

Abstract
Purpose:
The activating mutation AKT1E17K occurs in ~7% of ER+ metastatic breast cancer (MBC). We report, from a multipart, first-in-human, Phase I study (NCT01226316), tolerability and activity of capivasertib, an oral AKT inhibitor, as monotherapy or combined with fulvestrant in expansion cohorts of AKT1E17K-mutant ER+ MBC patients.
Patients and Methods:
Patients with an AKT1E17K mutation, detected by local (NGS) or central (plasma-based BEAMing) testing, received capivasertib 480 mg bid, 4 days on, 3 days off, weekly or 400 mg bid combined with fulvestrant at the labeled dose. Study endpoints included safety, objective response rate (ORR; RECIST v1.1), progression-free survival (PFS) and clinical benefit rate at 24 weeks (CBR24). Biomarker analyses were conducted in the combination cohort.
Results:
From October 2013 to August 2018, 63 heavily pretreated patients received capivasertib (20 monotherapy, 43 combination). ORR was 20% with monotherapy, and within the combination cohort was 36% in fulvestrant-pretreated and 20% in fulvestrant-naïve patients, although this latter group may have had more aggressive disease at baseline. AKT1E17K mutations were detectable in plasma by BEAMing (95%, 41/43), ddPCR (80%, 33/41) and NGS (76%, 31/41). A ≥50% decrease in AKT1E17K at cycle 2 day 1 was associated with improved PFS. Combination therapy appeared more tolerable than monotherapy (most frequent grade ≥3 adverse events: rash [9% vs 20%], hyperglycemia [5% vs 30%], diarrhea [5% vs 10%]).
Conclusions:
Capivasertib demonstrated clinically meaningful activity in heavily pretreated AKT1E17K-mutant ER+ MBC patients, including those with prior disease progression on fulvestrant. Tolerability and activity appeared improved by the combination.
Keywords: Capivasertib, fulvestrant, AKT, ER-positive, breast cancer
Introduction
Estrogen-receptor-positive (ER+), HER2-negative (HER2−) breast cancer is the most common subtype of metastatic breast cancer (MBC), accounting for >400,000 deaths worldwide every year (1, 2). The incorporation of inhibitors of mTOR and CDK4/6 into endocrine therapy has led to substantial improvements in patient outcomes (3-8). However, once endocrine-therapy-refractory disease inevitably develops, chemotherapy remains the only approved option, and little progress has been made for this phase of illness. Given the successes of genomically selected therapy in other solid tumors harboring driver alterations (9, 10), widescale efforts to identify therapeutically actionable genomic subsets of breast cancer have been undertaken (11-15).
The PI3K pathway is one of the most commonly activated signaling pathways in ER+ breast cancer (16). The efficacy of an isoform-selective PI3K inhibitor in PIK3CA-mutant ER+ HER2− MBC was recently demonstrated in a Phase III study (17), providing proof of concept that this pathway is therapeutically targetable in this clinical context. While PIK3CA mutations represent the most common mechanism of PI3K pathway activation, in an estimated 7% of ER+ breast cancers, pathway activation can occur through mutation in AKT1 (15), predominantly AKT1E17K (~80%). In such cases, signaling is constitutively activated through pathologic localization of AKT1 to the plasma membrane (18-20). Although, in the largest comparative analysis of matched AKT1-mutant and wild-type ER+ MBC patients, there did not appear to be significant differences in terms of overall survival or duration on endocrine- and CDK4/6 inhibitor therapy, patients with AKT1-mutant disease were, however, noted to have significantly longer durations on MTOR inhibitor therapy (21), indicative of the potential therapeutic relevance of this alteration in breast cancer. Moreover, AKT1E17K-mutant tumors may not be amenable to PI3K inhibitors owing to their PI3K-independent mechanism of AKT activation (15, 22-27). As such, patients harboring AKT1E17K mutations represent a genomic subset of ER+ MBC in need of unique therapeutic approaches.
Capivasertib (AZD5363) is an oral, potent, selective ATP-competitive pan-AKT kinase inhibitor (28). We previously explored the efficacy of capivasertib monotherapy in patients with advanced solid tumors harboring an AKT1E17K mutation, including 20 patients with ER+ MBC, whereby the objective response rate (ORR) was 20% and median progression-free survival (PFS) was 5.5 months (29). Consistent with this observation, similar capivasertib monotherapy efficacy was recently reported in the AKT1-mutant arm of the NCI-MATCH study in multiple solid tumors, including ER+ MBC (30).
As observed with isoform-selective PI3K inhibitors, preclinical data with capivasertib suggests that efficacy in ER+ breast cancer may be limited in part by a compensatory increase in ER-dependent gene transcription, suggesting that combination strategies may be required to maximize therapeutic efficacy in this subtype (31-33). Accordingly, preclinical models suggest synergistic efficacy when capivasertib is combined with fulvestrant, an ER antagonist and degrader approved for the treatment of ER+ MBC (32). Therefore, to clinically explore the hypothesis that simultaneous inhibition of AKT and ER would enhance antitumor efficacy in AKT1E17K-mutant ER+ breast cancer, we amended the prior Phase I study to include a multicohort expansion of the combination of capivasertib and fulvestrant.
Here we present the safety, efficacy and biomarker analysis for the combination of capivasertib and fulvestrant in ER+, AKT1E17K-mutant MBC. To provide additional clinical context, final results for capivasertib monotherapy in ER+, AKT1E17K-mutant MBC are also presented.
Methods
Study Design and Participants
The protocol started as the first-in-human, multipart, Phase I, dose- and schedule-finding study of capivasertib. Following identification of a recommended Phase II dose, the safety and efficacy of capivasertib was further explored in multiple molecularly and histologically defined Phase I expansion cohorts recruited at study centers worldwide. Results of the initial dose escalation, pharmacodynamic cohort, and monotherapy efficacy in patients with advanced solid tumors, as well as those with activating PIK3CA or AKT1 mutations, have previously been reported (29, 34). The study start date was December 2010 and the estimated completion date is December 2019 (ClinicalTrials.gov, NCT01226316).
Here we report the results of capivasertib plus fulvestrant in patients with advanced ER+ breast cancer with AKT1E17K mutations, including patients without prior fulvestrant therapy (fulvestrant-naïve cohort) and those who received prior fulvestrant (fulvestrant-pretreated cohort; Figure 1). Updated and final efficacy data of capivasertib monotherapy in ER+ AKT1E17K-mutant breast cancer are also included.
Figure 1. Study Design of the ER+ AKT1-Mutant Breast Cancer Patient Cohorts.
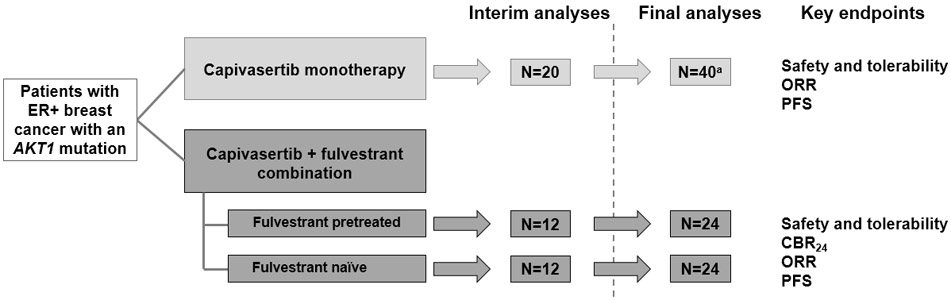
The breast cancer cohorts were part of a larger open-label, multipart, Phase I study of the first-in-human evaluation of oral capivasertib in patients with advanced solid malignancies. These Phase I expansion cohorts were non-randomized; the monotherapy cohort enrolled first, followed by the combination therapy cohort. Protocol-specified analyses planned for each study part: For monotherapy, analyses were planned after 20 patients were followed up for 12 weeks/withdrawn from the study. For combination therapy, interim analysis was planned after 12 patients in each cohort were followed up for 24 weeks/withdrawn from the study, and final analysis was planned after up to 24 patients in total in each cohort were followed up for 24 weeks/withdrawn from the study. aUp to 120. CBR24, clinical benefit rate at 24 weeks; ER, estrogen receptor; ORR, objective response rate; PFS, progression-free survival.
Eligible patients had histologically confirmed ER+, HER2− MBC with progressive measurable disease (according to Response Evaluation Criteria in Solid Tumors [RECIST] v1.1) that was refractory to standard therapies or for which no standard therapies exist, and they harbored an AKT1E17K tumor mutation. Qualifying AKT1E17K mutations were identified either through local testing, as routinely obtained at participating sites, or via a central plasma-based analysis using the OncoBEAM™ BEAMing (beads, emulsification, amplification, and magnetics) assay with previously described methods (11). Specifically, local testing employed various next-generation sequencing (NGS)-based assays, in accordance with local standard practice without any threshold for positivity mandated by AstraZeneca for enrollment. Central plasma-based BEAMing analysis with the OncoBEAM™ assay used a 0.02% threshold of analyzed AKT1 copies containing the E17K mutation for positivity (35). Further inclusion criteria included age 18 years or older and an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1. Key exclusion criteria included active central nervous system metastases, prior treatment with catalytic AKT inhibitors (prior exposure to all other agents in the PI3K/AKT/mTOR pathway, including allosteric AKT inhibitors, was allowed), and clinically significant abnormalities of glucose metabolism, defined by any of the following criteria: i) diagnosis of diabetes mellitus type 1 or 2 (irrespective of management); ii) baseline fasting glucose value of ≥7 mmol/L (fasting is defined as no calorific intake for at least 8 hours); and iii) glycated hemoglobin (HbA1c) >8% (>64 mmol/mol).
All patients provided written informed consent, and the study was performed in accordance with the Declaration of Helsinki, Good Clinical Practice, and the AstraZeneca policy on bioethics (36).
Procedures
Monotherapy patients were treated with capivasertib at the previously determined recommended Phase II monotherapy dose of 480 mg (34), administered orally, twice daily (bid) for 4 days on followed by 3 days off, repeated weekly. A treatment cycle was defined as 3 weeks. In the combination cohorts, capivasertib was administered at the previously determined recommended Phase II combination therapy dose of 400 mg bid, 4 days on, 3 days off, repeated weekly, in addition to fulvestrant at the labeled dose (37).
Response assessments were performed by computed tomography (CT) or magnetic resonance imaging (MRI) every two cycles for 24 weeks, then every 12 weeks until disease progression, death, or withdrawal. Safety was assessed throughout the study period and until day 28 after discontinuation of study treatment according to the National Cancer Institute’s Common Terminology Criteria for Adverse Events (CTCAE) v4.0. Adverse events were coded with the Medical Dictionary for Regulatory Activities (MedDRA) v19.1.
Blood was collected at every study visit for analysis of tumor-derived, cell-free DNA (cfDNA). AKT1E17K mutation status was assessed in tumor tissue by local testing and/or in cfDNA by central testing using BEAMing (OncoBEAM™, Sysmex Inostics, Baltimore, MD, USA) (38) and droplet digital polymerase chain reaction technology (ddPCR) with an allele-specific assay for both the mutant and the wild-type allele (39). Central NGS was performed retrospectively on tumor tissue when available, by FoundationOne (40), and on cfDNA using a hybrid capture-based panel covering 300 genes (AZ300).
Outcomes
The primary study endpoint was safety and tolerability of capivasertib in combination with fulvestrant. Secondary endpoints included: ORR, defined as a confirmed partial response (PR) or complete response (CR); duration of response (DOR), defined as the time from confirmed objective response to disease progression or death; PFS, defined as the time from the first day of treatment to disease progression or death; and clinical benefit rate at 24 weeks (CBR24), defined as disease response (PR or CR) or stabilization for ≥24 weeks. Responses were investigator assessed according to RECIST v1.1 and required confirmation. Patients who discontinued prior to their first response assessment were considered non-evaluable for best overall response and non-responders by intent-to-treat analysis.
Statistical Analysis
All analyses were conducted according to the protocol and statistical analysis plan (and were as previously reported for the monotherapy cohort) (29). Although the primary endpoint throughout this multipart Phase I study remained safety and tolerability, the sample size of the expansion cohort reported here was determined with the aim of detecting a signal of efficacy, should one exist, using CBR24. The capivasertib and fulvestrant combination cohorts underwent protocol-specified analyses, conducted independently for each (fulvestrant-naïve and fulvestrant-pretreated) cohort, when 12 patients (at interim analysis) and 24 (at final analysis) per cohort were evaluable for CBR24 (Figure 1). The sample size was determined based on pre-specified target values for CBR24 of 65% and 40% for fulvestrant-naïve and fulvestrant-pretreated patients, respectively (with 24 patients per cohort, there would be a 90% chance of at least 13 and 7 clinical benefit responses, respectively). At interim analysis, enrollment to the fulvestrant-naïve cohort halted, while the fulvestrant-pretreated cohort continued and completed accrual. Eight subsequent patients who were being screened at the time of closing each cohort were permitted to enroll, leading to a total of 16 and 28 patients in the fulvestrant-naïve and fulvestrant-pretreated cohorts, respectively. Final analysis occurred on August 7, 2018, when all 44 patients had had the opportunity to reach 24 weeks of treatment. All patients who received at least one dose of capivasertib (n=44) were evaluable for safety. Efficacy data are reported for 43 AKT1E17K-mutant patients and exclude one patient enrolled with a non-E17K mutation (AKT1E40K).
DOR and PFS were estimated using the Kaplan–Meier method. Patients without a progression event as of the analysis date were censored at the last known assessment. Post hoc analyses of endocrine-sensitive and -pretreated subpopulations and of patients treated with ≤2 or ≥3 prior lines of chemotherapy for MBC were performed. Patients were defined as sensitive to prior endocrine therapy if they had ≥24 months of endocrine therapy before recurrence in the adjuvant setting and/or a response or stabilization for ≥6 months of endocrine therapy for advanced disease. Exploratory biomarker analyses investigated the association between cfDNA response, defined as >50% decrease in AKT1E17K-mutant copies/mL plasma from baseline to cycle 2 day 1, and radiographic response. All analyses were done with SAS v9.04.
Results
Patient Characteristics
Sixty-three AKT1E17K-mutant ER+ MBC patients received capivasertib either as monotherapy (n=20) or in combination with fulvestrant (n=43; Table 1). The majority of combination therapy patients were enrolled based on AKT1E17K mutation detection by local laboratory testing of tumor tissue (77%, 33/43), with the remaining (n=10) patients enrolled through central laboratory plasma testing. Among patients who received the combination, 28 were previously fulvestrant pretreated and 15 were fulvestrant naïve. Median age was 57 years (range: 38–76). Most patients had visceral disease at enrollment (87%) and were heavily pretreated. Overall, 91% of patients had received prior chemotherapy, 35% mTOR inhibitors, and 24% CDK4/6 inhibitors for metastatic disease. Only 54% of patients exhibited sensitivity to prior endocrine therapy, defined by at least 24 months of endocrine therapy before recurrence in the adjuvant setting and/or a response or stabilization for at least 6 months of endocrine therapy for advanced disease. However, caution should be exercised in interpreting this seemingly low rate of endocrine therapy sensitivity compared with rates of ~80% reported in pivotal Phase III trials conducted in ER+ MBC patients (5, 41), given the retrospective and exploratory nature of this analysis. In combining both monotherapy and combination therapy cohorts, certain differentiating baseline characteristics were apparent between the fulvestrant-naïve (n=21) and fulvestrant-pretreated (n=42) patients. Specifically, a high proportion of fulvestrant-naïve patients were treated with first-line chemotherapy in the metastatic setting (38% vs 12%, respectively) and had received fewer total lines of endocrine therapy (median 2 vs 4, respectively).
Table 1.
Baseline Characteristics in Patients With ER+ HER2− AKT1E17K-Mutant Metastatic Breast Cancer
| Capivasertib Monotherapy |
Capivasertib + Fulvestrant (N=43)a |
All Capivasertib-Treated Patients (N=63)a |
||||
|---|---|---|---|---|---|---|
| Breast-Specific Cohort (N=20) |
Fulvestrant Naïve (n=15) |
Fulvestrant Pretreated (n=28) |
Fulvestrant Naïve (n=21) |
Fulvestrant Pretreated (n=42) |
Total (n=63)a |
|
| Median age, years (range) | 57 (38–71) | 58 (42–76) | 56 (40–73) | 57 (39–76) | 57 (38–73) | 57 (38–76) |
| Female gender, n (%) | 20 (100) | 15 (100) | 28 (100) | 21 (100) | 42 (100) | 63 (100) |
| Race, n (%) | ||||||
| White | 16 (80) | 9 (60) | 18 (64) | 12 (57%) | 31 (74) | 43 (68) |
| Asian | 1 (5) | 6 (40) | 5 (18) | 7 (33%) | 5 (12) | 12 (19) |
| Black | 2 (10) | 0 | 1 (4) | 1 (5%) | 2 (5) | 3 (5) |
| Other/missing | 1 (5) | 0 | 4 (14) | 1 (5%) | 4 (10) | 5 (8) |
| WHO/ECOG performance status, n (%) | ||||||
| 0 | 10 (50) | 4 (27) | 10 (36) | 8 (38) | 16 (38) | 24 (38) |
| 1 | 10 (50) | 11 (73) | 18 (64) | 13 (62) | 26 (62) | 39 (62) |
| Hormone receptor statusb | ||||||
| ER+ and PR+, n (%) | 14 (70) | 11 (73) | 23 (82) | 15 (71) | 33 (79) | 48 (76) |
| ER+ and PR−, n (%) | 5 (25) | 4 (27) | 5 (18) | 5 (24) | 9 (21) | 14 (22) |
| HER2−, n (%) | 20 (100) | 15 (100) | 28 (100) | 21 (100) | 42 (100) | 63 (100) |
| Visceral disease, n (%) | 20 (100) | 12 (80) | 23 (82) | 18 (86) | 37 (88) | 55 (87) |
| Median number of prior anticancer regimens, n (range)c | ||||||
| Total | 7 (3–14) | 4 (1–7) | 6 (2–12) | 5 (1–7) | 7 (2–14) | 6 (1–14) |
| Chemotherapy | 4 (0–6) | 2 (0–5) | 2 (0–6) | 3 (0–5) | 3 (0–6) | 3 (0–6) |
| Endocrine therapy | 4 (0–7) | 1 (0–4) | 4 (2–6) | 2 (0–4) | 4 (1–7) | 3 (0–7) |
| Number of prior endocrine therapies, n (%)c | ||||||
| 1 | 1 (5) | 6 (40) | 0 | 6 (29) | 1 (2) | 7 (11) |
| 2 | 4 (20) | 5 (33) | 5 (18) | 8 (38) | 6 (14) | 14 (22) |
| ≥3 | 14 (70) | 2 (13) | 23 (82) | 4 (19) | 35 (83) | 39 (62) |
| Prior endocrine therapyc | ||||||
| Aromatase inhibitor | 0 | 6 (40) | 8 (29) | 6 (29) | 8 (19) | 14 (22) |
| Tamoxifen | 0 | 3 (20) | 0 | 3 (14) | 0 | 3 (5) |
| Aromatase inhibitor and tamoxifen | 18 (90) | 4 (27) | 20 (71) | 9 (43) | 33 (79) | 42 (67) |
| Prior sensitivity to endocrine therapy, n (%)d | 11 (55) | 7 (47) | 16 (57) | 10 (48) | 24 (57) | 34 (54) |
| Prior chemotherapy for metastatic disease, n (%) | 19 (95) | 12 (80) | 26 (93) | 17 (81) | 40 (95) | 57 (91) |
| Chemotherapy as first-line therapy in the metastatic setting, n (%) | 5 (25) | 6 (40) | 2 (7) | 8 (38) | 5 (12) | 13 (21) |
| Prior CDK4/6 inhibitor, n (%) | 3 (15) | 1 (7) | 11 (39) | 2 (10) | 13 (31) | 15 (24) |
| Prior mTOR inhibitor, n (%) | 11 (55) | 2 (13) | 9 (32) | 4 (19) | 18 (43) | 22 (35) |
| Prior P13K inhibitor, n (%) | 1 (5) | 1 (7) | 4 (14) | 1 (5) | 5 (12) | 6 (10) |
Percentage calculated based on total N in each treatment group. In the monotherapy group, 6 patients were fulvestrant naïve and 14 fulvestrant pretreated.
Excludes one non-AKT1E17K patient, who was enrolled based on an AKT1E40K mutation detected by local NGS
Includes both primary and metastatic biopsy
Inclusive of adjuvant or metastatic therapies
Defined by at least 24 months of endocrine therapy before recurrence in the adjuvant setting and/or a response or stabilization for at least 6 months of endocrine therapy for advanced disease. ECOG, Eastern Cooperative Oncology Group; ER, estrogen receptor; HER2, human epidermal growth factor receptor 2; PR, progesterone receptor; WHO, World Health Organization.
Safety
Adverse events (AEs) causally linked to study treatment by the investigator are shown in Table 2. The most common all-grade AEs for the monotherapy cohort were diarrhea (65%), nausea (50%), hyperglycemia (45%), and vomiting (45%). Similarly, for the combination cohort, the most common AEs were diarrhea (59%), nausea (30%), maculopapular rash (21%), fatigue (18%), and hyperglycemia (18%). Grade ≥3 AEs attributed to study treatments were observed in 50% of patients in the monotherapy cohort, most commonly hyperglycemia (30%) and maculopapular rash (20%), and 21% of patients in the combination cohort, most commonly maculopapular rash (9%). AEs irrespective of causality are shown in Supplementary Table 1. No new safety signals were identified with the combination of fulvestrant.
Median duration of capivasertib exposure in the monotherapy cohort and combination cohort was 166 days (mean daily dose 870 mg) and 123 days (775 mg), respectively. In the monotherapy cohort, 13 (65%) patients required dose interruption, 7 (35%) dose reduction, and 1 (5%) discontinuation as a result of a treatment-related AE (confusion). In the combination cohort, 19 (43%) patients required dose interruption, 4 (9%) dose reduction, and 5 (11%) discontinuation because of an AE (Supplementary Table 2), three of which were treatment related (eosinophilic pneumonia, fatigue, and rash). There were no treatment-related or AE-attributable deaths in either cohort.
Efficacy Analyses
At the time of data cut-off, seven patients remained on therapy, the majority having discontinued because of disease progression (Supplementary Figure 1). Median follow-up (time to event) for all capivasertib-treated patients who were censored at the time of primary analysis was 8.1 months (range: 0–27.5). Efficacy in the monotherapy cohort and combination cohorts (overall and by prior fulvestrant therapy exposure) is shown in Table 3 and Figures 2 and 3. Among patients receiving combination therapy, ORR was 36% (95% CI: 19–56) in fulvestrant-pretreated patients and 20% (95% CI: 4–48) in fulvestrant-naïve patients. ORR in the monotherapy cohort was 20% (95% CI: 8–58). Across both monotherapy and combination cohorts (n=63), ORR was 33% (95% CI: 20–50) in fulvestrant-pretreated patients and 14% (95% CI: 3–36) in fulvestrant-naïve patients. Despite the numerically higher ORRs observed in the fulvestrant-pretreated patients, CBR24 was broadly similar across groups. Specifically, in the combination cohort, CBR24 was 50% (95% CI: 31–69) in fulvestrant-pretreated and 47% (95% CI: 21–73) in fulvestrant-naïve patients. Across both monotherapy and combination cohorts, CBR24 was 50% (95% CI: 34–66) in fulvestrant-pretreated and 43% (95% CI: 22–66) in fulvestrant-naïve patients.
Table 3.
Treatment Efficacy for Patients With ER+ HER2− AKT1E17K-Mutant Metastatic Breast Cancer
| Capivasertib Monotherapy |
Capivasertib + Fulvestrant Combination (N=43) |
All Capivasertib-Treated Patients (N=63) |
||||
|---|---|---|---|---|---|---|
| Breast-Specific Cohort (N=20) |
Fulvestrant Naïve (n=15) |
Fulvestrant Pretreated (n=28) |
Fulvestrant Naïve (n=21) |
Fulvestrant Pretreated (n=42) |
Total (n=63) |
|
| Objective responsea | ||||||
| ORR, % (95% CI) | 20 (8–58) | 20 (4–48) | 36 (19–56) | 14 (3–36) | 33 (20–50) | 27 (17–40) |
| Complete response, n (%) | 0 | 0 | 0 | 0 | 0 | 0 |
| Partial response, n (%) | 4 (20) | 3 (20) | 10 (36) | 3 (14) | 14 (33) | 17 (27) |
| DOR ≥6 months, n (%) | 2 (10) | 3 (20) | 8 (29) | 3 (14) | 10 (24) | 13 (21) |
| Stable disease 24 weeks, n (%) | 5 (25) | 4 (27) | 4 (14) | 6 (29) | 7 (17) | 13 (21) |
| Clinical benefit rate at 24 weeks, % (95% CI)b | 45 (23–69) | 47 (21–73) | 50 (31–69) | 43 (22–66) | 50 (34–66) | 48 (35–61) |
| Median PFS, months (95% CI) | 5.4 (3–7) | 5.6 (2–14) | 5.0 (3–8) | 5.4 (3–10) | 5.0 (4–7) | 5.4 (4–7) |
Response is based on investigator tumor assessments in accordance with RECIST v1.1 in patients with measurable disease.
Confirmed no fewer than 4 weeks after the criteria for response were initially met
Clinical benefit defined as confirmed best overall response of complete response, partial response, or stable disease for at least 24 weeks. CI, confidence interval; DOR, duration of response; ER, estrogen receptor; HER2, human epidermal growth factor receptor 2; ORR, objective response ratio; PFS, progression-free survival; RECIST, Response Evaluation Criteria in Solid Tumors.
Figure 2. Efficacy of Capivasertib Monotherapy in ER+ AKT1E17K-Mutant MBC (n=20).
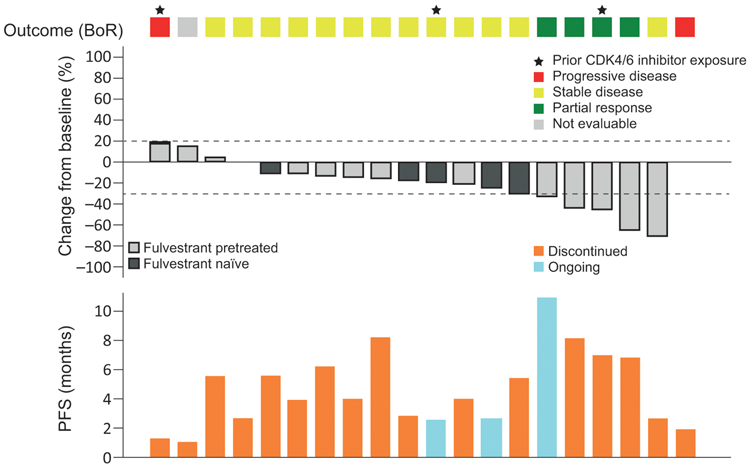
Plot based on patients with available RECIST data at baseline and at least one follow-up assessment. Investigator-assessed best percentage change from baseline was the change in the sum of longest diameters of target lesions. BoR, best objective response; ER, estrogen receptor; MBC, metastatic breast cancer; PFS, progression-free survival; RECIST, Response Evaluation Criteria in Solid Tumors.
Figure 3. Combined Efficacy and Biomarker Data From the Combination Therapy (Capivasertib + Fulvestrant) Cohort in ER+ AKT1E17K-Mutant MBC (n=43).

Best RECIST response and associated PFS integrated with genomic analyses for all 43 patients enrolled in the combination cohorts. Top to bottom: prior exposure to a CDK4/6 inhibitor; best objective response; best change from baseline in target lesion diameter according to RECIST v1.1; PFS in months; AKT1E17K mutation detection at baseline by various testing platforms (BEAMing, ddPCR, NGS) in tissue and/or ctDNA, and at C2D1 by ddPCR in ctDNA; and percentage change (≥50% decrease) in AKT1E17K-mutant copies in ctDNA by ddPCR measured on C2D1 of study treatment compared with baseline (C1D1). For 33 patients with somatic mutations detected in ctDNA by NGS, the AKT1E17K MAF, as well as the MAF from other key alterations, is presented together with the median MAF of all somatic mutations detected in each sample. Two patients lacked genomic data (not tested), and eight patients had no somatic mutations detected in their ctDNA samples by NGS, although they did by the more sensitive OncoBEAM™ and/or ddPCR assays and were deemed low shedders. Key co-occurring gene mutations detected by NGS analysis in ctDNA samples are indicated in the genomic heat map at the bottom of the figure. AF, allele frequency; C1D1, cycle 1 day 1; C2D1, cycle 2 day 1; ctDNA, circulating tumor DNA; ddPCR, droplet digital polymerase chain reaction; ER, estrogen receptor; FMI, Foundation Medicine, Inc; MAF, mutant allele fraction; MBC, metastatic breast cancer; NGS, next-generation sequencing; PFS, progression-free survival; RECIST, Response Evaluation Criteria in Solid Tumors.
To determine whether additional patient and treatment characteristics could further enrich for patients who experienced benefit from capivasertib, several exploratory post hoc subgroup analyses were conducted. Across monotherapy and combination therapy patients, ORR and CBR24 were numerically higher in patients who had received ≤2 prior lines of chemotherapy (35% and 62%, respectively) compared with those who had received ≥3 prior lines (22% and 38%, respectively; Supplementary Table 3). Analyses classifying patients based on prior endocrine therapy sensitivity were also conducted but did not clearly predict benefit of capivasertib-based therapy.
Exploratory Biomarker Analyses
Central biomarker assessments in the combination cohort utilized a variety of assays. Central tissue NGS was performed in 42% (18/43) of patients. BEAMing was used for mutation detection in plasma cfDNA collected at screening and detected the AKT1E17K mutation in 95% (41/43) of patients. ddPCR and broader NGS profiling were performed on plasma cfDNA collected on the first day of treatment (cycle 1 day 1), detecting the AKT1E17K mutation in 80% (33/41) and 76% (31/41) of patients tested, respectively (Figure 3). These data demonstrate that plasma-based analyses offer an additional diagnostic opportunity for AKT1E17K mutation testing (35). In this cohort, a ≥50% decrease from baseline at cycle 2 day 1 was associated with improved PFS (Supplementary Figure 2), similar to that previously demonstrated in the monotherapy cohort (29).
In 41 patients tested, the broader genetic profiling of plasma samples by NGS identified co-occurring alterations in ESR1 (n=10, almost all in fulvestrant-pretreated patients and all in those with a detectable AKT1 E17K mutation by plasma NGS), TP53 (n=8, predominantly in fulvestrant-naïve patients), MAP3K1 (n=4), PIK3CA (n=4) and FGFR1 (n=2) (Figure 3). In one of the PIK3CA-mutant cases, despite an AKT1E17K mutation being detected by BEAMing at a very low mutant allele fraction (MAF), the AKT1E17K mutation was not in fact detectable by NGS, indicative of the subclonality of the alteration in this patient (Supplementary Figure 3). Evidence of a subclonal AKT1E17K mutation could also be found in another patient, in whom the AKT1E17K mutation was detected by BEAMing and ddPCR but not by the NGS analysis that detected other somatic mutations in this patient. While 8 (20%) patients did not shed sufficient circulating tumor DNA for mutation detection by NGS (ie low shedders), in all other (31; 94%) patients, the AKT1E17K mutation was detected at a level around or above the median MAF indicative of the predominantly clonal nature of this alteration (Figure 3 and Supplementary Figure 3). In this limited sample set, no obvious pattern in AKT1E17K clonality or co-incident tumor mutations was associated with clinical outcome, although, potentially of interest, 6 of the 8 (75%) cases identified as low shedders by NGS had an objective response, and none of the patients whose tumors harbored a TP53 mutation achieved an objective response. While the sample size was small, the observed higher frequency of TP53 mutations in the fulvestrant-naïve compared with the fulvestrant-pretreated cohort (33% vs 12%, respectively) potentially supports the observation that this group had more aggressive disease biology (42). Equally, the identified TP53 mutations could be related to the greater degree of cytotoxic chemotherapy exposure in this cohort (43, 44). An integrated analysis of efficacy and genomic data is shown in Figure 3.
Discussion
In this multicohort Phase I study, we sequentially explored the safety and efficacy of the pan-AKT inhibitor capivasertib, initially alone and later in combination with fulvestrant, in ER+ AKT1E17K-mutant MBC patients. The safety profile was similar to that in prior reports (29, 34, 45), although combination therapy appeared better tolerated, likely because of the lower dose of capivasertib (400 mg bid 4 days on, 3 days off) administered with fulvestrant compared with the monotherapy dose of capivasertib (480 mg bid 4 days on, 3 days off), as suggested by the dose–response relationship observed for key capivasertib-related toxicities such as hyperglycemia (46).
Although the study was not designed to directly compare activity across groups, and noting that the fulvestrant-naïve (n=21) patients treated in this study may have had a more aggressive disease profile at baseline than those who were fulvestrant pretreated (n=42), optimal efficacy was nonetheless observed with combination therapy, specifically in fulvestrant-pretreated patients (ORR 36%; CBR24 50%). Taken together, these findings are encouraging, particularly given the heavily pretreated nature of the study population. There is also reason to believe that our data compare favorably with prior reports on molecular therapy in the clinic. For example, BELLE-3 evaluated fulvestrant, with or without the pan-PI3K inhibitor buparlisib, in mTOR-inhibitor-exposed patients, reporting, respectively, an ORR of 8% versus 2% and CBR24 of 25% versus 15% (47). This provides a useful benchmark for fulvestrant monotherapy following mTOR inhibitor exposure in a notably less pretreated (no more than one line of chemotherapy and no prior fulvestrant were permitted) population. Similarly, our data compare favorably with expected chemotherapy outcomes in endocrine-resistant patients (48).
The benefit of adding capivasertib to hormone therapy in AKT1E17K-mutant patients is consistent with preclinical data (32). More broadly, the role for co-targeting ER and PI3K pathway alterations has been demonstrated in multiple randomized, Phase III studies of PI3K inhibitors (17, 49). Furthermore, in the recently reported randomized Phase II FAKTION study, the addition of capivasertib to fulvestrant showed a significant improvement in PFS in a molecularly unselected, aromatase-inhibitor-pretreated but fulvestrant-naïve ER+ MBC population (50). Given the increasing genomic complexity of breast cancer as it advances through multiple lines of therapy, this recent trial report supports our observation and hypothesis, and others’, that earlier introduction of targeted therapies to a less clonally diverse disease is likely to be necessary to garner significant improvements in outcome in patients harboring these driver oncogenic alterations (14).
Acknowledging that only 24% of enrolled patients in this study received prior CDK4/6 inhibitors, agents that are now standard of care in combination with an aromatase inhibitor or fulvestrant in the first- or second-line setting, these data remain of interest. Outcomes of targeted therapy following CDK4/6 inhibitor exposure in ER+ MBC are largely unknown. However, preclinical models with acquired resistance to CDK4/6 inhibitors do indicate retained sensitivity to PI3K pathway inhibition combined with endocrine therapy (51, 52). Indeed, in SOLAR-1, the small subset of patients with prior CDK4/6 inhibitor exposure did still appear to derive benefit from the addition of apelisib to fulvestrant (17). Additionally, of interest, recent preclinical data have implicated PTEN loss, as a potential mechanism of resistance to CDK4/6 inhibitors, via increased AKT activation in vitro and in vivo (53), a hypothesis since observed in the clinic where enrichment of PTEN loss-of-function alterations has been described in tumor samples obtained after CDK4/6 inhibitor therapy (54). Intriguingly, in this context (PTEN-null models resistant to CDK4/6 inhibitors), AKT inhibition may in fact be superior to PI3K inhibition (53). It is also clear from preclinical work that constitutively active AKT induces resistance to PI3K inhibition in breast cancer cell lines, and, interestingly, increased AKT1 expression was identified in a very small cohort of biopsies collected post-treatment with alpelisib (55). Clinical data demonstrating a role for AKT1 mutations mediating resistance to anti-estrogens or CDK4/6 inhibitors are limited. A recent clinical series (n=57) noted an over-representation of PI3K pathway mutations (PIK3CA, AKT1, TSC2, and/or loss or truncation mutations of PTEN) among patients with a poor response to neoadjuvant letrozole (Pre-operative Endocrine Prognostic Index [PEPI] >4 and/or recurrence), although this was unlikely to be driven by AKT1, as none of the three AKT1-mutant cases in this report experienced a recurrence and two of the three were actually categorized in the responder group (PEPI <4 and no recurrence) (56). Additionally, an endocrine-therapy-exposed ER+ breast cancer dataset did not identify AKT1 mutations in tumors intrinsically resistant to letrozole; rather, AKT1 mutations were detected in those sensitive to the therapy (57, 58). In agreement with this, genomic profiling of a large (n=1501) cohort of endocrine-therapy-naïve versus endocrine-therapy-exposed ER+ breast cancers did not show any evidence of AKT1 mutations being associated with resistance to hormonal therapy (15). Finally, findings from a recent institutional dataset (n=58) have proposed activating events in AKT1 as a possible mechanism of resistance to therapy containing CDK4/6 inhibitors, along with in vitro data showing overexpression of AKT1 as conferring resistance to CDK4/6 inhibitors (50), although, thus far, this has not been observed in genomic analysis from the registration studies of these agents (59, 60). Moreover, genomic analysis of 348 ER+ breast cancers treated with CDK4/6 inhibitors, as well as comparative analysis of tumors before (n=838) versus after (n=221) CDK4/6 inhibitor therapy, along with paired analysis of tumors before versus after CDK4/6 inhibitor therapy (n=210), has not identified an association between AKT1 mutations and therapeutic resistance to CDK4/6 inhibitors (54, 61).
Our study has several important limitations. Firstly, this trial was not formally powered to compare efficacy across treatment groups. Secondly, although efficacy appeared most robust in fulvestrant-pretreated patients, it is noteworthy that the fulvestrant-naïve patients enrolled here appeared to be a subgroup with poorer prognosis. Given this, we cannot rule out the role that demographic imbalance between the groups driven by adverse patient selection factors may have played in the apparent difference in treatment outcomes. Thirdly, we do not know the extent to which the presence of an AKT1E17K mutation may influence the natural history or response to standard therapy for MBC. Despite this, recent analyses suggest that prognoses of AKT1E17K-mutant and wild-type MBC patients appear largely comparable, somewhat mitigating this concern (21). Finally, despite opening this study at 16 sites internationally, the rarity of this biomarker led to slow accrual (22 months to enroll 44 patients in the combination cohort), despite having central screening by BEAMing in plasma implemented, in addition to local testing.
In conclusion, this study demonstrates that AKT1E17K is a clinically relevant, valid target in ER+ breast cancer and that the AKT inhibitor capivasertib is tolerable and active as both monotherapy and in combination with fulvestrant, including in patients with prior fulvestrant resistance. We confirm that the majority of enrolled patients had detectable AKT1E17K in plasma at baseline and demonstrate the feasibility of enrollment based on centralized plasma screening for this rare genomic biomarker (35). With other genomic biomarkers such as PIK3CA mutations expected to become part of routine management paradigms over the coming years in breast cancer, these data have the prospect of becoming part of a rationale to incorporate other potentially actionable alterations in breast cancer, including ERBB2 and AKT1, into diagnostic testing algorithms and for the early identification of these alterations in the metastatic disease course (62, 63). Finally, data from this study, along with the FAKTION study, have provided the basis for a confirmatory Phase III study that will take into account populations with and without prior use of CDK4/6 inhibitors.
Supplementary Material
Table 2.
AEs Causally Linked to Study Treatment (>10% of Patients) and Grade ≥3 AEs (>2 Patients)
| AE by Preferred Term |
Capivasertib Monotherapy Breast-Specific Cohort (N=20) |
Capivasertib + Fulvestrant Combination (N=44)a |
Total (N=64)a |
|||
|---|---|---|---|---|---|---|
| All grades | Grade ≥3 | All grades | Grade ≥3 | All grades | Grade ≥3 | |
| Any AE (causally related to capivasertib), n (%) | 19 (95) | 10 (50) | 38 (86) | 9 (21) | 57 (89) | 19 (30) |
| Diarrhea | 13 (65) | 2 (10) | 26 (59) | 2 (5) | 39 (61) | 4 (6) |
| Nausea | 10 (50) | 0 | 13 (30) | 1 (2) | 23 (36) | 1 (2) |
| Hyperglycemia | 9 (45) | 6 (30) | 8 (18) | 2 (5) | 17 (27) | 8 (13) |
| Vomiting | 9 (45) | 0 | 7 (16) | 0 | 16 (25) | 0 |
| Fatigue | 8 (40) | 0 | 8 (18) | 1 (2) | 16 (25) | 1 (2) |
| Rash maculopapular | 6 (30) | 4 (20) | 9 (21) | 4 (9) | 15 (23) | 8 (13) |
| Decreased appetite | 3 (15) | 0 | 7 (16) | 1 (2) | 10 (16) | 1 (2) |
| Stomatitis | 4 (20) | 0 | 6 (14) | 0 | 10 (16) | 0 |
| Dry skin | 4 (20) | 0 | 3 (7) | 0 | 7 (11) | 0 |
| Abdominal pain | 4 (20) | 0 | 2 (5) | 0 | 6 (9) | 0 |
| Dizziness | 4 (20) | 0 | 2 (5) | 0 | 6 (9) | 0 |
| Pruritus | 3 (15) | 0 | 3 (7) | 0 | 6 (9) | 0 |
| Dry mouth | 4 (20) | 0 | 0 | 0 | 4 (6) | 0 |
Includes AEs with an onset date on or after the date of first dose and up to and including 28 days following the date of last dose of study. A patient is only counted once for each preferred term.
Includes one non-AKT1E17K patient excluded from the efficacy analyses, who was enrolled based on an AKT1E40K mutation detected by local NGS. AE, adverse event; NGS, next-generation sequencing.
Statement of translational relevance.
Early identification of the AKT1E17K genomic biomarker, coupled with a novel targeted and non-myeloablative agent, could enhance treatment options in AKT1E17K-mutant ER+ metastatic breast cancer. In this first-in-human, multipart, Phase I expansion study, capivasertib alone or in combination with fulvestrant was well tolerated and showed promising anticancer activity in such a patient population, including those with prior disease progression on fulvestrant. Tolerability and efficacy appeared marginally better with combination therapy, suggesting that combination AKT and ER inhibition is an effective targeted therapy approach for AKT1E17K-mutant ER+ metastatic breast cancer. Furthermore, our data provide a rationale for incorporating potentially actionable alterations in breast cancer into diagnostic testing algorithms for the early identification of these alterations in the metastatic disease course.
Acknowledgments
This study was sponsored by AstraZeneca. Capivasertib was discovered by AstraZeneca subsequent to a collaboration with Astex Therapeutics (and its collaboration with the Institute of Cancer Research and Cancer Research Technology Limited). U. Banerji acknowledges infrastructural funding from Cancer Research UK, Experimental Cancer Medicine Centre and Biomedical Research Centre grants, in addition to a National Institutes of Health Research Professorship award (RP-2016-07-028). All investigators at Memorial Sloan Kettering Cancer Center (L.M. Smyth, M. Scaltriti, B.S. Taylor, S. Chandarlapaty, J. Baselga, D.M. Hyman) wish to acknowledge the support of the NCI Cancer Center Support Grant (CCSG P30 CA08748). S. Chandarlapaty acknowledges support of the BCRF. We are grateful for the assistance of Neville Cope, Lucy Keeling, Jayantha Ratnayake and Carolina Salinas-de Souza for contributing to the preparation of this manuscript. We thank the patients and their carers who participated in this study. Medical writing assistance was provided by Martin Goulding, DPhil and Kristin Almond, PhD from Mudskipper Business Ltd, funded by AstraZeneca. Capivasertib (AZD5363) is an investigational medical product with no approved indication.
The study sponsor, AstraZeneca, provided organizational support, obtained data, performed the analyses, and had a role in data interpretation and writing of the manuscript. All authors had access to study data, and the corresponding author had final responsibility for the decision to submit the manuscript for publication.
Footnotes
Conflicts of interest
LMS has acted in a consultancy or advisory role for AstraZeneca and Roche Genentech and has received research funding from AstraZeneca, Puma Biotechnology, and Roche Genentech, travel or accommodation expenses from AstraZeneca, Pfizer, Puma Biotechnology, and Roche Genentech, and honoraria from Pfizer. KT has received research funding from Daiichi Sankyo, Pfizer, and Lilly. MO has received research funding from AstraZeneca, Boehringer Ingelheim, GSK, Immunomedics, Novartis, Puma Biotechnology, Roche Genentech, and Seattle Genetics and consultancy fees from AstraZeneca, Eisai, GP Pharma, Grünenthal, GSK, Novartis, Pierre-Fabre, Puma Biotechnology, Roche Genentech, and Seattle Genetics. EC has received consultancy fees from Lilly, Novartis, Pfizer, and Roche. IAM has participated in advisory boards (compensated) for Abbvie, AstraZeneca, Genentech, GSK, Lilly, Macrogenics, Novartis, Immunogenics, Pfizer, Puma Biotechnology, and Seattle Genetics and has received institutional research funding from Genentech, Novartis, and Pfizer. LB has acted in a consultancy or advisory role for AstraZeneca, Celgene, Eisai, Genomic Health, Ipsen, Lilly, Novartis, Pfizer, Pierre Fabre, and Roche and received research funding from Celgene, Genomic Health, and Novartis, honoraria from Lilly, Novartis, and Pfizer, and travel or accommodation expenses from Celgene, Pfizer, Ipsen, and Roche. UB has received research grants from AstraZeneca, Chugai, and Onyx Pharmaceuticals and consultancy fees from Astex and Novartis. MS has received research funds from Daiichi Sankyo, Immunomedics, Menarini Ricerche, Puma Biotechnology, and Targimmune, has participated in scientific advisory boards for Menarini Ricerche and Bioscience Institute, and is a cofounder of Medendi. BST has received honoraria and research funding from Genentech and participated in advisory boards for Boehringer Ingelheim and Loxo Oncology, a wholly owned subsidiary of Eli Lilly. SC has received a research grant from Daiichi Sankyo and consultancy fees from BMS, Context Therapeutics, Eli Lilly, Novartis, Revolution Medicines, and Sermonix Pharmaceutical. DMH reports stock ownership in Fount Therapeutics, has acted in a consultancy or advisory role for AstraZeneca, Bayer, Boehringer Ingelheim, Chugai Pharma, Eli Lilly, Genentech, and Pfizer, and has received research funding from AstraZeneca, Bayer, Loxo Oncology, and Puma Biotechnology and travel or accommodation expenses from Chugai Pharma and Genentech. HA, JA, AB, DC, CC, ECdB, AF, JH, JPOL, RM, RMc, MM, MP, VR, GS, and JB are employees of AstraZeneca. M-PS declared no competing interests.
Data from this manuscript were partially reported at the San Antonio Breast Cancer Symposium (SABCS), December 5–9, 2017, San Antonio, TX, USA.
Data-Sharing Statement
Data underlying the findings described in this manuscript may be obtained in accordance with AstraZeneca’s data sharing policy described at:
https://astrazenecagrouptrials.pharmacm.com/ST/Submission/Disclosure.
References
- 1.Gong Y, Liu YR, Ji P, Hu X, Shao ZM. Impact of molecular subtypes on metastatic breast cancer patients: a SEER population-based study. Sci Rep 2017;7:45411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.GLOBOCAN 2018. Breast cancer factsheet. 2019. Available at: http://gco.iarc.fr/today/data/factsheets/cancers/20-Breast-fact-sheet.pdf. [Google Scholar]
- 3.Baselga J, Campone M, Piccart M, Burris HA, III, Rugo HS, Sahmoud T, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med 2012;366:520–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hortobagyi GN, Stemmer SM, Burris HA, Yap YS, Sonke GS, Paluch-Shimon S, et al. Updated results from MONALEESA-2, a Phase III trial of first-line ribociclib plus letrozole versus placebo plus letrozole in hormone receptor-positive, HER2-negative advanced breast cancer. Ann Oncol 2018;29:1541–7. [DOI] [PubMed] [Google Scholar]
- 5.Turner NC, Slamon DJ, Ro J, Bondarenko I, Im SA, Masuda N, et al. Overall survival with palbociclib and fulvestrant in advanced breast cancer. N Engl J Med 2018;379:1926–36. [DOI] [PubMed] [Google Scholar]
- 6.Im SA, Lu YS, Bardia A, Harbeck N, Colleoni M, Franke F, et al. Overall survival with ribociclib plus endocrine therapy in breast cancer. N Engl J Med 2019;381:307–16. [DOI] [PubMed] [Google Scholar]
- 7.Goetz MP, Toi M, Campone M, Sohn J, Paluch-Shimon S, Huober J, et al. MONARCH 3: abemaciclib as initial therapy for advanced breast cancer. J Clin Oncol 2017;35:3638–46. [DOI] [PubMed] [Google Scholar]
- 8.Finn RS, Martin M, Rugo HS, Jones S, Im SA, Gelmon K, et al. Palbociclib and letrozole in advanced breast cancer. N Engl J Med 2016;375:1925–36. [DOI] [PubMed] [Google Scholar]
- 9.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011;364:2507–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hyman DM, Taylor BS, Baselga J. Implementing genome-driven oncology. Cell 2017;168:584–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Richardson AL, Iglehart JD. BEAMing up personalized medicine: mutation detection in blood. Clin Cancer Res 2012;18:3209–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Robson M, Im SA, Senkus E, Xu B, Domchek SM, Masuda N, et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N Engl J Med 2017;377:523–33. [DOI] [PubMed] [Google Scholar]
- 13.Litton JK, Rugo HS, Ettl J, Hurvitz SA, Goncalves A, Lee KH, et al. Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. N Engl J Med 2018;379:753–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bertucci F, Ng CKY, Patsouris A, Droin N, Piscuoglio S, Carbuccia N, et al. Genomic characterization of metastatic breast cancers. Nature 2019;569:560–4. [DOI] [PubMed] [Google Scholar]
- 15.Razavi P, Chang MT, Xu G, Bandlamudi C, Ross DS, Vasan N, et al. The genomic landscape of endocrine-resistant advanced breast cancers. Cancer Cell 2018;34:427–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lux MP, Fasching PA, Schrauder MG, Hein A, Jud SM, Rauh C, et al. The PI3K Pathway: Background and Treatment Approaches. Breast Care (Basel) 2016;11:398–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andre F, Ciruelos E, Rubovszky G, Campone M, Loibl S, Rugo HS, et al. Alpelisib for PIK3CA-mutated, hormone receptor-positive advanced breast cancer. N Engl J Med 2019;380:1929–40. [DOI] [PubMed] [Google Scholar]
- 18.Carpten JD, Faber AL, Horn C, Donoho GP, Briggs SL, Robbins CM, et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature 2007;448:439–44. [DOI] [PubMed] [Google Scholar]
- 19.Kim MS, Jeong EG, Yoo NJ, Lee SH. Mutational analysis of oncogenic AKT E17K mutation in common solid cancers and acute leukaemias. Br J Cancer 2008;98:1533–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012;490:61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smyth LM, Zhou Q, Nguyen B, Yu C, Lepisto EM, Arnedos M, et al. Characteristics and outcome of AKT1 E17K-mutant breast cancer defined through AACR GENIE, a clinicogenomic registry. Cancer Discov 2020;[Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brown JS, Banerji U. Maximising the potential of AKT inhibitors as anti-cancer treatments. Pharmacol Ther 2017;172:101–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chang MT, Asthana S, Gao SP, Lee BH, Chapman JS, Kandoth C, et al. Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat Biotechnol 2016;34:155–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hassan B, Akcakanat A, Holder AM, Meric-Bernstam F. Targeting the PI3-kinase/Akt/mTOR signaling pathway. Surg Oncol Clin N Am 2013;22:641–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rudolph M, Anzeneder T, Schulz A, Beckmann G, Byrne AT, Jeffers M, et al. AKT1 (E17K) mutation profiling in breast cancer: prevalence, concurrent oncogenic alterations, and blood-based detection. BMC Cancer 2016;16:622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yi KH, Lauring J. Recurrent AKT mutations in human cancers: functional consequences and effects on drug sensitivity. Oncotarget 2016;7:4241–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beaver JA, Gustin JP, Yi KH, Rajpurohit A, Thomas M, Gilbert SF, et al. PIK3CA and AKT1 mutations have distinct effects on sensitivity to targeted pathway inhibitors in an isogenic luminal breast cancer model system. Clin Cancer Res 2013;19:5413–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davies BR, Greenwood H, Dudley P, Crafter C, Yu DH, Zhang J, et al. Preclinical pharmacology of AZD5363, an inhibitor of AKT: pharmacodynamics, antitumor activity, and correlation of monotherapy activity with genetic background. Mol Cancer Ther 2012;11:873–87. [DOI] [PubMed] [Google Scholar]
- 29.Hyman DM, Smyth LM, Donoghue MTA, Westin SN, Bedard PL, Dean EJ, et al. AKT inhibition in solid tumors with AKT1 mutations. J Clin Oncol 2017;35:2251–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kalinsky K, Hong F, Mccourt CK, Sachdev JC, Mitchell EP, Zwiebel JA, et al. AZD5363 in patients (pts) with tumors with AKT mutations: NCI-MATCH subprotocol EAY131-Y, a trial of the ECOG-ACRIN Cancer Research Group (EAY131-Y). Eur J Cancer 2018;103(Suppl 1):e15. [Google Scholar]
- 31.Bosch A, Li Z, Bergamaschi A, Ellis H, Toska E, Prat A, et al. PI3K inhibition results in enhanced estrogen receptor function and dependence in hormone receptor-positive breast cancer. Sci Transl Med 2015;7:283ra51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ribas R, Pancholi S, Guest SK, Marangoni E, Gao Q, Thuleau A, et al. AKT antagonist AZD5363 influences estrogen receptor function in endocrine-resistant breast cancer and synergizes with fulvestrant (ICI182780) in vivo. Mol Cancer Ther 2015;14:2035–48. [DOI] [PubMed] [Google Scholar]
- 33.Toska E, Osmanbeyoglu HU, Castel P, Chan C, Hendrickson RC, Elkabets M, et al. PI3K pathway regulates ER-dependent transcription in breast cancer through the epigenetic regulator KMT2D. Science 2017;355:1324–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Banerji U, Dean EJ, Pérez-Fidalgo JA, Batist G, Bedard PL, You B, et al. A Phase 1 open-label study to identify a dosing regimen of the pan-AKT inhibitor AZD5363 for evaluation in solid tumors and in PIK3CA-mutated breast and gynecologic cancers. Clin Cancer Res 2018;24:2050–9. [DOI] [PubMed] [Google Scholar]
- 35.de Bruin EC, Whiteley JL, Corcoran C, Kirk PM, Fox JC, Armisen J, et al. Accurate detection of low prevalence AKT1 E17K mutation in tissue or plasma from advanced cancer patients. PLoS One 2017;12:e0175779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.AstraZeneca. Global policy: bioethics. 2016. Available at: https://www.astrazeneca.com/content/dam/az/PDF/2016/Bioethics_policy.pdf. [Google Scholar]
- 37.Jones RH, Carucci M, Casbard A, Butler R, Alchami F, Bale CJ, et al. Capivasertib (AZD5363) plus fulvestrant versus placebo plus fulvestrant after relapse or progression on an aromatase inhibitor in metastatic ER-positive breast cancer (FAKTION): A randomized, double-blind, placebo-controlled, phase II trial. J Clin Oncol 2019;37: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Diehl F, Li M, Kinzler KW, Vogelstein B, Dressman D. BEAMing: single-molecule PCR on microparticles in water-in-oil emulsions. Nat Methods 2006;3:551–9. [DOI] [PubMed] [Google Scholar]
- 39.Paoletti C, Schiavon G, Dolcem EM, Darga EP, Carr TH, Geradts J, et al. Circulating biomarkers and resistance to endocrine therapy in metastatic breast cancers: correlative results from AZD9496 oral SERD Phase I trial. Clin Cancer Res 2018;24:5860–72. [DOI] [PubMed] [Google Scholar]
- 40.Foundation Medicine. FoundationOne® CDx. 2019. Available at: https://www.foundationmedicine.com/genomic-testing/foundation-one-cdx. [Google Scholar]
- 41.Hortobagyi GN. Everolimus plus exemestane for the treatment of advanced breast cancer: a review of subanalyses from BOLERO-2. Neoplasia 2015;17:279–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meric-Bernstam F, Zheng X, Shariati M, Damodaran S, Wathoo C, Brusco L, et al. Survival outcomes by TP53 mutation status in metastatic breast cancer. JCO Precis Oncol 2018;2018:10.1200/PO.17.00245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Razavi P, Li BT, Brown DN, Jung B, Hubbell E, Shen R, et al. High-intensity sequencing reveals the sources of plasma circulating cell-free DNA variants. Nat Med 2019;25:1928–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Coombs CC, Zehir A, Devlin SM, Kishtagari A, Syed A, Jonsson P, et al. Therapy-Related Clonal Hematopoiesis in Patients with Non-hematologic Cancers Is Common and Associated with Adverse Clinical Outcomes. Cell Stem Cell 2017;21:374–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dean E, Banerji U, Schellens JHM, Krebs MG, Jimenez B, van Brummelen E, et al. A Phase 1, open-label, multicentre study to compare the capsule and tablet formulations of AZD5363 and explore the effect of food on the pharmacokinetic exposure, safety and tolerability of AZD5363 in patients with advanced solid malignancies: OAK. Cancer Chemother Pharmacol 2018;81:873–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Elvin P, Palmer A, Womack C, Tall M, Swales KE, Garrett MD, et al. Pharmacodynamic activity of the AKT inhibitor AZD5363 in patients with advanced solid tumors. J Clin Oncol 2014;32(15S):abst 2541. [Google Scholar]
- 47.Di LA, Johnston S, Lee KS, Ciruelos E, Lonning PE, Janni W, et al. Buparlisib plus fulvestrant in postmenopausal women with hormone-receptor-positive, HER2-negative, advanced breast cancer progressing on or after mTOR inhibition (BELLE-3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 2018;19:87–100. [DOI] [PubMed] [Google Scholar]
- 48.Cortes J, O'Shaugnessy J, Loesch D, Blum JL, Vahdat LT, Petrakova K, et al. Eribulin monotherapy versus treatment of physician's choice in patients with metastatic breast cancer (EMBRACE): a phase 3 open-label randomised study. Lancet 2011;377:914–23. [DOI] [PubMed] [Google Scholar]
- 49.Baselga J, Dent SF, Cortés J, Im Y-H, Diéras V, Harbeck N, et al. Phase III study of taselisib (GDC-0032) + fulvestrant (FULV) v FULV in patients (pts) with estrogen receptor (ER)-positive, PIK3CA-mutant (MUT), locally advanced or metastatic breast cancer (MBC): primary analysis from SANDPIPER. J Clin Oncol 2018;36(18 Suppl):abst LBA1006. [Google Scholar]
- 50.Jones RH, Carucci M, Claire A, Butler R, Alchami F, Bale CJ, et al. Capivasertib (AZD5363) plus fulvestrant versus placebo plus fulvestrant after relapse or progression on an aromatase inhibitor in metastatic ER-positive breast cancer (FAKTION): a randomized, double-blind, placebo-controlled, Phase II trial. J Clin Oncol 2019;37(15 Suppl):abst 1005. [Google Scholar]
- 51.O'Brien NA, McDermott MSJ, Conklin DF, Gaither A, Luo T, Ayala R, et al. Targeting activated PI3K/mTOR signaling overcomes resistance to CDK4/6-based therapies in preclinical ER+ breast cancer models. Cancer Res 2019;79(13 Suppl):abst 3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.O'Brien NA, Conklin D, Luo T, Ayala R, Issakhanian S, Kalous O, et al. Anti-tumor activity of the PI3K/mTOR pathway inhibitors alpelisib (BYL719) and everolimus (RAD001) in xenograft models of acquired resistance to CDK-4/6 targeted therapy. Cancer Res 2017;77(13 Suppl):abst 4150. [Google Scholar]
- 53.Costa C, Wang Y, Ly A, Hosono Y, Murchie E, Walmsley CS, et al. PTEN Loss Mediates Clinical Cross-Resistance to CDK4/6 and PI3Kalpha Inhibitors in Breast Cancer. Cancer Discov 2020;10:72–85. [DOI] [PubMed] [Google Scholar]
- 54.Razavi P Molecular profiling of ER+ metastatic breast cancers to reveal association of genomic alterations with aquired resistance to CDK4/6 inhibitors. J Clin Oncol 2019;37:37s. [Google Scholar]
- 55.Le X, Antony R, Razavi P, Treacy DJ, Luo F, Ghandi M, et al. Systematic Functional Characterization of Resistance to PI3K Inhibition in Breast Cancer. Cancer Discov 2016;6:1134–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Guerrero-Zotano AL, Stricker TP, Formisano L, Hutchinson KE, Stover DG, Lee KM, et al. ER(+) Breast Cancers Resistant to Prolonged Neoadjuvant Letrozole Exhibit an E2F4 Transcriptional Program Sensitive to CDK4/6 Inhibitors. Clin Cancer Res 2018;24:2517–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Giltnane JM, Hutchinson KE, Stricker TP, Formisano L, Young CD, Estrada MV, et al. Genomic profiling of ER(+) breast cancers after short-term estrogen suppression reveals alterations associated with endocrine resistance. Sci Transl Med 2017;9: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wander SA, Cohen O, Gong X, Johnson GN, Buendia-Buendia J, Lloyd M, et al. The genomic landscape of intrinsic and acquired resistance to cyclin-dependLloyd,M.ent kinase 4/6 inhibitors (CDK4/6i) in patients with hormone receptor-positive (HR+)/HER2− metastatic breast cancer (MBC). Cancer Res 2020;DOI: 10.1158/1538-7445.SABCS19-PD2-09: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.O'Leary B, Cutts RJ, Liu Y, Hrebien S, Huang X, Fenwick K, et al. The Genetic Landscape and Clonal Evolution of Breast Cancer Resistance to Palbociclib plus Fulvestrant in the PALOMA-3 Trial. Cancer Discov 2018;8:1390–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Goetz MP, Beck T, Campone M, Hurvitz S, Im S-A, Johnston S, et al. Efficacy of abemaciclib based on genomic alterations detected in baseline circulating tumor DNA from the MONARCH 3 study of abemaciclib plus nonsteroidal aromatase inhibitor. Cancer Res 2020;80: [Google Scholar]
- 61.Li Z, Razavi P, Li Q, Toy W, Liu B, Ping C, et al. Loss of the FAT1 Tumor Suppressor Promotes Resistance to CDK4/6 Inhibitors via the Hippo Pathway. Cancer Cell 2018;34:893–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hyman DM, Piha-Paul SA, Won H, Rodon J, Saura C, Shapiro GI, et al. HER kinase inhibition in patients with HER2− and HER3-mutant cancers. Nature 2018;554:189–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Condorelli R, Mosele F, Verret B, Bachelot T, Bedard PL, Cortes J, et al. Genomic alterations in breast cancer: level of evidence for actionability according to ESMO Scale for Clinical Actionability of molecular Targets (ESCAT). Ann Oncol 2019;30:365–73. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.