

Abstract
Coronavirus disease of 2019 (COVID-19), which originated in China in 2019, shows mild cold and pneumonia symptoms that can occasionally worsen and result in deaths. SARS-CoV-2 was reported to be the causative agent of the disease and was identified as being similar to SARS-CoV, a causative agent of SARS in 2003. In this review, we described the phylogeny of SARS-CoV-2, covering various related studies, in particular, focusing on viruses obtained from horseshoe bats and pangolins that belong to Sarbecovirus, a subgenus of Betacoronavirus. We also describe the virological characteristics of SARS-CoV-2 and compare them with other coronaviruses. More than 30,000 genome sequences of SARS-CoV-2 are available in the GISAID database as of May 28, 2020. Using the genome sequence data of closely related viruses, the genomic characteristics and evolution of SARS-CoV-2 were extensively studied. However, given the global prevalence of COVID-19 and the large number of associated deaths, further computational and experimental virological analyses are required to fully characterize SARS-CoV-2.
Keywords: COVID-19, Coronavirus, Comparative genomics, SARS-CoV-2, Viral evolution
Background
On December 12, 2019, an epidemic of acute respiratory syndrome in humans started in the city of Wuhan, Hubei province, central China [1–4]. The causative agent of the symptom was found to be a novel coronavirus (CoV), of which genome is phylogenetically similar to that of the severe acute respiratory syndrome (SARS) CoV (SARS-CoV) [1–4]. Because of that, World Health Organization (WHO) named the symptoms coronavirus disease 19 (COVID-19) [5], and the Coronaviridae Study Group of the International Committee on Taxonomy of Viruses (ICTV) named the novel CoV as SARS-CoV-2 [6]. In this review, we noted characteristics of SARS-CoV-2 compared to those of other CoVs.
Main text
Phylogeny of SARS-CoV-2
SARS-CoV-2 is a member of the coronavirus family (Coronaviridae). The family Coronaviridae is a relatively large family that includes a variety of viral species. The coronavirus family is divided into two subfamilies: Letovirinae and Orthocoronavirinae [7]. SARS-CoV-2 is classified as an orthocoronavirus subfamily member. The orthocoronavirus subfamily is further divided into four genera: Alphacoronavirus, Betacoronavirus, Gammacoronavirus, and Deltacoronavirus [7]. In addition, the genus Betacoronavirus is reported to be divided into five subgenera: Sarbecovirus, Hibecovirus, Nobecovirus, Merbecovirus, and Embecovirus [7, 8].
The maximum likelihood (ML) tree based on amino acid sequences of open reading frame 1ab (ORF1ab) indicated the phylogenetic relationship of various CoVs shown in Fig. 1. The phylogenetic tree was constructed from 61 viruses belonging to the orthocoronavirus subfamily. More than 100 CoVs were isolated from various mammalian and avian species, and the CoVs shown in Fig. 1 are representatives selected by the authors to illustrate diversity of CoVs, of which complete genomes are available in public databases excluding an unclassified coronavirus found in Tropidophorus sinicus (Chinese waterside skink). The Guangdong Chinese water skink CoV was used as an outgroup in Fig. 1, which was the only CoV found in reptiles other than mammals and birds [12]. SARS-CoV-2, along with SARS-CoV and Middle East respiratory syndrome (MERS)-CoV, is classified in the genus Betacoronavirus. SARS-CoV-2 and SARS-CoV belong to the subgenus Sarbecovirus, accompanying various CoVs found in bats, in particular from horseshoe bats (genus Rhinolophus).
Fig. 1.
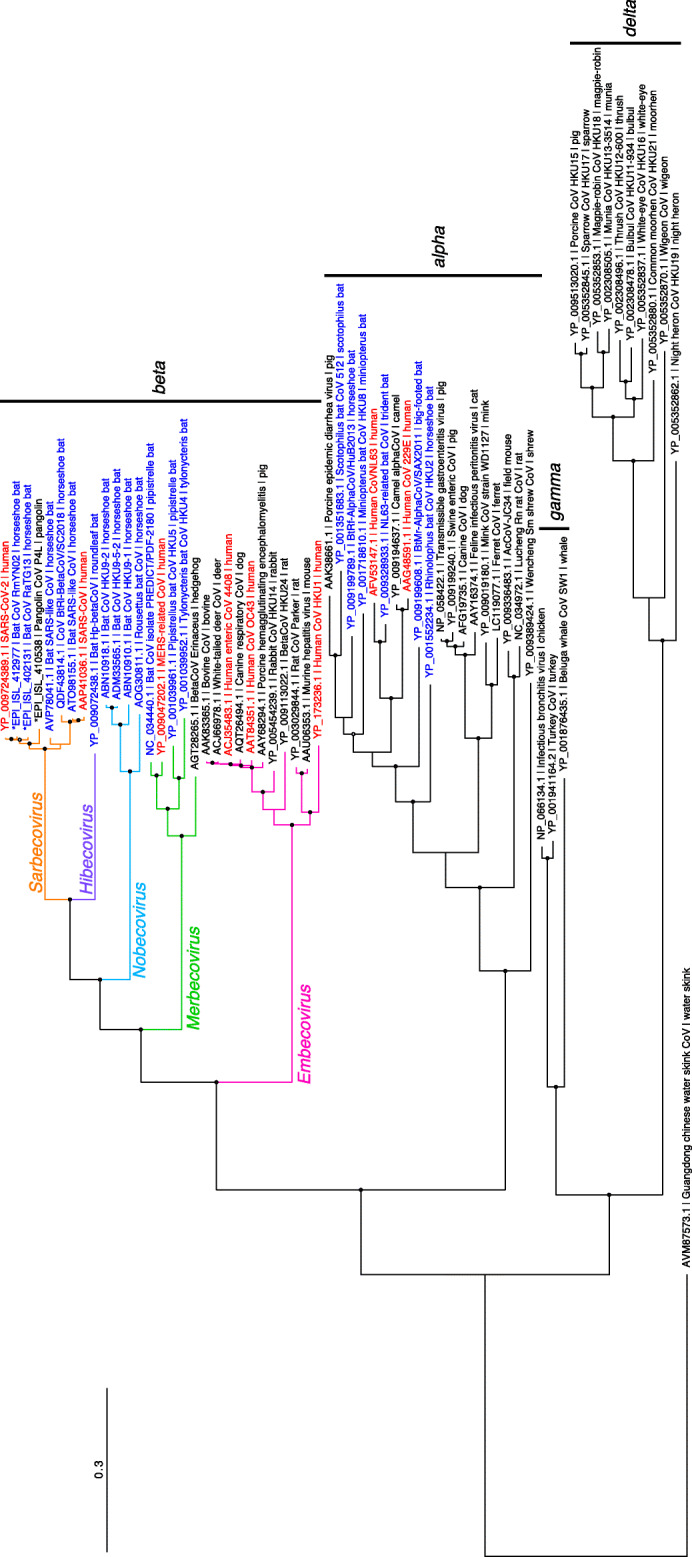
Phylogeny of orthocoronaviruses. Maximum likelihood (ML)-based phylogenetic tree of 61 orthocoronaviruses. Partial amino acid sequences of ORF1ab were used for the analysis. We generated the multiple alignment of the sequences using L-INS-i of MAFFT version 7.453 [9], and the amino acid substitution model LG+I+G was selected using ProtTest3 [10]. Based on the model, we constructed an ML tree using RAxML-NG [11] applying 1000 bootstrapping tests. GenBank or GISAID (that was indicated by asterisk (*)) accession number, strain name, and host of each virus are indicated for each branch terminal. CoVs obtained from humans or bats are shown in red or blue, respectively. A black or open circle corresponds to bootstrap values ≥ 95% or ≥ 80%, respectively. The scale is shown in the upper left
In addition to SARS-CoV-2, SARS-CoV, and MERS-CoV, there are four other CoVs that cause common cold symptoms in humans: human CoV (HCoV) HKU1 and HCoV OC43, belonging to the genus Betacoronavirus, and HCoV 229E and HCoV NL63, belonging to the Alphacoronavirus. Although there are few reported cases, human enteric coronaviruses (HECV) that cause diarrhea in humans belong to the Betacoronavirus genus. Viruses closely related to HCoV HKU1 are present in rodents, and HECV is closely related to CoVs isolated from even-toed animals (bovine and deer). These data indicate that these HCoVs were derived from CoVs of domestic animals and small animals such as rodents. There are multiple types of CoVs in non-human animals, and it is undeniable that coronaviral transmissions from domestic, companion, and wild animals to humans might have occurred many times without people realizing it.
The phylogenetic relationship of SARS-CoV-2 with other closely related CoVs belonging to subgenus Sarbecovirus is illustrated in Fig. 2. Note that entire genomic sequences were used for this phylogenetic analysis. CoVs which are the most closely related to the SARS-CoV-2 are Bat CoVs, in particular strains RmYN02 [14] and RaTG13 [4], both of which are isolated from horseshoe bats (genus Rhinolophus). Further, CoVs found in Malaysian pangolins are the next closest to SARS-CoV-2 as well. These observations are also indicated by Fig. 1, which is based on partial amino acid sequences of the ORF1ab gene. As shown in the Fig. 2, most of the CoVs belonging to subgenus Sarbecovirus were found in horseshoe bats or other bat species. Therefore, although we still do not know the direct origin of SARS-CoV-2, it is highly possible that CoV(s) belonging to Sarbecovirus in horseshoe bats could be the origin of SARS-CoV-2.
Fig. 2.

Phylogeny of CoVs belonging to Sarbecoronavirus. ML-based phylogenetic tree of 41 CoVs belonging to Sarbecoronavirus including SARS-CoV-2. Whole genome sequences were used for the analysis. We generated the multiple alignment of the sequences using L-INS-i of MAFFT version 7.453 [9], and the nucleotide substitution model GTR+I+G was selected using ModelTest-NG [13]. Based on the model, we constructed an ML tree using RAxML-NG [11], applying 1000 bootstrapping tests. Please see Fig. 1 legend for the details of this figure
Phenotypic features and genomic structures of SARS-CoV-2
The phenotypic features of CoVs are as follows. The viral particles are spherical, 100 to 120 nm in diameter, with envelopes derived from the host cell membrane. CoVs were named “coronaviruses” because they are characterized by spike protein projections on the surface of the viral particles (about 20 nm in length), and their shape resembles a crown (corona) under electron microscopy. Those features are embodied in SARS-CoV-2 [1].
The genome structure of CoVs is a non-segmented, positive-sense single-stranded RNA (+ssRNA). The genome size ranges from 27 to 32 kb: a cap structure at the 5′ end followed by a reader sequence of about 70 bases, several ORFs coding various proteins, and a non-translated region including a poly-A sequence at the 3′ end. Figure 3 shows the genomic structure of SARS-CoV-2 (29.9 kb). For the ORFs from the 5′ end, a region of about 20 kb corresponds to the two ORFs (ORF1a and ORF1b). ORF1a and ORF1b encode 11 and 5 non-structural proteins: nsp1 to nsp11 and nsp12 to 16, respectively. ORF1a is translated directly from the genomic RNA; however, expression of ORF1b requires a − 1 ribosomal frameshift near the end of ORF1, resulting in a single ORF1ab polypeptide. Downstream from the ORF1ab, there are ORFs encoding a few to more than ten structural/non-structural proteins. The common structural proteins of CoV subfamily viruses are nucleocapsid (N), spike (S), membrane (M), and envelope (E) proteins. The S protein is responsible for both binding to receptors expressed on the cell membranes of susceptible cells and membrane fusion. The M and E proteins are involved in the assembly and budding of viral particles. CoVs also code various non-structural proteins in ORF1ab as well as in other ORFs, in particular near the 3′ end, although the details of the exact genes in the SARS-CoV-2 genome are still unclear mainly due to overlapping genes encoded in a different coding frame as illustrated in Fig. 3.
Fig. 3.

Genomic structure of SARS-CoV-2. Schematic genomic structure of SARS-CoV-2 was shown based on the SARS-CoV-2 Wuhan-Hu-1 (NCBI Reference Sequence ID: NC_045512.2). The scale was shown on the top. Each ORF was illustrated based on the NCBI annotation of NC_045512.2, and a rectangle filled with black corresponds to a structural protein. The number in parentheses is the length of amino acid sequence (aa, amino acid). A gene name as well as rectangle colored in light blue was a hypothetical ORF which is not annotated NC_045512.2 currently. ORF3b is based on Konno et al. [15], and the others are based on Davidson et al. [16] and Jungreis et al. [17]
The SARS-CoV-2 genome shares nucleotide identity to the genomes of Bat CoV RaTG13 (96%) [4], Bat CoV RmYN02 (93%) [14], Pangolin CoV (90%) [18–20], SARS-CoV (80%) [4], and MERS-CoV belonging to Merbecovirus (50%) [21]. However, the nucleotide identity varied greatly depending on genes as well as genomic loci [4, 14, 18–22]. For example, the receptor-binding domain of S genes of SARS-CoV-2 is very similar to that of Pangolin CoVs, rather than those of Bat CoVs RaTG13 and RmYN02 [14, 18], while a polybasic (furin) cleavage site, which is one of the prominent features of SARS-CoV-2 [23, 24], was found only in Bat CoV RmYN02 among CoVs belonging to the subgenus Sarbecovirus [14]. ORF1ab of SARS-CoV-2 is quite similar to that of Bat CoV RmYN02 rather than that of RaTG13 [14]. Those complex genomic features could be a consequence of inter-viral recombination [25]. With respect to the differences in each gene of Sarbecovirus, it was reported that ORF3b differs greatly in length among viruses belonging to the Sarbecovirus genus, including SARS-CoV-2 and SARS-CoV, and that these differences could contribute to differences in the anti-interferon activity [15]. Moreover, it was found that there are SARS-CoV-2 variants showing a longer ORF3b, which were isolated from two patients with severe diseases [15]. This observation may indicate an increased the ability of the longer ORF3b to suppress interferon induction in those patients.
Genome sequencing data analyses of SARS-CoV-2
SARS-CoV-2 information including genome sequencing data was collected in a database called GISAID (Global Initiative on Sharing All Influenza Data, https://www.gisaid.org) [26], which shares sequence data on potentially pandemic infectious viruses, as well as methods for sequencing and relevant geographic and clinical information. The GISAID database includes sequencing data that are not available in public nucleotide databases such as GenBank. As the name implies, this database was constructed at the time of the influenza A H1N1 2009 pandemic, but it covers SARS-CoV-2 in view of urgency. In the GISAID database, not only SARS-CoV-2 but also highly similar viral sequences such as CoVs isolated from bats and pangolins have been collected. Based on the viral sequences as well as geographical and sample collection information in the GISAID database, Nextstrain (https://nextstrain.org) [27] shares phylogenetic, geographical, and genomic analyses of SARS-CoV-2, illustrating the real-time evolution of SARS-CoV-2. Note that Nextstrain has been used to analyze the phylogeny of not only SARS-CoV-2 but also other pathogenic viruses that can potentially pose a public health threat. At the time of writing this article (May 28, 2020), 30,699 SARS-CoV-2 and closely related viral sequences are stored in the GISAID database, and 4308 SARS-CoV-2 genomes were analyzed in the Nextstrain. According to the Nextstrain, the number of substitutions in the SARS-CoV-2 genome was estimated at approximately 26 substitutions per year. Considering the size of SARS-CoV-2 genome (29.9 kb), the estimated evolutionary rate is approximately 0.90 × 10−3 substitution/site/year. The value of this evolutionary rate is similar when compared to other previously reported rates of SARS-CoV (0.80–2.38 × 10−3, Zhao et al.) [28], MERS-CoV (0.63–1.12 × 10−3) [29–31], and HCoV OC43 (0.43 × 10−3) [32]. To the best of our knowledge, the mutation rate (the number of substitutions per site per replication cycle) of SARS-CoV-2 has not been examined yet, but it could be lower than that other RNA viruses such as influenza viruses because the SARS-CoV-2 genome encodes a proofreading exoribonuclease called ExoN in nonstructural protein 14 (nsp14) of the ORF1b as it was reported in SARS-CoV [33].
We know that the evolution of coronaviruses occurs not only by nucleotide mutations but also by recombination. In particular, it has been suggested that the feline infectious peritonitis virus, which causes lethal infectious peritonitis in cats, was caused by recombination of a feline coronavirus with a canine coronavirus [34]. Furthermore, porcine infectious peritonitis virus transforms into porcine respiratory coronavirus (PRCV), which causes respiratory disease when a portion of the S protein is deficient [35]. In murine hepatitis virus (MHV), three amino acid mutations were found to be associated with demyelination and hepatitis [36].
No conclusions have been reached as to whether amino acid mutations are responsible for the difference in SARS-CoV-2 virulence, although certain nucleotide mutations are widely spread in the population. Tang et al. reported that the current coronavirus was divided into two genotypes (designated L and S) depending on an amino acid site 84 (S84L) of ORF8 gene [37]. When compared with closely related CoVs such as Bat CoV RaTG13 and Pangolin CoVs, the ancestral type of SARS-CoV-2 was thought to be S-type [37]. However, the L-type emerged in the beginning of the COVID-19 outbreak, and the current major type of SARS-CoV-2 widely spreading all over the world is L-type as of May 21, 2020 (https://nextstrain.org). Zhang et al. analyzed the clinical and immunological data from 326 confirmed cases of COVID-19 and compared them with viral genetic variation including the S84L mutation, but they could not find any association among them [38]. Korber et al. reported a mutation at an amino acid site 614 (D614G) of S protein that is currently dominant in Europe [39]. Since the S protein is essential in infecting cells and is a primary target for neutralizing antibodies, the mutations in the S protein could be related to the virulence; however, this hypothesis should be evaluated experimentally using reverse genetics. Although more than 5000 mutations accumulated in the SARS-CoV-2 population [40], there are no shreds of evidence currently supporting that SARS-CoV-2 genomes are separating into distinct genotypes during the evolution [41].
Conclusion
Although only about a half year has passed since a genome sequence of SARS-CoV-2 was shared in the GISAID database, more than 30,000 genomes are now available. Using the genome sequence data with closely related viral genome data, the genomic characteristics and evolution of SARS-CoV-2 were extensively studied. However, SARS-CoV-2 is still prevailing around the world and is causing many deaths. Further viral genomic and experimental virological analyses are required to characterize SARS-CoV-2.
Acknowledgements
We thank the GISAID database (https://www.gisaid.org), which shares sequence data of SARS-CoV-2 and related viral species. We also thank Editage (www.editage.com) for English language editing. Phylogenetic analyses in this work were performed in part on the NIG supercomputer at ROIS National Institute of Genetics and SHIROKANE at Human Genome Center (the Univ. of Tokyo).
Abbreviations
- aa
Amino acid
- CoV
Coronavirus
- COVID-19
Coronavirus disease 19
- E
Envelope
- HCoV
Human coronavirus
- HECV
Human enteric coronaviruses
- ICTV
International Committee on Taxonomy of Viruses
- M
Membrane
- ML
Maximum likelihood
- MERS
Middle East respiratory syndrome
- N
Nucleocapsid
- ORF
Open reading frame
- S
Spike
- SARS
Severe acute respiratory syndrome
- WHO
World Health Organization
Authors’ contributions
SN and TM conceived the concept of this review. SN conducted a phylogenetic analysis. SN and TM wrote the manuscript. Both authors read and approved the final manuscript.
Funding
This study was partially funded by JSPS KAKENHI Grants-in-Aid for Scientific Research on Innovative Areas 16H06429, 16K21723, and 19H04843 (to SN); AMED Research Program on Emerging and Re-emerging Infectious Diseases 19fk0108171s0101 (to SN); and 2020 Tokai University School of Medicine Research Aid (to SN).
Availability of data and materials
Phylogenetic data shown in this review are available upon request.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
The original publication contained an error in the main text related to the genus Betacoronavirus. The article has been updated.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
12/21/2020
An amendment to this paper has been published and can be accessed via the original article.
Contributor Information
So Nakagawa, Email: so@tokai.ac.jp.
Takayuki Miyazawa, Email: takavet@infront.kyoto-u.ac.jp.
References
- 1.Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020;382(8):727–733. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395:565–574. doi: 10.1016/S0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu F, Zhao S, Yu B, Chen YM, Wang W, Song ZG, et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579(7798):265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.WHO (World Health Organization): Novel Coronavirus (2019-nCoV) situation report – 22; https://www.who.int/docs/default-source/coronaviruse/situation-reports/20200211-sitrep-22-ncov.pdf. (Accessed 25 May 2020).
- 6.Coronaviridae Study Group of the International Committee on Taxonomy of Viruses The species severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol. 2020;5(4):536–544. doi: 10.1038/s41564-020-0695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.ICTV (International Committee on Taxonomy of Viruses): https://talk.ictvonline.org/ictv-reports/ictv_9th_report/positive-sense-rna-viruses-2011/w/posrna_viruses/222/coronaviridae. (Accessed 25 May 2020).
- 8.Woo PC, Huang Y, Lau SK, Yuen KY. Coronavirus genomics and bioinformatics analysis. Viruses. 2010;2(8):1804–1820. doi: 10.3390/v2081803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30(4):772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Darriba D, Taboada GL, Doallo R, Posada D. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics. 2011;27(8):1164–1165. doi: 10.1093/bioinformatics/btr088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kozlov AM, Darriba D, Flouri T, Morel B, Stamatakis A. RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics. 2019;35(21):4453–4455. doi: 10.1093/bioinformatics/btz305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shi M, Lin XD, Chen X, Tian JH, Chen LJ, Kun L, et al. The evolutionary history of vertebrate RNA viruses. Nature. 2018;556(7700):197–202. doi: 10.1038/s41586-018-0012-7. [DOI] [PubMed] [Google Scholar]
- 13.Darriba D, Posada D, Kozlov AM, Stamatakis A, Morel B, Flouri T. ModelTest-NG: a new and scalable tool for the selection of DNA and protein evolutionary models. Mol Biol Evol. 2020;37(1):291–294. doi: 10.1093/molbev/msz189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou H, Chen X, Hu T, Li J, Song H, Liu Y, et al. A novel bat coronavirus closely related to SARS-CoV-2 contains natural insertions at the S1/S2 cleavage site of the spike protein. Curr Biol. In press. [DOI] [PMC free article] [PubMed]
- 15.Konno Y, Kimura I, Uriu K, Fukushi M, Irie Y, Koyanagi Y, et al. SARS-CoV-2 ORF3b is a potent interferon antagonist whose activity is further increased by a naturally occurring elongation variant. bioRxiv. doi: 10.1101/2020.05.11.088179. [DOI] [PMC free article] [PubMed]
- 16.Davidson AD, Williamson MK, Lewis S, Shoemark D, Carroll MW, Heesom K, et al. Characterisation of the transcriptome and proteome of SARS-CoV-2 using direct RNA sequencing and tandem mass spectrometry reveals evidence for a cell passage induced in-frame deletion in the spike glycoprotein that removes the furin-like cleavage site. bioRxiv. doi: 10.1101/2020.03.22.002204. [DOI] [PMC free article] [PubMed]
- 17.Jungreis I, Sealfon R, Kellis M. Sarbecovirus comparative genomics elucidates gene content of SARS-CoV-2 and functional impact of COVID-19 pandemic mutations. bioRxiv. doi: 10.1101/2020.06.02.130955.
- 18.Xiao K, Zhai J, Feng Y, Zhou N, Zhang X, Zou JJ, et al. Isolation of SARS-CoV-2-related coronavirus from Malayan pangolins. Nature. In press. [DOI] [PubMed]
- 19.Lam TT, Shum MH, Zhu HC, Tong YG, Ni XB, Liao YS, et al. Identifying SARS-CoV-2 related coronaviruses in Malayan pangolins. Nature. In press. [DOI] [PubMed]
- 20.Zhang T, Wu Q, Zhang Z. Probable pangolin origin of SARS-CoV-2 associated with the COVID-19 outbreak. Curr Biol. 2020;30(7):1346–1351.e2. doi: 10.1016/j.cub.2020.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu C, Liu Y, Yang Y, Zhang P, Zhong W, Wang Y, et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm Sin B. In press. [DOI] [PMC free article] [PubMed]
- 22.Andersen KG, Rambaut A, Lipkin WI, Holmes EC, Garry RF. The proximal origin of SARS-CoV-2. Nat Med. 2020;26(4):450–452. doi: 10.1038/s41591-020-0820-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Coutard B, Valle C, de Lamballerie X, Canard B, Seidah NG, Decroly E. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antiviral Res. 2020;176:104742. doi: 10.1016/j.antiviral.2020.104742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function, and antigenicity of the SARS- CoV-2 spike glycoprotein. Cell. 2020;181(2):281–292.e6. doi: 10.1016/j.cell.2020.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li X, Giorgi EE, Marichann MH, Foley B, Xiao C, Kong XP, et al. Emergence of SARS-CoV-2 through recombination and strong purifying selection. bioRxiv doi: 10.1101/2020.03.20.000885. [DOI] [PMC free article] [PubMed]
- 26.Shu Y, McCauley J. GISAID: global initiative on sharing all influenza data - from vision to reality. Euro Surveill. 2017;22(13):30494. doi: 10.2807/1560-7917.ES.2017.22.13.30494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hadfield J, Megill C, Bell SM, Huddleston J, Potter B, Callender C, et al. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics. 2018;34(23):4121–4123. doi: 10.1093/bioinformatics/bty407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao Z, Li H, Wu X, Zhong Y, Zhang K, Zhang YP, et al. Moderate mutation rate in the SARS coronavirus genome and its implications. BMC Evol Biol. 2004;4:21. doi: 10.1186/1471-2148-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cotton M, Watson SJ, Kellam P, Al-Rabeeah AA, Makhdoom HQ, Assiri A, et al. Transmission and evolution of the Middle East respiratory syndrome coronavirus in Saudi Arabia: a descriptive genomic study. Lancet. 2013;382:1993–2002. doi: 10.1016/S0140-6736(13)61887-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cotton M, Watson SJ, Zumla AI, Makhdoom HQ, Palser AL, Ong SH, et al. Spread, circulation, and evolution of the Middle East respiratory syndrome coronavirus. mBio. 2014;5:e01062–e01013. doi: 10.1128/mBio.01062-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dudas G, Carvalho LM, Rambaut A, Bedford T. MERS-CoV spillover at the camel-human interface. Elife. 2018;7:e31257. doi: 10.7554/eLife.31257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vijgen L, Keyaerts E, Moës E, Thoelen I, Wollants E, Lemey P, et al. Complete genomic sequence of human coronavirus OC43: molecular clock analysis suggests a relatively recent zoonotic coronavirus transmission event. J. Virol. 2005;79:1595–1604. doi: 10.1128/JVI.79.3.1595-1604.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith EC, Blanc H, Surdel MC, Vignuzzi M, Denison MR. Coronaviruses lacking exoribonuclease activity are susceptible to lethal mutagenesis: evidence for proofreading and potential therapeutics. PLoS Pathog. 2013;9(8):e1003565. doi: 10.1371/journal.ppat.1003565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Terada Y, Matsui N, Noguchi K, Kuwata R, Shimoda H, Soma T, et al. Emergence of pathogenic coronaviruses in cats by homologous recombination between feline and canine coronaviruses. PLoS One. 2014;9:e106534. doi: 10.1371/journal.pone.0106534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rasschaert D, Duarte M, Laude H. Porcine respiratory coronavirus differs from transmissible gastroenteritis virus by a few genomic deletions. J Gen Virol. 1990;71:2599–2607. doi: 10.1099/0022-1317-71-11-2599. [DOI] [PubMed] [Google Scholar]
- 36.Das Sarma J, Fu L, Hingley ST, Lai MM, Lavi E. Sequence analysis of the S gene of recombinant MHV-2/A59 coronaviruses reveals three candidate mutations associated with demyelination and hepatitis. J Neurovirol. 2001;7:432–436. doi: 10.1080/135502801753170282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tang X, Wu C, Li X, Song Y, Yao X, Wu X, et al. On the origin and continuing evolution of SARS-CoV-2. Natl Sci Rev. In press. [DOI] [PMC free article] [PubMed]
- 38.Zhang X, Tan Y, Ling Y, Lu G, Liu F, Yi Z, et al. Viral and host factors related to the clinical outcome of COVID-19. Nature. In press. [DOI] [PubMed]
- 39.Korber B, Fischer WM, Gnanakaran S, Yoon H, Theiler J, Abfalterer W, et al. Spike mutation pipeline reveals the emergence of a more transmissible form of SARS-CoV-2. bioRxiv doi: 10.1101/2020.04.29.069054.
- 40.CoV-GLUE, http://cov-glue.cvr.gla.ac.uk (Accessed 25 May 2020).
- 41.MacLean OA, Orton RJ, Singer JB, Robertson DL. No evidence for distinct types in the evolution of SARS-CoV-2. Virus Evolution. 2020;6(1):veaa034. doi: 10.1093/ve/veaa034. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Phylogenetic data shown in this review are available upon request.