

Abstract
Our previous study of DNA methylation in the pediatric soft tissue tumor rhabdomyosarcoma (RMS) demonstrated that fusion-positive (FP) and fusion-negative (FN) RMS tumors exhibit distinct DNA methylation patterns. To further examine the significance of DNA methylation differences in RMS, we investigated genome-wide DNA methylation profiles in discovery and validation cohorts. Unsupervised analysis of DNA methylation data identified novel distinct subsets associated with the specific fusion subtype in FP RMS and with RAS mutation status in FN RMS. Furthermore, the methylation pattern in normal muscle is most similar to the FN subset with wild-type RAS mutation status. Several biologically relevant genes were identified with methylation and expression differences between the two fusion subtypes of FP RMS or between the RAS wild-type and mutant subsets of FN RMS. Genomic localization studies showed that promoter and intergenic regions were hypomethylated and the 3’ untranslated regions were hypermethylated in FP compared to FN tumors. There was also a significant difference in the distribution of PAX3-FOXO1 binding sites between genes with and without differential methylation. Moreover, genes with PAX3-FOXO1 binding sites and promoter hypomethylation exhibited the highest frequency of overexpression in FP tumors. Finally, a comparison of RMS model systems revealed that patient-derived xenografts most closely recapitulate the DNA methylation patterns found in human RMS tumors compared with cell lines and cell line-derived xenografts. In conclusion, these findings highlight the interaction of epigenetic changes with mutational alterations and transcriptional organization in RMS tumors, and contribute to improved molecular categorization of these tumors.
Keywords: Rhabdomyosarcoma, fusion protein, DNA methylation, expression, xenograft
Introduction
Rhabdomyosarcoma (RMS) is a family of pediatric soft tissue tumors associated with the skeletal muscle lineage.1 From a genetic perspective, RMS comprises two major subtypes, fusion-positive (FP) and fusion-negative (FN), which show substantial overlap with the histologic subtypes of alveolar and embryonal RMS, respectively. FP RMS is associated with 2;13 or 1;13 chromosomal translocations resulting in a PAX3-FOXO1 or PAX7-FOXO1 fusion, respectively, and henceforth referred to collectively as PAX-FOXO1.2, 3 The PAX-FOXO1 fusions encode potent transcription factors with increased activation of PAX3/PAX7 target genes and associated oncogenic functions.4–7 Genome-wide sequencing studies demonstrated that FP RMS has essentially no recurrent point mutations, while FN RMS displays numerous recurrent mutations, including frequent mutations in the RAS signaling pathway.8, 9
The critical role that epigenetic alterations, including DNA methylation, play in the development of cancers, such as RMS, is increasingly appreciated. DNA methylation influences the regulatory activity of the gene promoter and body as well as more distal enhancers.10–12 These epigenetic mechanisms regulate gene expression and contribute to tumor suppressor inactivation and oncogene activation in cancers. In RMS, genome-wide DNA methylation studies previously reported evidence for an association of DNA methylation patterns with histological subtype and clinical outcome.13–16
Although DNA methylation, genetic alterations, and gene expression in RMS tumors have been individually characterized, the relationship between these parameters is still unclear. In this study, we undertook an integrative analysis of DNA methylation, point mutations, and gene expression in 86 human RMS tumors. Our comparative analysis of these datasets highlights how the DNA methylation pattern reflects the interactions of DNA structural changes and transcription factors, such as PAX-FOXO1, with chromatin organization and thereby contributes to the tumor cells’ aberrant gene expression.
Materials and Methods
A more detailed description of our methods is available in Supplementary Methods.
Tissue, cell line and xenograft samples
A discovery cohort of 38 RMS tumors and a validation cohort of 48 RMS tumors were included in this study. The RMS tumors in the discovery cohort were received from the Children’s Oncology Group Biopathology Center, and a subset of these tumors (n=24, 63%) were previously described.15 RMS tumors in the validation cohort were described in a previous comprehensive genomic analysis.9 Normal de-identified fetal and neonatal skeletal muscle samples without a history or pathologic evidence of cancer were obtained post-mortem. Eleven RMS cell lines, 11 CDXs, and 14 PDXs were used in this study (Supplementary Table 1).
Fusion status testing
PAX3-FOXO1 or PAX7-FOXO1 fusion status was determined by reverse transcriptase-PCR17, 18 or fluorescence in situ hybridization for all samples without unambiguous embryonal RMS histology.19 Alternative fusion genes were determined by whole-genome or transcriptome sequencing.9
DNA methylation assay and data analysis
Genomic DNA extraction and bisulfite conversion were performed as previously described. 9, 15 Bisulfite-converted genomic DNA was analyzed using the Infinium HumanMethylation450 (HM450) BeadChip platform (Illumina). IDAT files from the HM450 array were processed using the R/Bioconductor packages minfi and methylumi. The data were normalized using the SWAN (subset-quantile within array normalization) algorithm.20
Mutation and gene expression analysis
For FN RMS samples in the discovery cohort, mutation status was determined by targeted sequencing.21, 22 For RMS tumors in the validation cohort, DNA mutation and RNA expression were previously determined using whole-genome or exome sequencing and RNA sequencing, respectively.9
Additional data analysis
Hierarchical clustering and principal component analysis (PCA) were performed as previously described.15 Fisher’s exact test was used to test for associations between methylation groups and genetic alterations. Enrichment of gene sets with PAX3-FOXO1 binding genes was also calculated using the Fisher’s exact test.23 An unpaired t test or one-way ANOVA was used to compare DNA methylation levels between category groups.
Immunohistochemistry
We obtained RMS tissue microarrays (TMAs) from the Children’s Hospital of Philadelphia and the Children’s Oncology Group, containing 18 and 39 FP RMS tumors respectively. Immunohistochemistry (IHC) was performed using a mouse monoclonal anti-CDKN1C antibody (p57 Kip2 Ab-6, Thermo Scientific).24
Results
DNA methylation profiling identifies novel subsets within FP and FN RMS subtypes
To confirm and extend our previous findings that FP and FN RMS subtypes exhibit distinct DNA methylation profiles,15 we studied genome-wide DNA methylation in a discovery cohort of 21 FP tumors (12 PAX3-FOXO1 and 9 PAX7-FOXO1) and 17 FN tumors using the HM450 array. Unsupervised hierarchical analysis clearly segregated the tumors into two distinct groups according to the presence or absence of PAX-FOXO1 (Figure 1A). A PCA confirmed this close association of methylation pattern and fusion status (Supplementary Figure 1A).
Figure 1. DNA methylation profiling in discovery and validation cohorts identifies molecular subsets in FP and FN RMS.
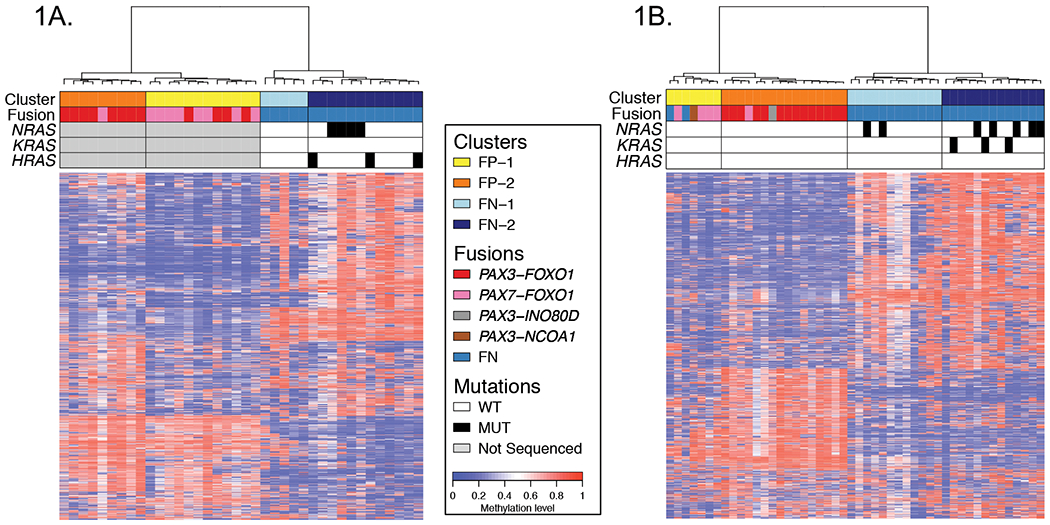
Heat maps for discovery (A) and validation (B) cohorts displaying the subsets of FP and FN RMS defined by DNA methylation. These displays are based on the top 1% most varied DNA probes across RMS tumors. Differences in the top 1% most varied DNA probes between the discovery and validation cohorts may contribute to the small differences in methylation patterns displayed for the RMS subsets. Fusion status and RAS mutation status are shown in the upper panel in addition to the methylation-defined subsets. Abbreviations: FN, fusion-negative; FP, fusion-positive; WT, wild-type; MUT, mutant-type.
To gain further insight into the DNA methylation patterns in RMS, we noted that our DNA methylation analysis identified two major subsets (FP-1 and FP-2) within the FP cluster, and two major subsets (FN-1 and FN-2) within the FN cluster (Figure 1A). For tumors within the FP cluster, we reasoned that these DNA methylation-based subclasses may correspond to the specific fusion subtype, either PAX3-FOXO1 or PAX7-FOXO1. Our evaluation of the association between these DNA methylation-defined subsets (FP-1 and FP-2) and the specific fusion subtype showed that the PAX3-FOXO1 and PAX7-FOXO1 fusions were significantly enriched in the FP-2 and FP-1 subsets, respectively (Figure 1A and Supplementary Table 2). We found that 33% of the tumors in FP-1 compared to 89% in FP-2 are PAX3-FOXO1-positive (P=0.024). In contrast, 67% of tumors in FP1 and only 11% of tumors in FP2 are PAX7-FOXO1-positive (P=0.024).
We next sought to investigate the significance of the methylation-defined subsets (FN-1 and FN-2) within the FN cluster. Owing to the high rate of recurrent RAS mutations in FN RMS, we investigated the relationship of these two FN subsets to RAS mutation status.8, 9 Targeted sequencing was used to elucidate HRAS, KRAS and NRAS mutation status in the 17 FN RMS tumors in the discovery cohort. A striking finding of this analysis is that RAS gene mutations were differentially distributed between the FN-1 and FN-2 subsets. Whereas none of the tumors in FN-1 contains a mutation in one of the three RAS genes, 58% of tumors in FN-2 harbor mutant RAS genes (P=0.044) (Figure 1A, Supplementary Table 2).
Validation of genetic differences between methylation-defined RMS subsets
To validate the genetic differences between the methylation-defined RMS subsets found in our discovery cohort, we generated DNA methylation profiles in an additional cohort of 21 FP tumors (14 PAX3-FOXO1, 5 PAX7-FOXO1, and 2 alternative fusions [PAX3-NCOA1 and PAX3-INO80D]) and 27 FN tumors using the HM450 array. Hierarchical clustering of the DNA methylation data in the validation cohort again yielded two distinct groups, one with all FP RMS tumors along with 2 FN tumors and the other with 25 of the 27 FN RMS tumors (Figure 1B). A PCA analysis showed that the two “discordant” FN tumors map in a region between the FP and FN clusters (Supplementary Figure 1B). This unsupervised analysis of the validation cohort also demonstrated two major subsets within the FP cluster (FP-1 and FP-2), and two major subsets within the FN cluster (FN-1 and FN-2). Our comparison of the PAX3-FOXO1 and PAX7-FOXO1 gene fusions in the two FP subsets confirms the significant associations between these two gene fusions and DNA methylation pattern in this validation cohort (PAX3-FOXO1, 0% in FP-1 versus 88% in FP-2, P<0.001; PAX7-FOXO1, 57% in FP-1 versus 6% in FP-2, P=0.017) (Supplementary Table 2). Comparison of the two methylation-defined FN subsets in the validation cohort also confirms the statistically significant association between RAS mutation status and DNA methylation pattern (mutant RAS, 17% in FN-1 versus 62% in FN-2, P=0.041) (Figure 1B and Supplementary Table 2).
CDKN1C methylation and expression status differs between PAX3-FOXO1- and PAX7-FOXO1-positive RMS tumors
We further explored DNA methylation differences between fusion subtypes in FP RMS tumors. To increase the sample size, we combined the PAX3-FOXO1- and PAX7-FOXO1-positive tumors in the discovery and validation cohorts. Unsupervised analysis clearly exhibited the distinct DNA methylation patterns between PAX3-FOXO1- and PAX7-FOXO1-positive tumors (Supplementary Figure 2A). A subsequent supervised analysis (|Δ|≥0.25 and adjusted P-value <0.05) found 1688 probes that were significantly hypermethylated (corresponding to 184 promoter-hypermethylated genes) and only 97 probes that were significantly hypomethylated (corresponding to 6 promoter-hypomethylated genes) in PAX3-FOXO1- compared with PAX7-FOXO1-positive tumors (Figure 2A, Supplementary Table 3).
Figure 2. DNA methylation characteristics associated with PAX3-FOXO1- and PAX7-FOXO1-positive RMS tumors.
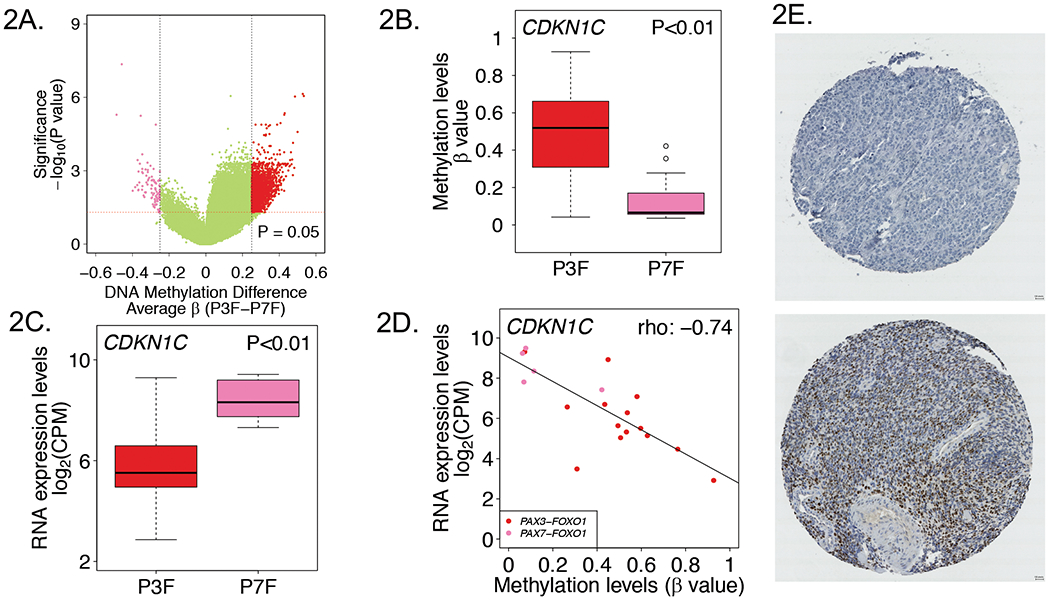
A, Volcano plot displaying differentially methylated probes between PAX3-FOXO1- and PAX7-FOXO1-positive RMS tumors. Red and pink dots denote hypermethylated and hypomethylated probes in PAX3-FOXO1- versus PAX7-FOXO1-positive tumors, respectively. CDKN1C promoter methylation (B) and RNA expression (C) levels in PAX3-FOXO1- and PAX7-FOXO1-positive RMS tumors. D, Correlation plot of DNA methylation versus RNA expression levels for CDKN1C in PAX3-FOXO1- and PAX7-FOXO1-positive RMS tumors. E, Representative CDKN1C immunohistochemical staining results in PAX3-FOXO1- and PAX7-FOXO1-positive tumors. Upper, CDKN1C negative staining in a PAX3-FOXO1-positive tumor; Lower, strong staining in a PAX7-FOXO1-positive tumor. Abbreviations: P3F, PAX3-FOXO1; P7F, PAX7-FOXO1.
Examination of the differentially methylated genes between the two FP subtypes identified CDKN1C, a gene with potential importance in RMS tumor biology.25 CDKN1C encodes a cyclin-dependent kinase and is located within an imprinted region at chromosomal region 11p15.5, which frequently shows loss of heterozygosity in RMS. CDKN1C is a putative tumor suppressor gene and aberrant promoter hypermethylation has been suggested as a mechanism for inactivation of one allele of this gene in RMS and other human cancers with 11p15.5 allelic loss. In our study, CDKN1C exhibited promoter hypermethylation in PAX3-FOXO1- compared to PAX7-FOXO1-positive tumors (4 probes and mean p-value=0.006, representative data for CpG site cg05090695 shown in Figure 2B). Based on our finding of differential methylation of CDKN1C between PAX3-FOXO1- and PAX7-FOXO1-positive tumors, we examined CDKN1C mRNA expression levels in these FP subtypes. This analysis revealed CDKN1C was significantly underexpressed in PAX3-FOXO1- compared to PAX7-FOXO1-positive tumors (~6.0-fold change, P<0.01) (Figure 2C). Furthermore, there is an inverse correlation between DNA methylation and RNA expression of CDKN1C in the FP RMS tumors (Spearman’s rho: −0.74, P<0.001) (Figure 2D).
To assess CDKN1C protein expression in PAX3-FOXO1 versus PAX7-FOXO1–positive RMS tumors, we performed immunohistochemistry on TMAs containing FP RMS tumors (Figure 2E). In the first TMA, we found a significant difference in CDKN1C protein expression between PAX3-FOXO1- and PAX7-FOXO1-positive tumors (P<0.05); CDKN1C expression was absent or weak in 33.3%, moderate in 33.3%, and strong in 33.3% of PAX3-FOXO1-positive cases, whereas all PAX7-FOXO1-positive tumors (100%) showed strong CDKN1C expression. The difference in CDKN1C protein expression between the two fusion subtypes was further validated in a second set of RMS TMAs; CDKN1C staining was absent or weak in 88.9%, moderate in 7.4% and strong in 3.7% of PAX3-FOXO1-positive cases compared to expression that was absent or weak in 41.7%, moderate in 41.7% and strong in 16.7% of PAX7-FOXO1-positve cases (P<0.01) (Supplementary Table 4).
DNA methylation in RAS mutant and wild-type FN RMS tumors
To further investigate DNA methylation differences between RAS mutant and wild-type FN RMS tumors, we combined FN RMS tumors from the discovery and validation cohorts. Unsupervised analysis confirmed the presence of the distinct methylation-defined FN-1 and FN-2 subsets, corresponding to the RAS wild-type- and mutant-enriched categories, respectively (Supplementary Figure 2B). A supervised analysis (|Δβ|≥0.25 and P-value <0.05) of DNA methylation differences identified 117 hypermethylated CpG sites (corresponding to 28 promoter-hypermethylated genes) and 77 hypomethylated CpG sites (corresponding to 7 promoter-hypomethylated genes) in RAS mutant compared to RAS wild-type FN RMS tumors (Figure 3A, Supplementary Table 5).
Figure 3. DNA methylation characteristics associated with RAS mutant and wild-type FN RMS tumors.
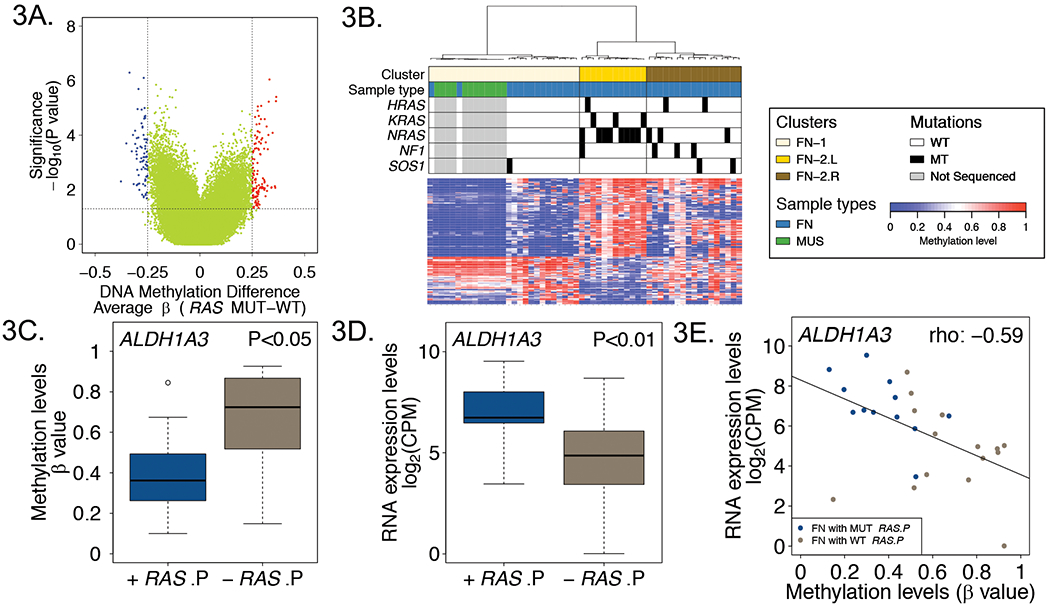
A, Volcano plot displaying differentially methylated probes between RAS mutant and wild-type FN RMS tumors. Red and blue dots denote hypermethylated and hypomethylated probes in RAS mutant versus wild-type tumors, respectively. B, Heat map comparing DNA methylation of normal fetal and neonatal skeletal muscle with RAS mutant and wild-type FN tumors based on differentially methylated probes between RAS mutant and wild-type FN tumors. Mutation events are represented by black rectangles in the upper panel. Promoter DNA methylation (C) and RNA expression (D) levels of ALDH1A3 in RAS pathway mutant and wild-type FN RMS tumors. E, Correlation plot of DNA methylation versus RNA expression levels for ALDH1A3 in RAS pathway mutant and wild-type FN RMS tumors. Abbreviations: WT, wild-type; MUT, mutant-type; +RAS.P, FN tumors with RAS pathway gene mutations; -RAS.P, FN tumors without RAS pathway gene mutations.
To extend our previous finding that the DNA methylation pattern in skeletal muscle is more similar to FN than FP RMS tumors, we next compared DNA methylation patterns in these FN RMS subsets with normal fetal and neonatal muscle samples, which were also profiled on the HM450 array platform.15 We performed a clustering analysis of FN tumors and muscle samples based on the differentially methylated CpG sites in RAS mutant versus wild-type FN tumors (Figure 3B). This analysis defined two main subgroups, which closely correspond to the RAS wild-type-enriched FN-1 and RAS mutant-enriched FN-2 subsets. This FN-1 subset contained 56% of RAS wild-type tumors and all 12 normal muscle samples, whereas the FN-2 subset contained all RAS mutant and the remaining 44% of RAS wild-type tumors. This analysis demonstrates that normal developing muscle has a DNA methylation pattern more similar to the RAS wild-type-enriched subset than the RAS mutant-enriched subset.
Further examination of the DNA methylation pattern within the RAS mutant-enriched FN-2 subset revealed two smaller subgroups. The left subgroup (FN-2L) consists of 71% of RAS mutant tumors and no RAS wild-type tumors, whereas the right subgroup (FN-2R) consists of the remaining RAS mutant (29%) and RAS wild-type tumors (44%). To investigate why RAS wild-type tumors in this right subgroup exhibit DNA methylation similar to RAS mutant tumors, we postulated that additional genetic alterations in RAS pathway genes may occur in these RAS wild-type tumors. Mutations were studied in these cases, either as part of the original genome-wide RMS sequencing study or on a targeted sequencing platform. In support of our hypothesis, we found that mutations of two additional RAS pathway genes (NF1 and SOS1) were present at a high frequency in the RAS wild-type tumors in the FN-2R subgroup (42%) (Figure 3B). By considering mutations in all of these RAS pathway genes, the frequency of tumors harboring mutations in RAS pathway genes was significantly higher in the FN-2R subgroup (59%) than in the FN-1 subset (7%, P<0.01).
To evaluate whether differential methylation in FN tumors with mutant RAS pathway genes versus wild-type RAS pathway genes corresponds to gene expression differences, we performed differential methylation and differential expression analyses using DNA methylation data from the combined FN cohort and the available RNAseq data from our validation cohort. We identified 48 hypomethylated CpG sites (corresponding to 5 promoter-hypomethylated genes) and only 3 hypermethylated CpG sites in mutant RAS pathway- compared to wild-type RAS pathway-FN tumors (|Δβ|≥0.25 and adjusted P-value <0.05) (Supplementary Table 6). Examination of these differentially methylated genes highlighted ALDH1A3, a gene previously reported as a potential marker for cancer stem cells in embryonal RMS.26, 27 Our data showed that ALDH1A3 was promoter-hypomethylated in RAS pathway mutant versus wild-type FN tumors (2 probes and mean p-value=0.026, representative data for CpG site cg23191950 shown in Figure 3C). Furthermore, ALDH1A3 was overexpressed in RAS pathway mutant versus wild-type tumors (~4.6-fold change, P<0.01), and there was a significant negative correlation between DNA methylation and gene expression of ALDH1A3 (Spearman’s rho: −0.59, P=0.001) (Figure 3D, 3E).
Distribution of DNA methylation between FP and FN tumors across genomic features
Given that the HM450 array provides extensive genome-wide coverage of CpG sites, we next investigated the genomic distribution of DNA methylation in FP versus FN tumors, using the DNA methylation data from the validation cohort. Consistent with our previous findings, we observed that FP tumors show an overall significant decrease in global DNA methylation in comparison to FN tumors (Figure 4A, 4B).15 We further compared DNA methylation in FP versus FN tumors in four annotated genomic features: promoters, gene bodies, 3’ UTR regions and intergenic regions. Of interest, FP tumors show significantly lower DNA methylation in promoter and intergenic regions in comparison to FN tumors. In contrast, FP tumors display significantly higher DNA methylation in 3’ UTR regions compared with FN tumors, and there is no significant DNA methylation difference in the gene bodies.
Figure 4. Distribution of DNA methylation differences between FP and FN tumors across genomic features.
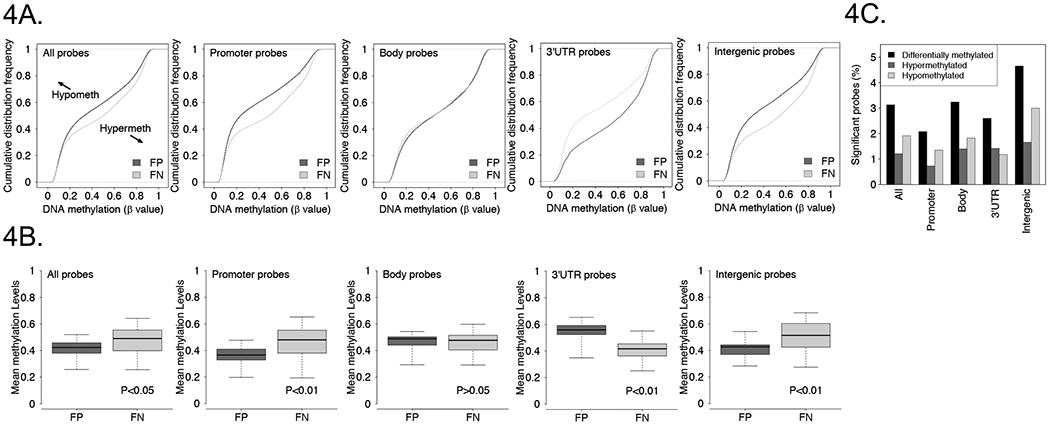
A, Cumulative distribution frequency plots of CpG methylation levels in FP and FN tumors based on genomic features. B, Overall DNA methylation level (mean β value) of FP and FN tumors based on genomic features. C, Frequency of differentially methylated CpG probes in FP versus FN tumors based on genomic features.
In a supervised analysis, (|Δβ|≥0.25 and adjusted P-value <0.05), we identified 12269 autosomal CpG sites that were differentially methylated between FP and FN RMS tumors. Further analysis of these differentially methylated CpG sites revealed that they were not evenly distributed with respect to the four annotated genomic features described above. These differentially methylated probes were enriched in intergenic regions and depleted in the promoter and 3’ UTR regions (Figure 4C). In addition, these differentially methylated CpG sites can be further divided into 4747 hypermethylated and 7522 hypomethylated probes in FP compared to FN tumors (Supplementary Table 7). We next examined the distribution of these hyper- and hypomethylated probes across the genomic features. The hypermethylated probes were enriched in FP tumors in the body, 3’ UTR, and intergenic regions and depleted in the promoters. In contrast, hypomethylated probes were enriched in FP tumors in the intergenic region and were depleted in the promoter, body and 3’ UTR regions (Figure 4C and Supplementary Table 8).
Association of promoter hypomethylation with expression of PAX3-FOXO1 targets
Although the PAX-FOXO1 transcription factor and DNA methylation both contribute to gene expression in FP tumors, it remains unclear whether the fusion protein and DNA methylation changes collaborate with each other, either directly or indirectly, to regulate gene expression. To investigate whether PAX-FOXO1 binding is associated with a distinct promoter methylation pattern in FP tumors, we used the list of PAX3-FOXO1 binding sites elucidated in a chromatin immunoprecipitation sequencing study23 to identify PAX3-FOXO1 targets among the genes that are differentially methylated between FP and FN tumors (Supplementary Table 9). We found that differentially methylated genes more often contained PAX3-FOXO1 binding sites, (7.8%) and thus were more often PAX3-FOXO1 targets than genes without differential methylation (4.9%) (P<0.01). Further, we found that genes with either promoter hypermethylation (10.1%) or hypomethylation (6.7%) were more often PAX3-FOXO1 targets than genes without differential methylation. Though the proportion of PAX3-FOXO1 target genes among genes with promoter hypomethylation is less compared to genes with promoter hypermethylation, there are overall more PAX3-FOXO1 target genes in the group with promoter hypomethylation (60 genes) than in the group with promoter hypermethylation (45 genes).
To investigate whether altered promoter methylation impacts on the expression of genes with PAX3-FOXO1 binding sites, we performed differential expression analysis between FP and FN tumors, and compared the presence of PAX3-FOXO1 binding sites with the expression status of genes with promoter hypomethylation or hypermethylation. We identified 1063 overexpressed genes and 1155 underexpressed genes in FP tumors compared to FN tumors (absolute fold-change >2 and adjusted P value <0.05) (Supplementary Table 10). Among the genes with promoter hypomethylation, PAX3-FOXO1 binding sites are significantly overrepresented in overexpressed genes (29.4%) compared to non-differentially expressed genes (5.0%) (P<0.01), whereas there is no significant difference in the distribution of PAX3-FOXO1 binding sites between underexpressed and non-differentially expressed genes (Supplementary Table 11).
To understand how promoter hypomethylation contributes to overexpression of genes with PAX3-FOXO1 binding sites, we compared the frequency of overexpression among genes categorized according to the presence or absence of PAX3-FOXO1 binding sites and the presence or absence of promoter hypomethylation. We observed that genes with PAX3-FOXO1 binding sites display a significantly higher frequency of overexpression compared with genes without PAX3-FOXO1 binding sites, regardless of the presence or absence of promoter hypomethylation (P<0.01) (Table 1). Further, genes with promoter hypomethylation displayed a significantly higher frequency of overexpression compared with genes without promoter hypomethylation, regardless of the presence or absence of PAX3-FOXO1 binding sites (P<0.01). In a comparison of the four groups categorized according to the PAX3-FOXO1 binding site and promoter hypomethylation status, the group of genes with both PAX3-FOXO1 binding sites and promoter hypomethylation exhibited the highest frequency of overexpression (54.3%). Although this combination is associated with the highest frequency of overexpression, the number of overexpressed genes in this category (25 genes) is relatively small. In contrast, the number of overexpressed genes in the category with PAX3-FOXO1 binding sites but without hypomethylation (178 genes) is 7-fold higher. Therefore, there still is a large number of genes for which the presence of PAX3-FOXO1 binding sites is sufficient to drive overexpression without accompanying promoter hypomethylation.
Table 1.
Frequency of overexpression in subsets defined by PAX3-FOXO1-binding sites and DNA hypomethylation.
| Frequency of overexpressed genesa |
|||
|---|---|---|---|
| With PAX3-FOXO1 binding sites | Without PAX3-FOXO1 Binding sites | ||
| + Hypomethylation | 25 / 46 (54.3%) | 60 / 455 (13.1%) | P<0.01b |
| − Hypomethylation | 178 / 804 (22.1%) | 660 / 11846 (5.6%) | P<0.01b |
| P<0.01c | P<0.01c | ||
Data is presented in the format of “No. of overexpressed genes / No. of total genes, %”.
Fisher’s exact test was performed by using non-differentially methylated genes as reference group.
RMS PDXs display DNA methylation pattern similar to RMS tumors.
It is currently unknown whether any RMS model systems faithfully recapitulate DNA methylation patterns found in RMS primary tumors. To address this issue, methylation patterns in 11 RMS cell lines, 11 cell line-derived xenografts (CDXs) and 14 patient-derived xenografts (PDXs) were compared to patterns in primary tumors (Supplementary Table 1). We performed a hierarchical clustering analysis of RMS primary tumors, cell lines, CDXs, and PDXs. This analysis identified a FP cluster and a FN cluster, which grouped all the samples based on fusion status, with the exception of two FN tumors described above (Figure 5A). Further examination of the FP and FN clusters showed that all cell lines and CDXs form one subset in each cluster whereas nearly all PDXs and primary tumors form a second subset. Of note, there is generally no difference in DNA methylation pattern between pairs of subcutaneous and intramuscular CDXs. Moreover, all pairs of cell line and corresponding CDX cluster together at the terminal branch of the dendrogram. The finding of two PDXs (RH41 and RH36) clustering with cell lines and CDXs suggests that PDXs may lose the native methylation pattern under some unknown circumstances. PCA analysis of the DNA methylation data corroborates the divergence between FP and FN RMS samples across the sample types and confirms that PDXs cluster with tumor samples whereas CDXs cluster with cell lines in either the FP or FN groups (Figure 5B).
Figure 5. Comparison of methylation profiles of RMS cell lines, CDXs, PDXs and patient tumors.
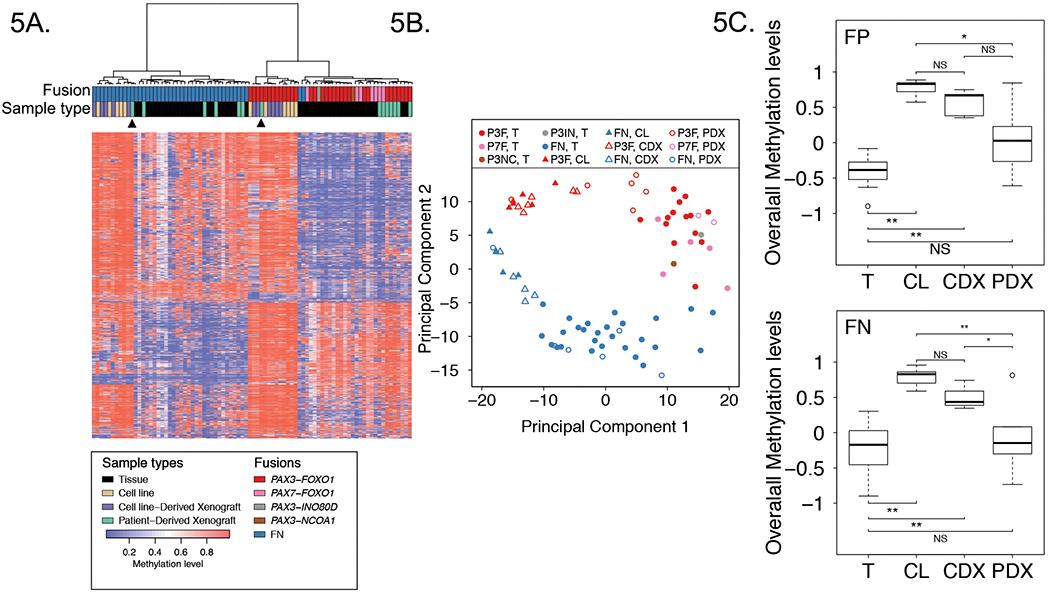
Heat map (A) and PCA (B) analyses comparing DNA methylation profiles of 11 RMS cell lines, 11 CDXs, 14 PDXs and 48 patient tumors in the discovery cohort based on top 1% most varied DNA methylation probes across patient tumors. Arrowheads from left to right denote RH36 PDX and RH41 PDX. C, Overall DNA methylation levels in RMS cell lines, CDXs, PDXs and patient tumors. Data for FP samples are shown above, and data for FN samples are shown below. The plots summarize the distribution of standardized average DNA methylation levels in each sample type based on the top 1% most varied probes across RMS tumors. Adjusted P values in FP samples were calculated using one way ANOVA with Games-Howell test, and adjusted P values in FN samples were calculated using one way ANOVA with Tukey-Kramer multiple comparison post hoc test. Abbreviations: P3F, PAX3-FOXO1; P7F, PAX7-FOXO1; P3IN, PAX3-INO80D; P3NC, PAX3-NCOA1; T, tumor; CL, cell line. *, P<0.05; **, P<0.01; NS, not significant.
Studies from other tumor types indicated that cancer cell lines are typically hypermethylated compared to tumor tissue.28, 29 An examination of the heat maps in our clustering analysis confirms this finding for RMS samples. In particular, the vast majority of the analyzed CpG sites are hypermethylated in the cell lines and CDXs in contrast to the wider distribution of hypo- and hypermethylation found in primary tumors and PDXs (Figure 5A).
Finally, we focused separately on the FP or FN groups and compared overall DNA methylation levels in the four sample types. We found that overall methylation levels in FP cell lines and CDXs are significantly higher than levels in primary tumors (adjusted P value < 0.01); in contrast, there is no statistical differences in overall methylation levels between PDXs and primary tumors (Figure 5C). A similar pattern is found in the FN samples.
Discussion
In this study, we used high-density arrays to investigate genome-wide DNA methylation in FP and FN RMS tumors, and identified patterns associated with genetically and biologically distinct subsets of RMS tumors. One important observation is that the PAX3-FOXO1- and PAX7-FOXO1-positive subsets are molecularly distinguished by DNA methylation. In contrast, a recent study indicated that all tumors positive for PAX3-FOXO1 or PAX7-FOXO1 fusions were grouped into two clusters (A1/A2), yet the two clusters (A1 and A2) did not coincide with the two different fusion subtypes.14 Several studies revealed different clinical features between patients with PAX3-FOXO1- and PAX7-FOXO1-positive tumors, including age, site, and outcome.18 Molecular differences that may support these clinical differences have been reported; the PAX7-FOXO1 fusion gene is usually amplified while the PAX3-FOXO1 fusion gene is rarely amplified, and amplification of the 12q13-q14 (CDK4) and 13q31 (MIR17HG) chromosomal regions are enriched in PAX3-FOXO1- and PAX7-FOXO1-positive tumors, respectively.30–32 However, transcriptomic classification has not been able to reveal robust gene expression differences in unsupervised analysis of PAX3-FOXO1- and PAX7-FOXO1-positive tumors.33 The different methylation patterns between PAX3-FOXO1- and PAX7-FOXO1-positive tumors shown in this study suggest that epigenetic differences may contribute to the distinct biology and clinical features in these two FP subsets, and may provide novel surrogate markers.
Our study demonstrates that FN RMS tumors can be readily classified into RAS mutant and wild-type-enriched subsets based on DNA methylation, pointing to a novel framework for molecular classification of FN RMS tumors. In contrast to a recent study suggesting that a methylation-defined subset of the embryonal RMS subtype is characterized by a high frequency of FGFR4/RAS/AKT pathway mutations, our study specifically highlights the enrichment of RAS mutations in a DNA methylation-defined subset of FN RMS tumors.14 Associations between RAS mutations and DNA methylation have been found in several cancer categories, such as lung and colorectal cancer.34,35 Our finding of different methylation patterns between RAS mutant and wild-type FN RMS tumors suggests several possible mechanisms. One possibility is that RAS mutations induce DNA methylation changes during RMS development. This explanation is consistent with our finding that RAS wild-type FN RMS is more similar than mutant RAS tumors to normal developing muscle. It is possible that mutant RAS-mediated regulation of EZH2 expression may be a prerequisite to subsequent DNA methylation changes.36 A second possibility is that these DNA methylation events are selected and interact with RAS mutations in a collaborative model of FN RMS tumorigenesis. Based on the findings that DNMT3A mutations cooperate with NRAS mutations in acute leukemia development,37 DNA methylation changes may result from synergistic interactions between a DNA methyltransferase and RAS mutations during RMS development.38, 39 A third possibility is that these DNA methylation patterns reflect the molecular differences between progenitor cells that give rise to these FN RMS subtypes.
Our study highlights that promoter DNA hypomethylation contributes to transcriptional activation of a subset of PAX-FOXO1 target genes. Genome-wide mapping studies of PAX3-FOXO1 binding sites revealed that only a small fraction of binding sites occur in promoter elements whereas most binding sites are within distal enhancers.23 It is proposed that the PAX-FOXO1 proteins mediate their transcriptional impact by recruiting other transcription factors and chromatin-binding proteins, such as BRD4, to form super-enhancers. Moreover, transcription may be driven by three-dimensional looping that brings PAX-FOXO1-associated super-enhancers to promoters and enables physical interaction of super-enhancer-bound proteins with promoters.40 As the lack of DNA methylation is fundamental to open chromatin states, we hypothesize that promoter DNA hypomethylation may coordinate with PAX-FOXO1 to drive target gene expression. In accord with this concept, we observed a significant depletion of DNA methylation in the promoter region in FP compared with FN RMS tumors. It should be noted that the association between PAX-FOXO1 binding sites and genes with differential promoter methylation reported in the current study was not identified in our previous study.15 This discrepancy is most likely due to use in the current study of the HM450 array, which has much higher promoter CpG site coverage than the HM27 DNA methylation array used in our previous study.
There is also a large number of genes with PAX3-FOXO1 binding sites that are overexpressed without accompanying promoter hypomethylation in FP compared to FN cases. This group of genes includes a small subset with promoter hypermethylation in FP cases (6%) and a larger subset in which there is no differential promoter methylation between FP and FN cases (~94%). For those overexpressed genes with promoter hypermethylation, it is postulated that 1) DNA hypermethylation may inhibit binding of repressive factors in the promoter region, or 2) overexpression may result from hypomethylation of alternative promoters. For those overexpressed genes without differential promoter methylation, these loci may be transcriptionally active in both FP and FN RMS and thus have the same DNA methylation status. In this scenario, the increased expression found in FP cases may be programmed by PAX3-FOXO1-induced chromatin changes that are independent of promoter methylation changes. Finally, though the present study focused on the relationship between PAX-FOXO1 and promoter methylation, we also speculate possible links between PAX-FOXO1 and DNA methylation in other regions including 5’ UTR, 3’ UTR and intergenic regions, as suggested by our finding of differing DNA methylation between FP and FN RMS tumors.
Our study shows that RMS PDXs have a global DNA methylation pattern that is highly representative of primary tumor samples, and that RMS cell lines and CDXs have a distinctly different DNA methylation signature. These results are concordant with reports in head and neck cancer and osteosarcoma.29, 41 This finding indicates that RMS PDXs are the most suitable models for investigating issues related to DNA methylation in RMS. As only a small fraction of DNA methylation events in cultured cell lines or CDXs mirror that in primary tumors, we caution using cell line models for dissecting the role of DNA methylation in RMS. In this regard, we attempted to further investigate DNA methylation changes associated with PAX3-FOXO1 expression by using a CRISPR/Cas9 editing strategy to inactivate PAX3-FOXO1 in a FP RMS cell line. Our preliminary data identified DNA methylation differences in subclones with PAX3-FOXO1 inactivation relative to control subclones. However, only a very small subset of the methylation changes in this cell line-based study were concordant with methylation differences between FP versus FN RMS tumors (unpublished data). This finding is consistent with the divergence of the methylation pattern in RMS cell lines from that in tumors, and suggests that further investigation of these issues should be performed in PDXs.
These findings highlight that systematic identification of DNA methylation events is revealing important epigenetic mechanisms involved in the molecular pathogenesis of cancers associated with gene fusions, such as PAX-FOXO1, as well as cancers associated with other oncogenic events, such as RAS mutations. Future studies of DNA methylation alterations in cancers such as RMS will therefore contribute to improved molecular classification of these tumors, and will provide a foundation for improved understanding of epigenetic regulation and the possible application of epigenetic-directed therapeutics in these cancers.
Supplementary Material
Novelty and Impact:
Our comprehensive integrative analysis of DNA methylation, genetic and gene expression alterations in the pediatric soft tissue cancer rhabdomyosarcoma revealed biologically distinct subsets in fusion-positive and fusion-negative rhabdomyosarcoma defined by DNA methylation pattern. Our analysis also demonstrates collaboration between the PAX3-FOXO1 fusion oncoprotein and promoter hypomethylation to regulate a subset of PAX3-FOXO1 transcriptional targets. These findings contribute to an improved molecular classification and better understanding of epigenetic regulation in rhabdomyosarcoma.
Acknowledgements
We thank Kris Ylaya, Jeffery Hanson, Holly S. Stevenson, Diana Corao, Mary Olanich, Belynda Hicks and Kristie Jones for technical assistance. We also thank the Children’s Oncology Group for providing specimens for this study.
Financial Support: This research was supported by the Intramural Research Program of the National Cancer Institute and the Joanna McAfee Childhood Cancer Foundation.
Footnotes
Disclosure of Potential Conflicts of Interest: The authors have no potential conflicts of interest to disclose.
References
- 1.Olanich ME, Barr FG. A call to ARMS: targeting the PAX3-FOXO1 gene in alveolar rhabdomyosarcoma. Expert Opin Ther Targets 2013;17: 607–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Galili N, Davis RJ, Fredericks WJ, Mukhopadhyay S, Rauscher FJ 3rd, Emanuel BS, Rovera G, Barr FG. Fusion of a fork head domain gene to PAX3 in the solid tumour alveolar rhabdomyosarcoma. Nat Genet 1993;5: 230–5. [DOI] [PubMed] [Google Scholar]
- 3.Davis RJ, D’Cruz CM, Lovell MA, Biegel JA, Barr FG. Fusion of PAX7 to FKHR by the variant t(1;13)(p36;q14) translocation in alveolar rhabdomyosarcoma. Cancer Res 1994;54: 2869–72. [PubMed] [Google Scholar]
- 4.Missiaglia E, Williamson D, Chisholm J, Wirapati P, Pierron G, Petel F, Concordet JP, Thway K, Oberlin O, Pritchard-Jones K, Delattre O, Delorenzi M, et al. PAX3/FOXO1 fusion gene status is the key prognostic molecular marker in rhabdomyosarcoma and significantly improves current risk stratification. J Clin Oncol 2012;30: 1670–7. [DOI] [PubMed] [Google Scholar]
- 5.Scheidler S, Fredericks WJ, Rauscher FJ 3rd, Barr FG, Vogt PK. The hybrid PAX3-FKHR fusion protein of alveolar rhabdomyosarcoma transforms fibroblasts in culture. Proc Natl Acad Sci U S A 1996;93: 9805–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Skapek SX, Anderson J, Barr FG, Bridge JA, Gastier-Foster JM, Parham DM, Rudzinski ER, Triche T, Hawkins DS. PAX-FOXO1 fusion status drives unfavorable outcome for children with rhabdomyosarcoma: a children’s oncology group report. Pediatr Blood Cancer 2013;60: 1411–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xia SJ, Barr FG. Analysis of the transforming and growth suppressive activities of the PAX3-FKHR oncoprotein. Oncogene 2004;23: 6864–71. [DOI] [PubMed] [Google Scholar]
- 8.Chen X, Stewart E, Shelat AA, Qu C, Bahrami A, Hatley M, Wu G, Bradley C, McEvoy J, Pappo A, Spunt S, Valentine MB, et al. Targeting oxidative stress in embryonal rhabdomyosarcoma. Cancer Cell 2013;24: 710–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shern JF, Chen L, Chmielecki J, Wei JS, Patidar R, Rosenberg M, Ambrogio L, Auclair D, Wang J, Song YK, Tolman C, Hurd L, et al. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov 2014;4: 216–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet 2012;13: 484–92. [DOI] [PubMed] [Google Scholar]
- 11.Yang X, Han H, De Carvalho DD, Lay FD, Jones PA, Liang G. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell 2014;26: 577–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Charlet J, Duymich CE, Lay FD, Mundbjerg K, Dalsgaard Sorensen K, Liang G, Jones PA. Bivalent Regions of Cytosine Methylation and H3K27 Acetylation Suggest an Active Role for DNA Methylation at Enhancers. Mol Cell 2016;62: 422–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mahoney SE, Yao Z, Keyes CC, Tapscott SJ, Diede SJ. Genome-wide DNA methylation studies suggest distinct DNA methylation patterns in pediatric embryonal and alveolar rhabdomyosarcomas. Epigenetics 2012;7: 400–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seki M, Nishimura R, Yoshida K, Shimamura T, Shiraishi Y, Sato Y, Kato M, Chiba K, Tanaka H, Hoshino N, Nagae G, Shiozawa Y, et al. Integrated genetic and epigenetic analysis defines novel molecular subgroups in rhabdomyosarcoma. Nat Commun 2015;6: 7557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun W, Chatterjee B, Wang Y, Stevenson HS, Edelman DC, Meltzer PS, Barr FG. Distinct methylation profiles characterize fusion-positive and fusion-negative rhabdomyosarcoma. Mod Pathol 2015;28: 1214–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tombolan L, Poli E, Martini P, Zin A, Romualdi C, Bisogno G, Lanfranchi G. NELL1, whose high expression correlates with negative outcomes, has different methylation patterns in alveolar and embryonal rhabdomyosarcoma. Oncotarget 2017;8: 33086–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barr FG, Smith LM, Lynch JC, Strzelecki D, Parham DM, Qualman SJ, Breitfeld PP. Examination of gene fusion status in archival samples of alveolar rhabdomyosarcoma entered on the Intergroup Rhabdomyosarcoma Study-III trial: a report from the Children’s Oncology Group. J Mol Diagn 2006;8: 202–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sorensen PH, Lynch JC, Qualman SJ, Tirabosco R, Lim JF, Maurer HM, Bridge JA, Crist WM, Triche TJ, Barr FG. PAX3-FKHR and PAX7-FKHR gene fusions are prognostic indicators in alveolar rhabdomyosarcoma: a report from the children’s oncology group. J Clin Oncol 2002;20: 2672–9. [DOI] [PubMed] [Google Scholar]
- 19.Dumont SN, Lazar AJ, Bridge JA, Benjamin RS, Trent JC. PAX3/7-FOXO1 fusion status in older rhabdomyosarcoma patient population by fluorescent in situ hybridization. J Cancer Res Clin Oncol 2012;138: 213–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maksimovic J, Gordon L, Oshlack A. SWAN: Subset-quantile within array normalization for illumina infinium HumanMethylation450 BeadChips. Genome Biol 2012;13: R44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brohl AS, Patidar R, Turner CE, Wen X, Song YK, Wei JS, Calzone KA, Khan J. Frequent inactivating germline mutations in DNA repair genes in patients with Ewing sarcoma. Genet Med 2017;19: 955–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chang W, Brohl AS, Patidar R, Sindiri S, Shern JF, Wei JS, Song YK, Yohe ME, Gryder B, Zhang S, Calzone KA, Shivaprasad N, et al. MultiDimensional ClinOmics for Precision Therapy of Children and Adolescent Young Adults with Relapsed and Refractory Cancer: A Report from the Center for Cancer Research. Clin Cancer Res 2016;22: 3810–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cao L, Yu Y, Bilke S, Walker RL, Mayeenuddin LH, Azorsa DO, Yang F, Pineda M, Helman LJ, Meltzer PS. Genome-wide identification of PAX3-FKHR binding sites in rhabdomyosarcoma reveals candidate target genes important for development and cancer. Cancer Res 2010;70: 6497–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Radujkovic A, Dietrich S, Andrulis M, Benner A, Longerich T, Pellagatti A, Nanda K, Giese T, Germing U, Baldus S, Boultwood J, Ho AD, et al. Expression of CDKN1C in the bone marrow of patients with myelodysplastic syndrome and secondary acute myeloid leukemia is associated with poor survival after conventional chemotherapy. Int J Cancer 2016;139: 1402–13. [DOI] [PubMed] [Google Scholar]
- 25.Roeb W, Boyer A, Cavenee WK, Arden KC. PAX3-FOXO1 controls expression of the p57Kip2 cell-cycle regulator through degradation of EGR1. Proc Natl Acad Sci U S A 2007;104: 18085–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Drummond CJ, Hanna JA, Garcia MR, Devine DJ, Heyrana AJ, Finkelstein D, Rehg JE, Hatley ME. Hedgehog Pathway Drives Fusion-Negative Rhabdomyosarcoma Initiated From Non-myogenic Endothelial Progenitors. Cancer Cell 2018;33: 108–24 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nakahata K, Uehara S, Nishikawa S, Kawatsu M, Zenitani M, Oue T, Okuyama H. Aldehyde Dehydrogenase 1 (ALDH1) Is a Potential Marker for Cancer Stem Cells in Embryonal Rhabdomyosarcoma. PLoS One 2015;10: e0125454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Suter CM, Norrie M, Ku SL, Cheong KF, Tomlinson I, Ward RL. CpG island methylation is a common finding in colorectal cancer cell lines. Br J Cancer 2003;88: 413–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hennessey PT, Ochs MF, Mydlarz WW, Hsueh W, Cope L, Yu W, Califano JA. Promoter methylation in head and neck squamous cell carcinoma cell lines is significantly different than methylation in primary tumors and xenografts. PLoS One 2011;6: e20584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barr FG, Duan F, Smith LM, Gustafson D, Pitts M, Hammond S, Gastier-Foster JM. Genomic and clinical analyses of 2p24 and 12q13-q14 amplification in alveolar rhabdomyosarcoma: a report from the Children’s Oncology Group. Genes Chromosomes Cancer 2009;48: 661–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Duan F, Smith LM, Gustafson DM, Zhang C, Dunlevy MJ, Gastier-Foster JM, Barr FG. Genomic and clinical analysis of fusion gene amplification in rhabdomyosarcoma: a report from the Children’s Oncology Group. Genes Chromosomes Cancer 2012;51: 662–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reichek JL, Duan F, Smith LM, Gustafson DM, O’Connor RS, Zhang C, Dunlevy MJ, Gastier-Foster JM, Barr FG. Genomic and clinical analysis of amplification of the 13q31 chromosomal region in alveolar rhabdomyosarcoma: a report from the Children’s Oncology Group. Clin Cancer Res 2011;17: 1463–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davicioni E, Anderson MJ, Finckenstein FG, Lynch JC, Qualman SJ, Shimada H, Schofield DE, Buckley JD, Meyer WH, Sorensen PH, Triche TJ. Molecular classification of rhabdomyosarcoma--genotypic and phenotypic determinants of diagnosis: a report from the Children’s Oncology Group. Am J Pathol 2009;174: 550–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Selamat SA, Chung BS, Girard L, Zhang W, Zhang Y, Campan M, Siegmund KD, Koss MN, Hagen JA, Lam WL, Lam S, Gazdar AF, et al. Genome-scale analysis of DNA methylation in lung adenocarcinoma and integration with mRNA expression. Genome Res 2012;22: 1197–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hinoue T, Weisenberger DJ, Lange CP, Shen H, Byun HM, Van Den Berg D, Malik S, Pan F, Noushmehr H, van Dijk CM, Tollenaar RA, Laird PW. Genome-scale analysis of aberrant DNA methylation in colorectal cancer. Genome Res 2012;22: 271–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Riquelme E, Behrens C, Lin HY, Simon G, Papadimitrakopoulou V, Izzo J, Moran C, Kalhor N, Lee JJ, Minna JD, Wistuba II. Modulation of EZH2 Expression by MEK-ERK or PI3K-AKT Signaling in Lung Cancer Is Dictated by Different KRAS Oncogene Mutations. Cancer Res 2016;76: 675–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lu R, Wang P, Parton T, Zhou Y, Chrysovergis K, Rockowitz S, Chen WY, Abdel-Wahab O, Wade PA, Zheng D, Wang GG. Epigenetic Perturbations by Arg882-Mutated DNMT3A Potentiate Aberrant Stem Cell Gene-Expression Program and Acute Leukemia Development. Cancer Cell 2016;30: 92–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen B, Liu X, Savell VH, Dilday BR, Johnson MW, Jenkins JJ, Parham DM. Increased DNA methyltransferase expression in rhabdomyosarcomas. Int J Cancer 1999;83: 10–4. [DOI] [PubMed] [Google Scholar]
- 39.Megiorni F, Camero S, Ceccarelli S, McDowell HP, Mannarino O, Marampon F, Pizer B, Shukla R, Pizzuti A, Marchese C, Clerico A, Dominici C. DNMT3B in vitro knocking-down is able to reverse embryonal rhabdomyosarcoma cell phenotype through inhibition of proliferation and induction of myogenic differentiation. Oncotarget 2016;7: 79342–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gryder BE, Yohe ME, Chou HC, Zhang X, Marques J, Wachtel M, Schaefer B, Sen N, Song Y, Gualtieri A, Pomella S, Rota R, et al. PAX3-FOXO1 Establishes Myogenic Super Enhancers and Confers BET Bromodomain Vulnerability. Cancer Discov 2017;7: 884–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guilhamon P, Butcher LM, Presneau N, Wilson GA, Feber A, Paul DS, Schutte M, Haybaeck J, Keilholz U, Hoffman J, Ross MT, Flanagan AM, et al. Assessment of patient-derived tumour xenografts (PDXs) as a discovery tool for cancer epigenomics. Genome Med 2014;6: 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.