

Abstract
Background:
Seviteronel was being developed by Innocrin Pharamceuticals as a selective cytochrome P450c17a (CYP17) 17,20-lyase (lyase) inhibitor and androgen receptor (AR) antagonist with activity against prostate cancer cells in vitro and in vivo. This open-label Phase II clinical study evaluated the tolerability and efficacy of seviteronel in patients with metastatic castration-resistant prostate cancer (mCRPC) previously treated with enzalutamide.
Experimental Design:
Patients with mCRPC who previously progressed on enzalutamide were divided into two cohorts based on prior exposure to docetaxel. Seviteronel was administered without routine oral steroids either twice daily with dose titration (450 mg) or once daily without dose titration (600 mg or 750 mg). The primary objective was to determine the rate of significant PSA response (i.e. decline of >50%) following 12 weeks of seviteronel.
Results:
Seventeen patients (median age: 71 [60 to 92]) were enrolled, with 8 patients having received prior docetaxel. Patients received a median of 2 cycles of treatment, with a majority of patients discontinuing treatment due to toxicity related to study drug. Most common adverse events included concentration impairment, fatigue, tremor and nausea. Despite changes in dosing, the study was closed prematurely due to magnitude of toxicity. One of seventeen patients (6%) had a significant PSA decline.
Conclusions:
Seviteronel was not generally well tolerated in patients with mCRPC who previously received enzalutamide and not associated with significant clinical reponses. Further investigation of single-agent seviteronel in this patient population is not warranted, however studies investigating seviteronel with low-dose dexamethasone are ongoing in patients with AR-postive tumors.
Keywords: mCRPC, acquired resistance, CYP17 inhibitor, androgen receptor, enzalutamide
MicroAbstract
This open label Phase II clinical study evaluated the safety and efficacy of seviteronel (dosed once or twice daily without oral steroids) in patients with mCRPC previously treated with enzalutamide. The study was terminated early due to sub-optimal dosing strategies and significant CNS toxicity. Further evaluation of seviteronel is not warranted in this patient population on the basis of limited tolerability and insufficient clinical activity.
INTRODUCTION
The recent introduction of highly potent and efficacious anti-hormonal therapies has improved the treatment landscape of patients with castration resistant prostate cancer (CRPC). Since 2011, the FDA has approved three anti-hormonal agents for the treatment of CRPC: abiraterone acetate (AA), enzalutamide (ENZ), and apalutamide (APA).1–6 AA is an irreversible and potent inhibitor of the 17,20-lyase activity of cytochrome P450c17a (CYP17), blocking downstream production of androgens. However, AA also potently inhibits the 17-alpha-hyroxylase activity of CYP17, requiring the co-administration of oral steroids (e.g. prednisone) to reduce upstream steroid accumulation, cortisol suppression, and mineralocorticoid excess.7–9 AA has shown a significant overall survival (OS) advantage in patients with metastatic CRPC (mCRPC).1,3 ENZ and APA are both second generation androgen receptor (AR) antagonists that have also shown an OS improvement in patients with CRPC, the latter only in patients with non-metastatic CRPC (nmCRPC).2,4–6,10
Despite favorable responses in patients with castration resistance, disease progression is inevitable following treatment with current second-generation anti-hormonal agents, often a result of acquired resistance mediated via the AR pathway.11 Increased intratumoral androgen biosynthesis, AR overexpression, AR splice variation, and AR point mutations are implicated in treatment resistance to potent AR antagonists and CYP17 inhibitors.12–17 The T878A and L702H mutations in the AR have been associated with resistance to AA therapy, conferred via AR pathway activation by progesterone/pregnenolone9 and prednisone,18,19 respectively. Notably, the F876L mutation converts ENZ and APA from AR antagonists into AR agonists in vitro, with several cases documented clinically.20,21 Numerous clinical trials have been initiated to investigate newer antihormonal agents (e.g. orteronel, darolutamide, EPI-506) aimed to overcome AR pathway-mediated acquired resistance associated with the currently approved agents.22
Seviteronel (INO-464) is an orally bioavailable, dual inhibitor of CYP17 lyase activity and the AR, with approximately 10-fold selectivity towards the CYP17 lyase over hydroxylase,23 and competitive inhibition of wild-type and mutated forms of the AR (e.g. T877A, F876L).24 The unique mechanism of action of seviteronel may offer a potential therapeutic option in the setting of prior AR-targeted treatment failure while sparing the use of concomitant steroids. Seviteronel was shown to be effective in several in vivo models using CRPC cell lines, including MR49F, MDA-PCA-133, and LNCaP (expressing the AR F876L, H874Y, T877A mutations respectively).24–26 Additionally, seviteronel was shown to be more potent than AA in established enzalutamide-resistant cell lines (e.g. C4–2, C4–2B, MR49C, MR49F).24,25
There is currently an unmet clinical need to improve treatments in the post-ENZ setting of mCRPC. Sequential use of AA following ENZ has shown minimal improvements in both progression-free survival (PFS) and OS due to AR-mediated cross-resistance,27 as evidenced by clinical biomarkers such as AR-V7 expression.11,13,14 The safety, tolerability, pharmacokinetics (PK), and, notably, preliminary clinical activity of seviteronel has been evaluated for both twice daily and once daily dosing regimens in patients with treatment-naïve and previously treated CRPC (NCT02012920, NCT02361086).28,29 Patients with PSA declines were observed on both studies, but limited seviteronel tolerability associated with twice daily dosing (e.g. frequent dose reductions, treatment discontinuations) ultimately led to 600 mg or 750 mg once daily dosing regimens.29 The current phase II study (NCT02130700) investigates the use of seviteronel in patients with progressive mCRPC who experienced disease progression following at least 3 months of ENZ monotherapy with and without prior exposure to cytotoxic chemotherapy.
PATIENTS AND METHODS
Study Population
Patients aged 18 years or older with progressive mCRPC previously treated with enzalutamide for greater than three 28-day cycles were eligible for this study. Progression was defined as either a minimum of two rising PSA levels at least one week apart, appearance of one or more new lesions on bone scan, or new or growing lesions on CT scan. Patients were required to have an ECOG status of ≤1 (≤2 allowed for patients post-chemotherapy) with adequate organ and marrow function, castrate levels of testosterone (<50 ng/dl, achieved via orchiectomy or continuous LHRH agonist/antagonist therapy), and have discontinued previous treatment at least 28 days prior to study entry.
Patients with an uncontrolled intercurrent illness, an HIV-positive diagnosis on combination antiretroviral therapy, active Hepatitis B or C infections, or a history of another invasive malignancy within the preceding 3 years were excluded from this study. No more than one prior course of cytotoxic chemotherapy was permitted, and only patients with prior cytotoxic chemotherapy may have received prior therapy with agents targeting CYP17 (e.g. abiraterone, galeterone, orteronel). Patients with adrenal insufficiency requiring daily hydrocortisone/prednisone or prior palliative radiation within 2 weeks of study entry were not eligible. Additionally, patients with known brain metastases or a history of seziures were excluded from this study.
Study Design
This was a Phase 2, open label study designed to explore the benefit of seviteronel in patients with mCRPC who have previously been treated with enzalutamide. The primary objective of this study was to determine the rate of significant PSA response as defined by a ≥ 50% decrease in baseline serum PSA after 12 weeks of seviteronel administered without routine oral steroids (per PCWG2 criteria).30 Patients were stratified into two cohorts: pre-docetaxel based chemotherapy or post-docetaxel based chemotherapy. In each cohort, a Simon optimal two-stage design was used, with alpha=0.10 and beta=0.10, to rule out a 5% response rate (p0=0.05) in favor of a targeted 25% PSA response rate (p1=0.25). The first stage of accrual would be 9 patients in each cohort (18 patients total, initially). A significant PSA response in 1 or more of 9 patients in a study arm would increase enrollment to a total of 24 patients in that arm, with 3 or more responses in 24 patients warranting further study. The secondary objective for this study was to determine the radiographic response and time to progression as per the modified RECIST 1.1 criteria.31
Treatment and Toxicity Evaluation
Patients initially received seviteronel 150 mg by mouth twice daily with titration in increments of 150 mg every two weeks to a final dose of 450 mg twice daily. After the results of a simultaneous trial became available,29 the protocol was amended to modify the dose and administration schedule of seviteronel to 750 mg by mouth once daily in an effort to improve tolerability. Seven patients were treated with the original dosing regimen followed by six patients who received seviteronel 750 mg by mouth once daily. Frequent dose reductions and treatment discontinuations led to an additional amendment, reducing the dose of seviteronel to 600 mg by mouth once daily for the remaining 4 patients enrolled on study.
Adverse events were classified and graded according to the National Cancer Institute Common Toxicity Criteria for Adverse Events (CTCAE version 4.0). Treatment was held for a Grade 3 adverse event that was possibly, probably, or definitely related to seviteronel until resolution to Grade 1 or baseline. Dose reductions were allowed for low grade adverse events at the discretion of the investigator. Re-escalation of the dose was not permitted and patients requiring more than 2 dose reductions permanently discontinued seviteronel. Seviteronel was permanently discontinued for a Grade 4 adverse event or a treatment delay of greater than 6 weeks.
Pharmacokinetic Analysis
To better understand the PK of seviteronel, a subset of men on this trial were given a single oral dose (600mg or 750mg) with food and had PK samples drawn out to 48 hours (hr) post dose on the first day of cycles 1 and 2. For these patients, the day 2 dose was withheld, with daily dosing resuming on day 3 of cycles 1 and 2 and continued through each 28-day cycle. Blood for PK measurements were drawn into sodium heparin (green top; BD Biosciences) tubes, processed into plasma immediately, and stored frozen until bioanalytical analysis. Seviteronel plasma concentrations were measured using a validated liquid chromatography-tandem mass spectrometric (LC-MS/MS) assay with a lower limit of quantitation (LLOQ) of 20 ng/mL. Pharmacokinetic parameters were calculated using noncompartmental methods using Phoenix WinNonlin v7.0 (Certara, Cary, NC).
Statistical Analyses
The Kaplan-Meier method was used to evaluate time to progression and overall survival, separately by cohort as well as overall. Analyses of progression were done by evaluating time to PSA or radiographic progression (whichever came first); the analysis censored each patient’s follow-up at their off-study date if they did not have either a PSA progression or radiographic progression noted on-study. The log-rank test was used to determine the statistical significance of the difference between pairs of Kaplan-Meier curves. The data cutoff for this analysis was February 8, 2019.
RESULTS
Patient Characteristics
A total of 17 patients with mCRPC were enrolled from April 2014 to August 2016. Baseline characteristics separated by cohort are presented in Table 1, with approximately half of the patients having previous exposure to docetaxel (N=9, 53%). Most patients presented with high risk prostate cancer at time of initial diagnosis as demonstrated by Gleason Score (i.e. Gleason Score ≥ 8; N=12, 71%) and metastatic disease with bone involvement at baseline (N=16, 94%). A majority of the patients had been treated previously with a first-generation androgen receptor antagonist (anti-androgens such as bicalutamide, flutamide, nilutamide; N=16, 94%) in addition to prior treatment with enzalutamide as required by the study eligibility criteria. Thirteen patients (76%) had been previously treated with immunotherapy, with 12 patients (71%) receiving either investigational anti-cancer vaccines (e.g. PROSTVAC, PANVAC, ME-TARP; N=8, 47%) or FDA-approved sipuleucel-T (N=4, 24%). Additionally, 5 patients (29%) had prior treatment with anti-angiogenic targeted agents, the most notable regimen comprising of bevacizumab, docetaxel and either thalidomide or lenalidomide (N=3, 18%).
Table 1.
Patient Demographics and Baseline Characteristics
| Pre-docetaxel | Post-docetaxel | Total | |
|---|---|---|---|
| N = 9 | N = 8 | N = 17 | |
| Age (years)# | 71 (60, 85) | 72 (65, 92) | 71 (60, 92) |
| Race | |||
| White | 7 (78) | 8 (100) | 15 (88) |
| Other | 2 (22) | 0 (0) | 2 (12) |
| Weight (kg)# | 96.5 (76.6,129.4) |
89.1 (74.3,112.7) |
93.0 (74.3,129.4) |
| ECOG Status | |||
| 0 | 2 (22) | 0 (0) | 2 (12) |
| 1 | 7 (78) | 7 (87.5) | 14 (82) |
| 2 | 0 (0) | 1 (12.5) | 1 (6) |
| Baseline PSA# | 36.13 (6.92, 69.23) |
75.97 (14.69,190) |
54.88 (6.92,190) |
| Site of Metastasis | |||
| Bone | 3 (33) | 5 (62.5) | 8 (48) |
| Lymph Node (LN) | 1 (11) | 0 (0) | 1 (6) |
| Bone and LN | 4 (45) | 0 (0) | 4 (24) |
| Bone and Visceral | 1 (11) | 3 (37.5) | 4 (24) |
| Gleason Score^ | |||
| 6 | 1 (11) | 1 (12.5) | 2 (12) |
| 7 | 1 (11) | 2 (25) | 3 (17) |
| 8–10 | 7 (78) | 5 (62.5) | 12 (71) |
| Prior Treatments* | |||
| Bicalutamide | 7 (78) | 7 (87.5) | 14 (82) |
| Nilutamide | 2 (22) | 3 (37.5) | 5 (29) |
| Flutamide | 3 (33) | 4 (50) | 7 (41) |
| Ketoconazole | 2 (22) | 2 (25) | 4 (24) |
| Immunotherapy+ | 7 (77) | 6 (75) | 13 (76) |
| Anti-angiogenic Therapy& | 0 (0) | 5 (62.5) | 5 (29) |
All values are reported as total (percent) per column with unless otherwise noted.
Reported as an average value and range.
Gleason Score that was reported at the time of diagnosis
Prior treatments other than enzalutamide or docetaxel
Treatments include: Anti-PD-1/PD-L1 antibodies, sipuleucel-T, PANVAC, PROSTVAC, and TARP
Treatment regimens containing the following agents: TRC-105, AMG386, thalidomide, lenalidomide, bevacizumab.
Clinical Response
Patients received a median of 2 cycles (range, 1 to 8 cycles), limiting clinical evaluation of PSA response and radiographic progression as per PCWG2, which recommends waiting at least 12 weeks before documenting response or progression.30 Of the 17 evaluable patients, 10 (59%) patients discontinued therapy due to adverse events, 4 (24%) patients due to disease progression, 2 (12%) patients because of physician discretion, and 1 (6%) patient due to intercurrent illness unrelated to study treatment. Of all patients, only one patient from the pre-chemotherapy cohort receiving 750 mg once daily met the primary objective (6%), with a maximal PSA decline of 88% achieved following 3 cycles of treatment and disease progression occurring after 6 months on-study. An additional patient in the pre-chemotherapy cohort, who received 750 mg once daily with dose reduction to 450 mg once daily during Cycle 1, exhibited a minimal decline in PSA (15%) after 11 weeks of treatment before discontinuing due to toxicity. All remaining patients had rising PSA values on study, with 8 of those patients having documented PSA progression per PCWG2. Only four of 17 (24%) patients underwent restaging for the indication of radiographic progression of disease, with only one patient having a secondary scan to confirm progression per PCWG2.30
Kaplan-Meier plots for time to PSA progression or radiographic progression and OS are provided in Figures 1 and 2, respectively. Median time to disease progression (mTDP), measured as either PSA progression or radiographic progression (whichever came first), was 3.5 months (95% CI: 2.2 to 3.6 months). Patients without previous chemotherapy had a mTDP of 3.6 months (95% CI: 3.2 to 4.6 months) and patients previously treated with docetaxel had a mTDP of 2.7 months (95% CI: 1.6 to 3.5 months); though the log-rank assessment demonstrated a statistically significant difference in mTDP between cohorts (p=0.0096), this finding was not interpreted to be clinically meaningful. Median overall survival (mOS) for all patients was 13.4 months (95% CI: 6.6 to 14.3 months). Patients with prior chemotherapy had a mOS of 13.0 months (95% CI: 5.2 to 14.1 months) whereas patients with no prior chemotherapy had a mOS of 14.3 months (95% CI: 5.6 to 27.6 months).
Figure 1. Time to PSA or Radiographic Progression.
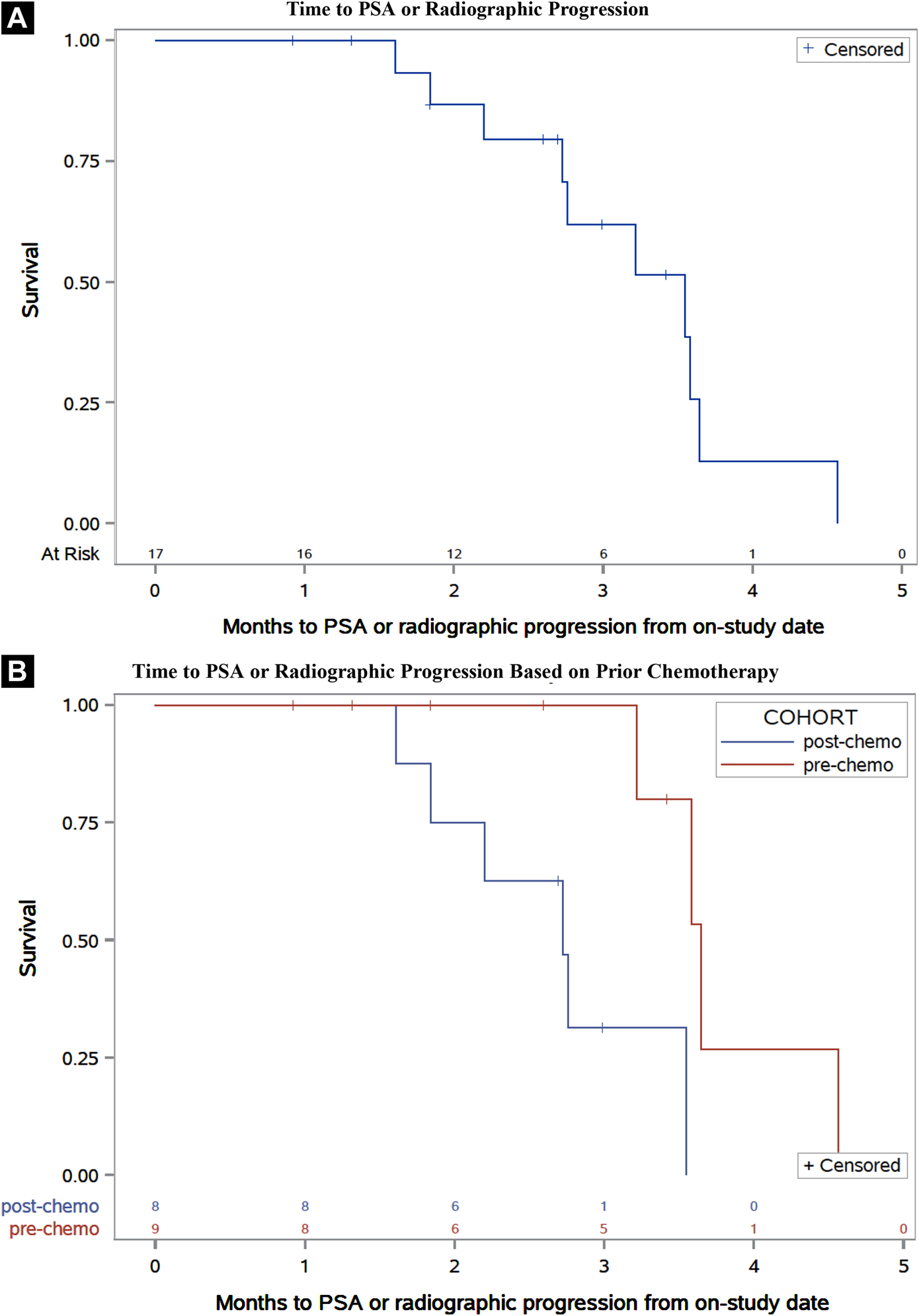
Kaplan-Meier plots of time to PSA or radiographic progression of the total population (A) and separated by patients with or without prior docetaxel therapy (B).
Figure 2. Median Overall Survival.
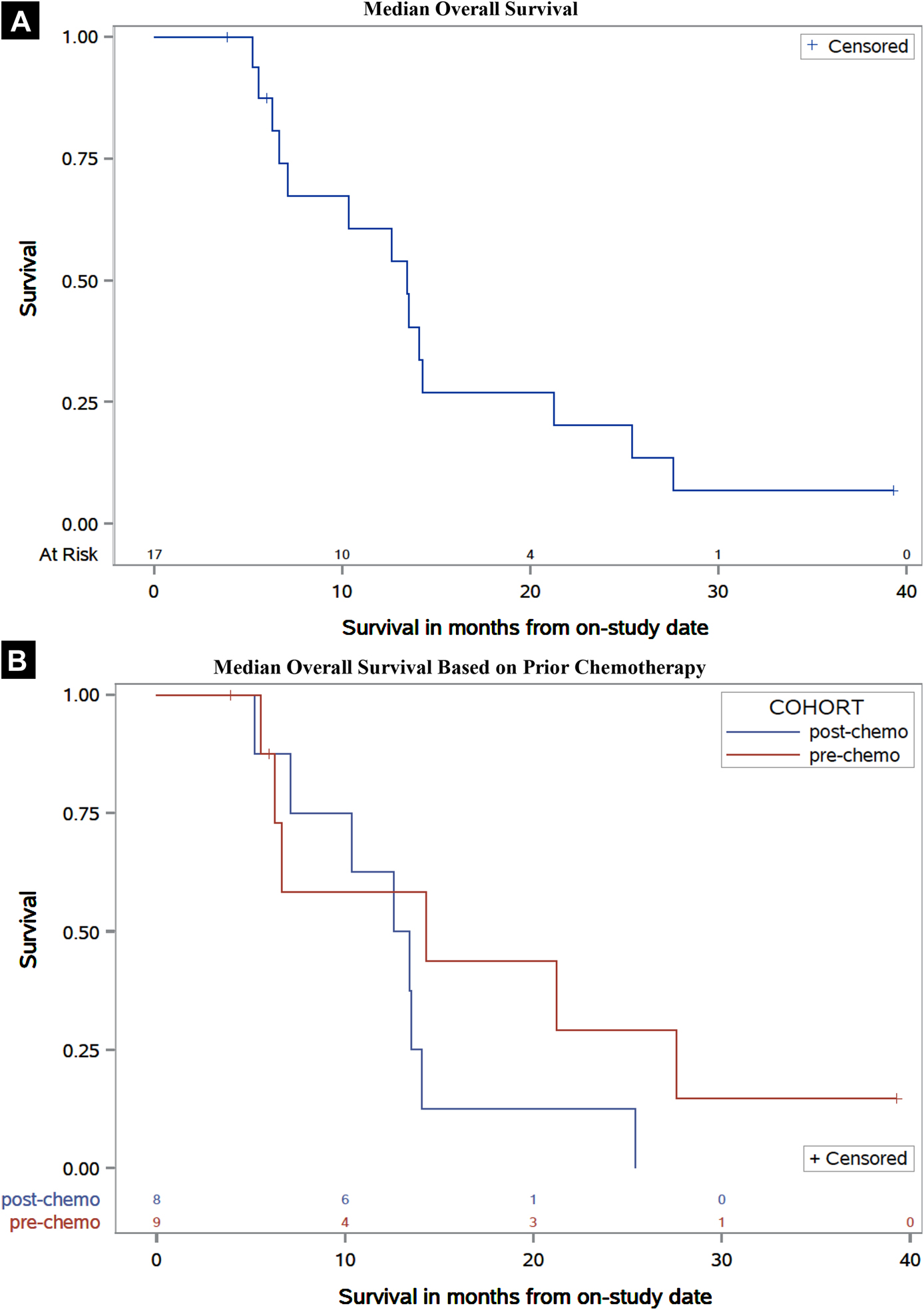
Kaplan-Meier plots of overall survival of the total population (A) and separated by patients with or without prior docetaxel therapy (B). Median potential follow-up was 37.4 months.
Toxicity
Grade 1, 2 and 3 adverse events that were probably, possibly or definitely related to study treatment that was reported in greater than 15% of the patients are listed in Table 2. The most common adverse events included concentration impairment, fatigue, tremor, and nausea, each of which each occurred in >50% of the patients. Reported Grade 3 adverse events at least possibly related to study treatment included concentration impairment, dizziness, nausea, hypotension, fall, and dehydration (not reported in Table 2, 2 patients [12%] experienced Grade 3 dehydration). No Grade 4 adverse events related to the study treatment were reported. Only one patient experienced two Grade 4 adverse events on study, respiratory failure and pneumonitis, that were attributed unrelated and unlikely, respectively, due to likely association with existing comorbidities. Supplemental Table 1 describes the incidence of the most common adverse events by dosing group. Toxicities that occurred in >50% of the all patients were mostly consistent across all dosing strategies with the exception of diarrhea, which was not reported in patients receiving 750 mg once daily. Ultimately, nine patients had CNS toxicities (most commonly concentration impairment and fatigue) that contributed to their treatment discontinuation.
Table 2.
Adverse Events with an Attribution of at Least Possible Occurring in Greater than 15% of Patients Who Received Study Treatment (N = 17) Based on National Cancer Institute Common Terminology Criteria for Adverse Events (Version 4.0)
| No. Patients (%) | ||||
|---|---|---|---|---|
| Adverse Event | All Grades (Gr 1–3) | Grade 1 | Grade 2 | Grade 3 |
| Concentration Impairment | 14 (82) | 9 (53) | 4 (24) | 1 (6) |
| Fatigue | 11 (65) | 3 (18) | 8 (47) | 0 (0) |
| Tremor | 10 (59) | 9 (53) | 1 (6) | 0 (0) |
| Nausea | 9 (53) | 7 (41) | 0 (0) | 2 (12) |
| Dizziness | 6 (35) | 3 (18) | 1 (6) | 2 (12) |
| Blurred Vision | 4 (24) | 3 (18) | 1 (6) | 0 (0) |
| Hypotension | 4 (24) | 1 (6) | 2 (12) | 1 (6) |
| Vomiting | 3 (18) | 3 (18) | 0 (0) | 0 (0) |
| Edema (limbs) | 3 (18) | 2 (12) | 1 (6) | 0 (0) |
| Fall | 3 (18) | 1 (6) | 1 (6) | 1 (6) |
| Gait Disturbance | 3 (18) | 3 (18) | 0 (0) | 0 (0) |
| Malaise | 3 (18) | 0 (0) | 3 (18) | 0 (0) |
| Presyncope | 3 (18) | 0 (0) | 3 (18) | 0 (0) |
In addition to dose modifications, alternative toxicity management strategies were attempted in a small number of patients (N=5). The precise mechanism of the CNS toxicities associated with seviteronel is unclear but hormonal or steroidal alterations were proposed as potential factors. As a result, some patients received estrogen supplementation (N=3) or prednisone (N=2), either prophylactically or after symptom development. No symptomatic improvement was noted with either approach.
Pharmacokinetics
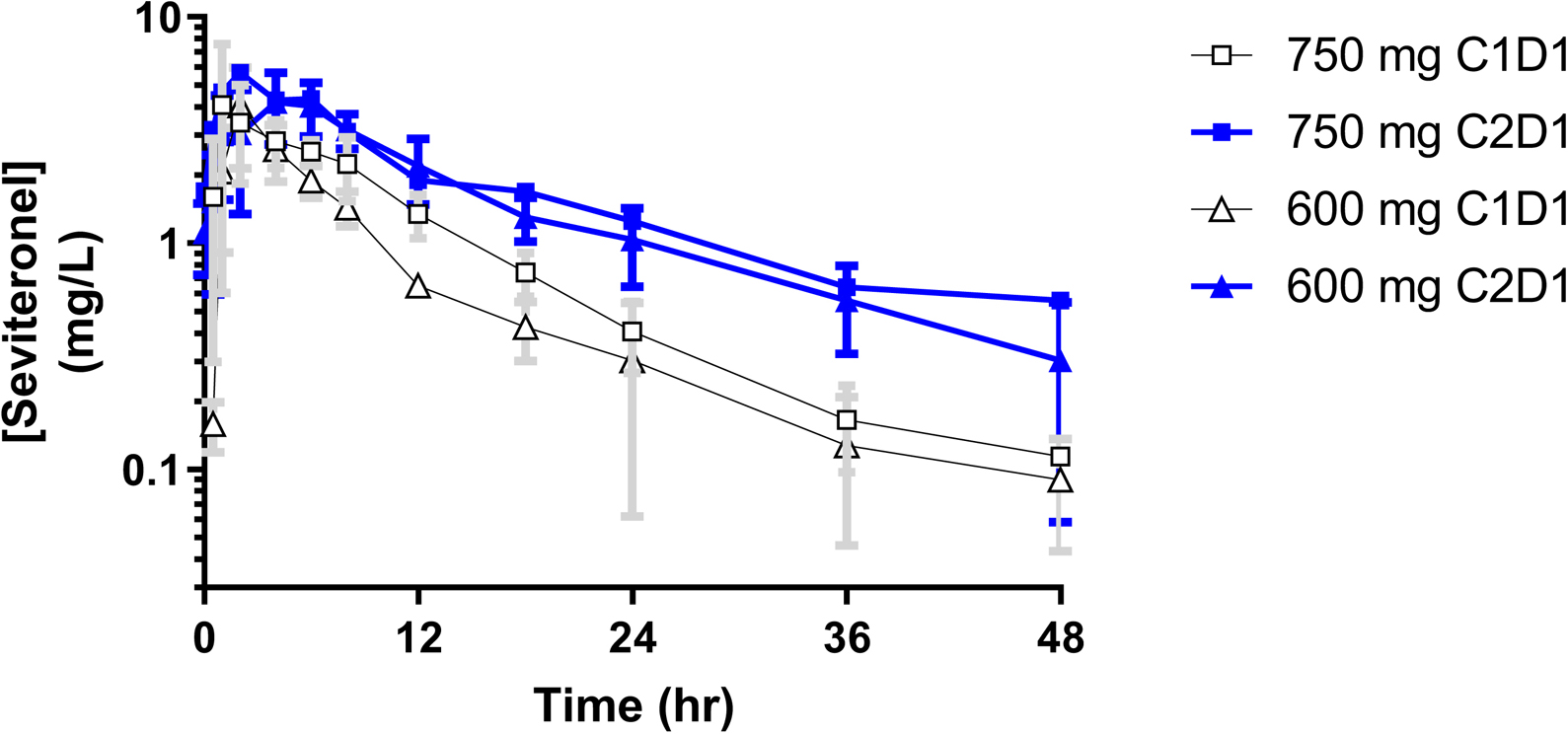
Of the 8 patients with PK samples obtained, 7 had a full PK time course in Cycle 1 and 6 patients had full time courses in both Cycle 1 and 2 for calculation of PK parameters. Seviteronel demonstrated an apparent monophasic elimination that began approximately 2–4 hrs post dose. Patients taking 750 mg demonstrated higher CMAX (p=0.50) and AUCINF (p=0.01) compared to those on 600 mg on C1D1, yet with comparable TMAX, clearance, and half-life (Table 3). Figure 3 depicts mean plasma concentration-time curves for each cycle on each dose level, where steady-state (Cycle 2 Day 1) levels are higher than those at first-dose due to extensive accumulation. Exposure-response analyses assessing the correlation of either CMAX or AUC to AE grade for concentration impairment, tremor, confusion, and nausea found no significant associations.
Table 3.
Pharmacokinetics of First Dose (Cycle 1) vs Steady-State (Cycle 2).
| 600 mg† | 750 mg⊥ | |||
|---|---|---|---|---|
| C1D1 (n=4) |
C2D1 (n=4)# |
C1D1 (n=3) |
C2D2 (n=l) |
|
| CMax1 (mg/L) | 4.07 ± 1.92 | 4.66 ± 1.16 | 5.11 ± 1.9 | 5.65 |
| Cmax/D2 (mg/L/mg) | 6.77 ± 3.20 | 7.76 ± 1.93 | 6.82 ± 2.54 | 7.53 |
| Tmax (hr) | 2.0 ± 0.0 | 5.00 ± 2.58 | 2.0 ± 1.7 | 2.0 |
| AUC (hr*mg/L) |
33.0 ± 4.33 | 55.4 ± 5.96 | 44.2 ± 2.15 | 63.5 |
| AUC/D (hr*mg/L/mg) |
55.1 ± 7.21 | 92.3 ± 9.94 | 58.9 ± 2.87 | 84.6 |
| T1/2(hr) | 16.4 ± 4.78 | 14.8 ± 5.94 | 10.5 ± 4.35 | 18.3 |
| CL/F3(L/hr) | 18.4 ± 2.50 | 10.9 ± 1.32 | 17.0 ± 0.84 | 11.8 |
| Vz/F (L) | 438 ± 146 | 229 ± 77.7 | 260 ± 121 | 312 |
Abbreviations: CMAX (maximum plasma concentration), AUC (area under the plasma concentration vs time curve extrapolated to infinity for “First”; AUCtau for “SS”),
Dose-normalized parameters
CL/F for “First” calculated as =Dose/AUCinf; CL/F for “SS” calculated as =Dose/AUCtau
t1/2 (half-life), Vz/F (apparent oral volume of distribution in the terminal phase; determined based on CL/F method used, i.e. Vz/F = CL/F/kel)
Patients 15, 16, and 17 all received 600mg on both C1D1 and C2D1; patient 14 was reduced to 450mg on C2D1 and this data was not included in this table.
Patient 10 is the lone patient given 750mg on both C1D1 and C2D1; patient 11 was reduced to 600mg on C2D1; patient 12 did not have C2D1 data available.
Figure 3. Mean Seviteronel Plasma Concentration vs Time Curves by Dose and Cycle.
DISCUSSION/CONCLUSIONS
Seviteronel, given both as twice-daily and once-daily regimens in the absence of oral steroid supplementation, was generally not well tolerated in this phase II study that assessed patients with mCRPC who previously progressed on enzalutamide. While the toxicity profile largely reflects what has been previously published in both patients with breast cancer32 and prostate cancer,29 the high prevalence of concentration impairment, per se, seen on this study has not been previously reported and was not expected based on preclinical toxicology data with this agent. It is worth noting, however, that the phase 1 study of seviteronel with once daily dosing was not devoid of CNS toxicity, with 71% of patients experiencing fatigue, 52% with dizziness and 33% with blurry vision, all of which could be different manifestations of CNS toxicity.29 In this study, fatigue was commonly associated with concentration impairment, and those adverse events could present with overlapping symptomatology. Concentration impairment occurred in 14 of 17 patients (82%), with Grade 2 or higher occurring in 5 of those patients and it contributed to treatment discontinuation in nine patients. Additionally, concentration impairment frequently resulted in dose reductions or treatment cessation. Other toxicities occurring in greater than 50% of this patient population included fatigue, tremor, and nausea, for which fatigue was previously reported at a similar frequency. This study did not find a correlation between drug exposure and adverse event severity, however, these analyses were limited by a small sample size. Although these patients often were previously treated with immunotherapy and angiogenesis inhibitors on clinical trials, it is unlikely that those treatments contributed to the CNS toxicity seen.
The most common toxicities observed with seviteronel, which included those with apparent CNS origin, are also found in patients experiencing adrenal glucocorticoid insufficiency.33–35 This suggests that minor CYP17 hydroxylase inhibition may be present with seviteronel administration and to aid in ameliorating those toxicities, the co-administration of the glucocorticoid mimetic dexamethasone was investigated in other new and ongoing breast and prostate cancer studies.29,32 Though no Grade 4 adverse events attributable to seviteronel were reported on this study, nine Grade 3 adverse events did occur, including concentration impairment and dizziness. Limited tolerability greatly impacted the analysis of clinical endpoints, including radiographic response and time to disease progression. Only one of 17 patients on study (6%) achieved the study’s primary endpoint of a PSA decline greater than 50%, although toxicity required multiple changes in dosing strategy which limits the ability to evaluate the potential efficacy of seviteronel in this study.
The PK analysis of seviteronel in the present study uncovered significant increases in AUC from first dose to steady-state. This phenomenon was observed with unpublished sponsor data when PK sampling stopped at 24 hr at first dose and 8 hr at steady-state, however this wasn’t reported in either of the previously published studies that only reported first dose PK.29,32 The data collected on this trial was sampled out to 48 hr in order to better estimate the elimination rate, which ultimately lead to a more accurate half-life estimate compared to previous analyses.29,32 With a mean first dose half-life of 13.9 hr (range 7.9 – 23.5 hr), seviteronel was expected to accumulate 43% above first-dose with once daily dosing until it reached steady-state (69.5 hr, or 2.9 days, to reach 97% steady-state). This half-life estimate was based on a 48-hr sampling window and considered to be more accurate than prior PK studies that only sampled to 24 hr on first dose or 8 hr at steady-state (T1/2 ~ 6–9 hr).29,32 Steady-state dose-normalized CMAX was 20% higher than Cycle 1 (means: 6.79 ug/L/mg vs 8.16 ug/L/mg; p=0.44). Dose-normalized steady-state AUCτ was on average 61% higher than first dose AUCINF (6 of 6 patients with available data had increases; 56.7 hr*ug/L/mg vs 91.3 hr*ug/L/mg; p=0.001). This significant accumulation (61% by AUC) to steady-state supports the toxicity profile, especially with persistent CNS events that take several half-lives to resolve.
Efforts to improve the seviteronel toxicity profile were ongoing through the duration of this study, as evidenced by two adjustments to the on-study dosing strategy. Drug accumulation was originally postulated as a contributing factor to increased toxicity, with higher trough concentrations mediating CNS related adverse events, prompting reduction in dosing frequency from twice daily to once daily. The separate phase I dose escalation study evaluating once daily dosing did not formally define a maximum tolerated dose (MTD), but suggested seviteronel could be given at 750 mg or 600 mg once daily.29 The adoption of once daily dosing of seviteronel 750 mg on the present study showed a minimal improvement in tolerability. Further investigation with 600 mg once daily provided a similar adverse event profile without a significant clinical response, suggesting that both doses for once daily administration were not truly viable, especially without oral steroid co-administration. It is worth noting that inter-individual variability in seviteronel disposition would not adequately explain these toxicities. Clinical characteristics such as body weight and prandial status, the latter of which significantly impacts abiraterone bioavailability,36 do not require clinically meaningful seviteronel dose modifications.37 Like enzalutamide, seviteronel exhibits low inter-individual variability in men with mCRPC, with only body weight having a minimal impact on seviteronel clearance.37,38
Currently available clinical data in patients with mCRPC post-ENZ or post-AA may suggest a limited or absent therapeutic window for both twice daily and once daily dosing of single-agent seviteronel. Initial investigations of seviteronel 450 mg given twice daily yielded several PSA declines of 50% or greater in 2 of 7 patients who had previously received ENZ,28 providing the initial rationale for the current study. Cumulative assessment of once daily dosing regimens in 28 patients with mCRPC previously treated with AA or ENZ is associated with only two clinical responses (7%), both at the 750 mg dose: one patient previously treated with AA with a PSA decline of greater than 30%,29 and the one clinical response reported in the current study. Total daily doses of 750 mg or greater appear to be associated with PSA declines in patients with mCRPC previously treated with AA or ENZ, however, tolerability has greatly limited the duration of treatment and potentially clinical response. It is possible that a tolerable and efficacious dose of seviteronel is not achievable for a majority of patients with mCRPC following anti-hormonal therapy or chemotherapy, at least by utilizing currently established dosing strategies in the absence of oral steroid co-administration, which was thought to be an important attribute of seviteronel in its clinic development.
Another potential contributing factor to the limited clinical response is exposure to prior lines of treatment. In patients receiving sequential lines of standard of care treatments (i.e. docetaxel, AA, ENZ and cabazitaxel), reported PSA response rates associated with 2nd, 3rd and 4th lines of treatment are 38%, 24%, and 16% respectively.27 When specifically analyzing sequences of ENZ followed by AA, two studies reported PSA response rates of 3% and 8%,39,40 which is similar to the currently evaluated treatment sequence of ENZ followed by seviteronel. PSA response rates in treatment naïve patients receiving seviteronel were 11% (N=26) and 33% (N=9) for twice daily dosing and once daily dosing respectively (daily cumulative doses of 600 mg or greater).28,29 Importantly, an association with increased response rates in treatment naïve patients compared to pre-treated patients (33% vs. 0%) was shown in the Phase I study evaluating once daily dosing.29 The only significant PSA decline in the present study occurred in a patient not previously treated with chemotherapy. Available clinical data does not support the utility of sevitronel’s unique mechanism of action in the post-ENZ setting. Though characterization of tumor alterations was not performed on this study, it is possible the role of AR point mutations with affinity to seviteronel was negligible, either due to a minimal role driving ENZ-resistant disease (e.g. T877A, H874Y)41 or limited prevelance based on previous clinical reports in patients with mCRPC post-ENZ (e.g. F876L).12,15,16,20 Moreover, seviteronel’s proposed specificity for the CYP17 lyase activity, proposed to mitigate mineralocorticoid excess associated with AA, was overshadowed by intolerable CNS toxicity. The inability of seviteronel to produce robust clinical responses may be indicative of the drug’s ineffectiveness to target acquired resistance in a generalized post-ENZ mCRPC patient population in addition to the drug’s sub-optimal dose density resulting from limited tolerability.
This trial provides a cautionary tale as the field of prostate cancer looks to target an androgen receptor pathway that has mutated or is otherwise resistant to standard AR-directed therapies. In developing next-generation AR-targeted therapy, heretofore underappreciate neurocognitive toxicity may be a significant limitation despite promising preclinical rationale, as it was with seviteronel. Preclinical studies have previously demonstrated the capability of ENZ and APA to penetrate the blood-brain barrier.42 Furthermore, this is not the first agent targeting the AR which has suggested neurotoxicity. There have been several studies suggesting that ADT has been associated with some degree of cognitive decline.43 Enzalutamide has also been associated with severe fatigue in some patients and even seizures.44 Notably, an episode of seizure activity coupled with limited anti-tumor activity during Phase I evaluation terminated the development of the novel anti-androgen, BMS-641988.45 As further AR-targeting strategies are investigated, great care needs to be taken to monitor for off-target neurologic toxicity.
In conclusion, none of the dosing strategies implemented for seviteronel administration in the present study were well tolerated by patients with mCRPC previously treated with ENZ. The limited tolerability coupled with a clinically insignificant response rate does not support further development of seviteronel, especially without oral steroid co-administration, in patients with mCRPC.
Supplementary Material
Clinical Practice Points.
Improving the efficacy of available treatments for patients with mCRPC following either anti-hormonal therapy or chemotherapy is an important objective currently under clinical investigation.
Seviteronel is an orally bioavailable, dual inhibitor of CYP17 lyase activity and the AR, proposed to limit toxicity associated with mineralocorticoid excess and target AR pathway mediated resistance following treatment with ENZ or AA in patients with mCRPC.
Seviteronel given via twice daily or once daily dosing in patients with mCRPC previously treated with ENZ and/or docetaxel was associated with dose limiting CNS toxicities and insignificant clinical response.
This study highlights the importance of appropriate dose selection and well-designed pharmacokinetic analyses, as AUC assessments demonstrated significant increases in drug exposure following one cycle of treatment compared to the first dose.
Clinical experience with seviteronel highlights the potential for dose-limiting neurocognitive toxicity often associated with the development of newer AR-targeted therapies, especially in the setting of acquired resistance.
Acknowledgements
We thank the nursing staff of National Cancer Institute and the fellows of the Genitourinary Malignancies Branch at National Cancer Institute for their care of our patients; Peraton for data management assistance. Most importantly, we appreciate the patients with cancer who enroll in investigational trials to advance the knowledge of this disease.
Grant Support
This work was supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Publisher's Disclaimer: Disclaimer
Publisher's Disclaimer: The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organization imply endorsement by the U.S. Government.
REFERENCES
- 1.de Bono JS, Logothetis CJ, Molina A, et al. : Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med 364:1995–2005, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Scher HI, Fizazi K, Saad F, et al. : Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med 367:1187–97, 2012 [DOI] [PubMed] [Google Scholar]
- 3.Ryan CJ, Smith MR, de Bono JS, et al. : Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med 368:138–48, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beer TM, Armstrong AJ, Rathkopf DE, et al. : Enzalutamide in metastatic prostate cancer before chemotherapy. N Engl J Med 371:424–33, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith MR, Saad F, Chowdhury S, et al. : Apalutamide Treatment and Metastasis-free Survival in Prostate Cancer. N Engl J Med 378:1408–1418, 2018 [DOI] [PubMed] [Google Scholar]
- 6.Rathkopf DE, Scher HI: Apalutamide for the treatment of prostate cancer. Expert Rev Anticancer Ther 18:823–836, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Attard G, Reid AH, A’Hern R, et al. : Selective inhibition of CYP17 with abiraterone acetate is highly active in the treatment of castration-resistant prostate cancer. J Clin Oncol 27:3742–8, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Attard G, Reid AH, Yap TA, et al. : Phase I clinical trial of a selective inhibitor of CYP17, abiraterone acetate, confirms that castration-resistant prostate cancer commonly remains hormone driven. J Clin Oncol 26:4563–71, 2008 [DOI] [PubMed] [Google Scholar]
- 9.Attard G, Reid AH, Auchus RJ, et al. : Clinical and biochemical consequences of CYP17A1 inhibition with abiraterone given with and without exogenous glucocorticoids in castrate men with advanced prostate cancer. J Clin Endocrinol Metab 97:507–16, 2012 [DOI] [PubMed] [Google Scholar]
- 10.Hussain M, Fizazi K, Saad F, et al. : Enzalutamide in Men with Nonmetastatic, Castration-Resistant Prostate Cancer. N Engl J Med 378:2465–2474, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Robinson D, Van Allen EM, Wu YM, et al. : Integrative clinical genomics of advanced prostate cancer. Cell 161:1215–1228, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Annala M, Vandekerkhove G, Khalaf D, et al. : Circulating Tumor DNA Genomics Correlate with Resistance to Abiraterone and Enzalutamide in Prostate Cancer. Cancer Discov 8:444–457, 2018 [DOI] [PubMed] [Google Scholar]
- 13.Antonarakis ES, Lu C, Wang H, et al. : AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med 371:1028–38, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Antonarakis ES, Lu C, Luber B, et al. : Clinical Significance of Androgen Receptor Splice Variant-7 mRNA Detection in Circulating Tumor Cells of Men With Metastatic Castration-Resistant Prostate Cancer Treated With First- and Second-Line Abiraterone and Enzalutamide. J Clin Oncol 35:2149–2156, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Azad AA, Volik SV, Wyatt AW, et al. : Androgen Receptor Gene Aberrations in Circulating Cell-Free DNA: Biomarkers of Therapeutic Resistance in Castration-Resistant Prostate Cancer. Clin Cancer Res 21:2315–24, 2015 [DOI] [PubMed] [Google Scholar]
- 16.Wyatt AW, Azad AA, Volik SV, et al. : Genomic Alterations in Cell-Free DNA and Enzalutamide Resistance in Castration-Resistant Prostate Cancer. JAMA Oncol 2:1598–1606, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu C, Lou W, Zhu Y, et al. : Intracrine Androgens and AKR1C3 Activation Confer Resistance to Enzalutamide in Prostate Cancer. Cancer Res 75:1413–22, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cai C, Balk SP: Intratumoral androgen biosynthesis in prostate cancer pathogenesis and response to therapy. Endocr Relat Cancer 18:R175–82, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boudadi K, Antonarakis ES: Resistance to Novel Antiandrogen Therapies in Metastatic Castration-Resistant Prostate Cancer. Clin Med Insights Oncol 10:1–9, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Joseph JD, Lu N, Qian J, et al. : A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discov 3:1020–9, 2013 [DOI] [PubMed] [Google Scholar]
- 21.Balbas MD, Evans MJ, Hosfield DJ, et al. : Overcoming mutation-based resistance to antiandrogens with rational drug design. Elife 2:e00499, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yoo S, Choi SY, You D, et al. : New drugs in prostate cancer. Prostate Int 4:37–42, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rafferty SW, Eisner JR, Moore WR, et al. : Highly-selective 4-(1,2,3-triazole)-based P450c17a 17,20-lyase inhibitors. Bioorg Med Chem Lett 24:2444–7, 2014 [DOI] [PubMed] [Google Scholar]
- 24.Norris JD, Ellison SJ, Baker JG, et al. : Androgen receptor antagonism drives cytochrome P450 17A1 inhibitor efficacy in prostate cancer. J Clin Invest 127:2326–2338, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Toren PJ, Kim S, Pham S, et al. : Anticancer activity of a novel selective CYP17A1 inhibitor in preclinical models of castrate-resistant prostate cancer. Mol Cancer Ther 14:59–69, 2015 [DOI] [PubMed] [Google Scholar]
- 26.Maity SN, Titus MA, Gyftaki R, et al. : Targeting of CYP17A1 Lyase by VT-464 Inhibits Adrenal and Intratumoral Androgen Biosynthesis and Tumor Growth of Castration Resistant Prostate Cancer. Sci Rep 6:35354, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Caffo O, De Giorgi U, Fratino L, et al. : Clinical Outcomes of Castration-resistant Prostate Cancer Treatments Administered as Third or Fourth Line Following Failure of Docetaxel and Other Second-line Treatment: Results of an Italian Multicentre Study. Eur Urol 68:147–53, 2015 [DOI] [PubMed] [Google Scholar]
- 28.de Bono JS, Pezaro CJ, Gillessen S, et al. : The oral CYP17-Lyase (L) inhibitor VT-464 in patients with CRPC. J Clin Oncol 33(suppl 7):abstr 187, 2015 [Google Scholar]
- 29.Gupta S, Nordquist LT, Fleming MT, et al. : Phase 1 Study of Seviteronel, a Selective CYP17 Lyase and Androgen Receptor Inhibitor, in Men with Castration-Resistant Prostate Cancer. Clin Cancer Res, 2018 [DOI] [PubMed] [Google Scholar]
- 30.Scher HI, Halabi S, Tannock I, et al. : Design and end points of clinical trials for patients with progressive prostate cancer and castrate levels of testosterone: recommendations of the Prostate Cancer Clinical Trials Working Group. J Clin Oncol 26:1148–59, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schwartz LH, Litiere S, de Vries E, et al. : RECIST 1.1-Update and clarification: From the RECIST committee. Eur J Cancer 62:132–7, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bardia A, Gucalp A, DaCosta N, et al. : Phase 1 study of seviteronel, a selective CYP17 lyase and androgen receptor inhibitor, in women with estrogen receptor-positive or triple-negative breast cancer. Breast Cancer Res Treat, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Charmandari E, Nicolaides NC, Chrousos GP: Adrenal insufficiency. Lancet 383:2152–67, 2014 [DOI] [PubMed] [Google Scholar]
- 34.Michels A, Michels N: Addison disease: early detection and treatment principles. Am Fam Physician 89:563–8, 2014 [PubMed] [Google Scholar]
- 35.DL L, AS F, DL K, et al. : Harrison’s Principles of Internal Medicine, 18th Ed New York, McGraw-Hill, 2012 [Google Scholar]
- 36.Szmulewitz RZ, Peer CJ, Ibraheem A, et al. : Prospective International Randomized Phase II Study of Low-Dose Abiraterone With Food Versus Standard Dose Abiraterone In Castration-Resistant Prostate Cancer. J Clin Oncol 36:1389–1395, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peer CJ, Schmidt KT, Kindrick JD, et al. : A population pharmacokinetic analysis of the oral CYP17 lyase and androgen receptor inhibitor seviteronel in patients with advanced/metastatic castration-resistant prostate cancer or breast cancer. Cancer Chemother Pharmacol 84:759–770, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gibbons JA, Ouatas T, Krauwinkel W, et al. : Clinical Pharmacokinetic Studies of Enzalutamide. Clin Pharmacokinet 54:1043–55, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Noonan KL, North S, Bitting RL, et al. : Clinical activity of abiraterone acetate in patients with metastatic castration-resistant prostate cancer progressing after enzalutamide. Ann Oncol 24:1802–7, 2013 [DOI] [PubMed] [Google Scholar]
- 40.Loriot Y, Bianchini D, Ileana E, et al. : Antitumour activity of abiraterone acetate against metastatic castration-resistant prostate cancer progressing after docetaxel and enzalutamide (MDV3100). Ann Oncol 24:1807–12, 2013 [DOI] [PubMed] [Google Scholar]
- 41.McCrea E, Sissung TM, Price DK, et al. : Androgen receptor variation affects prostate cancer progression and drug resistance. Pharmacol Res 114:152–162, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moilanen AM, Riikonen R, Oksala R, et al. : Discovery of ODM-201, a new-generation androgen receptor inhibitor targeting resistance mechanisms to androgen signaling-directed prostate cancer therapies. Sci Rep 5:12007, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nelson CJ, Lee JS, Gamboa MC, et al. : Cognitive effects of hormone therapy in men with prostate cancer: a review. Cancer 113:1097–106, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Beer TM, Armstrong AJ, Rathkopf D, et al. : Enzalutamide in Men with Chemotherapy-naive Metastatic Castration-resistant Prostate Cancer: Extended Analysis of the Phase 3 PREVAIL Study. Eur Urol 71:151–154, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rathkopf D, Liu G, Carducci MA, et al. : Phase I dose-escalation study of the novel antiandrogen BMS-641988 in patients with castration-resistant prostate cancer. Clin Cancer Res 17:880–7, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.