

Abstract
Purpose
Riboflavin and its cofactors are essential for cellular energy generation, responses to oxidative stress, and overall homeostasis. Retbindin is a novel retina-specific riboflavin binding protein essential for the maintenance of retinal flavin levels, but its function remains poorly understood. To further elucidate the function of retbindin in retinal health and disease, we evaluated its role in retinal degeneration in a cone-rod dystrophy model associated with the R172W mutation in the photoreceptor tetraspanin Prph2.
Methods
We performed structural, functional, and biochemical characterization of R172W-Prph2 mice with and without retbindin (Rtbdn−/−/Prph2R172W).
Results
Retbindin is significantly upregulated during degeneration in the R172W model, suggesting that retbindin plays a protective role in retinal degenerative diseases. This hypothesis was supported by our findings that R172W mice lacking retbindin (Rtbdn−/−/Prph2R172W) exhibit functional and structural defects in rods and cones that are significantly worse than in controls. Retinal flavin levels were also altered in the Rtbdn−/−/Prph2R172W retina. However, in contrast to the Rtbdn−/− retina which has reduced flavin levels compared to wild-type, Rtbdn−/−/Prph2R172W retinas exhibited elevated levels of riboflavin and the flavin cofactor FMN.
Conclusions
These results indicate that retbindin plays a protective role during retinal degeneration, but that its function is more complex than previously thought, and suggest a possible role for retbindin in protecting the retina from phototoxicity associated with unbound flavins. This study highlights the essential role of precisely regulated homeostatic mechanisms in photoreceptors, and shows that disruption of this metabolic balance can contribute to the degenerative process associated with other cellular defects.
Keywords: retinal diseases, flavin, peripherin 2, peripherin/RDS, retinal degeneration, mouse model
Retbindin is a novel retina-specific protein located in the interphotoreceptor matrix (IPM) at the tips of photoreceptor outer segments (OS).1,2 In the IPM metabolites and nutrients are exchanged between photoreceptors and the retinal pigment epithelium (RPE).3 Retbindin is secreted into the IPM and peripherally anchored to the plasma membrane via electrostatic forces, and we showed that mice lacking retbindin (Rtbdn−/−) exhibit retinal degeneration as well as defects in rod and cone function.2,4 Our previous studies using both degenerative models that lack rods but retain some cones and models in which GFP is knocked into cells that express retbindin have shown that retbindin expression is limited to rods.2,4 Retbindin shares significant homology with the riboflavin binding domain of chicken oviduct riboflavin binding protein (RBP).1,2 Subsequently, we determined that retbindin binds riboflavin in vitro2 and that flavin levels were reduced in Rtbdn−/− mice.4
Riboflavin is not synthesized by the body and is the precursor of flavin mononucleotide (FMN) and flavin adenine dinucleotide (FAD), essential enzymatic cofactors.5–7 FMN and FAD are involved in many processes, including antioxidant systems (where they are cofactors for glutathione reductase) and energy generation. Cellular energy production involves the use of FAD/FMN in the electron transport chain8 and during beta-oxidation of fatty acids.9 Retinal flavin levels are several-fold higher than blood levels,10 reflecting the extraordinarily high demand for energy in photoreceptors and retinal susceptibility to oxidative stress. Combined these observations have led us to hypothesize that retbindin may play a role in the ability of the retina to concentrate flavins.
Flavin levels are decreased in models of rapid retinal degeneration such as the rd1 and rd10.11 However, it is not clear what role retbindin or flavin metabolism may play during retinal degeneration. To begin evaluating this, here we examine the role of retbindin in a model of cone-rod dystrophy, the R172W peripherin 2 transgenic mouse model (Prph2R172W).12 Peripherin 2 is essential for the formation of rod and cone OSs.13,14 The R172W mouse model has been well characterized, and mimics many of the cone-rod dystrophy phenotypes seen in patients carrying the R172W mutation,12,15–18 including functional defects in cones and late-onset retinal degeneration.
Here, we report that retbindin levels are significantly upregulated in Prph2R172W retinas before and during degeneration. Furthermore, eliminating retbindin in the R172W model (Rtbdn−/−/Prph2R172W) exacerbated structural and functional degeneration associated with the R172W mutation. Interestingly, retinal flavin levels were elevated in the Rtbdn−/−/Prph2R172Wretinas. Our results clearly demonstrate that retbindin has a role during degeneration and suggest that the contribution of retinal flavins during degeneration is complicated.
Materials and Methods
Animals
Retbindin knockout (Rtbdn−/−) and R172W-Prph2 transgenic mice were generated and characterized as reported previously.2,4,12 The Prph2R172W mice on wild-type (WT) peripherin 2 background were cross-bred into the Rtbdn−/− background to generate the Rtbdn−/−/Prph2R172Wmice. WT, Rtbdn−/− and Prph2R172W mice were included for comparisons. All procedures were approved by University of Houston Institutional Animal Care and Use Committee (IACUC).
Animals are backcrossed onto our in-house WT line, a strain that was created by breeding FVB mice to C57BL/6, eliminating the rd1 and rd8 mutations and then inbreeding for over 10 generations. PCR genotyping confirmed that none of the mice used in this study carry the rd8 mutation. Animals were housed under 30- to 50-lux cyclic light 12 hours light:12 hours dark. Both sexes were equally included in the analyses. For sample collection, animals were euthanized using CO2 asphyxiation and retinas were collected as described before19 and immediately frozen in liquid nitrogen and stored at −80°C until used.
Protein Chemistry
Immunoblot analyses, as well as soluble IPM, cytoplasm/membrane fractionation, were performed as detailed previously.2 Antibodies used are described in the Table.
Table.
Antibodies
| Antigen | Antibody | Species | Source | Concentration |
|---|---|---|---|---|
| Retbindin | Anti-Rtbdn | Rbt PC | In-house2,54 | 1:500 (WB/IF) |
| Prph2 | RDS-2B7 | Ms MC | In-house | 1:1,000 (WB) |
| IRBP | Anti-IRBP | Rbt PC | Reference55 | 1:1,000 (WB) |
| GAPDH | Ab8245 | Ms MC | Abcam, Cambridge, MA, USA | 1:1,000 (WB) |
| beta-Actin-HRP | A3854 | Ms MC | Sigma-Aldrich, St. Louis, MO, USA | 1:50,000 |
| GFAP | MAB360 | Ms MC | Millipore,Burlington, MA, USA | 1:1,000 (IF) |
| FLAD1 | G-4, sc-376819 | Ms MC | Santa Cruz Biotechnology, Dallas, TX, USA | 1:1000 (WB) |
| Rabbit-HRP | AP187P | Goat PC | Sigma-Aldrich, St. Louis, MO, USA | 1:25,000 (WB) |
| Mouse-HRP | AP181P | Goat PC | Sigma-Aldrich, St. Louis, MO, USA | 1:25,000 (WB) |
| Alexa Fluor-anti rabbit 647 | A21245 | Goat PC | Thermo Fisher Scientific, Waltham, MA, USA | 1:1,000 (IF) |
| Alexa Fluor-anti mouse 555 | A31570 | Donkey PC | Thermo Fisher Scientific, Waltham, MA, USA | 1:1,000 (IF) |
| PNA Alexa Fluor 488 | L21409 | N/A | Thermo Fisher Scientific, Waltham, MA, USA | 1:500 (IF) |
| Isolectin B4 Alexa Fluor 568 | L21412 | N/A | Thermo Fisher Scientific, Waltham, MA, USA | 1:250 (IF) |
| DAPI (stain) | 62248 | N/A | Thermo Fisher Scientific, Waltham, MA, USA | 1:1,000 (IF) |
Soluble IPM, cytoplasmic and membrane fractions were prepared by placing intact freshly extracted postnatal day (P)30 retinas in phosphate buffered saline solution [PBS] (pH 7.2) containing protease inhibitors on ice for 15 minutes without agitation. The supernatant (soluble IPM fraction) was collected after centrifugation at 113g for 5 minutes with an Eppendorf centrifuge 5427R (Eppendorf International, Hamburg, Germany). The pellets were incubated in hypotonic buffer (PBS, pH 7.2 containing protease inhibitors) on ice for 15 minutes and then extracted with a hand-held motorized pestle tip homogenizer and centrifuged at 50,000g for 30 minutes. The supernatant (cytoplasmic fraction) was removed, and the pellet (membrane fraction) was resuspended/solubilized in PBS (pH 7.2) containing 1% Triton X-100 and protease inhibitors and then homogenized with sonication. After 1 hour's incubation at 4°C, samples were centrifuged at 16,128g for 5 minutes. The supernatant was considered to contain the membrane bound proteins.
Immunofluorescence
Eyes were fixed in Davidson's fixative (32% ethanol, 11% acetic acid, 2% formaldehyde),2 embedded in paraffin, and cut into 10-µm sections. After rehydration and antigen retrieval (10 mmol/L citrate buffer, pH 6.0), sections were blocked, probed for retbindin as previously described,2 and subsequently incubated with secondary antibodies. Antibodies are listed in the Table. Slides were mounted with Prolong Gold (no. P36934, Thermo Fisher Scientific, Waltham, MA, USA), and images were captured using Zeiss 800 LSM confocal system with an Airyscan detector and processed in Zen 2 lite software (Zeiss, Thornwood, NY, USA). Images in Figure 1B (upper panel) and Figure 5 were collected under epifluorescent conditions at original magnification × 40. Images in Figure 1B (lower panel) were captured at original magnification × 63 and shown are Airyscan processed collapsed planes from a confocal stack. Images in Figure 3B were collected at original magnification × 20 and are collapsed views of confocal images with six planes (1 µm each). For cone counts, sections were labeled with peanut agglutinin (PNA) and imaged 200 µm inferior and superior to the optic nerve. For each animal, the number of cones in the superior and inferior images was averaged to obtain a single value for that animal. N = 3 animals/genotype and age.
Figure 1.

Retbindin is upregulated in the Prph2R172W retinas. (A) Immunoblot analysis of retbindin under reducing conditions in five to six independent retinal extracts taken from P30 and P90 WT and Prph2R172W mice showing significant upregulation at both time points. Retbindin levels were measured densitometrically using Bio-Rad Image Lab v4.1 software and normalized to actin. Data are presented as mean ± SEM. *P < 0.05 and **P < 0.001 by two-way analysis of variance with Tukey's post hoc test. (B) Representative images of P30 retinal cross-sections taken from WT and Prph2R172W mice labeled for retbindin (red) with nuclei counterstained with DAPI (blue). OS, outer segment; IS, inner segment; ONL, outer nuclear layer; INL, inner nuclear layer. Scale bars: 5 µm. Images in B (upper panel) were collected under epifluorescent conditions at magnification × 40. Images in B (lower panel) were captured at magnification ×63 and shown are airyscan processed collapsed planes from a confocal stack. (C) Reducing SDS-PAGE western blot analysis of P30 WT and Prph2R172W retinal extracts separated into soluble IPM, membrane, and cytoplasmic fractions, probed with anti-Rtbdn, -IRBP, -GAPDH and -Prph2 antibodies.
Figure 5.

Rtbdn−/−/Prph2R172W retina shows altered pattern of GFAP labeling. Shown are representative images of retinal sections probed for GFAP (green) and DAPI (blue). Images were collected under epifluorescent conditions at original magnification × 40. Arrows highlight increased gliosis at P90. ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer. Scale bar: 10 µm.
Figure 3.

Photoreceptor degeneration is accelerated in the Rtbdn−/−/Prph2R172W. (A) Shown are light micrographs of retinal sections from WT, Rtbdn−/−, Prph2R172W, and Rtbdn−/−/Prph2R172W mice at P30 and P90. Scale bar: 50 µm. (B) Cone photoreceptors were labeled using PNA at P30 and P90. Shown are representative images taken at P90, 200 µm superior to the optic nerve head. Scale bar: 20 µm. Images in were collected at original magnification × 20 and are collapsed views of confocal images with six planes (1 µm each). (C) Photoreceptor nuclear count for each genotype was plotted across the retina as mean ± SEM. The cells were counted in the area covering 100 µm every 200 µm starting at the optic nerve head. N = 3 eyes/genotype. Asterisk indicates WT, Rtbdn−/−, and Prph2R172W compared to Rtbdn−/−/Prph2R172W. Plus signs indicate comparison between WT and Prph2R172W. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 by two-way analysis of variance with Tukey's post hoc comparison. (D) Cones were counted in images captured 200 µm inferior and superior of the ONH and averaged to give a value for each animal. N = 3 animals/genotype and age. *P < 0.05, *P < 0.01 for two-way analysis of variance with Tukey's post hoc comparison. OS, outer segment; IS, inner segment; ONL, outer nuclear layer; INL, inner nuclear layer; I, inferior; S, superior; ONH, optic nerve head.
Quantification of Cellular ATP Levels
The total levels of cellular ATP in freshly collected retinal samples were measured using Abcam (ab113849) luminescent ATP detection Assay kit as previously described.4 In brief, retinas were placed in 110 µl PBS (pH 7.4) and 1 × protease inhibitors and homogenized using sonication and centrifuged at 16, 128 × g for 5 minutes. Supernatant 100 µL for each sample was transferred to a single well of a 96-well and plate processed as per manufacturer's instructions. All of these procedures were performed under a dim red light. The plate was immediately placed in the microplate reader (Spectramax M5; Molecular Devices, Sunnyvale, CA, USA). The ATP levels in each sample were calculated based on luminescence values from a standard curve.
Electroretinography
Full-field ERG was performed at P30, P60, and P90 under scotopic and photopic conditions as described previously20,21 using UTAS system (LKC, Gaithersburg, MD, USA).
Light Histology and Morphometry
Eyes were collected, fixed, embedded and sectioned as described in.12 Slides with the sections along the optic nerve were stained with hematoxylin and eosin then mounted using Permount mounting medium (Thermo Fisher Scientific). Images were captured every 200 µm along the superior-inferior plane starting from the optic nerve using Zeiss Axioskop 50. Outer nuclear layer cells were counted within the areas enclosing 100 µm length every 200 µm.
Transmission Electron Microscopy
The superior hemisphere of the eyes was marked and then harvested, punctured at the ora serrata, and fixed in mixed aldehyde fixative (2% paraformaldehyde, 2% glutaraldehyde, 100 mmol/L cacodylate, 0.025% CaCl2 [pH 7.4]) for 2 hours. Cornea and lens were removed, and eye cups were returned back to the fixative and incubated at 4°C overnight. The globes were embedded in plastic resin, sectioned, and stained with osmium tetroxide as explained elsewhere.21,22 Ultrastructural imaging was performed as previously described.21
Fundus and Fluorescein Angiography
Fundus and fluorescein angiogram imaging was performed as described previously23 using Phoenix Micron IV system (Phoenix Research Laboratories, Pleasanton, CA, USA). After capturing bright field images, mice were intraperitoneally injected with clinical grade (Akorn Ak-fluor 10%) fluorescein at a dose of 0.01 mL/10 gm weight of the animal. Thirty seconds after injection, images were collected using the 451.5- to 486.5-nm excitation and 488-nm emission GFP filter. All of the animals were exposed to the same amount of light at same voltage and gain. Images were obtained using Discover-1.2 software.
Flavin Quantification by HPLC
Animals were fasted for 6 hours before euthanasia to prevent any feeding variability among animals. Retinas were collected and snap-frozen in liquid nitrogen and kept at −80°C. Microextraction and quantification of flavins (riboflavin, FAD, and FMN) were carried out as described11 using Waters HPLC system (Waters, Milford, MA, USA).
Statistical Analysis
Statistical analyses were done using one- or two-way analysis of variance with Tukey's post hoc comparison. Graphpad Prism v.8 was used for all analysis.
Results
Retbindin Levels are Upregulated in Prph2R172W Retinas
To begin evaluating whether retbindin is important during photoreceptor degeneration, we assessed its levels in retinal extracts harvested from the Prph2R172W mice. This mouse line carries endogenous wild-type Prph2 and a R172W mutant Prph2 allele, resulting in expression of R172W protein at levels equivalent to ∼40% of endogenous Prph2 levels.12 Previously we showed that overexpression of WT Prph2 is not toxic to the retina, but R172W animals exhibit gradual structural and functional retinal degeneration.12,15 Immunoblot analysis showed that retbindin levels were 2.5 fold higher in Prph2R172Wthan nontransgenic wild-type (WT) at P30 (Fig. 1A) and critically that levels continued to rise in Prph2R172W from P30 to P90, 3.7-fold higher than in WT at P90.
Our previous biochemical studies showed that retbindin is an extracellular protein, which co-localizes with the IPM marker wheat germ agglutinin (but not the disc component rhodopsin).2 Retbindin is normally found in two areas; the IPM between the apical edge of the OSs and the RPE, and surrounding the inner segments (IS).2 Immunofluorescence labeling at P30 demonstrated that Prph2R172W retinas exhibit labeling in these areas, but rather than a layer of labeling restricted to the apical OS/RPE junction, Prph2R172Wretinas exhibit retbindin labeling throughout the OS layer (Fig. 1B). We previously found that retbindin is an extracellular membrane associated component of the insoluble IPM. To determine whether this subcellular localization is preserved in the Prph2R172W, we fractionated retinas into the soluble IPM, retinal membranes, and retinal cytoplasm. The majority of retbindin in both the WT and Prph2R172W is found in the membrane fraction (Prph2 shown as a positive control), with no significant retbindin in the soluble IPM (IRBP as a positive control) (Fig. 1C). A small portion of total retbindin is found in the cytoplasmic fraction and interestingly, two retbindin bands are detected. These likely reflect newly synthesized immature (upper band) and mature retbindin (lower band) and suggests that retbindin undergoes posttranslational modification/processing.
Retbindin Ablation Exerts Negative Functional and Structural Effects on Prph2R172W Retina
The observed upregulation of retbindin in Prph2R172W retinas supported the idea that retbindin plays a role during retinal degeneration. To help understand whether this upregulation was a protective response (i.e., to help protect the retina in the face of a degenerative insult) or a contributor to degeneration, we backcrossed the Prph2R172W mice onto the Rtbdn−/− background. We conducted full-field scotopic and photopic electroretinography (ERG) to measure rod and cone function, respectively (Fig. 2A). Consistent with previous findings, elimination of retbindin (Rtbdn−/−) had no deleterious effects on retinal function up to P90.4 In contrast, eliminating retbindin in the Prph2R172W retina led to statistically significant reductions in both cone and rod responses at all three ages in comparison to WT and single mutants (Fig. 2B). Eliminating retbindin in Prph2R172W mutants had a larger effect on rod function than cone function; for example, at P30, rod function (scotopic a-wave) in the Rtbdn−/−/Prph2R172W was reduced 52% compared to the Prph2R172W, while cone function (photopic b-wave) was reduced by 21%. However, this difference may be due to the fact that the R172W mutation affects cones more than rods, and when compared to WT, the Rtbdn−/−/Prph2R172W had similar reductions in both rod and cone function (to 38% and 40% of WT, respectively at P90). Scotopic b-wave is a measure of second-order neuron signaling, and here we find that scotopic a- and b-waves were reduced fairly proportionally.
Figure 2.

Lack of retbindin in the Prph2R172W mutant causes defect in rod and cone functions. (A) Representative waveforms of scotopic and photopic ERG responses recorded from P30 WT, Rtbdn−/−, Prph2R172W, and Rtbdn−/−/Prph2R172W mice. (B, C) Average amplitudes of maximum scotopic a-wave (B) and photopic b-waves (C) plotted as mean ± SEM from mice of the indicated genotypes at P30, P60 and P90. N = 10 to 19 animals for each genotype/age. + indicates comparisons between WT and Rtbdn−/−/Prph2R172W as well as between Rtbdn−/− and Rtbdn−/−/Prph2R172W. Ampersand indicates comparisons between WT and Rtbdn−/−, asterisk indicates comparisons between WT and Prph2R172W, and yen symbol (¥) indicates comparison between Prph2R172W and Rtbdn−/−/Prph2R172W. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 by two-way analysis of variance with Tukey's post hoc comparison.
Histologic analyses showed no obvious signs of retinal degeneration at P30 in the Prph2R172W or the Rtbdn−/−; however, there is evident thinning of the outer nuclear layer (ONL), as well as the OS and IS layers in the Rtbdn−/−/Prph2R172Wretina (Fig. 3A). To quantify this degeneration, we counted ONL nuclei (95-97% rods) across the retina (Fig. 3C). ONL counts in WT, Rtbdn−/−, and Prph2R172W were not significantly different from each other at P30, but there was a ∼25% reduction in ONL nuclei in the Rtbdn−/−/Prph2R172W central retina compared to WT (Fig. 3C, left). At P90, this was even more pronounced, with ∼65% reduction in ONL cells in the Rtbdn−/−/Prph2R172W central retina compared to in WT. In striking contrast, the Rtbdn−/− still shows no degeneration at P90, and the Prph2R172W only shows a very small reduction in the ONL at P90 (Fig. 3c, right). Although retbindin is expressed only in rods, long-term retbindin deficiency led to rod and cone defects.2 Here we saw exacerbated cone functional deficits in the Rtbdn−/−/Prph2R172W retina, so we counted cone photoreceptors in the central retina labeled with peanut agglutinin (Figs. 3B,D). Similar to rods, we observed significant loss of cone cells in the Rtbdn−/−/Prph2R172W retinas at both P30 and P90 (Fig. 3D).
Ultrastructural examination showed that, unlike the WT and the single mutants that exhibited nicely aligned stacks of discs, most OSs in the Rtbdn−/−/Prph2R172W were highly disorganized (asterisks, Fig. 4A) and ISs were swollen (white arrowheads, Fig. 4A) at P30 and P90. This disorganization is clearer at higher magnification (Fig. 4B); some OS discs in the Rtbdn−/−/Prph2R172W retina are misaligned (blacked asterisks, Fig. 4B), shorter (black arrow heads, Fig. 4B) or totally disrupted (black arrows, Fig. 4B). OS disorganization is exacerbated at P90 where swirls are present in Rtbdn−/−/Prph2R172Wretinas (white arrowheads, Fig. 4B) next to fairly organized OSs (white arrows, Fig. 4B). In addition, RPE cells showed marked vacuolization in the Rtbdn−/−/Prph2R172W (black arrows, Fig. 4A). These vacuoles often contain membranous debris (Fig. 4A, inset), possibly undigested OS. However, in contrast to photoreceptor defects which are much more severe in the Rtbdn−/−/Prph2R172W than in WT or single mutants, RPE defects are seen in both double mutants and Prph2R172W, suggesting RPE changes may be primarily due to the R172W mutation.
Figure 4.
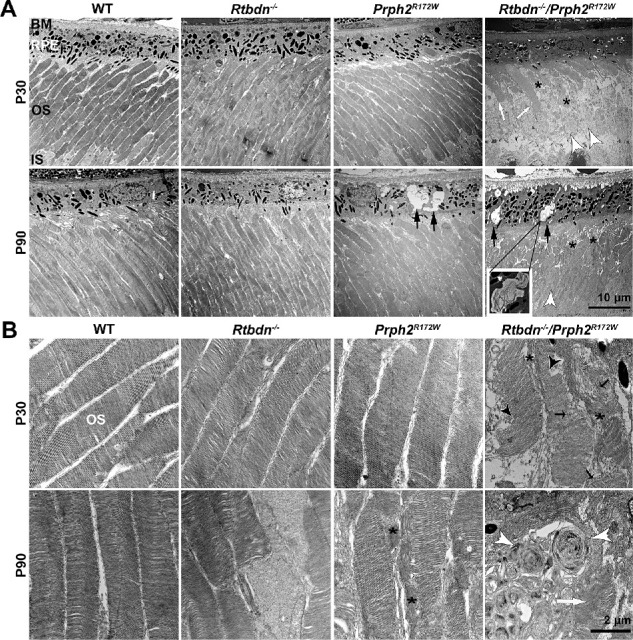
Retbindin ablation in the Prph2R172W retina causes ultrastructural changes. (A) Shown are representative EM images of OS and RPE for the indicated genotypes. Black arrows denote vacuoles present in the RPE. White arrows show OSs with well stacked disc. Asterisks point out the disorganized, degenerated OSs and white arrowheads mark the swollen ISs. Scale bar: 10 µm, original magnification × 5000. (B) Shown are representative EM images of OSs from the indicated genotypes. Black arrows denote disrupted OS discs and asterisks mark misaligned discs. White arrowheads show OS whorls whereas white arrow highlights more normal disc stacking. Black arrowheads point to shortened discs. Scale bar: 2 µm, original magnification × 25,000. BM, Bruch's membrane; RPE, retinal pigment epithelium; OS, outer segment; IS, inner segment.
Ablation of Retbindin in the Prph2R172W Retina Leads to Retinal Gliosis
One sign of retinal stress and gliosis24–26 is upregulation of glial fibrillary acidic protein (GFAP) in Müller cells. Reactive gliosis can be neuroprotective, but chronic gliosis can contribute to vascular and other retinal pathologies.27 At P30, GFAP was restricted to the nerve fiber layer as expected. However, by P90 GFAP labeling was increased in the Rtbdn−/−/Prph2R172W retina, with Müller cell labeling extending into the inner plexiform and inner nuclear layer, consistent with gliosis (Fig. 5, arrows).
Rtbdn−/−/Prph2R172W Mice Exhibit Fundus and Retinal Vascular Abnormalities
Humans and mice carrying the R172W mutation exhibit alterations in retinal vasculature with varying degrees of penetrance.15,17 To assess whether eliminating retbindin had any effect on retinal vasculature we performed fundus imaging and fluorescein angiography at multiple time points. There were no overt changes in fundus appearance or fluorescein angiogram at P90 (Fig. 6A), but by P240, Rtbdn−/−/Prph2R172W eyes start to exhibit changes (Figs. 6A, 6B). On fundus images, large degenerative patches become evident (black arrowheads, Fig. 6B) and correlate with areas where diffuse GFP signal from the choroid can be seen (white arrowheads, Fig. 6B). This phenotype is variable among age-matched mice (Fig. 6B). In P240 Rtbdn−/−/Prph2R172W animals, 12 of 13 animals showed neovascular tufts, and three of 13 exhibited splotchy degenerated areas (on fundus images). These phenotypes also occurred in the Prph2R172W, five of nine animals exhibited neovascular tufts and five of nine splotchy degenerative areas. In contrast, none (of seven) Rtbdn−/− mice exhibited these phenotypes at P240, and only one of seven wild-type mice had a very small number of neovascular tufts. To help better visualize these neovascular tufts, we labeled blood vessels on retinal flat mounts using isolectin B4 (red, Fig. 6C). Capillary tufts (white arrows) were observed in the central and peripheral areas of the Prph2R172W retinas, a phenotype that was worsened in Rtbdn−/−/Prph2R172W retinas.
Figure 6.

Rtbdn−/−/Prph2R172W mice exhibit fundus and vascular abnormalities. (A) Fundus images and fluorescein angiograms were captured from mice of the indicated genotypes at P90 and P240. (B) Shown are representative fundus and fluorescein angiograms from three additional Rtbdn−/−/Prph2R172W mice. White arrows show neovascular tufts, and white arrowheads denote leakage. (C) Shown are representative isolectin-B4 stained (red), flat mounted retinas from P240 WT, Rtbdn−/−, Prph2R172W, and Rtbdn−/−/Prph2R172W mice. Arrows indicate neovascular tufts.
Changes in Flavin Processing in the Rtbdn−/−/Prph2R172W
Riboflavin and its cofactors FMN and FAD are essential for retinal health,28–30 and flavin levels are significantly reduced in the Rtbdn−/− retina.4 We observe that at P30, riboflavin, FAD, and FMN levels are decreased in the Rtbdn−/− and Prph2R172W (compared to WT, Figs. 7A–C, left). P30 FMN levels are also reduced in the Rtbdn−/−Prph2R172W, however, riboflavin levels are elevated at P30 (compared to the Rtbdn−/−). This finding is more pronounced at P90 where both FMN and riboflavin levels are increased in the Rtbdn−/−Prph2R172W compared to Rtbdn−/− (Figs. 7A,B). Interestingly, while riboflavin and FMN levels are elevated in the Rtbdn−/−/Prph2R172W, FAD levels in the Rtbdn−/−/Prph2R172W retina remain reduced over time, similar to the Rtbdn−/− (Fig. 7C).
Figure 7.

Retinal flavins are altered in the Rtbdn−/−/Prph2R172W retina. (A–C) HPLC analysis was used to measure flavins (riboflavin [A], FMN [B], FAD [C]) in retinal samples collected from the mice of the indicated genotypes at P30 and P90. Data are presented as pmol/retina and plotted as mean ± SEM). N = 10 to 12 retinas for each genotype. (D) Immunoblot analysis was performed for FLAD1 under reducing conditions in retinal extracts from P90 WT, Rtbdn−/−, Prph2R172Wand Rtbdn-/-/Prph2R172W mice. N = 6 retinas/group. Band density was measured densitometrically on nonsaturated blots, normalized to actin, and plotted as mean ± SEM. (E) ATP was measured in retinas of the indicated genotypes at P30 and are presented in µmol/L/retina. N = 4 to 10 retinas per genotype. Data were plotted as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 by two-way analysis of variance (ANOVA) (A–C) or one-way ANOVA (D, E) with Tukey's post hoc comparison.
Within cells, FAD synthase (FLAD1) catalyzes the adenylation of FMN to form FAD. The increased levels of riboflavin and FMN (Figs. 7A, 7B) without concomitant increases in FAD (Fig. 7C) prompted us to ask whether FLAD1 protein levels were changed in the Rtbdn−/−/Prph2R172W retina. However, retinal FLAD1 levels were not significantly different in any group (Fig. 7D).
Rtbdn−/−/Prph2R172W Retina Harbors Increased ATP Levels
Apoptosis and energy metabolism are tightly coupled processes, and it is well known that deficiencies in energy metabolism can lead to visual dysfunction and neuronal degeneration.31 We previously found that elimination of retbindin led to reduced ATP levels in the retina.4 Therefore, ATP levels were measured in Rtbdn−/−/Prph2R172W retinas. ATP levels were reduced in the Rtbdn−/− as expected but were significantly elevated in the Rtbdn−/−/Prph2R172W(Fig. 7E).
Discussion
Eliminating retbindin in a model of inherited cone-rod dystrophy (R172W) caused exacerbation in degenerative phenotypes such as rod and cone function, rod and cone cell loss, and retinal gliosis. These phenotypes were much more severe in the Rtbdn−/−/Prph2R172W than either the Rtbdn−/− or Prph2R172W alone. Retbindin levels are significantly increased in the R172W model at P30, and these two factors combined suggest that retbindin plays a protective role for both rods and cones (though it is only expressed in rods2) during photoreceptor degeneration. Our results also suggest that retbindin is essential for retinal homeostasis. We hypothesized that one important function of retbindin is to facilitate enrichment of retinal flavins. In the Rtbdn−/−, riboflavin, FAD, and FMN levels are all reduced, consistent with this idea. This idea is also largely supported by data from the Prph2R172W line. Young Prph2R172W retinas have reduced flavin levels, possibly due to increased energy demands either to promote apoptosis or cell survival in a retina facing degenerative insults. However, in this model, retbindin levels are elevated, and by P90, riboflavin and FMN levels are back to WT levels.
Our current data also suggest an additional role for retbindin. Flavins are delivered to the retina via inner retinal blood vessels and the RPE,32,33 and potentially taken into retinal cells through riboflavin organic ion transporters RFVT1-3 (SLC52A1-A3).34,35 In the blood, flavins bind to albumin and immunoglobulins, and inside cells they bind as cofactors to the enzymes of the riboflavin proteome. This protein binding is essential; unbound flavins that are exposed to light (such as in the retina) undergo photoreduction and are converted to reactive species that can initiate lipid peroxidation and cellular toxicity.36–38 Thus it is logical to hypothesize that an additional function of retbindin is to bind flavins in the IPM to prevent this photoreduction-associated toxicity. Support for this hypothesis comes from our previous in vitro experiments showing that retbindin was capable of preventing light-induced riboflavin-associated cell death.39 This function may also explain why degeneration in the Prph2R172W was so exacerbated by removal of retbindin; indeed elevated flavin levels coupled with a lack of flavin binding proteins (i.e., retbindin) may translate to increased lipid peroxidation and oxidative stress-induced cellular toxicity.
However, analysis of flavin levels in the Rtbdn−/−/Prph2R172W adds a layer of complication. In spite of the absence of retbindin, both riboflavin and FMN are up at P90 in the Rtbdn−/−/Prph2R172W compared to the Rtbdn−/−. Why should introducing the Prph2 R172W mutation “rescue” the Rtbdn−/− phenotype of reduced flavin levels? One hypothesis is that the exacerbated retinal degeneration in the Rtbdn−/−/Prph2R172W might lead to increased flavin levels compared to the Rtbdn−/−. Energy is required during apoptotic cell death,40 which is occurring in Prph2-associated retinal degeneration, and flavins play a role in cellular energy generation.41 Yet this hypothesis is inconsistent with our previous findings. At P30, the rd1 and rd10 models both show decreases in flavin levels,11 and the amount degeneration we see at P90 in the Rtbdn−/−/Prph2R172W(at which point flavin levels are increased) is in between that seen at P30 in the rd1 and rd10.
Another interesting finding is that although FMN and riboflavin levels are elevated in the Rtbdn−/−/Prph2R172W and Prph2R172W compared to Rtbdn−/−, FAD levels remain reduced. This raises the question of whether FMN, specifically, is in high demand during the photoreceptor degeneration seen in the Prph2R172W. The majority of the enzymes that use flavins utilize FAD rather than FMN (84% vs. 16%35,42). However, FMN-dependent enzymes are involved in a wide variety of cellular processes, including energy generation in the electron transport chain; synthesis of methionine, coenzyme A, and tRNAs; and pyrimidine breakdown, among other functions. Further investigation will be needed to understand whether increased FMN levels during retinal degeneration are tied to increased demand for FMN as a cellular cofactor.
Support for the idea that there is increased demand for flavins as cofactors in energy generation in this degenerative model comes from our observation that there are elevated levels of total ATP in Rtbdn−/−/Prph2R172W retinas. Apoptosis is a highly regulated process which requires ATP at many different steps, and there are mixed findings regarding levels of ATP in tissues undergoing apoptosis.43–45 However, it has been shown that apoptotic signals can lead to necrotic cell death if there is insufficient ATP.46–48 Thus the increased ATP levels in the Rtbdn−/−/Prph2R172W retinas may reflect a mechanism to protect surrounding tissues,49 that is, by ensuring that apoptosis can be completed rather than defaulting to necrosis. Future studies may more precisely evaluate cell death mechanisms ongoing in these models.
One interesting observation is that the absence of retbindin in the Prph2R172W retina resulted in vascular pathologies such as the appearance of neovascular tufts. However, we did not observe vascular pathology at early ages, at timepoints when retinal function and photoreceptor structure were already significantly affected. This suggests that the vascular pathology is a secondary effect to retinal degeneration in the Rtbdn−/−/Prph2R172W retina. Early photoreceptor dysfunction in rats is predictive of ensuing neurovascularization,50 and other studies demonstrated that photoreceptor energy demand controls the vascular supply and drive vessel growth.51–53 Given the role of flavin-binding enzymes in cellular metabolism and the potential role of retbindin in flavin processing in the retina, it is logical to hypothesize that retinal metabolism is altered in Rtbdn−/− retinas during degeneration, thus accelerating associated vascular pathologies.
It has become increasingly apparent that metabolism and cellular bioenergetics are essential to cellular homeostasis and that aberrations in these processes play a major role in a widening circle of pathologies. As a result, understanding the mechanisms by which retinal nutrient balance is generated and maintained is absolutely essential. Our findings here support the importance of a clearer understanding of the mechanisms underlying retinal homeostasis: by removing retbindin and altering flavins in the retina, degeneration is accelerated in a model of inherited retinal disease. This study indicates that retbindin is essential for retinal health and suggests it may play a protective role during retinal degeneration. Our findings also raise a large number of questions about the role of flavins in retinal homeostasis and disease, about the role of retbindin as a flavin binding protein in the retina, and about potential other roles for retbindin. They also highlight unique tissue-specific biology. While flavins are essential cofactors in all cells, the unique, light-rich tissue microenvironment of the retina coupled with the high metabolic rate create a tissue with unique demands for flavin and flavin binding proteins. The study of retbindin is quite new, and there is much we still do not know about its function in the retina. This study advances our understanding of the role of retbindin in models of retinal pathology, and we look forward to future mechanistic studies to help further our understanding of its function. Our ultimate goal is to identify therapeutic targets related to retbindin that will help promote cellular homeostasis in a variety of disease states. With the current availability of tools for precisely measuring mitochondrial energetics and assessing metabolome profiles, coupled with these unique animal models, we have an unparalleled opportunity to explore retinal homeostasis at the most fundamental cellular level.
Acknowledgments
The authors thank Barb Nagel for her technical assistance.
Supported by grants from the National Institutes of Health (R01 EY026499-MIN, MRA, R01 EY10609-MIN, P20 GM125528-SMC).
Disclosure: A.M. Genc, None; M.S. Makia, None; T. Sinha, None; S.M. Conley, None; M.R. Al-Ubaidi, None; M.I. Naash, None
References
- 1. Wistow G, Bernstein SL, Wyatt MK, et al.. Expressed sequence tag analysis of human retina for the NEIBank Project: retbindin, an abundant, novel retinal cDNA and alternative splicing of other retina-preferred gene transcripts. Mol Vis. 2002; 8: 196–204. [PubMed] [Google Scholar]
- 2. Kelley RA, Al-Ubaidi MR, Naash MI. Retbindin is an extracellular riboflavin-binding protein found at the photoreceptor/retinal pigment epithelium interface. J Biol Chem. 2015; 290: 5041–5052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rohlich P. The interphotoreceptor matrix: electron microscopic and histochemical observations on the vertebrate retina. Exp Eye Res. 1970; 10: 80–86. [DOI] [PubMed] [Google Scholar]
- 4. Kelley RA, Al-Ubaidi MR, Sinha T, et al.. Ablation of the riboflavin-binding protein retbindin reduces flavin levels and leads to progressive and dose-dependent degeneration of rods and cones. J Biol Chem. 2017; 292: 21023–21034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jorns MS, Wang B, Jordan SP. DNA repair catalyzed by Escherichia coli DNA photolyase containing only reduced flavin: elimination of the enzyme's second chromophore by reduction with sodium borohydride. Biochemistry. 1987; 26: 6810–6816. [DOI] [PubMed] [Google Scholar]
- 6. Susin SA, Lorenzo HK, Zamzami N, et al.. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999; 397: 441–446. [DOI] [PubMed] [Google Scholar]
- 7. Entsch B, van Berkel WJ. Structure and mechanism of para-hydroxybenzoate hydroxylase. FASEB J. 1995; 9: 476–483. [DOI] [PubMed] [Google Scholar]
- 8. Kim HJ, Winge DR.. Emerging concepts in the flavinylation of succinate dehydrogenase. Biochim Biophys Acta. 2013; 1827: 627–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Thorpe C, Matthews RG, Williams CH Jr. Acyl-coenzyme A dehydrogenase from pig kidney. Purification and properties. Biochemistry. 1979; 18: 331–337. [DOI] [PubMed] [Google Scholar]
- 10. Batey DW, Eckhert CD.. Analysis of flavins in ocular tissues of the rabbit. Invest Ophthalmol Vis Sci. 1991; 32: 1981–1985. [PubMed] [Google Scholar]
- 11. Sinha T, Makia M, Du J, Naash MI, Al-Ubaidi MR. Flavin homeostasis in the mouse retina during aging and degeneration. J Nutr Biochem. 2018; 62: 123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ding XQ, Nour M, Ritter LM, Goldberg AF, Fliesler SJ, Naash MI. The R172W mutation in peripherin/rds causes a cone-rod dystrophy in transgenic mice. Hum Mol Genet. 2004; 13: 2075–2087. [DOI] [PubMed] [Google Scholar]
- 13. Jansen HG, Sanyal S.. Development and degeneration of retina in rds mutant mice: electron microscopy. J Comp Neurol. 1984; 224: 71–84. [DOI] [PubMed] [Google Scholar]
- 14. Reuter JH, Sanyal S.. Development and degeneration of retina in rds mutant mice: the electroretinogram. Neurosci Lett. 1984; 48: 231–237. [DOI] [PubMed] [Google Scholar]
- 15. Conley SM, Stuck MW, Burnett JL, et al.. Insights into the mechanisms of macular degeneration associated with the R172W mutation in RDS. Hum Mol Genet. 2014; 23: 3102–3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ekstrom U, Andreasson S, Ponjavic V, et al.. A Swedish family with a mutation in the peripherin/RDS gene (Arg-172-Trp) associated with a progressive retinal degeneration. Ophthalmic Genet. 1998; 19: 149–156. [DOI] [PubMed] [Google Scholar]
- 17. Michaelides M, Holder GE, Bradshaw K, Hunt DM, Moore AT. Cone-rod dystrophy, intrafamilial variability, and incomplete penetrance associated with the R172W mutation in the peripherin/RDS gene. Ophthalmology. 2005; 112: 1592–1598. [DOI] [PubMed] [Google Scholar]
- 18. Wells J, Wroblewski J, Keen J, et al.. Mutations in the human retinal degeneration slow (RDS) gene can cause either retinitis pigmentosa or macular dystrophy. Nat Genet. 1993; 3: 213–218. [DOI] [PubMed] [Google Scholar]
- 19. Winkler BS. The electroretinogram of the isolated rat retina. Vision Res. 1972; 12: 1183–1198. [DOI] [PubMed] [Google Scholar]
- 20. Cheng T, Peachey NS, Li S, Goto Y, Cao Y, Naash MI. The effect of peripherin/rds haploinsufficiency on rod and cone photoreceptors. J Neurosci. 1997; 17: 8118–8128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Conley SM, Stuck MW, Watson JN, Naash MI. Rom1 converts Y141C-Prph2-associated pattern dystrophy to retinitis pigmentosa. Hum Mol Genet. 2017; 26: 509–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Stricker HM, Ding XQ, Quiambao A, Fliesler SJ, Naash MI. The Cys214→Ser mutation in peripherin/rds causes a loss-of-function phenotype in transgenic mice. Biochem J. 2005; 388: 605–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Koirala A, Makkia RS, Cooper MJ, Naash MI. Nanoparticle-mediated gene transfer specific to retinal pigment epithelial cells. Biomaterials. 2011; 32: 9483–9493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lieth E, Barber AJ, Xu B, et al.. Glial reactivity and impaired glutamate metabolism in short-term experimental diabetic retinopathy. Penn State Retina Research Group. Diabetes. 1998; 47: 815–820. [DOI] [PubMed] [Google Scholar]
- 25. Hernandez MR, Miao H, Lukas T. Astrocytes in glaucomatous optic neuropathy. Prog Brain Res. 2008; 173: 353–373. [DOI] [PubMed] [Google Scholar]
- 26. Gallego BI, Salazar JJ, de Hoz R, et al.. IOP induces upregulation of GFAP and MHC-II and microglia reactivity in mice retina contralateral to experimental glaucoma. J Neuroinflammation. 2012; 9: 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Penn JS, Madan A, Caldwell RB, Bartoli M, Caldwell RW, Hartnett ME. Vascular endothelial growth factor in eye disease. Prog Retin Eye Res. 2008; 27: 331–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Venkataswamy G. Ocular manifestations of vitamin B-complex deficiency. Br J Ophthalmol. 1967; 51: 749–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wong-Riley MT. Energy metabolism of the visual system. Eye Brain. 2010; 2: 99–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McCormick DB. Two interconnected B vitamins: riboflavin and pyridoxine. Physiol Rev. 1989; 69: 1170–1198. [DOI] [PubMed] [Google Scholar]
- 31. Calaza KC, Kam JH, Hogg C, Jeffery G. Mitochondrial decline precedes phenotype development in the complement factor H mouse model of retinal degeneration but can be corrected by near infrared light. Neurobiol Aging. 2015; 36: 2869–2876. [DOI] [PubMed] [Google Scholar]
- 32. Kubo Y, Yahata S, Miki S, Akanuma SI, Hosoya KI. Blood-to-retina transport of riboflavin via RFVTs at the inner blood-retinal barrier. Drug Metab Pharmacokinet. 2017; 32: 92–99. [DOI] [PubMed] [Google Scholar]
- 33. Kubo Y, Miki S, Akanuma SI, Hosoya KI. Riboflavin transport mediated by riboflavin transporters (RFVTs/SLC52A) at the rat outer blood-retinal barrier. Drug Metab Pharmacokinet. 2019; 34: 380–386. [DOI] [PubMed] [Google Scholar]
- 34. Yonezawa A, Inui K. Novel riboflavin transporter family RFVT/SLC52: identification, nomenclature, functional characterization and genetic diseases of RFVT/SLC52. Mol Aspects Med. 2013; 34: 693–701. [DOI] [PubMed] [Google Scholar]
- 35. Balasubramaniam S, Christodoulou J, Rahman S. Disorders of riboflavin metabolism. J Inherit Metab Dis. 2019; 42: 608–619. [DOI] [PubMed] [Google Scholar]
- 36. Huvaere K, Cardoso DR, Homem-de-Mello P, Westermann S, Skibsted LH. Light-induced oxidation of unsaturated lipids as sensitized by flavins. J Phys Chem B. 2010; 114: 5583–5593. [DOI] [PubMed] [Google Scholar]
- 37. Song PS, Metzler DE.. Photochemical degradation of flavins. IV. Studies of the anaerobic photolysis of riboflavin. Photochem Photobiol. 1967; 6: 691–709. [DOI] [PubMed] [Google Scholar]
- 38. Treadwell GE, Cairns WL, Metzler DE. Photochemical degradation of flavins. V. Chromatographic studies of the products of photolysis of riboflavin. J Chromatogr. 1968; 35: 376–388. [DOI] [PubMed] [Google Scholar]
- 39. Kelley RA, Al-Ubaidi MR, Naash MI. Retbindin Is Capable of Protecting Photoreceptors from Flavin-Sensitized Light-Mediated Cell Death In Vitro. Adv Exp Med Biol. 2018; 1074: 485–490. [DOI] [PubMed] [Google Scholar]
- 40. Kushnareva Y, Newmeyer DD. Bioenergetics and cell death. Ann N Y Acad Sci. 2010; 1201: 50–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mansoorabadi SO, Thibodeaux CJ, Liu HW. The diverse roles of flavin coenzymes–nature's most versatile thespians. J Org Chem. 2007; 72: 6329–6342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lienhart WD, Gudipati V, Macheroux P. The human flavoproteome. Arch Biochem Biophys. 2013; 535: 150–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nomura K, Imai H, Koumura T, Arai M, Nakagawa Y. Mitochondrial phospholipid hydroperoxide glutathione peroxidase suppresses apoptosis mediated by a mitochondrial death pathway. J Biol Chem. 1999; 274: 29294–29302. [DOI] [PubMed] [Google Scholar]
- 44. Zamaraeva MV, Sabirov RZ, Maeno E, Ando-Akatsuka Y, Bessonova SV, Okada Y. Cells die with increased cytosolic ATP during apoptosis: a bioluminescence study with intracellular luciferase. Cell Death Differ. 2005; 12: 1390–1397. [DOI] [PubMed] [Google Scholar]
- 45. Perez de Lara MJ, Guzman-Aranguez A, de la Villa P, Diaz-Hernandez JI, Miras-Portugal MT, Pintor J. Increased levels of extracellular ATP in glaucomatous retinas: possible role of the vesicular nucleotide transporter during the development of the pathology. Mol Vis. 2015; 21: 1060–1070. [PMC free article] [PubMed] [Google Scholar]
- 46. Skulachev VP. Bioenergetic aspects of apoptosis, necrosis and mitoptosis. Apoptosis. 2006; 11: 473–485. [DOI] [PubMed] [Google Scholar]
- 47. Nicotera P, Melino G.. Regulation of the apoptosis-necrosis switch. Oncogene. 2004; 23: 2757–2765. [DOI] [PubMed] [Google Scholar]
- 48. Ankarcrona M, Dypbukt JM, Bonfoco E, et al.. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron. 1995; 15: 961–973. [DOI] [PubMed] [Google Scholar]
- 49. Perez de Lara MJ, Pintor J. Presence and release of ATP from the retina in an Alzheimer's disease model. J Alzheimers Dis. 2015; 43: 177–181. [DOI] [PubMed] [Google Scholar]
- 50. Akula JD, Hansen RM, Tzekov R, et al.. Visual cycle modulation in neurovascular retinopathy. Exp Eye Res. 2010; 91: 153–161. [DOI] [PubMed] [Google Scholar]
- 51. Fu Z, Lofqvist CA, Liegl R, et al.. Photoreceptor glucose metabolism determines normal retinal vascular growth. EMBO Mol Med. 2018; 10: 76–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sapieha P. Eyeing central neurons in vascular growth and reparative angiogenesis. Blood. 2012; 120: 2182–2194. [DOI] [PubMed] [Google Scholar]
- 53. Joyal JS, Gantner ML, Smith LEH. Retinal energy demands control vascular supply of the retina in development and disease: The role of neuronal lipid and glucose metabolism. Prog Retin Eye Res. 2018; 64: 131–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kelley RA, Al-Ubaidi MR, Naash MI. The potential role of flavins and retbindin in retinal function and homeostasis. Adv Exp Med Biol. 2016; 854: 643–648. [DOI] [PubMed] [Google Scholar]
- 55. Redmond TM, Wiggert B, Robey FA, et al.. Isolation and characterization of monkey interphotoreceptor retinoid-binding protein, a unique extracellular matrix component of the retina. Biochemistry. 1985; 24: 787–793. [DOI] [PubMed] [Google Scholar]