

Summary
The GATA‐type sexual development transcription factor NsdD has been implicated in virulence, secondary metabolism and asexual development in filamentous fungi. However, little is known about its function in the yeast‐to‐hypha transition and in microsclerotium formation. In the current study, the orthologous NsdD gene MrNsdD in the entomopathogenic fungus Metarhizium rileyi was characterized. Transcriptional analysis indicated that MrNsdD was involved in yeast‐to‐hypha transition, conidiation and microsclerotium formation. After targeted deletion of MrNsdD, dimorphic transition, conidiation, fungal virulence and microsclerotium formation were all impaired. Compared with the wild‐type strain, the ΔMrNsdD mutants were hypersensitive to thermal stress. Furthermore, transcriptome sequencing analysis revealed that MrNsdD regulated a distinct signalling pathway in M. rileyi during the yeast‐to‐hypha transition or microsclerotium formation, but exhibited overlapping regulation of genes during the two distinct developmental stages. Taken together, characterization of the MrNsdD targets in this study will aid in the dissection of the molecular mechanisms of dimorphic transition and microsclerotium development.
The significance of MrNsdD in dimorphic transition, conidium and microsclerotium formation. The MrNsdD mediated regulation of target genes involved in dimorphic transition and microsclerotium development. It provides valuable understanding the molecular mechanism of dimorphic transition and microsclerotium development.
Introduction
Entomopathogenic fungi are reported to cause high levels of mortality in biocontrol programmes for agricultural and forestry pests (Wang et al., 2016; Zhao et al., 2016). Metarhizium rileyi (previously Nomuraea rileyi) is a well‐known dimorphic entomopathogenic fungus that infects a range of lepidopterous pests (Fronza et al., 2017). The reproduction of M. rileyi in pests is via conidia, but the current conidium‐based mass production methods are not cost effective and require special growth conditions (stimulatory light and maltose) for conidiation, factors which jeopardize the commercial development of M. rileyi (de Faria and Wraight, 2007). To promote the commercialization of M. rileyi, the generation of melanized microsclerotia induced in liquid culture is an alternative production route for active propagules (Song et al., 2014).
The conidium development process in filamentous fungi involves several morphological changes. The most in‐depth characterization of the genetic basis of conidiation has been intensively investigated in the model fungi Aspergillus spp. and Neurospora crassa (Ogawa et al., 2010). As core regulatory elements, transcription factors play an important role in the transcriptional regulation of gene expression during conidiation (Park and Yu, 2012; Shelest, 2017). Studies on conidiation‐associated transcription factors have mainly included upstream transcription activators FlbB and FlbC and the negative regulator SfgA in A. nidulans (Wieser et al., 1994; Seo et al., 2006), and the positive regulator StuA in filamentous fungi, such as Acremonium chrysogenum and Ustilago maydis (García‐pedrajas et al., 2010; Hu et al., 2015). Previous work identified an NSD (never in sexual development) group of A. nidulans mutants that were unable to form any sexual structures (Han et al., 2001). The DNA‐binding domains of NsdC and NsdD in the group are diverse and operate independent regulatory mechanisms during fungal morphogenesis (Kim et al., 2009; Cary et al., 2012). Recent genetic studies revealed that the NsdD GATA‐type zinc finger transcription factor, a global regulator, is involved in the conidium formation in Aspergillus spp. (Cary et al., 2012; Lee et al., 2014). Moreover, it also has been implicated in hyphal growth, oxidative stress response, polysaccharide‐degradation by exoenzymes, biosynthesis of secondary metabolites and/or appressoria formation in Botrytis cinerea, Fusarium fujikuroi, Penicillium oxalicum and Sclerotinia sclerotiorum (Schumacher et al., 2014; Niehaus et al., 2017; He et al., 2018; Li et al., 2018). However, little is known about the function of Nsd proteins in the entomopathogenic fungi.
To investigate the regulatory mechanism of dimorphic transition and conidiation in M. rileyi, a comparative transcriptome analysis was used to dissect conidial development (Song et al., unpublished data). Results showed that the expression of a gene orthologous to NsdD (MrNsdD) was down‐regulated during the yeast‐to‐hypha transition and was up‐regulated during conidiation. In addition, previous comparative transcriptome analysis revealed that MrNsdD expression level was up‐regulated during M. rileyi microsclerotium formation (Song et al., 2013). These results suggested that MrNsdD may play a key role in dimorphic transition and in the development of both conidia and microsclerotia. However, the details of the transcriptional regulation mechanisms involved remain to be elucidated.
This study sought to elucidate the function of MrNsdD in conidiation, dimorphic transition and microsclerotia formation. These functional studies demonstrated that MrNsdD regulated dimorphic transition, conidiation, thermal stress tolerance, virulence and microsclerotia formation. Furthermore, transcriptome analysis showed that MrNsdD‐mediated regulation of target genes was involved in the dimorphic transition and in microsclerotia development in M. rileyi.
Results
Bioinformatic features and the generation of mutants
The MrNsdD ortholog (NCBI accession No. MN116705) was located in the genomic database of M. rileyi (Shang et al., 2016) and was 1454 bp long, including three introns, encoding a protein with a molecular mass of 120.2 kDa (http://expasy.org/tools/protparam.html). The protein encoded contained a GATA zinc finger DNA‐binding domain at the C‐terminus. The amino acid sequence of MrNsdD showed similarities (85–92% identity) to the NsdD protein of Metarhizium spp. (Hu et al., 2014; Shang et al., 2016) and a zinc finger protein (61% identity) of Beauveria bassiana (Kim et al., 2016). In terms of phylogeny, the sequences shared an identity of 53%–92% with NsdD proteins from other fungi (Fig. S1).
To examine the biological functions of MrNsdD in M. rileyi, gene deletion mutants and complemented transformants were generated, isolated and verified by polymerase chain reaction (PCR) and quantitative real‐time‐PCR (qPCR) analyses with primers in Table S1 (Fig. S2). The ΔMrNsdD mutants and complemented (ΔMrNsdD + NsdD) strains were subjected to further studies.
Function of MrNsdD in conidiation
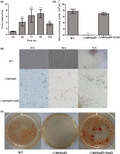
Compared with the wild‐type (WT, CQNr01 strain) and complemented strains, the ΔMrNsdD mutant displayed defects in conidial germination rate, dimorphic transition and conidiation capacity. When the tested strains were incubated on SMAY (Sabouraud maltose agar fortified with 1% (w/v) yeast extract) solid medium, the ΔMrNsdD mutant required a significantly longer time to achieve 50% conidial germination [mean ± standard error (SE) half‐maximal germination time] (GT50 = 8.6 ± 0.4 h) than WT (GT50 = 7.1 ± 0.4 h) and complemented (GT50 = 7.0 ± 0.3 h) strains (P < 0.01) (Fig. 1A). However, there were no significant differences in colony growth rate among the various strains (data not shown).
Fig. 1.
MrNsdD plays important roles in the asexual cycle of Metarhizium rileyi.
A. GT50 (mean time required for 50% germination) as an index of conidial germination rate.
B. Transcription of MrNsdD during conidiation. Aliquots (2.5 μl) of conidial suspensions (107 conidia ml−1) of the wild‐type (WT) strain were pipetted onto SMAY (Sabouraud maltose agar fortified with 1% (w/v) yeast extract) plates under continuous light at 25 °C for eight days. Stages of conidial development: conidia at initial inoculation time (day 0), blastospores (day 2), hyphal period (day 4), conidiation initiation (day 6) and conidia at start of maturation (day 8).
C. Images of various conidial suspensions on SMAY plates.
D. TT50 (median transition time required for 50% transition of blastospores to hyphae) for transition of blastospores to hyphae. TT50 was estimated using a probit analysis.
E. Statistical analysis of conidia yield. Error bars represent standard error. *P < 0.05, **P < 0.01 compared with the WT strain.
To examine the role of MrNsdD in conidia production, expression level was analysed by qPCR. Expression level analysis in the WT showed that MrNsdD was down‐regulated during blastospores formation (day 2) and hyphal vegetative growth (day 4), but was up‐regulated during the initiation of conidiation (day 6) and during conidial maturation (day 8) (Fig. 1B). These results indicated that MrNsdD may function in the yeast‐to‐hypha transition and during conidiation.
Phenotypic investigations showed that, by day 3, the yeast‐to‐hyphae transition was delayed in the ΔMrNsdD mutant compared with the WT and complemented strains (Fig. 1C), and the dimorphic transition rates were not unanimous in various concentrations of test strains. Further dimorphic transition analyses showed that there was a significant difference between the ΔMrNsdD mutant (mean ± SE, median transition time required for 50% transition of blastospores to hyphae, TT50 = 8.8 ± 0.4 days), and both the WT (TT50 = 7.2 ± 0.4 days) and complemented strains (TT50 = 6.9 ± 0.4 d) (P < 0.01) (Fig. 1D), suggesting important roles for MrNsdD in the dimorphic transition of M. rileyi.
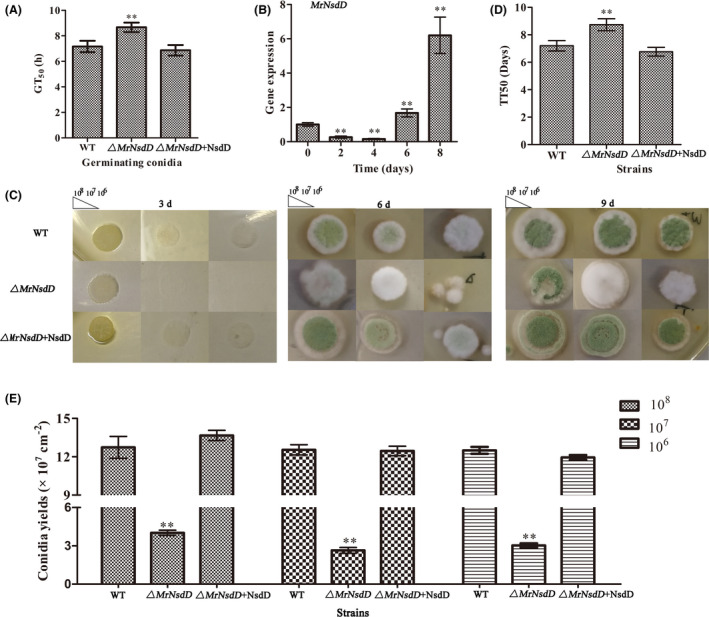
Conidial yields were quantified after 12‐days incubation. The yields were 68.5%– 80.0% lower in the ΔMrNsdD mutant, compared with the WT and complemented strains (P < 0.01) (Fig. 1E). The conidiophores on the WT and complemented strain colonies exhibited the radial architecture characteristic of M. rileyi, whereas, on the conidiophores of the ΔMrNsdD mutants, no radiating phialides were observed (Fig. 2A). Furthermore, the expression of several conidiation‐required genes for activation of fungal development was examined by transcriptional analysis of the mutant and the WT (Wieser et al., 1994; Zhou et al., 2018). The genes selected for analysis included the transcription factor gene MrFlbC, the hydrophobin gene (MrHyd1) crucial for conidial structure and hydrophobicity and the gene (MrCsp) encoding the conidial septation protein. As shown in Fig. 2B, the significantly reduced expression of each of these genes in the ΔMrNsdD mutant suggests that these down‐regulations could have caused the conidiation defects observed. Taken together, transcriptional and phenotypic analyses suggested that MrNsdD plays an important role in conidiation.
Fig. 2.
Conidiophore architecture and relative transcript abundance analysis of ΔMrNsdD mutant.
A. Effect of MrNsdD deletion on conidiophore architecture. For light microscope conidiophore observations, tested strains were grown for 6 days on SMAY solid medium. Scale bar, 20 mm.
B. Relative transcript levels of conidiation‐required genes in the 6‐day incubation period of the ΔMrNsdD mutant relative to the wild‐type (WT) strain. Error bars represent standard error. *P < 0.05, **P < 0.01 compared with WT strain.
Transcriptome sequencing analysis of MrNsdD‐mediated regulation of dimorphic transition
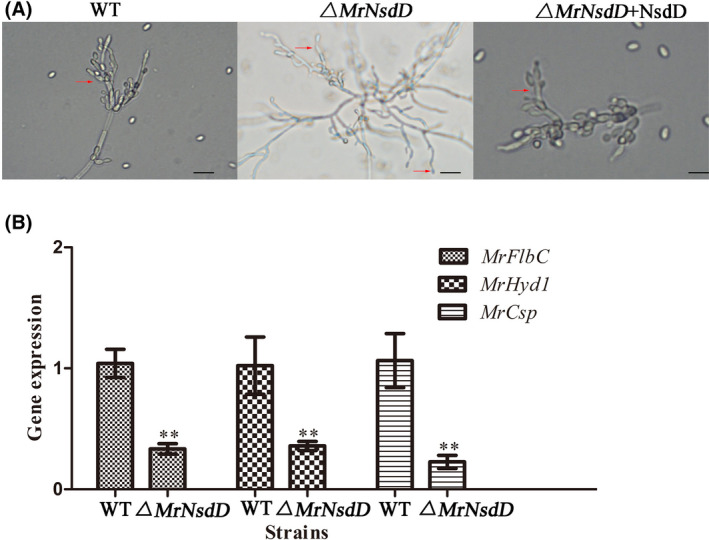
To investigate the MrNsdD‐mediated regulation of dimorphic transition, the transcriptomes of WT and the ΔMrNsdD mutant were compared over the 3‐d incubation period on SMAY medium. We considered genes to be differentially expressed when their fold change (FC) was ≥ 2 at a false discovery rate (FDR) < 0.01. Differentially expressed genes (DEGs) including 383 genes were up‐regulated, and 207 genes were down‐regulated in the ΔMrNsdD mutant (Fig. 3A). Among the 590 differentially expressed genes, four genes were expressed only in WT and three genes were expressed only in the ΔMrNsdD mutant (Table S2).
Fig. 3.
Comparative transcriptomic of genes related to dimorphic transition and microsclerotium development.
A. Differentially expressed genes (DEGs) in WT and the ΔMrNsdD mutant during dimorphic transition or microsclerotium development.
B. Overview of two distinct morphogenesis with respect to DEGs.
DEGs were functionally grouped into Gene Ontology (GO) classes including 35 functional categories (Fig. S3) and 19 clusters of orthologous groups (COG) (Fig. S4). A total of 105 DEGs were assigned to 20 Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment pathways. The top three pathways were biosynthesis of antibiotics (16.2%), carbon metabolism (11.7%) and oxidative phosphorylation (10.8%) (Fig. S5). And an overview of DEGs from two distinct development stages (dimorphic transition versus microsclerotium development) in the ΔMrNsdD mutant was established. Results would be described in the following section (Fig. 3B).
MrNsdD‐mediated regulation of microsclerotium development
Expression level of MrNsdD during microsclerotium development was analysed by qPCR. Compared with the vegetative growth stage (after 36 h), transcriptional expression of MrNsdD peaked during microsclerotium initiation and maturation (72–120 h) (Fig. 4A), indicating that MrNsdD might be involved in the regulation of microsclerotia formation.
Fig. 4.
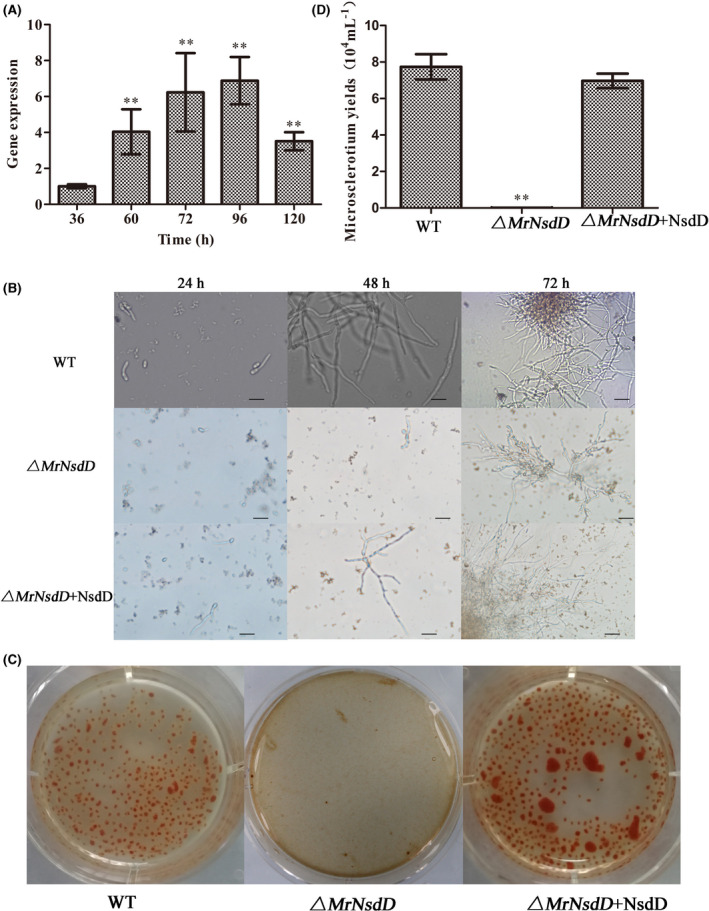
MrNsdD expression during microsclerotium development and phenotypic characterization of microsclerotium development in liquid amended medium (AM) culture.
A. Transcription of MrNsdD during microsclerotium development. Samples were collected during the following stages: blastospores (36 h), hyphal period (60 h), microsclerotium initiation (72 h), microsclerotium formation (96 h) and microsclerotium maturation (120 h).
B. Development of microsclerotium in AM. Scale bar: 50 μm.
C. Phenotypic characterization of microsclerotium formation. Conidial suspension of the tested strains was inoculated in AM and cultured for 6 days.
D. Microsclerotium yields of the tested strains. Error bars represent standard error. *P < 0.05, **P < 0.01, when compared with WT or the results at 36 h.
After 72 h of incubation in liquid amended medium (AM) culture, WT and complemented strains started to form microsclerotium, whereas minimal vegetative growth was observed by this time in the ΔMrNsdD mutant (Fig. 4B). After 144 h of incubation, the ΔMrNsdD mutant displayed a significant reduction in microsclerotium formation compared with that observed in the WT and complemented strains under the same culture conditions (Fig. 4C). There was no significant difference in microsclerotia biomass between the WT and complemented strains (data not shown), but the microsclerotium yield of the ΔMrNsdD mutant was negligible (Fig. 4C and D), indicating that MrNsdD was required for microsclerotia formation.
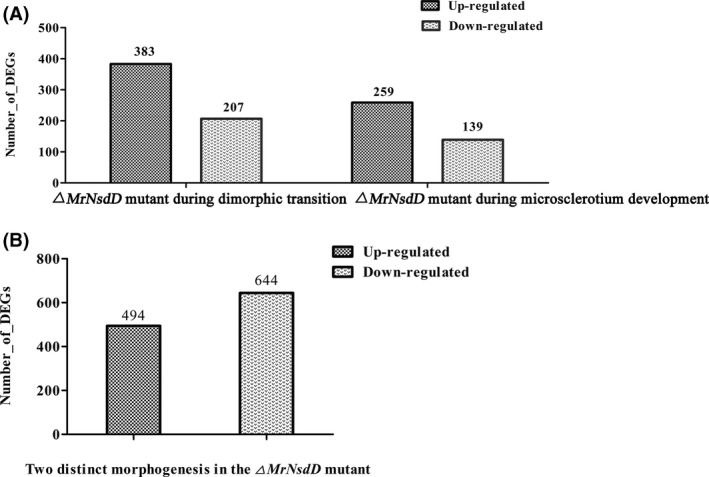
To identify target genes potentially regulated by MrNsdD during microsclerotium formation, comparative transcriptomics analysis was performed between the WT and the ΔMrNsdD mutant. Overall, DEGs showed that 259 genes were up‐regulated and 139 genes were down‐regulated in the ΔMrNsdD mutant (Fig. 3A). Among the 398 differentially expressed genes, 2 genes were expressed only in WT and 5 genes were expressed only in ΔMrNsdD mutants (Table S3).
DEGs were functionally grouped into GO classes including 35 functional categories (Fig. S6) and 19 COG clusters (Fig. S7). A total of 57 DEGs were assigned to 20 KEGG enrichment pathways (Fig. S8). The top three pathways were biosynthesis of antibiotics (20.1%), biosynthesis of amino acids (10.5%) and oxidative phosphorylation (8.8%) (Fig. S8).
Overview of transcriptomics data of two distinct development stages
To confirm the gene expression patterns from the comparative transcriptomics data, 15 genes from samples collected during dimorphic transition (Table S4) and 15 genes from samples collected during microsclerotium development (Table S5) were analysed by qPCR. Results showed that the transcriptomics data and the qPCR results were closely correlated (Fig. S9 and Tables S4 and S5).
Furthermore, compared with their expression during dimorphic transition in the ΔMrNsdD mutants, 494 genes were up‐regulated and 644 genes were down‐regulated during microsclerotium development in the ΔMrNsdD mutant (Fig. 3B). Among the 1138 differentially expressed genes, 11 genes were expressed only in microsclerotium development and 21 genes were expressed only in dimorphic transition (Table S6). The DEGs were functionally grouped into 39 GO functional categories (Fig. S10) and 21 COG clusters (Fig. S11). A total of 201 DEGs were assigned to 20 KEGG enrichment pathways. The top three pathways were biosynthesis of antibiotics (17.9%), carbon metabolism (11.9%) and biosynthesis of amino acids (9.9%) (Fig. S12).
To identify the key target genes regulated by MrNsdD in the two distinct developmental stages, several genes involved in the pathway of carbon metabolism and in peroxisome functions (Table S7) were analysed by qPCR. The carbon metabolism‐related genes included genes encoding phosphoserine phosphatase (MrPp), pyridoxal phosphate‐dependent enzyme (MrPpt), hexokinase‐1 (MrHe1), D‐3‐phosphoglycerate dehydrogenase 1 (MrDpd1) and O‐acetylserine sulfhydrylase (MrOas). The peroxisome‐related genes included genes encoding catalase 1 (MrCat1), sarcosine oxidase (MrSo), oligopeptide transporter protein (MrOtp), carnitine acetyl transferase (MrCatf), fructosyl‐amino acid oxidase (MrFaao), superoxide dismutase (MrSod) and peroxisomal biogenesis factor 2 (MrPbf2). In relation to dimorphic transition in the ΔMrNsdD mutant, MrDpd1, MrOas, MrPbf2 and MrFaao genes were all up‐regulated during microsclerotium development, whereas MrPp, MrPpt, MrHe1, MrCat1, MrSo, MrOtp, MrCatf and MrSod genes were down‐regulated (Fig. 5).
Fig. 5.
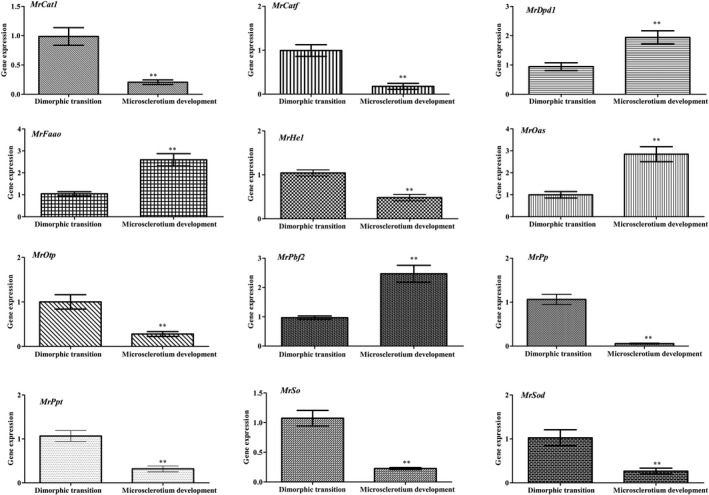
Overview of two distinct M. rileyi morphogenesis with respect to relative transcript analysis. Data were obtained from transcriptomic data of dimorphic transition vs microsclerotium development in the ΔMrNsdD mutant. Error bars represent standard error. *P < 0.05, **P < 0.01, significant differences compared to the data from the dimorphic transition phase.
Role of MrNsdD in tolerance to heat and oxidative stress
Overall, the heat tolerance of conidial germination, expressed as the incubation time at 45 ℃ required to reduce germination by 50% (GR50), was significantly lower in the ΔMrNsdD mutant (GR50 = 37.67 ± 3.32 min) than in the WT (GR50 = 51.58 ± 4.05 min) and complemented strains (GR50 = 53.67 ± 2.62 min) (P < 0.01) (Fig. 6A). These results suggested that MrNsdD was important for normal thermal stress response.
Fig. 6.
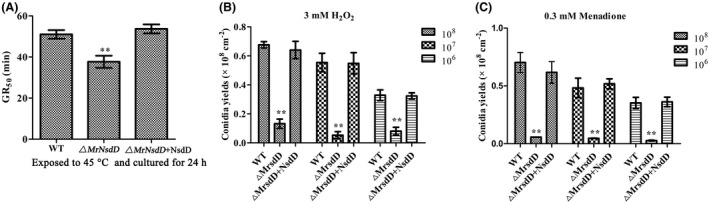
Effects of MrNsdD on heat and abiotic stress tolerance.
A. GR50 (the incubation time at 45 ℃ required to reduce germination by 50%) (min) for conidial tolerance to a heat stress at 45℃. Data were estimated using a probit analysis.
B, C. Conidial yields of tested strains with oxidative stress. Error bars represent standard error. *P < 0.05, **P < 0.01 compared with the WT strain.
To examine the role of the ΔMrNsdD mutant in tolerance to abiotic stresses, the strains tested were incubated on SMAY plates containing oxidative stress agents. Compared with the WT and complemented strains, under oxidative stress conditions, the conidial yields of the ΔMrNsdD mutant were severely affected, exhibiting 52–91% decreases (Fig. 6B and C). Compared with the conidial yields without abiotic stress, there is no conidium production loss in the null mutant with respect to oxidative stress.
Role of MrNsdD in virulence
To evaluate the role of the MrNsdD gene in determining fungal virulence, two methods of infection were used. Compared with the WT and the complemented strains, the half‐maximal lethal time (LT50), reflecting the activity of the ΔMrNsdD mutant towards Spodoptera litura third‐instar larvae, as a result of topical application was significantly delayed by 1.5 days (P < 0.01) and of injection infection was significantly delayed by 2.9 d (P < 0.01) (Fig. 7). The mean ± SE LT50 values for assessing the virulence of the WT strain were 6.2 ± 0.3 d using the topical bioassay and 4.3 ± 0.4 days using the injection bioassays, compared with LT50 values exhibited by the complemented strain of 6.3 ± 0.3 days from topical bioassay and 4.9 ± 0.2 days from the injection bioassays. These results showed that MrNsdD contributes to fungal virulence.
Fig. 7.
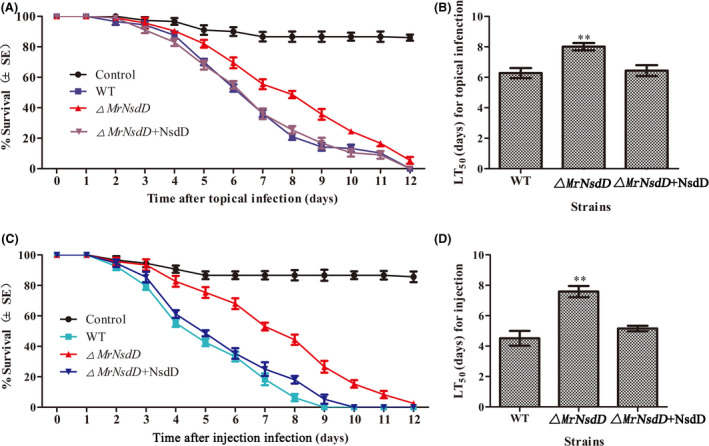
Insect bioassays. Insect survival after (A) topical application and (C) injection of conidia suspension of the tested strains. LT50 (half‐maximal lethal time) for the virulence of tested strains inoculated by (B) topical infection and (D) injection application assays. Error bars represent standard error. *P < 0.05, **P < 0.01, significant differences compared with the WT.
Discussion
NsdD plays pivotal roles in asexual and sexual development in fungi, but its functions in entomopathogenic fungi have not previously been characterized. In the current study, the function of MrNsdD was investigated in M. rileyi, revealing that knockout of MrNsdD resulted in decreased conidial and microsclerotium yield and increased hypersensitivity to thermal stress, compared with the WT parent.
NsdD in Aspergillus spp. and its orthologs, such as SUB‐1 in N. crassa, SsNsd1 in S. sclerotiorum, Csm1 in F. fujikuroi and PoxNsdD in P. oxalicum, had previously been confirmed as key negative regulators of conidiation (Ogawa et al., 2010; Park and Yu, 2012; Niehaus et al., 2017; He et al., 2018; Li et al., 2018). However, deletion of MrNsdD in M. rileyi delayed conidiation (Fig. 1E). Unlike the above‐mentioned filamentous fungi, M. rileyi has a special dimorphic lifestyle with yeast cells forming on SMAY medium for 2–4 days, before transformation into the filamentous form and conidium production after 6 days (Pendland and Boucias, 1997). Previous investigations had shown that the delay in making the dimorphic transition in regulatory gene mutants in M. rileyi resulted in decreased conidiation (Wang et al., 2018, 2019; Song et al., 2018a), whereas advancing the time of dimorphic transition in gene mutants increased conidial production (Song et al., 2018b). The delay in the dimorphic transition in the ΔMrNsdD mutant (Fig. 1C) negatively affected conidium production. On the other hand, the defective architecture of the conidiophore structure and the down‐regulation of expression of conidiation‐required genes (Fig. 2) in the ΔMrNsdD mutant could also be causes of delayed and reduced conidiation. These studies showed that different strategies existed for regulating conidiation via NsdD in different species of filamentous fungi.
The dimorphic transition is critical for the pathogenesis and lifecycle of dimorphic fungi in vitro and in vivo (Boyce and Adrianopoulos, 2015; Gauthier, 2015). However, the mechanisms by which the dimorphic transition occurs are not well defined. Previous investigations had confirmed that the dimorphic transition process was associated with increased reactive oxygen species (ROS) levels (Song et al., 2018b). Further transcriptomics investigations of the mutant found that the expression of many genes annotated as being related to ROS detoxification was up‐ or down‐regulated during the dimorphic transition in the ΔMrNsdD mutant (Figs S6 and S10).
Multicellular microsclerotia form following the aggregation of vegetative hyphae and are enclosed by a melanized rind layer, which plays a significant role in the survival and persistence of these infectious propagules in nature (Jackson et al., 2010; Song et al., 2013). The melanized M. rileyi microsclerotia, induced in liquid culture, also exhibited excellent stress tolerance and storability (Song et al., 2014). Interestingly, as with defective microsclerotium formation in S. sclerotiorum (Li et al., 2018) and sclerotium formation in Aspergillus flavus (Cary et al., 2012), the ΔMrNsdD mutants produced only a few microsclerotium (Fig. 4). A total of 398 DEGs were identified in the ΔMrNsdD mutant (Fig. 3), and, as in other fungi (Jackson et al., 2010; Cary et al., 2012; Li et al., 2018), the regulation of ROS scavenging and secondary metabolism by MrNsdD was observed (Table S5 and Fig. S8). A prevailing theory is that interplays between different transcription factors regulate microsclerotium differentiation (Song, 2018). Our previous study demonstrated that a bZIP transcription factor (MrAp1) plays an important role in mediating redox homoeostasis during microsclerotium development (Song et al., 2018a). Additionally, an APSES‐type transcription factor orthologous gene (MrApses) in M. rileyi was shown to be involved in the regulation of morphogenesis in ascomycete fungi (Mancera et al., 2015; Sarmiento‐Villamil et al., 2018) and was confirmed to mediate microsclerotium development (Xin et al., 2020). Further analysis found that the expression of the two transcription factors (MrAp1 and MrApses) was up‐regulated in the ΔMrNsdD mutant during microsclerotium development (Table S5 and Fig. S9) and the expression of MrNsdD was up‐regulated in two independent transcription factor deletion mutants (Song et al., 2018a; Xin et al., 2020), indicating that these three transcription factors (MrAp1, MrApses and MrNsdD) exhibited interplay regulation in microsclerotium development. Further bioinformatics analysis found that the three transcription factors co‐regulated the expression of 51 genes during microsclerotium development (Fig. S13). Further experiments are needed to elucidate how the interplay among these transcription factor genes regulates microsclerotium development.
The DEGs, identified from the transcriptomics data, might play an important role in the two distinct morphologies. Although the NsdD is a global regulator, there were distinct patterns of gene expression regulated by MrNsdD during the yeast‐to‐hypha transition and during microsclerotium formation (Fig. 5, Table S7). Detecting different carbon sources and utilizing them in the most efficient manner possible would require complex signalling networks (Huberman et al., 2018). Interestingly, the identification of DEGs from the carbon metabolism pathway led to the discovery that MrNsdD was involved in the metabolism of different carbon sources. Previous investigations had proposed that ROS were associated with both dimorphic transition and microsclerotium development (Song, 2018; Song et al., 2018a, 2018b). Peroxisomes are among the main organelles for ROS production in fungi. The DEGs associated with peroxisomes indicated that MrNsdD exhibited not only analogous functions but also diverse roles in regulation of expression. However, the detailed mechanisms by which regulation occurs need to be further studied.
Successful insect infection by entomopathogenic fungi is closely related to the infection formation, adaptation to the stress associated with the host and utilization of the nutrition from the host for proliferation (Chen et al., 2016; Wang et al., 2016; Zhao et al., 2016). Similar to results of studies on other fungi (Schumacher et al., 2014; Niehaus et al., 2017; Li et al., 2018), disruption of MrNsdD resulted in decreased virulence (Fig. 7). Unlike in B. cinerea and F. fujikuroi that ΔMrNsdD mutants were not hypersensitive to oxidative stress (Schumacher et al., 2014; Niehaus et al., 2017), one possible explanation for the decreased virulence is reduced dimorphic transition, as a slower growing fungus takes longer to kill insects.
In the present study, the regulatory roles of MrNsdD in the yeast‐to‐hypha transition, in conidium and microsclerotium development and in stress tolerance in M. rileyi were dissected, providing new insights into the control of fungal morphogenesis. Such systematic and transcriptome‐wide characterization can be effectively used to provide further information on the mechanisms governing the lifecycle of fungi.
Experimental procedures
Microbial strains and culture conditions
The M. rileyi strain CQNr01 (Engineering Research Center for Fungal Insecticides, Chongqing, China) was used as the WT in this study. Escherichia coli DH5α (Invitrogen, Shanghai, China) was used for plasmid propagation. Agrobacterium tumefaciens AGL‐1 (Invitrogen) was used for fungal transformations. M. rileyi cultures were routinely grown on SMAY solid medium. The ΔMrNsdD mutants were constructed by knocking out the MrNsdD gene and were screened for on SMAY medium supplemented with 450 μg ml−1 hygromycin B. Complemented mutants were isolated on modified Czapek‐Dox medium (40 g l−1 maltose, 2 g l−1 NaNO3, 1 g l−1 KH2PO4, 0.5 g l−1 MgSO4.7H2O, 0.5 g l−1 KCl, 0.01 g l−1 FeSO4.7H2O and 15 g l−1 agar) supplemented with 50 μg ml−1 sulfonylurea as described previously (Song et al., 2018a).
Genomic manipulation and analysis
The MrNsdD sequence obtained from previous transcriptomics analysis (Song et al., 2013) was used as the query to search through the M. rileyi genome database (NCBI accession No. AZHC00000000.1) (Shang et al., 2016). Orthologs were structurally compared through Blast analysis (http://blast.ncbi.nlm.nih.gov/Blast.cgi). Relationships were determined by construction of a phylogenetic tree with the neighbour‐joining method, using MEGA6 software (http://www.megasoftware.net).
To determine the functions of MrNsdD, gene deletion was performed by using Agrobacterium‐mediated transformation (Shao et al., 2015). The deletion plasmid consisted of a Pzp‐Ptrpc‐Hph‐Knock backbone, two 1.4‐kb flanking regions and a hygromycin‐trpC resistance cassette. The flanking regions were amplified by PCR, using NsLF/NsLR and NsRF/NsRR (Table S1), and treated with the restriction enzymes, and then ligated independently into Pzp‐Ptrpc‐Hph‐Knock. The deletion plasmid was then transformed into the WT for targeted gene deletion by homologous recombination. For complementation of the deletion mutants, a full‐length open reading frame with putative promoter and downstream sequences of MrNsdD was amplified and inserted into the complementation vector Pzp‐Sur‐cassette (Song et al., 2018a). Putative deletion or complemented mutants were screened on a selective medium and verified by PCR and qPCR (Song et al., 2018a).
Phenotypic analysis
Aliquots (2.5 μl) of conidial suspensions (106, 107, or 108 conidia ml−1) of tested strains were pipetted centrally onto SMAY plates (9‐cm diameter) and incubated at 25 °C under continuous light for 12 d. From 6 d onwards, three culture plugs were taken each day at 3‐days intervals using a borer. The aerial conidiation capacity of each strain was quantified according to previously described methods (Song et al., 2018b). Colony morphology was examined, and images were collected using a digital camera (60‐mm Macro lens, Canon Inc., Tokyo, Japan) and a light microscope. To assess the capacity for dimorphic transition, switching rates were assessed as previously described (Li et al., 2016). In brief, approximately 100 simple yeast cells of each strain were pipetted onto a SMAY plate and grown at 25 °C. Switching percentages at each of the indicated times were recorded, and TT50 was calculated.
Conidial suspensions (108 conidia ml−1) of the tested strains were inoculated onto SMAY plates at 25 ºC. After inoculation for 14 h, germination was assessed every 2 h, and the number of germinated conidia was counted under a light microscope (400×). Conidial thermotolerance was quantified as the GR50 after exposure to 45 °C in a water bath as described previously (Wang et al., 2013; Song et al., 2014). To assess the clonal growth rate, hyphal discs (6‐mm diameter) were taken from 3‐days‐old SMAY cultures, using a borer, and placed centrally onto SMAY plates and incubated for 12 days at 25 °C. The mean growth diameter of each colony after each stress period was estimated as a growth index for each strain.
To quantify oxidative stress tolerance, 2.5 μl aliquots conidial suspensions (106, 107 or 108 conidia ml−1) of each strain were pipetted onto SMAY plates supplemented with menadione (0.03 mM) or H2O2 (3 mM) for the oxidative stress assay. All plates were incubated at 25 °C under continuous light for 12 days, following which the conidial yields of each strain were determined as described previously (Song et al., 2018b).
To examine the role of MrNsdD in microsclerotium differentiation, 1 ml of 108 conidia ml−1 suspension of each strain was inoculated into liquid AM (40 g l−1 glucose, 2.5 g l−1 peptone, 5 g l−1 yeast extract, 4.0 g l−1 KH2PO4, 0.8 g l−1 CaCl2.2H2O, 0.6 g l−1 MgSO4.7H2O, 0.1 g l−1 FeSO4.7H2O, 37 mg l−1 CoCl2.6H2O, 16 mg l−1 MnSO4.H2O and 14 mg l−1 ZnSO4.7H2O) for microsclerotium production as described previously (Song et al., 2013). After 6 d of shaking incubation, biomass and microsclerotium yield were quantified as described previously (Song et al., 2018a). Microsclerotium morphology was observed and recorded using the digital camera and microscope set‐up described earlier.
Transcriptional activity and transcriptomics analysis
To quantify transcript abundance of specific genes associated with conidium development, a 2.5 μl aliquot of a conidial suspensions (107 conidia ml−1) of the WT strain was pipetted onto a SMAY plate, which was incubated at 25 °C, and samples were collected at 0, 2, 4, 6 and 8 days for total RNA extraction. For time‐specific expression patterns during microsclerotium formation, samples of WT inoculated into liquid AM and incubated at 25 °C were collected at 36, 60, 72, 96 and 120 h for total RNA extraction using the same method. Gene expression patterns were confirmed for samples of each strain cultured in AM for 72 h or on SMAY for 3 or 6 days. Total RNAs were reverse transcribed into complemented DNAs (cDNAs), using SuperScript Ⅱ Reverse Transcriptase (Invitrogen). Each cDNA was used as template for qPCR using SYBR Green (Invitrogen). Three replicates were performed, and the amplicons were used for melting curve analysis to check the amplification specificity. The transcripts of β‐tubulin (Mrtub) and translation elongation factor (Mrtef) genes were used as internal standards. Relative transcript level of each gene was calculated using the 2−ΔΔCt method (Vandesompele et al., 2002).
To further study the expression patterns of MrNsdD during dimorphic transition or microsclerotium development, transcriptome analysis of the WT and the ΔMrNsdD mutant cultured in AM for 72 h or on SMAY for 3 days was performed in triplicate. RNA samples, library construction and sequencing were performed on an Illumina HiSeq TM 2500 platform (BioMarker, Beijing, China). The target genes regulated by MrNsdD were quantified using the fragments per kilobase of exon per million mapped fragments method (Mortazavi et al., 2008). DEGs were performed with DESeq software (Ostlund et al., 2010). DEGs were annotated and assigned functional categories using MapMan annotation (Thimm et al., 2004). Raw sequence data have been deposited in the Beijing Institute of Genomics Genome Sequence Archive (accession number PRJCA001563).
Virulence assays
Conidial virulence was assessed on third‐instar S. litura larvae by immersion into 5 μl cottonseed oil of 1 × 106 conidia ml−1 suspension or injection with the aqueous conidial suspension as described previously (Song et al., 2018a). Thirty larvae were treated with three replicates per treatment group. Pure cottonseed oil and sterile water containing 0.01% Tween 80 without conidia were used as sham controls. All treated larvae were reared as described previously (Song et al., 2018a). The survival of larvae was monitored and recorded daily, and the LT50 was estimated by probit analysis using SAS version 9.1 software (Raymond, 1985).
Statistical analysis
All data from the studies, with three replicates per treatment group, were analysed using analysis of variance with SPSS 16.0 software (IBM, Armonk, NY, USA), with multiple pairwise comparisons being carried out using Duncan’s multiple range tests, with P ≤ 0.05 being considered to be significant. Graphs were prepared using GraphPad Prism 5 (GraphPad Software Inc., La Jolla, CA, USA).
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Fig. S1. Phylogenetic tree of MrNsdD protein.
Fig. S2. Confirmation of gene disruption and complementation.
Fig. S3. Gene ontology annotation of differentially expressed genes and all genes during dimorphic transition.
Fig. S4. Clusters of orthologous groups classifications of consensus sequence during dimorphic transition.
Fig. S5. Kyoto Encyclopedia of Genes and Genomes enrichment pathways of differentially expressed genes during dimorphic transition.
Fig. S6. Gene ontology annotation of differentially expressed genes and all genes during microsclerotium development.
Fig. S7. Clusters of orthologous groups classifications of consensus sequence during microsclerotium development.
Fig. S8. Kyoto Encyclopedia of Genes and Genomes enrichment pathways of differentially expressed genes during microsclerotium development.
Fig. S9. Quantitative real‐time‐PCR (qPCR) of the WT and the ΔMrNsdD mutants during (A) dimorphic transition and (B) microsclerotium development.
Fig. S10. Gene ontology annotation of differentially expressed genes and all genes in two developmental stages (dimorphic transition vs microsclerotium development) in the ΔMrNsdD mutant.
Fig. S11. Clusters of orthologous groups classifications of consensus sequence in two developmental stages (dimorphic transition vs microsclerotium development) in the ΔMrNsdD mutant.
Fig. S12. Kyoto Encyclopedia of Genes and Genomes enrichment pathways of consensus sequence in two developmental stages (dimorphic transition vs microsclerotium development) in the ΔMrNsdD mutant.
Fig. S13. Venn diagram showing the number of shared differentially expressed genes in the three fungal transcription factors.
Table S1. Oligonucleotide primers used in this study.
Table S2. Unique genes expressed only in WT or ΔMrNsdD mutant during dimorphic transition.
Table S3. Unique genes expressed only in WT or ΔMrNsdD mutant during microsclerotium development.
Table S4. Gene expression in the ΔMrNsdD mutant relative to WT during dimorphic transition.
Table S5. Gene expression in the ΔMrNsdD mutant relative to WT during microsclerotium development.
Table S6. Unique genes expressed only in microsclerotium development and dimorphic transition.
Table S7. Differentially expressed genes in the ΔMrNsdD mutant involved in carbon metabolism and peroxisome in two distinct development stages.
Acknowledgements
This research was supported financially by National Natural Science Foundation of China (No. 31701127), Science and Technology Project of Sichuan (2019YJ0407) and Luzhou (No. 2018‐JYJ‐32, 2019‐RCM‐94), and Foundation of Southwest Medical University (01‐00031114).
Microbial Biotechnology (2020) 13(5), 1489–1501
Funding information
This research was supported financially by National Natural Science Foundation of China (No. 31701127), Science and Technology Project of Sichuan (2019YJ0407) and Luzhou (No. 2018‐JYJ‐32, 2019‐RCM‐94), and Foundation of Southwest Medical University (01‐00031114).
References
- Boyce, K. J. , and Adrianopoulos, A. (2015) Fungal dimorphism: the switch from hyphae to yeast is a specialized morphogenetic adaption allowing colonization of a host. FEMS Microbiol Rev 39: 797–811. [DOI] [PubMed] [Google Scholar]
- Cary, J. W. , Harris‐Coward, P. Y. , Ehrlich, K. C. , Mack, B. M. , Kale, S. P. , Larey, C. , et al (2012) NsdC and NsdD affect Aspergillus flavus morphogenesis and aflatoxin production. Eukaryot Cell 11: 1104–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, X. X. , Xu, C. , Qian, Y. , Liu, R. , Zhang, Q. Q. , Zeng, G. H. , et al (2016) MAPK cascade‐mediated regulation of pathogenicity, conidiation and tolerance to abiotic stresses in the entomopathogenic fungus Metarhizium robertsii . Environ Microbiol 18: 1048–1062. [DOI] [PubMed] [Google Scholar]
- de Faria, M. R. , and Wraight, S. P. (2007) Mycoinsecticides and Mycoacaricides: a comprehensive list with worldwide coverage and international classification of formulation types. Biol Control 43: 237–256. [Google Scholar]
- Fronza, E. , Specht, A. , Heinzen, H. , and de Barros, N. M. (2017) Metarhizium (Nomuraea) rileyi as biological control agent. Biocontrol Sci Techn 2: 1–22. [Google Scholar]
- García‐pedrajas, M. D. , Baeza‐Montañez, L. , and Gold, S. E. (2010) Regulation of Ustilago maydis dimorphism, sporulation, and pathogenic development by a transcription factor with a highly conserved APSES domain. Mol Plant Microbe Interact 23: 211–222. [DOI] [PubMed] [Google Scholar]
- Gauthier, G. M. (2015) Dimorphism in fungal pathogens of mammals, plants, and insects. PLoS Pathog 11: e1004608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, K. H. , Han, K. Y. , Yu, J. H. , Chae, K. S. , Jahng, K. Y. , and Han, D. M. (2001) The nsdD gene encodes a putative GATA‐type transcription factor necessary for sexual development of Aspergillus nidulans . Mol Microbiol 41: 299–309. [DOI] [PubMed] [Google Scholar]
- He, Q. P. , Zhao, S. A. , Wang, J. X. , Li, C. X. , Yan, Y. S. , Wang, L. , et al (2018) Transcription factor PoxNsdD regulates the expression of genes involved in plant biomass‐degrading enzymes, conidiation and pigment biosynthesis in Penicillium oxalicum . Appl Environ Microbiol 84: e01039‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, X. , Xiao, G. H. , Zheng, P. , Shang, Y. F. , Su, Y. , Zhang, X. Y. , et al (2014) Trajectory and genomic determinants of fungal‐pathogen speciation and host adaptation. Proc Natl Acad Sci USA 111: 16796–16801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, P. , Wang, Y. , Zhou, J. , Pan, Y. , and Liu, G. (2015) AcstuA, which encodes an APSES transcription regulator, is involved in conidiation, cephalosporin biosynthesis and cell wall integrity of Acremonium chrysogenum . Fungal Genet Biol 83: 26–40. [DOI] [PubMed] [Google Scholar]
- Huberman, L. B. , Coradetti, S. T. , and Glass, N. L. (2018) Network of nutrient‐sensing pathways and a conserved kinase cascade integrate osmolarity and carbon sensing in Neurospora crassa . Proc Natl Acad Sci USA 25: 8665–8674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson, M. A. , Dunlap, C. A. , and Jaronski, S. T. (2010) Ecological considerations in producing and formulating fungal entomopathogens for use in insect biocontrol. Biocontrol 55: 129–145. [Google Scholar]
- Kim, H. R. , Chae, K. S. , Han, K. H. , and Han, D. M. (2009) The nsdC gene encoding a putative C2H2‐type transcription factor is a key activator of sexual development in Aspergillus nidulans . Genetics 182: 771–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, S. , Lee, S. J. , Nai, Y. S. , Yu, J. S. , Lee, M. R. , Yang, Y. T. , et al (2016) Characterization of T‐DNA insertion mutants with decreased virulence in the entomopathogenic fungus Beauveria bassiana . Appl Microbiol Biotechnol 100: 8889–8900. [DOI] [PubMed] [Google Scholar]
- Lee, M. K. , Kwon, N. J. , Choi, J. M. , Lee, I. S. , Jung, S. , and Yu, J. H. (2014) NsdD is a key repressor of asexual development in Aspergillus nidulans . Genetics 197: 159–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y. , Wang, Z. K. , Liu, X. E. , Song, Z. Y. , Li, R. , Shao, C. W. , et al (2016) Siderophore biosynthesis but not reductive iron assimilation is essential for the dimorphic fungus Nomuraea rileyi conidiation, dimorphism transition, resistance to oxidative stress, pigmented microsclerotium formation, and virulence. Front Microbiol 7: 931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J. T. , Mu, W. H. , Veluchamy, S. , Liu, Y. Z. , Zhang, Y. H. , Pan, H. Y. , et al (2018) The GATA‐type IVb zinc‐finger transcription factor SsNsd1 regulates asexual–sexual development and appressoria formation in Sclerotinia sclerotiorum . Mol Plant Pathol 19: 1679–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancera, E. , Porman, A. M. , Cuoman, A. M. , Bennett, R. J. , and Johnson, A. D. (2015) Finding a missing gene: EFG1 regulates morphogenesis in Candida tropicalis . G3‐Genes Genom Genet 5: 849–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi, A. , Williams, B. A. , McCue, K. , Schaeffer, L. , and Wold, B. (2008) Mapping and quantifying mammalian transcriptomes by RNA‐Seq. Nat Methods 5: 621–628. [DOI] [PubMed] [Google Scholar]
- Niehaus, E. M. , Schumacher, J. , Burkhardt, I. , Rabe, P. , Münsterkötter, M. , Güldener, U. , et al (2017) The GATA‐type transcription factor Csm1 regulates conidiation and secondary metabolism in Fusarium fujikuroi . Front Microbiol 8: 1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa, M. , Tokuoka, M. , Jin, F. J. , Takahashi, T. , and Koyama, Y. (2010) Genetic analysis of conidiation regulatory pathway in koji‐mold Aspergillus oryzae . Fungal Genet Biol 47: 10–18. [DOI] [PubMed] [Google Scholar]
- Ostlund, G. , Schmitt, T. , Forslund, K. , Kostler, T. , Messina, D. N. , Roopra, S. , et al (2010) InParanoid 7: new algorithms and tools for eukaryotic orthology analysis. Nucleic Acids Res 38: 196–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, H. S. , and Yu, J. H. (2012) Genetic control of asexual sporulation in filamentous fungi. Curr Oopin Microbiol 15: 669–677. [DOI] [PubMed] [Google Scholar]
- Pendland, J. C. , and Boucias, D. G. (1997) In vitro growth of the entomopathogenic hyphomycete Nomuraea rileyi . Mycologia 89: 66–71. [Google Scholar]
- Raymond, M. (1985) Presentation d’un programme d’analyse logprobit pour micro‐orinateur. Cah Orstom Entomol Med Parasitol 22: 117–121. [Google Scholar]
- Sarmiento‐Villamil, J. L. , García‐Pedrajas, N. E. , Baeza‐Montañez, L. , and García‐Pedrajas, M. D. (2018) The APSES transcription factor Vst1 is a key regulator of development in microsclerotium‐ and resting mycelium‐producing Verticillium species. Mol Plant Pathol 19: 59–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher, J. , Simon, A. , Cohrs, K. C. , Viaud, M. , and Tudzynski, P. (2014) The transcription factor BcLTF1 regulates virulence and light responses in the necrotrophic plant pathogen Botrytis cinerea . PLoS Genet 10: e1004040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo, J. A. , Guan, Y. , and Yu, J. H. (2006) FluG‐dependent asexual development in Aspergillus nidulans occurs via depression. Genetics 172: 1535–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang, Y. F. , Xiao, G. H. , Zheng, P. , Cen, K. , Zhan, S. , and Wang, C. S. (2016) Divergent and convergent evolution of fungal pathogenicity. Genome Biol Evol 8: 1374–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao, C. W. , Yin, Y. P. , Qi, Z. R. , Li, R. , Song, Z. Y. , and Wang, Z. K. (2015) Agrobacterium tumefaciens mediated transformation of the entomopathogenic fungus Nomuraea rileyi . Fungal Genet Biol 83: 19–25. [DOI] [PubMed] [Google Scholar]
- Shelest, E. (2017) Transcription factors in fungi: TFome dynamics, three major families, and dual‐specificity TFs. Front Genet 8: 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, Z. Y. (2018) Fungal microsclerotia development: essential prerequisite, influence factors, and molecular mechanism. Appl Microbiol Biotechnol 102(23): 9873–9880. [DOI] [PubMed] [Google Scholar]
- Song, Z. Y. , Yin, Y. P. , Jiang, S. S. , Liu, J. J. , Chen, H. , and Wang, Z. K. (2013) Comparative transcriptome analysis of microsclerotia development in Nomuraea rileyi . BMC Genom 14: 411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, Z. Y. , Yin, Y. P. , Jiang, S. S. , Liu, J. J. , and Wang, Z. K. (2014) Optimization of culture medium for microsclerotia production by Nomuraea rileyi and analysis of their viability for use as a mycoinsecticide. Biocontrol 59: 597–605. [Google Scholar]
- Song, Z. Y. , Yin, Y. P. , Lin, Y. L. , Du, F. , Ren, G. W. , and Wang, Z. K. (2018a) The bZip transcriptional factor activator protein‐1 regulates Metarhizium rileyi morphology and mediates microsclerotia formation. Appl Microbiol Biot 102: 4577–4588. [DOI] [PubMed] [Google Scholar]
- Song, Z. Y. , Yang, J. , Xin, C. Y. , Xing, X. R. , Yuan, Q. , Yin, Y. P. , et al (2018b) A transcription factor, MrMsn2, in the dimorphic fungus Metarhizium rileyi is essential for dimorphism transition, aggravated pigmentation, conidiation and microsclerotia formation. Microbial Biotechnol 11: 1157–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thimm, O. , Bläsing, O. , Gibon, Y. , Nagel, A. , Meyer, S. , Krüger, P. , et al (2004) MAPMAN: a user‐driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J 37: 914–939. [DOI] [PubMed] [Google Scholar]
- Vandesompele, J. , De Preter, K. , Pattyn, F. , Poppe, B. , Roy, N. V. , Paepe, A. D. , et al (2002) Accurate normalization of real‐time quantitative RT‐PCR data by geometric averaging of multiple internal control genes. Genome Biol 3: Research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J. , Zhou, G. , Ying, S. H. , and Feng, M. G. (2013) P‐type calcium ATPase functions as a core regulator of Beauveria bassiana growth, conidiation and responses to multiple stressful stimuli through cross‐talk with signalling networks. Environ Microbiol 15: 967–979. [DOI] [PubMed] [Google Scholar]
- Wang, J. B. , St Leger, R. J. , and Wang, C. S. (2016) Advances in genomics of entomopathogenic fungi. Adv Genet 94: 67–105. [DOI] [PubMed] [Google Scholar]
- Wang, Z. K. , Song, Z. Y. , Zhong, Q. , Du, F. , and Yin, Y. P. (2018) Adaption to stress via Pbs2 during Metarhizium rileyi conidial and microsclerotia development. World J Microbiol Biotechnol 34: 107. [DOI] [PubMed] [Google Scholar]
- Wang, Z. K. , Yang, J. , Xin, C. Y. , Xing, X. R. , Yin, Y. P. , Chen, L. , et al (2019) Regulation of conidiation, dimorphism transition, and microsclerotia formation by MrSwi6 transcription factor in dimorphic fungus Metarhizium rileyi . World J Microbiol Biotechnol 35: 46. [DOI] [PubMed] [Google Scholar]
- Wieser, J. , Lee, B. N. , Fondon, J. 3rd , and Adams, T. H. (1994) Genetic requirements for initiating asexual development in Aspergillus nidulans . Curr Genet 27: 62–69. [DOI] [PubMed] [Google Scholar]
- Xin, C. Y. , Zhang, J. P. , Nian, S. J. , Wang, G. X. , Wang, Z. K. , Song, Z. Y. , et al (2020) Analogous and diverse functions of APSES‐type transcription factors in the morphogenesis of the entomopathogenic fungus, Metarhizium rileyi . Appl Environ Microb 86: e02928‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, H. , Lovett, B. , and Fang, W. (2016) Genetically engineering entomopathogenic fungi. Adv Genet 94: 137–163. [DOI] [PubMed] [Google Scholar]
- Zhou, G. , Ying, S. H. , Hu, Y. , Fang, X. , Feng, M. G. , and Wang, J. (2018) Roles of three HSF domain‐containing proteins in mediating heat‐shock protein genes and sustaining asexual cycle, stress tolerance, and virulence in Beauveria bassiana . Front Microbiol 9: 1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Phylogenetic tree of MrNsdD protein.
Fig. S2. Confirmation of gene disruption and complementation.
Fig. S3. Gene ontology annotation of differentially expressed genes and all genes during dimorphic transition.
Fig. S4. Clusters of orthologous groups classifications of consensus sequence during dimorphic transition.
Fig. S5. Kyoto Encyclopedia of Genes and Genomes enrichment pathways of differentially expressed genes during dimorphic transition.
Fig. S6. Gene ontology annotation of differentially expressed genes and all genes during microsclerotium development.
Fig. S7. Clusters of orthologous groups classifications of consensus sequence during microsclerotium development.
Fig. S8. Kyoto Encyclopedia of Genes and Genomes enrichment pathways of differentially expressed genes during microsclerotium development.
Fig. S9. Quantitative real‐time‐PCR (qPCR) of the WT and the ΔMrNsdD mutants during (A) dimorphic transition and (B) microsclerotium development.
Fig. S10. Gene ontology annotation of differentially expressed genes and all genes in two developmental stages (dimorphic transition vs microsclerotium development) in the ΔMrNsdD mutant.
Fig. S11. Clusters of orthologous groups classifications of consensus sequence in two developmental stages (dimorphic transition vs microsclerotium development) in the ΔMrNsdD mutant.
Fig. S12. Kyoto Encyclopedia of Genes and Genomes enrichment pathways of consensus sequence in two developmental stages (dimorphic transition vs microsclerotium development) in the ΔMrNsdD mutant.
Fig. S13. Venn diagram showing the number of shared differentially expressed genes in the three fungal transcription factors.
Table S1. Oligonucleotide primers used in this study.
Table S2. Unique genes expressed only in WT or ΔMrNsdD mutant during dimorphic transition.
Table S3. Unique genes expressed only in WT or ΔMrNsdD mutant during microsclerotium development.
Table S4. Gene expression in the ΔMrNsdD mutant relative to WT during dimorphic transition.
Table S5. Gene expression in the ΔMrNsdD mutant relative to WT during microsclerotium development.
Table S6. Unique genes expressed only in microsclerotium development and dimorphic transition.
Table S7. Differentially expressed genes in the ΔMrNsdD mutant involved in carbon metabolism and peroxisome in two distinct development stages.