

Abstract
Mammalian cells stably maintain high levels of DNA methylation despite expressing both positive (DNMT3A/B) and negative (TET1–3) regulators. Here, we used wildtype and TET triple knockout human embryonic stem cells (ESCs), generated DNMT3-null as well as TET and DNMT3-null pentuple knockouts, and compared methylation patterns using whole genome bisulfite sequencing (WGBS). The greatest impact on global methylation levels was observed in DNMT3-deficient cells, including reproducible focal demethylation at thousands of normally methylated loci. This demethylation depends upon TET expression and only occurs when both DNMT3s are absent. Dynamic loci are enriched for hydroxymethylcytosine and overlap with subsets of putative somatic enhancers that are methylated in ESCs and can be activated upon differentiation. We observe similar dynamics in mouse ESCs that were less frequenct in epiblast stem cells (EpiSCs) and scarce in somatic tissues, suggesting a conserved pluripotency-linked mechanism. Taken together, our data reveal tightly regulated competition between DNMT3s and TETs at thousands of somatic regulatory sequences within pluripotent cells.
Main
Mammalian genomes generally display high levels of CpG methylation, with the exception of most CpG-dense promoter regions, as well as certain enhancers that are either developmentally poised, actively engaged or decomissioned1. Somatic DNA methylation landscapes are overall stably propagated, with alterations to these global patterns linked to aging and disease2–5. In contrast, developmental regulation of CpG methylation during cellular differentiation generally reflects focal reprogramming in response to transcription factor binding6.
The de novo DNA methyltransferases DNMT3A and DNMT3B are responsible for addition of a methyl group to generate 5-methylcytosine, while DNMT1 is largely responsible for propagating pre-established modifications over DNA replication. In contrast, the Ten-eleven translocation (TET1, TET2, and TET3) enzymes oxidize 5-methylcytosine to generate 5-hydroxymethylcytosine (5hmC) or further oxidized species7,8,9 that can then be either passively or enzymatically removed from DNA10,11.
Both de novo methylation and demethylation are essential for normal development12 and single knockouts result in embryonic (DNMT1, DNMT3B) or early postnatal (DNMT3A, TET3) lethality13–15. Single and combined knockouts of TET1 and TET2 are largely viable but present a wide range of abnormalities16. Loss of function studies have been instrumental in assigning specific roles for each enzyme. For instance, DNMT3B methylates satellite repeats and transcriptionally active gene bodies17,18. DNMT3A appears to act at the periphery of hypomethylated enhancers, canyons and at bivalent promoters19,20 where TET1 and TET2 act in opposition to maintain a hypomethylated state20–25. TET2 may also have additional functions to regulate transcriptional elongation across gene bodies26.
DNMT1, DNMT3A and DNMT3B are expressed in human ESCs, along with TET1, TET2 and TET3, which differs from mouse ESCs, where TET3 is typically not expressed. Human ESCs require the expression of catalytically active DNMT1 to remain viable, while knockout of DNMT3A and 3B gives rise to morphologically normal cells that slowly lose methylation over time18. Conversely, human ESCs that lack all three TET enzymes appear to maintain global methylation levels, but show focal gains and losses of methylation, including hypermethylation of bivalent promoters21. While both DNMT3 and TET-free scenarios have been well described, to date no study has directly compared the impact of losing all demethylating and methylating capabilities on cellular viability, transcription, and differentiation.
To examine the molecular consequences of removing all active regulators of DNA methylation, we generated pentuple knockout human ESCs deficient in both DNMT3A and 3B, as well as all three TETs. Altogether, we compared genome-wide methylation levels across fourteen different wildtype and knockout human ESC lines. Importantly, we find that TET enzymes are focally recruited to thousands of somatic enhancers throughout the genome that nonetheless remain highly methylated in pluripotent cells as long as either DNMT3 is present. Furthermore, we find that TETs have widespread activity throughout the genome that requires DNMT3 expression to maintain steady state methylation levels. Together, our results suggest both focal and global competition for each enzyme class.
Results
TETs drive global demethylation in the absence of DNMT3s
To complement prior studies that investigated DNMT or TET loss in human ESCs, we set out to derive the full range of knockouts and comprehensively assess the interplay between methylating and demethylating enzymes. To this end, we took wildtype (WT) HUES8 ESCs and used Cas9 with single-guide RNAs (sgRNAs) targeted to DNMT3A and DNMT3B to derive HUES8 double knockout (DKO) ESCs that lack all de novo methyltransferase activity. We also obtained previously generated HUES8 ESCs that were depleted of all TET activity; TET1−/−;TET2−/−;TET3−/− triple knockout (TKO) ESCs as well as quadruple knockout (QKO) ESCs which are derived from the TKO with additional knockout of DNMT3B21. We then established a new cell line by further mutating DNMT3A within the HUES8 QKO to generate DNMT3A−/−;DNMT3B−/−;TET1−/−;TET2−/−;TET3−/− pentuple knockout (PKO) ESCs (Fig. 1a, top and Extended Data Fig. 1a–c, Source Data Fig. 1). All knockout cell lines appeared morphologically normal and expressed self-renewal and pluripotency-associated genes, suggesting that DNMT3 and TET expression is not required within the undifferentiated state (Extended Data Fig. 1d–f). We then performed WGBS to investigate the impact on global DNA methylation patterns within these lines, recovering 6.7 million matched CpGs with ≥ 10× coverage across all samples. As expected, HUES8 WT showed high mean CpG methylation (WT = 0.78, Fig. 1a, bottom). Global methylation levels decreased by 0.09 (DKO = 0.69) within our DNMT3-null ESCs after 6 passages compared to a global mean increase of only 0.01 (TKO = 0.79) in our TET-null ESCs. Interestingly, when the de novo methyltransferases were knocked out in TET-null cells, we observed little to no change at passage 6 (QKO = 0.78, PKO = 0.77). These results suggest that the majority of DNA demethylation following DNMT3 depletion depends on TET activity.
Figure 1: Focal demethylation by TET enzymes in the absence of DNMT3s.
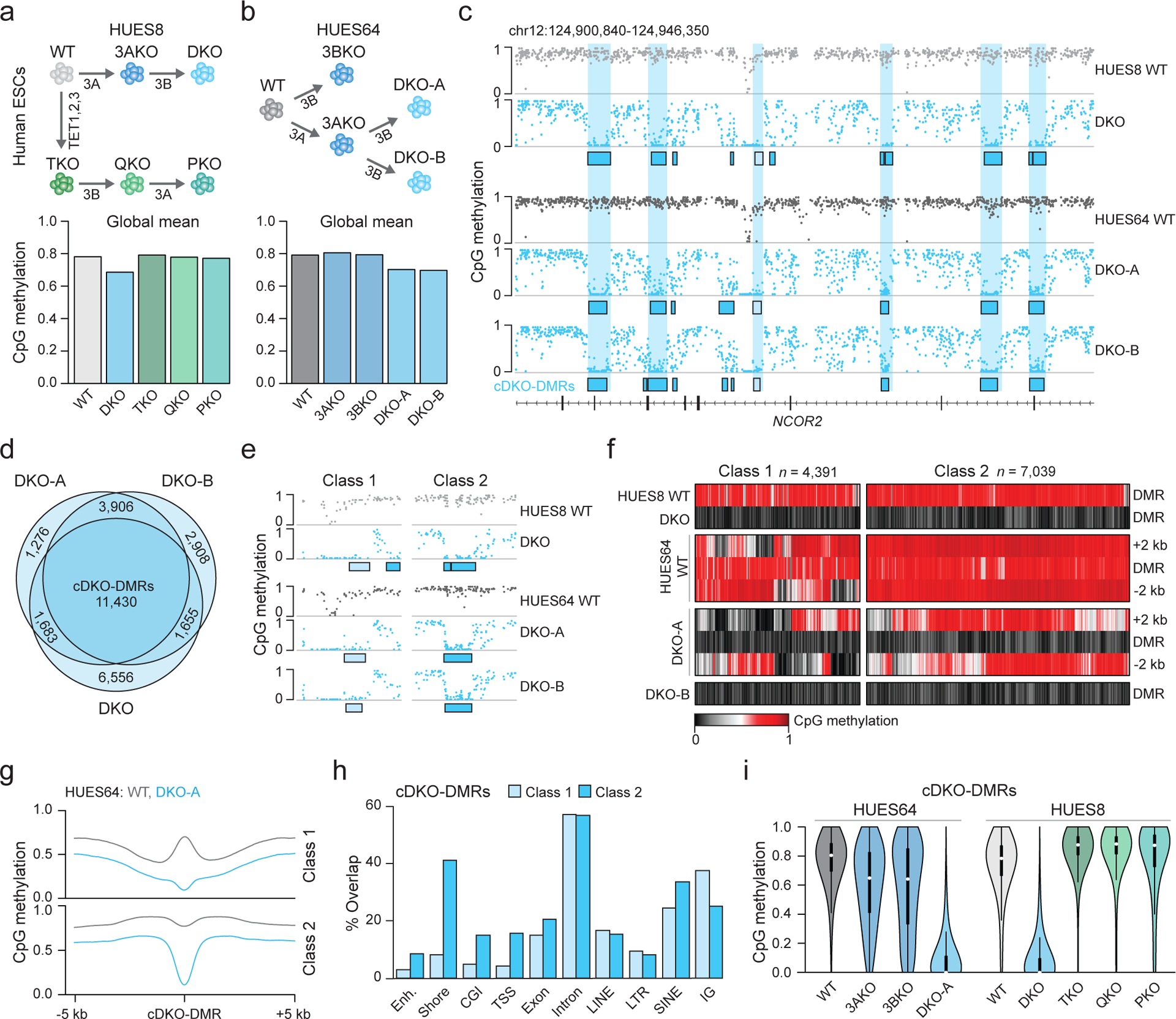
a) Schematic overview. Knockouts are shown under each arrow. 3A = DNMT3A, 3B = DNMT3B. Barplot shows the global mean methylation for each sample using matched CpGs (n = 6,708,067) generated by WGBS. DKO and PKO ESCs were passaged six times.
b) Schematic as in a. Barplot shows the global mean methylation for each sample (CpGs match panel a). DKO-A and DKO-B denote clone A and B (passage 3), respectively. 3AKO and 3BKO are from published datasets18.
c) Genome browser tracks displaying WT and DKO methylation levels, sample-specific DKO-DMRs (boxes) and shared cDKO-DMRs (shaded vertical bars).
d) The overlap between DKO-DMRs hypomethylated in each sample.
e) Genome browser track of selected DKO-DMRs shown in panel c. Class 1 (light shading) border already hypomethylated regions and class 2 (dark shading) are flanked by highly methylated DNA.
f) Heatmap of cDKO-DMRs including 2 kb on either side, separated by class.
g) Methylation composite plot of class 1 and class 2 cDKO-DMRs with 5 kb on either side.
h) The percentage of cDKO-DMRs that overlap with the shown genomic features. Categories are not exclusive. CGI = CpG island, TSS = transcription start site and IG = intergenic. Enhancers are defined from H1 ESCs32.
i) Violin plots for CpGs located within cDKO-DMRs (n = 214,732, 180,019, 138,424, 215,263, 203,504, 182,322, 202,028, 212,215, 145,869 CpGs respectively). Violin plots extend from the data minima to the maxima with the white dot indicating median, thick bar showing the interquartile range and thin bar showing 1.5× interquartile range.
Focal competition between DNMTs and TETs in pluripotent cells
To investigate this methylation loss further, we next sequenced WT and DKO cells from an additional human ESC line (HUES64), including two independent DKO clones (A and B) and compared methylation levels to previously derived18 single DNMT3A−/− (3AKO) and single DNMT3B−/− (3BKO) knockout data (Fig. 1b). The two different male WT ESCs appear comparable with similar global means (HUES64: 0.79 and HUES8: 0.78) and high correlation over 1-kb tiles (Pearson coefficient = 0.93, Extended Data Fig. 1g). Interestingly, while single DNMT3 knockouts at passage 22 remained highly methylated (3AKO = 0.81, 3BKO = 0.79), we again observed an almost immediate methylation decrease to 0.70 after only 3 passages for both HUES64 DKO clones (Fig. 1b). Upon closer inspection of the methylation patterns, we observed two distinct dynamic categories; a limited decrease (0.1–0.2) at ~10% of CpGs across the genome, as well as locally clustered CpGs that display near complete loss of methylation (Fig. 1c, Extended Data Fig. 2a). We were particularly intrigued by these extreme focal changes and proceeded to define differentially methylated regions using stringent criteria (DMRs; Δ > 0.6, P < 0.01, F-test). DKO-DMRs were remarkably consistent across our three DKO lines (Fig. 1c,d, P = 0, hypergeometric test). We therefore defined a “consensus” set of 11,430 cDKO-DMRs that were hypomethylated in all three samples (Supplementary Table 1). cDKO-DMRs maintain consistently low levels (mean WT = 0.749, DKO = 0.086), and average 688 bp in length. Approximately one third neighbored already hypomethylated regions in WT ESCs, such as CpG islands and transcription start sites (termed “class 1”) while the rest were located in otherwise highly methylated regions (intronic or intergenic) with distinct hypermethylated borders in DKO cells (termed “class 2”, Fig. 1e–h). Almost all cDKO-DMRs (93%) were redundantly methylated by either DNMT3A or 3B, and only became fully demethylated in the double knockout (Fig. 1i, Extended Data Fig. 2b). As these results strongly suggest an active demethylation mechanism, we studied methylation levels for cDKO-DMRs across the TET-depleted HUES8 cell lines (TKO, QKO and PKO). In the absence of TETs, the subsequent knockout of first DNMT3B and then DNMT3A did not result in cDKO-DMR demethylation (Fig. 1i). Even after 20 passages, cDKO-DMR methylation levels decreased only minimally in PKO cells (Extended Data Fig. 2c). Furthermore, the areas surrounding many class 1 cDKO-DMRs gain methylation in TKO cells, further implicating local TET activity in the focal demethylation (Extended Data Fig. 2d). Together, our data highlight a set of distinct and highly methylated regions in human pluripotent cells that rely on DNMT3A or 3B for their methylation and undergo rapid TET-mediated demethylation in their absence.
TETs display continuous activity at cDKO-DMRs
To verify that cDKO-DMRs depend on and continue to attract TET activity, we utilized PiggyBac transposition to reintroduce exogenous TET1 (the short isoform, hereafter referred to as TET1s), TET2 and TET3 into PKO cells and measured global methylation levels using WGBS (Fig. 2a). We observed a clear reduction in global mean methylation from 0.73 in PKO cells at passage 20 to 0.61, 0.59 and 0.54 after ectopically expressing each protein, while control cells transfected with only the transposase enzyme remained stable (mean = 0.70, Fig. 2b). Reintroducing any TET enzyme largely restored aberrantly hypermethylated regions in TKO cells and induced rapid cDKO-DMR demethylation, demonstrating that their targeting capability is highly redundant and preserved even after extended periods without TET-activity (Fig. 2c–e, Extended Data Fig. 2e). From a mechanistic standpoint, it is worth noting that the TET1s isoform lacks a CXXC domain for recruitment to unmethylated CpG dense regions27,28,29 yet we found no difference between TET1s and TET2 in a direct comparison, supporting a CXXC domain-independent recruitment mechanism (Extended Data Fig. 2f). Ongoing dynamics between TETs and their opposing enzymes at cDKO-DMRs is also supported by published ChIP-seq data, which show enrichment for both DNMT3B and TET1 in WT ESCs, although enrichment appears broader than the rather narrow and well-defined cDKO-DMRs (Extended Data Fig. 2g). Finally, we performed whole genome oxidative bisulfite sequencing (oxBS) and found significant local enrichment of 5hmC in WT cells (mean cDKO-DMRs = 0.07, background = 0.01, P = 2.2 × 10−16, two-sided t test, t = 231.8, df = 255,010, Fig. 2f, Extended Data Fig. 2h). Put together, these results confirm that TET enzymes are continuously recruited to thousands of loci throughout the genome and can direct rapid, focal demethylation if DNMT3 activity is absent.
Figure 2: Re-expression of TET enzymes demethylated PKO cDKO-DMRs.

a) TET1s, TET2 and TET3 were reintroduced into PKO cells using PiggyBac transposition. Control transfection was performed with only the transposase (PBase). A schematic diagram depicts the experimental design with the constructs shown below, w = weeks, Ins = insulator sequence.
b) WGBS-based mean methylation barplots for matched CpGs. P20 = passage 20.
c) Genome browser tracks across a 39-kb region of the LRP1 locus for HUES8 WT and seven of the modified HUES8 lines. Re-expression of TET1s, TET2 or TET3 in PKO cells causes global and specific loss of methylation at cDKO-DMRs (blue boxes).
d) Violin plots displaying methylation levels for CpGs within cDKO-DMRs (n = 176,669, 186,438, 155,912, 180,233, 128,709 CpGs respectively). Violin plots extend from the data minima to the data maxima with the white dot indicating median, thick bar showing the interquartile range and thin bar showing 1.5× interquartile range.
e) Composite plot of mean methylation levels for each class of cDKO-DMR, including 5 kb on either side for PKO cells at P20, control cells as well as the three TET rescue lines.
f) Hydroxymethylcytosine (5hmC) levels in WT HUES64 cells shown as distance from class 1 or class 2 cDKO-DMR center, CGIs or active ESC (H1) enhancers (enh).
A subset of cDKO-DMRs overlap with the 5’UTRs of LINEs
The robust and precise manner by which our cDKO-DMRs change their methylation state suggests that they may overlap with regulatory elements. Notably, a small subset includes the 5’ UTR of L1Hs or L1PA long interspersed nuclear elements (LINEs), which act as their functional promoter and generally display evolutionary age and CpG density-dependent demethylation following DNMT3 knockout (Fig. 3a,b, Extended Data Fig. 3a,b). These regions also show higher than expected 5hmC enrichment in WT ESCs, supporting continual TET recruitment (Fig. 3b). To investigate this further, we focused on the human-specific L1Hs elements and used the MinION platform from Oxford Nanopore to generate long reads (mean length: 8.9 kb) that enable unambiguous mapping and assessment of LINE-specific methylation states for HUES64 WT and DKO ESCs, as well as HUES8 WT, TKO and PKO ESCs (Extended Data Fig. 3c). In WT cells, we found that 7% L1Hs 5’UTRs (n = 207 full length covered in each sample) were already hypomethylated (Fig. 3c). Despite often not passing our stringent DMR criteria, 91% of WT methylated elements were demethylated following DNMT3A and DNMT3B knockout (Fig. 3c). Demethylation was dependent on TET expression, with only a small group showing loss in PKO cells (Fig. 3c). While this analysis allowed us to partition sequences that showed alternative methylation dynamics, we could not pinpoint any single-nucleotide polymorphism that was consistent with methylation state, suggesting a more complex mechanism behind DNMT/TET recruitment (Extended Data Fig. 3d,e). Of note, the 17 L1Hs 5’UTRs that remained methylated in DKO ESCs have a lower CpG density and GC content, which may indicate that higher CpG density favors TET-mediated recruitment and demethylation (Extended Data Fig. 3f). Cumulatively, it appears that LINE 5’UTRs are highly sensitive to DNMT3/TET expression and can switch methylation states rapidly, yet the methylated state is favored for most elements in DNMT3-expressing pluripotent cells.
Figure 3: TETs demethylate LINEs and somatic enhancers following DNMT3 loss.

a) The proportion of class 1 or class 2 cDKO-DMRs that overlap with selected features.
b) Genome browser tracks showing cDKO-DMR overlap within the 5’UTR of a human LINE (L1PA3) element. Fr = frontal. DKO-A is shown at passage 3 here and for all panels.
c) Heatmap displays methylation levels for L1Hs 5’UTRs as measured by Oxford Nanopore sequencing separated into groups by the methylation state. Loess-smoothed regression for methylation levels across full length L1Hs elements in each group are shown on the right, with the 5’UTR shaded in grey.
d) Genome browser tracks show extension of hypomethylation at an unmethylated active ESC enhancer following DNMT3 loss.
e) Heatmap of active ESC enhancer methylation levels, in HUES64 WT and DKO-A cells, including 5 kb on either side.
f) Genome browser tracks showing cDKO-DMR overlap with putative tissue-specific enhancers in the frontal lobe.
g) Binary heatmap of cDKO-DMR overlap with tissue-specific enhancers. Each row represents the enhancer set of a different tissue or cell type. Rows for H1 ESCs, frontal lobe and CD19+ B-cells are highlighted. Dark shading indicates overlap with a cDKO-DMR. Hierarchical clustering of rows and columns was performed using the complete linkage method.
h) Frontal lobe enhancers separated by their methylation status in WT, DKO-A and the frontal lobe.
i) Composite plots of H3K4me1 and H3K27ac enrichment across cDKO-DMRs in comparison to active ESC (H1) enhancers.
Somatic enhancers are targeted by DNMT3 and TET activity in pluripotent cells
While TETs can be recruited to LINE 5’UTRs30, they more frequently localize to promoters or active enhancers (Extended Data Fig. 4a), where they maintain the hypomethylated state23. Indeed 7% of cDKO-DMRs neighbor hypomethylated active enhancers, supporting previous evidence that DNMT3s protect enhancer boundaries from extended hypomethylation31,20 (Fig. 3a,d,e). Given a canonical role for TETs at active enhancers, we reasoned that cDKO-DMRs may also represent regulatory elements that only undergo demethylation in somatic cells. Strikingly, 85% of cDKO-DMRs overlap with at least one previously defined tissue-specific putative enhancer32 and are specifically demethylated in the associated cell type (Fig. 3a,f,g, Extended Data Fig. 4b,c; controlled background overlap ~35%). Interestingly, while cDKO-DMRs were almost always somatic enhancers, the inverse was not true: many somatic enhancers are either already hypomethylated in ESCs, remain highly methylated in their associated tissue, or do not display TET-dependent demethylation in ESCs despite losing methylation in other developmental contexts (Fig. 3h, Extended Data Fig. 4d). To explore DMR stability, we further passaged HUES64 DKO clone A cells and performed WGBS. By passage 28, we identified an increased number of 59,618 DKO-DMRs, 79% of which overlapped with putative tissue-specific enhancers (Extended Data Fig. 4e). Nonetheless, around one third of somatic enhancers remained methylated in late passage DKO cells (Extended Data Fig. 4f). To examine whether the ability to demethylate an enhancer is linked to developmental timing (when they become activated during development), we utilized a recently published dataset detailing enhancer activation and methylation dynamics over nine stages of pancreatic islet differentiation from HUES64 ESCs33 (Extended Data Fig. 4g). Interestingly, a similar proportion of enhancers at every stage of differentiation overlapped with DKO-DMRs, with a slightly higher frequency at more terminal differentiation stages (Extended Data Fig. 4g–i). Although speculative, this suggests that a subset of both embryonic and adult tissue-specific enhancers may exist in a state that depends on the continuous and opposing functions of DNMT3s and TETs.
Loss of DNA methylation at somatic enhancers in ESCs affects gene expression
We next wondered how TETs are recruited to this set of highly methylated somatic enhancers and what the impact of losing active DNMT3 recruitment could be. cDKO-DMRs have a greater than background CpG density (3.2%), yet this is not classifying since the majority of 1-kb tiles with matched CpG density do not lose methylation in DKO cells (Extended Data Fig. 5a,b). When we looked for associations with selected epigenetic features of cDKO-DMRs in WT ESCs, we found that they were generally more enriched for open chromatin and H3K4me1, but not H3K27ac, which is commonly associated with transcriptionally engaged “active” enhancers (Fig. 3i, Extended Data Fig. 5c–e). Although frequently overlapping, H3K4me1 enrichment was again not sufficient to predict cDKO-DMRs (Extended Data Fig. 5f). Further, only ~10% H3K4me1-enriched somatic enhancers were also enriched for H3K27me3, indicating absence of a classically “poised” chromatin state (Extended Data Fig. 5g). Furthermore, we found no enrichment of ESC enhancer-associated transcription factors, though we could confirm that these regions become bound by tissue-specific transcription factors after differentiation (Extended Data Fig. 5h–j). We then expanded our inquiry to other ESC-expressed TFs, including eight ChIP-seq datasets from WT HUES64 ESCs34 and 63 ChIP-seq datasets encompassing all ENCODE-probed H1 ESC factors (Extended Data Fig. 5k,l). However, we did not find any notable enrichment within cDKO-DMR boundaries, nor did we observe a consistent sequence motif for any known factor (Supplementary Table 2). Currently, we therefore hypothesize that TET recruitment may either depend upon a TF not included in the above set, noncoding RNA35, or could involve a more complex regulatory mechanism that remains to be elucidated.
Finally, we explored whether we could map DKO-DMRs to genes and assess potential regulatory roles. On average, cDKO-DMRs were located 48 kb from transcription start sites and could be assigned by proximity to 6,594 genes, which does not allow for meaningful pathway or interactome analysis. Instead, we performed RNA-seq to discern whether loss of methylation at these somatic enhancers affects gene expression. Indeed, late passage DKO cells significantly upregulated genes associated with differentiation (n = 2,455, FDR < 0.01, fold change ≥ 2), such as EOMES, SOX17, TGFB2, MSX2, SOX1 and SIX1 (Extended Data Fig. 5m, and Supplementary Table 3). Although we cannot distinguish direct from indirect effects, 526 of these genes already showed significant differential expression by passage 6. Distance to the nearest passage 28 DKO-DMR was significantly lower for genes that became deregulated and two thirds of differentially expressed genes were associated with a passage 28 DKO-DMR (Extended Data Fig. 5m,n). This indicates a potential link between demethylation of the targeted regulatory element to upregulated gene expression.
cDKO-DMRs are conserved and associated with pluripotency
To explore the conservation of this targeted dynamic methylation, we mutated Dnmt3a and Dnmt3b within mouse EpiSCs as these represent the closest developmental and molecular analog to human pluripotent cells (Extended Data Fig. 6a,b, Source Data Fig. 2). We performed WGBS for WT and DKO cells at passage 4 and identified 3,888 DMRs that showed similar properties to human DKO-DMRs, with 82% overlapping putative somatic enhancers36. While the number, precise sequence and co-ordinates did not directly map between species (as is expected for regulatory elements37, Extended Data Fig. 6c), mouse DKO-DMRs appeared in remarkably similar locations and in proximity to orthologous genes (Fig. 4a, Extended Data Fig. 6d,e). Thus, both the mechanism of methylation turnover at somatic enhancers and the regulatory logic of these regions appears largely conserved.
Figure 4: Targeted demethylation in the absence of DNMT3 appears conserved in mice.

a) Genome browser tracks for an orthologous locus in human ESCs and mouse EpiSCs show the conserved presence of DKO-DMRs following DNMT3 double knockout.
b) A comparison of DKO-DMRs identified in mouse DKO ESCs, DKO EpiSCs and a range of DKO-like Dnmt3a knockout mouse tissues. The number of DMRs derives from one experiment per condition. Homozygous Dnmt3a knockout mice (−/−) were generated by crossing Dnmt3a heterozygous mice (+/−). A representative photo of a WT and knockout mouse at day 23 is shown to highlight the expected runted phenotype in line with prior studies14. To the right, the number of DMRs identified for each cell type or each tissue is shown. Pluri. = Pluripotent.
As somatic cells also express DNMTs and TETs (Extended Data Fig. 7a,b), we were curious whether the DNMT3/TET interplay at somatic enhancers is exclusive to pluripotency, or whether it is preserved after differentiation. We therefore examined a range of systems that lack DNMT3 activity under various stages of developmental potential. These include mouse DKO ESCs cultured in serum/LIF, which represent a more naïve pluripotent state than the EpiSCs, and remain highly methylated (Extended Data Fig. 8a,b, Source Data Fig. 3). We also examined tissues from homozygous Dnmt3a−/− mice under the premise that most somatic cells do not express catalytically active DNMT3B38,39 and therefore loss of DNMT3A would create “DKO-like” cells that lack de novo methyltransferase activity (Fig. 4b). We then performed WGBS on WT and DKO ESCs at passage 4 as well as brain, colon, liver and lung tissue from 8 day old WT and Dnmt3a−/− mice (Fig. 4b). Compared to EpiSCs, we identified 10-fold more DKO-DMRs for ESCs (n = 63,971) and 10-fold fewer in somatic tissues (brain = 146, colon = 511, liver = 318, lung = 252, Extended Data Fig. 8c–g). The dynamic regulation of cDKO-DMRs therefore appears restricted to pluripotent cell states, with progressively fewer TET-targeted loci as development proceeds. In each case, 73–82% of the DMRs overlapped with mouse tissue-specific enhancers as defined previously36. However, somatic DKO-DMRs preferentially favor the class 1 subgroup located near CpG islands and shores, suggesting that, after differentiation, TETs largely function to preserve the boundary of already unmethylated regions (Extended Data Fig 8h,i). To complement our mouse data, we reanalyzed WGBS data from HUES64 WT and 3AKO human ESCs differentiated into post-mitotic motor neurons40, where cells lose protein expression of DNMT3B by day 2 to generate the “DKO-like” state in viable 3AKO cells (Extended Data Fig. 8j). Although cells had been without DNMT3 activity for ~12 days, cDKO-DMRs remained highly methylated despite continuous TET1–3 expression, further hinting that the key players involved in this process may be exclusively expressed in the pluripotent state (Extended Data Fig. 8k,l).
TET enzymes also demethylate broadly throughout the genome
After establishing that DNMTs and TETs regulate a subset of somatic enhancers in a pluripotency-specific manner, we returned to the more gradual background demethylation observed in both the DKO and PKO contexts (Extended Data Fig. 2a,c). To avoid confounding our global measurements with the targeted DNMT3 or TET activity described above, we excluded all low stringency DMRs identified between any pair of samples from further analysis (n = 238,497, Extended Data Fig. 9a–d) as well as all somatic enhancer regions32. Notably, DNMT3 loss in the presence or absence of TETs still resulted in global average methylation decreases of 0.0028 and 0.0037 per passage respectively (Fig. 5a, Extended Data Fig. 9e,f). Subsequently, we estimate the fidelity of DNA methylation patterns to be 99.75 and 99.64 per cell cycle (estimated using 6 days/passage and 4 days/passage with population doubling time of 28.8 h and 24 h for DKO and PKO cells respectively).
Figure 5: TET enzymes also have widespread global activity.

a) Mean methylation for matched CpGs (n = 7,905,649) in DKO and PKO ESCs. Background CpGs represent CpGs outside of defined enhancers or cDKO-DMRs.
b) Methylation difference over passaging for DKO and PKO cells (left and center) and WT hydroxymethylcytosine (5hmC) levels (right). CpGs are separated according to replication timing taken from previous repli-BS-seq data: S1 is early replicating and enriched for euchromatin, while S6 is late replicating and enriched for heterochromatin.
c) 5hmC levels in WT cells across different genomic features, CGI = CpG island, IG = intergenic.
d) Genome browser tracks of representative high and low 5hmC enrichment in WT cells. Genomic regions with higher 5hmC in WT lose more methylation in DKO cells.
e) For 1-kb tiles with different mean 5hmC levels, the difference in methylation between WT and DKO, TKO and WT or PKO and DKO cells is displayed. Boxes display the interquartile range while the bold line shows the median and whiskers extend to 1.5× the interquartile range.
f) Human ESCs were differentiated into motor neurons (MNs) and cultured for 60 days (d). WGBS and oxBS were performed at d16 and d60 and the respective methylation and 5hmC levels across different genomic features are displayed. TSS = transcription start site.
g) Genome browser tracks display methylation and 5hmC levels for MNs at d16 and d60. An increase in 5hmC is observed over the transcribed FOXP1 gene body, but also within the nearby intergenic region.
Methylation loss within heterochromatin has been suggested to emerge from low DNMT1 fidelity and mitotic division2. In support of this model, we observed preferential loss of methylation within heterochromatic regions for PKO cells that only express DNMT1, however, we also see a large decrease in euchromatic regions for DKO cells, implying additional contributions from global TET activity (Fig. 5b, Extended Data Fig. 9g). Upon closer inspection, we note low 5hmC signal distributed throughout the genome of WT ESCs, including at inert intergenic DNA, that exists at higher levels within euchromatin (Fig. 5b,c). Accordingly, methylation in regions with high WT 5hmC levels decrease the most in the absence of de novo DNMTs (Fig. 5d,e).
We next differentiated WT HUES64 ESCs to post-mitotic motor neurons, where 5hmC cannot be passively diluted over division, to more specifically track TET engagement and catalytic activity. After 60 days of culture, 5hmC levels were enriched within gene bodies (particularly for expressed genes) and intergenic loci, with the greatest increase ocurring within inert chromatin, supporting widespread oxidation by TETs (Fig. 5f,g, Extended Data Fig 10a–c). Therefore, in motor neurons as well as ESCs, TETs can oxidize methylcytosine broadly throughout the genome. This activity results in continuous turnover of cytosine modification and a global methylation decrease when not countered by DNMT3A or DNMT3B. Given this paradigm, one might expect ESCs to show a global methylation increase upon TET loss, as this would shift the balance in the favor of DNMT3 activity. Counterintuitively, and as previously noted in cancer cells41, a slight decrease was also observed in cells lacking TETs compared to matched WT (Extended Data Fig. 10d). Although speculative, this may be associated with the increased growth rate of TKO cells compared to WT, which could also impact methylation maintenance (Extended Data Fig. 10e).
To understand whether competing activity of DNMT and TET can contribute to methylation heterogeneity, as previously suspected for H3K4me1-enriched enhancers42, we performed hairpin bisulfite sequencing to measure symmetrical cytosine methylation on CpG dyads of the same molecule. We found fewer hemi-methylated dyads in the absence of de novo DNMTs and TETs (4.48% in PKO compared to 8.89% in WT cells), indicating that DNMT and TET act together to create epigenetic variation at single loci (Extended Data Fig. 10f–j). Furthermore, the stochasic nature of TET activity throughout the genome also contributes to methylation heterogeneity within populations of cells (Extended Data Fig. 10k,l).
Discussion
Here we report a comprehensive analysis of a series of human ESC lines devoid of DNMT3 and/or TET activity. We find that TETs are focally recruited to thousands of somatic enhancers that are highly methylated in the pluripotent state, which results in local 5hmC enrichment and targeted demethylation in the absence of countering DNMT3 activity.
The TET-dependent focal loss of methylation at these somatic elements upon loss of DNMT3 activity is intriguing for many reasons, including their conservation and relative stability. For example, after 28 passages, DNMT3 DKO cells accumulate additional DKO-DMRs, but also preserve the local architecture of immediately demethylated regions, i.e. DMRs maintain distinct borders without further erosion. This suggests an initially dynamic switch that is well defined in cis and can be maintained over extended periods. It remains to be determined what factors are responsible for this targeted recruitment of TETs in ESCs. We found that the three TET enzymes can act redundently and that recruitment appears to be independent of the TET CXXC domain. Although motif as well as preliminary TF binding enrichment analyses did not elucidate a clear candidate recruiting factor, we cannot rule out potential TF-mediated recruitment in general as many factors are yet to be interrogated by ChIP-seq in ESCs and have unknown binding motifs. Whatever the ultimate mechanism, it is most likely to be pluripotency-associated, as we observe no such loss of methylation at these loci in somatic cells similarly depleted of DNMT3 activity. It is also possible that there is no specific recruitment factor. Previous work shows that all three TETs can be ubiquitinated by CLR4, which is predicted to induce a conformational change that promotes direct TET binding to DNA43,44. It is therefore also possible that post-translational modification or unknown mechanisms may regulate TET recruitment or activity at certain target sites. Proteomics and biochemical strategies may be useful to explore these additional possibilities.
The focal competition between DNMTs and TETs at enhancers with somatic functions suggests a novel type of epigenetically regulated state within pluripotent cells. Around one third of somatic enhancers are known to attract analagous competing activities, but are instead kept in an unmethylated state in ESCs that is instructed by histone modifications45–47. Here, we now describe that another third remain highly methylated despite a similar recruitment of DNMT3s and TETs. For this set, which we could only identify by direct comparison between ESCs lacking DNMT3 expression with and without TETs present, the central question becomes why thousands of somatic enhancers actively recruit TET enzymes despite a default methylated state.
We hypothesize that this strategy could facilitate rapid switching of methylation states that may be required to eliminate DNA methylation efficiently during early cell fate transitions. For instance, active regulation may support rapid changes in cell cycle dependent control that are required for exit from pluripotency towards the early germ layers48,49. Simultaneously, these activities would also enable robust activation upon exposure to the appropriate differentiation signals. Based on our limited expression analysis, we speculate that keeping these regions methylated in pluripotent cells may be important to prevent binding of ESC-expressed or early developmental TFs that would otherwise lead to unwanted expression of differentiation associated genes, or conversely that methylated or hydroxymethylated cytosines may support binding of certain TFs50. Carefully designed experiments will be required to further tease these possible models apart and assign biological importance of these sites.
We further note that the pluripotent-specific nature of DNA methylation-based enhancer regulation found in our study suggests that some epigenetic states may be more robustly controlled within certain developmental windows than in others. Counterintuitively, the dynamic interplay of opposing DNMT and TET activities at these sites may ensure that they are more faithfully maintained in a DNA methylated state during a period where they might otherwise influence the developmental outcome. In this context, it will be interesting to explore whether the similar recruitment observed at LINEs represents a hijacked feature of somatic enhancer regulation to trigger activation during pluripotency-related stages, such as the germline, where DNMT3 activity is acutely downregulated. This early shift in genomic regulation would allow temporal L1Hs upregulation prior to alternative means of suppression (such as by H3K27 methylation51). Such cooption of an endogenous, pluripotency-specific mechanism would be analogous to what has been described for endogenous retroviruses52,53.
We also observe genome-wide activity of DNMT3s and TETs that causes a methylation shift after their loss. At this broader scale, we found increased TET-dependent DNA demethylation within euchromatin, which intuatively suggests that methylated cytosine located in open chromatin is more amenabe to stochastic oxidation. It seems reasonable that targeted TET activity at DKO-DMRs is desirable, while global demethylation represents an unavoidable off-target activity due to the proximity of enzymes to their substrate. Of course, background oxidation comes at a high cost that requires continuous de novo activity to maintain a highly methylated mammalian genome. From this view, a better understanding of these counteracting forces will be also relevant to understand various disease states. For example, DNMT3A is frequently mutated in cancer54 and impaired in several neurodegenerative diseases55, two somatic contexts where catalytic DNMT3B is not expressed and cells therefore lack all de novo activity.
Taken together, our results highlight focal competition between opposing DNA methylating and demethylating enzymes that uncovers yet another unique layer of epigenetic regulation within pluripotent cells.
Online methods
We have complied with all relevant ethical regulations. All human ESC work was approved by Harvard University’s ESCRO and the mouse work by the IACUC (#28–21). Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Cell culture
Human ESCs were grown in feeder-free conditions using Geltrex (Thermo Fisher Scientific) and mTeSR (STEMCELL Technologies) and split every 4–5 days using StemPro Accutase Cell Dissociation Reagent (Thermo Fisher Scientific). Mouse ESCs were cultured on mitomycin-C-treated MEFs in standard conditions (knockout DMEM containing 15% fetal bovine serum, 100 μM non-essential amino acids, 1 mM L-Glutamine, 1× penicillin-streptomycin, 0.1 mM 2-Mercaptoethanol, and 103 units of ESGRO LIF) and split every 3 days using TrypLE (Thermo Fisher Scientific). E3 GOF18-EGFP male EpiSCs (a generous gift from the Schöler laboratory) were cultured feeder-free as described previously56 (MEF-CM consisting of knockout DMEM supplemented with 20% knockout serum replacement, 100 μM non-essential amino acids, 1 mM L-Glutamine, 1× penicillin-streptomycin, 10 ng/μl FGF2, 0.1 mM 2-Mercaptoethanol) and were split in clumps every 3 days using collagenase type IV (STEMCELL Technologies). All lines were regularly tested and found negative for mycoplasma contamination.
Generation of Dnmt3a/3b double knockout ESCs and EpiSCs
To create Dnmt3a and Dnmt3b double knockout mouse ESCs and EpiSCs, we targeted both genes simultaneously by transfecting the cells with px458 (Addgene plasmid #48138) containing guides targeting the highly conserved PC motif in the catalytic domains in ESCs, and guides targeting both the PC motif as well as early coding exons in EpiSCs (Supplementary Table 4). Knockouts were verified by genotyping and Western blot. Mouse ESCs were transfected using the Amaxa 4D nucleofector X-Unit (Lonza) according to manufacturer’s guidelines, and EpiSCs were transfected using Xfect Transfection Reagent (Clontech) according to manufacturer’s guidelines.
Nanopore Sequencing and data processing
Ultra-high molecular weight genomic DNA (gDNA) was extracted from cells using a phenol-chloroform extraction. gDNA was then sheared to an average length of 20-kb fragments using g-TUBEs (Covaris) following manufacturer’s recommendations. Sheared gDNA was then size selected using the BluePippin High Pass Plus Gel Cassette (Sage Science). Sheared and purified gDNA was then used to prepare Nanopore libraries using the Ligation Sequencing Kit 1D (SQK-LSK108) following manufacturer’s guidelines. All MinION sequencing runs were processed using the Nanopype pipeline57 (v0.9.0). The basecaller Guppy (v3.1.5) was used with the r9.4.1 fast configuration. Quality filtering was disabled for any basecalling. Alignments were made against human genome hg38 using NGMLR58 (v0.2.7) with the ont parameter preset. Based on the same alignments, genome-wide methylation tracks were obtained from nanopolish (v0.11.0)59 and structural variations were called using Sniffles58 (v1.0.10). Nanopolish was used with default parameters while filtering for CpGs with > 5× coverage. Sniffles was configured to report variations of at least 500 bp, supported by at least five reads from read fragments longer than 2 kb (−l 500 −s 5 −r 2000). From the Sniffles output, L1Hs elements were identified as homozygous, heterozygous or missing and only those that were homozygous and at least 6 kb in length were used for subsequent analysis.
Generation of HUES8 mutant lines
PKO cells were obtained by disrupting the DNMT3A catalytic domain (exon 19) in HUES8 QKO cells using CRISPR-Cas9 (sgRNA listed in Supplementary Table 4). The sgRNA was cloned into the pSPCas9n(BB)-GFP (Addgene px461) vector and transfected into HUES8 QKO cells with FuGene HD (Promega) followed by sorting for green fluorescent protein (eGFP) signal for selection. After 10–14 days of culture, single cell clones were divided in a 1:3 ratio for maintenance and validation of gene editing respectively.
HUES8 DNMT3A−/−;DNMT3B−/− double knockout mutants were derived by first targeting DNMT3A using the same method and sgRNA described for PKO cells above. After confirming the DNMT3A knockout, the DNMT3B catalytic domain (exon 19) was targeted using three different DNMT3B gRNAs (Supplementary Table 4) cloned into the px458 vector and transiently transfected into the HUES8 DNMT3A−/− cells. To assess correct knockout mutants, the PCR fragments around each sgRNA target region from individual clones were subcloned into pCR™Blunt II-TOPO® vector (Invitrogen) followed by Sanger sequencing with M13 primers or sequencing primers. In addition, western blot analyses were performed with whole cell lysate.
TET piggybac plasmid construction
The coding sequences for FLAG-TET2-P2A-GFP (GenScript, custom synthesis of TET2 (NM_001127208.2) in pcDNA3.1(+)-P2A-eGFP vector) and FLAG-HA-TET3 (Addgene #49446) were excised from their plasmids by restriction digest of 5 μg plasmid with ApaI and AflII, or KpnI-HF and XbaI respectively (all NEB, 1 μl each was digested at 37°C for 120 min). Subsequently, the corresponding fragments were gel-purified (Qiagen).
The target vector pPiggyBac MCS (a modified version of plasmid #SPB-007, Transposagen, with additional cloning sites) was digested in the same manner and additionally dephosphorylated (NEB, 1 μl AP, 37°C for 20 min) prior to gel purification. The coding sequences were cloned into pPiggyBac MCS using the Quick Ligation kit (NEB) in a ratio of 1:3 and transformed into Escherichia coli (NEB stbl TET2, DH5α (Meissner laboratory) for TET3). Plasmids were isolated (Qiagen) and the correct sequence of the constructs was confirmed by Sanger sequencing.
The T2A-BFP sequence was obtained from pU6-sgRNA EF1Alpha-puro-T2A-BFP (Addgene #60955) by PCR amplification using Gibson primers with overhangs for pPiggyBac-TET3 digested with XbaI (Supplementary Table 4). After gel purification, the T2A-BFP amplicon was inserted into pPiggyBac-TET3 using the NEBuilder HiFi DNA Assembly Master Mix (NEB) in a ratio of 1:5 (50°C, 30min) and transformed into E. coli (DH5α; Meissner laboratory). Plasmids were isolated and the correct sequence of the constructs was confirmed by Sanger sequencing.
The TET1 coding sequence was obtained by PCR amplification of pIRES-hrGFP II-TET1 (Addgene #83568) using Gibson primers to create overhangs for pPiggyBac MCS digested with XbaI (Supplementary Table 4). As we wanted the shorter TET1 isoform, the primers amplify only the necessary sequence and introduces an additional ATG. After gel purification, the TET1 amplicon was inserted into pPiggyBac MCS using the NEBuilder HiFi DNA Assembly Master Mix (NEB) in a ratio of 1:5 and transformed into E. coli (DH5α; Meissner laboratory). Plasmids were isolated and the correct sequence of the constructs was confirmed by Sanger sequencing. One of the confirmed clones of pPiggyBac TET1 was then digested with XbaI and assembled with the P2A-mCherry amplicon (obtained by PCR amplification from pDnmt3b1-P2A-mCherry60) using the NEBuilder HiFi DNA Assembly Master Mix (NEB) in a ratio of 1:5 and transformed into E. coli (DH5α; Meissner laboratory). Plasmids were isolated and the correct sequence of the constructs was confirmed by Sanger sequencing.
TET rescue experiments
TET1, TET2 or TET3 transposons described above were introduced using the piggyBac system, by co-transfection with a plasmid encoding the transposase (TransposagenBio). We used a 3:1 molar ratio of transposon to transposase 3:1, transfecting 70% confluent PKO cells using the Lipofectamin 3000 reagent (Invitrogen) according to the manufacturer’s instructions. After overnight incubation of the DNA complex on the cells, the cell monolayers were washed with PBS and incubated with mTeSR-1 containing RevitaCell supplement (Gibco) for two more days. Successful integration of each TET enzyme in PKO cells was assessed by fluorescence-activated cell sorting (FACS) and positive cells were placed back into culture for expansion. About 5–7 weeks after transfection, positive cells were collected by FACS followed by snap freezing the cell pellets. For a control, we transfected 70% confluent PKO cells with the transposase only. These cells were also sorted and plated in low density to mirror the rescue experiments.
Hairpin bisulfite sequencing
WT and PKO ESCs were arrested in G2/M phase with 2 μg/ml Nocodazole for 16 hours. Accutase (STEMCELL Technologies) was used to detach cells, which were then snap frozen prior to gDNA extraction using the Quick-DNA universal kit (Zymo Research). Following this, 3 μg DNA was sheared to average fragment size of 200 bp using Covaris Microtube and S2 ultrasonicator (Covaris). After shearing, DNA was concentrated to 50 μl in water with 1.8 volumes of Agencourt AMPure XP beads (Beckman Coulter) and was prepared for Illumina sequencing using Kapa LTP library preparation kit (Kapa Biosystems). We modified a previously described hairpin adapter for bisulfite sequencing61 by including a 12-bp unique molecular identifier (UMI) with methylated cytosine bases (Integrated DNA Technologies, Supplementary Table 4). Barcoded fork-adapters for Illumina sequencing (NEXTflex bisulfite-seq barcodes) were purchased from Bio Scientific. After post-ligation purification with 0.7 volumes of AMPure XP beads, biotinylated DNA molecules were pulled-down on 50 μl Dynabeads MyOne Streptavidin C1 beads (Thermo Fisher Scientific). Libraries were detached from C1 beads by heating them in 20 μl 10 mM Tris pH 7.5 buffer at 95 °C for 10 minutes. After separating beads on a magnet, DNA in supernatant was bisulfite converted using the EpiTect Fast Bisulfite Kit (Qiagen) extending both 60°C incubation steps to 20 min each. DNA was then subjected to 20 cycles of amplification using primers provided with Bio Scientific adapters and Kapa Uracil+ amplification master mix (Kapa Biosystems). Amplified DNA was loaded on 2% pre-cast gel E-gelEX (Thermo Fisher Scientific) and fragments of size 500–650 bp were selected by gel extraction using Zymoclean Gel DNA recovery kit (Zymo Research). Final libraries were sequenced with 300 bp paired-end reads on the Illumina Miseq platform. Fastqs were aligned to the hg19 human genome, then reads were filtered to eliminate those with identical barcodes. Next, we used HBS-tools62 for adapter trimming and methylation dyad calling.
Western blot in human cell lines
Western blot analysis was performed with whole cell lysate. The cell monolayer was rinsed three times with ice-cold PBS prior to collecting cells with a cell lifter (VWR) and pelleting by centrifugation. The cell pellets were lysed in 20 mM HEPES pH8.0, 100 mM NaCl, 1 mM EDTA and 1% Tween20 containing protease inhibitor cocktail (ROCHE) on ice for 30 minutes, sonicated, and cell debris was cleared by centrifugation. The protein concentration of the supernatant was quantified using the micro BCA protein assay kit (Pierce). About 70–100 μg of total protein was separated on 4–12% SDS-PAGE gel (Invitrogen) and transferred to PVDF membranes (GE Healthcare) followed by incubation with anti-rabbit-DNMT3A antibody (Abcam ab188470 lot:GR224165–13) and anti-rabbit-GAPDH antibody (Cell Signaling 2118 lot: 10) as a loading control. All blots were developed using the ECL reaction kit (Bio-Rad) according to the manufacturer’s instructions.
Western Blot in mouse cell lines
Cells were collected and washed twice in PBS before being resuspended in cell lysis buffer (25 mM HEPES pH7.6, 5 mM MgCl2, 25 mM KCl, 0.05 mM EDTA, 10% Glycerol, 0.1% IGEPAL, 1× Roche protease inhibitor, 1 mM DTT). The cell lysate was spun for 5 minutes at 1,500 rpm to pellet the nuclei. Nuclei were washed once to remove cell debris (10 mM HEPES pH7.6, 3 mM MgCl2, 100 mM KCl, 0.01 mM EDTA, 10% glycerol, 1× Roche protease inhibitor, 1 mM DTT). Nuclei were spun down at 3,000 g for 5 minutes. Nuclei were then resuspended in 150 μl RIPA buffer and vortexed for 20 minutes at 4°C. This mixture was spun at 12,000 rpm for 15 minutes and the supernatant was collected. 20 μl of 4× LDS buffer was then added to 60 μl of supernatant and denatured at 72°C for 10 minutes. Western blots were performed with anti-Dnmt1 (1:1,000 dilution, Abcam ab87654 lot: GR3194562–7), anti-Dnmt3a (1:2,000, Abcam ab188470 lot: GR224165–2), anti-Dnmt3b (1:500, Abcam ab176166 lot: GR3199224–3), anti-histone H3 (Abcam ab1791 lot: GR293197–1) and anti-GAPDH (Cell Signaling Technology mAb#2118L lot: 10) antibodies and imaged using HRP chemiluminescence.
Dnmt3a knockout mice
As Dnmt3a knockout mice die at around four weeks after birth14, we crossed heterozygous Dnmt3a knockout mice in which the Dnmt3a catalytic domain, PC and ENV motifs were replaced with a cassette containing an IRES-lacZ gene and a neomycin-resistance gene (B6;129S4-Dnmt3a<tm1Enl>) to obtain homozygous offspring. Eight days after birth, we collected colon, lung and liver tissue for whole genome bisulfite sequencing. Mice were genotyped using primers specific for the wildtype or mutant locus (Supplementary Table 4).
Motor neuron differentiation and culture
HUES64 WT human ESCs were cultured in mTeSR medium (Stem Cell Technologies), which was replenished daily. Once > 75% confluence had been reached, colonies were dissociated using Accutase (Innovative Cell Technologies). hESCs were then maintained in mTeSR with 10 μM ROCK inhibitor (Y-27632, DNSK International) for 24 h before being cultured in mTeSR alone. At > 70% confluence, differentiation was initiated. For 14 days, cells were maintained in a 1:1 ratio of Neurobasal and DMEM/F12 medium supplemented with N2, B27, GlutaMax, and non-essential amino acids (N2B27; Thermo Fisher, Life Technologies). Additionally, medium contained 1 μM Retinoic Acid (RA, Sigma) and 1 μM Smoothened Agonist (SAG, DNSK International). For the first six days, cells were administered dual-SMAD inhibitors SB-431542 (10 μM, DNSK International) and LDN-193189 (100 nM, DNSK International). For the remaining eight days, cells were treated with 5 μM Notch inhibitor DAPT (DNSK International) and 4 μM VEGF/FGF inhibitor SU5402 (DNSK International). Medium with small molecules was refreshed daily. At day 14, cells were dissociated using TrypLE Express with DNase I (Thermo Fisher, Worthington) and plated at a density of 400,000 cells/well in a 12-well plate. Neurons were maintained in Neurobasal medium supplemented with N2, B27, GlutaMax, and non-essential amino acids (NBM). Medium was additionally supplemented with neurotrophic factors BDNF, CNTF, GDNF (10 ng/ml, R&D systems), and AA (200 ng/ml, Sigma). For the first 24 hours, neurons were maintained in 2% Hyclone FBS (VWR) with Y-27632 ROCK inhibitor. EdU (10 μM, Thermo Fisher) was administered from days 14–21. Half the volume of medium with small molecules was refreshed every two to three days. For collection, neurons were washed twice with PBS and gently lifted from the wells using a cell scraper. Neurons were then pelleted and snap frozen until sample processing.
Whole Genome Bisulfite Sequencing
DNA was extracted using the Zymo Quick-DNA Universal Kit, and bisulfite conversion was carried out using the Qiagen Epitect Fast kit, both according to the manufacturer’s instructions. The eluted DNA was processed immediately using the Accel-NGS Methyl-seq DNA library kit (Cat# DL-ILMMS-12, Swift Biosciences) following the manufacturer’s recommendations. PCR products were cleaned up using the Agencourt AMPure XP system (Beckman Coulter Cat# A63881). The absence of adapter-dimers was confirmed using an Agilent Bioanalyzer. We then sequenced the libraries on the HiSeq4000, NovaSeq S2 or Hiseq2500 generating 150 bp paired-end reads.
Whole genome oxBS
Genomic DNA from HUES64 cells was extracted using the Quick-DNA universal kit (Zymo Research). 400 ng DNA was sheared to average fragment size of 200 bp using S2 focused-ultrasonicator (Covaris). After shearing, DNA was concentrated to 50 μl in water with 1.8 volumes of Agencourt AMPure XP beads (Beckman Coulter) and was processed with TrueMethyl Whole Genome kit (Cambridge Epigenetix, now available from Tecan) in hydroxymethylation mode up to the library preparation step. The library construction module was replaced with the Accel-NGS Methyl-Seq kit (Swift Biosciences) following the manufacturer’s recommendations.
WGBS/oxBS data processing
Raw sequencing reads were trimmed using cutadapt and aligned against human (hg19) or mouse (mm9) genome using BSMAP. Duplicate reads were removed using Picard tools. Methylation calls were made using MOABS mcall module and only CpGs covered with at least 10 reads were considered for further analysis. To generate 5hmC levels, the methylation state derived from WGBS (mC + 5hmC) was subtracted from the methylation state derived from oxBS (mC) for CpGs with coverage of at least 10 reads.
Data analysis
For comparison to genomic features, BedTools was used to intersect methylation data within CpG islands63, coding features (RefSeq), H1 ESC-defined typical and super enhancers32 and repeats (RepeatMasker).
Chromatin modification peaks or bam files were taken from ENCODE (H1 ESCs) and used to compare overlap or local enrichment. We also used all available bam files from ChIP-seq transcription factor experiments performed in H1 cells to study the enrichment at cDKO-DMRs. Here, we used the software Homer64 to create tag directories for each bam file, then used Homer findMotifsGenome.pl to determine the enrichment at features of interest.
Violin plots, boxplots, heatmaps, composite plots and scatter plots were all created in R. IGV was used to visualize CpG-specific methylation levels throughout the genome and create browser images.
RNA sequencing
RNA-seq libraries were prepared using the TrueSeq RNA Sample Prep v2 HS Protocol (Illumina), followed by 50 bp paired-end sequencing on the NextSeq500 Sequencing System (Illumina). RNA sequencing data were aligned using STAR aligner followed by stringtie v1.3 to the hg19 genome. Log2 TPM values were used to plot expression levels in R.
Statistics
For WGBS data analysis, to identify discrete regions regulated by TETs or DNMTs, we performed DMR calling by using a CpG-specific F-test. For stringent DMRs, we merged regions where at least 8/10 CpGs had P value < 0.01 with a difference of ≥ 0.6, and for reduced stringency we used 5/8 CpGs with P value < 0.01 and difference ≥ 0.25. DMRs were merged if within a maximum distance of 1 kb. After calling per sample DKO-DMRs in our three clones HUES64 DKO-A, DKO-B and HUES8 DKO, we identified n = 17,684, 19,715 and 20,456 DKO-DMRs respectively. We then merged these into one region list (causing overall DMRs per sample to decrease in the Venn diagram seen in Figure 1d). From this list, a region was determined as demethylated if the mean methylation level was below 0.2. Consensus cDKO-DMRs had mean methylation below 0.2 in all three samples.
To compare mouse Dnmt3a−/− DKO-DMRs, all tissue-specific DKO-DMRs were combined, then a region was considered as demethylated if the difference between WT and Dnmt3a−/− was at least 0.2.
For RNA-seq data analysis, the R package edgeR was used. “glmQLFit” was used to fit a quasi-likelihood negative binomial generalized log-linear model to count data and then “glmQLFTest” was used to calculate P values and Bonferroni-corrected FDR values for each gene. Genes with FDR < 0.01 and a log fold change of at least two were selected as differentially expressed. We also filtered out genes where both groups had log count values < 2.5, which was considered no change in expression even though this can derive highly significant P values.
All t tests were 2-sided. Note that the software R outputs the minimal P value 2.2 × 10−16 so where stated, the true P value may in fact be smaller.
Extended Data
Extended Data Fig. 1. Characterization of pentuple knockout ESCs.
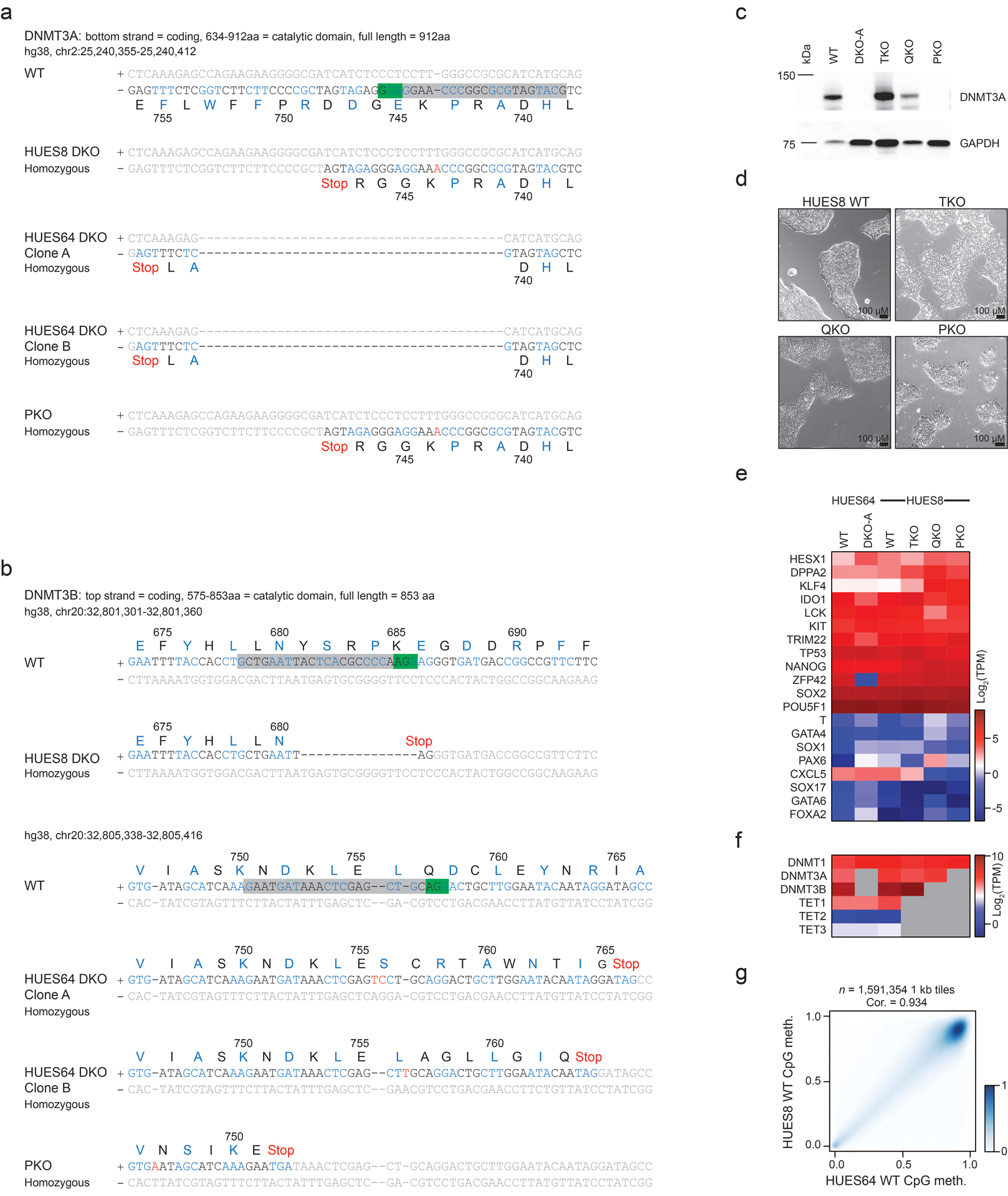
a) Sequencing data showing the mutations introduced into the DKO and PKO ESCs for DNMT3A. Green box = PAM sequence, grey box = sgRNA sequence. Codons are colored black or blue, grey text denotes non-coding DNA. The respective amino acid (aa) single letter name and sequence number is displayed below the DNA.
b) Sequencing data for DNMT3B, as described in a. A different sgRNA was used to target DNMT3B in HUES8 ESCs compared to HUES64.
c) Western blot for DNMT3A in HUES8 WT, HUES64 DKO clone A, HUES8 TKO, QKO and PKO cells. GAPDH is shown as the loading control. Full blots in Source Data Fig. 1. PKO cells were assessed for DNMT3A expression by Western blot six times with consistent results.
d) HUES8 lines at 10X magnification showing that all KO cell lines appear morphologically normal. These images are representative of a full 10 cm culture dish over weeks of passaging.
e) Heatmap showing RNA sequencing data as log2 TPM expression values for a set of pluripotency and self-renewal associated genes (highly expressed), as well as a subset of differentiation associated genes (low expression). DKO-A was collected at passage 6 and PKO at passage 12.
f) Heatmap showing RNA sequencing data as log2 TPM expression values DNMTs and TETs, grey boxes depict genes that are knocked out in each sample.
g) Comparison of global methylation levels between HUES64 and HUES8 wildtype cells where intensity of blue shading indicates density of data points. The Pearson correlation coefficient (cor.) is displayed.
Extended Data Fig. 2. Characterization of DKO-DMRs and TET rescue in PKO cells.
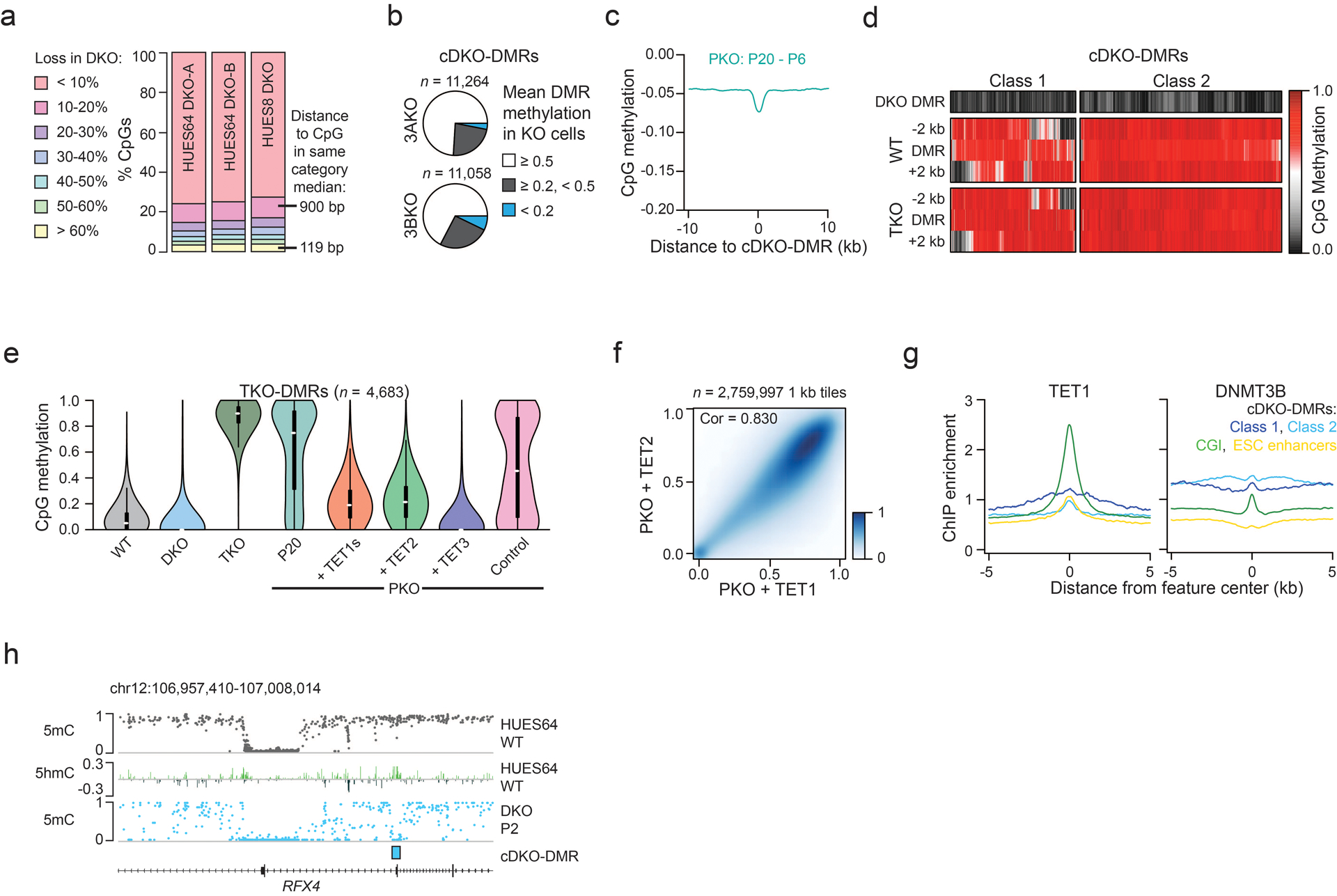
a) Bar chart showing the proportion of CpGs with varying methylation loss in DKO cells compared to WT. CpGs that lost >60% methylation were generally located in closer proximity compared to CpGs that lost 10–20% methylation.
b) The proportion of cDKO-DMRs with respective mean methylation levels in single DNMT3A−/− (3AKO) or DNMT3B−/− (3BKO) knockout ESCs.
c) Composite plot showing methylation difference between passage (P) 20 and 6 in PKO cells as distance from cDKO-DMR. There is a small background loss (−0.045) with a slightly greater focal decrease (−0.07), but methylation levels stay generally high.
d) The methylation level in HUES64 DKO-A, HUES64 WT and HUES8 TKO cells including the 2 kb either side is displayed. For regions where the neighboring 2 kb is hypomethylated in WT cells (class 1), almost every region shows an increase in methylation following TET loss.
e) Violin plots showing methylation at TKO-DMRs across different HUES8 lines. We used stringent parameters to define TKO-DMRs that aberrantly gain methylation upon loss of TET expression (also described later in Extended Data Fig. 9a). Methylation levels were rescued by re-expression of TET. Violin plots extend from the data minima to the maxima, white dot indicates median, thick bar shows the interquartile range and thin bar shows 1.5x interquartile range.
f) Methylation levels across 1 kb tiles for PKO ESCs rescued with either TET1s or TET2. Intensity of blue shading indicates density of data points. Pearson correlation coefficient (cor) is displayed.
g) ChIP-seq enrichment (from Ref 21) for TET1 and DNMT3B over CpG islands (CGI), H1-specific enhancers and cDKO-DMRs.
h) Representative browser tracks displaying methylation levels in WT and DKO cells as well as 5hmC levels in WT ESCs. Increased 5hmC is observed over the cDKO-DMR and at the border of the hypomethylated CGI, where TETs are known to localize.
Extended Data Fig. 3. LINE 5’UTRs are actively demethylated.
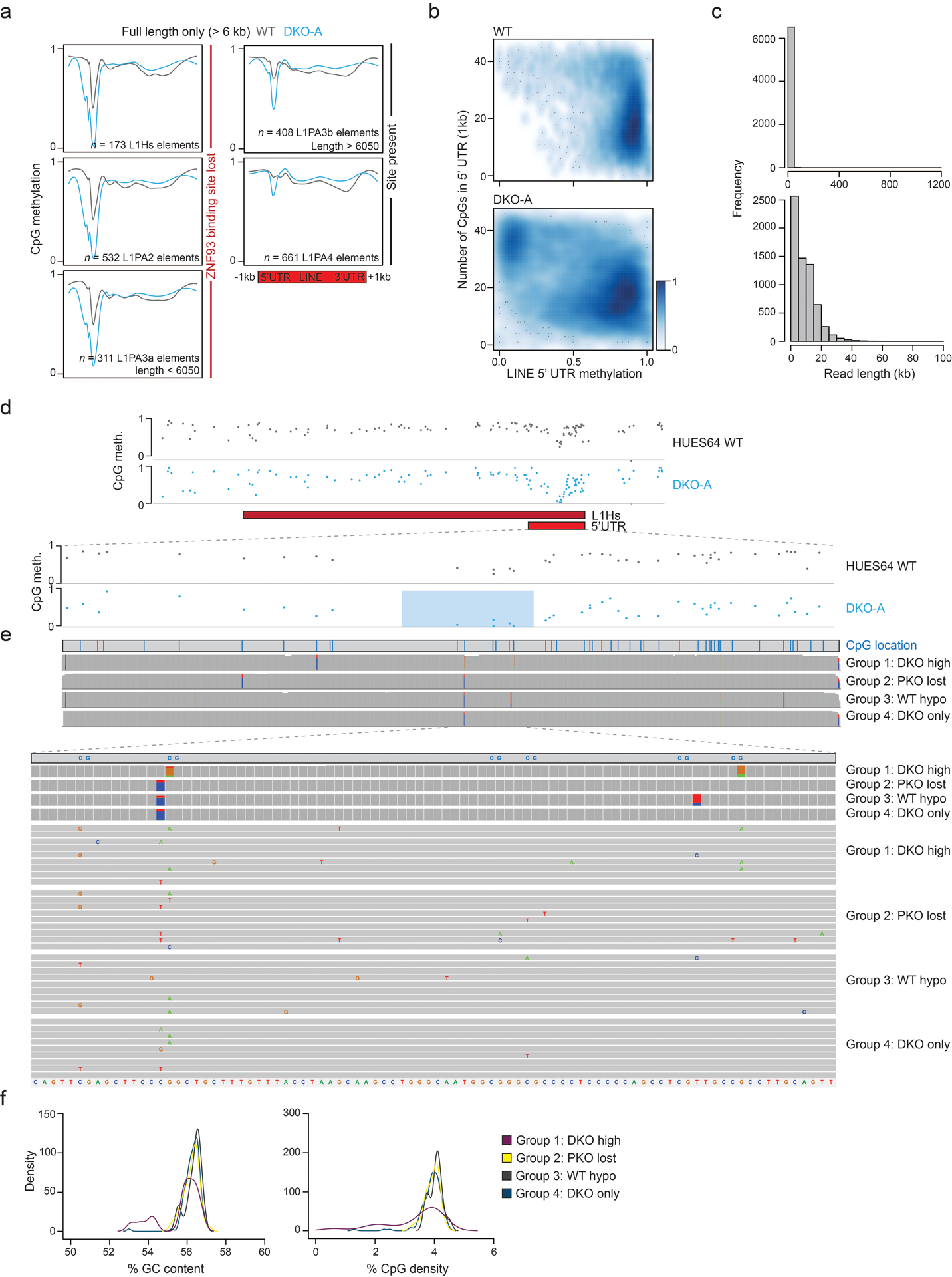
a) Methylation composite plots for WT and DKO-A cells over LINE L1 elements (oriented with 5’UTR to the left). L1Hs are the youngest and L1PA4 the oldest class displayed. Methylation at the 5’UTR of younger elements appears more sensitive to DNMT3 loss. In particular, sensitivity appears associated with ZNF93 binding site loss within the 5’UTR.
b) Smoothed scatter plots display the relationship between WT or DKO methylation level and CpG density within the 5’UTR, for all LINE L1 elements where intensity of blue shading indicates density of data points. Lower CpG density elements tend to retain methylation after DNMT3 loss while CpG dense 5’UTRs lose methylation.
c) Histogram of read lengths generated from Nanopore sequencing for HUES64 WT cells. Top = all reads, bottom = reads < 100 kb.
d) Example of CpG methylation across a group 4 L1Hs element (defined in Figure 3c) showing the focal decrease within the 5’UTR. Below, just the 5’UTR is displayed and CpGs with the most methylation loss following DNMT3 knockout are highlighted with a blue box.
e) Each L1Hs 5’UTR was aligned to a consensus sequence. The CpG locations within this sequence are illustrated by vertical blue lines. Below, a summary for genetic variants at each position is shown for each group of L1Hs (defined in Figure 3c). Colored boxes display the proportion of bases that vary from the consensus sequence. For each group, nine example 5’UTR sequences are displayed, zoomed in on the six CpGs highlighted in d. Each element is represented as a grey bar with colored letters indicating variations from the consensus sequence (shown at the bottom of the panel). None of the groups exhibit a unique common variant.
f) Density plots showing the distribution of 5’UTR GC content and CpG density for each group of L1Hs, from Figure 3c.
Extended Data Fig. 4. cDKO-DMRs overlap with tissue-specific enhancers.
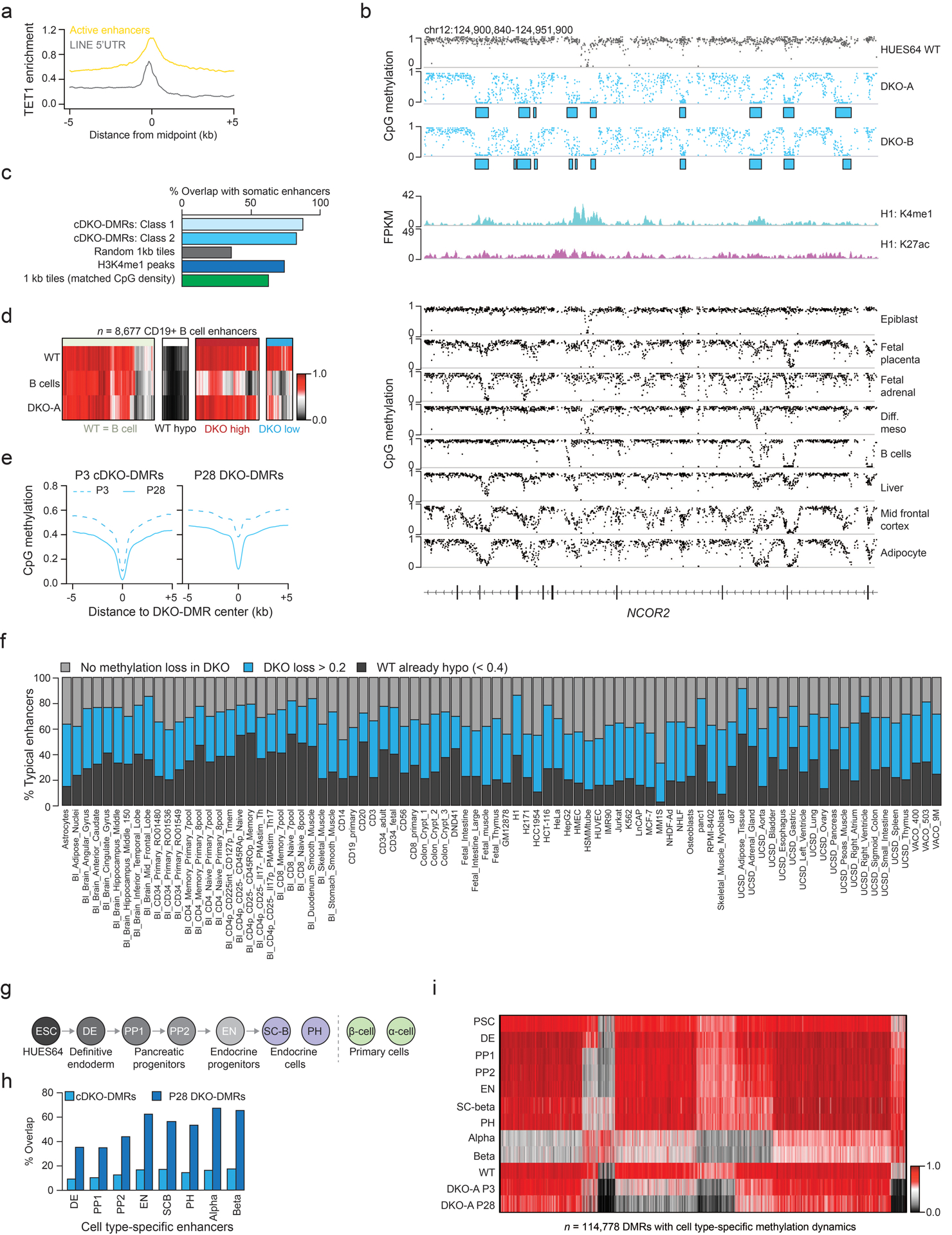
a) TET1 enrichment (from Ref 21) for LINE 5’UTRs and active H1 typical enhancers showing a greater enrichment at enhancers.
b) A region within the gene NCOR2 that has many cDKO-DMRs. These lose methylation in different somatic tissues as shown below. H3K27ac and H3K4me1 tracks display ENCODE data derived from H1 ESCs. Methylation tracks are from Ref 6.
c) The percentage of overlap with putative somatic enhancers (defined in Ref 32) for class 1 and class 2 cDKO-DMRs, an equal number of same-sized randomly selected regions, H3K4me1 peaks in ESCs (H1) and 1 kb tiles with matched CpG density to cDKO-DMRs (3.1–3.3%).
d) B cell enhancers (defined in Ref 32) separated by methylation levels in WT ESCs, DKO ESCs and B cells.
e) Composites showing methylation levels in passage (P) 3 and 28 DKO ESCs for P3 and P28 DKO-DMRs.
f) For 86 different previously defined putative enhancer sets32, the stacked bar plots display the proportion that are already hypomethylated in WT ESCs, lose methylation in DKO cells (WT – DKO difference > 0.2) or remain methylated in DKO cells. For this analysis, P28 DKO was used.
g) Schematic of in vitro pancreatic islet cell differentiation (from Ref 33).
h) The proportion of cDKO-DMRs or P28 DKO-DMRs that overlap with each set of cell type specific enhancers. Enhancers were previously defined33.
i) For regions defined as showing dynamic methylation changes during differentiation to beta islet cells33, methylation levels are shown for these cell types as well as HUES64 WT and DKO-A ESCs.
Extended Data Fig. 5. Chromatin states and TF interaction at cDKO-DMRs.
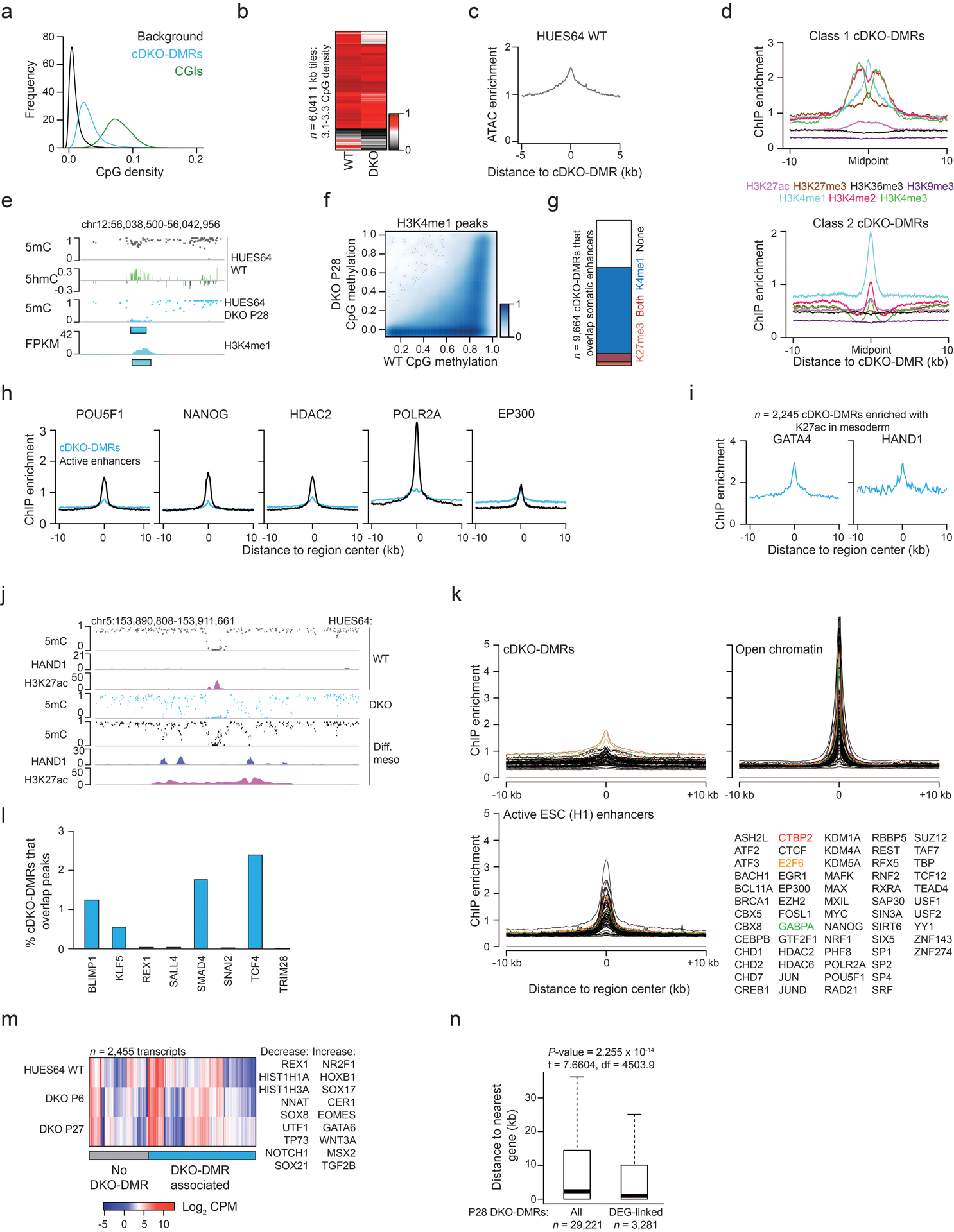
a) CpG density for random background regions, cDKO-DMRs and CGIs.
b) For 1 kb tiles with CpG density matching cDKO-DMRs (3.1–3.3%), the mean methylation in HUES64 WT and DKO-A is shown. Methylation loss is not dependent on CpG density.
c) ATAC-seq signal enrichment for cDKO-DMRs.
d) Enrichment for chromatin modifications (ENCODE, H1 ESCs) as distance from cDKO-DMRs.
e) A representative locus showing enrichment of H3K4me1 and 5hmC at a cDKO-DMR. P = passage.
f) Methylation levels for ENCODE-defined H3K4me1 peaks (≥3 CpGs, methylation ≥0.1 in WT ESCs, n = 71,665) for WT and DKO cells at P28. Intensity of blue shading indicates density of data points. Many regions remain methylated indicating that H3K4me1 enrichment does not predict demethylation.
g) For cDKO-DMRs that overlap with putative somatic enhancers, the proportion that are enriched with H3K4me1 and H3K27me3 is displayed. Overlap with both marks indicates a poised chromatin state that is more common for already hypomethylated poised enhancers.
h) ChIP-seq enrichment (ENCODE, H1 ESCs) for different enhancer-associated factors at cDKO-DMRs and active ESC enhancers.
i) HAND1 (GSM1505812) and GATA4 (GSM1505644) enrichment in HUES64 differentiated mesoderm (Ref 34) for cDKO-DMRs that show H3K27ac (GSM1505669) enrichment in mesoderm (i.e. enhancer-like chromatin state).
j) Browser tracks displaying regions that lose methylation while gaining H3K27ac and transcription factor (HAND1) binding upon differentiation to mesoderm. The same CpGs also lose methylation in DKO ESCs. ChIP-seq data is from Ref 34.
k) Enrichment for TF binding (ENCODE, H1 ESCs) at cDKO-DMRs, open DNA (based on HUES64 ATAC-seq) and active ESC enhancers is displayed. The most enriched factors at cDKO-DMRs (CTBP2, E2F6 and GABPA) show only low enrichment that could be confounded by the more open chromatin at cDKO-DMRs. No factor showed unique enrichment that would infer them as candidate factors for TET/DNMT recruitment.
l) The percentage of cDKO-DMRs that overlap with different TF binding sites, defined by ChIP-seq peak data (Ref 34). Overlap is consistently low.
m) Expression levels for differentially expressed genes between WT and DKO with examples to the right. P = passage. Each P28 DKO-DMR was assigned to its nearest gene, and those with a DKO-DMR assigned are highlighted with the blue bar.
n) The distribution of absolute distance to the nearest gene for P28 DKO-DMRs. “DEG-linked” refers to DKO-DMRs that have been assigned to a differentially expressed gene by proximity. Statistics refer to a 2-sided T-test. For the box plots: median is shown in bold, box displays interquartile range, whiskers extend to 1.5x the interquartile range.
Extended Data Fig. 6. Generation of DNMT3A and 3B knockout mouse EpiSCs.
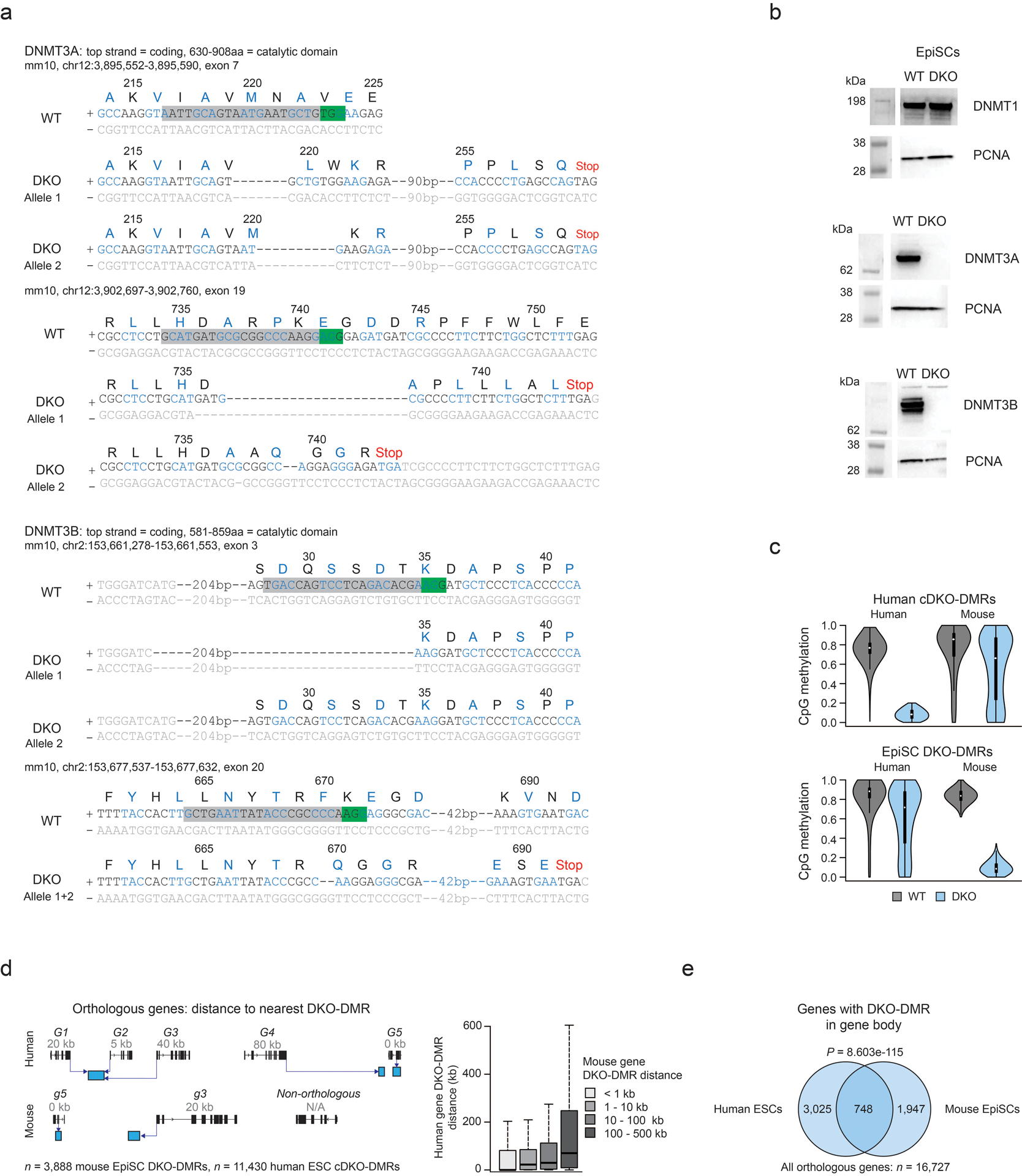
a) Sequencing data showing the Dnmt3a and Dnmt3b mutations introduced into mouse DKO EpiSCs. Two different sgRNAs were used for each gene. Green box = PAM sequence, grey box = sgRNA sequence. Codons are colored black or blue, grey text denotes non-coding DNA. The respective single letter amino acid (aa) and protein sequence number is displayed above the DNA.
b) Western blots showing protein levels for DNMT1, DNMT3A and DNMT3B in WT and DKO EpiSCs. PCNA is used as a loading control. Full blots in Source Data Fig. 2. Western blots were performed twice with consistent results.
c) Violin plots display methylation levels for cDKO-DMRs and EpiSC DKO-DMRs in WT and DKO cells for each species. The UCSC LiftOver tool was used to map DKO-DMRs between species. Violin plots extend from the data minima to the maxima with the white dot indicating median, thick bar showing the interquartile range and thin bar showing 1.5x interquartile range.
d) Schematic depicting how orthologous genes shared between human and mouse were assigned to the nearest DKO-DMR (left). Each gene was given a number (in kb) denoting the distance to the nearest DKO-DMR either up- or down-stream. Box plots for genes that were binned based in their distance to the nearest DKO-DMR in EpiSCs (shown in legend key), displaying the distance to the nearest DKO-DMR in human ESCs (right). A matched trend between distances in mouse and human cells in evident. For the box plots: the median is shown in bold, the box displays interquartile range and whiskers extend to 1.5 times the interquartile range.
e) Venn diagram showing the overlap of orthologous genes that have a DKO-DMR within the gene body (i.e. 0 kb away, as shown in panel d) in mouse EpiSCs and human ESCs. This overlap was highly significant (hypergeometric test).
Extended Data Fig. 7. DNMT and TET expression in somatic tissues.
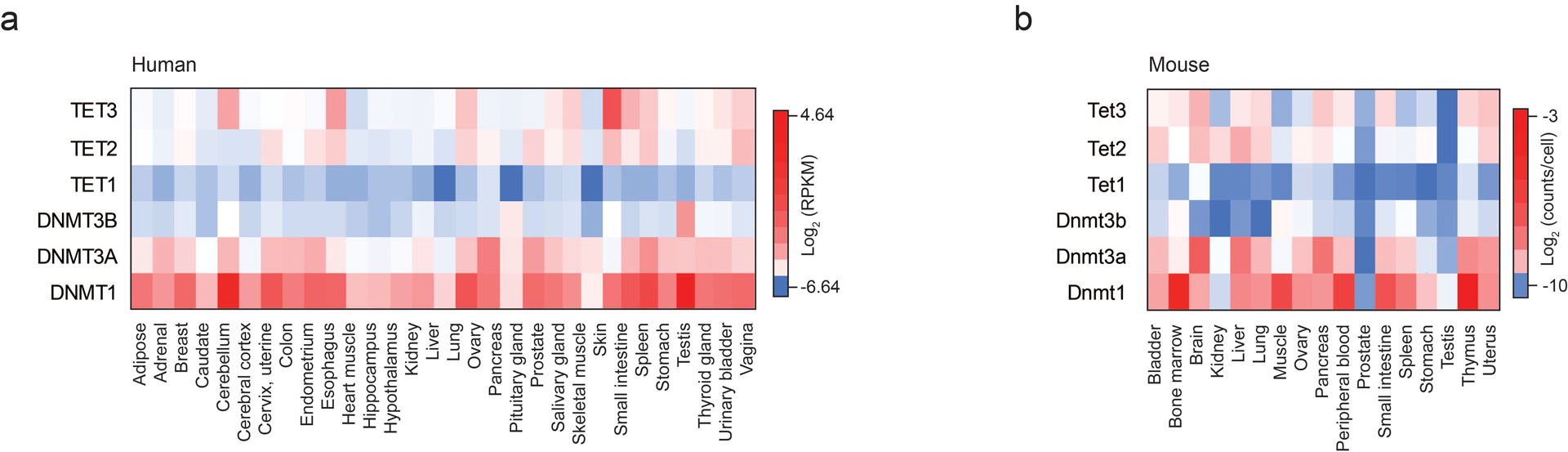
a) Heat map showing Log2 RPKM for DNMTs and TETs across human somatic tissues, based on data from the human protein atlas (ebi.ac.uk)
b) Heat map showing Log2 transcript counts per cell for murine somatic tissues, taken from single cell mouse atlas data65.
Extended Data Fig. 8. DKO-DMRs are associated with pluripotency.
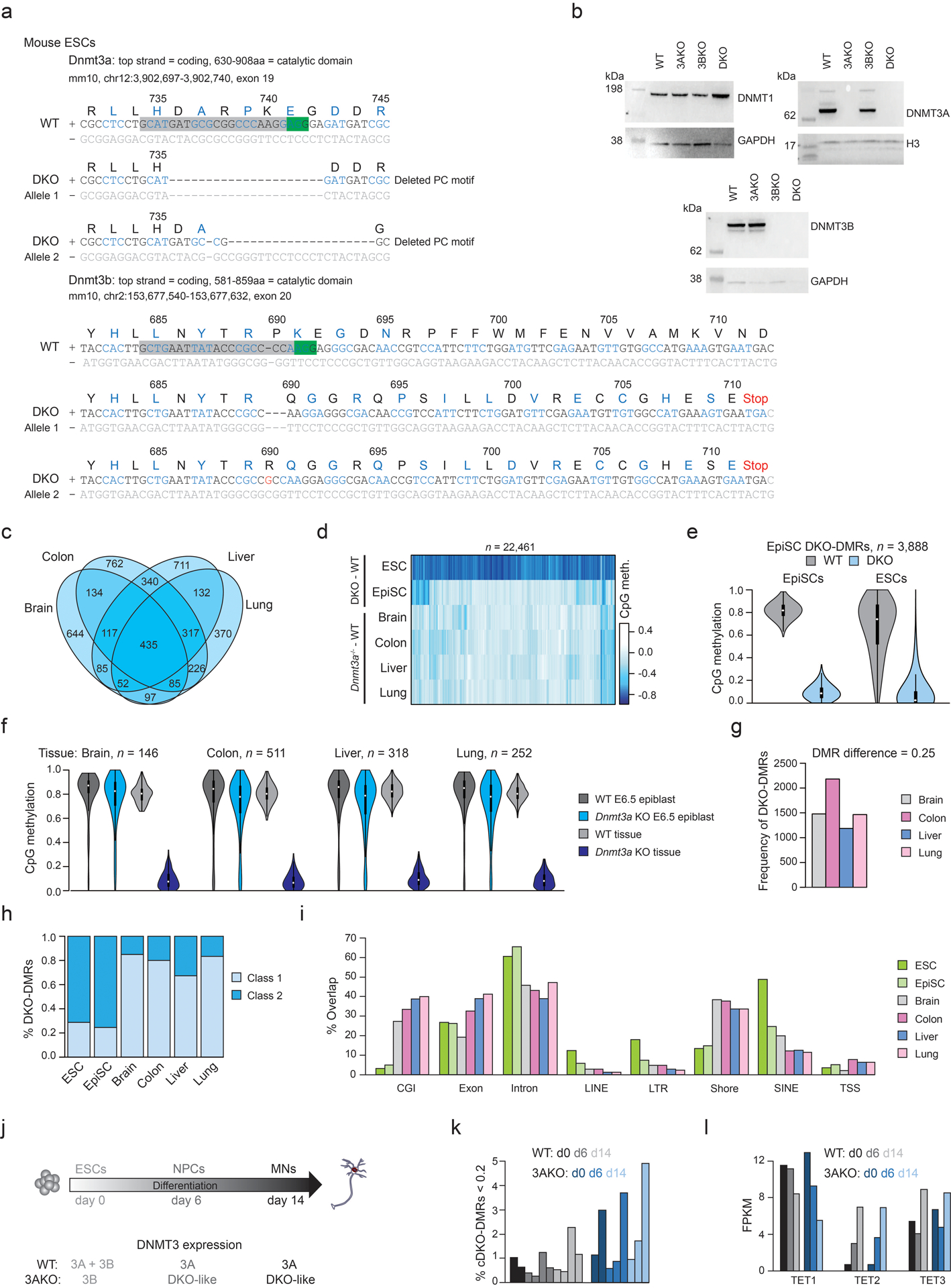
a) Sequence mutations introduced into the DKO ESCs. Green box = PAM sequence, grey box = sgRNA. Codons are colored black or blue, grey text denotes non-coding DNA. Amino acid (aa) and protein sequence number are displayed above the DNA.
b) Western blots for WT, DNMT3A−/− (3AKO), DNMT3B−/− (3BKO) and double knockout (DKO) mouse ESCs. GAPDH or H3 were used as loading controls. Full blots in Source Data Fig. 3. Western blots were performed three times with consistent results.
c) Overlap between DKO-DMRs called in each Dnmt3a−/− tissue.
d) The difference in methylation between WT and Dnmt3a−/− tissues, ESCs or EpiSCs.
e) Methylation levels for EpiSC DKO-DMRs in EpiSCs and ESCs. Violin plots extend from the data minima to the maxima, white dot indicates median, thick bar shows the interquartile range and thin bar shows 1.5x interquartile range.
f) Methylation levels for tissue-specific DKO-DMRs in WT and Dnmt3a−/− E6.5 epiblast and 8 day old tissues. Somatic DKO-DMRs were fully methylated in Dnmt3a−/− embryos following implantation due to DNMT3B expression hence must have lost methylation at a later stage. Violin plots extend from the data minima to the maxima, white dot indicates median, thick bar shows the interquartile range and thin bar shows 1.5x interquartile range.
g) The number of DKO-DMRs identified when using a reduced stringency of 0.25 differential methylation instead of 0.6 to compensate for mixed cell types. We identified more DKO-DMRs (1,186–2,182) but still many less than in ESCs, and this may include false positives.
h) The percentage of DKO-DMRs that fall into class 1 or 2 (described in Figure 1).
i) The overlap of DKO-DMRs with genomic features. Categories are not exclusive. CGI = CpG island, TSS = transcription start site.
j) Schematic showing the differentiation of human ESCs to motor neurons (MNs) via neuronal progenitor cells (NPCs). For WT and 3AKO ESCs expression of DNMT3A and DNMT3B is displayed across the time course. After day two, 3AKO cells are “DKO-like” as they do not express either DNMT3.
k) The percentage of cDKO-DMRs with mean methylation <0.2 in each sample during differentiation. Only 1.3% and 2.4% of cDKO-DMRs lost methylation for WT and 3AKO MNs respectively. WGBS is from Ref 39.
l) Expression of TET1–3 in WT and 3AKO ESCs during differentiation. RNA-seq is from Ref 39.
Extended Data Fig. 9. Identification of DMRs across human ESCs.

a) Schematic illustrating how DMR calling was performed between each pair of samples. The number of DMRs that gain methylation (Incr.) in the cell type shown to the right is located above the arrow, and the number of DMRs that lose methylation (Decr.) is shown below the arrow. As we identified only few regions for other comparisons that met the same criteria as for DKO-DMRs (stringent DMRs; black) we also used less stringent criteria (Δ > 0.25) to define DMRs (blue). P = passage.
b) CpG methylation of TKO-, QKO- and PKO-DMRs detected with lower stringency as defined in panel a. The n is lower than shown in panel a due to drop-out from requiring 10x coverage per region.
c) Location of the DMRs shown in b. Features are not exclusive. Enh. = enhancer, CGI = CpG island, TSS = transcription start site, IG = intergenic. As expected and previously reported in Ref 21, the majority of DMRs that occur in the absence of TETs were CGI promoters or shores of developmental genes that may be more susceptible to hypermethylation due to lack of transcriptional activity.
d) Low stringency TKO-DMRs are grouped by their methylation status in QKO and PKO cells. Of the TKO-DMRs, 23% lost the aberrantly gained methylation upon KO of DNMT3B (in QKO), 21% only upon further loss of DNMT3A (PKO) and 56% remained highly methylated despite loss of both DNMT3s pointing to the importance of TET activity in keeping these loci demethylated.
e) The difference in methylation over passaging across each genomic feature.
f) The location of DMRs identified between PKO at P6 and P20. Categories are not exclusive.
g) CpG methylation levels for a representative early-replicating region, where methylation in DKO cells decreases the most, (left) and a representative late-replicating region, where PKO cells show the strongest methylation loss (right).
Extended Data Fig. 10. Broad TET activity results in genome-wide 5hmC enrichment and methylation heterogeneity.

a) Mean 5hmC level in HUES64 differentiated motor neurons (MN) over gene bodies grouped into categories according to their expression status, d = day. The median is shown in bold, the box displays interquartile range and whiskers extend to 1.5x the interquartile range.
b) The difference (d60 – d16) in 5hmC vs. methylation for MNs at different genomic features. CGI = CpG island, TSS = transcription start site, IG = intergenic.
c) Transcript expression for genes involved in TET-mediated demethylation and base excision repair pathways.
d) For genomic features, the difference in methylation between ESCs is displayed.
e) Cell counts for HUES8 WT and TKO cells over 5d culture. Center points show the mean and error bars display the standard deviation for three independent cell culture replicates.
f) Schematic showing how stochastic turnover of cytosine modification can lead to intercellular heterogeneity. mC = methylcytosine (black circle), 5hmC = hydroxymethylcytosine (grey circle), C = unmethylated cytosine (white circle).
g) Representative browser tracks showing the methylation status for CpGs detected using hairpin bisulfite sequencing (BS; top) and WGBS (bottom). Each CpG covered by hairpin-BS is colored according to dyad methylation state.
h) Proportion of CpG dyads that were methylated, unmethylated or hemi-methylated in arrested HUES64 WT or PKO ESCs.
i) For regions enriched with different chromatin marks (ENCODE, H1 ESCs), the proportion of CpG dyads with each methylation status is displayed.
j) The percentage of CpG dyads that were hemi-methylated according to location within each genomic feature. TE = active typical enhancer.
k) Single cell RRBS data showing the percentage of unmethylated CpGs per cell. The violin plot extends from the data minima to the maxima where each dot is a single cell. Of CpGs that are normally methylated in bulk WGBS (≥ 0.9), we observed that 6.6% ±3.37 showed zero methylation in individual cells. These may have been targeted by TETs and are likely an underestimate as 5hmCs cannot be detected with our RRBS based approach.
l) The proportion of CpGs with zero, intermediate (not exactly zero or one) or one methylation status for arrested WT ESCs. In bulk populations, ESC arrest removes replication-associated heterogeneity and causes most CpGs to become fully methylated. However, 25% remain intermediate66. The density plot shows methylation loss for each category of CpGs with intermediate CpGs showing the greatest loss suggesting they may be more frequently targeted by TETs hence TET activity may contribute to intermediate methylation levels in bulk populations.
Supplementary Material
Acknowledgements
We would like to thank members of the Meissner laboratory for their support in particular H. Kretzmer and S. Grosswendt for critical reading of the manuscript as well as R. Karnik for initial processing of the mouse Dnmt3a knockout data. We would also like to thank Danwei Huangfu and her team for providing the TKO and QKO HUES8 cells. We also acknowledge the Max Planck Institute for Molecular Genetics sequencing core. E.K. is a Les Turner ALS Research Center Investigator and a New York Stem Cell Foundation – Robertson Investigator and part of this work was supported by NIH grant NINDS/NIA R01NS104219. A.G. and A.M. are supported by NIGMS grant P01GM099117 and A.M. is supported by the Max Planck Society.
Footnotes
Competing interests statement
The authors declare no competing interests.
Data availability
Data have been deposited in the Gene Expression Omnibus (GEO) under accession GSE126958. Other published datasets used in this study include: HUES64 DNMT3A single knockout ESCs (GSM1545005), DNMT3B single knockout ESCs (GSM1545006) and mouse WT ESCs (GSM2339908).
References
- 1.Jadhav U et al. Extensive Recovery of Embryonic Enhancer and Gene Memory Stored in Hypomethylated Enhancer DNA. Molecular cell 74, 542–554.e545, doi: 10.1016/j.molcel.2019.02.024 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhou W et al. DNA methylation loss in late-replicating domains is linked to mitotic cell division. Nature genetics 50, 591–602, doi: 10.1038/s41588-018-0073-4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jin Z & Liu Y DNA methylation in human diseases. Genes & diseases 5, 1–8, doi: 10.1016/j.gendis.2018.01.002 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klutstein M, Nejman D, Greenfield R & Cedar H DNA Methylation in Cancer and Aging. Cancer research 76, 3446–3450, doi: 10.1158/0008-5472.can-15-3278 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Gangisetty O, Cabrera MA & Murugan S Impact of epigenetics in aging and age related neurodegenerative diseases. Frontiers in bioscience (Landmark edition) 23, 1445–1464 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Ziller MJ et al. Charting a dynamic DNA methylation landscape of the human genome. Nature 500, 477–481, doi: 10.1038/nature12433 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tahiliani M et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science (New York, N.Y.) 324, 930–935, doi: 10.1126/science.1170116 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ito S et al. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 466, 1129–1133, doi: 10.1038/nature09303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ito S et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science (New York, N.Y.) 333, 1300–1303, doi: 10.1126/science.1210597 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.He YF et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science (New York, N.Y.) 333, 1303–1307, doi: 10.1126/science.1210944 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maiti A & Drohat AC Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: potential implications for active demethylation of CpG sites. The Journal of biological chemistry 286, 35334–35338, doi: 10.1074/jbc.C111.284620 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith ZD & Meissner A DNA methylation: roles in mammalian development. Nature Reviews Genetics 14, 204, doi: 10.1038/nrg3354 (2013). [DOI] [PubMed] [Google Scholar]
- 13.Li E, Bestor TH & Jaenisch R Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 69, 915–926 (1992). [DOI] [PubMed] [Google Scholar]
- 14.Okano M, Bell DW, Haber DA & Li E DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99, 247–257 (1999). [DOI] [PubMed] [Google Scholar]
- 15.Gu TP et al. The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature 477, 606–610, doi: 10.1038/nature10443 (2011). [DOI] [PubMed] [Google Scholar]
- 16.Dawlaty MM et al. Combined deficiency of Tet1 and Tet2 causes epigenetic abnormalities but is compatible with postnatal development. Developmental cell 24, 310–323, doi: 10.1016/j.devcel.2012.12.015 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baubec T et al. Genomic profiling of DNA methyltransferases reveals a role for DNMT3B in genic methylation. Nature 520, 243–247, doi: 10.1038/nature14176 (2015). [DOI] [PubMed] [Google Scholar]
- 18.Liao J et al. Targeted disruption of DNMT1, DNMT3A and DNMT3B in human embryonic stem cells. Nature genetics 47, 469–478, doi: 10.1038/ng.3258 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Manzo M et al. Isoform-specific localization of DNMT3A regulates DNA methylation fidelity at bivalent CpG islands. The EMBO journal 36, 3421–3434, doi: 10.15252/embj.201797038 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jeong M et al. Large conserved domains of low DNA methylation maintained by Dnmt3a. Nature genetics 46, 17–23, doi: 10.1038/ng.2836 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Verma N et al. TET proteins safeguard bivalent promoters from de novo methylation in human embryonic stem cells. Nature genetics 50, 83–95, doi: 10.1038/s41588-017-0002-y (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu H et al. Dual functions of Tet1 in transcriptional regulation in mouse embryonic stem cells. Nature 473, 389–393, doi: 10.1038/nature09934 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu F, Liu Y, Jiang L, Yamaguchi S & Zhang Y Role of Tet proteins in enhancer activity and telomere elongation. Genes & development 28, 2103–2119, doi: 10.1101/gad.248005.114 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang L et al. TET2 coactivates gene expression through demethylation of enhancers. Science Advances 4, eaau6986, doi: 10.1126/sciadv.aau6986 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hon, Gary C et al. 5mC Oxidation by Tet2 Modulates Enhancer Activity and Timing of Transcriptome Reprogramming during Differentiation. Molecular cell 56, 286–297, doi: 10.1016/j.molcel.2014.08.026 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang Y et al. Distinct roles of the methylcytosine oxidases Tet1 and Tet2 in mouse embryonic stem cells. Proceedings of the National Academy of Sciences of the United States of America 111, 1361–1366, doi: 10.1073/pnas.1322921111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu Y et al. Genome-wide Regulation of 5hmC, 5mC, and Gene Expression by Tet1 Hydroxylase in Mouse Embryonic Stem Cells. Molecular cell 42, 451–464, doi: 10.1016/j.molcel.2011.04.005 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ko M et al. Modulation of TET2 expression and 5-methylcytosine oxidation by the CXXC domain protein IDAX. Nature 497, 122–126, doi: 10.1038/nature12052 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang W et al. Isoform Switch of TET1 Regulates DNA Demethylation and Mouse Development. Molecular cell 64, 1062–1073, doi: 10.1016/j.molcel.2016.10.030 (2016). [DOI] [PubMed] [Google Scholar]
- 30.de la Rica L et al. TET-dependent regulation of retrotransposable elements in mouse embryonic stem cells. Genome biology 17, 234 (2016). <>. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rinaldi L et al. Dnmt3a and Dnmt3b Associate with Enhancers to Regulate Human Epidermal Stem Cell Homeostasis. Cell stem cell 19, 491–501, doi: 10.1016/j.stem.2016.06.020 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Hnisz D et al. Super-enhancers in the control of cell identity and disease. Cell 155, 934–947, doi: 10.1016/j.cell.2013.09.053 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alvarez-Dominguez JR et al. Circadian Entrainment Triggers Maturation of Human In Vitro Islets. Cell stem cell 26, 108–122.e110, doi: 10.1016/j.stem.2019.11.011 (2020). [DOI] [PubMed] [Google Scholar]
- 34.Tsankov AM et al. Transcription factor binding dynamics during human ES cell differentiation. Nature 518, 344–349, doi: 10.1038/nature14233 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guallar D et al. RNA-dependent chromatin targeting of TET2 for endogenous retrovirus control in pluripotent stem cells. Nature genetics 50, 443–451, doi: 10.1038/s41588-018-0060-9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shen Y et al. A map of the cis-regulatory sequences in the mouse genome. Nature 488, 116, doi:10.1038/nature1124310.1038/nature11243https://www.nature.com/articles/nature11243#supplementary-informationhttps://www.nature.com/articles/nature11243#supplementary-information (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cheng Y et al. Principles of regulatory information conservation between mouse and human. Nature 515, 371–375, doi: 10.1038/nature13985 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huntriss J et al. Expression of mRNAs for DNA methyltransferases and methyl-CpG-binding proteins in the human female germ line, preimplantation embryos, and embryonic stem cells. Molecular reproduction and development 67, 323–336, doi: 10.1002/mrd.20030 (2004). [DOI] [PubMed] [Google Scholar]
- 39.Gordon CA, Hartono SR & Chédin F Inactive DNMT3B Splice Variants Modulate De Novo DNA Methylation. PLOS ONE 8, e69486, doi: 10.1371/journal.pone.0069486 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ziller MJ et al. Dissecting the Functional Consequences of De Novo DNA Methylation Dynamics in Human Motor Neuron Differentiation and Physiology. Cell stem cell 22, 559–574.e559, doi: 10.1016/j.stem.2018.02.012 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.López-Moyado IF et al. Paradoxical association of TET loss of function with genome-wide DNA hypomethylation. Proceedings of the National Academy of Sciences 116, 16933–16942, doi: 10.1073/pnas.1903059116 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rulands S et al. Genome-Scale Oscillations in DNA Methylation during Exit from Pluripotency. Cell Systems 7, 63–76.e12, doi: 10.1016/j.cels.2018.06.012 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nakagawa T et al. CRL4(VprBP) E3 ligase promotes monoubiquitylation and chromatin binding of TET dioxygenases. Molecular cell 57, 247–260, doi: 10.1016/j.molcel.2014.12.002 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu C et al. CRL4 Complex Regulates Mammalian Oocyte Survival and Reprogramming by Activation of TET Proteins. Science (New York, N.Y.) 342, 1518–1521, doi: 10.1126/science.1244587 (2013). [DOI] [PubMed] [Google Scholar]
- 45.Creyghton MP et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proceedings of the National Academy of Sciences 107, 21931–21936, doi: 10.1073/pnas.1016071107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nguyen MLT, Jones SA, Prier JE & Russ BE Transcriptional Enhancers in the Regulation of T Cell Differentiation. Frontiers in Immunology 6, doi: 10.3389/fimmu.2015.00462 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rada-Iglesias A et al. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470, 279, doi:10.1038/nature0969210.1038/nature09692https://www.nature.com/articles/nature09692#supplementary-informationhttps://www.nature.com/articles/nature09692#supplementary-information (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pauklin S, Madrigal P, Bertero A & Vallier L Initiation of stem cell differentiation involves cell cycle-dependent regulation of developmental genes by Cyclin D. Genes & development 30, 421–433, doi: 10.1101/gad.271452.115 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yiangou L et al. Cell cycle regulators control mesoderm specification in human pluripotent stem cells. The Journal of biological chemistry 294, 17903–17914, doi: 10.1074/jbc.RA119.008251 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yin Y et al. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science (New York, N.Y.) 356, eaaj2239, doi: 10.1126/science.aaj2239 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Walter M, Teissandier A, Pérez-Palacios R & Bourc’his D An epigenetic switch ensures transposon repression upon dynamic loss of DNA methylation in embryonic stem cells. eLife 5, e11418, doi: 10.7554/eLife.11418 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kunarso G et al. Transposable elements have rewired the core regulatory network of human embryonic stem cells. Nature genetics 42, 631–634, doi: 10.1038/ng.600 (2010). [DOI] [PubMed] [Google Scholar]
- 53.Schmidt D et al. Waves of retrotransposon expansion remodel genome organization and CTCF binding in multiple mammalian lineages. Cell 148, 335–348, doi: 10.1016/j.cell.2011.11.058 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Spencer DH et al. CpG Island Hypermethylation Mediated by DNMT3A Is a Consequence of AML Progression. Cell 168, 801–816.e813, doi: 10.1016/j.cell.2017.01.021 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cirulli ET et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science (New York, N.Y.) 347, 1436–1441, doi: 10.1126/science.aaa3650 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Greber B et al. Conserved and divergent roles of FGF signaling in mouse epiblast stem cells and human embryonic stem cells. Cell stem cell 6, 215–226, doi: 10.1016/j.stem.2010.01.003 (2010). [DOI] [PubMed] [Google Scholar]
- 57.Giesselmann P, Hetzel S, Müller F-J, Meissner A & Kretzmer H Nanopype: A modular and scalable nanopore data processing pipeline. Bioinformatics, doi: 10.1093/bioinformatics/btz461 (2019). [DOI] [PubMed] [Google Scholar]
- 58.Sedlazeck FJ et al. Accurate detection of complex structural variations using single-molecule sequencing. Nat Methods 15, 461–468, doi: 10.1038/s41592-018-0001-7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Simpson JT et al. Detecting DNA cytosine methylation using nanopore sequencing. Nat Methods 14, 407–410, doi: 10.1038/nmeth.4184 (2017). [DOI] [PubMed] [Google Scholar]
- 60.Galonska C et al. Genome-wide tracking of dCas9-methyltransferase footprints. Nature Communications 9, 597, doi: 10.1038/s41467-017-02708-5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhao L et al. The dynamics of DNA methylation fidelity during mouse embryonic stem cell self-renewal and differentiation. Genome research 24, 1296–1307, doi: 10.1101/gr.163147.113 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sun M-A, Velmurugan KR, Keimig D & Xie H HBS-Tools for Hairpin Bisulfite Sequencing Data Processing and Analysis. Adv Bioinformatics 2015, 760423–760423, doi: 10.1155/2015/760423 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Illingworth R et al. A novel CpG island set identifies tissue-specific methylation at developmental gene loci. PLoS biology 6, e22, doi: 10.1371/journal.pbio.0060022 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Heinz S et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Molecular cell 38, 576–589, doi: 10.1016/j.molcel.2010.05.004 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Han X et al. Mapping the Mouse Cell Atlas by Microwell-Seq. Cell 172, 1091–1107.e1017, doi: 10.1016/j.cell.2018.02.001 (2018). [DOI] [PubMed] [Google Scholar]
- 66.Charlton J et al. Global delay in nascent strand DNA methylation. Nature structural & molecular biology 25, 327–332, doi: 10.1038/s41594-018-0046-4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.