

Abstract
Purpose
Diabetic retinopathy (DR) is a leading cause of visual impairment. Müller cells in DR are dysfunctional due to downregulation of the inwardly rectifying potassium channel Kir4.1. Metformin, a commonly used oral antidiabetic drug, is known to elicit its action through 5′ adenosine monophosphate-activated protein kinase (AMPK), a cellular metabolic regulator; however, its effect on Kir4.1 channels is unknown. For this study, we hypothesized that metformin treatment would correct circadian rhythm disruption and Kir4.1 channel dysfunction in db/db mice.
Methods
Metformin was given orally to db/db mice. Wheel-running activity, retinal levels of Kir4.1, and AMPK phosphorylation were determined at study termination. In parallel, rat retinal Müller cell line (rMC-1) cells were treated using metformin and 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) to assess the effect of AMPK activation on the Kir4.1 channel.
Results
The wheel-running activity of the db/db mice was improved following the metformin treatment. The Kir4.1 level in Müller cells was corrected after metformin treatment. Metformin treatment led to an upregulation of clock regulatory genes such as melanopsin (Opn4) and aralkylamine N-acetyltransferase (Aanat). In rMC-1 cells, AMPK activation via AICAR and metformin resulted in increased Kir4.1 and intermediate core clock component Bmal-1 protein expression. The silencing of Prkaa1 (gene for AMPKα1) led to decreased Kir4.1 and Bmal-1 protein expression.
Conclusions
Our findings demonstrate that metformin corrects abnormal circadian rhythm and Kir4.1 channels in db/db mouse a model of type 2 diabetes. Metformin could represent a critical pharmacological agent for preventing Müller cell dysfunction observed in human DR.
Keywords: Müller cell dysfunction, circadian rhythm, AMPK
Diabetes is a progressive metabolic disorder, advanced by complex interactions among various environmental and genetic influences. Diabetic retinopathy (DR) is observed in one-third of the diabetic population1 and is a major complication of diabetes, affecting visual function and acuity.2 DR continues to be one of the major causes of blindness, contributing to 4.8% of the visually impaired population worldwide.3,4 The multifaceted and multifactorial trait of DR is a major hurdle in developing treatments for initial stages of DR. The progression of DR is gradually gaining appreciation as a disease of the neurovascular unit, and several studies propose that diabetes-induced variations in retinal neurons and glia lead to early clinically manifested vascular injury.5
Müller cells are the principal glial cells of the retina, spanning and enclosing the depth of retinal neurons.6 Müller cells have an array of ion channels, ligand receptors, and transmembrane transporter molecules.7 Specialized inwardly rectifying potassium (Kir) channels regulate potassium conductance in Müller cells. Kir4.1, a weak rectifying potassium channel, is expressed predominately in Müller cells and contributes to both inward and outward potassium currents.8,9 Kir4.1 manifests in a polarized arrangement in Müller cells, apparent at proximal endfoot regions, subretinal spaces, and perivascular regions.6 Müller cells, in part, maintain retinal osmoregulation, and it has been suggested that diabetes dampens retinal Kir4.1 expression and causes Müller cell swelling.6,7 Thus, any alteration in Kir4.1 activity greatly disturbs the functionality of Müller cells.
Müller cells have an internal signaling pathway that prevents osmotic swelling, and an osmotic imbalance results in the release of adenosine triphosphate (ATP) by Müller cells.10,11 This change is sensed by 5′ adenosine monophosphate-activated protein kinase (AMPK), which is activated through an increase in AMP/ATP. AMPK represents a critical cellular metabolic sensor that balances fuel metabolism in response to energy demand and supply.12 AMPK is known to be activated by various conditions such as hypoxia, ischemia, and glucose reduction.13
Circadian clocks synchronize with daily light/dark cycles, causing gene regulation rhythmicity. In mammals, the suprachiasmatic nucleus (SCN) is the central clock regulated by light; it, in turn, synchronizes individual peripheral clocks present in organs.14 Previous studies have demonstrated that the retina has an independent circadian clock mediated by photopigments, melanopsin, and dopamine.15 The retinal clock regulates photoreceptor survival16 and ganglion cell viability during aging,17 in addition to being critical for retinal functions, such as phagocytosis, disk shedding, and corneal thickness maintenance. AMPK, at the core of the nutrient response, is known to phosphorylate and destabilize core clock genes and alter circadian rhythm.18 Kir4.1 in Müller cells exhibits a diurnal rhythm, and disruption of that rhythm dampens Kir4.1 expression, causing Müller cell dysfunction.19 However, it remains unknown how the circadian clock influences AMPK and Kir4.1 function.
The purpose of this study was to determine whether AMPK activation by metformin, a common antidiabetic drug, protects against Kir4.1 channel loss in Müller cells. Using an animal model of type 2 diabetes (i.e., db/db mice), we assessed wheel-running activity and studied the impact of metformin on Kir4.1 expression in retinal Müller cells. This animal model allowed us to investigate diabetes, circadian rhythms, and retinal function under controlled conditions. We observed that metformin treatment of diabetic mice improved their wheel-running activity and corrected Kir4.1 levels in retinal Müller cells. Our study further reveals the critical role of the clock gene Bmal-1 in modulating AMPK signal and regulating Kir4.1 channels is Müller cells.
Methods
Animals
The animal studies were performed according to the National Institutes of Health Guiding Principles for the Care and Use of Animals and the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. The protocol was approved by the Institutional Animal Care and Use Committee (IACUC) at Indiana University. Male, 10- to 12-week-old BKS db (Leprdb/db) transgenic mice with a C57BLKS/J genetic background and the control BKS db/m (Leprdb/m) mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). The mice were housed in pairs (for db/db mice, due to their weight) or four animals per cage (for db/m mice) on a cob bedding. The bedding of the diabetic mice was changed frequently due to polyuria. The mice were maintained at normal room temperature under a 12-hour light/12-hour dark cycle in the animal care facilities of Indiana University. The welfare of the animals as regularly assessed by the veterinary staff of the Laboratory Animal Resource Center of Indiana University.
Locomotor Activity in Circadian Cabinets
The wheel-running activity was determined in circadian cabinets (Actimetrics, Wilmette, IL, USA). These cabinets are light tight, filtered, and fully ventilated. The built-in connectors were adapted to record temperature, humidity, light levels, and locomotor activity. The Leprdb/db mice were randomly divided into two groups. One group of mice received metformin (164 mg/kg) through their drinking water for 14 days; the metformin was dissolved at a concentration of 0.5 mg/ml. The db/db mice, on average, drank ∼15 ml of water (15.2 ± 3.3 ml), and their average weight was 46.08 ± 7.8 g; therefore, the daily dose of metformin was 164 ± 40 mg/kg/day. The other groups received plain water. The Leprdb/m mice served as controls, and their wheel-running activity was recorded. Actimetrics ClockLab software was used to analyze the diurnal rhythm.
Euthanasia of Mice
Per the IACUC protocol, the mice were euthanized by CO2 intoxication at the same time in the afternoon, between 1 PM and 2 PM. CO2 was delivered at an acceptable low rate of 10 to 30% volume displacement per minute.
Immunostaining
The retinal tissues were fixed onto slides with 4% paraformaldehyde. The retinal sections were blocked in 2% goat serum in staining buffer for 2 hours at room temperature. Primary antibodies—anti-Kir4.1 (1:200) anti-glutamine synthetase 1 (GS-1) (1:200)—were incubated overnight at 4°C. The secondary antibody (Alexa Fluor 488 Goat Anti-Rabbit and Alexa Fluor 555 Goat Anti-Mouse; Thermo Fisher Scientific, Waltham, MA, USA) were incubated for 2 hours. VECTASHIELD Antifade Mounting Medium with DAPI (Vector Laboratories, Burlingame, CA, USA) was added, and a coverslip was placed and sealed with nail varnish. Immunostained retinal sections were then observed under a fluorescence microscope.
Cell Culturing of Rat Retinal Müller Cells
The rat retinal Müller cell line (rMC-1) was generously gifted by Vijay Sarthy, Northwestern University, and Timothy S. Kern, University of California, Irvine. The rMC-1 cells were authenticated in series of tests: (1) short tandem repeat analysis, and (2) contamination of interspecies and mycoplasma (IDEXX BioAnalytics, Columbia, MO, USA). The rMC-1 cells were cultured in low-glucose (1000 mg/L) Dulbecco's modified Eagle's medium containing 10% fetal bovine serum, 1% antibiotic/antimycotic (100×), and 1% l-glutamine. rMC-1 cells were seeded (2.1 × 106 cells per T75 flask) and incubated overnight in media (vide supra) at 37°C to allow attachment to the poly-d-lysine-coated surface. When 70% confluency had been reached the following day, the cells attached to the T75 flask were detached using trypsin-EDTA (0.05%) and further cultured (vide supra) into T75 flasks at a splitting ratio of 1:3. Each T75 flask was then transferred into a six-well plate (0.3 × 106 cells/ well) and incubated overnight for further in vitro studies.
Prkaa1 siRNA
The rMC-1 cells were transfected with 5-nM Prkaa1 siRNA or negative control silencer control siRNA (Ambion; Life Technologies, Carlsbad, CA, USA) using Invitrogen Lipofectamine RNAiMax Transfection Reagent (Thermo Fisher Scientific) according to the manufacturer's instructions, and the cells were cultured for a further 24 hours.
AICAR and Metformin Treatment
The rMC-1 cells were treated with 500-µM 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) or 1-mM metformin for 5, 15, and 30 minutes.
Protein Analysis via Western Blotting
Cells were lysed in 50 µl RIPA buffer (Thermo Fisher Scientific) as per the manufacturer's protocol. Protein concentrations were estimated using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific) and was read using a Synergy H1 Hybrid Reader (BioTek Instruments, Winooski, VT, USA). Equal amounts of proteins (30 µg) were loaded and separated on 4% to 12% Novex Bis-Tris gel (Life Technologies). Proteins were transferred to a polyvinylidene difluoride membrane, which was blocked with 4% BSA (Sigma-Aldrich, St. Louis, MO, USA) in Tris-buffered saline and 0.1% Tween 20 (TBST; Sigma-Aldrich). Blocked membranes were then incubated overnight at 4°C with primary antibodies: rabbit-anti-Kir4.1 (1:2000), rabbit-anti-Bmal-1 (1:2000), and mouse-anti-α-tubulin (1:5000). Following the overnight incubation, membranes were incubated for 1 hour with secondary antibodies: goat anti-rabbit IgG (H+L) horseradish peroxidase (HRP) (1:2000) and goat anti-mouse IgG (H + L) HRP (1:100) obtained from Thermo Fisher Scientific and Santa Cruz Biotechnology (Dallas TX, USA). Following washing in TBST, Pierce ECL plus western blotting substrate (Thermo Fisher Scientific) was utilized to ensure adequate exposure for detection. The bands were visualized using Typhoon FLA 9500 (GE Healthcare Bio-Sciences, Pittsburgh, PA, USA). The images were then processed using ImageJ (National Institutes of Health, Bethesda, MA, USA). Each experiment was repeated five times (biological replicates), and the western blot intensities were represented in bar charts as integrated optical density.
Statistical Analysis
The data values are expressed as the mean ± SEM of an independent biological data point unless otherwise specified. Following are the number of mice in each group: db/m (n = 12), db/db (n = 9), and db/db + metformin (n = 8). The statistical significance was considered to be P < 0.05. Results were compared using unpaired t-tests for two groups or using either the Brown–Forsythe and Welch ANOVA test followed by unpaired t-tests or the Kruskal–Wallis ANOVA followed by a Dunn's test for multiple comparisons, using Prism 8.3.0 (GraphPad Software, La Jolla, CA, USA). The details of the respective statistical tests are provided in the figure legends.
Due to individual behavioral variation, the daily wheel-running activity for each mouse from day 8 to day 14 (after a washout period of 1 week) was used for statistical analysis. The sample size for wheel-running activity, therefore, represents the number of mice in each group × 7 days. The statistical analysis was performed using the Brown–Forsythe and Welch ANOVA test followed by unpaired t-tests with Welch's correction or the Dunnett T3 multiple comparison test.
Results
Metformin Treatment Corrects Wheel-Running Activity in db/db Mice
Overall, the metformin treatment was well tolerated by the mice, who showed no adverse effects. Metformin had no effect on body mass, insulin levels, or glycated hemoglobin (Table 1). The db/db mice are known to exhibit a disturbed circadian rhythm. The assessment of wheel-running activity using circadian rhythm cabinets helps in the measurement of circadian rhythms. Using this tool, we investigated whether or not metformin treatment would correct circadian rhythm dysfunction in db/db mice. The db/m mice exhibited a rhythmic pattern of wheel-running consistent with the nocturnal nature of rodents—that is, an increase in wheel running during the night (lights off) and little to no wheel-running during the day (lights on). The wheel-running of db/db mice was sporadic and showed no consistent pattern. The metformin treatment improved the overall wheel-running, and there was an improvement in the rhythmic pattern of wheel running (Fig. 1A).
Table 1.
Metabolic Parameters
| Parameter, Mean ± SEM | db/m Mice | n | db/db Mice | n | db/db + Met Mice | n |
|---|---|---|---|---|---|---|
| Body mass (g) | 32.61 ± 0.86 | 9 | 51.26 ± 3.71** | 5 | 49.69 ± 3.07*** | 8 |
| Insulin (ng/ml) | 0.77 ± 0.11 | 10 | 5.52 ± 2.48 | 7 | 4.25 ± 1.47 | 11 |
| Glycated hemoglobin (%) | 3.92 ± 0.13 | 9 | 8.13 ± 0.73** | 7 | 7.98 ± 0.65*** | 11 |
Brown–Forsythe and Welch ANOVA followed by unpaired t-test; **P < 0.01, ***P < 0.001 versus db/m.
Figure 1.

Metformin corrects circadian rhythm in db/db mice. (A) Representative actograms show the wheel-running activity of db/m and db/db mice under regular light and dark conditions. (B) Bar chart shows the total wheel-running activity of db/m mice (n = 61), db/db mice (n = 42), and metformin-treated db/db mice (n = 55). ****P < 0.0001 (Brown–Forsythe and Welch ANOVA test followed by unpaired t-test).
Using ClockLab software, we performed a series of quantitative assessments of circadian parameters. Overall, the metformin treatment improved total wheel-running activity, as demonstrated by an increase in the number of revolutions per day for the metformin-treated group (Fig. 1B). When the activity pattern was further subdivided into light and dark phase, there was an improvement in revolutions for the metformin treatment for both phases (Table 2).
Table 2.
Activity Patterns
| Revolutions/Day, Mean ± SEM | db/m Mice | n | db/db Mice | n | db/db + Met Mice | n |
|---|---|---|---|---|---|---|
| Total activity | 21,303 ± 1305 | 61 | 632.1 ± 140*** | 42 | 1322 ± 271# | 55 |
| Light activity | 1207 ± 257 | 61 | 70.21 ± 14*** | 42 | 339.4 ± 61### | 55 |
| Dark activity | 20,096 ± 1273 | 61 | 562.1 ± 130**** | 42 | 1285 ± 230### | 55 |
For db/db compared to db/m, ***P < 0.001, ****P < 0.0001. For db/db compared to db/db + Met, ###P < 0.001, #P < 0.05.
Metformin Treatment Corrects Kir4.1 in db/db Mice
To determine the protective effect of metformin treatment on retinal Kir4.1 level, we stained retinal sections from db/m and db/db mice with Kir4.1 (green) to visualize the prominence of inwardly-rectifying potassium channels and GS-1 (red) for the identification of Müller cells. The isotype control for GS-1 did not exhibit non-specific staining (Supplementary Fig. S1). Kir4.1 staining of the retinal sections of db/m mice near the blood vessels, internal limiting membrane, and outer limiting membrane was consistent with previous reports.7,20 The db/db mice demonstrated a decrease in both Kir4.1 and GS-1 staining; however, there was no apparent change in Müller cell numbers. There was an increase in Kir4.1 staining in the metformin-treated db/db mice, around the blood vessels, outer plexiform layer, and outer limiting membrane (Fig. 2).
Figure 2.

Metformin treatment upregulates Kir4.1 channels in db/db mice. Retinal sections were stained with GS-1 (red) and Kir4.1 (green). The db/db retinas demonstrated an overall decrease in Kir4.1 channels near the blood vessels (white arrows) and outer limiting membrane (small arrow head). The metformin treatment corrected Kir4.1 levels in db/db mice (n = 9) near the blood vessels. Inset shows images at higher magnification. Scale bar: 20 µm. GLC, ganglion cell layer; IPL, inner plexiform layer; INL, inner nuclear layer; OPL, outer plexiform layer; ONL, outer nuclear layer; PR, photoreceptor layer.
Metformin Treatment Corrects the Retinal Clock
In order to gain insight into the involvement of the retinal clock in correcting the overall rhythm of db/db mice, we studied mRNA levels of three key components of the retinal clock: arylalkylamine N-acetyltransferase (AANAT), melanopsin (Opn4), and tyrosine hydroxylase (Th). The db/db mice showed a significant decrease in AANAT (Fig. 3A) and Opn4 (Fig. 3B); however, a decrease observed in Th did not reach statistical significance (Fig. 3C). The metformin treatment significantly corrected the lower levels of AANAT and Opn4, and the levels of Th in db/db mice remained unchanged. (Figs. 3A–3C).
Figure 3.
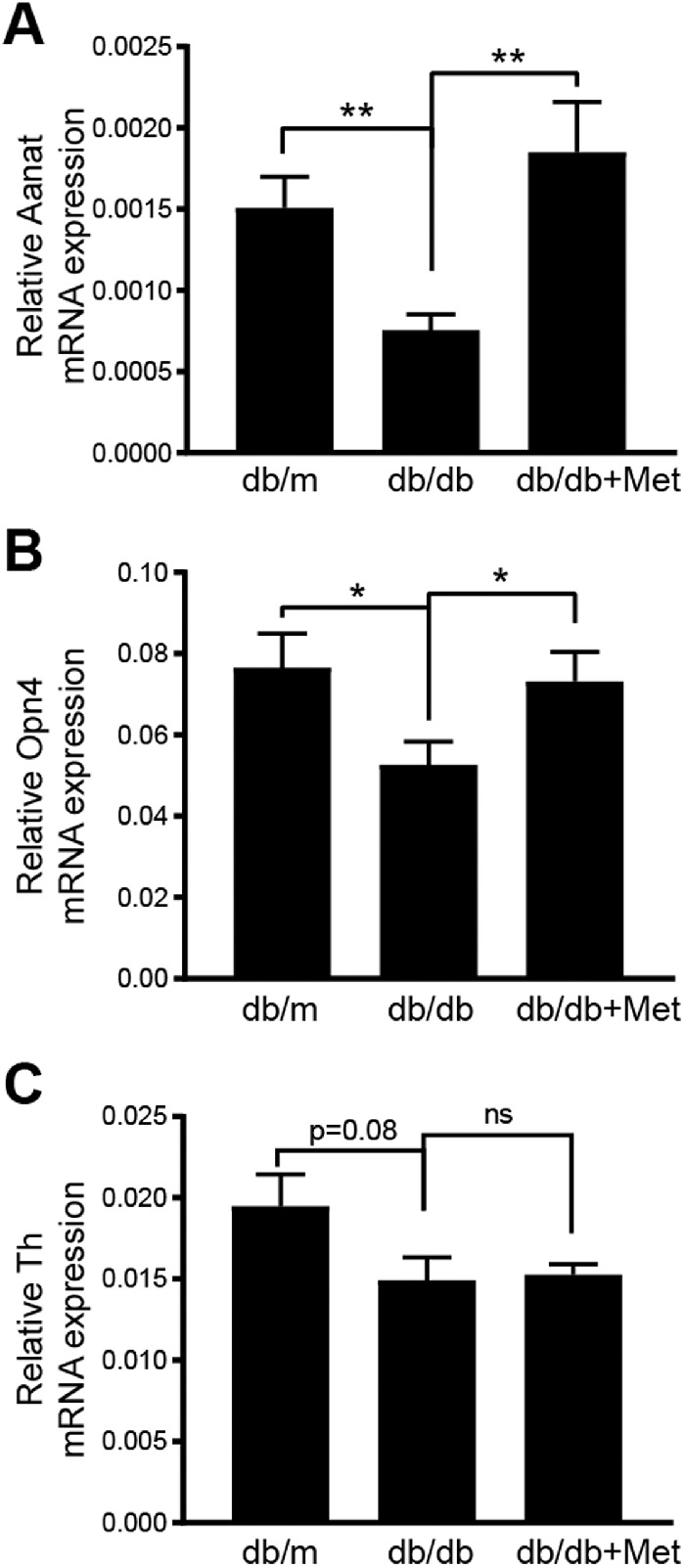
Metformin treatment corrects retinal clock. Bar chart shows mRNA levels of (A) arylalkylamine N-acetyltransferase (AANAT), (B) melanopsin 4 (Opn-4), and (C) tyrosine hydroxylase (Th) after treatment with metformin (db/m mice, n = 12; db/db mice, n = 9; db/db + metformin mice, n = 8). *P < 0.05, **P < 0.01 (Brown–Forsythe and Welch ANOVA test followed by unpaired t-test).
Metformin Treatment Regulates Kir4.1 via AMPK
Metformin indirectly mediates AMPK phosphorylation through inhibition of complex 1 of the mitochondrial respiratory chain, which leads to an increased AMP:ATP ratio.21,22 To test whether metformin has an effect on Müller cells, we treated rMC-1 cells with metformin in vitro. We performed a time–response study of 5 to 30 minutes. We detected a significant increase in Kir4.1 protein expression at 15 minutes (Figs. 4A, 4B). Next, we used a specific activator of AMPK, AICAR, to determine whether the Kir4.1 increase is AMPK dependent. We treated rMC-1 cells with AICAR in vitro and performed a time–response study of 5 to 30 minutes. An increase in Kir4.1 was observed at all time points, but we observed a significant increase in Kir4.1 protein expression at 15 minutes, similar to the effect of metformin (Figs. 4C, 4D) as compared to the untreated control. To further establish the role of AMPK in regulating Kir4.1, we performed a loss-of-function study using siRNA for AMPK (Prkaa1). The AMPK siRNA successfully reduced Prkaa1 mRNA levels (Fig. 4E). We treated the rMC-1 cells with Prkaa1 siRNA and evaluated the protein expression of Kir4.1. Transfection of rMC-1 cells with Prkaa1 siRNA substantially decreased Kir4.1 protein levels (Figs. 4F, 4G)
Figure 4.
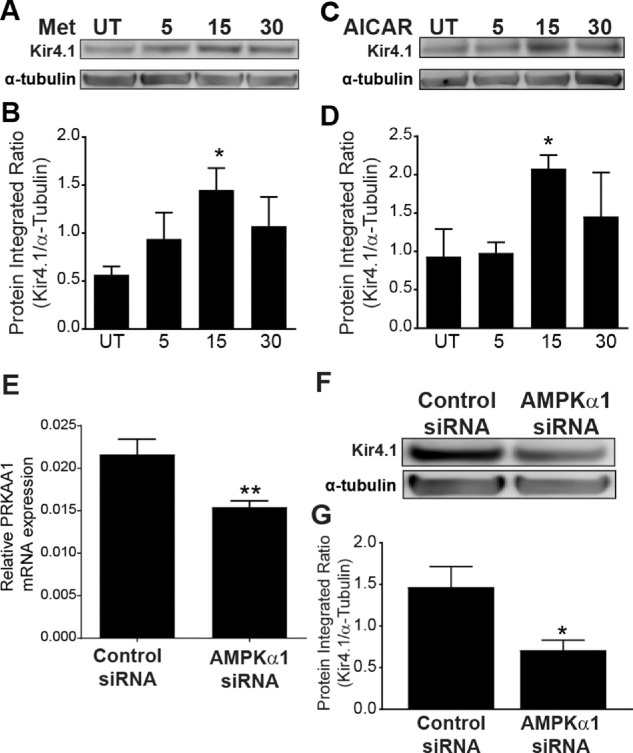
AMPK regulates Kir4.1 in rat Müller cells. The cultured rMC-1 cells were treated with metformin, AICAR, or vehicle (UT, untreated) at different time intervals. The western blots show an increase in Kir4.1 levels in rMC-1 cells treated with (A) metformin and (C) AICAR. The bar charts show integrated optical density data for (B) metformin and (D) AICAR treatment (n = 5; *P < 0.05). The rMC-1 cells were transfected with AMPKα1 (Prkaa1) siRNA or a scrambled control siRNA. (E) Bar chart shows mRNA levels of Prkaa1 after siRNA silencing (n = 6; **P < 0.01, unpaired t-test). A representative western blot (F) and its respective bar chart (G) show a decrease in Kir4.1 protein expression when AMPK (Prkaa1) is silenced (n = 5; *P < 0.05, Kruskal–Wallis ANOVA followed by Dunn's test).
Clock Gene Bmal-1 Is a Mediator of Kir4.1
To understand the relevance of circadian rhythm to AMPK and Kir4.1, we sought to study the circadian clock gene, Bmal-1. We have previously reported that the expression of Kir4.1 in Müller cells is under clock regulation through the Bmal-1 gene.19 To test whether AMPK activation influences Bmal-1 levels, we treated rMC-1 cells with metformin and AICAR, and the protein expression of Bmal-1 was evaluated. With the metformin treatment (Figs. 5A, 5B), there was a significant increase in Bmal-1 at 15 minutes, and for the AICAR treatment (Figs. 5C, 5D) there was a significant increase in Bmal-1 at all time points. To further understand the influence of AMPK on Bmal-1, we silenced AMPK in rMC-1 cells with Prkaa1 siRNA, and Bmal-1 protein expression was assessed. Prkaa1 siRNA transfection decreased Bmal-1 protein expression (Figs. 5E, 5F).
Figure 5.

The clock gene Bmal-1 is an important regulator of AMPK in Müller cells. rMC-1 cells were treated with metformin, AICAR, or vehicle (UT, untreated), and the Bmal-1 levels were determined using western blots. The representative images of western blots and bar charts show upregulation of Bmal-1 after treatment with (A, B) metformin (n = 4) or (C, D) AICAR (n = 5; *P < 0.05, Kruskal–Wallis ANOVA followed by Dunn's test). (E) AMPKα1 siRNA-transfected rMC-1 cells show a decrease in Bmal-1. (F) The bar chart shows the quantification of integrated optical density (n = 5).
Metformin Treatment of db/db Mice Upregulates Kir4.1 via AMPK and Bmal-1
Next, we tested the hypothesis that AMPK–Bmal-1 activation has a protective effect on Kir4.1 levels in db/db mice. We analyzed the protein expression of AMPK and Kir4.1, as well as the phosphorylation of AMPK-α1, on threonine 172. The db/db mice showed a decrease in total AMPK, and there was a decrease in phosphorylated AMPK-α1 (Fig. 6A). The protein levels of Bmal-1 (Fig. 6B) and Kir4.1 (Fig. 6C) were decreased in db/db mice, and the metformin treatment corrected the reduced levels of these proteins in db/db mice.
Figure 6.
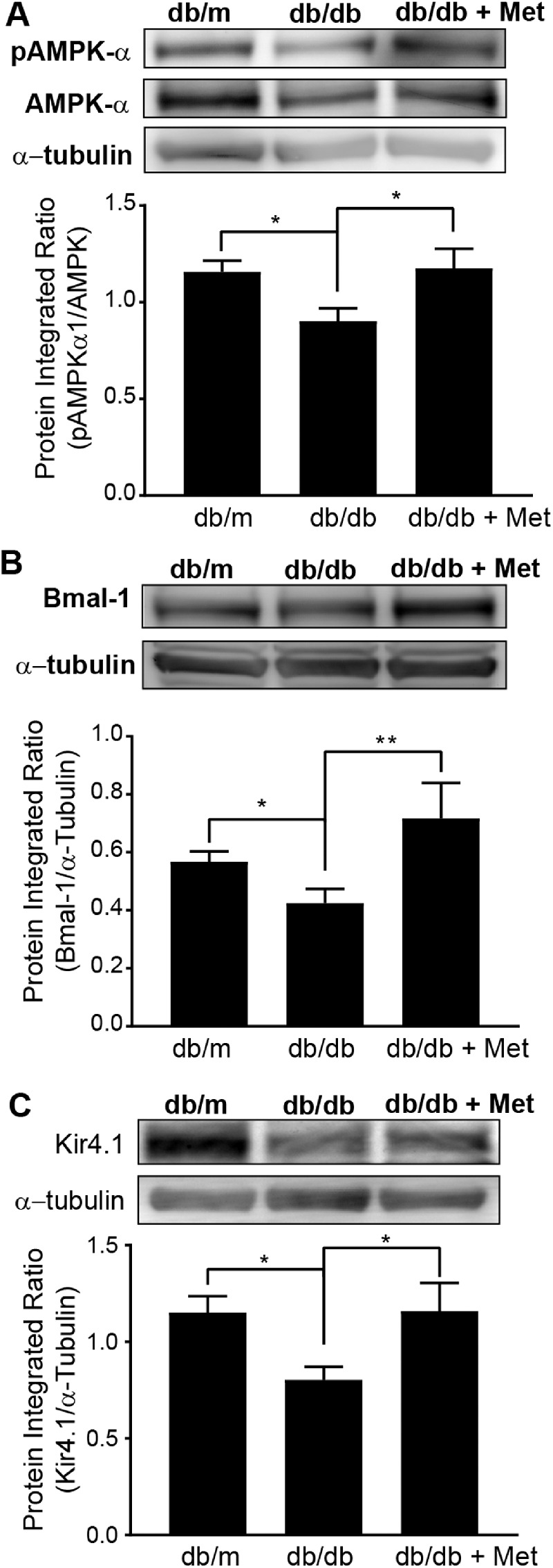
Metformin treatment of db/db mice upregulates Kir4.1 via AMPK. Representative western blots show the protein expression and the respective bar charts show the quantification of the following: (A) PhosphoAMPKt172 and total AMPK (control mice, n = 6; db/db mice, n = 6; db/db + Met mice, n = 5; *P < 0.05, Kruskal–Wallis ANOVA followed by Dunn's test). (B) Bmal-1 (control mice, n = 4; db/db mice, n = 4; db/db + Met mice, n = 4; *P < 0.05, **P < 0.01, Kruskal–Wallis ANOVA followed by Dunn's test). (C) Kir4.1 in metformin-treated db/db mice (control mice, n = 8; db/db mice, n = 8; db/db + Met mice, n = 5; *P < 0.05, Kruskal–Wallis ANOVA followed by Dunn's test).
Discussion
We previously found that DR is associated with dysregulated diurnal rhythm.23 Diabetes causes damage to retinal potassium conductance, resulting in a drastic decrease in Kir4.1 expression.6,7 It has also been found that the induction of diabetes inactivates AMPK in the retina.24 AMPK activity is a crucial element of the central clock in the suprachiasmatic nucleus, as it produces a regulatory circadian rhythm response for the metabolic system and gene expression.25 Aligning with these studies, we observed that metformin treatment of db/db mice corrects the central circadian rhythm, modifying the target retinal clock genes and Kir4.1 channel.
The db/db mice that received metformin exhibited nearly equivalent body weight and glycated hemoglobin and insulin levels at the study termination. We did not see an expected change in glycated hemoglobin after metformin treatment. We believe this may be attributed to the shorter duration of this study; typically, a change in glycated hemoglobin is observed 2 to 3 months after initiation of metformin treatment. It is noteworthy that the changes that we observed in our study cannot be attributed to a systemic decrease in blood glucose but rather are due to the non-glucose-mediated effects of metformin.
Retinal neurodegeneration is recognized to be an initial pathway involved in the pathogenesis of DR, contributing to microcirculatory events that arise as part of DR.26 Müller cells are principal glial cells of the retina and are central, columnar “micro units” of retinal neurons.6 A characteristic of mature Müller cells is their strong potassium conductance, and Kir4.1 is a predominant potassium channel that is involved in correcting potassium concentrations due to its polarized arrangement in Müller cells.8,27 In our studies, metformin treatment improved Kir4.1 levels near the blood vessels and outer limiting membrane; strikingly, Kir4.1 levels on the internal limiting membrane decreased, supporting the possibility of delocalization or altered distribution of Kir4.1 channels reported previously.20
Metformin, a widely prescribed type 2 diabetes drug, promotes skeletal muscle glucose uptake.28,29 Metformin is known to activate AMPK in an indirect manner via inhibition of mitochondrial complex I. AMPK is required for its lipid-lowering and insulin-sensitizing action. Because the interaction between AMPK and Kir4.1 potassium conductance is crucial to osmotic balance, we aimed to observe the interactive roles of AMPK and Kir4.1. Previous studies have associated Müller cell swelling with the induction of osmotic stress. A decrease in potassium conductance is related to the downregulation of both gene and protein expression of Kir4.1.30 AMPK, especially AMPKα1, has been known to be activated under osmotic stress conditions.31 AICAR is a cell-permeable precursor that phosphorylates to imidazole monophosphate, mimicking AMP, which directly acts as an agonist for AMPK activation.28,32 The induction of AICAR confirms the collaborative role of AMPK and Kir4.1. We observed that Kir4.1 protein expression increased when AMPK was activated through its agonist, AICAR, suggesting that AMPK plays a protective role in the pathogenesis of DR.28
We found that the activation of AMPK by metformin resulted in a significant increase in Kir4.1 protein expression, providing evidence for the correlation of AMPK and Kir4.1 in the maintenance of Müller cells. In contrast, Prkaa1 siRNA silences AMPK and reduces Kir4.1 expression, confirming the connection between Kir4.1 and AMPK. In a previous study, mouse embryonic fibroblast cells were cultured in normal glucose and treated with AICAR. The circadian period was lengthened compared to the standard (low) glucose medium.33 It would have been interesting to study similar culturing conditions for rMC-1 cells; however, we were primarily interested in establishing the effect of AMPK activation under standard glucose conditions, so our studies were limited to low-glucose conditions.
Circadian rhythms are synchronized to the 24-hour period of the Earth's rotation, and light serves as the major synchronizing agent.34 Diabetes is linked with circadian rhythm disruption of the central and peripheral circadian clock, and it disrupts the circadian regulation of core clock genes occurring in the retina. The retina is an extremely cyclic tissue, with metabolic and inflammatory demands fluctuating considerably due to the influence of light and dark cycles. Altered synchronization in association with retinal demands in diabetes could attribute to DR pathogenesis.14 In our study, we detected the diminishing effect of Kir4.1 specific to Müller cells in diabetic mice; however, it is known that db/db mice are leptin receptor resistant and are known to possess circadian rhythm dysfunction. Leptin plays a vital role in the signaling properties of the brain that control energy and metabolism.35 Therefore, it is likely that the protective effect of metformin may have a central mechanism involving leptin signaling, which was not investigated here but our study paves the way for future studies in this direction.
The circadian rhythm mechanism is closely regulated to the interlinked expression of specific clock regulatory genes.36 Binding of the CLOCK:Bmal-1 unit to the E-boxes of CRY and PER activates Cry and Per promoters, respectively, causing CRY and PER protein translation during the late hours of the day and early hours of the night.37 CRY and PER complex bind to the CLOCK:Bmal-1 unit, creating a negative feedback loop and inhibiting its own transcription of Cry and Per mRNA. AANAT stability is regulated by cAMP, and its transcription is controlled directly by the circadian clock via the presence of an E-box in the promoter region.38 We previously reported that the Kncj10 promoter gene for Kir4.1 exhibits an E-box, and Bmal may serve as a regulator of Kir4.1 protein expression.19,38 It is noteworthy that, in our studies, metformin treatment upregulated gene expression for melanopsin mRNA, opn4. The melanopsin is expressed by intrinsically photosensitive retinal ganglion cells; these non-image-forming photoreceptor cells communicate directly with the SCN, as well as with other areas of brain cells.39 Although dopamine has been reported to play an important role in regulating the retinal clock, we did not see a beneficial effect of metformin on tyrosine hydroxylase.40 Thus, in our studies, we speculate that metformin treatment produced a physiologically significant effect by increasing Kir4.1 via the AMPK–Bmal pathway and correcting the retinal clock and overall daily rhythm in db/db mice. However, long-term studies are necessary to establish causal relationships with the development of DR.
Despite these limitations, our studies suggest that metformin treatment is protective for the principal inwardly rectifying potassium-conducting channel, Kir4.1, in Müller cells. When AMPK is mediated through AICAR and metformin, Kir4.1 protein expression is increased, with Bmal-1 playing an intermediary role. Our results suggest that AMPK is a crucial therapeutic target in preventing Müller cell dysfunctionality in DR.
Supplementary Material
{kind=link}
Acknowledgments
The authors thank Jeffrey S. Elmendorf, PhD, Department of Cellular and Integrative Physiology, Center for Diabetes and Metabolic Diseases, Indiana University, for thoughtful discussions on this project. The authors also thank Peiyi Zhang and Chong Gu, PhD, at Purdue Statistical Consulting Service, Purdue University, for consultations and assistance concerning data analysis.
Supported by a Pilot and Feasibility award from the Center for Diabetes and Metabolic Diseases, Indiana University, and by a National Institutes of Health National Eye Institute award (EY027779).
Disclosure: A. Alex, None; Q. Luo, None; D. Mathew, None; R. Di, None; A.D. Bhatwadekar, None
References
- 1. Lee R, Wong TY, Sabanayagam C. Epidemiology of diabetic retinopathy, diabetic macular edema and related vision loss. Eye Vis (Lond). 2015; 2: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zheng Y, He M, Congdon N. The worldwide epidemic of diabetic retinopathy. Indian J Ophthalmol. 2012; 60: 428–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fong DS, Sharza M, Chen W, Paschal JF, Ariyasu RG, Lee PP. Vision loss among diabetics in a group model Health Maintenance Organization (HMO). Am J Ophthalmol. 2002; 133: 236–241. [DOI] [PubMed] [Google Scholar]
- 4. Marshall SM, Flyvbjerg A. Prevention and early detection of vascular complications of diabetes. BMJ. 2006; 333: 475–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Antonetti DA, Klein R, Gardner TW. Diabetic retinopathy. N Engl J Med. 2012; 366: 1227–1239. [DOI] [PubMed] [Google Scholar]
- 6. Bringmann A, Pannicke T, Grosche J, et al.. Müller cells in the healthy and diseased retina. Prog Retin Eye Res. 2006; 25: 397–424. [DOI] [PubMed] [Google Scholar]
- 7. Pannicke T, Iandiev I, Wurm A, et al.. Diabetes alters osmotic swelling characteristics and membrane conductance of glial cells in rat retina. Diabetes. 2006; 55: 633–639. [DOI] [PubMed] [Google Scholar]
- 8. Kofuji P, Biedermann B, Siddharthan V, et al.. Kir potassium channel subunit expression in retinal glial cells: implications for spatial potassium buffering. Glia. 2002; 39: 292–303. [DOI] [PubMed] [Google Scholar]
- 9. Takumi T, Ishii T, Horio Y, et al.. A novel ATP-dependent inward rectifier potassium channel expressed predominantly in glial cells. J Biol Chem. 1995; 270: 16339–16346. [DOI] [PubMed] [Google Scholar]
- 10. Wurm A, Lipp S, Pannicke T, et al.. Endogenous purinergic signaling is required for osmotic volume regulation of retinal glial cells. J Neurochem. 2010; 112: 1261–1272. [DOI] [PubMed] [Google Scholar]
- 11. Krügel K, Wurm A, Linnertz R, et al.. Erythropoietin inhibits osmotic swelling of retinal glial cells by Janus kinase-and extracellular signal-regulated kinases1/2-mediated release of vascular endothelial growth factor. Neuroscience. 2010; 165: 1147–1158. [DOI] [PubMed] [Google Scholar]
- 12. Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011; 13: 1016–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ruderman NB, Carling D, Prentki M, Cacicedo JM. AMPK, insulin resistance, and the metabolic syndrome. J Clin Invest. 2013; 123: 2764–2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang Q, Tikhonenko M, Bozack SN, et al.. Changes in the daily rhythm of lipid metabolism in the diabetic retina. PLoS One. 2014; 9: e95028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sakamoto K, Liu C, Kasamatsu M, Pozdeyev NV, Iuvone PM, Tosini G. Dopamine regulates melanopsin mRNA expression in intrinsically photosensitive retinal ganglion cells. Eur J Neurosci. 2005; 22: 3129–3136. [DOI] [PubMed] [Google Scholar]
- 16. Ogilvie JM, Speck JD. Dopamine has a critical role in photoreceptor degeneration in the rd mouse. Neurobiol Dis. 2002; 10: 33–40. [DOI] [PubMed] [Google Scholar]
- 17. Baba K, Pozdeyev N, Mazzoni F, et al.. Melatonin modulates visual function and cell viability in the mouse retina via the MT1 melatonin receptor. Proc Natl Acad Sci USA. 2009; 106: 15043–15048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jordan SD, Lamia KA. AMPK at the crossroads of circadian clocks and metabolism. Mol Cell Endocrinol. 2013; 366: 163–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hassan I, Luo Q, Majumdar S, Dominguez JM, Busik JV, Bhatwadekar AD. Tumor necrosis factor alpha (TNF-α) disrupts Kir4. 1 channel expression resulting in Müller cell dysfunction in the retina. Invest Ophthalmol Vis Sci. 2017; 58: 2473–2482. [DOI] [PubMed] [Google Scholar]
- 20. Curtis T, Hamilton R, Yong P-H, et al.. Müller glial dysfunction during diabetic retinopathy in rats is linked to accumulation of advanced glycation end-products and advanced lipoxidation end-products. Diabetologia. 2011; 54: 690–698. [DOI] [PubMed] [Google Scholar]
- 21. Kim J, Yang G, Kim Y, Kim J, Ha J.. AMPK activators: mechanisms of action and physiological activities. Exp Mol Med. 2016; 48: e224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shaw RJ, Lamia KA, Vasquez D, et al.. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005; 310: 1642–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Busik JV, Tikhonenko M, Bhatwadekar A, et al.. Diabetic retinopathy is associated with bone marrow neuropathy and a depressed peripheral clock. J Exp Med. 2009; 206: 2897–2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang J, Xi X, Widness M, Gao L. Activation of AMP-activated protein kinase blocks hyperglycemia-induced apoptosis in retinal cells. Invest Ophthalmol Vis Sci. 2004; 45: 3230. [Google Scholar]
- 25. Um J-H, Pendergast JS, Springer DA, et al.. AMPK regulates circadian rhythms in a tissue-and isoform-specific manner. PLoS One. 2011; 6: e18450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Villarroel M, Ciudin A, Hernández C, Simó R. Neurodegeneration: an early event of diabetic retinopathy. World J Diabetes. 2010; 1: 57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nagelhus EA, Horio Y, Inanobe A, et al.. Immunogold evidence suggests that coupling of K + siphoning and water transport in rat retinal Müller cells is mediated by a coenrichment of Kir4.1 and AQP4 in specific membrane domains. Glia. 1999; 26: 47–54. [DOI] [PubMed] [Google Scholar]
- 28. Zhou G, Myers R, Li Y, et al.. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001; 108: 1167–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hayashi T, Hirshman MF, Kurth EJ, Winder WW, Goodyear LJ. Evidence for 5′ AMP-activated protein kinase mediation of the effect of muscle contraction on glucose transport. Diabetes. 1998; 47: 1369–1373. [DOI] [PubMed] [Google Scholar]
- 30. Iandiev I, Uckermann O, Pannicke T, et al.. Glial cell reactivity in a porcine model of retinal detachment. Invest Ophthalmol Vis Sci. 2006; 47: 2161–2171. [DOI] [PubMed] [Google Scholar]
- 31. Barnes K, Ingram JC, Porras OH, et al.. Activation of GLUT1 by metabolic and osmotic stress: potential involvement of AMP-activated protein kinase (AMPK). J Cell Sci. 2002; 115: 2433–2442. [DOI] [PubMed] [Google Scholar]
- 32. Hardie DG, Carling D. The AMP‐activated protein kinase: fuel gauge of the mammalian cell? Eur J Biochem. 1997; 246: 259–273. [DOI] [PubMed] [Google Scholar]
- 33. Lamia KA, Sachdeva UM, DiTacchio L, et al.. AMPK regulates the circadian clock by cryptochrome phosphorylation and degradation. Science. 2009; 326: 437–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zee PC, Attarian H, Videnovic A. Circadian rhythm abnormalities. Continuum (Minneap Minn). 2013; 19: 132–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Laposky AD, Bradley MA, Williams DL, Bass J, Turek FW. Sleep-wake regulation is altered in leptin-resistant (db/db) genetically obese and diabetic mice. Am J Physiol Regul Integr Comp Physiol. 2008; 295: R2059–2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Green CB, Takahashi JS, Bass J. The meter of metabolism. Cell. 2008; 134: 728–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ye R, Selby CP, Chiou Y-Y, Ozkan-Dagliyan I, Gaddameedhi S, Sancar A. Dual modes of CLOCK:BMAL1 inhibition mediated by Cryptochrome and Period proteins in the mammalian circadian clock. Genes Dev. 2014; 28: 1989–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tosini G, Chaurasia SS, Michael Iuvone P. Regulation of arylalkylamine N‐acetyltransferase (AANAT) in the retina. Chronobiol Int. 2006; 23: 381–391. [DOI] [PubMed] [Google Scholar]
- 39. Hattar S, Liao H-W, Takao M, Berson DM, Yau K-W. Melanopsin-containing retinal ganglion cells: architecture, projections, and intrinsic photosensitivity. Science. 2002; 295: 1065–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Korshunov KS, Blakemore LJ, Trombley PQ. Dopamine: a modulator of circadian rhythms in the central nervous system. Front Cell Neurosci. 2017; 11: 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.