

Abstract
Background:
Previously, we showed that abnormal levels of bioactive lipids in bronchoalveolar lavage fluid (BALF) from infants with cystic fibrosis (CF) correlated with early structural lung damage.
Method:
To extend these studies, BALF bioactive lipid measurement by mass spectrometry and chest computed tomography (CT, combined with the sensitive PRAGMA-CF scoring method) were performed longitudinally at 2-year intervals in a new cohort of CF children (n=21, aged 1–5 yrs).
Results:
PRAGMA-CF, neutrophil elastase activity, and myeloperoxidase correlated with BALF lysolipids and isoprostanes, markers of oxidative stress, as well as prostaglandin E2 and combined ceramide precursors (Spearman’s Rho >0.5; P<0.01 for all). Multiple protein agonists of inflammation and tissue remodeling, measured by Olink protein array, correlated positively (r=0.44–0.79, p <0.05) with PRAGMA-CF scores and bioactive lipid levels. Notably, levels of lysolipids, prostaglandin E2 and isoprostanes at first BALF predicted the evolution of PRAGMA-CF scores 2 years later. In wild-type differentiated primary bronchial epithelial cells, and in CFTR-inducible iCFBE cells, treatment with a lysolipid receptor agonist (VPC3114) enhanced shedding of pro-inflammatory and pro-fibrotic proteins.
Conclusions:
Together, our findings suggest that bioactive lipids in BALF correlate with and possibly predict structural lung disease in CF children, which supports their use as biomarkers of disease progression and treatment efficacy. Furthermore, our data suggest a causative role of airway lysolipids and oxidative stress in the progression of early CF lung disease, unveiling potential therapeutic targets.
Keywords: Lysophosphatidic acid, lysolipid receptor, EGFR, ADAM17, Amphiregulin, oxidative stress
INTRODUCTION
Progressive lung disease characterized by airway obstruction, bacterial infection, and inflammation is the main cause of morbidity in cystic fibrosis (CF) patients. Structural lung anomalies are readily detected by chest computed tomography (CT) in CF infants within a few months of birth [1]. Mucus obstruction and bacterial infections are not the sole causes of inflammation in early CF lung disease. Indeed, knockout of the CF transmembrane conductance regulator (CFTR) is sufficient to cause inflammatory lung disease in ferrets, even in the absence of bacterial infection [2]. In addition, analysis of bronchoalveolar lavage fluid (BALF) from CF infants, show elevated neutrophil count, neutrophil elastase (NE), and interleukin 8 (IL-8) levels in the absence of detectable bacterial infection [3].
Another explanation for the pro-inflammatory environment in CF lungs could be abnormal bioactive lipid metabolism. Numerous clinical and preclinical studies, outlined below and in the discussion, support the existence of a close relationship between CF lung disease and abnormal lipid metabolism [4, 5]. Lysophosphatidic acid (LPA) is known to enhance the pro-inflammatory response in the presence of infection through specific receptors [6]. CFTR-deficient mice have abnormal ceramide and sphingosine metabolism of lipids in the airways, correlating with inflammation in the absence of infection [7, 8]. Also, CF mouse and human lung epithelia show accumulation of long-chain ceramides compared to very long chain ceramides, which is linked to lung inflammation [8–11]. Initial apparent discrepancies concerning enhanced or [12] or reduced total ceramide [13] levels in CF tissue could be resolved by comparing the different analysis procedures used [13]. Furthermore, interventions targeting sphingosine and ceramide metabolism reduce lung inflammation in CFTR-deficient mice [8, 14].
Because abnormal lipid metabolism is linked to inflammation in CF models, we expect that it could play a role in shaping the clinical lung outcome in CF infants. Indeed, in a previous cross-sectional study conducted on an Australian cohort of CF infants (AREST-CF), we observed that ceramide precursors, ceramide chain length ratio, and lipid markers of oxidative stress (isoprostanes and lysolipids) were correlated to neutrophilic inflammation in the airways [15]. However, that study did not investigate the association with future disease outcomes or causative effects in model systems.
Here, we hypothesized that abnormal bioactive lipids would contribute to the escalation of lung inflammation, leading to lung damage, in a cohort of infants with CF monitored longitudinally by chest CT and bronchoalveolar lavage. Furthermore, we investigated the effect of lysolipids on inflammatory signaling by control and CF epithelial cells in culture. Together, our data indicate an association between abnormal airway bioactive lipid profile, inflammation, and structural lung disease early in the life of CF patients.
METHODS
Human subjects.
Children with CF were enrolled in an early disease monitoring program (I-BALL) at the Erasmus MC - Sophia children’s hospital (Rotterdam, The Netherlands). CF was diagnosed by neonatal screening and/or positive sweat test and DNA analysis [16]. As part of this monitoring program, chest CT scans and BALF were collected at the ages of 1, 3, and 5 years in 23 children. In 21 patients, two consecutive time points (1 and 3, or 3 and 5) were collected. The institutional review board of the Erasmus MC approved the study, and all parents signed informed consent. The I-BALL study is registered on Clinicaltrials.gov (identifier: NCT02907788).
Bronchoscopy and BALF collection.
Bronchoscopy was performed under general anesthesia. Three aliquots of 1 ml/kg saline were instilled and aspirated into the right middle lobe, and one in lingula or most affected lobe. All BALF samples were immediately put on ice in separate aliquots. For clinical determination of cell count and microbiology, a pooled portion of all aliquots of BALF was used. Cell count was done by counting and classifying 300 cells, and the percentages of total leukocyte and myeloid subsets were reported. The second and third aliquots from the right middle lobe were used for the assessment of inflammatory biomarkers. After centrifugation, pelleted cells were collected and fixed. Fixed cells and cell-free supernatants were stored at −80°C until batch analysis.
Chest CT and scoring.
Chest CT scans were collected without anesthesia at the same visit as the bronchoscopy using a Siemens SOMATOM® Force ultra-fast scanner. Free-breathing scans were acquired for each patient. The PRAGMA-CF CT scoring method was used to score structural lung damage on chest CT scans [17]. The PRAGMA-CF scoring method gives a quantitative measure of percentage bronchiectasis (%Bx) and total disease score (%Dis) as a portion of the total lung. %Dis includes bronchiectasis, mucus plugging, atelectasis and bronchial wall thickening. CT images were scored in one batch by a single investigator. Both repeatability and reproducibility were tested using intra- and intercorrelation coefficient by scoring a random selection of scans by the same or a separate investigator, respectively.
Lipidomics.
To quantify airway bioactive lipids, cell-free BALF supernatants were analyzed using an HPLC-MS/MS platform. An in-depth explanation of the method is reported in a previous publication by our group [15]. In brief, BALF samples were randomized and analyzed together with quality control, calibration, and blank samples. Lipids were analyzed on different platforms for positively charged, negatively charged, oxylipins, and oxidative stress-related lipids. By integration of assigned peaks, expressed as absolute concentration relative to deuterated internal standards, data were calibrated for analysis.
Soluble mediator quantification.
For soluble mediator measurements, cell-free BALF was used. NE activity was measured using a fluorometric assay with a specific elastase substrate (Z-Ala-Ala-Ala-Ala-)2RH110 (Catalog #600610, Cayman Chemicals, Ann Arbor, Michigan, USA), per manufacturer’s protocol. Myeloperoxidase (MPO) activity and protein abundance were measured using detection by immunocapture as previously reported [4]. We used the ELISA portion of these data which report total MPO protein; it was previously shown from the activity step of this assay that MPO in early CF BALF is active [4]. All BALF samples analyzed yielded significant data above background, which were included in regression analysis.
Human bronchial epithelial culture.
Control (non-CF) bronchial epithelial cells (HBEC) were obtained from lung resections at the Erasmus MC and differentiated on permeable inserts at air-liquid interface (ALI), as described in our previous work [18]. Non-CF HBEC cells were treated with LPA receptor agonist (VPC31143, 10 μM, Avanti Polar Lipids, Alabaster, AL, USA) for 8 hours. Culture medium from the basal side was collected and used for analysis. In other experiments, CFTR-inducible immortalized airway cells iCFBE [19] were obtained from Dr. Guido Veit, McGill University, Montreal, Canada and cultured at ALI, as described in [18], and used as described above.
Protein array.
Cell-free BALF from patients and medium from the basal side of HBEC-ALI cell cultures were analyzed using a Fluidigm-based protein array (Olink Proteomics, Uppsala, Sweden). The array simultaneously measures in 96 × 10 μl samples a set of 92 protein markers involved in inflammation and tumorigenesis. In brief, targeted proteins were bound by oligonucleotide-labeled antibody probes. Using microfluidic real-time PCR, the DNA sequence was detected and quantified [20]. Protein markers were excluded if their level was below the detection level in more than 20% of the samples.
Statistical analysis.
The non-parametric Spearman test was used for correlation analysis of given lipid levels with current and future clinical outcomes. To reduce the number of variables and preserve statistical power, principal component analysis (PCA) was used to generate compound parameters [21]. By applying the PCA algorithm, we calculated compound variables that explains over 60% variability summarizing correlated primary variables [22]. Samples were analyzed in same batch as previous publication and therefore grouping and weight were determined using both cohorts as described in a previous publication [15]. Prism Graphpad version 8 and RStudio analytics 3.6.1 software were used for analysis.
RESULTS
Clinical characteristics of enrolled patients.
This study enrolled 23 children with CF aged 1 (n=5) and 3 (n=18) years. Out of these 23, 21 had another chest CT scan at ages 3 (n=5) and 5 (n=16), respectively. Clinical data are summarized in Table 1, with full details of individual patients included in Suppl. Table 1. Three out of the 23 patients were diagnosed as CF-screen positive, inconclusive diagnosis (CFSPID). Excluding these three patients from our statistical analysis did not alter results noticeably, therefore they were included in all analyses. Quantitative total chest CT scores (%Dis [17]) at the time of the first BALF collection ranged from 0.2–5.4% (Table 1). Nine out of 23 patients had no bronchiectasis at time of BALF. Four of these nine patients developed detectable bronchiectasis in the next two years. Using a sensitive fluorescence assay (see methods) all patients had detectable NE levels in the BALF allowing a correlation analysis of the full cohort, in contrast to previous studies using an ELISA based colorimetric assay [1] . %Dis score positively correlated with NE and MPO activity and negatively with alveolar macrophage percentage (Table 2). PRAGMA-CF scores and pathology cell counts did not differ significantly based on sex, mutation, pancreatic status, bacterial culture results, or antibiotic treatment.
Table 1. Patient characteristics.
Patient characteristics of a full cohort of 23 patients. For numerical data, the median and range are reported. For categorical data, the number of patients, with the percentage of the full cohort, are reported. Age, gender, pancreatic status, mutation, culture results, antibiotics at the time of bronchoscopy, and pathology cell counts are acquired retrospectively from medical records of the patients. PRAGMA-CF %Dis and %Bx are measured quantitatively using the PRAGMA-CF scoring methods. CFTR mutations and individual data are listed in Table S1. PRAGMA-CF scores and pathology cell counts do not differ significantly when grouped based on gender, mutation, pancreas status, culture results, or antibiotic treatment. *“PRAGMA-CF after two years” contained 21 patients. Two patients did not receive a chest CT scan.
| Patient characteristics n=23 | ||||
| Age | 1 year | 5 | 22% | |
| 3 year | 18 | 78% | ||
| Gender | Male | 8 | 34.78% | |
| Female | 15 | 65.22% | ||
| Pancreas status | Insufficient | 19 | 82.61% | |
| Sufficient | 4 | 17.39% | ||
| Mutation | F508Del homozygote | 12 | 52.17% | |
| F508Del heterozygote | 10 | 43.48% | ||
| Other mutation | 1 | 4.35% | ||
| Culture result | Positive | 8 | 34.78% | |
| Negative | 15 | 65.22% | ||
| Positive | 1 | 4.35% | ||
| Negative | 22 | 95.65% | ||
| Antibiotics at time of bronchoscopy | Yes | 21 | 91.30% | |
| No | 2 | 8.70% | ||
| Pulmozyme at time of bronchoscopy | Yes | 7 | 30.40% | |
| No | 16 | 69.60% | ||
| PRAGMA-CF | %Disease score | 1.75% | (0.16–5.39) | |
| %Bronchiectasis | 0.07% | (0–0.62) | ||
| PRAGMA-CF after two years* | %Disease score | 1.27% | (0–9.09) | |
| %Bronchiectasis | 0.43% | (0–2.77) | ||
| Pathology cell count | %Neutrophils | 24% | (4–71) | |
| %Macrophages | 62% | (26–91) | ||
| %Lymphocytes | 5% | (2–20) | ||
| %Eosinophils | 0% | (0–3) | ||
Table 2. Correlation of chest CT score with inflammatory biomarkers.
Reporting Spearman rho correlation values of chest CT score with inflammation outcomes. Asterisks show p-values (*P < 0.05, ** P < 0.01, *** P < 0.001). PRAGMA-CF %Dis, %Bx, and %Dis after two years are measured quantitively using the PRAGMA-CF scoring methods. NE and MPO are measured using cell-free supernatant from BALF. BALF lymphocytes, eosinophils, alveolar macrophages, and neutrophil percentages are acquired as described in methods.
| PRAGMA %Bx | PRAGMA %Dis after 2 years | N.Elastase (mU/ml) | MPO (μg/ml) | Lymphocytes | Eosinophils | Alveolar macrophages | Neutrophils | |
|---|---|---|---|---|---|---|---|---|
| PRAGMA %Dis | 0.29 | 0.49* | 0.66*** | 0.69*** | 0.12 | 0.21 | −0.45* | 0.37 |
| PRAGMA %Bx | 0.26 | 0.32 | 0.12 | −0.18 | 0.34 | −0.18 | 0.21 | |
| PRAGMA %Dis after 2 years | 0.3 | 0.58** | 0.18 | 0.11 | −0.33 | 0.17 | ||
| N.Elastase (mU/ml) | 0.64** | 0.15 | 0.21 | −0.61** | 0.52* | |||
| MPO (ng/ml) | 0.045 | 0.18 | −0.75*** | 0.67*** | ||||
| Lymphocytes | 0.05 | −0.04 | −0.12 | |||||
| Eosinophils | −0.26 | 0.21 | ||||||
| Alveolar macrophages | −0.97*** |
Lipidomic analysis.
Lipidomic analysis was performed by mass spectrometry on BALF as described previously, focusing on positive lipids and oxidative stress panel, since significant correlations between early CF pathology and lipid species were observed in these categories in particular [15]. Bioactive lipid families including lysophosphatidic acid (LPA), lysophosphatidylcholines (LPC), phosphatidylcholines (PC), sphingolipids, isoprostanes, and prostaglandins (PG) were measurable in all BALF samples studied (Suppl Figure S1). To circumvent the loss of statistical power caused by the presence of a large number of compounds a given bioactive lipid family, we used dimensional reduction by PCA to reduce the number of variables within a bioactive lipid family to a single compound variable [15]. A heatmap of all compound lipid families shows robust correlation coefficients of the principal component eigenvectors with related individual lipids (Suppl Figure S1).
Lysolipids, sphingolipids and isoprostanes correlate with early structural lung disease.
PRAGMA-CF scores correlated with the compound lipid variables (Table 3), and the exclusion of patients with a mild phenotype (Suppl Table S1) did not alter the correlations significantly. Notably, %Dis scores correlated with both lysolipid principal components, LPC and LPA, the sphingosine principal component, comprising the ceramide precursors sphingomyelin, sphinganine, and sphingosine. The lipidomic platform also measured multiple PG species and their peroxidized isoprostane derivatives, which are considered markers of oxidative stress. Both the pro-inflammatory prostaglandin PGE2 and its peroxidized derivative 8-iso-PGE2 correlated with %Dis chest CT scores (Table 3). By contrast, the correlation between the phospholipid principal component (PC), a measure of the total phospholipid content of the BALF and %Dis was not significant. In addition, none of the principal components correlated significantly with the low bronchiectasis scores (%Bx). These data confirm and extend the results obtained previously by our group on a similar, independent cohort of Australian infants with CF [15].
Table 3. Correlation of lipids with chest CT score and inflammatory biomarkers.
Reporting Spearman rho correlation values of lipids with %Dis score and inflammatory biomarkers. Lipid levels from patient BALF are measured using an HPLC-MS/MS platform. LPA, LPC, ceramide precursors, and PC are summarized in a compound variable calculated using PCA. 8isoPGE1, 8isoPGE, and PGE2 levels are measured on the same platform. (* P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001), the absence of asterisks means the correlation did not reach the p-value threshold of 0.05.
| LPA | LPC | Ceramide precursors | PC | 8iso PGE1 | 8iso PGE2 | PGE2 | |
|---|---|---|---|---|---|---|---|
| PRAGMA %Dis | 0.58** | 0.58** | 0.73*** | 0.19 | 0.38 | 0.59** | 0.53** |
| PRAGMA %Bx | 0.3 | 0.31 | 0.27 | 0.01 | 0.2 | 0.31 | 0.27 |
| PRAGMA %Dis after 2 years | 0.42 | 0.49* | 0.45* | 0.05 | 0.51* | 0.35 | 0.47* |
| Lymphocytes | 0.27 | 0.2 | 0.2 | 0.63** | 0.03 | 0.12 | 0.28 |
| Eosinophils | 0.49* | 0.32 | 0.24 | 0.08 | −0.21 | 0 | 0.35 |
| Alveolar macrophages | −0.55** | −0.56** | −0.56** | −0.06 | −0.47* | −0.51* | 0.53* |
| Neutrophils | 0.44* | 0.45* | 0.47* | −0.02 | 0.39 | 0.44* | 0.39 |
| NE (mU/ml) | 0.73*** | 0.77**** | 0.79**** | 0.46* | 0.61** | 0.69*** | 0.76**** |
| MPO (ng/ml) | 0.78**** | 0.76**** | 0.81**** | 0.15 | 0.48* | 0.72*** | 0.7*** |
Bioactive lipids correlate with CF lung neutrophil infiltration and activity.
Next, we sought to determine whether bioactive lipids detected in the BALF would relate to the inflammatory status of the airways. Here, BALF relative cell count was used as a measure of inflammation in the airways (Table 1). Isoprostanes, PGE2, and the LPA, LPC, and ceramide precursor principal components all correlated significantly with BALF percentages of neutrophil (positively) and macrophages (negatively), whereas the compound phospholipid parameter did not (Table 3). Moreover, we identified strong correlations between compound LPA, LPC, and ceramide precursors, isoprostanes and PGE2 with NE and MPO activities in BALF supernatant (Table 3), suggesting a potential relationship between the inflammatory profile and bioactive lipids in the CF lung.
Pro-inflammatory cytokines are elevated in patients with high PRAGMA-CF score and bioactive lipid levels.
To further investigate the relationship between bioactive lipid levels and lung disease, protein markers of inflammation and tissue remodeling were measured in BALF supernatant using a protein array. Of the 96 included mediators, 62 were reliably measured in the whole patient cohort. Correlation scores between these 62 markers and PRAGMA-CF scores, BALF lipid levels, BALF neutrophil and macrophage percentages, and neutrophil enzyme (NE, MPO) activities are presented in a heat map (Figure 1). Almost all detectable markers on this array correlated positively with PRAGMA-CF score and BALF NE and MPO activities, and negatively with BALF macrophage percentage. Importantly, most protein mediators correlated with bioactive lipid levels, in particular, lysolipids (LPA and LPC), ceramide precursors and isoprostanes.
Figure 1. Protein array correlation heatmap.
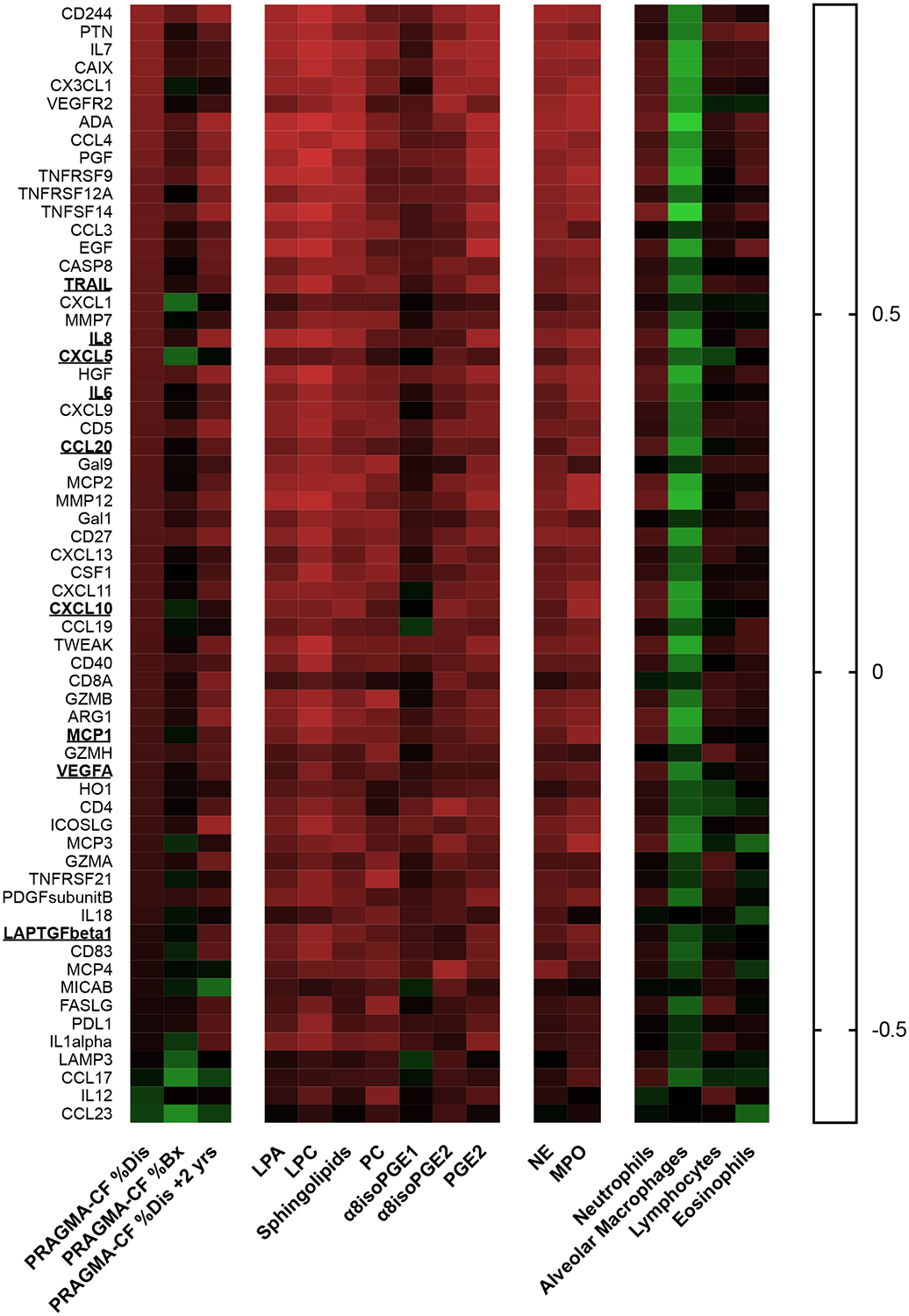
Heatmap showing Spearman rho values of proteins measured in BALF by protein array analysis (Olink INF/ONC) array, with PRAGMA-CF scores, lipid values, and inflammation outcomes. Underlining indicates eight markers that are secreted by primary airway cells (HBEC-ALI, INF array), elevated by the LPA agonist VPC31143 (Table S2), and also correlate with lipids and inflammation markers.
Bioactive lipids in BALF predict structural lung disease progression.
The LPC and ceramide precursor compound variables measured in BALF correlated significantly with %Dis score measured 2 years later (Table 3). PGE2 also correlated with %Dis two years after the first bronchoscopy. The correlation with isoprostane 8-iso-PGE1 was significant, but its related species 8-iso-PGE2 was not (Table 3). The LPA compound variable, correlated as well but did not quite reach the significance cut-off in this cohort (Spearman Rho 0.42, P-value = 0.06). By contrast, the PC principal component did not correlate with %Dis score. In addition, the bronchiectasis score (%Bx) measured two years post-BALF collection did not correlate with any of the measured lipid compound variables. Together, these results suggest the presence of a relationship between certain bioactive lipids and progression of lung damage in infants with CF.
Treatment with an LPA agonist enhances cytokine and growth factor shedding in human bronchial epithelial cells in vitro.
To provide insights into the potential role of the lysophospholipid receptor (LPAn) signaling pathway in CF lung disease, we treated healthy differentiated primary human bronchial epithelial cells (HBEC) grown at air-liquid interface (ALI) with an LPA1–3 agonist (VPC31143). After activation with VPC31143, the basal medium was collected and compared to that from control untreated HBEC-ALI cells (N=3), using a targeted protein array. Thirty out of the ninety-two markers were detected in all samples, of which eighteen were expressed 2-fold higher or more in VPC31143-treated compared to control HBEC-ALI cells (Table S2). Several of these proteins were detected in the CF BALF as well and correlated with BALF LPA, isoprostane, and PRAGMA-CF (Figure 1 and Table S2). VPC31143 treatment also strongly activated ADAM17-dependent shedding of the EGF receptor (EGFR) agonist and growth factor amphiregulin (AREG) in normal HBEC-ALI (Figure 2A), as well as in immortalized CFTR-deficient airway cells (iCFBE, [23] (Figure 2B). Induction of CFTR function with doxycycline in iCFBE reduced basal AREG shedding, as shown previously [18], and substantially decreased the stimulation by VPC31143. Furthermore, in EGF-starved HBEC-ALI, stimulation with VPC31143 induced a relocation of the EGF receptor antigen from a cytoplasmic endocytic pool (Figure 2C) to a lateral membrane-bound position (Figure 2D). This is consistent with a change from a ligand free monomeric intracellular state, to a membrane-bound state [24] .This suggests a potential cross-talk between EGFR/ADAM17/AREG and LPAn receptor systems in airway epithelial cells, enhancing pro-fibrotic and pro-inflammatory signaling, as has been observed in other cellular models [25]. A figure sketching the most relevant reported relationships between lipid species, signaling proteins, and lung disease is presented in Suppl. Figure S2.
Figure 2. Activation of the EGFR/ADAM17/AREG axis in airway cells by lysolipids.
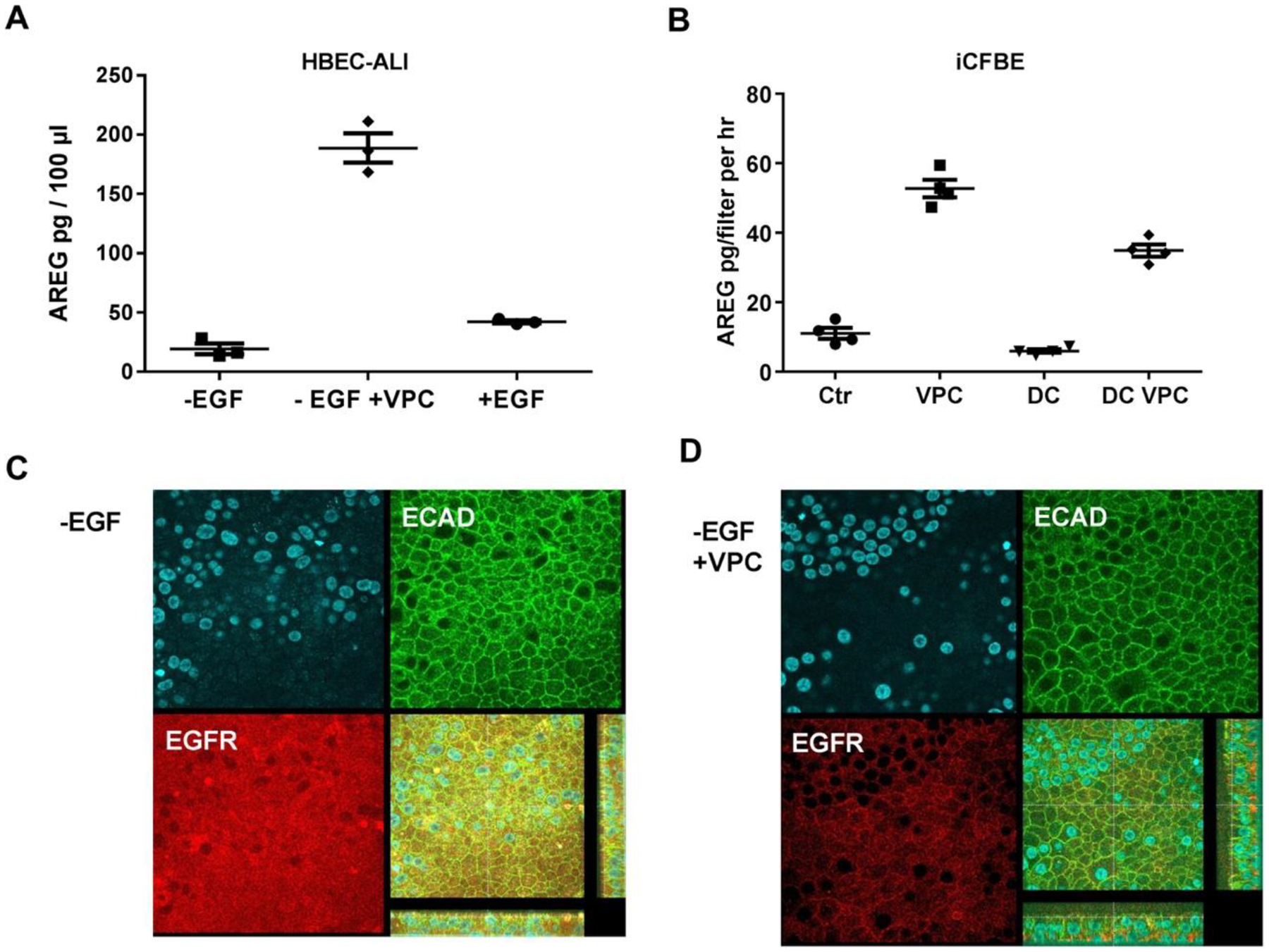
A: Differentiated normal bronchial epithelial cells (HBEC-ALI) were cultured in the absence of EGF for 24 hrs, to reduce the activity of EGFR/ADAM17 dependent AREG shedding to basal levels [23]. Subsequently, a vehicle control (-EGF), or 10 μM of the LPA1–3 receptor agonist VPC31114 (-EGF +VPC), or EGF, were added for 8 hours. Basal medium was collected after 24 hrs, and AREG was measured by ELISA. B: Immortalized CF epithelial cells carrying a doxycycline-inducible wtCFTR gene (iCFBE) [19], cultured on filter inserts at the air-liquid interface as described previously [18], were treated with PLA receptor agonist (VPC) or vehicle (Ctr). In parallel, cells were treated with doxycycline for 24 hrs to induce CFTR expression (DC), before treatment with VPC. Basal media were collected and analyzed by ELISA. C,D: Human HBEC-ALI cells cultured in the absence of EGF as in A, treated with vehicle (C) or VPN31114 (D) were fixed and stained with a nuclear stain (DAPI, blue, upper left), and antibodies against the lateral membrane marker E-cadherin (ECAD, green), and the EGFR receptor (EGFR) as described in [18]. The lower right panels show the merged fluorescence signals, plus the z-stacks (apical membrane towards the panel, with the focal planes used, yellow lines). Data show a marked and CFTR-dependent increase of AREG shedding, upon LPA receptor agonist treatment of differentiated polarized airway cells, and relative relocalization of EGFR antigen from the cytoplasm to the lateral membrane, consistent with recruitment of an inactive form to the plasma membrane.
Discussion
In this study we showed that multiple species of lysolipids, sphingolipids, isoprostanes and prostaglandins measured in BALF using HPLC-MS/MS correlated with structural lung damage in CF infants measured by quantitative chest CT analysis. We found that children with high BALF bioactive lipid levels and PRAGMA-CF scores also displayed high PRAGMA-CF scores 2 years later, suggesting that they could contribute to or serve as risk factors for the progression of early CF lung disease. Additionally, we found that BALF MPO was associated with structural damage two years later. While this is not strictly a predictive result because PRAGMA %Dis was largely unchanged in this period, it does suggest that the damage associated with these proteins becomes firmly established, underscoring importance of early interventions.
BALF lipids correlated with inflammatory markers, namely relative neutrophil and macrophage percentages, NE and MPO enzymatic activities, and a large panel of cytokines and growth factors (Figure 2) that together control pro-inflammatory and tissue remodeling signaling (Figure S2). The contribution of the individual signaling molecules remains to be established. This confirms and significantly extends our previous data showing a correlation of isoprostanes and lysolipids with PRAGMA-CF scores, neutrophilaemia and IL-8 obtained in a cross-sectional study with the independent Australian AREST-CF cohort [15]. At the time of BALF we did not observe a significant correlation between the low bronchiectasis scores and any of our parameters. Because only four out of nine patients developed detectable bronchiectasis after BALF, we also could not establish a correlation between our lipid markers and the progression of bronchiectasis. A correlation between LPC and %Dis was also found by our group in a partially overlapping cohort, using an untargeted metabolomics approach [4]. Aside from these previous publications from our group, to our knowledge no other study explored the potential correlations in CF infant airways between structural lung disease and a comprehensive panel of bioactive lipids. A recent publication compared BALF lipids of CF children, children with protracted bacterial bronchitis, and healthy controls [26]. That study showed that total phospholipid species, the main components of surfactant, are not markedly different in CF, and confirmed that LPC was significantly higher in CF children compared to non-CF children, consistent with our previous and current study. Together, these data illustrate the value of monitoring BALF lipids to study the mechanism and progression of early CF lung disease. However, future studies with a longer longitudinal surveillance and a larger cohort of CF infants will be needed to validate the monitoring of BALF active lipids as disease biomarkers.
In both this study and a previous study [15], enhanced lysolipid signaling correlated with early CF lung disease. The source of these lysolipids could be activated myeloid cells, or enhanced activity of the phospholipase cPLA2 in CF epithelia [27]. The potential effect of enhanced lysolipids on the progression of CF lung disease is illustrated by our in vitro data. In normal primary airway epithelial cells grown at ALI, we observed a marked increase of submucosal pro-inflammatory and tissue remodeling signaling by the lysolipid receptor agonist VPC31143. Basolateral ADAM17/EGFR-dependent shedding of the growth factor AREG, and secretion of several cytokines involved in neutrophil and macrophage recruitment and activation were strongly enhanced by VPC31143 in both primary normal and immortalized CF epithelial cells (Table S2, Figure S2). This is consistent with previous studies showing the role of lysophosphatidic acid (LPA) in promoting a pro-inflammatory response in human airway epithelial cells and in mouse models [6, 28]. Moreover, we showed here that BALF LPA correlates with extracellular NE and MPO, consistent with increased neutrophil transmigration and degranulation, as previously shown in blood neutrophils [29]. Together, these results suggest a potential role of mucosal lysolipids, and in particular LPA species, in inducing myeloid inflammation in early CF lung disease by triggering epithelial LPAn receptors.
In addition to LPA, the BALF ceramide precursor principal component, representing sphingosine, sphinganine, and sphingomyelin, had strong correlations with %Dis and inflammatory markers in this cohort. Though not known for their biological activity, their derivatives, including ceramides and sphingosine phosphate, are reportedly linked to CF lung disease, and therapeutic options are being investigated [7–9, 11]. In our previous work, the ratio of long to very long chain ceramides (LCC/VLVV) was significantly higher in CF compared to non-CF conditions, and correlated with %Dis [15]. It is hypothesized that the imbalance of ceramide species with respect to chain length modulates the activity of signaling receptors, which can be addressed by treatment with the retinoid fenretinide [11, 13]. Moreover, amitriptyline, an inhibitor of the rate limiting enzyme in ceramide production, sphingomyelinase, decreased the development of P. aeruginosa-induced pneumonia and inflammation in CF mutant mice [30] and showed an improvement of FEV1 and bodyweight of CF patients after one year of therapy [31]. However, it is not obvious how the sphingosine parameter in cell-free BALF relates to specific target enzymes and downstream substrate pools in the various contributing cell populations. Future research involving multi-omics studies of advanced cellular models (“lung on a chip”) could shed further light on this and help us to explore therapeutic options.
Isoprostanes are mainly generated by non-enzymatic oxidation of polyunsaturated fatty acids (PUFA) and their derivatives and are therefore considered markers of free radical production, which in the BALF from inflamed lung has multiple sources, epithelial and myeloid. Reid et al. reported elevated isoprostane (8isoPGE2a) levels in sputum of adult CF patients during exacerbations, correlating with lung function (FEV1) [5]. We found that BALF isoprostane levels in CF infants correlated with chest CT scores and BALF inflammation markers (Table 3, Figure 1), extending and confirming our previous results in a different, cross-sectional cohort [15], and establishing isoprostanes in BALF as biomarkers for oxidative stress in early CF lung disease. In a parallel study, we observed that markers of oxidative stress and downstream pro-inflammatory signaling are enhanced in CF compared to non-CF HBEC-ALI, suggesting that this is an inherent trait of CF airways [23].
While this study provides insights in the role of active biolipids in promoting early CF lung disease, there are several important limitations to mention. First, BALF analysis reflects the activity of multiple cell types over a large proximal to distal area. Thus, the cellular origin of the molecules measured, the sequence of events and their exact roles in pathogenesis are still unclear. Further clinical studies and experimental investigation in animal models and advanced cell culture models are needed to provide more insight into this key issue. Second, while the present longitudinal CF infant cohort yielded new insights, its size is limited. A healthy control group would have been ideal, however submitting healthy children to BAL collection and chest CT on a regular basis is unethical.
In conclusion, abnormal lipid metabolism in CF airways likely contributes to amplified inflammatory signals generated by CF airway epithelia, possibly starting at birth, creating a vicious circle of inflammation and lung damage. Measuring lipid levels may be clinically useful for identifying high-risk patients. Since structural lung damage occurs in the first years of life, early intervention in this cascade at the lipid level could prove beneficial. In fact, in CF mutant mice, intervention on the S1P metabolic pathway reduced mucus production and inflammation [15]. Intervention at the epithelial LPAn receptors would be an option, multiple antagonists are being developed or on the market (BMS-986020, BMS-986234, NCT01766817), none of which are in trial for CF thus far. Future studies are required to explore this therapeutic space.
Supplementary Material
Highlights.
Abnormal lipid metabolism correlates with early CF lung disease and inflammation
Bioactive lipids and lung damage correlate with cytokines and growth factors in BALF
Bioactive lipids including lysolipids correlate with future structural lung damage
Lysolipid receptor agonist enhances cytokine shedding by airway epithelial cells
Acknowledgments
We sincerely thank Elrozy R. Andrinopoulou, registered biostatistician from the department of statistics in Erasmus MC for initial guidance in statistical analysis, Badies Manai, research nurse, for recruitment of patients and data management, at McGill University, Montreal, Canada: Drs Guido Veit and Gergely Lukacs for providing the CFTR inducible iCFBE cells and Dr John Hanrahan for providing primary bronchial epithelial cells.
Funding:
Supported By: NIH R01HL126603; ERARE INSTINCT; NCFS HITCF-1.
ABBREVIATIONS
- ADAM
a disintegrase and metalloproteinase
- ALI
air-liquid interface
- BAL
bronchoalveolar lavage
- BALF
bronchoalveolar lavage fluid
- Bx
bronchiectasis
- CF
cystic fibrosis
- CFSPID
cystic fibrosis Screen Positive, Inconclusive Diagnosis
- CFTR
cystic fibrosis transmembrane conductance regulator
- CT
computed tomography
- Dis
disease score
- EGFR
epidermal growth factor receptor
- GSH
glutathione
- HBEC
human bronchial epithelial culture
- HPLC-MS/MS
high-performance liquid chromatography-mass spectrometry
- iCFBE
immortalized Cystic fibrosis bronchial epithelial cells
- IL-8
Interleukin 8
- LPA
lysophosphatidic acid
- LPC
lysophosphatidylcholine
- MPO
myeloperoxidase
- NE
neutrophil elastase
- PC
phosphatidylcholine
- PCA
Principal component analysis
- PG
prostaglandin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statement.
All authors state no conflict of interest.
REFERENCES
- 1.Sly PD, et al. , Lung disease at diagnosis in infants with cystic fibrosis detected by newborn screening. American journal of respiratory and critical care medicine, 2009. 180(2): p. 146–152. [DOI] [PubMed] [Google Scholar]
- 2.Rosen BH, et al. , Infection is not required for mucoinflammatory lung disease in CFTR-knockout ferrets. American journal of respiratory and critical care medicine, 2018. 197(10): p. 1308–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Khan TZ, et al. , Early pulmonary inflammation in infants with cystic fibrosis. American journal of respiratory and critical care medicine, 1995. 151(4): p. 1075–1082. [DOI] [PubMed] [Google Scholar]
- 4.Chandler JD, et al. , Myeloperoxidase oxidation of methionine associates with early cystic fibrosis lung disease. European Respiratory Journal, 2018: p. 1801118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reid DW, et al. , Oxidative stress and lipid-derived inflammatory mediators during acute exacerbations of cystic fibrosis. Respirology, 2007. 12(1): p. 63–69. [DOI] [PubMed] [Google Scholar]
- 6.Zhao J, et al. , Lysophosphatidic acid receptor 1 modulates lipopolysaccharide-induced inflammation in alveolar epithelial cells and murine lungs. American Journal of Physiology-Lung Cellular and Molecular Physiology, 2011. 301(4): p. L547–L556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Teichgräber V, et al. , Ceramide accumulation mediates inflammation, cell death and infection susceptibility in cystic fibrosis. Nature Medicine, 2008. 14(4): p. 382–391. [DOI] [PubMed] [Google Scholar]
- 8.Veltman M, et al. , Correction of lung inflammation in a F508del CFTR murine cystic fibrosis model by the sphingosine-1-phosphate lyase inhibitor LX2931. American Journal of Physiology-Lung Cellular and Molecular Physiology, 2016. 311(5): p. L1000–L1014. [DOI] [PubMed] [Google Scholar]
- 9.Karandashova S, et al. , Neutrophil elastase correlates with increased sphingolipid content in cystic fibrosis sputum. Pediatric pulmonology, 2018. 53(7): p. 872–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tabazavareh ST, et al. , Lack of sphingosine causes susceptibility to pulmonary Staphylococcus aureus infections in cystic fibrosis. Cellular Physiology and Biochemistry, 2016. 38(6): p. 2094–2102. [DOI] [PubMed] [Google Scholar]
- 11.Garić D, et al. , Biochemistry of very-long-chain and long-chain ceramides in cystic fibrosis and other diseases: The importance of side chain. Progress in Lipid Research, 2019. 74: p. 130–144. [DOI] [PubMed] [Google Scholar]
- 12.Teichgräber V, et al. , Ceramide accumulation mediates inflammation, cell death and infection susceptibility in cystic fibrosis. Nature medicine, 2008. 14(4): p. 382. [DOI] [PubMed] [Google Scholar]
- 13.Garić D, et al. , Fenretinide differentially modulates the levels of long-and very long-chain ceramides by downregulating Cers5 enzyme: evidence from bench to bedside. Journal of Molecular Medicine, 2017. 95(10): p. 1053–1064. [DOI] [PubMed] [Google Scholar]
- 14.Guilbault C, et al. , Cystic fibrosis fatty acid imbalance is linked to ceramide deficiency and corrected by fenretinide. American journal of respiratory cell and molecular biology, 2009. 41(1): p. 100–106. [DOI] [PubMed] [Google Scholar]
- 15.Scholte BJ, et al. , Oxidative stress and abnormal bioactive lipids in early cystic fibrosis lung disease. Journal of Cystic Fibrosis, 2019. [DOI] [PubMed] [Google Scholar]
- 16.Dankert-Roelse JE, et al. , Newborn blood spot screening for cystic fibrosis with a four-step screening strategy in the Netherlands. Journal of Cystic Fibrosis, 2019. 18(1): p. 54–63. [DOI] [PubMed] [Google Scholar]
- 17.Rosenow T, et al. , PRAGMA-CF. A quantitative structural lung disease computed tomography outcome in young children with cystic fibrosis. American journal of respiratory and critical care medicine, 2015. 191(10): p. 1158–1165. [DOI] [PubMed] [Google Scholar]
- 18.Stolarczyk M, et al. , Extracellular oxidation in cystic fibrosis airway epithelium causes enhanced EGFR/ADAM17 activity. American Journal of Physiology-Lung Cellular and Molecular Physiology, 2018. 314(4): p. L555–L568. [DOI] [PubMed] [Google Scholar]
- 19.Veit G, et al. , Proinflammatory cytokine secretion is suppressed by TMEM16A or CFTR channel activity in human cystic fibrosis bronchial epithelia. Mol Biol Cell, 2012. 23(21): p. 4188–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Assarsson E, et al. , Homogenous 96-Plex PEA Immunoassay Exhibiting High Sensitivity, Specificity, and Excellent Scalability. PLoS ONE, 2014. 9(4): p. e95192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Joliffe I and Morgan B, Principal component analysis and exploratory factor analysis. Statistical methods in medical research, 1992. 1(1): p. 69–95. [DOI] [PubMed] [Google Scholar]
- 22.Greenacre M and Primicerio R, Multivariate analysis of ecological data. 2014: Fundacion BBVA. [Google Scholar]
- 23.Stolarczyk M, et al. , Extracellular oxidation in cystic fibrosis airway epithelium causes enhanced EGFR/ADAM17 activity. Am J Physiol Lung Cell Mol Physiol, 2018. 314(4): p. L555–L568. [DOI] [PubMed] [Google Scholar]
- 24.Tanaka T, et al. , Ligand-activated epidermal growth factor receptor (EGFR) signaling governs endocytic trafficking of unliganded receptor monomers by non-canonical phosphorylation. Journal of Biological Chemistry, 2018. 293(7): p. 2288–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Panupinthu N, et al. , Self-reinforcing loop of amphiregulin and Y-box binding protein-1 contributes to poor outcomes in ovarian cancer. Oncogene, 2014. 33(22): p. 2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seidl E, et al. , Lavage lipidomics signatures in children with cystic fibrosis and protracted bacterial bronchitis. Journal of Cystic Fibrosis, 2019. [DOI] [PubMed] [Google Scholar]
- 27.Dif F, et al. , Critical role of cytosolic phospholipase A2α in bronchial mucus hypersecretion in CFTR-deficient mice. European Respiratory Journal, 2010. 36(5): p. 1120–1130. [DOI] [PubMed] [Google Scholar]
- 28.Cummings R, et al. , Protein Kinase Cδ Mediates Lysophosphatidic Acid-induced NF-κB Activation and Interleukin-8 Secretion in Human Bronchial Epithelial Cells. 2004. 279(39): p. 41085–41094. [DOI] [PubMed] [Google Scholar]
- 29.Tou J.-s. and Gill JS, Lysophosphatidic acid increases phosphatidic acid formation, phospholipase D activity and degranulation by human neutrophils. Cellular signalling, 2005. 17(1): p. 77–82. [DOI] [PubMed] [Google Scholar]
- 30.Becker KA, et al. , Acid Sphingomyelinase Inhibitors Normalize Pulmonary Ceramide and Inflammation in Cystic Fibrosis. American Journal of Respiratory Cell and Molecular Biology, 2010. 42(6): p. 716–724. [DOI] [PubMed] [Google Scholar]
- 31.Adams C, et al. , Long-Term Pulmonal Therapy of Cystic Fibrosis-Patients with Amitriptyline. 2016. 39(2): p. 565–572. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.