

Abstract
Primary brain tumor patients often experience neurological, cognitive, and depressive symptoms that profoundly affect quality of life. The DNA alkylating agent, temozolomide (TMZ), along with radiation therapy forms the standard of care for glioblastoma (GBM) – the most common and aggressive of all brain cancers. Numerous studies have reported that TMZ disrupts hippocampal neurogenesis and causes spatial learning deficits in rodents; however, the effect of TMZ on mature hippocampal neurons has not been addressed. In this study, we examined the mitochondrial-mediated mechanisms involving TMZ-induced neural damage in primary rat neural stem/progenitor cells (NSC) and hippocampal neurons. TMZ inhibited mtDNA replication and transcription of mitochondrial genes (ND1 and Cyt b) in NSC by 24 h, whereas the effect of TMZ on neuronal mtDNA transcription was less pronounced. Transmission electron microscopy imaging revealed mitochondrial degradation in TMZ-treated NSC. Acute TMZ exposure (4 h) caused a rapid reduction in dendritic branching and loss of postsynaptic density-95 (PSD95) puncta on dendrites. Longer TMZ exposure impaired mitochondrial respiratory activity, increased oxidative stress, and induced apoptosis in hippocampal neurons. The presented findings suggest that NSC may be more vulnerable to TMZ than hippocampal neurons upon acute exposure; however long-term TMZ exposure results in neuronal mitochondrial respiratory dysfunction and dendritic damage, which may be associated with delayed cognitive impairments.
Keywords: Chemotherapy, Mitochondria, Neural stem/progenitor cells, Hippocampal neurons, Oxidative stress, Neurotoxicity, Temozolomide
1. Introduction
Chemotherapy-related cognitive impairments (CRCI) have been described for various chemotherapeutic agents in patients with CNS (central nervous system) and non-CNS tumors (Taphoorn and Klein, 2004; Winocur et al., 2018). Primary brain tumor patients often experience neurological, cognitive, and depressive symptoms. A meta-analysis of depression in cancer patients revealed that with a prevalence rate of 28%, brain cancer is the subtype most strongly associated with depression (Krebber et al., 2014). The majority (85%) of patients experience neurological complications, although, for over 30% of long-term GBM survivors, the cognitive impairments persist throughout life (Steinbach et al., 2006; Hottinger et al., 2009). The effect of chemotherapy on cognitive sequelae in brain tumor patients is challenging to assess in this patient population due to tumor-specific factors that alter neurocognitive function, such as inflammation and tumor location. Additionally, age, genetic background, treatment related-factors such as cranial radiation, surgical resection, adjuvant treatment, duration, and dosing, further contribute to the impairment of neurocognitive function (Allen and Loughan, 2018; Abrey, 2012).
Cranial radiation therapy (RT) for treatment of CNS tumors has been widely shown to suppress long-term potentiation (LTP) in the dentate gyrus of the hippocampus (Garthe et al., 2009; Wu et al., 2012); deplete neural stem progenitor/cells (NSC) and neurons, and provoke morphological damage to surviving neurons (Parihar and Limoli, 2013), which can result in progressive cognitive impairment (Nolen et al., 2016; Acharya et al., 2011). The DNA alkylating agent temozolomide (TMZ), along with RT forms the standard of care for GBM following tumor resection. GBM is the most common and aggressive of all high-grade gliomas, accounting for approximately 80% of all malignant primary brain tumors (Agnihotri et al., 2013). TMZ targets nuclear DNA (nDNA) and generates nDNA adducts that induce cell cycle arrest and cell death by apoptosis. TMZ also alters mtDNA and respiratory function in glioma (Di et al., 2016; Oliva et al., 2010). However, its effect on NSC and non-proliferating neural cells such as neurons, is not clear.
Reports of TMZ-induced neural toxicity have primarily focused on the loss of NSC in the rodent hippocampus. TMZ is used as an experimental tool to deplete adult rodent neurogenesis; these studies have found its application to be associated with depression (Egeland et al., 2017), anxiety (Pereira-Caixeta et al., 2018), and impaired spatial and contextual discrimination in mice (Niibori et al., 2012; Garthe et al., 2009). To date, no studies have investigated changes in hippocampal neuronal integrity or neural mitochondrial function after TMZ exposure. NSC, hippocampal neurons, and the excitatory synapses they carry, play a critical role in hippocampal processes, including learning and memory (Schmidt-Hieber et al., 2004; Snyder et al., 2001); therefore, it is plausible that neuronal damage may also contribute to the observed deficits following TMZ exposure.
Mitochondrial dysfunction, elevated oxidative stress, and dendritic spine remodeling are hallmarks of neuronal toxicity in a variety of neurodegenerative disorders (Lomeli et al., 2017; Lin and Beal, 2006; Schon and Manfredi, 2003). The brain is highly susceptible to oxidative stress as it has a high metabolic demand and low antioxidant capacity compared to other organs (Commandeur et al., 1995). Neuronal dynamic processes such as synaptic plasticity, neurotransmission, and membrane ion gradients are highly dependent upon ATP generated by the mitochondrial electron transport chain (ETC) (Raefsky and Mattson, 2017). Mitochondrial respiratory dysfunction and oxidative stress are associated with chemotherapy-related cognitive impairments provoked by various chemotherapeutic agents such as cisplatin (Lomeli et al., 2017; Chiu et al., 2018), cyclophosphamide (Oboh and Ogunruku, 2010; Acharya et al., 2015), doxorubicin (Keeney et al., 2018; Tangpong et al., 2007), and methotrexate (Hess and Khasawneh, 2015). Given the known effects of TMZ on mitochondrial function in glioma (Di et al., 2016; Oliva et al., 2010), and its association with depression and NSC depletion (Egeland et al., 2017; Gong et al., 2011), we hypothesized that TMZ induces hippocampal mitochondrial dysfunction and oxidative stress. We examined the in-vitro effects of TMZ on mitochondrial DNA integrity and transcription in NSC and neurons. Additionally, we examined the effects of TMZ on neuronal mitochondrial respiratory function, oxidative stress, and resulting changes in neuronal morphology and survival.
2. Materials and methods
2.1. Standard protocol approvals
All experiments conformed to the National Institutes of Health guidelines. Hippocampal neuron and NSC cell cultures were generated using Sprague Dawley rats (Charles River Laboratories) following the guidelines established by the Institutional Animal Care and Use Committee of the University of California, Irvine (UCI). Institutional Review Board (IRB) approval was obtained at the UCI Medical Center and Children’s Hospital of Orange County (CHOC) for the isolation and expansion of human-derived NSC (SC27) (Schwartz et al., 2003).
2.2. Dissociated hippocampal neuron and NSC cultures
Dissociated hippocampal neuron cultures were prepared from Sprague Dawley pups on postnatal day 0 (P0) of either sex, as described previously (Andres et al., 2013, 2014). Cultures were seeded at a density of 400–600 cells/mm2 on 12 mm coverslips (Thermo Scientific) pre-coated with 0.2 mg/ml poly-D-lysine (Sigma Aldrich) and initially maintained in Neurobasal Medium (NBM) with B-27 (Invitrogen) at 37 °C and 5% CO2. On the third day in-vitro (3 DIV), cultures were treated with 5 μM arabinoside-cytosine (Sigma Aldrich) to inhibit glial proliferation and refreshed biweekly with conditioned media (NBM with B-27 preconditioned for 24 h over 1- to 3-week-old glial cell cultures). Neurons were used for experiments on 17–24 days in-vitro (17–21 DIV).
Rat NSC were isolated from the hippocampi of embryonic day 19 (E19) Sprague Dawley pups and cultured as described (Andres et al., 2014). The cells were plated onto T25 flasks pre-coated with 20 μg/mL fibronectin (Invitrogen) and maintained in Knockout™ DMEM/F-12 with 2 mM GlutaMAX™ Supplement, 20 ng/mL bFGF, 20 ng/mL EGF, and 2% StemPro® Neural supplement (Gibco). Passage 6–10 NSC were used for experiments.
Human NSC (SC27) were derived from post-mortem cortical brain tissue of premature neonates and cultured as previously described (Schwartz et al., 2003). Briefly, following tissue harvest, tissue was diced and digested by incubation at 37 °C for 20–40 min in DMEM/F12 containing 10% FBS, 2.5 U/mL papain (Worthington, Freehold, NJ), and 1 U/mL neutral protease (Roche, Indianapolis, IN). Cortical tissue homogenates were plated onto 5 μg/mL fibronectin-coated dishes in primary growth medium composed of DMEM/F-12 containing 10% BIT 9500 (Stem Cell Technologies), 40 ng/mL bFGF, 20 ng/mL EGF, and 20 ng/mL platelet-derived growth factor-AB (PDGF-AB; Peprotech). After 24 h in culture, the media was refreshed, and adherent cell populations were expanded for three to four passages. Immunocytochemistry and RT-PCR analysis confirmed the expression of NSC markers, including doublecortin, nestin, nucleostemin, SOX2, glial fibrillary acidic protein (GFAP), and Ki67 (Schwartz et al., 2003). SC27 cells were cultured on 10 μg/mL fibronectin-coated T75 flasks in DMEM/F-12 (Gene Clone), containing 1% BIT9500 (Stem Cell Technologies),10 μg/mL Gentamycin (MP Biomedicals), 10 μg/mL Cipro-floxacin (TEVA), 2.5 μg/mL Amphotericin B (Fisher Scientific), 100 μg/mL Pen/Strep (Gibco), 292 μg/mL L-glutamine (Gibco), 20 ng/mL bFGF, and 20 ng/mL EGF. SC27 cultures were passaged when confluent using Nonenzymatic Cell Dissociation Solution (Sigma Aldrich).
2.3. Temozolomide application in-vitro
In-vitro, TMZ (Sigma Aldrich) was made into a 100 mM stock solution by dissolving in DMSO (Sigma Aldrich). TMZ was diluted to a final concentration as specified in the figures in the respective culture medium of each cell type. The control group was exposed to an equal volume of DMSO at the same time-points. The doses used in our study were based on the TMZ doses used in other in-vitro studies of GBM. In the literature, in-vitro studies routinely use 10–500 μM TMZ, as TMZ-sensitive GBM cell lines have an IC50 < 500 μM (Perazzoli et al., 2015), whereas patient-derived low-grade and high-grade GSC lines have TMZ IC50 > 500 μM (Gong et al., 2011; Perazzoli et al., 2015).
2.4. Transmission electron microscopy (TEM)
In-vitro, SC27 (passage 9) cultures were treated with TMZ or DMSO of equal volume (control) for 48 h. The cells were washed with PBS, collected, and fixed for TEM in glutaraldehyde at room temperature. The samples were sent to the University of California, Irvine Pathology Services Core Facility for TEM processing, and imaging. The TEM images were graded blinded to treatment. Percentages of mitochondria type for each treatment group were determined. Normal mitochondria with dense staining of the inner matrix and intact cristae were classified as Type I. Abnormal swollen mitochondria with fragmented cristae were classified as Type II. Lastly, Type III comprised of vesicular mitochondria lacking dense staining of the inner matrix and disorganized cristae. A total of 645 mitochondria were counted in the control group, and 437 mitochondria in the TMZ group. A total of 15, 2 μm images were quantified per group.
2.5. MtDNA qPCR assay
The PCR assay was a modification of Santos et al. (2006). Total DNA was purified from cell samples using the Qiagen Genomic Tip and Genomic DNA Buffer Set Kit (Qiagen, Valencia, CA, USA). For SC27 cells, a small mitochondrial fragment (221-bp) was amplified and standardized to a 13.5-kb fragment from the nuclear-encoded gene β-globin. A mitochondrial fragment (235-bp) was amplified and standardized to a 12.5-kb fragment from the nuclear-encoded gene, Clusterin (TRPM-2), in rat hippocampal neurons and NSC. PCR products were normalized to control levels. The sequences for human β-globin (13.5-kb) and mtDNA fragment (221-bp) primer sets used are: β-globin forward 5′-CGAGTAAGAGACCATTGTGGCAG-3′, reverse 5′-GCACTGGCTTTAGGAGTTGGACT-3′; mtDNA fragment forward 5′-CCCCACAAACCCCATTACTAAACCCA-3′ and reverse 5′-TTTCATCATGCGGAGATGTTGGA-3′. The sequences for rat TRPM-2 (12.5-kb) and mtDNA fragment (235-bp) primer sets used are: TRPM-2 forward 5′-AGACGGGTGAGACAGCTGCACCTTTTC-3′, reverse 5′-CGAGAGCATCAAGTGCAGGCATTAGAG-3′; mtDNA fragment forward 5′-CCTCCCATTCATTATCGCCGCCCTTGC-3′, reverse 5′-GTCTGGGTCTCCTAGTAGGTCTGGGAA-3′.
2.6. Cell viability assay (MTT)
Rat NSC were seeded in 96-well plates at a concentration of 104 cells/well. The cells were incubated with 100 μM, 200 μM, or 500 μM TMZ, 24 h post-seeding. After 7 days, cell viability was measured by MTT assay (Roche). 20 μl of 5 mg/ml MTT tetrazolium salt dissolved in PBS, pH 7.4, was added to each well, and the plate was incubated for 5 h. The medium was aspirated from each well, and 200 μl of DMSO was added to each well to dissolve the insoluble formazan salts. Absorbance was measured at 570 nm using a BioRad 680 plate reader. Each treatment group contained 5 replicates, in 3 independent experiments. Cell viability was normalized to the control group.
2.7. Quantitative RT-PCR assay
The assay was performed as described previously (Lomeli et al., 2017). Total RNA was extracted using RNeasy Mini Kit (Qiagen), and cDNA was generated using the iScript™ cDNA Synthesis Kit (Bio-Rad). The sequences for ND1 gene expression levels were normalized to those of 18S rRNA. The sequences for rat ND1 primer sets were: forward 5′-CACCCCCTTATCAACCTCAA-3′; reverse 5′-ATTTGTTTCTGCGAGGG TTG-3′. Cyt b gene expression levels were normalized to those of 18S rRNA. The sequences for rat Cyt b and 18S rRNA primer sets were: Cyt b forward 5′-CGAAAATCTACCCCCTATT-3′, reverse 5′-GTGTTCTACTGGTTGGCCTC-3′; 18S rRNA forward 5′-TCAATCTCGGGTGGCTGAACG-3′, reverse 5′-GGACCAGAGCGAAAGCATTTG-3′. All primers were ordered from IDT, Integrated Device Technology, Inc, Coralville, Iowa, USA.
2.8. Seahorse XF24 cellular bioenergetic analysis
The Seahorse XF24 Extracellular Flux Analyzer (Agilent Technologies) was used to determine the bioenergetic profile of intact neurons (Lomeli et al., 2017). Oxygen consumption rates (OCR) were measured in adherent hippocampal neurons using the Cell Mito Stress Kit (Agilent Technologies). Dissociated hippocampal neurons were plated at a density of 5 × 104 cells/well in 0.1 mg/mL poly-D-lysine coated XF24 cell culture microplates. Baseline rates were measured at 37 °C three times before the sequential injection of oligomycin (2 μM), FCCP (1 μM), and rotenone (1 μM) plus antimycin A (1 μM). Basal OCR levels were determined by subtracting the non-mitochondrial respiration rate from the last rate measurement before the oligomycin injection. Maximum respiration was calculated by subtracting the non-mitochondrial respiration rate from the maximum rate measurement after FCCP injection. Three OCR measurements were taken after the addition of each inhibitor. All measurements were normalized to protein content per well using the DC protein assay (Bio-Rad). OCR data was collected using the Wave software (Agilent Technologies) and analyzed using the XF Cell Mito Stress Kit Report generator.
2.9. Immunocytochemistry (ICC)
The 24 well-plates containing coverslips with cultured neurons were placed into ice slush. Neurons were fixed with ice-cold 4% paraformaldehyde (PFA, Fisher Scientific) in PBS, pH 7.4, for 12 min. The following antibodies were used: mouse anti-PSD95 1:4000 (Thermo Fisher MA1–046) and rabbit anti-cleaved Caspase-9 (Cell Signaling 9507) 1:100. Anti-PSD95 was diluted in blocking buffer (3% bovine serum albumin, 0.1% Triton-X in PBS, pH 7.4) and anti-cleaved caspase-9 was diluted in blocking buffer (5% donkey serum and 0.3% Triton-X in PBS, pH 7.4) overnight at 4 °C. The next day coverslips were washed with PBS and incubated in the appropriate secondary antibodies conjugated to Alexa Fluor 488 at 1:500 or Alexa Fluor 568 at 1:400 (Invitrogen) at room temperature for 1.5 h.
2.10. Image analysis of dendritic branches and spines
Dendritic spines were visualized by using ICC for PSD95, a reliable marker of mature synapses (Andres et al., 2013). Dendritic branching was evaluated using Sholl analysis. Total dendritic length was measured, and the number of intersections between branches and the concentric circles at increasing 20 μm segments from the soma was quantified. For PSD95 quantification, each individual puncta was considered a separate spine, and counts were not adjusted for puncta size. PSD95 puncta density was quantified as the number of PSD95 puncta per 20 μm of dendrite length, comparing dendrites of the same order (Andres et al., 2014). Severe dendritic injury such as beading was identified in the 500 μM TMZ (24 h) group, which prevented analysis.
2.11. CellROX oxidative stress quantification
Neurons plated on 12 mm coverslips were incubated in culture medium containing 5 μM CellROX Green Reagent (Life Technologies) for 20 min at 37 °C and 5% CO2 (Lomeli et al., 2017). After incubation, cells were fixed as described above and washed with PBS before mounting. Cells were processed for imaging with DAPI Fluoromount G (Southern Biotech) mounting medium. Confocal microscopy, Zeiss LSM700, was used to generate neuronal images. 10 μm z-series (2.5 μm steps) images were captured at 20X (NA 0.8) spanning across entire neurons. The relative fluorescence intensity of the CellROX green probe was quantified by ImageJ. The experiment was repeated twice, with similar results. Each experiment included 3 sister coverslips per treatment group. 5 images were analyzed per coverslip, for a total of 800–1200 cells per treatment group.
2.12. Terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL assay)
The TUNEL assay was performed using the NeuroTACS™ In Situ Apoptosis Detection Kit (Trevigen, Inc. Gaithersburg MD). Following 12 min fixation with 4% PFA in PBS, pH 7.4, neurons were incubated in NeuroPore™ for 2 h at 4 °C. The assay was performed as described in the manufacturer’s protocol. Neurons were imaged by light microscopy using an Olympus BX43 light microscope at 10X magnification.
2.13. Systematic analysis and statistical considerations
Each experiment included 2–3 replicates per treatment group. In the imaging experiments, neurons were sampled equally from each coverslip. For PSD95 puncta quantification, 4 dendrites from 2 separate neurons were sampled per coverslip for each treatment group. Images for PSD95 puncta analysis and Sholl analyses were generated using confocal microscopy, Zeiss LSM 510 (Oberkochen, Germany). 3 μm z-series (0.5 μm step) images were captured from distinct non-over-lapping dendrites and extended at least 100 μm from the soma at 63X (NA 1.4) using an oil-immersion objective. Analyses of all treatment groups were performed using one-way ANOVA or two-way repeated-measures (RM)-ANOVA or one-way ANOVA, followed by Bonferroni’s post-hoc multiple comparisons test. Significance levels were set at 0.05, and data are presented as mean ± SEM. Data were analyzed using GraphPad Prism 5.0 Software. All imaging and quantification were performed blinded to experimental conditions.
3. Results
3.1. Temozolomide alters human NSC morphology in-vitro
NSC (SC27) are sensitive to TMZ, as single-dose application of 200 μM TMZ kills 50% of cells after 7 days. In contrast, this treatment has minimal effect on low-grade and high-grade derived glioma stem-like cells (GSC) (Gong et al., 2011). To investigate the effect of TMZ on NSC mitochondria, NSC were exposed to 500 μM TMZ for 48 h, and morphology was examined by transmission electron microscopy (TEM) (Fig. 1). Normal mitochondria were observed in the control (Fig. 1A, B). After 48 h TMZ exposure, mitochondrial degradation and vacuolization were observed (Fig. 1C, D). Quantification of mitochondrial type revealed that TMZ treatment reduced the percentage of healthy mitochondria (Type I) and increased the percentage of abnormal swollen mitochondria with fragmented cristae (Type II) as compared to the control NSC group, there was no significant difference in Type III damaged mitochondria between the groups (Fig. 1E).
Fig. 1.
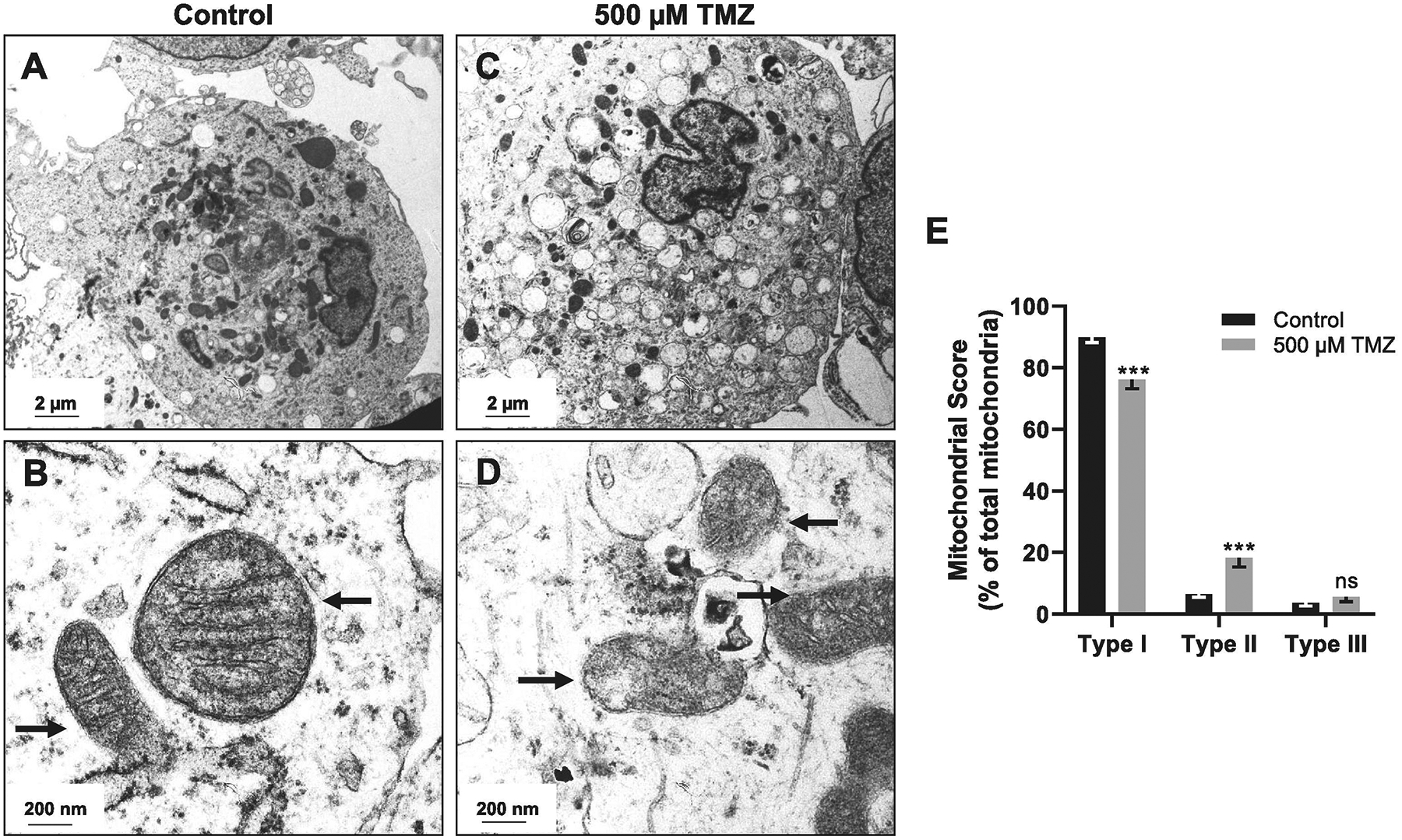
Temozolomide (TMZ) induced mitochondrial degradation in cultured NSC (SC27). (A) Transmission electron microscopy (TEM) of control SC27 showed (B) intact normal mitochondria (arrows). (C) SC27 treated with 500 μM TMZ showed (D) mitochondrial degradation (arrows) at 48 h. (E) Quantification of mitochondrial morphology. Graphs represent mean ± SEM of 15 images. *** p < 0.001, ns = not significant.
3.2. Temozolomide inhibits mtDNA synthesis in NSC
Temozolomide (TMZ) targets genomic DNA by its addition of methyl groups to purine residues. The most common lesions produced are methylation at the N7 and O6 sites on guanine, and O3 on adenine (Lee, 2016). We hypothesized that NSC mtDNA might be vulnerable to damage by TMZ due to its lack of histones and proximity to the electron transport chain (ETC), which makes it susceptible to oxidative damage. Also, mtDNA lesions may be less efficiently repaired compared to those of nDNA due to the absence of the nucleotide excision repair pathway in mitochondria (Larsen et al., 2005). To explore the effect of TMZ on NSC mitochondria, we examined the effect of TMZ on SC27 mtDNA replication. To compare mtDNA integrity, a 13.4-kb mtDNA fragment from control and TMZ-treated SC27 cells was amplified by qPCR. The PCR products were resolved by agarose gel electrophoresis (Fig. 2A). TMZ decreased the amount of full-length 13.4-kb mtDNA product amplified compared to control cells. We next used a quantitative PCR-based assay to compare the amplification of a mtDNA fragment to amplification of a nDNA fragment (Lomeli et al., 2017; Santos et al., 2006). TMZ produced a preferential decrease in amplification of NSC mtDNA as compared with a nuclear gene (β-Globin) (Fig. 2B). TMZ (200 μM) significantly reduced mtDNA amplification in SC27 cells, F(2,6) = 18.42, p = 0.0027.
Fig. 2.
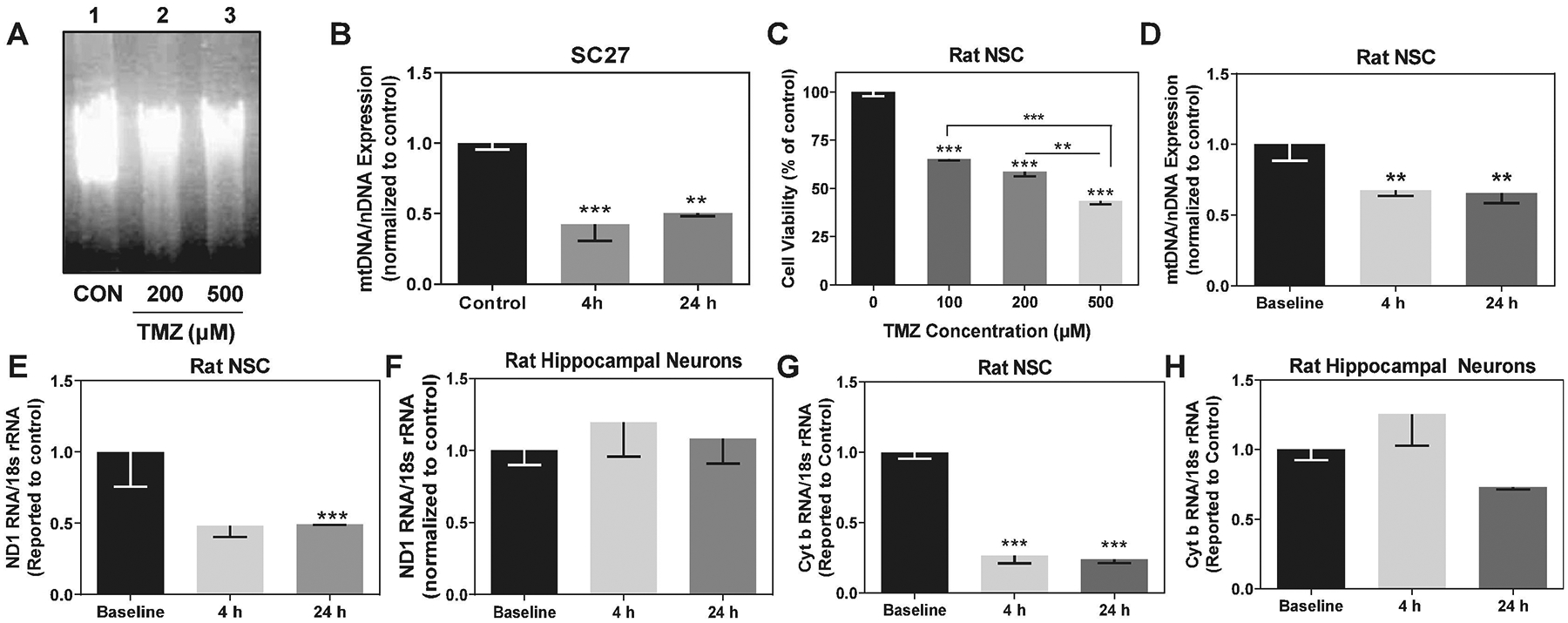
Temozolomide affects mtDNA integrity and impairs transcription of NADH dehydrogenase 1 and Cytochrome b in NSC but not cultured hippocampal neurons. (A) A representative agarose gel of a 13.4-kb mtDNA fragment amplified by quantitative PCR from genomic DNA of SC27 cells treated with 200 μM TMZ (lane 2), and 500 μM TMZ (lane 3) for 72 h. Lane 1 is control. (B) 200 μM TMZ caused a marked decrease in SC27 mtDNA levels. qPCR demonstrates selective reduction in amplification of a 221-bp mtDNA fragment compared to a nuclear gene (β-globin). (C) TMZ causes a dose-dependent decrease in viability of cultured rat NSC after 72 h as measured by MTT assay. (D) 500 μM TMZ decreased amplification of an intact rat 235-bp mtDNA fragment compared to a nuclear gene (Clusterin) in NSC. (E) Quantification of NADH dehydrogenase 1 mRNA levels (ND1) in rat NSC and (F) primary hippocampal neurons treated with 500 μM TMZ. (G) Quantification of mitochondrial encoded Cytochrome b (Cyt b) mRNA levels in rat NSC and (H) primary hippocampal neurons treated with 500 μM TMZ. Graphs represent mean ± SEM of 2–3 replicates. * p < 0.05, ** p < 0.01, *** p < 0.001.
Human NSC (SC27) are sensitive to TMZ and provide a relevant in-vitro model to examine the effects of chemotherapy on neural populations. However, to compare the effect of TMZ on the hippocampus (NSC and neurons), we sought to examine whether rat hippocampal NSC derived from the dentate gyrus (DG) would have comparable sensitivity to TMZ as human NSC. Rat NSC were exposed to clinically-relevant doses of TMZ for 7 days, and cell survival was as assessed by MTT assay. TMZ induced a dose-dependent decrease in cell viability (Fig. 2C). The TMZ IC50 in rat NSC is 366 μM, which is comparable to the IC50 (200 μM TMZ) in human NSC, 7 days after TMZ exposure (Gong et al., 2011). We continued our subsequent studies using cultured rat NSC and hippocampal neurons, which we have used to examine the effects cisplatin in a rat model (Lomeli et al., 2017; Andres et al., 2014). TMZ reduced the amplification of rat NSC mtDNA compared to a nDNA fragment (Clusterin). High-dose TMZ (500 μM) reduced mtDNA amplification at 4 h to 67.8% ± 2.4%, and at 24 h to 65.6% ± 4.1%, F(2,6) = 16.35, p = 0.0037 (Fig. 2D).
3.3. Temozolomide inhibits transcription of mitochondrial genes in NSC and hippocampal neurons
We next examined the effect of TMZ on the transcription of NADH dehydrogenase subunit 1 (ND1) and Cytochrome b (Cyt b), two mitochondrial-encoded genes associated with the mitochondrial ETC, in rat NSC and hippocampal neurons. ND1 is a mitochondrial-encoded protein subunit of complex I, the largest of the five complexes of the ETC. Defects involving complex I and III of the ETC have been associated with mutations in Cyt b (Acin-Perez et al., 2004; Budde et al., 2000). Studies in human and mouse cell lines harboring deleterious mutations in Cyt b have shown that an intact complex III is required for the assembly and respiratory activity of complex I, suggesting a structural association between these two complexes (Blakely et al., 2005). We examined whether TMZ impaired transcription of ND1. 500 μM TMZ markedly reduced ND1 transcription levels in rat NSC to 49.2% ± 0.3% (p = 0.0003), at 24 h (Fig. 2E). Strikingly, TMZ had no significant effect on ND1 levels in hippocampal neurons compared to the untreated control (Fig. 2F).
Cyt b is a major subunit of the catalytic core of complex III of the ETC and is the only component of complex III that is mtDNA encoded (Anderson et al., 1981). We assessed Cyt b transcription in rat NSC at 0 h, 4 h, and 24 h post-treatment with 500 μM TMZ (Fig. 2G). TMZ had a comparable reduction in Cyt b RNA levels at 4 h to (26.7% ± 5.7%) and 24 h (24.1% ± 2.8%) post-treatment, F(2,5) = 75.15, p = 0.0002. In contrast, 500 μM TMZ resulted in a modest decrease to 73.01% ± 1.5% in neuronal Cyt b levels at 24 h post-treatment, although not significant (F(2,4) = 2.11, p = 0.2367) (Fig. 2H). These results suggest that the NSC mitochondrial genome might be more sensitive to TMZ than that of hippocampal neurons.
3.4. Temozolomide reduces mitochondrial respiratory rates in cultured hippocampal neurons
To investigate if TMZ treatment may result in altered mitochondrial respiratory activity, we measured oxygen consumption rates (OCR) in hippocampal neurons using the Seahorse XF24 extracellular flux analyzer. OCR is a measure of oxidative phosphorylation. We examined the effect of 250 μM TMZ (Fig. 3A, B, C, D) and 500 μM TMZ (Fig. 3E, F, G, H) on neuronal mitochondrial respiration after 24 h and 48 h of treatment. 250 μM TMZ decreased basal OCR levels at 48 h, although not significantly (Fig. 3B). At the higher dose of 500 μM, TMZ significantly reduced basal OCR levels 48 h post-treatment (p = 0.0011) (Fig. 3F). 250 μM TMZ decreased neuronal mitochondrial maximum respiration at 24 h (p = 0.0486) (Fig. 3C), and at the high dose 500 μM TMZ (F(2,15) = 11.09, p = 0.0011) (Fig. 3G). 500 μM TMZ decreased the spare respiratory capacity (F(2,15) = 7.741, p = 0.0049), but not at the lower, 250 μM TMZ dose (Fig. 3D,H). The decrease in maximum respiration and spare respiratory capacity suggests that TMZ may impair the capacity of neurons to increase oxidative phosphorylation to meet the ATP demand under stressful energetically demanding conditions.
Fig. 3.
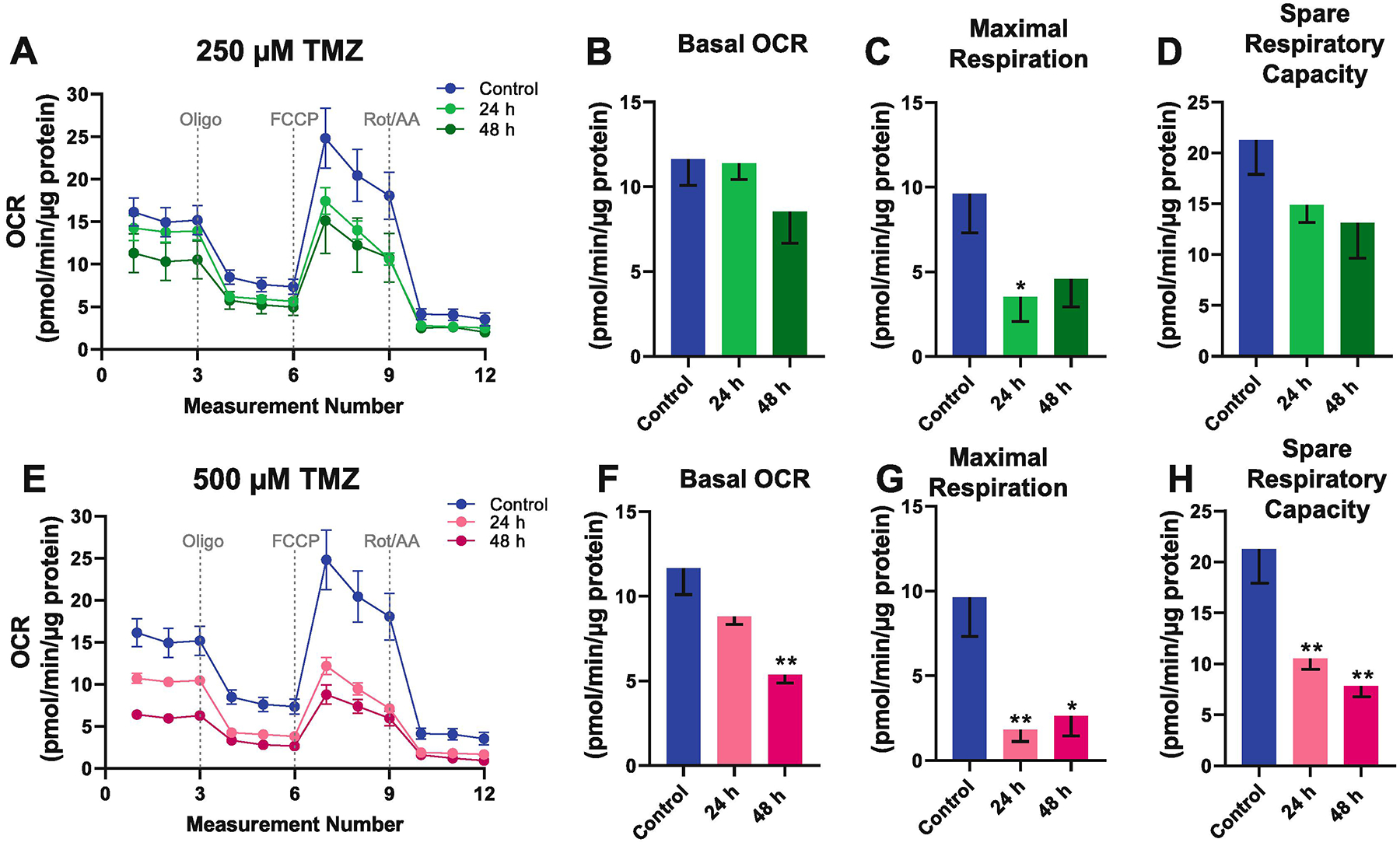
Temozolomide has a dose-dependent decrease of mitochondrial respiratory function in cultured hippocampal neurons. The Seahorse X24 Flux Analyzer was used to measure neuronal Oxygen Consumption Rate (OCR) using the Cell Mito Stress Kit. Mitochondrial bioenergetic profile of cultured hippocampal neurons treated with (A) 250 μM TMZ and (E) 500 μM TMZ for 24 h and 48 h. Quantitative analysis of OCR levels reveals that 250 μM TMZ had a minimal effect on neuronal (B) basal OCR, (C) maximal respiratory rate, and (D) spare respiratory capacity. In contrast, 500 μM TMZ exerts a significant reduction of neuronal (F) basal OCR, (G) maximal respiratory rate, and (H) spare respiratory capacity. Maximum respiration was calculated by subtracting the non-mitochondrial respiration rate from the maximum rate measurement after FCCP injection. Spare respiratory capacity was calculated by subtracting the basal respiration rate from the maximum rate measurement after FCCP injection. Graphs represent mean ± SEM, n = 6 replicates per group. * p < 0.05, ** p < 0.01, *** p < 0.001.
3.5. Temozolomide alters neuronal dendritic structures
Exposure to radiation and various chemotherapeutic agents has been shown to damage neuronal morphology (Parihar and Limoli, 2013; Acharya et al., 2015; Andres et al., 2014; Wu et al., 2017; Manchon et al., 2016; Seigers et al., 2013; Latzer et al., 2016). To examine the effect of TMZ on dendritic structures, we measured the number of dendritic branch points from the soma in hippocampal neurons treated with graded doses of TMZ (100 μM, 250 μM, and 500 μM) for 4 h and 24 h and immunolabeled for postsynaptic density-95 (PSD95) (Fig. 4A). TMZ exposure reduced neuronal dendritic branch points 4 h after treatment (Fig. 4B), with a more pronounced reduction in dendritic complexity evident at the highest TMZ dose (500 μM), and after 24 h (Fig. 4C). Repeated measures (RM) two-way ANOVA revealed an effect of interaction between TMZ dose and distance from soma for 4 h (F(33,220) = 3.599, p < 0.0001) and 24 h (F(33,220) = 2.629, p < 0.0001). Notably, 500 μM TMZ induced a rapid reduction in dendritic branching at 4 h post-treatment. However the effects of TMZ on mtDNA-encoded genes (Fig. 2F, H) and mitochondrial respiratory function (Fig. 3E, F) are slower, only becoming apparent 48 h after treatment, suggesting that neuronal damage provoked by TMZ is slow, but progressive and morphological damage may precede mitochondrial dysfunction.
Fig. 4.
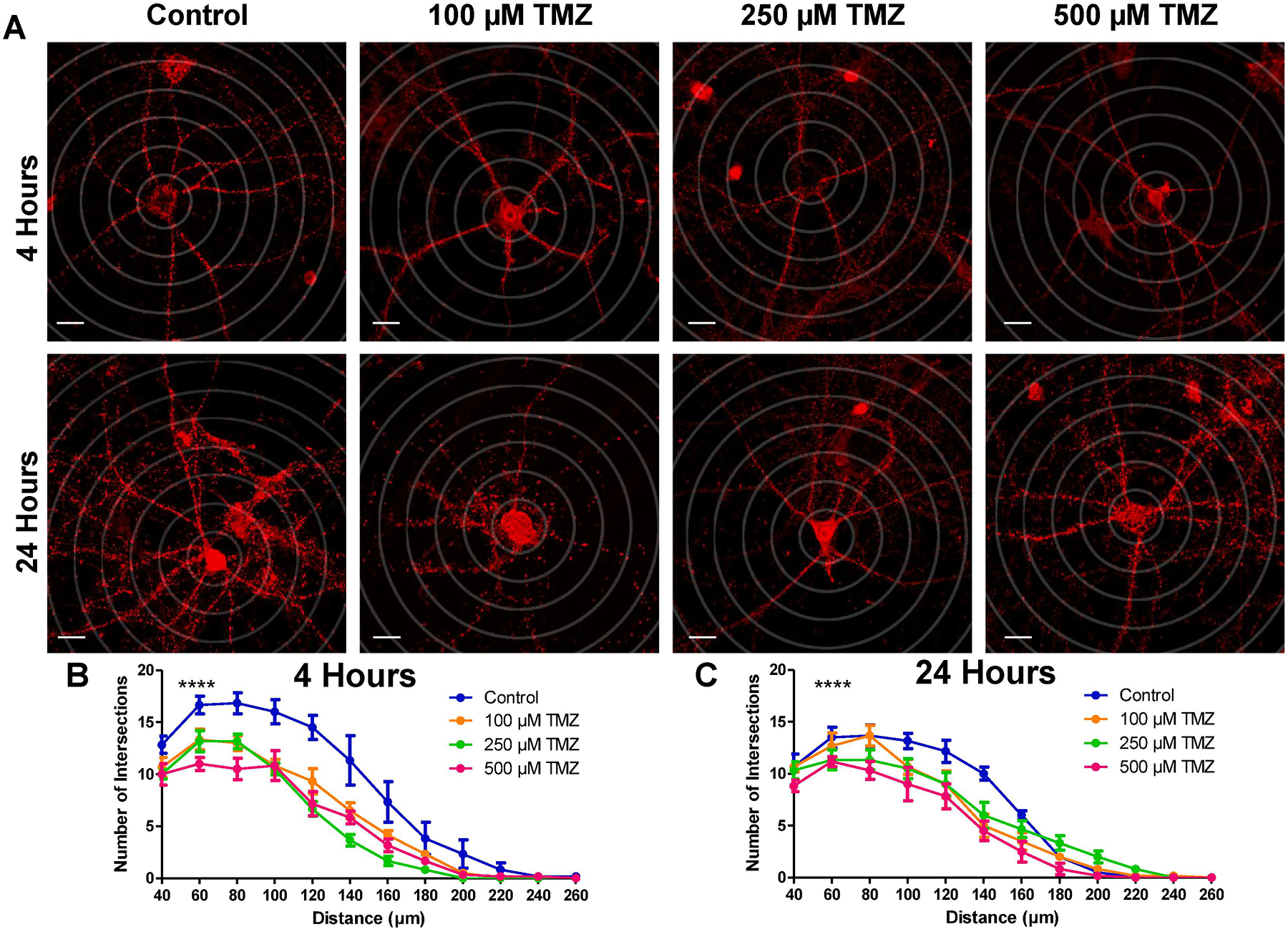
In-vitro treatment with graded doses of Temozolomide reduces dendritic branching of cultured hippocampal neurons. (A) Representative images of hippocampal neurons treated with TMZ and immunolabeled for postsynaptic density-95 (PSD95). Neurons are superimposed over concentric Sholl circles (20 μm increments). (B,C) Quantification of dendritic branches at intersecting points with Sholl circles at increasing distance from the soma after (B) 4 h and (C) 24 h of treatment shows that TMZ reduces dendritic complexity at both timepoints. Data are presented as mean ± SEM, **** p < 0.0001, (n = 6 neurons per group). Scale bars, 20 μm. Red, PSD95.
3.6. Temozolomide induces a loss of postsynaptic density-95 (PSD95) puncta in a dose- and time-dependent fashion
After assessing TMZ-induced changes in dendritic arborization, we sought to examine whether TMZ alters the number of PSD95 puncta found on dendritic branches, as a correlate measure of dendritic spine density (Andres et al., 2014; Andres et al., 2013). PSD95 is a scaffolding protein found at excitatory synapses; it anchors synaptic proteins, including NMDA and AMPA receptors, and potassium channels (Newpher and Ehlers, 2009; Sturgill et al., 2009). TMZ had an early (4 h) reduction in PSD95 puncta (F(3,48) = 23.30, p < 0.0001) (Fig. 5A). 100 μM TMZ did not reduce PSD95 puncta compared to control (p > 0.05) at 4 h, but at 24 h this dose significantly reduced PSD95 compared to control (p < 0.001) and had a comparable effect to 250 μM at 24 h (Fig. 5A, B). TMZ induced more extensive damage at 24 h, as loss of PSD95 was more pronounced (100 μM and 250 μM) and resulted in dendritic beading at the highest dose, 500 μM, which prevented PSD95 quantification in this treatment group (F(2,32) = 67.02, p < 0.0001) (Fig. 5B). This loss of PSD95 occurs concurrently with a reduction in dendritic branching.
Fig. 5.
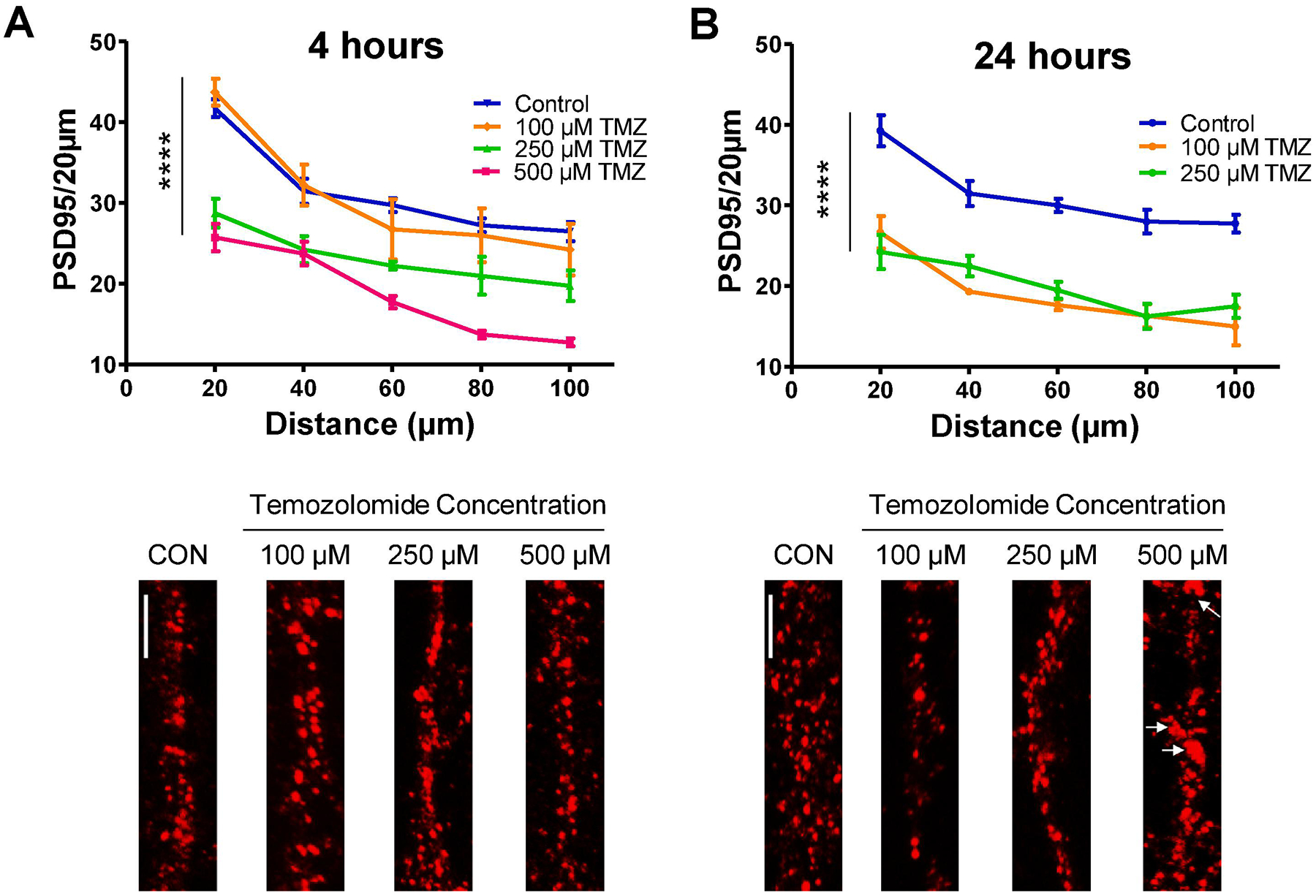
Temozolomide causes a dose- and time-dependent loss of PSD95 puncta in cultured hippocampal neurons. (A) Graph and representative images of dendrites immunolabeled for postsynaptic density-95 (PSD95) depict a dose-dependent reduction in PSD95 puncta after 4 h exposure. (B) The loss of PSD95 is more prominent after 24 h exposure. Dendritic beading (arrows) prevented quantification out to 100 μm from the soma for the 500 μM TMZ dose at 24 h, and therefore was omitted from the analysis. Scale bars, 5 μm. Data are presented as mean ± SEM, **** p < 0.0001, (n = 4 neurons per group). Scale bars, 20 μm. Red, PSD95.
3.7. Temozolomide induces oxidative stress in hippocampal neurons
TMZ-induced DNA damage increases reactive oxygen species (ROS) production, which contributes to apoptosis in glioma (Zhang et al., 2010; Oliva et al., 2011). We examined oxidative stress production in hippocampal neurons treated with TMZ (250 μM, 500 μM) for 7 days. Neurons were incubated with CellROX fluorescent probe for 20 min, and ROS production was quantified by measuring the fluorescence intensity of confocal images (Fig. 6A). TMZ significantly increased oxidative stress levels at 250 μM, and 500 μM compared to control levels (F(4,68) = 4.132, p = 0.0047), there was no statistical difference between low-dose and high-dose TMZ (Fig. 6B). The antioxidant N-acetylcysteine (NAC) was used as an ROS inhibitor, and 100 μM Menadione was used as a positive control to induce oxidative stress. Compared to studies examining TMZ-induced ROS production in glioma using similar doses of TMZ (250 μM), we found that ROS induction in neurons is slower (Zhang et al., 2010; Resende et al., 2018). In gliomas, ROS production peaked 72 h post-treatment, whereas, in our neuronal studies, a shorter duration of TMZ exposure did not result in significantly higher ROS levels compared to control (data not shown). These results suggest that TMZ related changes in neural structures may result in delayed complications that are not evident acutely after treatment.
Fig. 6.
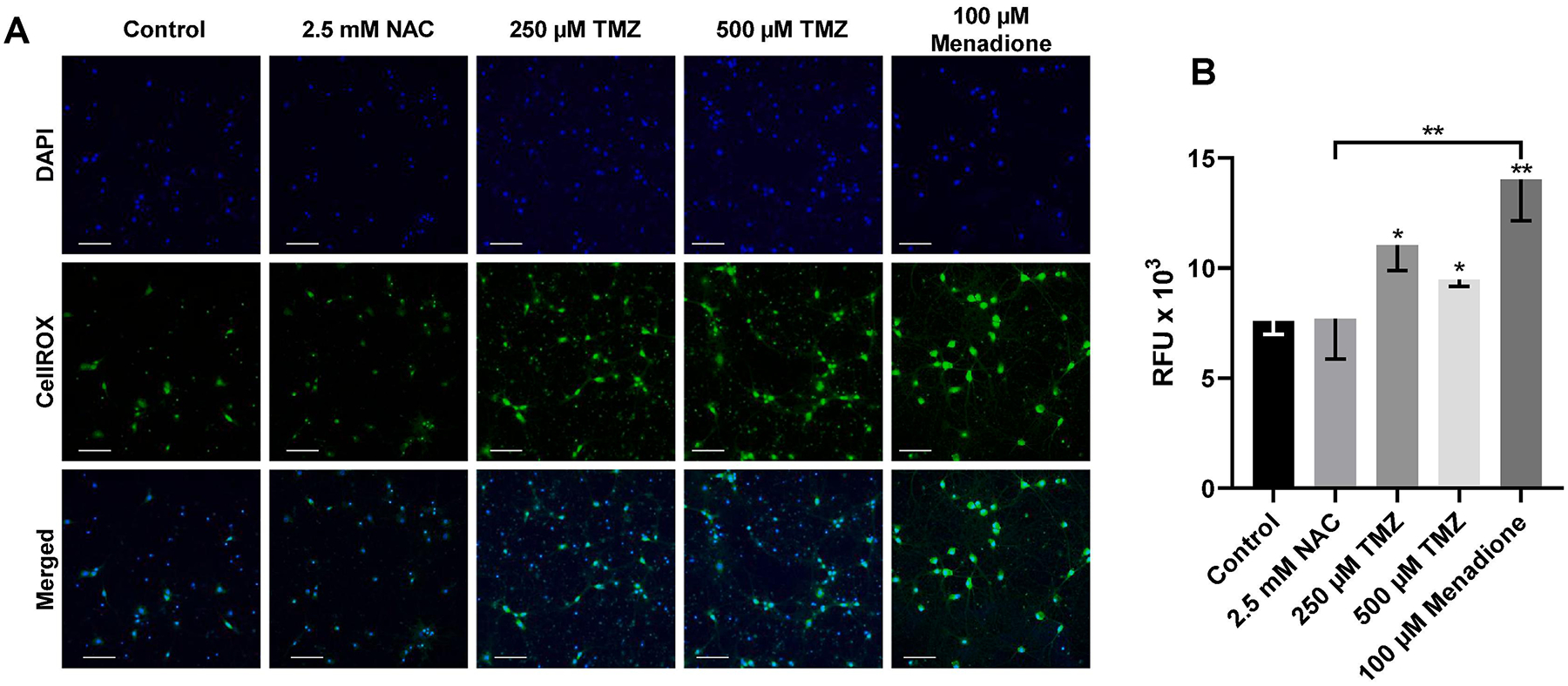
Temozolomide increases oxidative stress in cultured hippocampal neurons. (A) Rat hippocampal neurons (21 DIV) were exposed to 250 μM and 500 μM TMZ for 7 days. (B) Quantification of relative fluorescence intensity of CellROX probe as a measure of oxidative stress in TMZ treated neurons. ROS inhibitor, 2.5 mM N-acetylcysteine (NAC), 24 h treatment was used as a negative control. 100 μM Menadione, 24 h treatment, served as a positive control of oxidative stress. Data are presented as mean ± SEM, * p < 0.05, **p < 0.01, three sister coverslips per treatment. 5 images per coverslip. Scale bars, 100 μm. Blue, DAPI; Green, CellROX Oxidative Stress.
3.8. Temozolomide induces active caspase-9 expression and apoptotic cell death in hippocampal neurons
Next, to examine the effect of TMZ on neuronal apoptotic cell death, we quantified cleaved caspase-9 expression in hippocampal neurons 7 days after TMZ exposure (Fig. 7A, B). Upon cleavage, caspase-9 recruits and activates caspase −3 and −7, which culminates in apoptosis via the mitochondrial intrinsic cell death pathway (Boatright and Salvesen, 2003). TMZ exposure resulted in a dose-dependent increase in caspase-9 expression 7 days after treatment (F(2,6) = 11.35, p = 0.0091). High dose TMZ (500 μM TMZ) increased caspase-9 activation compared to control (p = 0.0075).
Fig. 7.
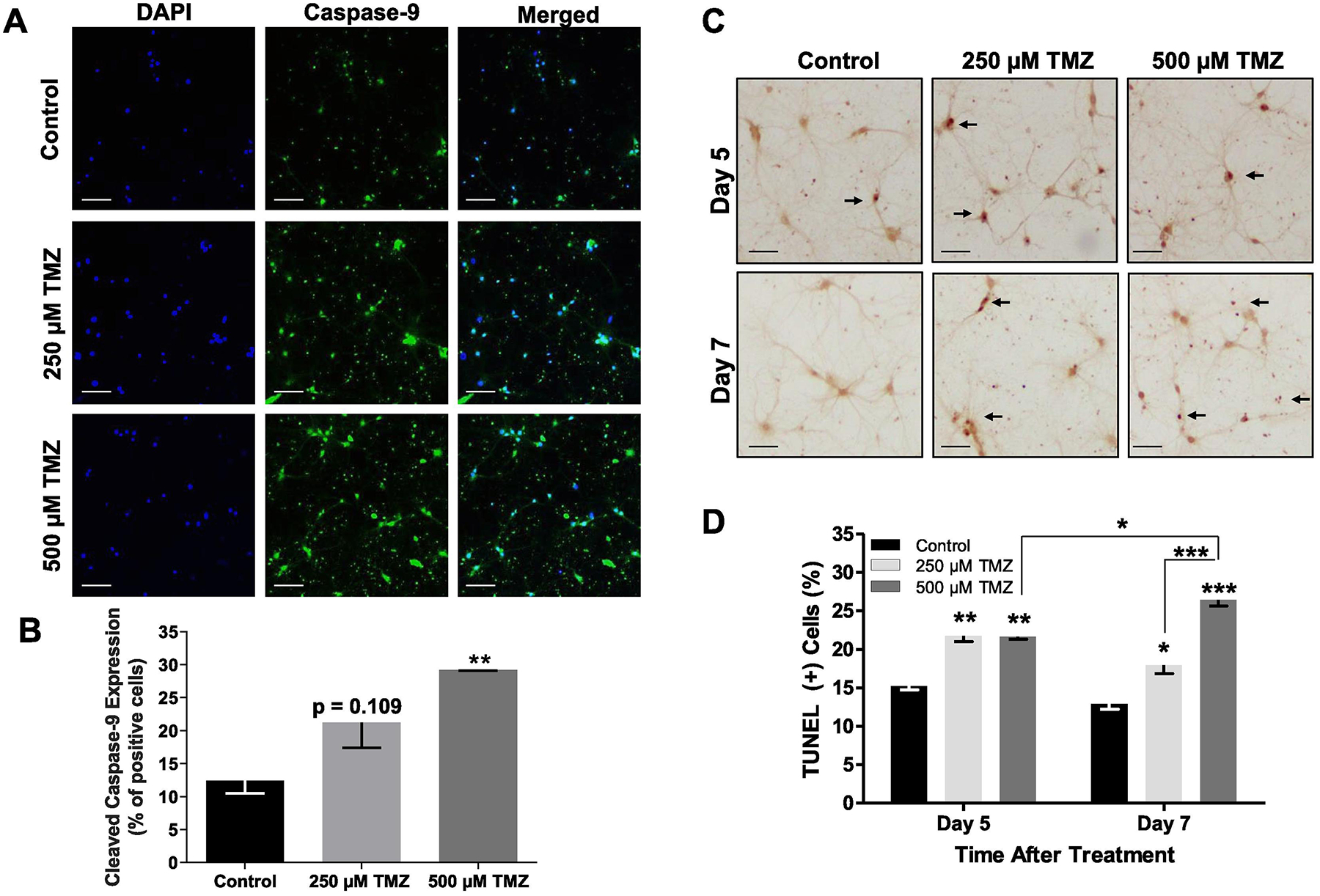
Temozolomide increases apoptosis in cultured hippocampal neurons. (A) Rat primary hippocampal neurons (21 DIV) were exposed to 250 μM and 500 μM TMZ for 7 days. (B) TMZ induced a dose-dependent increase in cleaved caspase-9 expression 7 days after treatment. (C) Images of TUNEL positive hippocampal neurons (24 DIV) exposed to 250 μM and 500 μM TMZ for 5 days, and 7 days. (D) Quantification of TUNEL(+) (nuclear dark brown staining) neurons shows that TMZ increased apoptotic neuronal death at both doses and timepoints. High dose TMZ (500 μM) further increased apoptotic cell death 7 days after treatment. Scale bars, 100 μm. Data are presented as mean ± SEM, * p < 0.05, ** p < 0.01, *** p < 0.001, n = 2–3 sister coverslips per group, 10 images per coverslip. Blue, DAPI; green, cleaved caspase-9; brown; TUNEL.
We also detected apoptosis by TUNEL assay, in neurons treated with TMZ (250 μM, 500 μM) for 5 and 7 days (F(2,10) = 73.04, p < 0.0001) (Fig. 7C, D). At 5 days, both TMZ doses caused a similar induction in apoptosis, 21.7% ± 0.74% and 21.6% ± 0.31%, respectively compared to control, 15.2% ± 0.48% (Fig. 7D). At 7 days, we observed a dose-dependent increase in apoptosis, 17.95% ± 1.09% and 26.42% ± 0.79%, respectively compared to control, 12.92% ± 0.79%. There was an increase in apoptotic cell death in neurons exposed to high-dose TMZ, 500 μM for 5 days compared to 7 days (p < 0.05), which was not evident at the lower dose. These results suggest that TMZ-induced damage in neurons is dose-dependent, with higher doses resulting in more pronounced mitochondrial respiratory dysfunction, dendritic damage, and cell death.
4. Discussion
The cognitive impairments and mood disorders associated with glioma-directed treatment are of grave concern for the increasing number of survivors. As the standard of care treatment for glioblastoma, TMZ is widely used, yet its effects on normal brain structures and function are understudied. Here we identified mitochondrial damage in cultured NSC and hippocampal neurons as a mechanism underlying TMZ-induced neurotoxicity. We examined mitochondrial morphology by TEM and found that TMZ induced mitochondrial degradation and vacuolization in human NSC. TMZ exposure reduced the amplification of a long, 13.4-kb mtDNA fragment and exerted a selective reduction in mtDNA amplification compared to nDNA in human NSC. Similarly, TMZ resulted in a comparable decrease in cell viability and a decrease in mtDNA amplification in rat NSC. Acute TMZ exposure significantly decreased ND1 and Cyt B mRNA transcription, two genes that encode components of complex I and complex III of the ETC respectively, which is indicative of damage to NSC mtDNA integrity elicited by TMZ. Although neuronal mtDNA was less sensitive to TMZ than NSC mtDNA, high dose TMZ impaired mitochondrial respiration and decreased the spare respiratory capacity. Since excitatory synapses on dendritic spines mediate learning-related plasticity, we examined the effects of TMZ on the morphology of hippocampal neurons. TMZ reduced dendritic complexity and synaptic protein PSD95 puncta density in a time- and dose-dependent manner. These neuronal morphological changes occurred at time-points prior to the observed respiratory dysfunction, which suggests hippocampal toxicity. Chronic exposure to TMZ increased oxidative stress in hippocampal neurons, culminating in neuronal apoptosis seven days after treatment. Alarmingly, NSC (human and rat) are more sensitive to TMZ than glioma cell lines. Comparison of the viability of various human NSC lines to low-grade and high-grade patient-derived glioma stem-like cells (GSC) demonstrate that GSC are resistant to 200 μM TMZ, which kills 50% of NSC (Gong et al., 2011). In contrast, mature neurons are less vulnerable to TMZ compared to NSC. 500 μM TMZ resulted in a 26.42% ± 0.79% decrease in neuronal cell viability (Fig. 7D), compared to a 43.47% ± 1.8% decrease in NSC viability seven days after treatment (Fig. 2C).
Similar to other DNA damaging agents such as cisplatin, TMZ induces mitochondrial dysfunction, apoptosis of neural cells, and morphological damage in surviving neurons. Using the same in-vitro system, cultured rat NSC and hippocampal neurons, we found that neurons were much more sensitive to cisplatin at doses lower than those used clinically (0.1–1 μM), which induced dendritic damage and apoptosis at earlier time-points compared to TMZ, whereas NSC were less sensitive (Andres et al., 2014). These results highlight the observations that some chemotherapeutic agents have higher neurotoxic potential than others, but also that distinct neural lineages have unique susceptibilities to chemotherapeutic agents (Dietrich et al., 2006). Also, these agents may have varying acute and chronic effects on neural cell populations in distinct microenvironments, which can result in delayed neurological complications observed in patients, including cognitive dysfunction, white and grey matter changes, and progressive myelin disruption.
Clinical studies have shown that TMZ contributes to anxiety, depression, and cognitive impairments in glioma patients, as well as those with non-CNS malignancies. A phase 1 dose-escalation study of TMZ for the treatment of metastatic melanoma (excluding patients with CNS metastasis) reported anxiety and depression in 59% and 35% of patients, respectively (Agarwala and Kirkwood, 2003). Longitudinal MRI to assess structural brain changes in newly diagnosed GBM patients treated with RT plus systemic TMZ revealed significant and progressive decreases in whole brain volume, cortical gray matter loss, and ventricular dilation (Prust et al., 2015). A progressive loss of white matter integrity was observed in the subventricular zone (SVZ), a region rich in NSC. As gray matter loss and ventricular dilation are strongly associated with neurocognitive decline in neurodegenerative diseases such as Alzheimer’s disease (Karas et al., 2003; Madsen et al., 2015), the incidence of these structural alterations in patients receiving chemoradiation may be indicative of changes in neurocognitive function. Of note, the onset of significant morphologic brain changes coincided with the time of RT completion, six weeks after the combined therapy. Although it is difficult to parse out the individual and synergistic contributions of RT and chemotherapy in these studies, the changes observed followed a delayed time course, resulting in progressive neurodegeneration weeks after the initial exposure (Prust et al., 2015).
In addition to TMZ, glioma patients may also receive adjuvant therapies, such as bevacizumab, an anti-VEGF monoclonal antibody, which has been associated with neurotoxicity. The FDA approved bevacizumab for recurrent GBM in 2017. In patients with recurrent GBM, treatment with bevacizumab has been shown to induce brain atrophy compared to control group patients who received the standard of care treatment but did not receive bevacizumab (Bag et al., 2015). Bevacizumab has also been linked to neurocognitive decline in newly diagnosed patients (Gilbert et al., 2014). Investigation of the effects of bevacizumab on the morphology and survival of dissociated rat hippocampal cell cultures revealed that long-term exposure to bevacizumab resulted in delayed (20–30 days post-treatment) reductions in dendritic length, without reducing neuronal or glial cell viability. In contrast, cortical neurons and glia are much more sensitive to bevacizumab, as cell viability was reduced 30 days post-treatment (Latzer et al., 2016). While many commonly used cancer therapies induce cognitive dysfunction, various cell types have distinct sensitives to chemotherapeutic agents, and damage to multiple neural lineages and blood vasculature may contribute to the neurocognitive impairments and mood changes experienced by cancer survivors.
To our knowledge, our study is the first to identify mitochondrial damage as a mechanism associated with TMZ-induced neurotoxicity in NSC and neuronal cultures. Other studies have examined the neurotoxic effects of TMZ on primary rat cortical microglia (Vairano et al., 2004), organotypic rat corticostriatal (Norregaard et al., 2012), and entorhinal-hippocampal slice cultures (Eyupoglu et al., 2006) and found that at clinically relevant doses, 10–500 μM, TMZ does not decrease cell viability following acute 24–72 h exposure. Collectively, these studies show that in agreement with the present study, TMZ may have chronic delayed effects on the normal brain, which may augment damage provoked by RT and other anti-cancer agents used during cancer treatment. Additional studies examining the chronic long-term effects of TMZ on neurons, oligodendrocytes, NSC, and other neural subtypes are warranted.
5. Conclusions
In summary, we demonstrated that mitochondrial dysfunction is a mechanism underlying TMZ-induced neurotoxicity in cultured NSC and hippocampal neurons. TMZ induced mitochondrial damage in NSC and neurons. Concomitant with the mitochondrial respiratory dysfunction in neurons was a loss of PSD95 puncta and decreased dendritic branch length, which is indicative of morphological damage. More extended exposure to TMZ increased oxidative stress and reduced neuronal viability. TMZ induced more pronounced decrements in NSC mtDNA integrity and viability compared to neurons, suggesting that NSC are more vulnerable to TMZ than neurons. TMZ may have various acute and chronic effects on mitochondria that affect neural function and survival, which may contribute to the neurological complications experienced by GBM patients.
Acknowledgments
The authors thank Adrienne Andres for assistance with the PSD95 immunocytochemistry experiments, and Vivek Abraham for technical help with Sholl analysis.
Financial support
This work was supported by the National Institute for Neurological Diseases and Stroke Award (NINDS/NIH) [NS072234], the National Center for Advancing Translational Sciences, NIH [UL1 TR001414], and the UCI Cancer Center Award [P30CA062203] from the National Cancer Institute. The NIH MBRS-IMSD training grant [GM055246] and the NINDS/NIH pre-doctoral fellowship [NS082174] provided support for N. Lomeli. The National Institute of Environmental Health Sciences of the NIH supported D.C. Pearre with grant T32CA060396, and its contents of the project described are solely the responsibility of the authors and do not necessarily represent the official views of the NIEHS or NIH.
Abbreviations:
- TMZ
temozolomide
- GBM
glioblastoma
- NSC
neural/stem progenitor cells
- ROS
reactive oxygen species
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick -end labeling
Footnotes
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.mito.2020.02.001.
References
- Taphoorn MJ, Klein M, 2004. Cognitive deficits in adult patients with brain tumours. Lancet Neurol. 3, 159–168. [DOI] [PubMed] [Google Scholar]
- Winocur G, Johnston I, Castel H, 2018. Chemotherapy and cognition: International cognition and cancer task force recommendations for harmonising preclinical research. Cancer Treat. Rev 69, 72–83. [DOI] [PubMed] [Google Scholar]
- Krebber AM, Buffart LM, Kleijn G, et al. , 2014. Prevalence of depression in cancer patients: a meta-analysis of diagnostic interviews and self-report instruments. Psychooncology 23, 121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbach JP, Blaicher HP, Herrlinger U, et al. , 2006. Surviving glioblastoma for more than 5 years: the patient’s perspective. Neurology 66, 239–242. [DOI] [PubMed] [Google Scholar]
- Hottinger AF, Yoon H, DeAngelis LM, Abrey LE, 2009. Neurological outcome of long-term glioblastoma survivors. J. Neurooncol 95, 301–305. [DOI] [PubMed] [Google Scholar]
- Allen DH, Loughan AR, 2018. Impact of cognitive impairment in patients with gliomas. Semin. Oncol. Nurs 34, 528–546. [DOI] [PubMed] [Google Scholar]
- Abrey LE, 2012. The impact of chemotherapy on cognitive outcomes in adults with primary brain tumors. J. Neurooncol 108, 285–290. [DOI] [PubMed] [Google Scholar]
- Wu PH, Coultrap S, Pinnix C, et al. , 2012. Radiation induces acute alterations in neuronal function. PLoS ONE 7, e37677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parihar VK, Limoli CL, 2013. Cranial irradiation compromises neuronal architecture in the hippocampus. Proc. Natl. Acad. Sci. U.S.A 110, 12822–12827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolen SC, Lee B, Shantharam S, et al. , 2016. The effects of sequential treatments on hippocampal volumes in malignant glioma patients. J. Neurooncol 129, 433–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acharya MM, Christie LA, Lan ML, et al. , 2011. Human neural stem cell transplantation ameliorates radiation-induced cognitive dysfunction. Cancer Res. 71, 4834–4845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agnihotri S, Burrell KE, Wolf A, et al. , 2013. Glioblastoma, a brief review of history, molecular genetics, animal models and novel therapeutic strategies. Archivum immunologiae et therapiae experimentalis 61, 25–41. [DOI] [PubMed] [Google Scholar]
- Di K, Lomeli N, Wood SD, Vanderwal CD, Bota DA, 2016. Mitochondrial Lon is over-expressed in high-grade gliomas, and mediates hypoxic adaptation: potential role of Lon as a therapeutic target in glioma. Oncotarget 7, 77457–77467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva CR, Nozell SE, Diers A, et al. , 2010. Acquisition of temozolomide chemoresistance in gliomas leads to remodeling of mitochondrial electron transport chain. J. Biol. Chem 285, 39759–39767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egeland M, Guinaudie C, Du Preez A, et al. , 2017. Depletion of adult neurogenesis using the chemotherapy drug temozolomide in mice induces behavioural and biological changes relevant to depression. Transl. Psychiatry 7, e1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira-Caixeta AR, Guarnieri LO, Medeiros DC, et al. , 2018. Inhibiting constitutive neurogenesis compromises long-term social recognition memory. Neurobiol. Learn. Mem 155, 92–103. [DOI] [PubMed] [Google Scholar]
- Niibori Y, Yu TS, Epp JR, Akers KG, Josselyn SA, Frankland PW, 2012. Suppression of adult neurogenesis impairs population coding of similar contexts in hippocampal CA3 region. Nat. Commun 3, 1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garthe A, Behr J, Kempermann G, 2009. Adult-generated hippocampal neurons allow the flexible use of spatially precise learning strategies. PLoS ONE 4, e5464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt-Hieber C, Jonas P, Bischofberger J, 2004. Enhanced synaptic plasticity in newly generated granule cells of the adult hippocampus. Nature 429, 184–187. [DOI] [PubMed] [Google Scholar]
- Snyder JS, Kee N, Wojtowicz JM, 2001. Effects of adult neurogenesis on synaptic plasticity in the rat dentate gyrus. J. Neurophysiol 85, 2423–2431. [DOI] [PubMed] [Google Scholar]
- Lomeli N, Bota DA, Davies KJA, 2017. Diminished stress resistance and defective adaptive homeostasis in age-related diseases. Clin. Sci. (London, England: 1979) (131), 2573–2599. [DOI] [PubMed] [Google Scholar]
- Lin MT, Beal MF, 2006. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443, 787. [DOI] [PubMed] [Google Scholar]
- Schon EA, Manfredi G, 2003. Neuronal degeneration and mitochondrial dysfunction. J. Clin. Investig 111, 303–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Commandeur JN, Stijntjes GJ, Vermeulen NP, 1995. Enzymes and transport systems involved in the formation and disposition of glutathione S-conjugates. Role in bioactivation and detoxication mechanisms of xenobiotics. Pharmacol. Rev 47, 271–330. [PubMed] [Google Scholar]
- Raefsky SM, Mattson MP, 2017. Adaptive responses of neuronal mitochondria to bioenergetic challenges: roles in neuroplasticity and disease resistance. Free Radical Biol. Med 102, 203–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomeli N, Di K, Czerniawski J, Guzowski JF, Bota DA, 2017. Cisplatin-induced mitochondrial dysfunction is associated with impaired cognitive function in rats. Free Radical Biol. Med 102, 274–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu GS, Boukelmoune N, Chiang ACA, et al. , 2018. Nasal administration of mesenchymal stem cells restores cisplatin-induced cognitive impairment and brain damage in mice. Oncotarget 9, 35581–35597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oboh G, Ogunruku OO, 2010. Cyclophosphamide-induced oxidative stress in brain: protective effect of hot short pepper (Capsicum frutescens L. var. abbreviatum). Exp. Toxicol. Pathol 62, 227–233. [DOI] [PubMed] [Google Scholar]
- Acharya MM, Martirosian V, Chmielewski NN, et al. , 2015. Stem cell transplantation reverses chemotherapy-induced cognitive dysfunction. Cancer Res. 75, 676–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeney JTR, Ren X, Warrier G, et al. , 2018. Doxorubicin-induced elevated oxidative stress and neurochemical alterations in brain and cognitive decline: protection by MESNA and insights into mechanisms of chemotherapy-induced cognitive impairment (“chemobrain”). Oncotarget 9, 30324–30339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tangpong J, Cole MP, Sultana R, et al. , 2007. Adriamycin-mediated nitration of manganese superoxide dismutase in the central nervous system: insight into the mechanism of chemobrain. J. Neurochem 100, 191–201. [DOI] [PubMed] [Google Scholar]
- Hess JA, Khasawneh MK, 2015. Cancer metabolism and oxidative stress: Insights into carcinogenesis and chemotherapy via the non-dihydrofolate reductase effects of methotrexate. BBA Clin. 3, 152–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong X, Schwartz PH, Linskey ME, Bota DA, 2011. Neural stem/progenitors and glioma stem-like cells have differential sensitivity to chemotherapy. Neurology 76, 1126–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz PH, Bryant PJ, Fuja TJ, Su H, O’Dowd DK, Klassen H, 2003. Isolation and characterization of neural progenitor cells from post-mortem human cortex. J. Neurosci. Res 74, 838–851. [DOI] [PubMed] [Google Scholar]
- Andres AL, Gong X, Di K, Bota DA, 2014. Low-doses of cisplatin injure hippocampal synapses: a mechanism for ‘chemo’ brain? Exp. Neurol 255, 137–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andres AL, Regev L, Phi L, et al. , 2013. NMDA receptor activation and calpain contribute to disruption of dendritic spines by the stress neuropeptide CRH. J. Neurosci 33, 16945–16960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perazzoli G, Prados J, Ortiz R, et al. , 2015. Temozolomide resistance in glioblastoma cell lines: implication of MGMT, MMR, P-glycoprotein and CD133 expression. PLoS ONE 10, e0140131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos JH, Meyer JN, Mandavilli BS, Van Houten B, 2006. Quantitative PCR-based measurement of nuclear and mitochondrial DNA damage and repair in mammalian cells. Methods Mol. Biol. (Clifton, NJ) 314, 183–199. [DOI] [PubMed] [Google Scholar]
- Lee SY, 2016. Temozolomide resistance in glioblastoma multiforme. Genes Diseases 3, 198–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen NB, Rasmussen M, Rasmussen LJ, 2005. Nuclear and mitochondrial DNA repair: similar pathways? Mitochondrion 5, 89–108. [DOI] [PubMed] [Google Scholar]
- Acin-Perez R, Bayona-Bafaluy MP, Fernandez-Silva P, et al. , 2004. Respiratory complex III is required to maintain complex I in mammalian mitochondria. Mol. Cell 13, 805–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budde SM, van den Heuvel LP, Janssen AJ, et al. , 2000. Combined enzymatic complex I and III deficiency associated with mutations in the nuclear encoded NDUFS4 gene. Biochem. Biophys. Res. Commun 275, 63–68. [DOI] [PubMed] [Google Scholar]
- Blakely EL, Mitchell AL, Fisher N, et al. , 2005. A mitochondrial cytochrome b mutation causing severe respiratory chain enzyme deficiency in humans and yeast. FEBS J. 272, 3583–3592. [DOI] [PubMed] [Google Scholar]
- Anderson S, Bankier AT, Barrell BG, et al. , 1981. Sequence and organization of the human mitochondrial genome. Nature 290, 457. [DOI] [PubMed] [Google Scholar]
- Wu L, Guo D, Liu Q, et al. , 2017. Abnormal development of dendrites in adult-born rat hippocampal granule cells induced by cyclophosphamide. Front. Cell. Neurosci 11, 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manchon JFM, Dabaghian Y, Uzor N-E, Kesler SR, Wefel JS, Tsvetkov AS, 2016. Levetiracetam mitigates doxorubicin-induced DNA and synaptic damage in neurons. Sci. Rep 6, 25705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seigers R, Schagen SB, Van Tellingen O, Dietrich J, 2013. Chemotherapy-related cognitive dysfunction: current animal studies and future directions. Brain Imaging and Behavior 7, 453–459. [DOI] [PubMed] [Google Scholar]
- Latzer P, Schlegel U, Theiss C, 2016. Morphological changes of cortical and hippocampal neurons after treatment with VEGF and bevacizumab. CNS Neurosci. Ther 22, 440–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newpher TM, Ehlers MD, 2009. Spine microdomains for postsynaptic signaling and plasticity. Trends Cell Biol. 19, 218–227. [DOI] [PubMed] [Google Scholar]
- Sturgill JF, Steiner P, Czervionke BL, Sabatini BL, 2009. Distinct domains within PSD-95 mediate synaptic incorporation, stabilization, and activity-dependent trafficking. J. Neurosci 29, 12845–12854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang WB, Wang Z, Shu F, et al. , 2010. Activation of AMP-activated protein kinase by temozolomide contributes to apoptosis in glioblastoma cells via p53 activation and mTORC1 inhibition. J. Biol. Chem 285, 40461–40471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva CR, Moellering DR, Gillespie GY, Griguer CE, 2011. Acquisition of chemoresistance in gliomas is associated with increased mitochondrial coupling and decreased ROS production. PLoS ONE 6, e24665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resende FFB, Titze-de-Almeida SS, Titze-de-Almeida R, 2018. Function of neuronal nitric oxide synthase enzyme in temozolomide-induced damage of astrocytic tumor cells. Oncol. Lett 15, 4891–4899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boatright KM, Salvesen GS, 2003. Mechanisms of caspase activation. Curr. Opin. Cell Biol 15, 725–731. [DOI] [PubMed] [Google Scholar]
- Dietrich J, Han R, Yang Y, Mayer-Pröschel M, Noble M, 2006. CNS progenitor cells and oligodendrocytes are targets of chemotherapeutic agents in vitro and in vivo. J. Biol 5 22–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwala SS, Kirkwood JM, 2003. Temozolomide in combination with interferon alpha-2b in patients with metastatic melanoma: a phase I dose-escalation study. Cancer 97, 121–127. [DOI] [PubMed] [Google Scholar]
- Prust MJ, Jafari-Khouzani K, Kalpathy-Cramer J, et al. , 2015. Standard chemoradiation for glioblastoma results in progressive brain volume loss. Neurology 85, 683–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karas GB, Burton EJ, Rombouts SA, et al. , 2003. A comprehensive study of gray matter loss in patients with Alzheimer’s disease using optimized voxel-based morphometry. NeuroImage 18, 895–907. [DOI] [PubMed] [Google Scholar]
- Madsen SK, Gutman BA, Joshi SH, et al. , 2015. Mapping ventricular expansion onto cortical gray matter in older adults. Neurobiol. Aging 36 (Suppl. 1), S32–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bag AK, Kim H, Gao Y, et al. , 2015. Prolonged treatment with bevacizumab is associated with brain atrophy: a pilot study in patients with high-grade gliomas. J. Neurooncol 122, 585–593. [DOI] [PubMed] [Google Scholar]
- Gilbert MR, Dignam JJ, Armstrong TS, et al. , 2014. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med 370, 699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vairano M, Graziani G, Tentori L, Tringali G, Navarra P, Russo CD, 2004. Primary cultures of microglial cells for testing toxicity of anticancer drugs. Toxicol. Lett 148, 91–94. [DOI] [PubMed] [Google Scholar]
- Norregaard A, Jensen SS, Kolenda J, et al. , 2012. Effects of chemotherapeutics on organotypic corticostriatal slice cultures identified by a panel of fluorescent and immunohistochemical markers. Neurotox. Res 22, 43–58. [DOI] [PubMed] [Google Scholar]
- Eyupoglu IY, Hahnen E, Trankle C, et al. , 2006. Experimental therapy of malignant gliomas using the inhibitor of histone deacetylase MS-275. Mol. Cancer Ther 5, 1248–1255. [DOI] [PubMed] [Google Scholar]