

Abstract
Frequent GNAQ and GNA11 mutations in uveal melanoma (UM) hyperactivate the MEK-ERK signaling pathway, leading to aberrant regulation of cyclin dependent kinases (CDK) and cell cycle progression. MEK inhibitors (MEKi) alone show poor efficacy in UM, raising the question of whether downstream targets can be vertically inhibited to provide long-term benefit. CDK4/6 selective inhibitors are FDA-approved in ER-positive breast cancer patients in combination with ER antagonists/aromatase inhibitors. We determined the effects of MEKi plus CDK4/6 inhibitors (CDK4/6i) in UM. In vitro, palbociclib, a CDK4/6i, enhanced the effects of MEKi via downregulation of cell cycle proteins. By contrast, in vivo CDK4/6 inhibition alone led to cytostasis and was as effective as MEKi plus CDK4/6i treatment at delaying tumor growth. RNA sequencing revealed upregulation of the oxidative phosphorylation (OxPhos) pathway in both MEKi-resistant tumors and CDK4/6i-tolerant tumors. Furthermore, oxygen consumption rate was increased following MEKi + CDK4/6i treatment. IACS-010759, an OxPhos inhibitor, decreased UM cell survival in combination with MEKi + CDK4/6i. These data highlight adaptive upregulation of OxPhos in response to MEKi + CDK4/6i treatment in UM and suggest that suppression of this metabolic state may improve the efficacy of MEKi plus CDK4/6i combinations.
Keywords: Uveal melanoma, MEK inhibitor, CDK4/6 inhibitor, Oxidative phosphorylation
Introduction
UM is the most common intraocular malignant tumor in adults. Unfortunately, up to 50% of UM patients who are treated for their primary tumors will ultimately succumb to metastatic disease (1). The liver is the main site for metastatic dissemination. Currently, there are no FDA-approved treatments for metastatic UM. Compared to the recent breakthroughs in the treatment of cutaneous melanoma, therapeutic strategies that are effective for advanced UM remain an unmet need.
Activating mutations in alpha subunits of the heterotrimeric G proteins GNAQ and GNA11 are found in >90% of UM (2–5). The encoded mutant forms of Gαq and Gα11 signal to several pathways, including MEK-ERK1/2 and YAP/TAZ (6–8). In a Phase II trial (NCT01143402), the MEK inhibitor, selumetinib, had a 15% overall response rate and improved median progression-free survival (PFS) compared to standard chemotherapy (15.9 weeks vs 7 weeks), but overall survival was not significantly improved (9). The SUMMIT Phase III trial analyzing the addition of selumetinib to dacarbazine was stopped early since it showed only a 1 month improvement of median PFS (10). Thus, MEKi may be a component of therapeutic approaches for advanced UM, but rational, effective combinatorial approaches are needed.
Constitutive MEK-ERK1/2 signaling upregulates cyclin D1 which binds to and activates CDK4/6, allowing for enhanced cell cycle progression. Three selective CDK4/6 inhibitors (palbociclib, ribociclib, and abemaciclib) are currently FDA-approved for ER+ breast cancer patients in combination with ER antagonists (11,12). We have shown in cutaneous melanoma that concurrent inhibition of MEK and CDK4/6 leads to enhanced apoptosis and tumor regressions, with complete responses in some tumors (13,14). The combination of ribociclib with the MEKi, binimetinib in NRAS-mutant metastatic melanoma patients improved response rates by more than two-fold compared to binimetinib alone (15).
In this study, we tested the hypothesis that the efficacy of MEKi would be enhanced by combination with CDK4/6i in UM. The effects of single agents and combinations were tested on human UM cells in vitro and in vivo. MEK and CDK4/6 targeting led to adaptive upregulation of OxPhos, a metabolic pathway previously implicated in resistance to BRAF and MEK inhibitors in cutaneous melanoma (16,17). Treatment with an OxPhos inhibitor improved the efficacy of MEKi + CDK4i in vitro and in vivo. Together, our data identify a metabolic adaptive tolerance mechanism of MEK plus CDK4/6 targeting and provide a potential strategy to overcome such adaptation.
Materials and Methods
Cell lines -
UM001 and UM004 were derived from human UM metastases and were established at Thomas Jefferson University (18,19). OMM1.3 cells were obtained from Dr. Bruce Ksander’s laboratory in 2014 whereas 92.1 cells were obtained from Dr. Martine Jager’s laboratory. WM3618F was obtained from Rockland Immunochemicals, Inc. (Pottstown, PA). UM001, UM004, OMM1.3, and WM3618F were confirmed to harbor the Q209P mutation in GNAQ by Sanger sequencing. 92.1 has the Q209L mutation in GNAQ, which was also confirmed. Culture conditions for UM001, UM004, OMM1.3, and 92.1 are described in previous reports (8,19–21). WM3618F was cultured in MCDB 153 medium (pH 7.6) containing 5% heat-inactivated FBS, 100 mM CaCl2, 50 IU penicillin, and 50 μg/mL streptomycin. Cell lines were tested for mycoplasma contamination every two months using the MycoScope Kit (Genlantis, San Diego, CA) and analyzed by STR for authentication.
Inhibitors -
Trametinib and PD0325901 (22) were purchased from SelleckChem (Houston, TX). Palbociclib was from Pfizer (New York, NY). IACS-010759 was purchased from ChemieTek (Indianapolis, IN). All drugs were dissolved according to manufacturers’ instructions.
Western blot analysis -
Protein lysates were prepared in Laemmli sample buffer, separated by SDS-PAGE, and proteins were transferred to PVDF membranes. Immunoreactivity was detected using HRP-conjugated secondary antibodies (EMD Millipore; Burlington, MA) and chemiluminescence substrate (Bio-Rad; Hercules, CA) on a Versadoc Imaging System (Bio-Rad). Primary antibodies were as follows: phospho-Rb (S780; #9307), RB (#9309), phospho-ERK1/2 (#9101), ERK1/2 (#9102), Cleaved PARP (#9541), and HSP90 (#4877), all purchased from Cell Signaling (Danvers, MA). Cyclin A1 (sc-751) and cyclin D1 (sc-718) antibodies were purchased from Sigma-Aldrich (St. Louis, MO).
Cell viability assay (MTT) -
Each cell line was cultured in 96-well plates at 2x103 cells per well with indicated inhibitors. Viable cells were measured at day 4. Thiazolyl Blue Tetrazolium Bromide (Sigma-Aldrich) was added to growth medium, incubated for 4 hours at 37°C, and solubilized overnight with equal volumes of 10% sodium dodecyl sulfate/0.1N HCl. A 96-well plate reader, Multiskan™ Spectrum (Thermo Fisher Scientific; Waltham, MA) was used to measure absorbance at 570 nM.
Reverse Phase Protein Array (RPPA) analysis -
Cells were treated with the indicated inhibitors for 48 hours, then lysed for RPPA analysis, as reported previously (21). Comparisons were performed between conditioned samples using the two-sample t-test method with 1,000 permutations. Multiple hypothesis test corrections were calculated and antibodies with a Storey q-value <0.05 and a fold ratio >2 were considered significant, unless noted otherwise. Calculations were performed in Matlab® (v2017b) using the mattest and mafdr functions.
Seahorse assays -
Cell Mito Stress tests were purchased from Seahorse Bioscience (Billerica, MA) and utilized on a XF24 Analyzer (Seahorse Bioscience) to measure oxygen consumption rate (pMoles/minute). Cells were seeded in XF24 cell culture microplates (Seahorse Bioscience, #100777-004) and treated with DMSO (control), PD0325901, palbociclib, or PD0325901 plus palbociclib. After 24 hours, the drug-laced media were removed and the cells were incubated with Seahorse XF base medium containing 1 mM pyruvate, 2 mM glutamine, and 10 mM glucose for 1 hour in a non-CO2 incubator at 37°C. To manipulate cell metabolism in the test, oligomycin (1 μM), carbonyl cyanide-4 (trifluoromethoxy) phenylhydrazone (FCCP) (1 μM) and rotenone/antimycinA (Rot/AA, 0.5μM) were injected. Oxygen consumption rate (OCR) was measured after each injection. The experiment was repeated 3-4 times. Data were normalized to protein concentration and analyzed using the Agilent Seahorse XF report generator.
Incucyte measurements -
Cells were treated as indicated and percentages of cell confluency in the culture wells were measured every 2 hours using the Incucyte® Live Cell Analysis Imaging System (Sartorius, Germany).
Annexin-V staining -
After the indicated treatments, cells were trypsinized, washed twice with PBS, and resuspended in binding buffer containing 1:20 Annexin V-APC (BD Pharmingen; Franklin Lakes, NJ) and 0.2 mg/mL propidium iodide (Life Technologies; Carlsbad, CA) for 15 minutes. Apoptotic cells were analyzed by flow cytometry on the BD FACSCelesta™ (BD Biosciences; Bedford, MA). Data were analyzed using the FlowJo software (Ashland, OR).
RNA sequencing -
RNA sequencing data were aligned to the GRCh38 human reference genome using Star aligner (23). RSEM (24) was used to quantify gene and transcript level expression. Gene differential expression analysis was performed using DESeq2 (25). GSEA (26,27) was used to determine enriched pathways in the MSigDB Hallmark gene set collection (28). Enrichment plots were generated using the ggplot2 package (v3.1.0 https://ggplot2.tidyverse.org/) in R (v3.5.1 http://www.R-project.org/).
Animal studies -
Experiments were approved by the Institutional Animal Care and Use Committee (IACUC) and performed at the Thomas Jefferson University animal facility that is accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care and has a full-time veterinarian. Athymic nude (nu/nu) (homozygous, 6-8 weeks) mice were purchased from The Jackson Laboratory (Bar Harbor, ME). The animals were injected subcutaneously with 5x106 UM001. When tumor xenografts were established (100-200 mm3), the animals were fed either control chow (n=4) or chow laced with MEKi (PD0325901) (n=3), CDK4/6i (palbociclib) (n=3), or the combination of MEKi and CDK4/6i (n=3). Palbociclib in both the single agent and combination arms was dosed in an intermittent schedule: 3 weeks on, 1 week off. Tumor volume was calculated by digital caliper measurements and the formula: volume = length x (width2/2). To detect Ki67 or cleaved caspase 3 staining, tumor samples were extracted after the animals were sacrificed, fixed in buffered formalin (1:10) for 24 hours, then embedded in paraffin for immunohistochemical staining using a Ki67 (BD Pharmingen) or cleaved caspase 3 (Cell Signaling) antibodies.
Statistical analysis of tumor growth -
Nonparametric rank-based longitudinal two-factor model with interaction (29,30) was fitted using the R package nparLD (31). In such model, the treatment group classification is considered a whole-plot factor and the day is considered a sub-plot factor. This nonparametric approach effectively changes the response variables from the actual tumor volumes into their ranks within the entire data set. The difference between treatment groups is tested in terms of the interaction effect (interaction between treatment group and day), which is equivalent to testing the difference in Day slopes between treatment group in a linear mixed effects model. To ensure that all animals have the same number of observations (requirement for using the R package nparLD), the observation days were limited to 98 and 4 longitudinal observations for 2 additional control animals were linearly interpolated using the available observations before and after.
Results
Enhanced effects of MEKi plus CDK4/6i combination in UM cell lines
To determine whether the combination of MEK plus CDK4/6 targeting is more efficacious than inhibition of either target alone, a panel of UM cell lines were treated with a MEKi (trametinib) alone, a CDK4/6i (palbociclib) alone, or the combination of both drugs. The combination of MEK plus CDK4/6 inhibition led to further reduction in cell viability compared to single agents in three out of the five cell lines tested: UM001 and UM004 (metastatic-derived UM cell lines), and 92.1 (primary tumor derived UM cell line) (Fig. 1A). Reverse Phase Protein Analysis (RPPA) revealed downregulation of cell cycle related proteins such as the CDK4/6 substrates, phospho-RB and FOXM1, with single agent palbociclib treatment and with combination treatment (Fig. 1B). By contrast, apoptotic proteins were not modulated (Supplemental Fig. 1). The modulation of downstream targets of MEKi and CDK4/6i were validated by Western blotting, which also confirmed a lack of induction of apoptotic markers (i.e. cleaved PARP) (Fig. 1C). Together, these results suggest that the combination of MEK plus CDK4/6 inhibition leads to cell cycle arrest via inhibition of downstream cell cycle proteins but does not induce apoptosis in vitro.
Figure 1. Enhanced effects of combined MEK and CDK4/6 inhibition in UM cell lines.
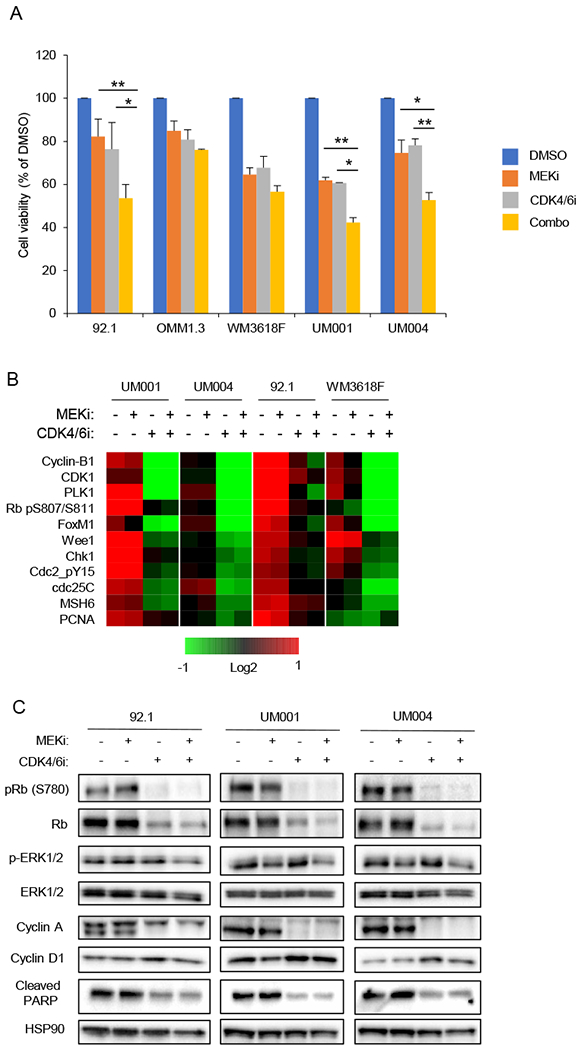
A MTT cell viability assays of UM cell lines treated with MEKi (trametinib, 5 nM) alone, CDK4/6i (palbociclib, 0.5 μM) alone, or the combination (trametinib + palbociclib) for 7 days (*p<0.05, **p<0.005) (n=3). B Downregulation of cell cycle proteins following MEKi + CDK4/6i treatment. UM cell lines treated with single agent or combination of MEKi (PD0325901, 5nM) and CDK4/6i (palbociclib, 0.5 μM) for 48 hours. Heatmap of median centered log2-transformed average group expression RPPA data for antibodies both differentially expressed (Storey q-value < 0.05, ratio >50%) and up or downregulated in at least two cell lines between combination or DMSO treated samples. C Western blot analysis of UM cell lines treated with single agent or combination of MEKi (PD0325901, 5 nM) and CDK4/6i (palbociclib, 0.5 μM) for 48 hours. Representative blots from triplicate experiments are shown.
MEK plus CDK4/6 targeting leads to tolerant tumors in vivo
The effects of MEKi, CDK4/6i, and the combination were further evaluated in vivo using subcutaneous xenografts of UM001 (21,32). Nude mice with UM001 xenografts were treated with control chow, MEKi (PD0325901), CDK4/6i (palbociclib), or MEKi plus CDK4/6i chow. Palbociclib was dosed in an intermittent schedule of 3 weeks on, 1 week off, in order to mirror dosing utilized in ongoing clinical trials (33). Consistent with the clinical findings (9), UM tumors treated with MEKi alone eventually progressed and acquired resistance, although tumors were smaller compared to controls (Fig. 2A, Supplemental Fig. 2). Surprisingly, tumors treated with CDK4/6i alone were growth arrested; similar results were observed with MEKi plus CDK4/6i combination treatment (Fig. 2A, Supplemental Fig. 2). Ki67 staining demonstrated that CDK4/6i treatment (with or without MEKi) arrested most tumor cells, which was not seen with MEKi or control treatment (Fig. 2B). These data suggest that CDK4/6 targeting in UM leads to cytostasis. Additionally, positive staining for cleaved caspase 3 was observed in tumors treated with MEKi plus CDK4/6i, indicating apoptosis with the combination treatment (Supplemental Fig. 3).
Figure 2. CDK4/6 inhibition in the metastatic UM line, UM001, leads to cytostatic tumors.
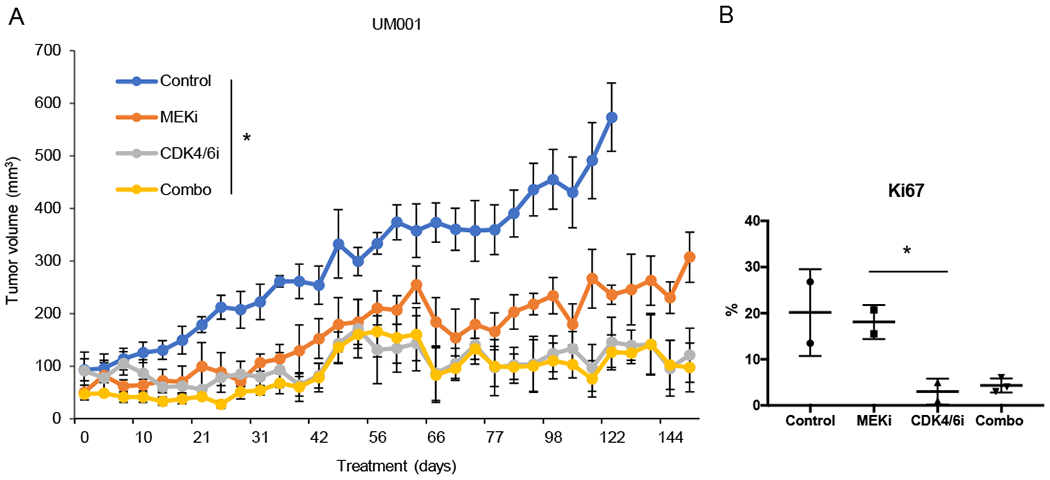
A UM001 xenografts were treated with control, MEKi (PD0325901), CDK4/6i (palbociclib), or the combination (combo) (*p=0.02) (n=3). Palbociclib was dosed intermittently, 3 weeks on, 1 week off. B Quantification of Ki67 positive tumor cells by IHC staining (*p<0.05).
OxPhos is enriched in both MEKi-resistant tumors and CDK4/6i-tolerant tumors
Neither MEKi nor CDK4/6i caused tumor regression, hence, we determined the mechanisms underlying resistance (MEKi-R) and tolerance (CDKi-T) by performing RNA-seq on the tumors harvested after treatment with control, MEKi, or CDK4/6i for 151 days. We described the cells as tolerant as they are similar to drug persister cells that have been described previously and are cells that survive treatment but do not proliferate actively until treatment is removed (34,35). PCA and sample similarity analysis revealed that the tumors grouped by treatment arms (Supplemental Fig. 3). We determined genes that were significantly up- or downregulated in MEKi-R or CDK4/6i-T groups compared to controls (CTL1, CTL2) (Fig. 3A; gene names and expression values are provided in Supplemental Table 1). Gene set enrichment analysis (GSEA) demonstrated significant enrichment in oxidative phosphorylation (OxPhos) gene sets in both MEKi-R and CDKi-T tumors (Fig. 3B and C). CDKi-T tumors also showed upregulation of genes related to immunogenic effects such as IFN-α response, inflammatory response and complement signaling (Fig. 3B). Since enrichment of OxPhos may provide a therapeutic vulnerability for UM treated with MEKi and/or CDK4/6i, we further analyzed the top ranked genes within the OxPhos pathway that are shared between MEKi-R and CDK4/6i-T tumors. Mitochondrial-related genes such as COX6C, CPT1A, and vacuolar ATPase were ranked highly (Fig. 3D).
Figure 3. Enrichment of OxPhos signature in CDK4/6i-tolerant (CDK4/6i-T) and MEKi-resistant (MEKi-R) tumors.
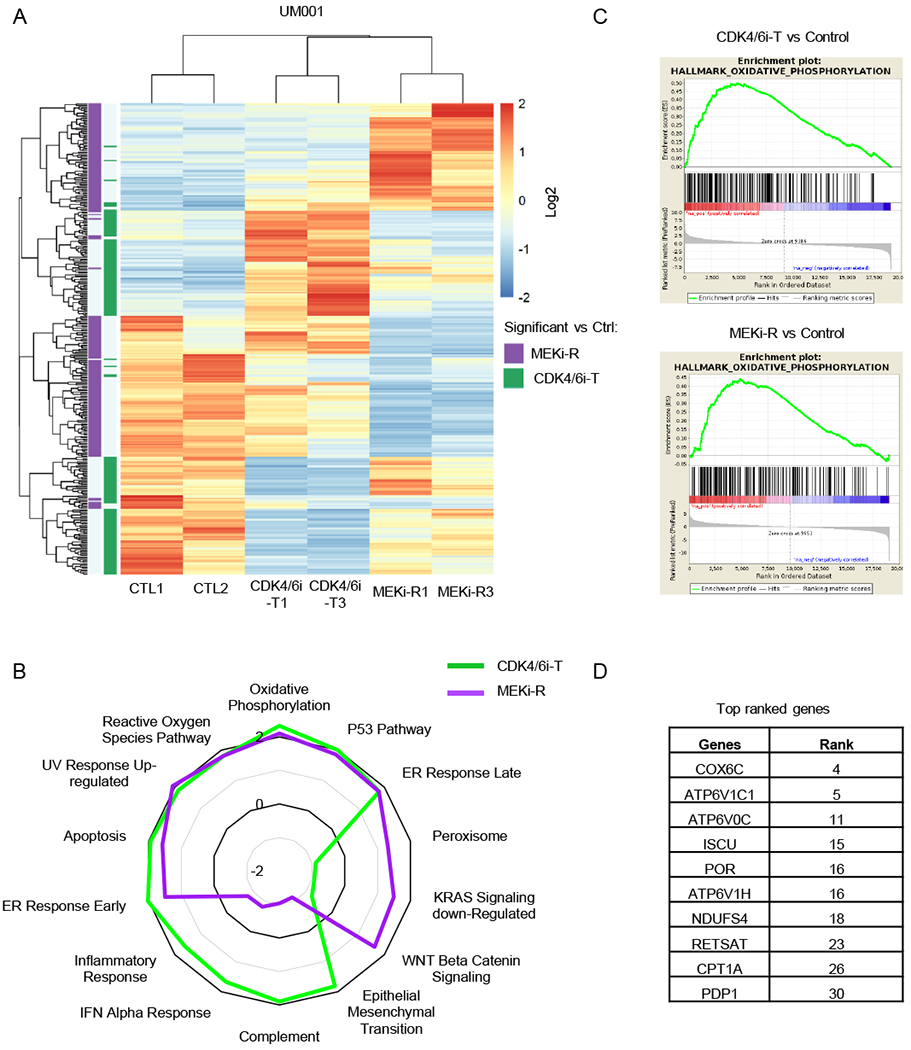
A Heatmap of unsupervised hierarchical clustering of samples based on genes differentially expressed between CDK4/6i-T and control (CTL1, CTL2) (green) or MEKi-R and control (CTL1, CTL2) (purple) (BHFDR < 0.05 and absolute log2(FC) > 1). B Radar plot showing normalized enrichment score (NES) values for CDK4/6i-T or MEKi-R vs control samples for the most commonly enriched gene sets as well as most variable between CDK4/6i-T and MEKi-R. C Gene set enrichment plots of the top-ranked Hallmark OxPhos gene set for comparison of CDK4/6i-T (top) or MEKi-R (bottom) to control samples. D Top ranked genes within the OxPhos pathway in both CDK4/6i-T and MEKi-R samples.
Upregulation of OxPhos by MEK and CDK4/6 targeting is functionally significant in UM
To determine whether the adaptive or acquired phenotypes observed were due to metabolic rewiring, we carried out Seahorse analysis. We transiently treated UM001 and WM3618F cells in vitro with DMSO (control), MEKi, CDK4/6i, or MEKi + CDK4/6i for 24 hours. Interestingly, for UM001 cells, single agent treatments led to increased oxygen consumption rate (OCR), and combination of the two drugs further accentuated this effect (Fig. 4A). There was a similar effect in a second cell line, WM3618F (Fig. 4B).
Figure 4. Induction of metabolic activity by CDK4/6 and MEK inhibition is functional and can be targeted with an OxPhos inhibitor.
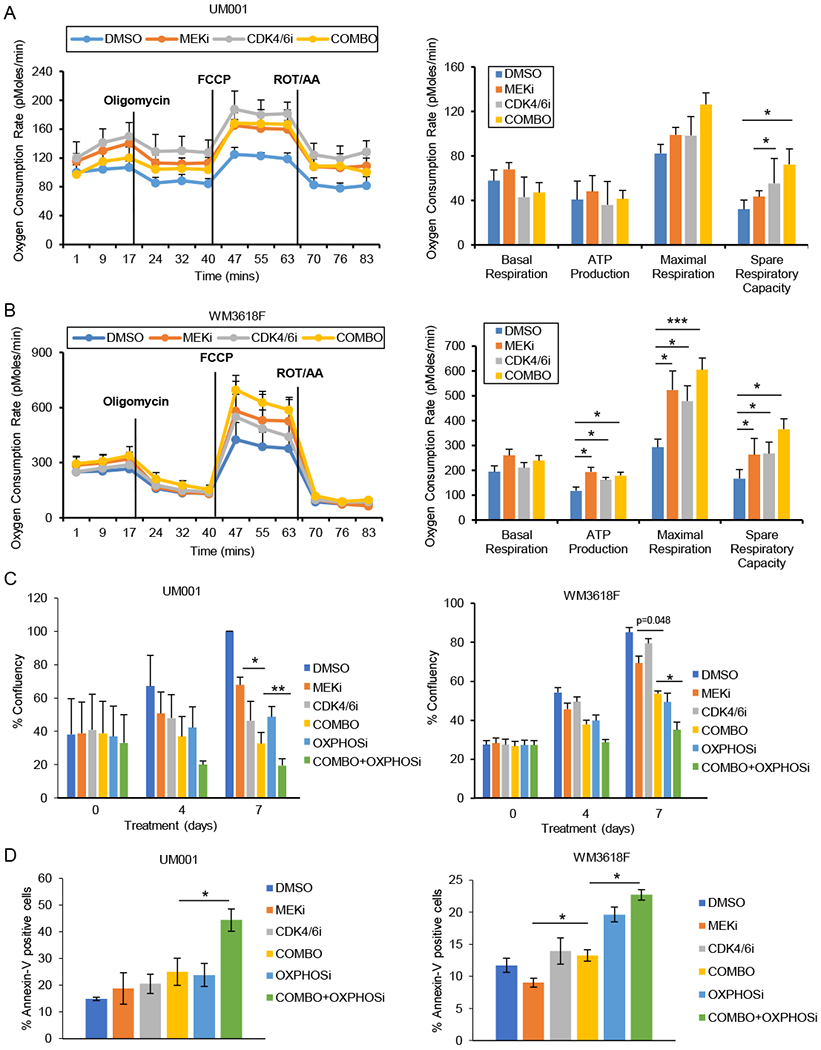
A OCR of UM001 treated with control (DMSO), MEKi (PD0325901, 5 nM), CDK4/6i (palbociclib, 0.5 μM), or the combination (PD0325901 + palbociclib) (n=3). B as A, but WM3618F cells were utilized. C Incucyte analysis of UM001 and WM3618F cell lines treated with control (DMSO), MEKi (PD0325901, 5 nM), CDK4/6i (palbociclib, 0.5 μM), OxPhosi (IACS-010759, 50 nM), MEKi + CDK4/6i (combo), or MEKi + CDK4/6i + OxPhosi (combo + OxPhosi) (*p=0.05, **p<0.005) (n=3). D Percentage annexin-V positive cells following treatment of UM001 and WM3618F with control (DMSO), MEKi (PD0325901, 5 nM), CDK4/6i (palbociclib, 0.5 μM), OxPhosi (IACS-010759, 50 nM), MEKi + CDK4/6i (combo), or MEKi + CDK4/6i + OxPhosi (combo + OxPhosi) for 48 hours (*p=0.05) (n=3).
To test whether the upregulation of OxPhos by MEK and CDK4/6 targeting has functional significance, we utilized IACS-010759, a small molecule inhibitor of complex I of the mitochondrial electron transport chain that reduces OCR in melanoma and other cell types (17) and is currently being evaluated in clinical trials (NCT02882321, NCT03291938) (36–38). Single agent treatment with IACS-010759 had moderate growth inhibitory effects on UM001 and WM3618F cells, but combination treatment with MEKi + CDK4/6i significantly decreased cell viability compared to IACS-010759 alone or MEKi + CDK4/6i (Fig. 4C). Treatment effects were reversible (Supplemental Fig. 4). As apoptosis was observed with MEKi and CDK4/6i treatment in vivo (Supplemental Fig. 5), we examined annexin V staining in cells in vitro treated with OxPhosi. We observed a significant increase in the percentage of annexin-V positive cells with the combination of OxPhosi + MEKi + CDK4/6i compared to combo (MEKi plus CDK4/6i) (Fig. 4D). Together, our data suggest that OxPhos inhibition may overcome the pro-survival effects of metabolic rewiring in UM cells treated with MEK inhibitors plus CDK4/6 inhibitors.
Discussion
New effective therapeutic strategies are a critical unmet clinical need for advanced UM. Despite the high frequency of GNAQ and GNA11 mutations in UM that enhance MEK-ERK1/2 signaling, MEKi are not clinically effective in patients, perhaps due to activation of additional downstream or parallel pathways. Here, we have evaluated the effects of targeting CDK4/6, which previously has shown positive combinatorial effects with MEKi in NRAS-mutant cutaneous melanoma tumors (13,14). Our study shows that CDK4/6i caused cell cycle arrest in UM cell lines in vitro and in vivo, but there were no significant combinatorial effects with MEKi. Tolerance to CDK4/6i and resistance to MEKi in UM cell lines and tumors were associated with increased OxPhos. Notably, our preliminary studies support the rationale to evaluate OxPhos inhibition further in this cancer type.
Multiple selective inhibitors of CDK4/6 have gained FDA-approval/breakthrough therapy designation in ER+/HER2− breast cancer (33). Trials in melanoma have utilized CDK4/6i alone and in combination with MEKi, but their use still needs to be optimized (39). For example, the combination of CDK4/6i + MEKi (ribociclib plus binimetinib) was tested in mutant NRAS cutaneous melanoma patients and yielded encouraging results (43% overall response rate) (40). In a phase II trial enrolling 22 patients with a 28-day regimen of continuous MEKi and intermittent CDK4/6i, there was a 41% overall response rate, 82% disease control rate, and 6.7-month median PFS (41). Here, we present the first investigation of CDK4/6i in a pre-clinical model of UM. In vitro, we found that CDK4/6 inhibition further reduced the growth of UM cell lines compared to MEKi alone. These effects were associated with inhibition of cell cycle progression but did not induce apoptosis. MEKi and CDK4/6i combination effects are likely not additive as CDK4/6 activation has been shown to be downstream of MEK pathway (20). By contrast, combination MEKi plus CDK4/6i enhanced cleaved caspase 3 levels in vivo and UM001 xenografts were non-responsive to MEKi, perhaps due to stromal resistance mechanisms (19,20). CDK4/6i alone or the combination of MEKi with CDK4/6i was more effective at reducing tumor volumes, although neither caused tumor regression. Interestingly, these data contrast with previous findings in cutaneous melanoma xenograft models, in which CDK4/6i alone caused moderate delays in tumor growth and the combination of MEKi plus CDK4/6i caused tumor regressions (13,14,42). Including more animals to each treatment arm may increase statistical significance between CDK4/6i alone and the combo arm and/or the different effects of combo or CDK4/6i alone may become more evident after discontinuing treatment.
The cytostasis following CDK4/6i in vivo treatment led us to examine potential mechanisms of this drug tolerant state and our analysis implicated the OxPhos metabolic pathway. In pancreatic cancer, CDK4/6 inhibition enhances glycolysis and OxPhos, as well as increases mitochondrial mass (43). Several studies in the cutaneous melanoma setting have identified up-regulation of OxPhos in response to targeted therapies that is associated with the expression of a transcription factor, PGC1α, a master regulator of OxPhos genes (44,45). Adaptive up-regulation of PGC1α has also been implicated in resistance to BRAF inhibitor therapy in BRAF-mutant cutaneous melanoma, but is yet to be analyzed in the setting of GNAQ/11 mutant UM (16,46). Thus, metabolic adaptations may be common across melanomas in response to targeted therapies.
Furthermore, we sought to examine whether the adaptive OxPhos response is functionally relevant. IACS-010759 is a potent inhibitor of electron transport chain complex I that is currently being evaluated in Phase I clinical trials (NCT02882321, NCT03291938) (36). Although, we did not show effects on OCR in uveal melanoma cells in this study, we have previously shown that IACS-010759 reduces OCR in cutaneous melanoma cells (17). Importantly, the addition of IACS-010759 to CDK4/6i + MEKi decreased cell growth and enhanced apoptosis, suggesting that direct OxPhos inhibition should be further analyzed as an approach to optimize targeted therapy treatment in UM. Besides directly targeting the electron transport chain machinery, future studies can investigate the possibility of targeting OxPhos by modulating its regulators. For example, inhibition of mTORC1/2 has been shown to decrease PGC1α expression and inhibit OxPhos (16). Therefore, an alternative therapeutic approach to targeting OxPhos would be mTOR inhibition in concert with CDK4/6i, although there remain tolerability concerns with mTOR inhibitors (47). In summary, our study provides an initial evaluation of CDK4/6i in UM. Our findings highlight potential tumor intrinsic adaptive mechanisms to CDK4/6i and approaches to optimize CDK4/6i-based therapeutic strategies.
Supplementary Material
Acknowledgments and Financial Support
This work is supported by grants to A.E. Aplin from NIH/NCI (R01 CA182635), a Melanoma Research Foundation Established Investigator Award, the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation and the Dr. Ralph and Marian Falk Medical Research Trust to A.E. Aplin and J. Aguirre-Ghiso. J.L.F. Teh was supported by an AACR-Ocular Melanoma Foundation Fellowship. M.A. Davies receives funding support from 2T32CA009666-21 and the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation. The Sidney Kimmel Cancer Center Flow Cytometry, Translational Pathology and Meta-Omics core facilities are supported by NCI Support Grant (P30 CA056036). The RPPA studies were performed at the Functional Proteomics Core Facility at The University of Texas MD Anderson Cancer Center, which is supported by a NCI Cancer Center Support Grant (P30 CA16672).
Conflict of interest: A.E. Aplin and J.L.F. Teh have ownership interest in patent number 9880150. A.E. Aplin reports receiving a commercial research grant from Pfizer Inc. (2013-2017), and has consulted for SpringWorks Therapeutics and Fortress Biotech within the last 3 years. M.A. Davies has been a consultant to Novartis, Array, Roche/Genentech, GSK, Sanofi-Aventis, and has been the PI of research grants to his institution from GSK, Sanofi-Aventis, Oncothyreon, and AstraZeneca. J.A. Aguirre-Ghiso is a Founder and Scientific Advisor for HiberCell LLC and has engaged in consulting with Eli Lilly in the last 3 years.
References
- 1.Collaborative Ocular Melanoma Study G. Assessment of metastatic disease status at death in 435 patients with large choroidal melanoma in the Collaborative Ocular Melanoma Study (COMS): COMS report no. 15. Arch Ophthalmol 2001; 119:670–6. [DOI] [PubMed] [Google Scholar]
- 2.Decatur CL, Ong E, Garg N, Anbunathan H, Bowcock AM, Field MG, et al. Driver Mutations in Uveal Melanoma: Associations With Gene Expression Profile and Patient Outcomes. JAMA Ophthalmol 2016; 134:728–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Van Raamsdonk CD, Bezrookove V, Green G, Bauer J, Gaugler L, O’Brien JM, et al. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature 2009; 457:599–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Robertson AG, Shih J, Yau C, Gibb EA, Oba J, Mungall KL, et al. Integrative analysis identifies four molecular and clinical subsets in uveal melanoma. Cancer Cell 2017; 32:204–20 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Van Raamsdonk CD, Griewank KG, Crosby MB, Garrido MC, Vemula S, Wiesner T, et al. Mutations in GNA11 in uveal melanoma. N Engl J Med 2010; 363:2191–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yu FX, Luo J, Mo JS, Liu G, Kim YC, Meng Z, et al. Mutant Gq/11 promote uveal melanoma tumorigenesis by activating YAP. Cancer Cell 2014; 25:822–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chua V, Lapadula D, Randolph C, Benovic JL, Wedegaertner PB, Aplin AE. Dysregulated GPCR signaling and therapeutic options in uveal melanoma. Mol Cancer Res 2017; 15:501–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lapadula D, Farias E, Randolph CE, Purwin TJ, McGrath D, Charpentier TH, et al. Effects of Oncogenic Galphaq and Galpha11 Inhibition by FR900359 in Uveal Melanoma. Mol Cancer Res 2019; 17:963–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carvajal RD, Sosman JA, Quevedo JF, Milhem MM, Joshua AM, Kudchadkar RR, et al. Effect of selumetinib vs chemotherapy on progression-free survival in uveal melanoma: a randomized clinical trial. JAMA 2014; 311:2397–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carvajal RD, Piperno-Neumann S, Kapiteijn E, Chapman PB, Frank S, Joshua AM, et al. Selumetinib in Combination With Dacarbazine in Patients With Metastatic Uveal Melanoma: A Phase III, Multicenter, Randomized Trial (SUMIT). J Clin Oncol 2018; 36:1232–9. [DOI] [PubMed] [Google Scholar]
- 11.Finn RS, Martin M, Rugo HS, Jones S, Im SA, Gelmon K, et al. Palbociclib and Letrozole in Advanced Breast Cancer. N Engl J Med 2016; 375:1925–36. [DOI] [PubMed] [Google Scholar]
- 12.Goetz MP, Toi M, Campone M, Sohn J, Paluch-Shimon S, Huober J, et al. MONARCH 3: Abemaciclib As Initial Therapy for Advanced Breast Cancer. J Clin Oncol 2017; 35:3638–46. [DOI] [PubMed] [Google Scholar]
- 13.Teh JL, Purwin TJ, Greenawalt EJ, Chervoneva I, Goldberg A, Davies MA, et al. An in vivo reporter to quantitatively and temporally analyze the effects of CDK4/6 inhibitor-based therapies in melanoma. Cancer Res 2016; 76:5455–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kwong LN, Costello JC, Liu H, Jiang S, Helms TL, Langsdorf AE, et al. Oncogenic NRAS signaling differentially regulates survival and proliferation in melanoma. Nat Med 2012; 18:1503–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schuler MH, Ascierto PA, Leon De Vos FYF, Postow MA, Van Herpen CML, Carlino MS, et al. Phase 1b/2 trial of ribociclib+binimetinib in metastatic NRAS-mutant melanoma: Safety, efficacy, and recommended phase 2 dose (RP2D). J Clin Oncol 2017; 15:959. [Google Scholar]
- 16.Gopal YN, Rizos H, Chen G, Deng W, Frederick DT, Cooper ZA, et al. Inhibition of mTORC1/2 overcomes resistance to MAPK pathway inhibitors mediated by PGC1alpha and oxidative phosphorylation in melanoma. Cancer Res 2014; 74:7037–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gopal YN, Gammon S, Prasad R, Knighton B, Pisaneschi F, Roszik J, et al. A Novel Mitochondrial Inhibitor Blocks MAPK Pathway and Overcomes MAPK Inhibitor Resistance in Melanoma. Clin Cancer Res 2019; 25:6429–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yoshida M, Selvan S, McCue PA, DeAngelis T, Baserga R, Fujii A, et al. Expression of insulin-like growth factor-1 receptor in metastatic uveal melanoma and implications for potential autocrine and paracrine tumor cell growth. Pigment Cell Melanoma Res 2014; 27:297–308. [DOI] [PubMed] [Google Scholar]
- 19.Cheng H, Terai M, Kageyama K, Ozaki S, McCue PA, Sato T, et al. Paracrine effect of NRG1 and HGF drives resistance to MEK Inhibitors in metastatic uveal melanoma. Cancer Res 2015; 75:2737–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng H, Chua V, Liao C, Purwin TJ, Terai M, Kageyama K, et al. Co-targeting HGF/cMET Signaling with MEK Inhibitors in Metastatic Uveal Melanoma. Mol Cancer Ther 2017; 16:516–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chua V, Orloff M, Teh JL, Sugase T, Liao C, Purwin TJ, et al. Stromal fibroblast growth factor 2 reduces the efficacy of bromodomain inhibitors in uveal melanoma. EMBO Mol Med 2019; 11:pii: e9081. doi: 10.15252/emmm.201809081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thompson N, Lyons J. Recent progress in targeting the Raf/MEK/ERK pathway with inhibitors in cancer drug discovery. Curr Opin Pharmacol 2005; 5:350–6. [DOI] [PubMed] [Google Scholar]
- 23.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013; 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 2011; 12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 2003; 34:267–73. [DOI] [PubMed] [Google Scholar]
- 27.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005; 102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 2015; 1:417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brunner E, ML P. Nonparametric Methods in Factorial Designs. Statistical Papers 2001; 42:1–52. [Google Scholar]
- 30.Brunner E, Domhof S, F L. Nonparametric Analysis of Longitudinal Data in Factorial Experiments. John Wiley & Sons, New York: 2002; [Google Scholar]
- 31.Noguchi K, Gel YR, Brunner E, Konietschke F. nparLD: An R Software Package for the Nonparametric Analysis of Longitudinal Data in Factorial Experiments. Journal of Statistical Software 2012; 50:1–23.25317082 [Google Scholar]
- 32.Kageyama K, Ohara M, Saito K, Ozaki S, Terai M, Mastrangelo MJ, et al. Establishment of an orthotopic patient-derived xenograft mouse model using uveal melanoma hepatic metastasis. J Transl Med 2017; 15:145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Finn RS, Crown JP, Lang I, Boer K, Bondarenko IM, Kulyk SO, et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol 2015; 16:25–35. [DOI] [PubMed] [Google Scholar]
- 34.Chisholm RH, Lorenzi T, Lorz A, Larsen AK, de Almeida LN, Escargueil A, et al. Emergence of drug tolerance in cancer cell populations: an evolutionary outcome of selection, nongenetic instability, and stress-induced adaptation. Cancer Res 2015; 75:930–9. [DOI] [PubMed] [Google Scholar]
- 35.Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010; 141:69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Molina JR, Sun Y, Protopopova M, Gera S, Bandi M, Bristow C, et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat Med 2018; 24:1036–46. [DOI] [PubMed] [Google Scholar]
- 37.Zhang L, Yao Y, Zhang S, Liu Y, Guo H, Ahmed M, et al. Metabolic reprogramming toward oxidative phosphorylation identifies a therapeutic target for mantle cell lymphoma. Sci Transl Med 2019; 11:pii: eaau1167. doi: 10.26/scitranslmed.aau. [DOI] [PubMed] [Google Scholar]
- 38.Vangapandu HV, Alston B, Morse J, Ayres ML, Wierda WG, Keating MJ, et al. Biological and metabolic effects of IACS-010759, an OxPhos inhibitor, on chronic lymphocytic leukemia cells. Oncotarget 2018; 9:24980–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee B, Sandhu S, McArthur G. Cell cycle control as a promising target in melanoma. Curr Opin Oncol 2015; 27:141–50. [DOI] [PubMed] [Google Scholar]
- 40.Sosman J, Kittaneh M, Lolkema M, Postow M, Schwartz G, Franklin C, et al. A phase 1b/2 study of LEE011 in combination with binimetinib (MEK162) in patients with NRAS-mutant melanoma: early encouraging clinical activity. J Clin Oncol 2014; 32:9009. [Google Scholar]
- 41.Van Herpen C, Postow MA, Carlino MS, Kalkavan H, Weise A, Amaria RN, et al. A phase 1b/2 study of ribociclib (LEE011; CDK4/6 inhibitor) in combination with binimetinib (MEK162; MEK inhibitor) in patients with NRAS-mutant melanoma. Euro J Cancer 2015; 2015:S663. [Google Scholar]
- 42.Teh JLF, Cheng PF, Purwin TJ, Nikbakht N, Patel P, Chervoneva I, et al. In Vivo E2F Reporting Reveals Efficacious Schedules of MEK1/2-CDK4/6 Targeting and mTOR-S6 Resistance Mechanisms. Cancer Discov 2018; 8:568–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Franco J, Balaji U, Freinkman E, Witkiewicz AK, Knudsen ES. Metabolic Reprogramming of Pancreatic Cancer Mediated by CDK4/6 Inhibition Elicits Unique Vulnerabilities. Cell Rep 2016; 14:979–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Haq R, Shoag J, Andreu-Perez P, Yokoyama S, Edelman H, Rowe GC, et al. Oncogenic BRAF regulates oxidative metabolism via PGC1alpha and MITF. Cancer Cell 2013; 23:302–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vazquez F, Lim JH, Chim H, Bhalla K, Girnun G, Pierce K, et al. PGC1alpha expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell 2013; 23:287–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baenke F, Chaneton B, Smith M, Van Den Broek N, Hogan K, Tang H, et al. Resistance to BRAF inhibitors induces glutamine dependency in melanoma cells. Mol Oncol 2016; 10:73–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Martelli AM, Buontempo F, McCubrey JA. Drug discovery targeting the mTOR pathway. Clin Sci (Lond) 2018; 132:543–68. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.