

Abstract
Protein folding quality control in cells requires the activity of a class of proteins known as molecular chaperones. Heat shock protein-90 (Hsp90), a multidomain ATP driven molecular machine, is a prime representative of this family of proteins. Interactions between Hsp90, its co-chaperones, and client proteins have been shown to be important in facilitating the correct folding and activation of clients. Hsp90 levels and functions are elevated in tumor cells. Here, we computationally predict the regions on the native structures of clients c-Abl, c-Src, Cdk4, B-Raf and Glucocorticoid Receptor, that have the highest probability of undergoing local unfolding, despite being ordered in their native structures. Such regions represent potential ideal interaction points with the Hsp90-system. We synthesize mimics spanning these regions and confirm their interaction with partners of the Hsp90 complex (Hsp90, Cdc37 and Aha1) by Nuclear Magnetic Resonance (NMR). Designed mimics selectively disrupt the association of their respective clients with the Hsp90 machinery, leaving unrelated clients unperturbed and causing apoptosis in cancer cells. Overall, selective targeting of Hsp90 protein-protein interactions is achieved without causing indiscriminate degradation of all clients, setting the stage for the development of therapeutics based on specific chaperone:client perturbation.
Keywords: Heat shock protein (Hsp90), Protein-protein interaction inhibitors, Molecular Recognition, Cochaperones, Protein Dynamics
Graphical Abstract
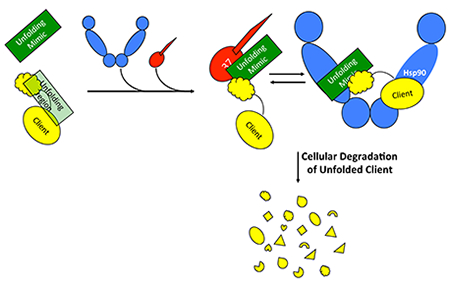
A novel, fully rational computational approach predicts the unfolding/unstable regions of proteins. Designed synthetic mimics of such regions block protein interactions with chaperones in cells, and induce apoptosis in cancer cells.
The Hsp90 family of molecular chaperones plays key roles in cell proteostasis by balancing the folding, activation, and turnover of a diverse set of client proteins, many of which fundamental for cancer development. Proteins of the Hsp90 family (Hsp90 in the cytosol, Grp94 in the ER and Trap1 in mitochondria) are homodimers with each individual chain consisting of three globular domains, the N-terminal (NTD), Middle and C-terminal (CTD). X-ray, SAXS and kinetic measurements have led to a mechanistic model in which global conformational modulations are triggered by ATP: ATP binding at the N-terminal domain [1] shifts the chaperone to a partially closed, and then into an asymmetric closed conformation that is significantly strained at the Middle:CTD interface, the region involved in client binding[2]. Upon ATP hydrolysis, strain is relieved through rearrangement of the client binding-site residues, driving structural changes in the client [3]. In this framework, ATP hydrolysis is coupled to client remodeling. For these reasons, pharmacological intervention on Hsp90-controlled processed has largely relied on the use of ATP-competitive inhibitors 4]. Hsp90 inhibition by ATP competitive inhibitors determines, however, the indiscriminate depletion of all Hsp90 clients, ultimately causing the upregulation of the heat shock response which ends up protecting cancer cells from apoptosis and causing toxicity [4g, 5].
The formation of client:Hsp90 complexes is a critical step in the regulation of specific client activities. Specificity in the selection of clients that lack sequence and structural homology [6] is acquired through recruiter cochaperones that provide the essential recognition/discrimination elements [7]. In this framework, Hsp90, its cochaperones and the clients engage in multicomponent assemblies, stabilized by dynamic protein-protein interactions (PPIs). Co-chaperones such as Cdc37 control the entry of kinases and other clients into the chaperone cycle [8],[9],[10], while other cochaperones, such as Aha1, provide additional layers of regulation by modulating the rates of ATP hydrolysis[11]. No specific structural elements or surface characteristics have been proposed as Hsp90-binding determinants [12],[13],[14]. Critical to Hsp90 mechanisms is that the interactions involved are conformationally heterogeneous, short-lived and relatively weak, with different clients interacting in distinct ways [15],[16],[17],[18].
These observations unveil a new opportunity for the design of client-selective chemical tools based on the idea that perturbing the (weak) interactions with (any of) the members of the chaperone assembly can impair a client’s folding. The lack of consensus binding motifs suggests the possibility to target unique interaction surfaces in order to specifically disrupt key client:chaperone or client:co-chaperone interactions.
Here, we predicted interaction interfaces of different clients, based only on the structure of the isolated client, as the rational basis for the design of selective peptide inhibitors of protein-protein interactions in chaperone complexes. The design is based on a novel computational method we recently developed for the prediction of locally unstable substructures in proteins. Here, the unstable substructures do not coincide with intrinsically disordered regions or coils (which may indeed be thermodynamically stable); rather they represent protein regions that, despite being ordered in the native state, have a high probability/tendency to undergo local unfolding and support conformational transitions leading to conformational heterogeneity [19]. These particular substructures thus represent potential ideal points of interaction with the Hsp90 machinery [2d, 20]. This knowledge is translated into the development of peptides spanning the predicted interaction sites with the aim to engage different constituents of the Hsp90 complex (Hsp90, Cdc37, Aha1). The ability of the designed molecules to bind to their chaperone complex members was confirmed by NMR. The peptides were cell permeable and selectively interfered with the association of their respective clients with the Hsp90 chaperone machinery, ultimately causing apoptosis in cancer cells. To the best of our knowledge, our work represents the first example in which ab initio, physics-based characterization of protein stability is leveraged for the selective chemical targeting of chaperone:client interactions in multicomponent complexes. This is achieved without significant indiscriminate inhibition or degradation of all clients, setting the stage for the definition of the pharmacophoric requirements for the development of PPI targeting molecules with therapeutic potential. We suggest that this approach can be extended to block the folding of otherwise undruggable proteins, providing a novel opportunity to block the pathologic activities of biomolecules for which no drug has been developed.
Computational design and NMR-characterization.
The Hsp90 system acts on clients late in their folding pathway and associates with substrates in which large parts of the domains are already folded in their native conformation. We therefore hypothesize that the chaperone system targets client substructures with minimal structural stability in the native state. From the physico-chemical point of view, locally-unstable substructures are characterized by distinct energetic properties as they are not involved in major intramolecular stabilizing interactions with other regions of the protein. Minimal intramolecular coupling, in turn, favors local instability and structural variations, distinctive properties of local unfolding.
To predict the location of minimally coupled, locally unstable substructures, we used the MLCE method [19b, 21] [see SI Methods]. We first tested our approach on the Hsp90 client Abelson leukemia (c-Abl) kinase protein. Two consensus sequences, labeled respectively A01 and A02 (Figure 1, Table 1), were designed as potential interaction epitopes by applying MLCE to MD simulations of c-Abl. A01 represents a conformational epitope localized at the N-lobe at the border with the C-lobe, spanning parts of the Gly-rich loop, β1, β2 and β3 strands and the αC helix (Figure 1). To achieve optimal spanning of predicted interaction regions, we linked the different constituent subparts by the addition of a number of glycine residues approximating the average distance between the respective terminals calculated from MD simulations (see Table 1). The second epitope, A02, is linear, located at the C-lobe, and spans the αG-helix preceded by the flexible αF loop. An 19F-modified phenyl alanine was site-specifically introduced into A01 and A02 to allow the characterization of the binding to members of the Hsp90 chaperone complexes (Hsp90, Cdc37, Aha1) by Fluorine Nuclear Magnetic Resonance (19F NMR) [22],[23] (see Table 1). Human Serum Albumin (HSA) was used as a control for non-specific binding.
Figure 1.
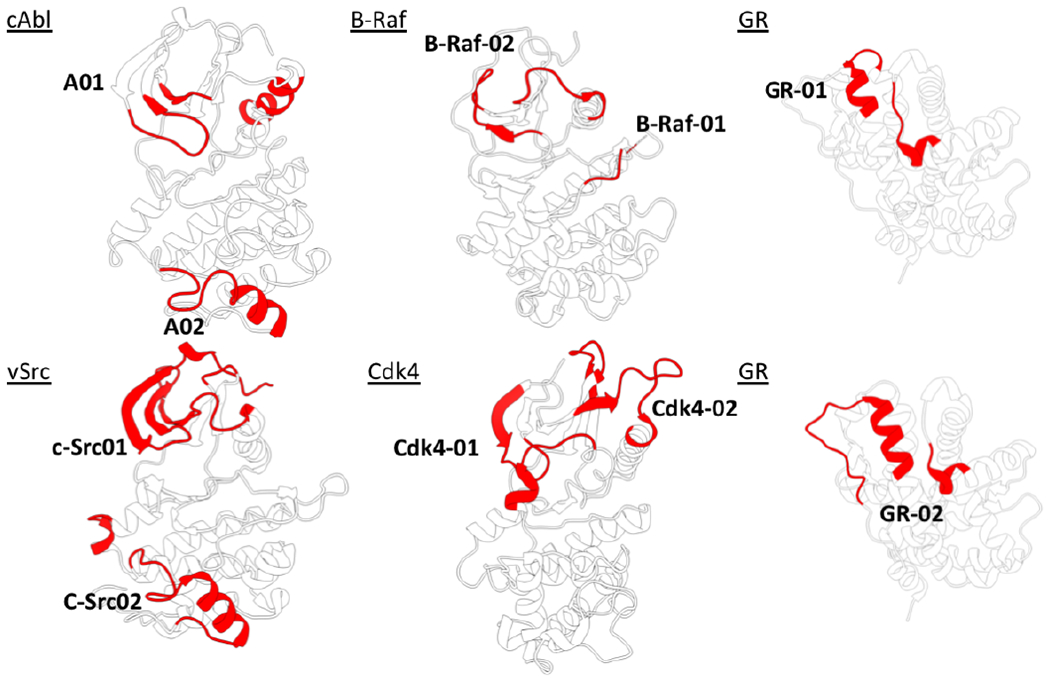
The 3D structures and names of the proteins analyzed in the paper. The regions colored in red are the substructures predicted to undergo unfolding and to be points of interaction with the members of the Hsp90 chaperone machinery.
Table 1.
Sequences of the mimics of chaperone binding regions.
| Protein | Mimic Name | Sequence[a,b] |
|---|---|---|
| c-Abl | A01 | (248)LGGGQFGEVY(265)-GG-(267)VAVKTL(274)-GGG-(282)EFLDEAAVMK(291) |
| A02 | FGG(438)SPYPGIDLSQVYELLEK(454) | |
| B-Raf | B-Raf-01 | GYSTKPQLA(79)GGG(39)NVTAPTPQ(46)G-(163)F-QHSGS(158) |
| B-Raf-02 | (22)FGTVYKGKW(29)GGG(71)GF-STKPQLA(79)GGG(39)NVTAPTPQ(46) | |
| Cdk4 | Cdk4-01 | (83)CATSRTDRE(91)G(45)PNGGGGGGGLPIST(58)GG(172)FQMALTPVV(180) |
| Cdk4-02 | (13)PVAEIGVGAYG(23)GG(43)RVP(45)GG(172)FQMALTPVV(180) | |
| cSrc | cSrc-01 | (21)LGQGCFG(27)G(46)KPGTMSP(52)GG(79)EEP(81)GG(12)RESL(15)G(34)WNGTT(38) |
| cSrc-02 | (100)GEMGK(104)GG(206)KGRVPYPGMVNREVLDQVERGFRM(229) | |
| GR | GR-01 | (635)TLPC(638)GG(556)TWRIMT(561)G(747)IEFPEMLA(754) |
| GR-02 | (545)YAGYDSSVPDSTWRIMTTLNM(565)GG(749)FPEMLA(754) | |
The numbering in square brackets reports the numbering in the original pdb file, reported in the first column.
The bold underlined F indicates fluorinated phenylalanine.
First, we tested the peptides individually at 10 μM in the presence and in absence of full length human Hsp90 by 19F R2 filter experiments. The 19F NMR signals of A01 and A02 decrease in the presence of Hsp90, indicating peptide-protein binding (Figure 2a, traces 1 and 2). Interestingly A01 and A02 continue to bind to Hsp90 even when they are tested together, suggesting that they do not compete with each other and interact with different sites. Next, we ran 19F-NMR interaction experiments testing 10 μM of each cAbl peptide against Aha1 and Cdc37. A01 showed the ability to bind Cdc37, while showing no binding to Aha1. We also checked the binding of 40 μM A01 and 40 μM A02 with 10 μM HSA: importantly, neither A01 nor A02 showed interaction with HSA (see Supporting Information Figure SI1).
Figure 2.
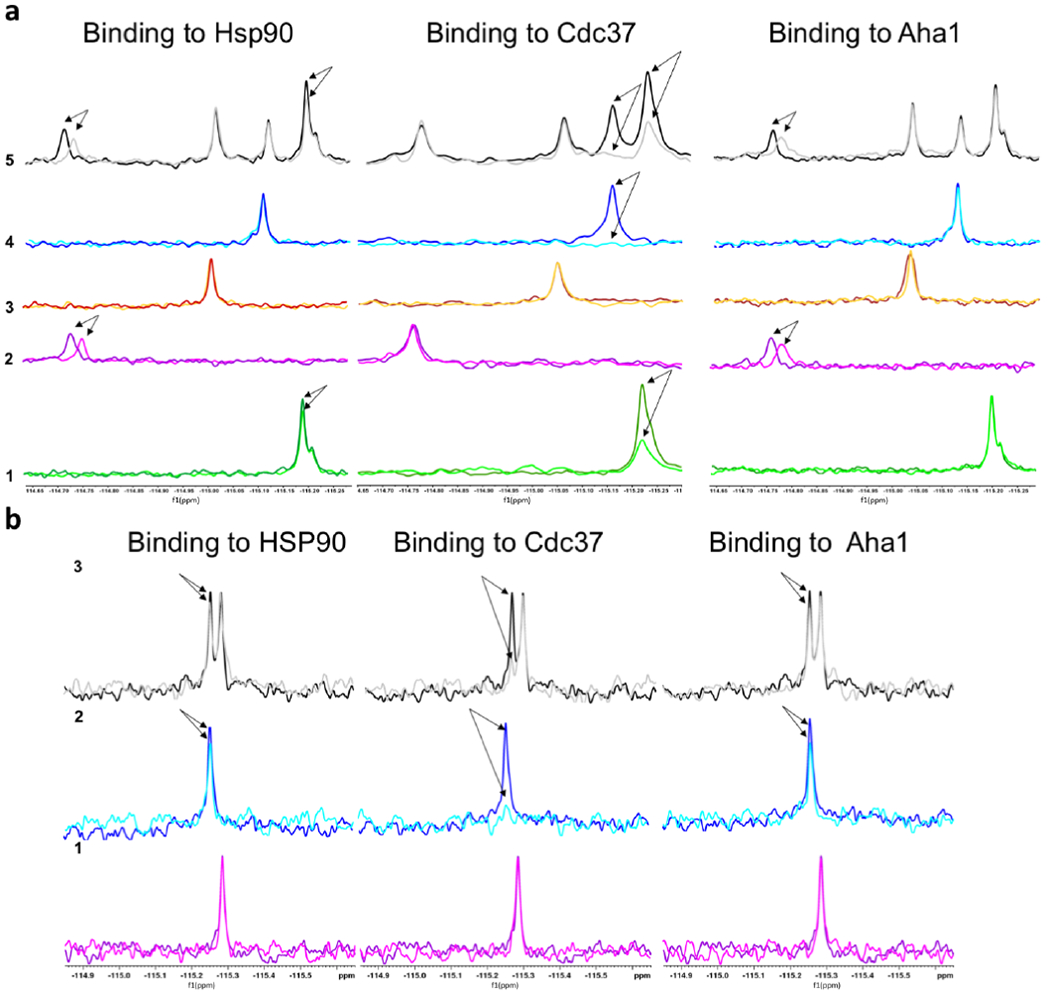
(a) 19F NMR experiments showing the binding of Abl and Braf peptides to the proteins of the Hsp90 chaperone system. The subpanels indicate the interactions of (1) Peptide A01; (2) A02; (3) B-Raf01; (4) B-Raf02; (5) all peptides are mixed together and added to the solution containing the protein. The NMR signals show no variation compared to the situation with the single peptides, indicating that there is no competition among the different sequences. (b) 19F NMR experiments showing the binding of GR peptides to the proteins of the Hsp90 chaperone system. (1) Peptide GR-01; (2) GR-02; (3) both peptides are mixed together and added to the solution containing the protein. The NMR signals show that GR-02 binds to all the protein, but the binding effect is higher on CDC37. No significant difference are observed in the binding of GR-02 in presence of GR-01.
To investigate the generality of our strategy, we next designed mimics of the chaperone-binding regions of oncogenic kinase B-Raf and extended MLCE-predictions to glucocorticoid receptor (GR), a protein with structure, substrate and functions that are completely unrelated to kinases. In B-Raf, two conformational epitopes were predicted, labeled B-Raf-01 and B-Raf-02 (Figure 1, Table 1), located in the N-lobe of the kinase at the border with the C-lobe. Interestingly, while the 19F NMR signals of peptides B-Raf-01 and B-Raf-02 (Figure 2a, traces 3 and 4) do not show any difference in the presence of Hsp90, indicating no or very weak interaction, B-Raf-02 binds to Cdc37 (Figure 2a, trace 4). Importantly, no interactions are detected with HSA (see Supporting Information Figure SI1).
In GR[24], two substructures were predicted (Figure 1, Table 1). Interestingly, they correspond to the region that Kirschke et al. [25] previously observed to engage the Hsp90 complex. 19F-NMR confirmed binding of synthetic mimics of GR unfolding regions to the members of the chaperone complex. GR-02, in particular, binds to all the tested proteins, with the highest effect on Cdc37 (Figure 2, trace 2).
Because NMR data supported the viability of our prediction and design approach, we extended our analysis to oncogenic kinases Cdk4 and c-Src for further testing in cancer cells. In Cdk4, the unfolding regions localize mainly in the N-lobe, corresponding to the region undergoing unfolding in the Agard cryoEM structure [20a]. In c-Src, an additional epitope traces the G4 helix in the C-lobe (see Figure 1, Table 1).
Effect of c-Abl peptides on Hsp90 chaperone complex in human cells.
To demonstrate the impact of the peptides based on c-Abl–Hsp90 interactions, we treated HEK293 cells with A01 and A02. We demonstrated a decrease of c-Abl protein levels and dissociation of c-Abl from Hsp90, consistent with interference of Hsp90 chaperoning by A01 and/or A02 (Figure 3a). Importantly, neither the related kinase c-Src nor the client kinases Cdk4 or Akt showed a defect in stability or activity, indicating the specificity of the A01 and A02 designed peptide epitopes for c-Abl (Figure 3a).
Figure 3.
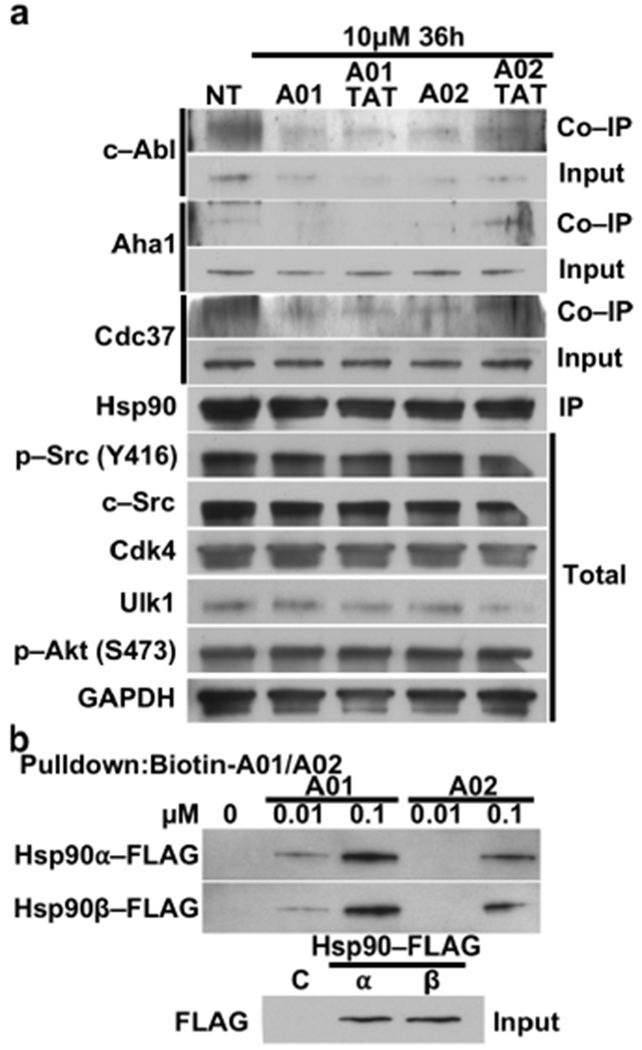
(a) HEK293 cells were treated with 10μM A01 or A02 with and without TAT. After 36 hours, lysate was prepared from the cells followed by immunoprecipitation of endogenous Hsp90. Inputs and co-immunoprecipitated proteins were evaluated by immunoblotting. GAPDH was used as a loading control. NT = no treatment. (b) Hsp90α-FLAG or Hsp90β-FLAG were overexpressed in HEK293 cells. Hsp90 binding to biotinylated A01 and A02 was examined by immunoblotting.
We have previously published that c-Abl phosphorylates Aha1-Y223 and promotes its binding to Hsp90 [26]. A01 and A02 treatment decreased Aha1-Hsp90 complex formation, demonstrating that the peptide-mediated dissociation of c-Abl and Hsp90 elicits a functional consequence (Figure 3a). We next tested whether A01 and A02 preferred a particular isoform of Hsp90, the constitutively expressed Hsp90β or the stress-inducible Hsp90α. Streptavidin-pulldown of the biotinylated peptides shows A01 binds with higher affinity to both isoforms of Hsp90 but neither peptide demonstrates isoform specificity (Figure 3b). Taken together we have demonstrated that our c-Abl–mimic peptides can disrupt the specific interaction of this client kinase with Hsp90 in cells.
Physiological impact of Hsp90 client-based peptides.
Our data with the c-Abl-based inhibitors prompted us to examine the impact of additional peptides spanning the predicted interaction regions of other clients (B-Raf, Cdk4, c-Src and GR) on their respective client:chaperone interactions. Treating HEK293 cells with increasing concentrations of these peptides led to dissociation of the clients from Hsp90 (Figure 4a–d). We confirmed that these peptides labeled with fluorescein can readily enter HEK293 cells (Figure 4e). We further examined the effect of these peptides in cancer cells. We treated the clear cell renal cell carcinoma cell line 786-O with B-Raf, Cdk4, and c-Src-based inhibitors. All the mimicking peptides, except c-Src02, and induced apoptosis, as revealed by the induction of the apoptotic marker cleaved caspase-3 (Figure 4f). Finally, we have also treated the 786-O cells with GR-based inhibitors (GR-01 and GR-02) and showed GR dissociation from Hsp90 (Figure 4g). Taken together, these data indicate that the designed peptides have the ability to enter cells and dissociate client proteins from Hsp90. They also demonstrate biological activity as they induce apoptosis in a cancer cell line.
Figure 4.
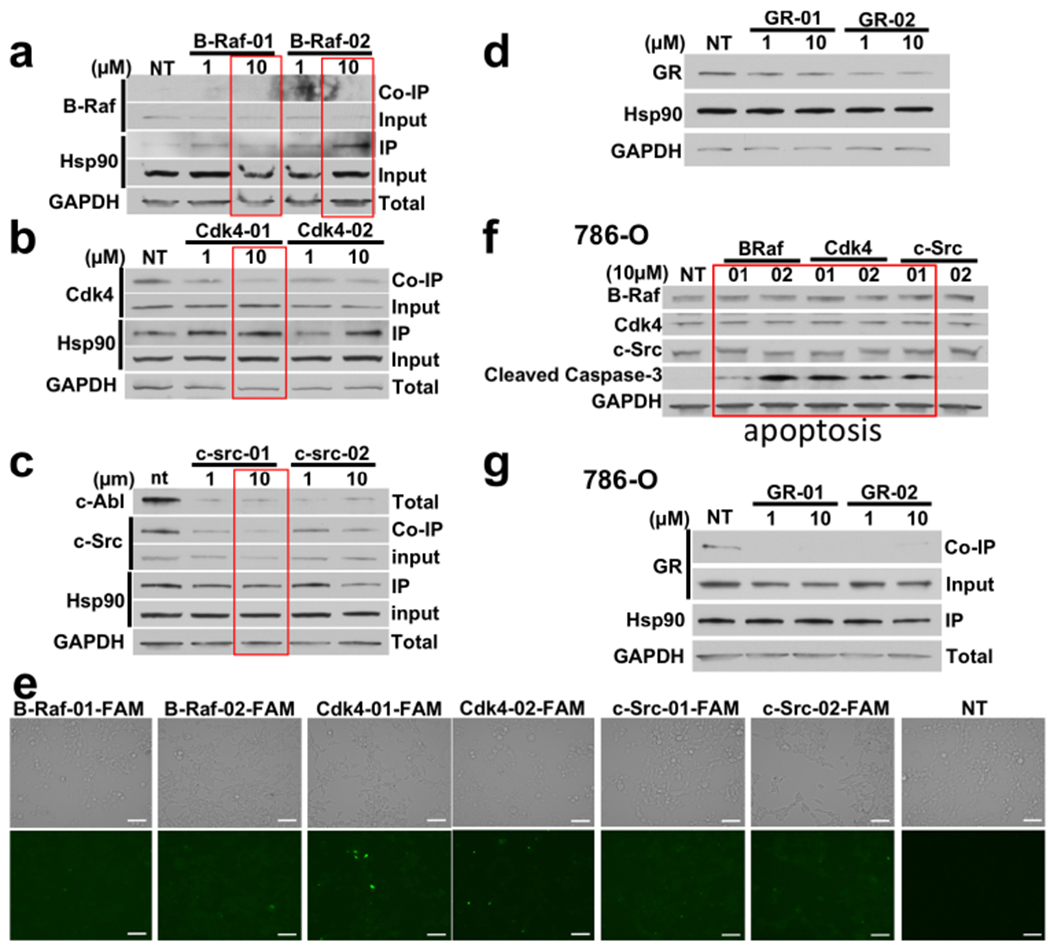
(a) Endogenous Hsp90 was immunopreciptated from HEK293 cells treated with the indicated amounts of B-Raf peptides. Hsp90 binding to B-raf was evaluated by immunoblotting. (b) Binding of Hsp90 and Cdk4 was evaluated by immunoblotting after treatment with the indicated concentrations of Cdk4-01 and Cdk4-02 and immunoprecipitation of endogenous Hsp90. (c) Binding of Hsp90 and c-Src was evaluated by immunoblotting after treatment with the indicated concentrations of c-Src-01 and c-Src-02 and immunoprecipitation of endogenous Hsp90. (d) HEK293 cells were treated with the indicated amounts of GR-01 or GR-02 then examined for total GR protein by immunoblotting. (e) Representative microscopy images of drug uptake in HEK293 cells treated with 10μM FAM-labeled peptides for 24h. Scale bar = 100μm. NT = non-treated (f) Apoptosis in cancer cells was evaluated by immunoblotting for the apoptotic marker cleaved caspase-3 in 786-O cells treated with the kinase peptide mimics. (g) Apoptosis in cancer cells was evaluated by immunoblotting for the apoptotic marker cleaved caspase-3 in 786-O cells treated with the kinase peptide mimics.
Discussion and Conclusions.
We have developed an integrated approach combining ab initio design, synthesis and cellular testing to advance the mechanistic investigation of protein stability. We leveraged this strategy to the design molecules that selectively interfere with Hsp90-mediated protein folding. Our data show that predicted locally unstable regions from the native structures of clients can define the preferential points of interaction with members of the Hsp90 system, and can be mimicked by synthetic peptide agents spanning such regions. These rationally designed chemical tools induce selective dissociation and/or degradation of cognate clients in cells.
Here, we introduce a novel concept of selective targeted protein depletion based on the interference with the protein-protein interactions (PPI) that underpin Hsp90-mediated folding processes. Binding of the synthetic peptide agents to Hsp90 and/or its cochaperones Cdc37 and Aha1 interferes with the formation of the complexes that control protein conformational maturation [27],[7],[28],[18]. It is worth noting that the affinities between Hsp90, cochaperones and clients are weak and only when all the components are correctly assembled do the complexes become functional. The client-mimicking PPI inhibitors can specifically target Hsp90 and/or Cdc37/Aha1, disrupt the binding of the clients to the chaperone complex and significantly reduce their cellular levels, perhaps due to their degradation.
At the atomic level, the EM structure from the Agard lab shows the N- and C- lobes of Cdk4 in complex with Hsp90-Cdc37 are completely separated, with the hinge region including the ɑC helix largely unfolded [20a]. Importantly, the location of the hinge largely overlaps with the substructures at the border between the N- and C-lobes that we predict to be most prone to unfolding (Figure 5). Our model is thus supported by and further corroborates EM-based observations. If the Hsp90 dependency of a kinase is linked to the tendency of the N-lobe and C-lobe to separate in an open state, the interaction surfaces are not random, but should correspond to energetically uncoupled regions that support the conformational reorganization leading to the separation of the two domains. Furthermore, existing data on GR point to regions that are reshaped during the formation of chaperone complexes and that significantly overlap with our designs [25].
Figure 5.
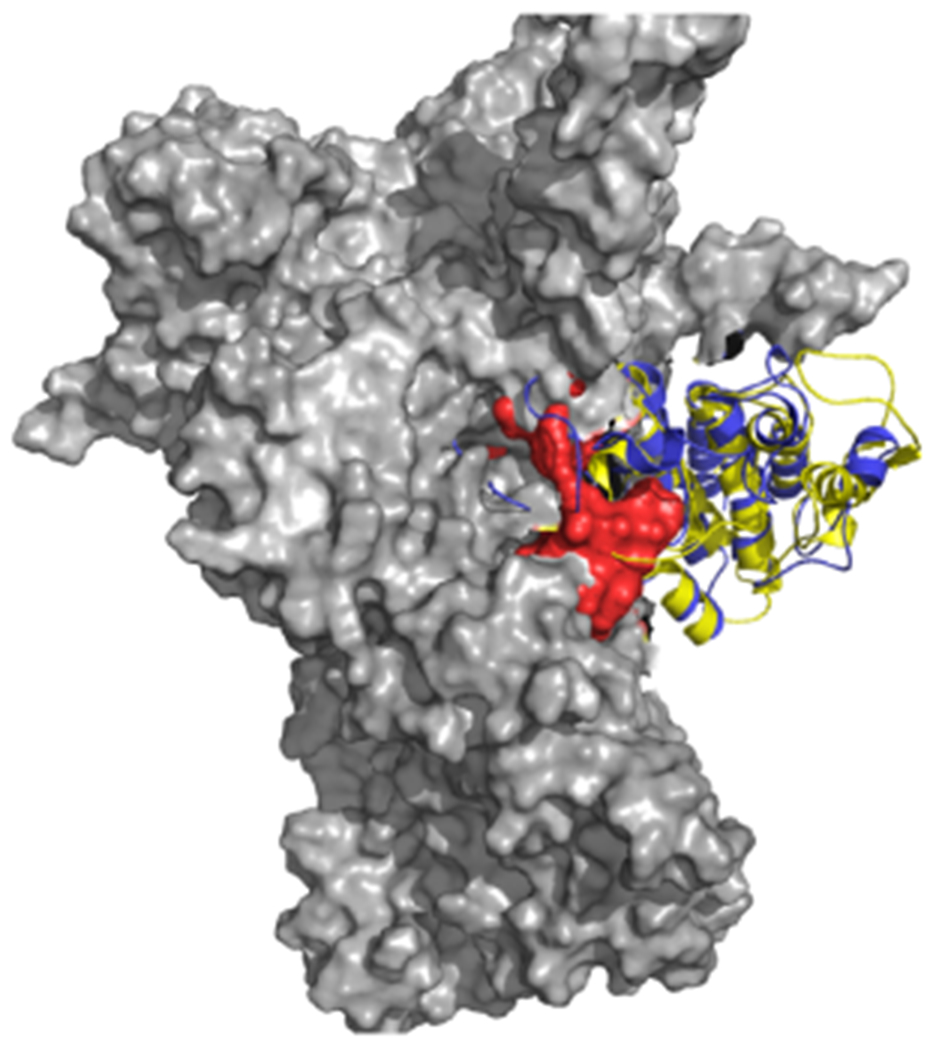
Structural superposition of the Cdk4 structure in complex with Hsp90 and Cdc37 in the original Cryo-EM structure (5FWK) with the structure of the Abl kinase (yellow). For clarity only Hsp90 (gray surface) and Cdk4 (blue) are displayed. The Cdk4 is superimposed to Abl (yellow) by overlapping the C-lobe. The red van der Waals spheres represent the predicted Hsp90 binding region, which appears to optimally trace the surface of Hsp90 and overlap with the unfolding part of Cdk4.
To our knowledge, our work represents the first example of selective chemical targeting of Hsp90 PPIs without significant inhibition or degradation of all chaperone clients. The notion that selective clearance of oncogenic clients can be achieved through perturbation of Hsp90 PPIs has implications for both mechanistic studies and for future therapeutic applications. Mechanistically, selective degradation of specific oncoproteins can be used as a complement to molecular biology to investigate their relevance in signaling pathways. In summary, amino-acid stretches with minimal intra-client coupling can be the drivers of selective degradation of specific Hsp90 client proteins. Synthetic mimics of these regions could possibly have advantages over ATP-competitive Hsp90 inhibitors. The latter compounds unselectively target the entire spectrum of Hsp90 client proteins, causing their destabilization. This lack of selectivity is frequently associated to toxicity, which limit the clinical application of those agents. In contrast, the perturbation of the Hsp90 PPIs with specific clients by our molecules enables a controlled modulation of chaperone networks. Finally, we suggest that the discovery of molecular entities that block chaperone mediated folding mechanisms can provide new opportunities for the development of drugs targeting otherwise undruggable proteins.
Supplementary Material
Acknowledgements
GC acknowledges funding from AIRC (Associazione Italiana Ricerca sul Cancro) through grant IG 20019.This work was partly supported with funds from National Institute of General Medical Sciences of the National Institutes of Health under Award Number R01GM124256 (M.M.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. This work was also supported by SUNY Upstate Medical University, Upstate Foundation (M.M) and Carol M. Baldwin Breast Cancer Research Fund grant (M.M.).
Footnotes
Publisher's Disclaimer: This manuscript has been accepted after peer review and appears as an Accepted Article online prior to editing, proofing, and formal publication of the final Version of Record (VoR). This work is currently citable by using the Digital Object Identifier (DOI) given below. The VoR will be published online in Early View as soon as possible and may be different to this Accepted Article as a result of editing. Readers should obtain the VoR from the journal website shown below when it is published to ensure accuracy of information. The authors are responsible for the content of this Accepted Article.
References
- [1].a) Lavery LA, Partridge JR, Ramelot TA, Elnatan D, Kennedy MA, Agard DA, Mol. Cell 2014, 53, 330–343;; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Richter K, Muschler P, Hainzl O, Buchner J, J. Biol. Chem 2001, 276, 33689–33696;; [DOI] [PubMed] [Google Scholar]; c) Richter K, Buchner J, Cell 2006, 127, 251–253;; [DOI] [PubMed] [Google Scholar]; d) Hessling M, Richter K, Buchner J, Nat. Struct. Mol. Biol 2009, 16, 287–293; [DOI] [PubMed] [Google Scholar]; e) Li J, Buchner J, Biomed. J 2013, 36, 106–117. [DOI] [PubMed] [Google Scholar]
- [2].a) Genest O, Reidy M, Street TO, Hoskins JR, Camberg JL, Agard DA, Masison DC, Wickner S, Mol. Cell 2013, 49, 464–473; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Shiau AK, Harris SF, Southworth DR, Agard DA, Cell 2006, 127, 329–340; [DOI] [PubMed] [Google Scholar]; c) Krukenberg KA, Street TO, Lavery LA, Agard DA, Q. Rev. Biophys 2011, 44, 229–255; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Verba KA, Agard DA, Trends in biochemical sciences 2017, 42, 799–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].a) Partridge JR, Lavery LA, Elnatan D, Naber N, Cooke R, Agard DA, eLife 2014, 10.7554/eLife.03487; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Elnatan D, Betegon M, Liu Y, Ramelot T, Kennedy MA, Agard DA, eLife 2017, 6, e25235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Vilenchik M, Solit D, Basso A, Huezo H, Lucas B, He H, Rosen N, Spampinato C, Modrich P, Chiosis G, Chem. and Biol 2004, 11, 787–797; [DOI] [PubMed] [Google Scholar]; b) Chiosis G, Curr. Top. Med. Chem 2016, 16, 2727–2728; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Shrestha L, Patel HJ, Chiosis G, Cell. Chem. Biol 2016, 23, 158–172; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Workman P, Clarke PA, Al-Lazikani B, Oncotarget 2016, 7, 3658–3661; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Neckers L, Workman P, Clin Cancer Res 2013, 18; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Neckers L, Blagg B, Haystead T, Trepel JB, Whitesell L, Picard D, Cell Stress and Chaperones 2018; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Neckers L, Trepel JB, Clin Cancer Res 2014, 20, 275–277. [DOI] [PubMed] [Google Scholar]
- [5].a) Trepel JB, Mollapour M, Giaccone G, Neckers L, Nat. Rev. Cancer 2010, 10, 537–549; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Garcia-Carbonero R, Carnero A, Paz-Ares L, Lancet Oncology 2013, 14, E358–E369; [DOI] [PubMed] [Google Scholar]; c) Butler LM, Ferraldeschi R, Armstrong HK, Centenera MM, Workman P, Mol. Cancer Res 2015, 13, 1445; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Hong DS, Banerji U, Tavana B, George GC, Aaron J, Kurzrock R, Cancer Treatment Reviews 2013, 39, 375–387. [DOI] [PubMed] [Google Scholar]
- [6].a) Taipale M, Jarosz DF, Lindquist S, Nat Rev Mol Cell Biol. 2010, 11, 515–528; [DOI] [PubMed] [Google Scholar]; b) Taipale M, Krykbaeva I, Koeva M, Kayatekin C, Westover KD, Karras GI, S L., Cell 2012, 150, 987–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Röhl A, Rohrberg J, Buchner J, Trends Biochem. Sci 2013, 38, 253–262. [DOI] [PubMed] [Google Scholar]
- [8].Caplan AJ, Mandal AK, Theodoraki MA, Trends. Cell. Biol 2007, 17, 87–92. [DOI] [PubMed] [Google Scholar]
- [9].Karnitz LM, Felts SJ, Sci. STKE 2007, 2007:pe2022. [DOI] [PubMed] [Google Scholar]
- [10].Keramisanou D, Aboalroub A, Zhang Z, Liu W, Marshall D, Diviney A, Randy W Larsen R Landgraf I. Gelis, Mol. Cell 2016, 62, 260–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zuehlke A, Johnson JL, BIOPOLYMERS 2010, 93 211–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].a) Citri A, Harari D, Shohat G, Ramakrishnan P, Gan J, Lavi S, Eisenstein M, Kimchi A, Wallach D, Pietrokovski S, Yarden Y, J. Biol. Chem 2006, 281, 14361–14369; [DOI] [PubMed] [Google Scholar]; b) Prince T, Matts RL, J. Biol. Chem 2004, 279, 39975–39981. [DOI] [PubMed] [Google Scholar]
- [13].Scroggins BT, Prince T, Shao J, Uma S, Huang W, Guo Y, Yun B-G, Hedman K, Matts RL, Hartson SD, Biochemistry 2003, 42, 12550–12561. [DOI] [PubMed] [Google Scholar]
- [14].Xu WP, Mollapour M, Prodromou C, Wang SQ, Scroggins BT, Palchick Z, Beebe K, Siderius M, Lee MJ, Couvillon A, Trepel JB, Miyata Y, Matts R, Neckers L, Mol. Cell 2012, 47, 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Pricer R, Gestwicki JE, Mapp AK, Accounts of chemical research 2017, 50, 584–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Cesa LC, Mapp AK, Gestwicki JE, Front. Bioeng. Biotechnol 2015, 3, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Thompson AD, Dugan A, Gestwicki JE, Mapp AK, ACS Chem. Biol 2012, 7, 1311–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Schopf FH, Biebl MM, Buchner J, Nature Reviews Molecular Cell Biology 2017, 18, 345–360. [DOI] [PubMed] [Google Scholar]
- [19].a) De Mori GMS, Micheletti C, Colombo G, J. Phys. Chem. B 2004, 108, 12267–12270; [Google Scholar]; b) Morra G, Colombo G, Proteins: Struct. Funct. and Bioinf 2008, 72, 660–672; [DOI] [PubMed] [Google Scholar]; c) Scarabelli G, Morra G, Colombo G, Biophys. J 2010, 98, 1966–1975; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Morra G, Meli M, Colombo G, J. Chem. Theory Comput 2018, 14, 5992–6001. [DOI] [PubMed] [Google Scholar]
- [20].a) Verba KA, Wang RY, Arakawa A, Liu Y, Shirouzu M, Yokoyama S, Agard DA, Science 2016, 352, 1542–1547; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Paladino A, Marchetti F, Ponzoni L, Colombo G, J. Chem. Theory Comput 2018, 14, 1059–1070. [DOI] [PubMed] [Google Scholar]
- [21].a) Marchetti F, Capelli R, Rizzato F, Laio A, Colombo G, J. Phys. Chem. Lett 2019, 10, 1489–1497; [DOI] [PubMed] [Google Scholar]; b) Peri C, Gagni P, Combi F, Gori A, Chian M, Longhi R, Cretich M, Colombo G, ACS Chem. Biol 2013, 8, 397–404 [DOI] [PubMed] [Google Scholar]
- [22].Dalvit C, Flocco M, Veronesi M, Stockman BJ, Comb. Chem. & HTS 2002, 5, 605–611. [DOI] [PubMed] [Google Scholar]
- [23].Dalvit C, Fagerness PE, Hadden DTA, Sarver RW, Stockman BJ, J. Am. Chem. Soc 2003, 125, 7696–7703. [DOI] [PubMed] [Google Scholar]
- [24].Sabbagh JJ, Cordova RA, Zheng D, Criado-Marrero M, Lemus A, Li P, Baker JD, Nordhues BA, Darling AL, Martinez-Licha C, Rutz DA, Patel S, Buchner J, Leahy JW, Koren J, Dickey CA, Blair LJ, ACS Chem. Biol 2018, 13, 2288–2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kirschke E, Goswami D, Southworth D, Griffin PR, Agard DA, Cell 2014, 157, 1685–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Dunn DM, Woodford MR, Truman AW, Jensen SM, Schulman J, Caza T, Remillard TC, Loiselle D, Wolfgeher D, Blagg BSJ, Franco L, Haystead TA, Daturpalli S, Mayer MP, Trepel JB, Morgan RML, Prodromou C, Kron SJ, Panaretou B, Stetler-Stevenson WG, Landas SK, Neckers L, Bratslavsky G, Bourboulia D, Mollapour M, Cell Reports 2015, 12, 1006–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Zierer BK, Rubbelke M, Tippel F, Madl T, Schopf FH, Rutz DA, Richter K, Sattler M, Buchner J, Nat. Struct. Mol. Biol 2016, 23, 1020–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Li J, Richter K, Buchner J, Nat. Struct. Mol. Biol 2011, 18, 61–66. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.