introduction
Since the first discovery of monoclonal antibodies (MAbs) in 19751 scientists have recognized their potential to fulfill the promise of Ehrlich’s magic bullet2 by becoming the vehicle for the specific delivery of drugs to sites of disease. While simple enough in concept, reduction-to-practice has been beset with multiple obstacles, including consideration of the MAb alone as a potential therapeutic agent. Original animal-derived MAbs, most notably from rodent sources, were found to be immunogenic when used in humans with functioning immune systems and recombinant protein technology to produce chimeric, and later humanized, MAb proteins had to be discovered3 and developed over many years4 before therapeutic application of even unsubstituted or ‘naked’ MAbs could reach clinical fruition. Most notably, clinical successes were achieved with alemtuzumab (anti-CD52) and rituximab (anti-CD20) against B-cell chronic lymphocytic leukemia (B-CLL) and non-Hodgkin’s lymphoma, respectively. Later, limited expansion to use of naked MAbs against solid tumors in clinical settings was achieved with approvals for trastuzumab (1998; breast cancer), cetuximab (2004; head and neck and colorectal cancers) and panitumumab (2006; metastatic colorectal cancer). Although many non-cancer-directed naked MAb clinical applications have been described, MAbs directed against tumor-associated antigens have been somewhat limited in their success rate and current research efforts are more focused on naked MAbs as immune checkpoint inhibitors.5
In addition to the multiple issues with MAb protein development it was realized very early on that naked MAbs against tumor-associated antigens did not offer a significant therapeutic effect even against established preclinical animal xenograft models of human cancer. It was probable that most MAbs would need to be coupled with a toxic entity such as a biological toxin, chemotherapy drug or a penetrating particle-emitting radionuclide to see a marked anti-tumor effect, thereby using the MAb as a delivery vector for the therapeutic agent. Whereas biological toxins could be extremely potent they usually come with unacceptable normal tissue toxicities and immunogenicity, still not fully solved to this day. Radionuclides have the significant theoretical advantage that they would not need to bind all tumor cells in a growth or be cellularly internalized to exert toxic effects against targeted tumors, but they engender significant and somewhat unavoidable off-target radiolytic effects against normal tissues. Chemotherapy drugs, however, could be readily obtained through manufacture and coupled to MAbs by known chemistries to produce well-defined MAb-drug conjugates for targeting specific cancer cell epitopes.
A Brief Overview of Therapeutics with Monoclonal Antibody Drug Conjugates
MAb immunoglobulin G (IgG)s with molecular weights in the 150,000 Dalton region have multiple lysyl- and carboxyl- groups that one might consider for conjugation of drug haptens, as well as oxidizable carbohydrate limited to their Fc constant regions and intra- and inter-disulfide cystine bonds capable of mild reduction to generate reactive cysteinyl- thiol groups. As such, chemists have multiple choices as to where to append drug moieties. While a full review is outside of the scope of this review, suffice it to say that drug conjugation sites can be scattered over the MAb molecule or more specifically targeted away from the all-important MAb-antigen binding site, and all possible substitution sites have now been investigated. In practice, both site-specific and non-site-specific conjugation approaches have shown some level of success, although early work often did not account for the further the need for the drug to become freed from the MAb protein, once targeted, in order to exert its toxic effect, and the thus the nature of the chemical linkage between MAb and drug also had to be addressed.6,7 Once understood, and despite reasonable-to-good preclinical therapy results in mouse tumor xenograft models of human cancers, clinical results proved much more disappointing. This was primarily due to the much lower amounts of MAb-drug (typically 0.001 to 0.1 percent of the injected dose per gram of tumor) seen to be targeted to human tumors in a clinical setting and off-target toxic effects due to high levels of MAb-drug conjugates in normal tissues. To address the low drug localization issue, much more potent drugs than standard chemotherapy drugs were thought to be necessary for clinical MAb-drug conjugates with the prototypical conjugate gemtuzumab ozogamicin, comprising a humanized anti-CD33 MAb conjugate and a super toxic calicheamicin analog, approved for acute myeloid leukemia as early as year 2000 (Figure 1).8 In a further successful development the toxic maytansinoid analog emtansine coupled to trastuzumab was approved for second-line breast cancer therapy in 20139 and work on newer calicheamicin analogs continues.10
Figure 1.

A calicheamicin-MAb conjugate showing the complexity of the ene-diyne system trigger, with the complex carbohydrate below the fused rings. The reducible disulfide acts as the trigger mechanism and is linked to the MAb via the latter’s lysine residues, a short-chain linker and a hydrazone moiety (which is also cleavable under mildly acidic conditions).
In parallel with these successful developments, research on newer generations of MAb-drug conjugates have focused on more defined conjugation techniques incorporating highly toxic drug analogs, MAb-site-specific drug conjugation sites, fully defined linker chemistries designed to release drugs in defined manners and the use of ever-more advanced molecular biology techniques for MAb recombination and redesign. A first example of the new sophistication is brentuximab vedotin targeting CD30 on Hodgkin’s lymphoma and comprised of a chimeric MAb linked to the microtubule disrupting agent monomethyl-auristatin E (MMAE) via a protease-cleavable linker, which is cleaved upon cellular internalization (Figure 2).11 Various iterations and improvements have been made and continue to be made, including addition of solubilizing linkers/addends containing poly(ethylene)glycol chains of various lengths, and extension of MAb-auristatin analogs (Aur0101) to solid tumor model application.12
Figure 2.

Structure of a brentuximab vedotin MAb-drug conjugate. The drug is coupled to the MAb via a reduced MAb cysteinyl-thiol group appended via a maleimide linkage on a six-carbon chain. At the other extreme is the self-immolative linker, which is protease cleaved after internalization of the conjugate, leading to breakage of the ester bond and release of the potent vedotin MMAE free drug analog (bottom).
Solubilizing agents for conjugates have often been found necessary as the more toxic drugs are most often highly hydrophobic and can often cause MAb aggregation and precipitation problems, an issue that had to be addressed even with the ‘moderately toxic’ chemotherapy-approved SN-38 irinotecan metabolite (Figure 3).13
Figure 3.

Active irinotecan derivative SN-38 coupled to a MAb with a cleavable linker at the heterocycle’s 20 ring oxygen position designed to produce the free active drug upon bond cleavage. The remaining linker consists of a solubilizing 7-PEG unit to counter the free drugs inherent hydrophobicity leading to a maleimide used for coupling to MAb cysteinyl- thiol groups.
Recent promising preclinical xenograft therapy results have employed the super-toxic DNA cross-linking agents based on pyrrolobenzodiazepines (PBDs) linked to MAbs in a number of ways, including a cleavable peptide and more simply as a disulfide, and notably using an engineered cysteinyl- residue inserted into a specific position of the re-engineered MAb heavy chains (Figure 4).14
Figure 4.

Structure of a MAb conjugate of the pyrrolobenzodiazepine DNA-cross-linking drug, attached to a MAb via cleavable peptide linker. In this instance the entire conjugate is to be internalized into target cells and undergo lysosomal degradation to release the active free drug.
When considering standard ADC therapies against the newer ADC near infrared photoimmunotherapy (NIR-PIT) approach, several factors may be borne in mind: 1) Whereas at first glance NIR-PIT adds the light activation source as an added complexity, upon reconsideration of the multiple involved factors this is a relatively minor change and may even be advantageous. In NIR-PIT, active, potent drug is only produced at the light source and non-targeted MAb-dye conjugate is essentially non-toxic. 2) Standard ADCs, especially later-generation super-toxic drug conjugates, will always have inherent off-target toxicity in-built into the conjugate that limits dosages and may be unpredictable. For all the various ADC strategies briefly outlined above, the advanced chemistries, linkers, drugs and molecular biology employed on MAb-drug conjugates over the past 30 years, it remains true that if a targeted epitope is present on normal cells and a very small dose of the MAb-drug conjugate reaches its intended target, then the MAb-drug conjugate will be extremely toxic, and the therapeutic index will probably remain very adverse. 3) Standard ADCs will require internalization into the target tumor cells and then drug release from the MAb for their efficacy, which is a secondary and complex process to initial epitope binding. NIR-PIT with a MAb-dye conjugate has been demonstrated to only need to achieve binding to the outer surface epitope and then to be activated with near-infra-red (NIR) light to cause potent cell-surface toxicity to the targeted cells. 4) It is highly questionable whether all or even most cells in a tumor growth can be targeted by a MAb ADC and then be sufficiently internalized for effective drug action, and, in theory, this appears necessary without secondary effects such as immune system activation which ADC NIR-PIT induces. 5) Potent ADCs almost invariably use very hydrophobic drugs, somewhat incompatible with buffered MAbs, and require significant developmental efforts to produce MAb conjugates. The MAb-drug NIR-PIT agents described herein are very simple to prepare, water-soluble and stable, non-aggregated conjugates. Finally, it can be noted due to space restrictions, that we have not touched on discussion of numerous other possible ADC variants such as those involving polymeric carriers, prodrugs, bispecific targeting, liposomes, etc. Nevertheless, in whatever format ADCs are considered, the above general issues persist, and it is notable that after all the work by extremely talented multi-disciplinary scientists over 30+ years only very few ADCs have reached clinical fruition, and those mostly against non-solid tumors. It is our intent in the subsequent discussions to offer ADC NIR-PIT as an improved paradigm for delivery of effective tumor ADC therapy using targeted MAb-drug conjugates comprising NIR dyes along with their subsequent photoactivation.
MAb-IR700 Near Infrared Photoimmunotherapy
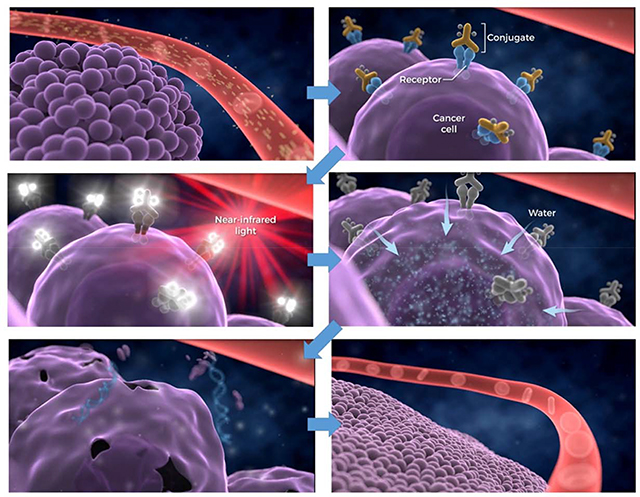
Near infrared photoimmunotherapy was first described and proposed as a new way to treat cancers after the observation that near infrared light was able to kill cancer cells membrane-bound by MAb conjugates (trastuzumab and panitumumab) of the NIR activatable dye, IR dye 700DX (IR700) (Figure 5), while conjugates that were not cell surface-bound showed no effects.15
Figure 5.

The active ester-linker-IR700 dye structure, with the ester being used to couple to MAb lysine residues. Note that the aromatic, essentially flat, phthalocyanine ring is counter-balanced by the oxysilicopropyl-aminotripropyl sulfonic acid groups held above and below the aromatic core. The stereochemistry prevents pi stacking of more than one aromatic ring even at multi-substitution on an antibody, thereby alleviating both quenching and aggregation of the dye and MAb, respectively. The six appended sulfonic acid groups and the two tetra-amino groups render the dye zwitterionic at physiological pH and the resulting MAb-IR 700 conjugate highly water-soluble.
Enabling this finding was the earlier discovery and description of the IR700 dye which unique combination of properties include, water solubility, non-aggregation even at high dye-to-protein ratios, no non-specific protein binding, photochemical stability in normal light, and high extinction coefficients and moderate fluorescent quantum yields.16 With absorbance and emission maxima near 700 nm [extinction coefficient 165,000; absorption maxima 689 nM, emission maxima 700 nm] the dye’s fluorescence properties are conveniently remote from the closest blood absorption maxima around 600 nm and water absorption maxima over 900 nm. Prior to development of this dye most organic dyes, like standard drugs, suffered from one or more of the drawbacks mentioned above, while alternative non-organic compositions such as quantum dots have good fluorescence properties but are not at all compatible with clinical use.
Most organic dyes, like most potent anti-cancer drugs are very hydrophobic species making their use with water-soluble MAbs very problematic in terms of development and production, and often leading to aggregation issues, both alone and as MAb conjugates, by virtue of their inherent tendency toward hydrophobic π-stacking. Interestingly for IR700, from the structural chemistry viewpoint the presence on the phthalocyanine-bound central silicon of the axial quaternary amine and sulfonic acid arms that present as zwitterionic charges under physiological conditions was cited by the developers as the reason for lack of aggregation seen with IR700, while its ready water solubility made development of MAb-IR700 conjugates at clinical scale much simpler than most MAb-drug conjugates and contributed to the rapid translation of the conjugates into clinical trials (vide infra). MAb conjugates of IR700 require no linker and are prepared readily with human/humanized/chimeric MAbs at low dye ester to MAb reaction ratios (around 5:1) to yield consistent conjugate substitution ratios of around 4:1, with no loss of binding activity or subsequent solubility problems, aggregation and low levels of self- and conditional quenching. The ready availability of the MAb and the IR700 dye together with the easy, high yield, aqueous conjugation and purification conditions led to rapid scale-up of processes from the initial milligram laboratory stage though to gram amounts and then kilogram clinical production levels. IR700 dye conjugates have low background autofluorescence, excellent photostability, brightness, a good excitation/emission window in the near-infra-red and are tolerant of salt. Clearly, the development of the structurally remarkable phthalocyanine dye sulfonic acids, originally developed for in vitro assay purposes, proved to be a necessary pre-requirement for subsequent NIR-PIT development.
In the first laboratory demonstrations of NIR-PIT efficacy in in vitro cellular studies it was found that target cellular toxicity was only effected if all the following conditions were met: 1) Cells must have MAb-IR700 bound at their surface membrane, unbound MAb-IR700 had no effect; 2) Cells bearing bound MAb-IR700 must be irradiated with wavelength-appropriate laser/LED light; non-irradiated cells showed no toxicity; 3) Unbound MAb-IR700 showed no toxicity to surrounding cells. In other words, all components and binding conditions had to be present in order to observe the toxic effect. Initial tests on Panitumumab-IR700 and Trastuzumab-IR700 on target-positive A431 and 3T3/HER2 cell lines, respectively, demonstrated identical therapeutic effects due to NIR-PIT although the former MAb showed significant therapeutic effect against A431 as a naked MAb. With extension to first animal therapy studies Panitumumab-IR700 showed significant tumor growth inhibition and life extension in A431 tumor xenograft-bearing mice.15 Since the first demonstration of NIR-PIT preclinical efficacy further studies have demonstrated positive results in multiple xenograft and other models of major cancers.17–23
With regard to the light source, initial preclinical NIR-PIT therapies in tumor-bearing mice were performed with 50 J/cm2 of surface illumination using a 690 nm LED lamp (power: 50–70 mW/cm2; wavelength spectrum 690 nm +− 20 nm).24 Although the light power was low and the light spectrum was not well matched to the excitation spectrum of IR700 that leading to low excitation efficacy, heat deposition in tissue was noted. In more recent preclinical studies and in clinical trials, NIR-PIT therapies were performed at 20–50 J/cm2 of surface illumination using a 690 nM Argon or diode laser system (wavelength spectrum 690 nm ± 4 nm) at 150 mW/cm2, following safety recommendations in consultation with the US FDA. This semi-optimized light spectrum was determined to be reasonably well matched to the IR700 excitation spectrum and led to high excitation efficacy and low heat deposition into tissue.25 For treating deeply located tumors, we found could reach virtually any parts of the body by employing a fiber optic diffuser inserted through a 20G injection needle,26 or alternatively, an endoscopic system with typical exposure to 100 J/cm of excitation light.27
Insights into NIR-PIT Mechanism(s) of Action
It is apparent that the near-IR radiation emitted by the NIR-PIT method will be much less toxic than non-targeted irradiation from gamma or X-rays, whether delivered by MAbs or using other modalities and we also propose that NIR-PIT will prove much less toxic than application of other ADCs. Nevertheless, there are some possible toxicities to remain aware of, for instance from local generation of heat, which can be part of the NIR-PIT mechanism of action. There is some likelihood of thermal injuries, particularly to skin, resulting from use of very high laser power settings (> 600 mW/cm2) and in this regard the finding that lower power at longer duration with the same total energy delivered could eliminate this effect is important.28–30 Both increasing light dose power settings and times along with increasing MAb-dye concentrations have been shown to positively impact the effects of NIR-PIT so NIR-PIT may be successfully performed at higher Mb-dye and lower laser power settings in order to achieve similar effects. Although there is a limit to the desirable level of light exposure it has been shown that repeat light dosing schedules are practical, often superior to single light doses, and such doses can be administered as early as 3 hours after a prior dose.28
The effect of partial heating effects as a contributor to NIR-PIT mechanism-of-action is hard to gauge. Originally, low light power settings were shown to effect successful NIR-PIT both in vitro and in vivo. Membrane effects leading to necrotic death seemed paramount with blebbing observed soon after NIR-PIT therapy quickly followed by cell membrane rupture (Figure 6).
Figure 6.

Outline of the general mechanism believed to be the major contributor to the NIR-PIT therapy effect. After conjugate binding and NIR light exposure cell membranes become compromised and absorption of water results in membrane blebbing and eventual disintegration.
Whereas reactive oxygen species are often identified for light initiating therapies, addition of sodium azide to test mixtures of MAb-IR700-bearing target cells, NIR-PIT therapy could not be completely abrogated by this potent reactive oxygen inhibitor.15 More recent studies have shown that NIR-PIT effects occur in as little as one minute after light exposure of MAb-IR700-targeted cells and even at this point, irreversible morphological changes have taken place in the affected cell mebranes.29 It has been shown that the IR700 dye readily fluoresces upon light absorption and emission but undergoes decomposition with loss of one and then both axial sulfonic acid arms above and below the plane of the phthalocyanine ring core (Figure 5) in a light-dose dependent manner. The remaining core phthalocyanine then becomes non-fluorescent and aggregated both on the MAb and at the bound cellular membrane, which we believe causes profound membrane distortions leading to swelling due to water internalization, and then quickly to the cell membrane blebbing and rupture observed microscopically. Further support for this type of mechanism was gained using low coherent quantitative phase microscopy after treatment of target cells with NIR-PIT which showed that cell rupture took place near peripheral binding sites of the MAb-dye conjugate, possibly because cell-bound conjugate aggregation upon light exposure induces changes in surface tension and thereby allow cells to become leaky (Figure 7).30
Figure 7.

Proposed mechanism of action of near infrared photoimmunotherapy. a) MAb-IR700 bound to cell surface tumor membrane by antigen binding is irradiated with near infrared light, b) The activation and emission of the dye induces loss of axial sulfonic acid-rich sidearms, c) Having lost its solubilizing sidearms the phthalocyanine ring becomes very hydrophobic and may induce aggregation and precipitation directly on MAb bound at the cell surface. In turn, this compromises membrane fidelity causing water influx into the cell and the subsequent blebbing and cell lysis.
A Brief Note on Photodynamic Therapy Versus Near Infrared Photoimmunotherapy
In passing it may be appropriate to mention photodynamic therapy, a modality dating back > 40 years with descriptions of efficacy against cutaneous and other accessible tumors using administration of hematoporphyrin derivatives followed by light exposure in the 630 nm range.31 This research ultimately led to the FDA approval of PHOTOFRIN (porfimer sodium) in 1995 for palliation of esophageal and endobronchial cancer due to blockages resulting from tumor growth. Treatment for esophageal involves administration of a 2mg/kg dose of porfimer followed 40–50 hours later with a laser light dose of 300 J/cm2. PHOTOFRIN is not a single chemical entity, being an oligomeric structure containing up to eight porphyrin ring units linked in various ratios by either ester of ether linkages (Figure 8).
Figure 8.

Core structure of the PHOTOFRIN small MW polymeric mixture. Wavy line bonds can link to the next porphyrin subunit by either ether or ester bonds, in varying ratios. Chiral centers are indeterminate in designation. There is one free carboxyl residue on polymer-central porphyrins and two on each end-terminal porphyrin.
Aside from composition heterogeneity, lack of target specificity due to non-specific binding and aggregation, the primary problem with PDT that has stopped its further adoption is the non-specific toxicity to normal tissues, not only from the laser but from ambient light meaning that patients would have to spend long periods in darkened conditions post-PDT treatment. The major differences between NIR-PIT and PDT are summarized in Table 1, and other than the use of a light activation step, the two modalities have little in common.
Table 1.
Comparison of NIR-PIT with the PDT method.
| Near Infrared Photoimmunotherapy (NIR-PIT) | Photodynamic Therapy (PDT) |
|---|---|
| Structurally defined high molecular weight antibody conjugate | Mixture of low molecular weight porphyrins |
| Specifically targets cells via a cell-surface-binding antibody | Relies on non-specific accumulation of dye in tissues after hydrophobic binding to plasma proteins. |
| Resistant to quenching effects due to 3D structure | Oxygen concentration dependent and quenched readily on non-specific albumin binding |
| Acts on the targeted cell surface only after binding | Relies on absorption of dyes internally into cells throughout the body |
| No normal, untargeted, cell activity | Remains inside cancer cells longer than normal cells. Normal cells allowed to partially clear dye prior to laser application. https://www.cancer.gov/about-cancer/treatment/types/surgery/photodynamic-fact-sheet |
| Acts on only cells specifically bound by antibody-dye conjugate. Limited to no off-target tissue effects. | Acts on all surrounding tissues containing dye. Substantial effects on surrounding tissues. |
| Damages and compromises conjugate-bound outer cell membranes | Damages cells by intracellular radical formation and damage to intracellular membranes and mitochondria |
| Cetuximab-IR700DX dose = 640 mg/m2 | Porfimer dose 2 mg/kg. Laser light at 40–50 hours followed by recommended debridement and second light dose at 96–120 hours. |
| Wavelength of light 690–700 nm | Wavelength of light 630 nm |
| Extinction coefficient, ε =165,000 M−1cm−1 | ε > 200 times lower than IR700DX |
| Clinical light dose 50 J/cm2; 100 J/cm | Light dose of 200 or 300 J/cm2 |
| Light exposure around 5 minutes | Light exposure 8 minutes 20 seconds |
| Systemic treatment | Local treatment |
| Curative intent on widely disseminated cancers | Palliative. Porfimer sodium only approved agent for endobronchial and esophageal cancers for relief of blockages due to tumor growth. |
| Insertion of laser into tumor recommended, if possible, to reduce normal tissue exposure and damage | |
| Inflammation a sign of a strong immune response | Inflammation may be fatal |
| High risk of possible ocular damage | |
| Patients susceptible to severe side-effects due to normal daylight, light bulbs etc. acting on dye located in/near all skin. Effects last for six weeks. |
Clinical Development and Early Clinical Results
A comparison of the anti-EGFR antibodies Panitumumab and Cetuximab for NIR-PIT when conjugated with IR700 indicated that the latter was superior in preclinical A431 xenograft and MDAMB468-luc orthotopic animal models, primarily due a longer circulatory half-time.32 Despite this, and the fact that the former MAb is humanized while the latter is chimeric, Cetuximab was chosen as the first MAb to undergo clinical development as a IR700 conjugate. This is because while both cetuximab and panitumumab are approved and available as naked MAbs for ready formulation in a manufacturing process the former is approved specifically for squamous cell tumors of the head and neck and offers a possible additive/synergistic contribution from ADCC, antibody-dependent complement cytotoxicity, along with basic blocking of the EGF receptor. New treatments for this indication are in especially great need due to the lack of options and the morbidities involved while the ready access to the EGFR positive lesions in disseminated form, wherein a broadly spread ‘field’ cancerization is often observed in patients, led us to believe that NIR-PIT may have a unique ability to prove itself in this indication.
At the time of writing Cetuximab-IR700 (ASP-1929; formerly RM-1929) has successfully passed phase 2a clinical safety and efficacy testing in advanced squamous cell head and neck cancer patients in the setting of locoregional, recurrent disease. In this 30 patient group the treatment was generally well tolerated and demonstrated clinically meaningful activity at an overall response rate of 43% comprised of five complete responders (16.7%) and nine partial responders (30 %) together with a disease control rate of 86.7% (26/30).33 Results have led to initiation of a definitive 275-patient Phase III multi-center clinical trial, currently recruiting patients (NCT03769506), comparing NIR-PIT using an infusion of 640 mg/m2 of Cetuximab-IR700 followed 24 h later with 690 nM light treatment for 5 minutes at 50 J/cm2 or 100 J/cm, with physician’s choice standard-of-care comprising cetuximab, methotrexate or docetaxel, using overall survival and progression-free survival as end-points.34
Latest Approaches and Future Directions
More recent preclinical research has investigated described MAb-IR700 NIR-PIT targeting of tissues thought to support and/or enable tumor growth such as tumor neovasculature35 and fibroblast cells.36 With respect to oral cancer, a recently published article described preclinical NIR-PIT targeting of the CD44 epitope on ‘stem-like’ cancer cells as an approach to address tumor heterogeneity.37 This work was done in syngeneic models as will future work on investigations into the potential effects of NIR-PIT on host immune systems in the setting of immune competent animal models. Immune checkpoint therapy is currently under active investigation in combination with various chemotherapies with the toxicity of the latter intended to ‘jump-start’ immune responses in patients who fail to respond to immune checkpoint therapy alone, the chemotherapy arms still entail some compromise of the patient immune system, up to and even including its abrogation in extreme applications. In a first study, a combination of anti-CD44 MAb-IR700 NIR-PIT followed by anti-PD-1 antigen-blocking therapy was shown to halt tumor growth in animal syngeneic models with induction of newly primed multi-clonal antigen T-cell responses, probably via tumor-adjacent dendritic cell activation after NIR-PIT-instigated antigen release.38 Perhaps, most significantly, mice that had their tumors cleared by the combination therapy were shown to resist secondary challenge by the same tumor line, indicating a highly desirable memory effect.
Experiments on combination of NIR-PIT with checkpoint inhibition strategies would offer significant advantages due to non-compromised immune systems in model systems. More generally, due to the rapid therapeutic mechanism with NIR-PIT, it should be feasible to observe ready, specific and near-instant tumor bed activation toward follow-on immune checkpoint inhibitor administration. To achieve more efficient but safer host immune-activation than systemic administration of immune-checkpoint inhibitors, locally depleting immune-suppressor cells in tumor beds using ADC NIR-PIT against CD25 for negative regulatory T-cells,39 or CXCR2 or CTLA-4 for myeloid derived suppressor cells, could minimize adverse autoimmune side effects. Immune-suppressor cell targeting NIR-PIT could be performed simultaneously with cancer-cell targeting NIR-PIT by using cocktail of Mab-IR700s against a cancer specific antigen and a suppressor cell marker. In comparison, chemotherapy administration paired with immune checkpoint targeting is a ‘blind event’ whose dose effects can only be gauged empirically on a patient-by-patient basis and its effects can take weeks-to-months to become fully apparent. NIR-PIT offers an opportunity both through fluorescent imaging and almost immediate quantitative estimation of the NIR-PIT effect on antigen release and immune cell activation to appraise the potential suitability of follow-on or concomitant checkpoint inhibitor treatments.
Acknowledgements
This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. This research was also supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research (ZIA BC011513).
References
- 1.Köhler G and Milstein C (1975) Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 256, 495–497. [DOI] [PubMed] [Google Scholar]
- 2.Strebhardt K and Ullrich A (2008) Paul Ehrlich’s magic bullet concept: 100 years of progress. Nature Rev. 8, 473–480. [DOI] [PubMed] [Google Scholar]
- 3.Reichmann L, Clark M, Waldmann H and Winter G (1988) Reshaping human antibodies for therapy. Nature. 332, 323–327. [DOI] [PubMed] [Google Scholar]
- 4.Kunert R and Reinhart D (2016) Advances in recombinant antibody manufacturing. Appl. Microbiol. Biotechnol. 100, 3451–3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Darvin P, Toor S, Nair VS and Elkord E. (2018) Immune checkpoint inhibitors: recent progress and potential biomarkers. Exp. Mol. Med. 50, 165–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang HM and Reisfeld RA (1988) Doxorubicin conjugated with a monoclonal antibody directed to a human melanoma-associated proteoglycan suppresses the growth of established tumor xenografts in nude mice. Proc. Natl. Acad. Sci. U.S.A. 85, 1189–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Trail PA, Willner D, Lasch SJ, Henderson AJ, Hofstead S, Casazza AM, Firestone RA, Hellström I and Hellström KE (1993) Cure of xenografted human carcinomas by BR96-doxodubicin immunoconjugates. Science. 261, 212–215. [DOI] [PubMed] [Google Scholar]
- 8.Damle NK and Frost P (2003) Antibody-targeted chemotherapy with immunoconjugates of calicheamicin. Curr. Opin. Pharmacol. 3, 386–390. [DOI] [PubMed] [Google Scholar]
- 9.Verma S, Miles D, Gianni L, Krop IE, Welslau M, Baselga J, Pegram M, Do-Youn Oh, Diéras V. Guardino, et al. (2016) Trastuzumab emtansine for HER2-positive advanced breast cancer. N. Engl. J. Med. 367, 1783–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shor B, Gerber H-P and Sapra P. (2015) Preclinical and clinical development of inotuzumab-ozogamicin in hematological malignancies. Molecul. Immunol. 67, 107–116. [DOI] [PubMed] [Google Scholar]
- 11.Levengood MR, Zhang X, Hunter JH, Emmerton KK, Miyamoto JB, Lewis TS and Senter PD (2017) Orthogonal cysteine protection enables homogeneous multi-drug antibody conjugates. Angew. Chem. Int. Ed. Engl. 56, 733–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Damelin M, Bankovich A, Bernstein J, Lucas J, Chen L, Williams S, Park A, Aguilar J, Ernstoff E, Charati M et al. (2017) A PKT7-targeted antibody-drug conjugate reduces tumor-initiating cells and induces sustained tumor regressions. Sci. Trans. Med. 9, https://stm.sciencemag.org/content/9/372/eaag2611 [DOI] [PubMed] [Google Scholar]
- 13.Cardillo TM, Govindan SV, Zalath MB, Rossi DL, Wang Y, Chang C-H and Goldenberg DM (2018) IMMU-140, a novel SN-38 antibody-drug conjugate targeting HLA-DR, mediates dual cytotoxic effects in hematological cancers and malignant melanoma. Mol. Cancer Ther. 17, 150–160. [DOI] [PubMed] [Google Scholar]
- 14.Pillow TH, Schutten M, Yu SF-, Ohri R., Sadowsky J., Poon KA, Solis W, Zhong F, Del Rosario G, Go MAT et al. (2017) Modulating therapeutic activity and toxicity of pyrrolobenzodiazepine antibody-drug conjugates with self-immolative disulfide linkers. Mol. Cancer Ther. 16, 871–878. [DOI] [PubMed] [Google Scholar]
- 15.Mitsunaga M, Ogawa M, Kosaka N, Rosenblum LT, Choyke PL and Kobayashi H (2011) Cancer cell-selective in vivo near infrared photoimmunotherapy targeting specific membrane molecules. Nat. Med. 17, 1685–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peng X, Draney DR, Volcheck WM, Bashford GR, Lamb DT, Grone DL, Zhang Y and Johnson CM (2006) Phthalocyanine dye as an extremely photostable and highly fluorescent near-infrared labeling agent. In: Optical Molecular Probes for Biomedical Applications, edited by Achilefu Samuel, Bonhop Darryl J., Raghavachari Ramesh, Proceedings of SPIE 6097, 60970E 10.1117/12.669173. https://www.spiedigitallibrary.org/conference-proceedings-of-spie/6097/1/Phthalocyanine-dye-as-an-extremely-photostable-and-highly-fluorescent-near/10.1117/12.669173.full?SSO=1 [DOI] [Google Scholar]
- 17.Sato K, Hanaoka H, Watanabe R, Nakajima T, Choyke PL and Kobayashi H (2015) Near-infra-red photoimmunotherapy in the treatment of disseminated peritoneal ovarian cancer. Mol. Cancer Ther. 14, 141–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Watanabe R, Hanaoka H, Sato K, Nagaya T, Harada T, Mitsunaga M, Kim I, Paik CH, Wu AM, Choyke PL et al. (2015) Photoimmunotherapy targeting prostate-specific membrane antigen: Are antibody fragments as effective as antibodies? J. Nucl Med. 56, 140–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maawy AA, Hiroshima Y, Zhang Y, Heim R, Makings L, Garcia-Guzman M, Luiken GA, Kobayashi H, Hoffman RM and Bouvet M (2015) Near infra-red photoimmunotherapy with anti-CEA-IR700 results in extensive tumor lysis and a significant decrease in orthotopic mouse models of pancreatic cancer. PLoS One. doi: 10.1371/journal.pone.0121989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sato K, Nagaya T, Choyke PL and Kobayashi H (2015) Near infrared photoimmunotherapy in the treatment of pleural disseminated NSCLC: Preclinical experience. Theranostics. 5, 698–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nagaya T, Sato K, Harada T, Nakamura Y, Choyke PL and Kobayashi H (2015) Near infrared photoimmunotherapy targeting EGFR positive triple negative breast cancer: Optimizing the conjugate-light regimen. PLoS One. doi: 10.1371/journal.pone.0136829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nagaya T, Nakamura Y, Sato K, Harada T, Choyke PL and Kobayashi H (2016) Near infrared photoimmunotherapy of B-cell lymphoma. Mol. Oncol. 10, 1404–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nagaya T, Nakamura Y, Okuyama S, Ogata F, Maruoka Y, Choyke PL and Kobayashi H (2017) Near-infrared photoimmunotherapy targeting prostate cancer with prostate-specific membrane antigen (PSMA) antibody. Mol Cancer Res. 15, 1153–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakajima T, Sato K, Hanaoka H, Watanabe R, Harada T, Choyke PL and Kobayashi H (2014) The effects of conjugate and light dose on photo-immunotherapy induced cytotoxicity. BMC Cancer, doi: 10.1186/1471-2407-14-389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Okuyama S, Nagaya Y, Ogata F, Maruoka Y, Sato K, Nakamura Y, Choyke PL and Kobayashi H (2017) Avoiding thermal injury during near-infra-red photoimmunotherapy (NIR-PIT): The importance of NIR light power density. Oncotarget. 8, 113194–113201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Okuyama S, Nagaya T, Sato K, Ogata F, Maruoka Y, Choyke PL and Kobayashi HK (2018) Interstitial near-infrared photoimmunotherapy: effective treatment areas and light doses needed for use with fiber optic diffusers. Oncotarget. 9, 1159–11169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nagaya T, Okuyama S, Ogata F, Maruoka Y, Choyke PL and Kobayashi HK (2018) Endoscopic near infrared photoimmunotherapy using a fiber optic diffuser for peritoneal dissemination of gastric cancer. Cancer Sci. 109, 1902–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ogata F, Nagaya T, Nakamura Y, Sato K, Okuyama S, Maruoka Y, Choyke PL and Kobayashi H (2017) Near infra-red photoimmunotherapy: a comparison of light dosing schedules. Oncotarget. 8, 35069–35075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sato K, Ando K, Okuyama S, Moriguchi S, Ogura T, Totoki S, Hanaoka H, Nagaya T, Kokawa R, Takahura H et al. (2018) Photoinduced ligand release from a silicon phthalocyanine dye conjugated with monoclonal antibodies: A mechanism of cancer cell toxicity after near-infra-red photoimmunotherapy. ACS Cent. Sci. 4, 1559–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ogata F, Nagaya T, Okuyama S, Maruoka Y, Choyke PL, Yamauchi T and Kobayashi H (2017). Dynamic changes in the cell membrane on three-dimensional low coherent quantitative phase microscopy (3D-LC-PQM) after treatment with near infrared photoimmunotherapy. Oncotarget, doi: 10.18632/oncotarget.22223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dougherty TJ, Kaufman JE, Goldfarb A, Weishaupt KR, Boyle D and Mittleman A (1978) Photoradiation therapy for the treatment of malignant tumors. Cancer Res. 38, 2628–2635. [PubMed] [Google Scholar]
- 32.Sato K, Watanabe R, Hanaoka H, Nakajima T, Kim I, Paik CH, Choyke PL and Kobayashi H (2014) Photoimmunotherapy: Comparative effectiveness of two monoclonal antibodies targeting the epidermal growth factor receptor. Mol. Oncol. 8, 620–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cognetti DM, Johnson JM, Curry JM, Mott F, Kochuparambil T, McDonald D, Fidler MJ, Stensen K, Vasan NR, Razaq M et al. (2019) Results of a phase 2a, multicenter, open-label, study of RM1929 photoimmunotherapy (PIT) in patients with locoregional, recurrent head and neck squamous cell carcinoma (rHNSCC). J. Clin. Oncol (suppl; abstr 6014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Biel MA, Gillenwater AM, Cognetti DM, Johnson JM, Argiris A and Tahara M (2019) A global phase III multicenter, randomized, double-arm, open trial of ASP-1929 photoimmunotherapy versus physician’s choice standard of care for treatment of patients with locoregional, recurrent head and neck squamous cell carcinoma (rHNSCC) J. Clin Oncol. (suppl; abstr TPS6094). [Google Scholar]
- 35.Nishimura T, Mitsunaga M, Ito K, Kobayashi H and Saruta M (2019) Cancer neovasculature-targeted near-infrared photoimmunotherapy (NIR-PIT) for gastric cancer: different mechanisms of phototoxicity compared to cell-membrane targeted NIR-PIT. Gastric Cancer. July 13 doi: 10.1007/s10120-019-00988-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Watanabe S, Noma K, Ohara T, Kashima H, Sato H, Kato T, Urano S Katsube R, Hashimoto Y, Tazawa H et al. (2019) Photoimmunotherapy for cancer-associated fibroblasts targeting fibroblast activation protein in human esophageal squamous cell carcinoma. Cancer Biol. Ther. June 11:1–15. doi: 10.1080/15384047.2019.1617566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nagaya T, Nakamura Y, Okuyama S, Ogata F, Maruoka Y, Choyke PL, Allen C and Kobayashi H (2017) Syngeneic mouse models of oral cancer are effectively targeted by anti-CD44-based NIR-PIT. Mol. Cancer Res. 15, 1667–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nagaya T, Friedman J, Maruoka Y, Ogata F, Okuyama S, Clavijo PE, Choyke PL, Allen C and Kobayashi H (2019) Host immunity following near-infrared photoimmunotherapy is enhanced with PD-1 checkpoint blockade to eradicate established antigenic tumors. Cancer Immunol. Res 7, 401–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sato K, Sato N, Xu B, Nakamura Y, Nagaya T, Choyke PL, Hasegawa Y and Kobayashi H (2016) Spatially selective depletion of tumor-associated regulatory T cells with near-infrared photoimmunotherapy. Sci. Trans. Med. August 17, 8(352):352ra110. [DOI] [PMC free article] [PubMed] [Google Scholar]