

Summary:
Cytokine-inducible SH2-containing protein (CIS, encoded by the gene CISH) is a key negative regulator of IL-15 signaling in natural killer (NK) cells. Here, we developed human CISH-knockout (CISH−/−) NK cells using an induced pluripotent stem cell-derived NK cell (iPSC-NK cell) platform. CISH−/− iPSC-NK cells demonstrate increased IL-15 mediated JAK-STAT signaling activity. Consequently, CISH−/− iPSC-NK cells exhibit improved expansion and increased cytotoxic activity against multiple tumor cell lines when maintained at low cytokine concentrations. CISH−/− iPSC-NK cells display significantly increased in vivo persistence and inhibition of tumor progression in a leukemia xenograft model. Mechanistically, CISH−/− iPSC-NK cells display improved metabolic fitness characterized by increased basal glycolysis, glycolytic capacity, maximal mitochondrial respiration, ATP-linked respiration and spare capacity respiration mediated by mTOR signaling that directly contributes to enhanced NK cell function. Together, these studies demonstrate CIS plays a key role to regulate human NK cell metabolic activity and thereby modulate anti-tumor activity.
Graphical Abstract
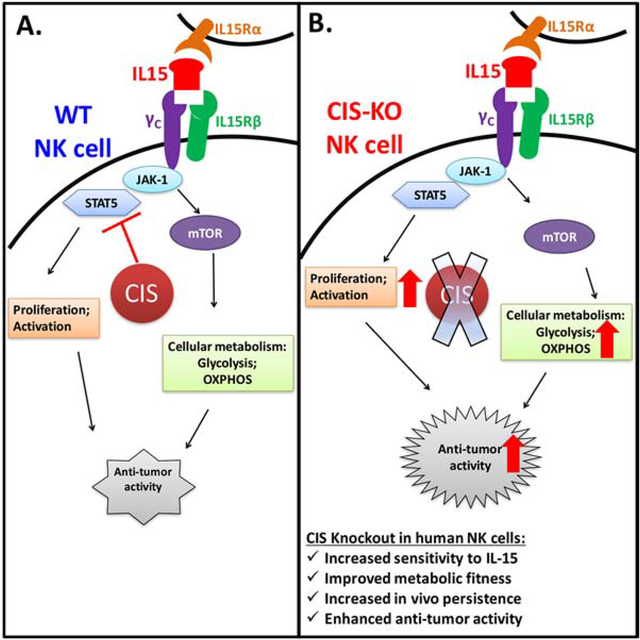
eTOC blurb:
CISH normally inhibits IL-15 signaling in natural killer (NK) cells. Here, Zhu and colleagues delete CISH expression in NK cells derived from human induced pluripotent stem cells (iPSCs). CISH−/− iPSC-derived NK cells demonstrate improved killing of tumor cells that is directly attributable to a more efficient metabolic profile.
Introduction:
Natural killer (NK) cells are an immune effector cell population with an intrinsic ability to kill virus-infected and tumor cells without prior antigen sensitization (Hodgins et al., 2019; Miller and Lanier, 2019; Morvan and Lanier, 2016; Zhu et al., 2018). NK cell-mediated cytotoxicity is regulated by integrating signals from a repertoire of activating and inhibitory receptors(Miller and Lanier, 2019). Activated NK cells can kill target cells through different mechanisms, including release of preformed cytolytic granules and proinflammatory cytokines as well as trigging cell apoptosis through death-receptor pathways by inducing death ligands such as FAS ligand and TRAIL(Miller and Lanier, 2019; Morvan and Lanier, 2016).
Clinical trials have shown allogeneic primary NK cells from peripheral blood (PB-NK) and umbilical cord blood (UCB-NK) to be safe without causing significant toxicity such as cytokine release syndrome (CRS), neurotoxicity or graft-versus-host disease (GVHD) (Dolstra et al., 2017; Geller et al., 2011; Miller and Lanier, 2019; Miller et al., 2005; Romee et al., 2016). While allogeneic NK cells have been shown to possess potent anti-AML activity, their efficacy for treating solid tumors has been limited (Hodgins et al., 2019; Miller and Lanier, 2019). One of the major reasons is adoptively transferred primary NK cells typically persist in vivo for a relatively short period of time limiting their anti-tumor efficacy. Unlike autologous CAR-T cell-based therapies that can persist and remain functional for months or years (Porter et al., 2015), allogeneic NK cells typically survive only a few weeks in the adoptive transfer setting (Geller et al., 2011; Miller et al., 2005). Therefore, engineering NK cells to improve their in vivo persistence would likely be beneficial to improve their anti-tumor activity. Human induced pluripotent stem cells (iPSCs) can be precisely genetically-modified at a clonal level using both viral and non-viral methods and then efficiently differentiated to produce mature NK cells (iPSC-NK cells)(Knorr et al., 2013; Li et al., 2018). Therefore, iPSCs provide an important platform to produce human NK cells with improved anti-tumor activity (Li et al., 2018; Zhu et al., 2020; Zhu et al., 2018). Indeed, previous studies have expressed novel NK cell-specific CARs that mediate improved killing of ovarian cancer cells in vitro and in vivo(Li et al., 2018). Additionally, stabilized expression of CD16 in iPSC-NK cells leads to improved antibody-dependent cell-mediated cytotoxicity (ADCC) and improved killing of both hematologic malignancies and solid tumors(Zhu et al., 2020).
IL-15 stimulates multiple NK cell functions including differentiation, proliferation, activation and survival (Huntington et al., 2009; Ranson et al., 2003). In clinical trials, IL-15 has been shown to stimulate NK cell proliferation and IL-15 levels correlated with in vivo expansion of infused NK cells in patients (Miller et al., 2018; Romee et al., 2018; Wrangle et al., 2018). However, high doses of IL-15 can cause toxicities (Floros and Tarhini, 2015; Miller et al., 2018). Therefore, increasing the sensitivity of NK cells to IL-15 to enable lower treatment doses of this cytokine provides an attractive approach to improve the anti-tumor activity of NK cells while eliminating the need for high doses of supplemental cytokines.
CIS is the first member identified in the suppressor of cytokine signaling (SOCS) protein family (other members: SOCS 1–7)(Hilton et al., 1998). SOCSs are induced by cytokines and in turn inhibit cytokine signaling, forming a classic negative feedback loop(Delconte et al., 2016; Putz et al., 2017). All SOCS proteins contain SOCS box sequence motif which enable the SOCS to function as adaptors for an E3 ubiquitin ligase complex and direct SOCS-interacting proteins to proteasomal degradation(Zhang et al., 2015). CIS, SOCS1 and SOCS3 have all been shown to bind directly to JAK1, inhibiting JAK-STAT signaling activation(Delconte et al., 2016; Kershaw et al., 2013). In mice, deletion of Cish augments IL-4 and IL-2 signaling in CD4+ T cells and increases sensitivity to IL-15 in NK cells, leading to better control of experimental tumor metastasis (Delconte et al., 2016; Putz et al., 2017). The same group showed CIS might be important for human NK cell immunity by deletion of CISH in human primary NK cells(Rautela et al., 2018), though another group that used CRISPR/Cas-mediated knock-out of CISH in primary human NK cells did not find improved anti-tumor activity(Pomeroy et al., 2018). Cish has also been reported to be induced by TCR stimulation in CD8+ T cells and inhibits their anti-tumor activity(Palmer et al., 2015). In humans, variants of CISH are associated with susceptibility to infectious diseases(Khor et al., 2010; Sun et al., 2014).
Cellular metabolism is now recognized to play a key role in regulating the function and differentiation of immune cells(Mah and Cooper, 2016; Marcais et al., 2017; O’Brien and Finlay, 2019; Pearce et al., 2013). Immune cells undergo dynamic metabolic shifts to support their activity and recent studies have shown this is specifically true in the case of NK cell function(Gardiner, 2019; Kobayashi and Mattarollo, 2017; Poznanski and Ashkar, 2019; Wegiel et al., 2018). For example, IFN-γ production via activating NK receptors requires glucose-driven oxidative phosphorylation(Keppel et al., 2015). Another study showed that the mammalian target of rapamycin complex 1 (mTORC1), a key regulator of cellular metabolism, is robustly stimulated in activated NK cells and is required for the production of IFN-γ(Donnelly et al., 2014). Two separate groups demonstrated mTOR could be activated by IL-15 and is essential for IL-15-mediated NK cell proliferation during development and activation(Mao et al., 2016; Marais et al., 2014). In addition, both aberrant glucose metabolism(Cong et al., 2018) and lipid accumulation(Michelet et al., 2018) in NK cells lead to dysfunction. However, there has been limited data showing that improving the metabolic fitness of NK cells mediates improved human NK cell function(Kobayashi and Mattarollo, 2017; Poznanski and Ashkar, 2019).
We hypothesized that deletion of CISH in human NK cells would improve their anti-tumor activities, similar to the approach that blocking the critical immune checkpoints such as PD-1 or CTLA-4. To test our hypothesis, we leveraged the iPSC-NK cell platform to knockout CISH in human NK cells at the clonal level. By utilizing iPSCs to derive gene-deleted NK cells, we ensure 100% of cells have the deleted gene, compared to studies that have used the CRISPR/Cas9 system to knockout genes in human PB-NK cells which results in lower and more variable amounts of gene deletion and must be repeated with every donor cell population(Pomeroy et al., 2019). We demonstrate that CISH−/− iPSC-NK cells have improved expansion, enhanced anti-tumor activity and persistence in vitro and in vivo. Notably, deletion of CISH in human NK cells leads to augmented single cell polyfunctionality, a profile that has been demonstrated to positively contribute to clinical outcome in CAR-T cells (Rossi et al., 2018). In addition, we show that CISH−/− iPSC-NK cells exhibited improved metabolic fitness which is mediated by mTOR signaling pathway. More importantly, the increased metabolic fitness directly contributes to improved anti-tumor function in CISH−/− iPSC-NK cells.
Results and Discussion:
Generation of CISH-KO NK cells from human iPSCs
Previous studies from our lab demonstrate that human iPSCs and human embryonic stem cells (hESCs) can be routinely differentiated into NK cells with phenotype and function similar to PB-NK cells(Knorr et al., 2013; Zhu and Kaufman, 2019). Here, we first examined CIS expression during different stages of NK cell differentiation. CIS expression was not detected in either undifferentiated iPSCs or early stage hematopoietic progenitor cells (CD34+ cells isolated at day 6 of differentiation) (Supplemental Figure S1A). However, CIS is induced by either IL-2 or IL-15 in iPSC-derived NK cells in a time-dependent manner, similar to PB-NK cells (Figures 1A, B). iPSC provides a unique platform to do genetic modifications at the clonal level and to then derive large numbers (clinical scale) of genetically engineered NK cells produced from a standardized starting cell source(Knorr et al., 2013; Li et al., 2018; Zhu and Kaufman, 2019). To explore the role of CIS in human NK cells, we utilized this platform and knocked out CISH in human iPSCs by CRISPR/Cas9 technology (Figure 1C). We used a pair of gRNAs located in direct and complementary strand targeting exon 3 of the CISH gene right after the start codon (exon 2) (Figure 1C). This strategy was reported to enhance genome editing specificity(Ran et al., 2013). We then selected 57 single clones and identified a CISH−/− clone with frame-shift mutations in both alleles (Figures 1D, E). All the mutations were within the range of the gRNA targeting region. CISH−/− iPSCs grow normally and maintain the typical phenotype and karyotype of undifferentiated iPSCs (Supplemental Figures S1B, C, D), indicating CIS does not regulate the maintenance of undifferentiated human pluripotent stem cells. We then derived human CISH−/− iPSC-NK cells from clonal CISH−/− iPSCs and confirmed the loss of CIS expression in these cells by immunoblotting (Figure 1E).
Figure 1. Generation of CISH-KO NK cells from human iPSCs.
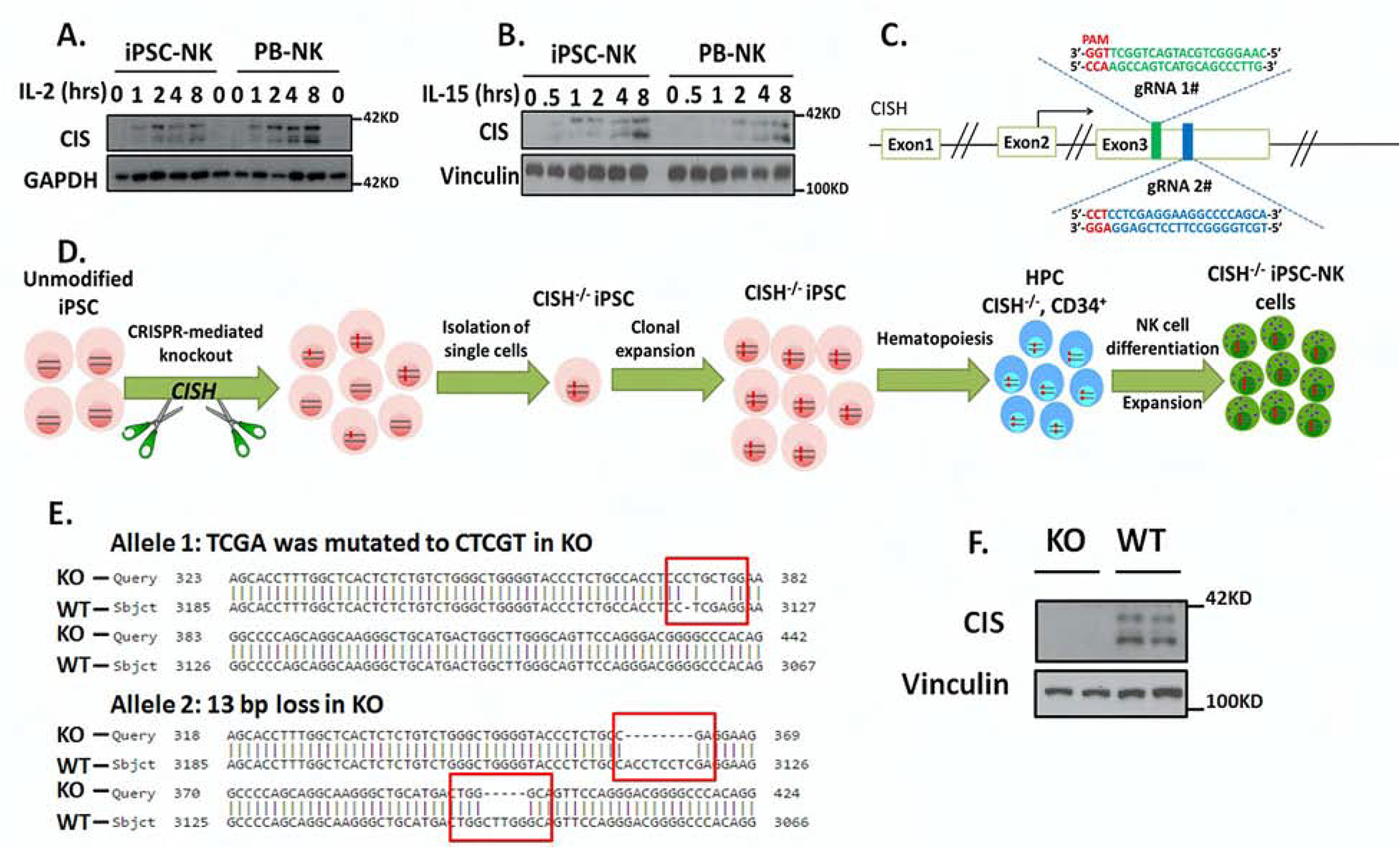
A–B. To demonstrate normal CIS expression iPSC-NK and PB-NK cells were incubated without cytokines for 8hrs and then simulated with (A) 100 U/ml IL-2 or (B) 10ng/ml IL-15 for the indicated times, then CIS expression was analyzed by immunoblotting (IB). GAPDH was used as loading control. C. Scheme of CRISPR/Cas9-mediated CISH KO using two guide RNAs (gRNA) located in direct and complementary strand targeting exon 3 of the CISH gene. D. Schematic representation for deriving clonal CISH−/− iPSC-NK cells from human iPSC. CRISPR/Cas9 mediated CISH KO was performed in WT iPSC, followed by identification of CISH−/− iPSC at clonal level. After clonal expansion, CISH−/− iPSC were differentiated to CD34+ hematopoietic progenitor cells through hematopoiesis and then CISH−/− iPSC-NK cells through NK cell differentiation using the method previously reported. E. Comparison of sequence in CISH KO clone (obtained by Sanger sequencing) with CISH WT sequence (exon3, from 3067 to 3185) by Basic Local Alignment Search Tool (BLAST) showing frame shift mutations (red rectangle) in both alleles. All mutations occurred in 2 gRNA targeted region. F. WT-iPSC-NK cells and CISH−/− iPSC-NK cells were simulated with 10 ng/ml IL-15 for 8 hours and CIS expression evaluated by IB. Vinculin was used as loading control. Data at A, B and F were repeated in 3 separate experiments.
CIS is an intracellular protein that negatively regulates IL-15 signaling and has been shown to be an important checkpoint to regulate NK cell mediated anti-tumor immunity in mice(Delconte et al., 2016). These murine studies demonstrated that deletion of Cish rendered NK cells hypersensitive to IL-15 and Cish−/− mice were resistant to multiple cancer metastases in vivo(Delconte et al., 2016). However, the impact of CIS on human NK cell function is less well defined. A key challenge preventing a better understanding of this protein’s role in human NK cell biology has been establishing methodology to consistently and efficiently genetically modify human NK cells, especially when deleting intracellular components like CIS. In this study, we used a well-defined system to produce homogeneous and well-characterized human iPSC-derived NK cells(Knorr et al., 2013; Li et al., 2018; Zhu and Kaufman, 2019).
Deletion of CISH in human iPSCs does not affect hematopoiesis but delays NK cell differentiation in vitro
To investigate the role of CIS during hematopoietic development, we analyzed hematopoietic progenitor cell surface antigens (CD31, CD34, CD43 and CD45) at day 6 of differentiation of WT-iPSCs and CISH−/− iPSCs. The development of hematopoietic progenitor cells was similar in both populations (~85% CD34+CD31+ cells and ~40–50% were CD34+CD43+) (Figures 2A–B), indicating loss of CIS does not affect early human hematopoietic differentiation. We then investigated the effect of deleting CISH during in vitro NK cell differentiation. After 3 weeks in NK cell differentiation conditions, WT-iPSCs produced ~ 40% CD45+CD56+ NK cells; however, the CISH−/− iPSCs produced only ~ 5% CD45+CD56+ NK cells at this early time point (Figure 2C). While NK cell differentiation was typically fully complete with >90% CD45+CD56+ NK cells after 4 weeks using WT iPSCs, the CISH−/− iPSC-cells only produced ~10% CD45+CD56+ NK cells at week 4 (Figure 2C). However, by week 5, these CISH−/− iPSC- NK cell cultures contained >80% CD45+CD56+ NK cells (Figures 2C–D). Notably, the yield of NK cells produced by WT-iPSCs and CISH−/− iPSCs are comparable by week 5 (typically ~1×107 CD45+CD56+ NK cells from 1×106 starting iPSCs). These results demonstrate deletion of CIS does lead to a developmental delay of NK cell differentiation, though phenotypically normal NK cells are routinely produced.
Figure 2. Deletion of CISH in human iPSCs do not affect hematopoiesis but delays NK cell differentiation in vitro.
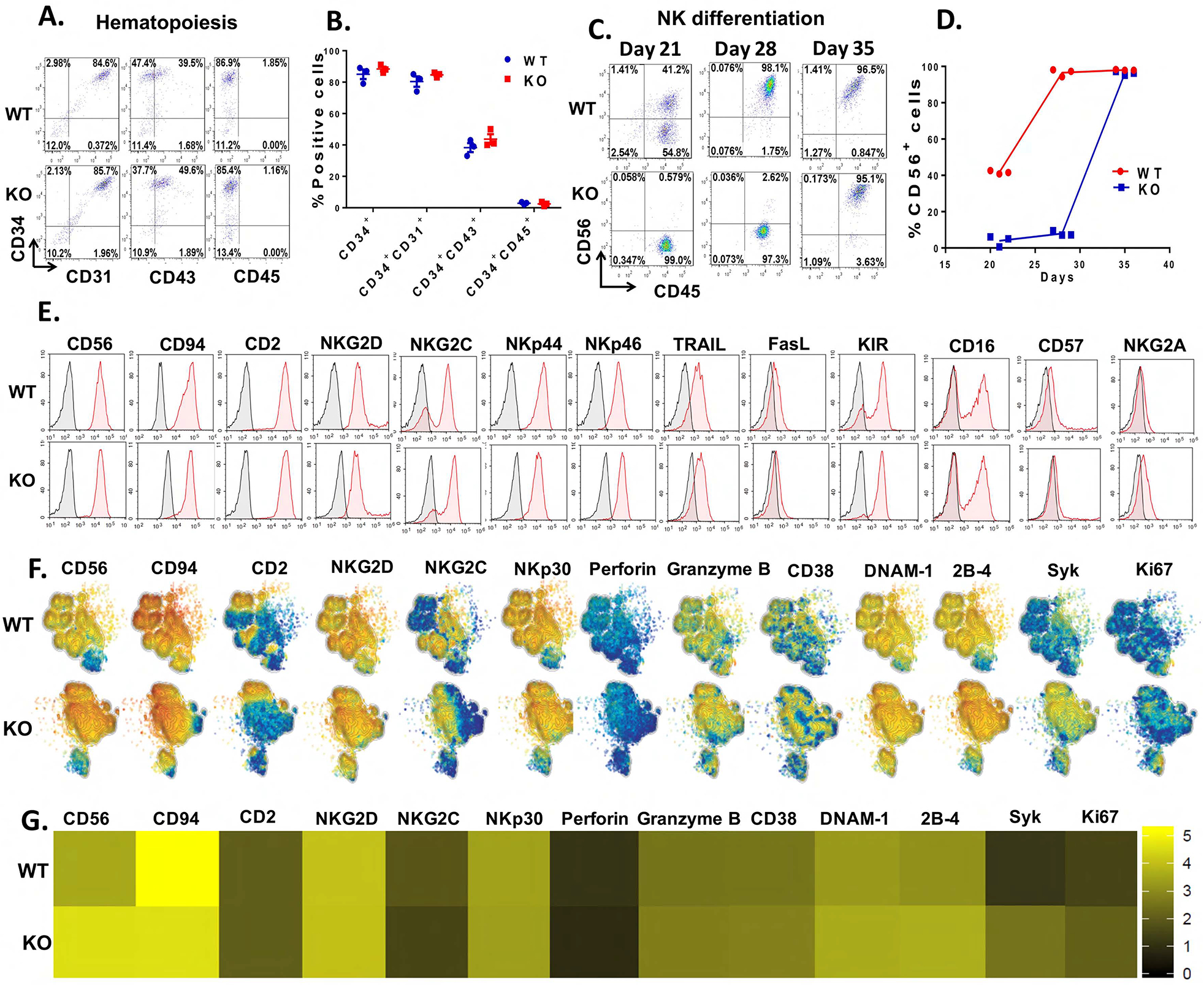
A. Analysis of hematopoietic progenitor cell markers (CD31, CD34, CD43 and CD45) in day 6 EBs generated from WT-iPSCs and CISH-KO iPSCs by flow cytometry. Data was repeated independently in 3 separate experiments. B. Quantification of the percentage of positive cells shown in A. C. Analysis of CD45+CD56+ NK cells during NK cell differentiation. Day 6 EBs generated from WT-iPSCs or CISH-KO iPSCs were incubated in NK differentiation media and analyzed by flow cytometry at 21, 28, and 35 day time points. D. Quantification of CD56+ cells shown derived at each time point. E. WT-iPSC-NK and CISH−/− iPSC-NK cells analyzed by flow cytometry for CD56 other indicated typical NK cell surface receptors. Data in A–E was repeated independently in 3 separate experiments. F. Mass cytometry analysis of WT-iPSC-NK and CISH−/− iPSC-NK cells. Individual t-SNE maps show the expression of selected NK cell surface and intracellular markers. Color indicates signal intensity, ranging from low (blue) to high (red). G. Heatmap of the marker expression showed in F. Color scale shows the expression level with yellow representing higher expression and black representing lower expression.
While CISH−/− iPSC-derived NK cells have similar phenotype as WT-iPSC-derived NK cells, they did take longer to develop in vitro. One possible explanation for this delayed NK cell differentiation when knocking out CISH in iPSCs is that the in vitro differentiation protocol (including cytokine concentrations) was developed using unmodified iPSC and might not be optimal for CISH−/−iPSCs, as these cells have improved sensitivity to cytokine stimulation that could affect differentiation pathways or kinetics of differentiation. Also, while we demonstrate the CISH−/− iPSCs have a normal karyotype, we also cannot exclude the possibility of sub-chromosomal somatic mutations could affect the rate of NK cell differentiation.
CISH−/− iPSC-NK cells are phenotypically mature
Next, to study the effect of CISH KO on NK phenotype, we analyzed expression of NK cell markers by flow cytometry (Figure 2E). CISH−/− iPSC-NK cells consist of a homogeneous population of CD56+ NK cells that co-express typical NK cell surface antigens including CD16, NKp44, NKp46, NKG2D, NKG2A, KIR, TRAIL and FasL (Figure 2E), similar to WT-iPSC-NK cells. In addition, CISH−/− iPSC-NK cells and WT-iPSC-NK cells similarly express NK cell maturation markers such as CD94, CD2, CD57 and NKG2C (Figure 2 E), indicating CISH−/− iPSC-NK cells are phenotypically mature and CISH KO does not affect NK cell phenotype and maturation. To further characterize CISH−/− iPSC-NK cells, we used mass cytometry (CyToF) and a panel of 37 antibodies against inhibitory, activating and homing receptors, as well as intracellular activation markers (Supplemental Table 1). As shown in the t-distributed stochastic neighbor-embedding (tSNE) algorithm map (Figure 2 F), CISH−/− iPSC-NK cells express typical NK cell surface markers similar to WT iPSC-NK cells. Interestingly, some markers associated with NK cell, proliferation and activation cytotoxicity are increased in CISH−/− iPSC-NK cells, including Ki67, DNAM-1, 2B4, Syk (Figure 2F, G). We then expanded CISH−/− iPSC-NK cells using irradiated K562-IL21-4-1BBL cells and IL-2 as previously utilized by our group and others (Denman et al., 2012; Knorr et al., 2013). Under these culture and expansion conditions, CISH−/− iPSC-NK cell expansion and cytolytic activity was similar to WT iPSC-NK cells (Supplemental figure 2 A–C). In addition, degranulation and cytokine production (Supplemental figure 2 D) upon stimulation are similar among WT iPSC-NK cells, CISH−/− iPSC-NK and PB-NK cells under these culture and expansion conditions. Taken together, these data demonstrate that CISH−/− iPSC-NK cells are phenotypically mature and have normal proliferative capacity under highly stimulated culture conditions.
CISH−/− iPSC-NK cells display better expansion and function compared with WT iPSC-NK cells under low cytokine concentration
Due to the role of CIS in inhibiting IL-15-mediated NK functions, we hypothesized that CISH−/− iPSC-NK cells may have better expansion and function under ‘stimulus-lacking’ (IL-15-limiting) conditions. We therefore maintained WT iPSC-NK cells and CISH−/− iPSC-NK cells either without cytokines or at a low concentration of IL-15 (1 ng/ml, compared to the normal 10 ng/ml IL-15 conditions(Felices et al., 2018; Wagner et al., 2017)) for 3 weeks. WT iPSC-NK cells were unable to survive when maintained at the low concentration of IL-15 or without cytokines (Figures 3A–B). In contrast, CISH−/− iPSC NK cells were able to expand for 3 weeks (>10-fold expansion) at the low concentration of IL-15 (Figure 3A). Similar findings were observed using a low concentration of IL-2 (10 U/ml, compared to the normal 50–100 U/ml(Knorr et al., 2013; Li et al., 2018; Wagner et al., 2017)) (Figure 3B). We then evaluated anti-tumor activity using cytotoxicity assays against K562, MOLM-13 (2 myeloid leukemia lines) and SKOV-3 (ovarian cancer line). Notably, after culturing with 1 ng/ml of IL-15 for 3 weeks, CISH−/− iPSC NK cells still maintained potent anti-tumor activity and exhibited better killing activity against tumor targets in both short-term (4 hours) and long-term (>20 hours) cytotoxicity assays than WT-iPSC-NK cells (Figure 3 C–F). Furthermore, we used NK cell degranulation (indicated by cell surface expression of CD107a) and IFN-γ expression as parameters for NK cell function. Consistent with cytotoxicity results, WT-iPSC-NK cells expressed minimal amounts of both CD107a and IFN-γ when stimulated with the tumor cells, indicating loss of function when cultured long term with a low concentration of cytokine (Figure 3 G–I). In contrast, CISH−/− iPSC NK cells exhibited significantly increased cytotoxic granule release (CD107a expression) and IFN-γ production (Figures 3G–I) when stimulated. To further extend this finding, we cultured WT-iPSC-NK cells, CISH−/− iPSC NK cells and PB-NK cells from 2 donors (PB-NK-1#, PB-NK-2#) either with low concentration of IL-15 (1ng/ml) or normal concentration of IL-15 (10 ng/ml) for 3 and 7 days and evaluated their killing activity against K562 cells (Figure S3). After culture with low IL-15 for 7 days, CISH−/− iPSC NK cells show better killing activity compared with WT-iPSC-NK cells or PB-NK cells (Figure S3B). However, no significant difference in killing activity were observed when cultured with normal IL-15 for 3 and 7 days (Figure S3 C, D). Together, these results demonstrate that deletion of CISH in NK cells leads to improved survival and function when cytokine stimulation is limiting.
Figure 3. CISH−/− iPSC-NK cells have better expansion and functions in vitro compared with WT iPSC-NK cells.
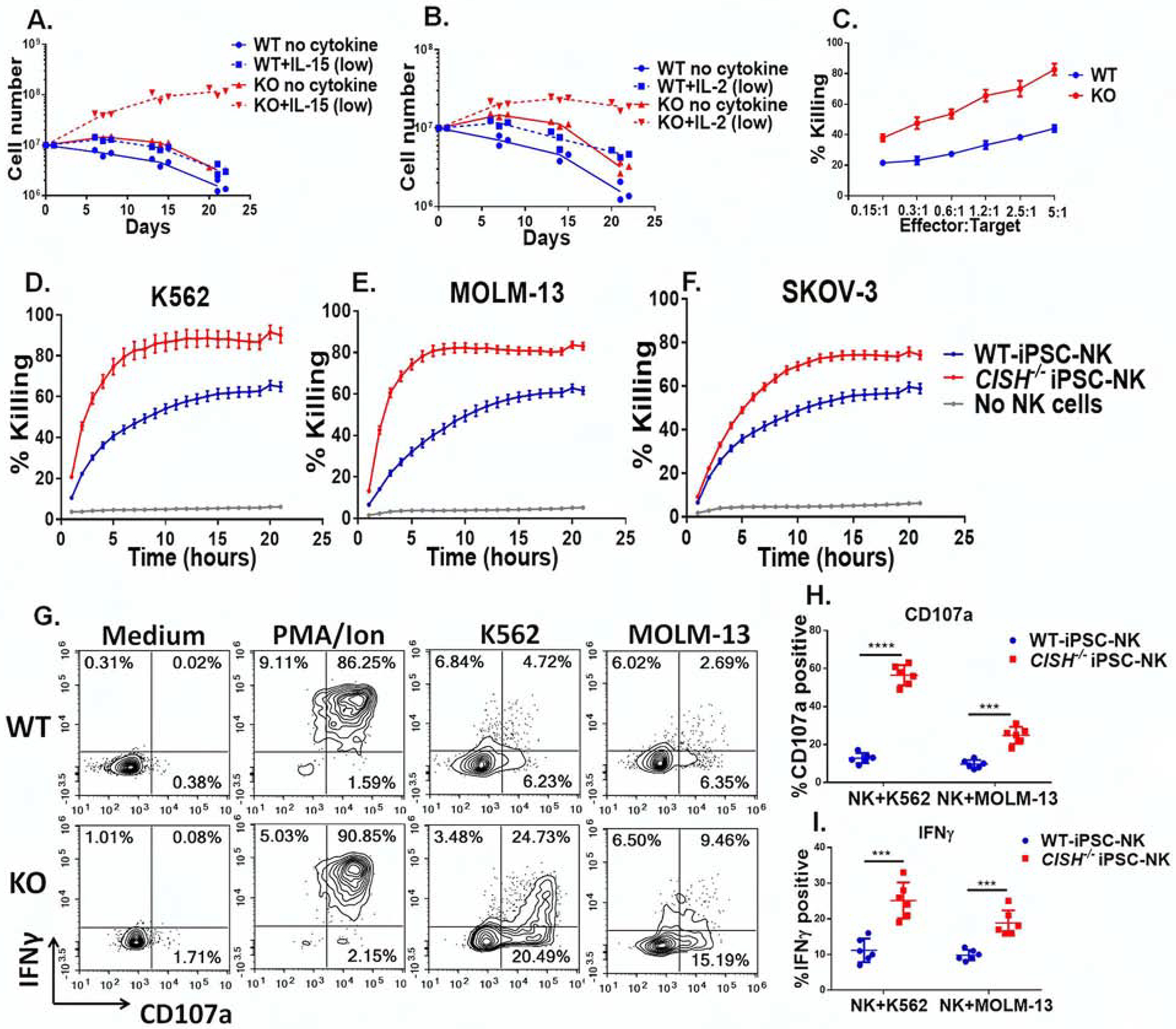
A&B Growth curve of CISH−/− iPSC-NK cells and WT iPSC-NK cells with or without a low concentration of IL-15 (A. 1ng/ml) and IL-2 (B. 10 U/ml). Data was repeated independently in 3 separate experiments. C. 4 hour cytotoxicity assay using CISH−/− iPSC-NK and WT iPSC-NK cells against K562 cells after 3 weeks of culture in low concentration IL-15 as determined using CellEvent™ Caspase-3/7 Green Flow Cytometry Assay. D–F. Killing against K562 (D), MOLM-13 (E) and SKOV-3 (F) cells was quantified over an extended time course using the IncuCyte real-time imaging system at E:T=1:1. G. CISH−/− iPSC-NK cells and WT iPSC-NK cells were maintained at low IL-15 for 3 weeks and production of CD107α and IFNγ in response to K562 and MOLM-13 cells was measured. CISH−/− iPSC-NK cells and WT iPSC-NK cells were left unstimulated or stimulated with a 1:1 ratio of target cells and stained for CD107a and IFNγ 4 hours later. Quantification of CD107a (H, n=6) and IFNγ (I, n=6) after stimulation with K562 or MOLM-13 cells. Paired t test was used to do the comparisons. ***P < 0.001, ****P < 0.0001. Data at C–F, H and I were shown as mean ± SD. Data at C-I were repeated in 3 separate experiments.
CISH−/− iPSC-NK cells display improved anti-tumor activity in vivo
To evaluate the anti-tumor activity of CISH−/− iPSC-NK cells in vivo, we assessed their killing of MOLM-13 AML cells in a mouse xenograft tumor model. Mice were injected intravenously (iv) with luciferase-expressing MOLM-13 cells and the next day received a single iv injection of 1×107 WT or CISH−/− iPSC-NK cells (Figure 4A). As in previous studies(Li et al., 2018), IL-2 was dosed every other day for 21 days to promote in vivo NK cell survival and expansion. Tumor growth was monitored by bioluminescent imaging (BLI) (Figure 4B). Treatment with NK cells significantly reduced tumor burden and CISH−/− iPSC-NK cells mediated significantly better anti-tumor activity when compared to treatment using WT iPSC-NK cells (Figures 4B–C). This led to markedly improved survival of mice treated with the CISH−/− iPSC-NK cells. 3 out of 5 mice treated with CISH−/− iPSC-NK cells had complete tumor clearance and long-term (>100 days) survival versus 0 of 5 mice treated with WT-iPSC-NK cells (Figure 4D). Moreover, we investigated the in vivo persistence of NK cells by examining peripheral blood for the presence of human NK cells (hCD56+ cells). On day 7, the number of CISH−/− iPSC-NK cells was significantly higher (38.2±4.3 compared to 12.0±2.8 cells/ul, p<0.001) in circulation than WT iPSC-NK cells (Figure 4E, F). Next, we performed a separate study to compare the in vivo persistence and homing of WT iPSC-NK cells, CISH−/− iPSC-NK cells and PB-NK cells (Supplemental figure 4). Again, CISH−/− iPSC-NK cells show significantly better persistence in peripheral blood in comparison with WT iPSC-NK cells or PB-NK cells at day 7 (Supplemental figure 4 A, B). Moreover, at day 14, CISH−/− iPSC-NK cells exhibited better persistence in peripheral blood, spleen and homing to bone marrow (Supplemental figure 4C–H). Together, these studies demonstrate that CISH−/− iPSC-NK cells have improved anti-tumor activity and persistence in vivo.
Figure 4. CISH−/− iPSC-NK cells demonstrate improved persistence and better anti-tumor activity in vivo.
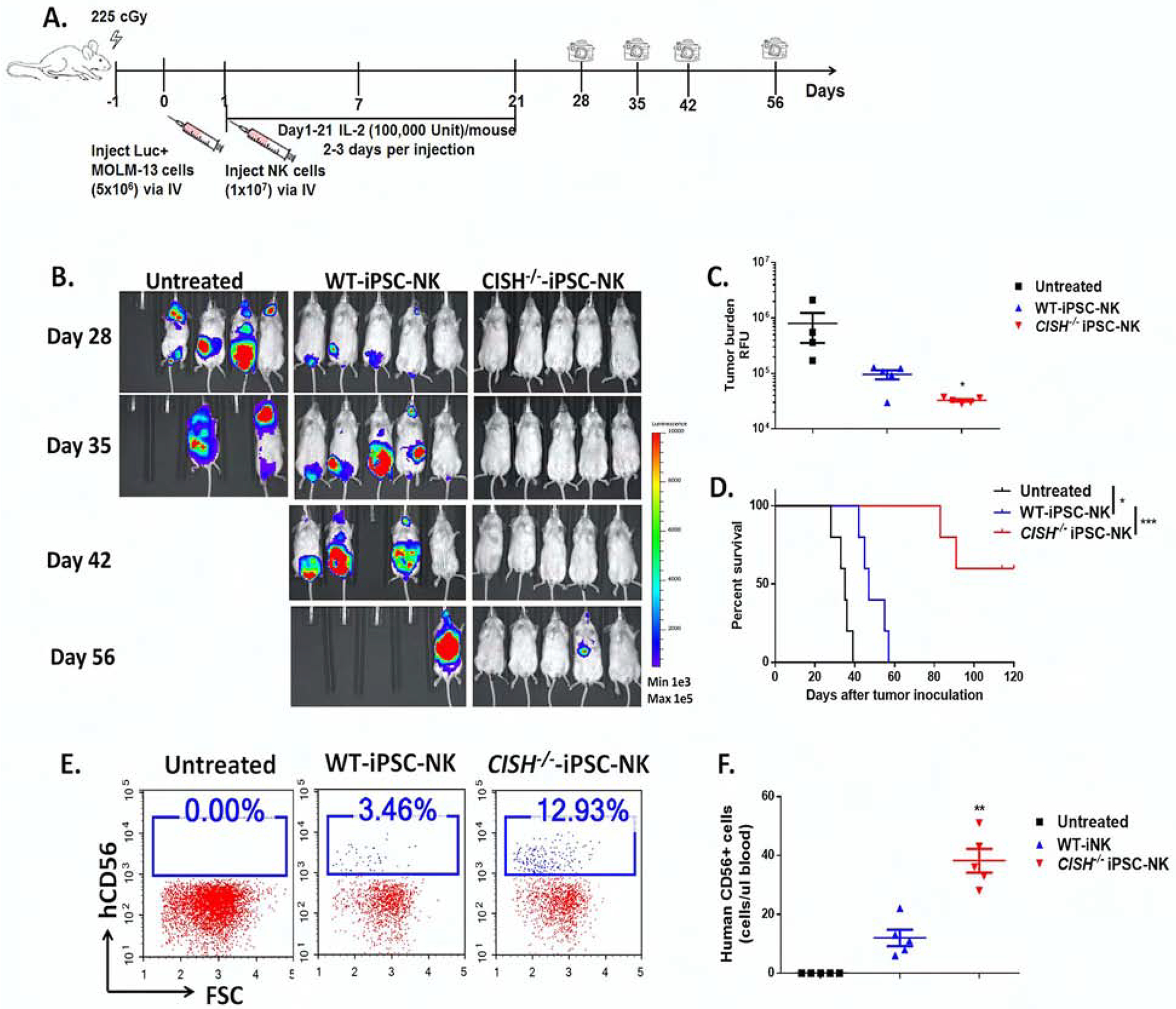
A. Diagram of in vivo treatment scheme. NSG mice were inoculated iv with 5×106 MOLM-13 cells expressing the firefly luciferase gene. 1 day after tumor injection, mice were either left untreated or treated with 1×107 WT-iPSC-NK or CISH−/− iPSC-NK cells. NK cells were supported by weekly injections of IL-2 for 3 weeks, and IVIS imaging was done to track tumor load. B. IVIS images showing progression of tumor burden. C. Tumor burden at day 28 was quantified and shown as mean ± SD. Statistics by one way ANOVA test; *p<0.05. D. Kaplan-Meier curve demonstrating survival of the experimental groups. Statistics: two-tailed Log-rank test, WT-iPSC-NK vs untreated, *p<0.05; WT-iPSC-NK vs CISH−/− iPSC-NK, ***p<0.001. E. Representative flow cytometric plot of human CD56+ cells in population from mice peripheral blood 7 days after NK cell treatment. F. Quantification of human CD56+ cells in peripheral blood (number of hCD56+ cells/ul blood) at Day 7, shown as mean ± SD. Statistics by one way ANOVA test; **p<0.01.
CISH−/− iPSC NK cells show increased IL-15 signaling activation
To further investigate the molecular mechanisms that mediate improved function of CISH−/− iPSC-NK cells, we performed RNA sequencing on WT iPSC-NK cells and CISH−/− iPSC-NK cells. Surprisingly, more than 6000 differentially expressed genes (the threshold of differential expression genes is: padj < 0.05) were identified in CISH−/− iPSC-NK cells (Figure 5A). Gene ontology (GO) enrichment analysis suggested that the top up-regulated signaling pathways in CISH−/− iPSC-NK cells were associated with leukocyte differentiation, proliferation, activation and cytokine secretion (Figure 5B and Table S2). These findings confirmed a role for CIS to regulate NK cell differentiation and this finding is consistent with our data that deletion of CISH delayed NK cell in vitro differentiation. Notably, more than 60 genes involved in positive regulation of lymphocyte activation were significantly up-regulated in CISH/− iPSC-NK cells compared with WT iPSC-NK cells (Figure 5C). We next analyzed IL-15 signaling in CISH−/− iPSC-NK cells. Kyoto Encyclopedia of Genes and Genome (KEGG) analysis of RNA sequencing data showed that the JAK-STAT pathway (IL-15 downstream pathway) is one of the most up-regulated signaling pathways in CISH−/− iPSC-NK cells (Figure 5D). Expression of 34 genes involved in the JAK-STAT signaling pathway was significantly up-regulated in CISH−/− iPSC-NK cells compared with WT iPSC-NK cells (Figure 5D). More specific analysis of cell signaling pathways demonstrated markedly increased phosphorylation of IL-15-stimulated JAK1 tyrosine phosphorylation, STAT3 and STAT5 in CISH−/− iPSC NK cells (Figure 5E). mTOR signaling activity which can be stimulated by IL-15 in NK cells (ref), is also up-regulated in CISH−/− iPSC NK cells as indicated by increased level of phosphorylated S6 (pS6) and phosphorylated S6K1 (pS6K1) (Figure 5F). Expression of CD122, a subunit shared by the receptors for IL-2 and IL-15, was also increased in CISH−/− iPSC NK cells with or without IL-15 stimulation (Figure 5G–H). This increase in expression contributes to the increase in IL-15 signaling. IL-15 stimulation leads to down-regulation of CD122 in both WT and CISH−/− iPSC NK cells (Figure 5G–H). Together, these data suggest that increased expression of genes involved in the lymphocyte and IL-15 signaling activation pathways in CISH−/− iPSC-NK cells provides a molecular mechanism for enhanced NK cell function.
Figure 5. CISH−/− iPSC NK cells show increased IL-15 signal activation.
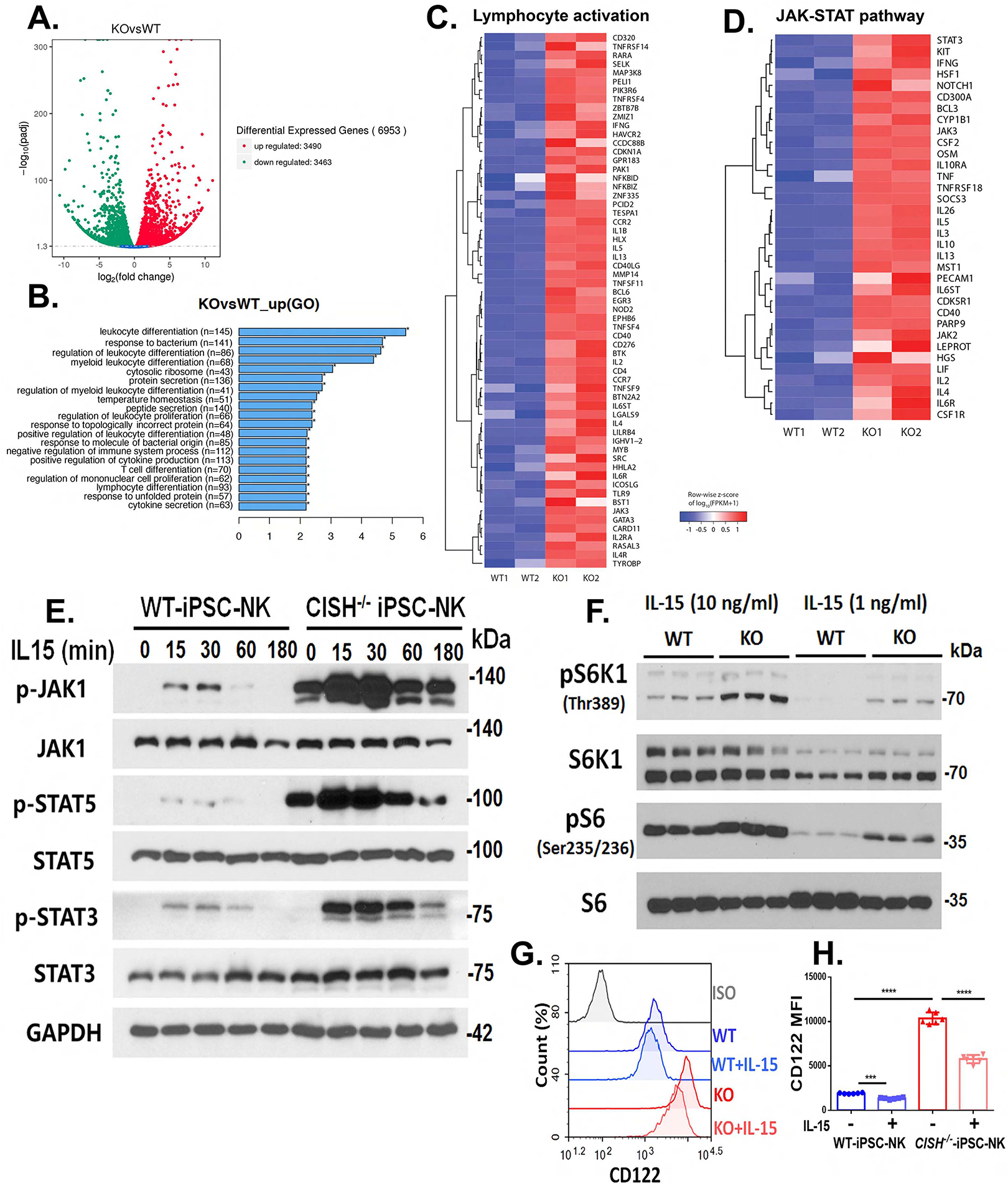
A–D. Differential expression gene between WT iPSC-NK and CISH−/− iPSC-NK cells was analyzed by RNA sequencing. A. Volcano diagram of differential expression genes. The threshold of differential expression genes is: padj < 0.05. B. Gene ontology (GO) enrichment analysis. Top 20 significantly enriched cellular compares, molecular function and biological process in CISH−/− iPSC-NK cells were shown. C. Heatmap view of expression of 61 genes involved in regulating lymphocyte activations in WT iPSC-NK and CISH−/− iPSC-NK cells. D. Heatmap view of the JAK-STAT pathway gene expression in WT iPSC-NK and CISH−/− iPSC-NK cells. E. Immunoblot analysis of WT iPSC-NK and CISH−/− iPSC-NK cells incubated without cytokines for 24 hrs and then simulated with 1 ng/ml IL-15 for the indicated times. F. Immunoblot analysis of mTOR downstream activation (pS6 and pS6K1) in WT iPSC-NK and CISH−/− iPSC-NK cells cultured in media with 10 ng/ml or 1 ng/ml IL-15 for 24 hrs. G. Representative flow cytometry plot of IL-2Rβ (CD122) surface expression on WT iPSC-NK, CISH−/− iPSC-NK cells cultured in media with or without IL-15 (1ng/ml) for 24 hrs. H. Quantification of CD122 expression (n=6). MFI: mean fluorescence intensity. Statistics: One way ANOVA, ***P=0.0002, ****P<0.0001. Data at E, G and H were repeated in 3 separate experiments and data at F were repeated in 2 separate experiments.
CISH−/− iPSC-NK cells show increased single-cell polyfunctionality
A recent study identified a subset of polyfunctional T cells in CAR T-cell products that produce ≥2 cytokines upon stimulation with CD19 antigen using high-content single-cell multiplex cytokine analysis(Rossi et al., 2018). Notably, pre-specified polyfunctionality strength index (PSI) manufactured CAR-T cells were associated with improved clinical responses and reduced toxicities in non-Hodgkins lymphoma patients(Rossi et al., 2018; Xue et al., 2017). Given the key role of IL-15 to regulate NK cell expansion and functionality, we examined if the increase in IL-15 signaling activation in CISH−/−-iPSC-NK cells similarly led to increased single-cell polyfunctional profiles, thus contributing to increased cytotoxicity of these cells. We evaluated polyfunctionality of CISH−/−-iPSC-NK cells, WT iPSC-NK cells and PB-NK cells (from 2 donors) using the same high-content single-cell multiplex cytokine analysis used in the CAR-T cell studies(Rossi et al., 2018; Xue et al., 2017). Cells were incubated at a low concentration of IL-15 (1 ng/ml) overnight and then simulated with IL-12 (10 ng/ml)/IL-18 (100 ng/ml) to elicit cytokine production. The polyfunctional profile of iPSC-NK cells and PB-NK cells upon stimulation with IL-12/IL-18 was dominated by effector molecules including Granzyme B, IFNγ, MIP-1α, Perforin and TNFα (Supplemental figure 5). Notably, CISH−/− -iPSC-NK cells showed the highest polyfunctional response with approximately 5% of total cells (~25% of all cytokine producing NK cells) being polyfunctional, whereas only about 1% to 2% of the WT iPSC-NK cells and PB-NK cells were polyfunctional (Supplemental figure 5). Moreover, the PSI was increased >10 fold in CISH−/−-iPSC-NK cells compared to WT-iPSC-NK cells and PB-NK cells. These results demonstrate that CISH−/−-iPSC-NK cells have better single-cell polyfunctionality in comparison with WT-iPSC-NK and PB-NK cells.
Deletion of CISH in human iPSC-NK cells improves metabolic fitness
Activated NK cells up-regulate the rate of glucose-driven glycolysis and oxidative phosphorylation (OxPhos) to provide energy to drive the pathways necessary for their effector functions (Kobayashi and Mattarollo, 2017; Mah and Cooper, 2016). Since IL-15 can exert important effects on NK cell metabolism(Felices et al., 2018; Marais et al., 2014), we compared the glycolytic and mitochondrial function of WT-iPSC-NK, CISH−/− iPSC-NK and PB-NK cells after incubation with a low concentration of IL-15 for 3 and 7 days using previously described Seahorse assays(Felices et al., 2018). Interestingly, after 3 days culture in low IL-15, glycolytic rate was slightly increased in CISH−/− iPSC-NK cells in comparison with both WT-iPSC-NK and PB-NK cells, which was reflected through an increase in basal glycolysis (measured before the addition of rotenone/antimycin A) (Figures 6A, B), and an increase in glycolytic capacity elucidated by subtracting the rate of glycolysis before and after addition of 2-deoxy-D-glucose (2DG) (Figures 6A, B). After 3 days’ culture in low IL-15, OxPhos was also increased in CISH−/− iPSC-NK cells compared with WT-iPSC-NK cells and PB-NK cells (Figure 6 C). Maximal respiration was significantly increased in CISH−/− iPSC-NK cells compared with WT-iPSC-NK cells and PB-NK cells (Figure 6D). ATP-linked respiration which was calculated from subtraction of basal respiration and respiration after addition of oligomycin, and mitochondrial spare respiratory capacity (SRC) measured after addition of carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone (FCCP) were modestly increased or show trend of increase in CISH−/− iPSC-NK cells (Figure 6D). While after 7 days’ culture with low IL-15, both glycolytic rate (Figure 6E, F) and OxPhos (Figure 6G, H) were dramatically increased in CISH−/− iPSC-NK cells compared with WT-iPSC-NK cells and PB-NK cells.
Figure 6. Deletion of CISH in human iPSC-NK cells improve metabolic fitness.
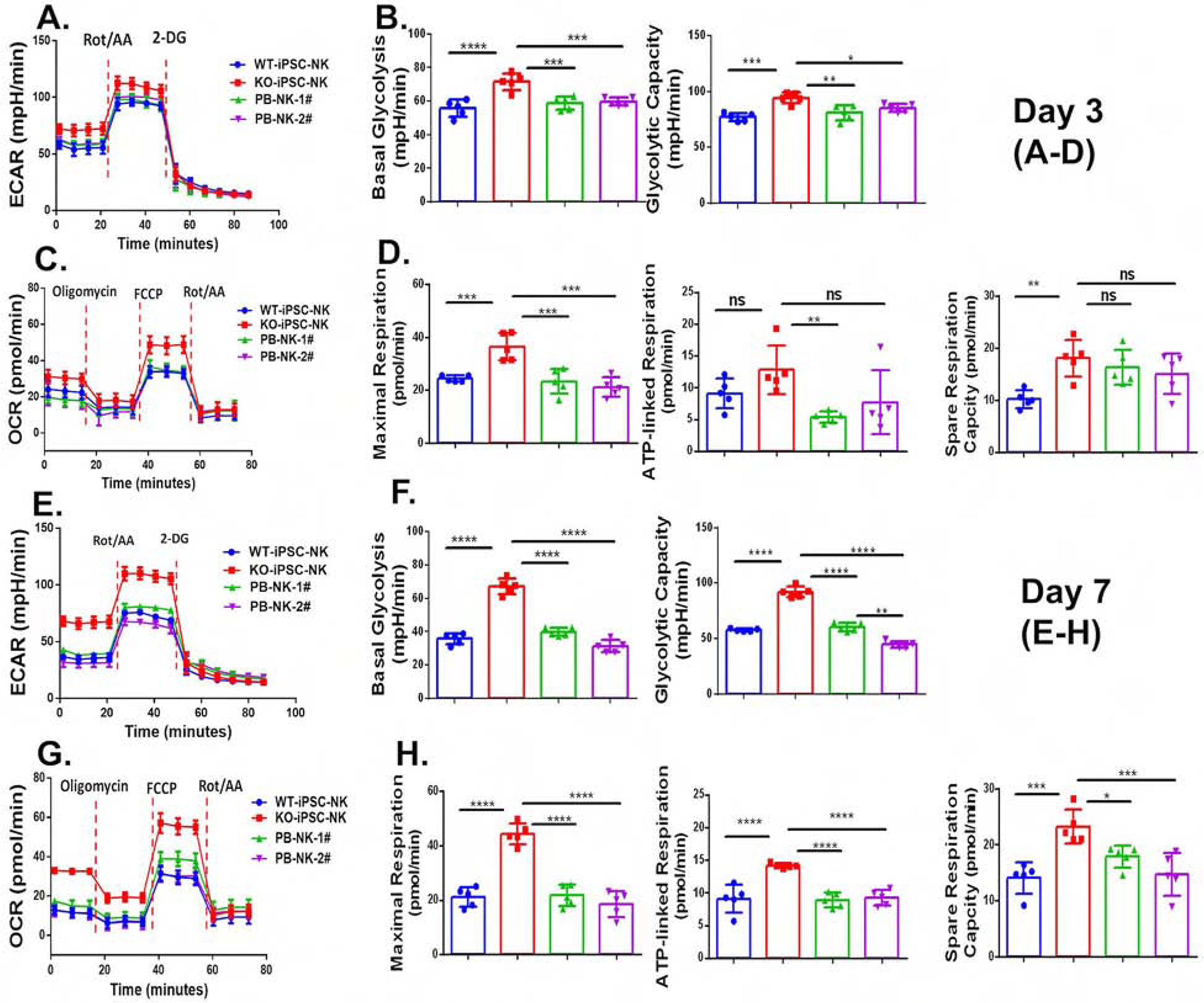
WT-iPSC-NK cells, CISH−/− iPSC-NK-cells and PB-NK cells from 2 donors (PB-NK-1# and PB-NK-2#) were incubated with a low concentration of IL-15 (1ng/ml) for 3 days (A–D) or 7 days (E–H). A. The extracellular acidification rate, (ECAR) was measured in real time in an XFe96 analyzer after injection of rotenone/antimycin A (Rot/AA) and 2-deoxy-D-glucose (2DG). B. Graphical analysis of Basal Glycolysis (left) and Glycolytic Capacity (right) derived from A (n=6). C. The oxygen consumption rate (OCR) was measured after injection of oligomycin, Carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP), and Rot/AA. D. Graphical analysis of Maximal Respiration (left), ATP-linked Respiration (middle) and Spare Capacity Respiration (SRC, right) derived from C (n=6). E. ECAR was measured. F. Graphical analysis of Basal Glycolysis (left) and Glycolytic Capacity (right) derived from E (n=6). G. OCR was measured. D. Graphical analysis of Maximal Respiration (left), ATP-linked Respiration (middle) and SRC (right) derived from G (n=6) Data were shown as mean ± SD and were repeated in 3 separate experiments. One way ANOVA was used to do all comparisons. **p<0.01, ***p<0.001, ****p<0.0001. ns: p>0.05.
Glucose-driven glycolysis and oxidative metabolism have been shown to be required for NK cell anti-tumor and anti-viral effector functions(Assmann et al., 2017; Donnelly et al., 2014; Keppel et al., 2015; Poznanski and Ashkar, 2019). This suggests that the increased IFN-γ production and cytotoxic activity of CISH−/− iPSC-NK cells was supported by increased glycolysis and OxPhos. Notably, SRC has been shown to be pivotal in memory T cell formation and could be used as an indicator of metabolic fitness(Pearce et al., 2013). These data demonstrate that CISH−/− iPSC-NK cells have improved metabolic fitness that can mediate potentially enhanced functions.
Improved metabolic fitness in CISH−/− iPSC-NK cells is mediated by mTOR signaling and contributes to enhanced function
While the importance of cellular metabolism on immune cell function is now well established, there has been more limited data on the ability to improve or optimize human NK cell-mediated activity via improved metabolic fitness of NK cells(Kobayashi and Mattarollo, 2017). We therefore tested whether improved metabolic fitness in CISH−/− iPSC-NK cells contributes to enhanced cytotoxicity. We used rapamycin (rapa), an inhibitor of mammalian target of rapamycin complex 1 (mTORC1), and key regulator of cellular metabolism(Donnelly et al., 2014; Marais et al., 2014). Rapa treatment decreased proliferation of both CISH−/− iPSC-NK cells and WT-iPSC NK cells (Figure 7A). Interestingly, treatment of CISH−/− iPSC-NK cells with rapa led to both decreased glycolysis and mitochondrial respiration (Figure 7 B–E). Rapa completely neutralized the improved glycolysis and OxPhos in CISH−/− iPSC-NK cells, bringing the metabolic rate to a level similar to WT-iPSC-NK cells (Figures 7B–E). Notably, rapa did not affect either glycolysis or mitochondrial respiration of WT-iPSC NK cells (Figure 7B–E). Indeed, treating with rapa slightly increased the ATP-linked aspiration rate in WT-iPSC-NK cells (Figure 7E). This indicates that the metabolic rate in WT-iPSC-NK cells under low cytokine concentration is reduced to a level that could not be further decreased by inhibition of mTOR signaling.
Figure 7. Improved metabolic fitness in CISH−/− iPSC-NK cells is mediated by mTOR signaling pathway and contributes to improved function.
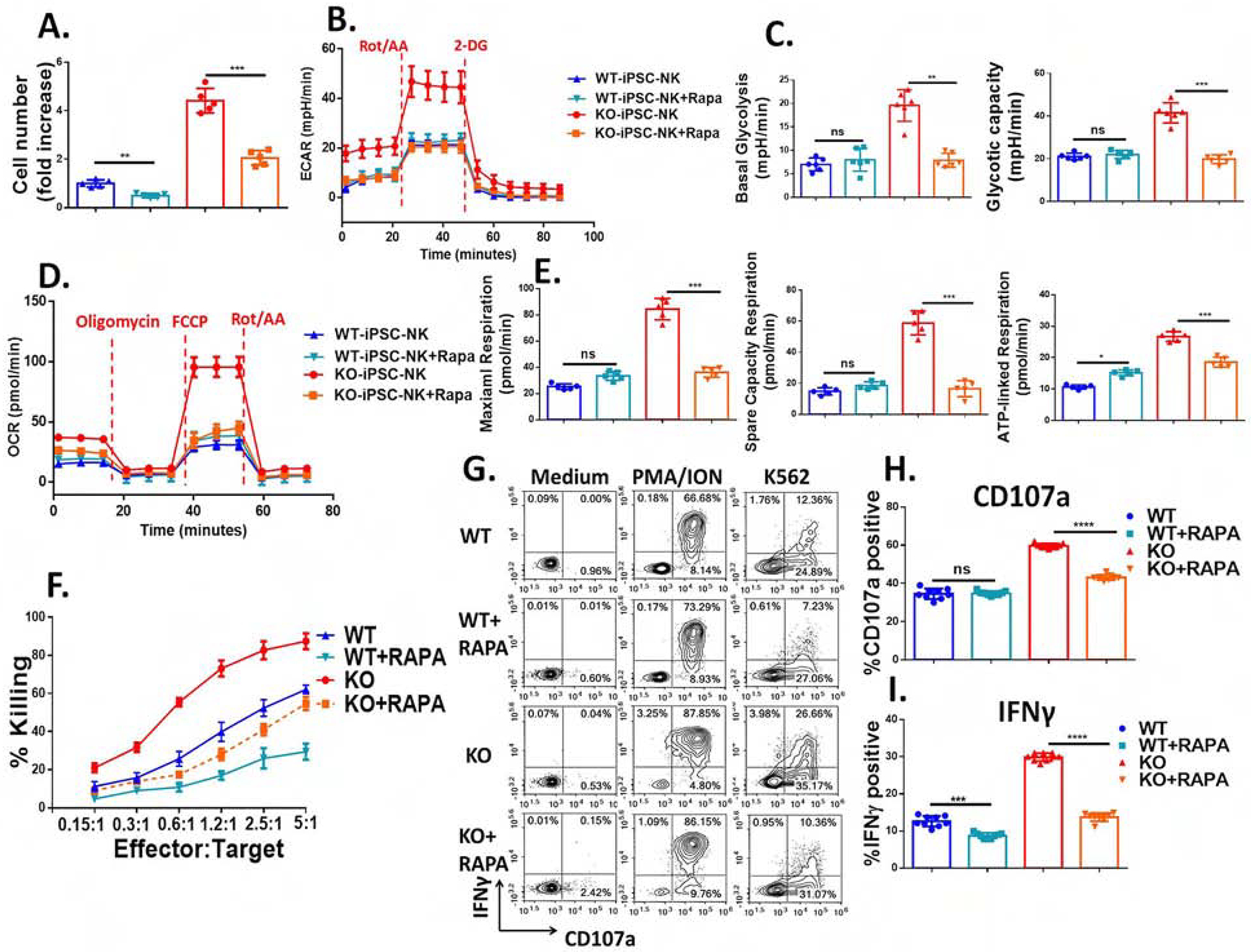
WT-iPSC-NK cells and CISH−/−-iPSC-NK-cells were incubated at low IL-15 (1 ng/ml) with or without Rapamycin (100 ng/ml) for 7 days. A. Quantification of Rapamycin’s effects upon NK cell proliferation. B. Rapamycin decreases the ECAR of CISH−/−-iPSC-NK-cells to a level similar to that observed in WT-iPSC-NK cells regardless of rapamycin’s addition. ECAR was measured in real time in an XFe96 analyzer after injection of Rot/AA and 2-DG. C. Graphical analysis of Basal Glycolysis (left) and Glycolytic capacity (right) derived from B (n=6). D. OCR was measured after injection of oligomycin, FCCP and Rot/AA. E. Graphical analysis of Maximal Respiration (left), Spare Capacity Respiration (middle) and ATP-linked Respiration (right) derived from D (n=6). F. After 7 days incubation with or without Rapamycin, killing against K562 cells was evaluated using CellEvent™ Caspase-3/7 Green Flow Cytometry Assay. G. CISH−/− iPSC-NK cells and WT iPSC-NK cells produce CD107a and IFNγ in response to K562 with or without rapamycin for 7 days (low dose IL-15, 1 ng/ml). CISH−/− iPSC-NK cells and WT iPSC-NK cells were left unstimulated or stimulated with a 1:1 ratio of target cells and stained for CD107a and IFNγ 4 hours later. Quantification of CD107a (H, n=6) and IFNγ (I, n=6) after stimulation with K562 or MOLM-13 cells. Data were shown as mean ± SD and were repeated in 3 separate experiments. Paired t test was used to do all comparisons. *P<0.05, **P<0.01, ***P < 0.001, ****P < 0.0001.
To investigate if improved metabolic fitness in CISH−/− iPSC-NK cells directly contributes to enhanced cytotoxicity, we evaluated cytotoxicity after treatment with rapa. Rapa-mediated decrease of glycolysis and mitochondrial respiration in CISH−/− iPSC-NK cells resulted in decreased cytotoxicity (Figure 7F), degranulation (Figures 7G, H) and cytokine production (Figures 7G, I). These results demonstrate that the improved metabolic fitness in CISH−/− iPSC-NK cells is mediated by the mTOR pathway and directly contributes to enhanced NK cell functions.
Using human stem cell-derived NK cell as a platform, we demonstrated that deletion of CIS in NK cells increased IL-15 mediated JAK-STAT pathway signaling activity, similar to their functions in mouse. We further demonstrated that human CISH−/− NK cells exhibited improved mTOR-mediated metabolic fitness which directly contributes to the improved anti-tumor activity. Furthermore, we showed that deletion of CISH dramatically increases in vivo persistence of NK cells after adoptive transfer, as well as improved single-cell polyfunctionality measured by production of multiple effector cytokines which might also contribute to their improved anti-tumor functions.
Cellular metabolism is now recognized to play a key role regulating the function and differentiation of immune cells(Gardiner, 2019; O’Brien and Finlay, 2019; Pearce et al., 2013). Immune cells undergo dynamic metabolic shifts to support their activity including overcoming the suppressive tumor microenvironment (TME) that changes the metabolism of immune cells - a process that inhibits the activity of these cells(Wegiel et al., 2018). The metabolic profile of CAR-T cells is known to play key role in their in vivo proliferation and function(Kawalekar et al., 2016). Multiple studies show that metabolic activity also regulates the cytolytic ability of NK cells(Gardiner, 2019; Kobayashi and Mattarollo, 2017; O’Brien and Finlay, 2019; Poznanski and Ashkar, 2019). Therefore, identifying key cellular and molecular mechanisms that regulate immune cell metabolism provides an important strategy to engineer immune cells to overcome the immune suppressive TME. IL-15 can regulate NK cell metabolism but its effects are complicated and controversial, depending on time and dose. Treating NK cells with high dose IL-15 (100 ng/ml) for 72 hours increases both glycolysis and OxPhos(Marais et al., 2014). Notably, continuous treatment of NK cells with IL-15 (10 ng/ml) for 9 days lead to NK cell exhaustion, impaired metabolism and reduced cytotoxicity(Felices et al., 2018). The same group reported that human adaptive NK cells (CD3−CD56dimCD57+NKG2C+) exhibited increased mitochondrial respiration which contributes to the increased IFNγ production in these cells(Cichocki et al., 2018). Here, we increased sensitivity of NK cells to IL-15 stimulation by removing a negative regulator, CIS. When challenged with an extremely low concentration of IL-15 (1 ng/ml), which is 10 times lower than normal concentration(Felices et al., 2018; Wagner et al., 2017) for up to 3 weeks, CISH−/− iPSC-NK cells exhibited increased expansion and improved function which we attributed to improved metabolic fitness in comparison with WT iPSC-NK cells. These results show that long term administration of IL-15 can be beneficial to NK cell metabolism and therefore function when the dose is optimized and attenuated. Cryopreservation is key for developing off-the-shelf cell therapy products. Recent studies indicated that metabolic pathways play a key role in cell cryopreservation process(Fu et al., 2019) and IL-15 activated cytokine-induced killer (CIK) cells were able to maintain phenotype and cytotoxicity potential after long-term cryopreservation(Bremm et al., 2019). Whether knocking out CISH can improve cryopreservation of NK cells needs further investigations.
These studies demonstrate that CIS is a key regulator of human NK cell survival and anti-tumor activity. Deletion of CIS does not affect in vitro maintenance of undifferentiated human iPSC and hematopoietic differentiation but delays in vitro NK cell differentiation. CISH−/− iPSC-NK cells have an improved metabolic profile and exhibit increased expansion and enhanced cytotoxicity when maintained at a low concentration of IL-15 in vitro. When tested in a human AML xenograft tumor model, CISH−/− iPSC-NK cells exhibited improved in vivo persistence and anti-tumor activities in comparison with WT-iPSC-NK cells. Mechanistically, we found that the enhanced anti-tumor function could be attributed to the increased single cell polyfunctionality and mTOR-mediated metabolic fitness.
Recent advances in NK cell-based adoptive immunotherapy, including the development of novel NK cell-specific CARs to enhance iPSC-derived NK cell-targeted anti-tumor activity(Li et al., 2018) and stabilizing CD16 expression to boost antibody dependent cellular cytotoxicity (ADCC), have yielded some exciting results(Zhu et al., 2020). These experimental studies are being translated into clinical therapies with iPSC-derived NK cells now in clinical trials for treatment of refractory malignancies (clinicaltrials.gov, NCT03841110 and NCT04023071). These phase 1 trials include use of un-engineered iPSC-derived NK cells combined with checkpoint inhibitors for treatment of refractory solid tumors, as well as iPSC-derived NK cells engineered to express a non-cleavable, high-affinity CD16 to mediate improved antibody dependent cell-mediated cytotoxicity (ADCC)(Zhu et al., 2020; Zhu et al., 2018). A trial using iPSC-NK cells that express an NK cell-specific CAR is also planned(Li et al., 2018). Together, this advancement of iPSC-derived NK cells into clinical trials highlights the ability for iPSCs to provide a unique platform to create NK cells with one or multiple genetic modifications (such as combination of CIS-deletion and CAR modification) to produce NK cells with improved metabolic fitness and enhanced anti-tumor functions(Zhu et al., 2018).
In summary, we report that silencing of an intracellular immune checkpoint in NK cells increase their single cell polyfunctionality and improve their metabolic fitness, leading to enhanced functions. These studies support clinical validation of blocking this intracellular immune checkpoint and improving metabolic fitness to enhance NK cell mediated anti-tumor activities for adoptive immunotherapy. We believe that metabolic and cytokine response reprogramming through CISH knock out can be combined with other exciting advances in NK cell immunotherapy including tumor target recognition (CAR and hnCD16) and immune checkpoint blockade amongst others to produce an all-in-one NK cell product for next generation immunotherapy.
STAR Methods:
RESOURCE AVAILABILITY:
Lead Contact
• Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dan S Kaufman (dskaufman@ucsd.edu)
Materials Availability Statement
• This study did not generate new unique plasmids/mouse lines/reagents
Data and Code Availability Statement
• The published article includes all datasets generated or analyzed during this study
• RNA sequencing data is available in GEO via accession number: GSE150155
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse Strain:
8–10 weeks’ female NOD/SCID/γc−/− (NSG) mice (Jackson Laboratories, n=5 per group) were used for in vivo experiments. After tumor cell inoculation, mice were randomly assigned to experimental groups. Mice were sacrificed when loss of ability to ambulate was observed. All mice were housed, treated, and handled in accordance with the guidelines set forth by the University of California, San Diego Institutional Animal Care and Use Committee and the National Institutes of Health’s Guide for the Care and Use of Laboratory Animals.
Cells:
hiPSC were generated from umbilical cord blood CD34+ cells (from female donor) and cultured as previously described (Knorr et al., 2013; Zhu and Kaufman, 2019). K562 (leukemia cell line established from a female patient with chronic myelogenous leukemia) and SKOV-3 (Ovarian cancer cell line derived from female patient with adenocarcinoma) were obtained from American Type Culture Collection (ATCC, Manassas, Virginia, U.S.). MOLM-13 (leukemia cell line derived from male patient with AML) cells were obtained from the DSMZ (Braunschweig, Germany). Primary human mononuclear cells were isolated through density gradient centrifugation from an apheresis product (San Diego Blood Center, from both female and male donors).
METHOD DETAILS
Cell culture
hiPSC were passaged using Accutase (STEMCELL Technologies, Vancouver, Canada, Cat. 07920) at a ratio of 1:4 to 1:10 on Matrigel (Corning, NY, U.S. Cat. 354277) coated plates, and cells were not allowed to reach full confluency prior to passaging (Knorr et al., 2013; Zhu and Kaufman, 2019). SKOV-3 were sub-cultured according to ATCC recommendations. K562 and MOLM-13 cells were maintained in RPMI 1640 (Thermo Fisher Scientific, Waltham, MA, 11875085) with 10% FBS. MOLM-13 cells were engineered to stably express luciferase and green fluorescent protein (GFP) using a pKT2-IRES-GFP:zeo plasmid in conjunction with a SleepingBeauty cassette (Addgene, Cambridge, Massachusetts) by nucleofection as previously described (Li et al., 2018).
Generation, identification and karyotyping of clonal CISH−/− iPSC
Deletion of CISH in iPSCs was done using CRISPR/Cas9 technology. We designed a pair of gRNAs targeting exon 3 of CISH gene: crRNA-1#: CAAGGGCTGCATGACTGGCT, crRNA-2#: TGCTGGGGCCTTCCTCGAGG; DNA templates for gRNA were synthesized and cloned into pGS-gRNA (GenScript, Inc). pSpCas9 (GenScript PX165, 2 μg), pGS-gRNA-1# (1 μg) and gRNA-2# (1 μg) were co-transfected to iPSC using Nucleofector™ 2b (Lonza, AAB-1001) with Human Stem Cell Nucleofector™ Kit (Lonza, VPH-5012). 24 hours after transfection, cells were selected with puromycin (2.5 ug/ml) (Sigma P8833) for 3 days. Then cells were seeded to 96-well plate at concentration of 1 cell/ml (100 μl per well) for single clone selection. Mutations in CISH gene were identified by Sanger sequencing using the following primer sets: 5’- CCAGCCAGAGGTCATGAAAC, 5’- ACCAGATTCCCGAAGGTAGG. Identified CISH KO clones were verified as single-cell clone by amplifying the target region (same primer sets as sequencing) and TA cloning. Karyotype characterization was done using twenty G-banded metaphase cells by Cell Line Genetics Inc.
Derivation and expansion of NK cells from WT and CISH−/− iPSCs
The derivation of NK cells from iPSCs has been previously described (Zhu and Kaufman, 2019). Briefly, 8,000 iPSCs were seeded in 96-well round-bottom plates with APEL media containing 40 ng/ml human Stem Cell Factor (SCF), 20 ng/ml human Vascular Endothelial Growth Factor (VEGF), 20 ng/ml recombinant human Bone Morphogenetic Protein 4 (BMP-4), and 10 μM rho kinase inhibitor (ROCK inhibitor, Y27632, Sigma). After 6 days of hematopoietic differentiation, spin embryoid bodies (EBs) were then directly transferred into each well of uncoated 6-well plates under NK cell differentiation conditions. NK cell differentiation conditions have previously been reported supplementing media with 5 ng/mL IL-3 (first week only), 10 ng/mL IL-15, 20 ng/mL IL-7, 20 ng/mL SCF, and 10 ng/mL flt3 ligand for 21–35 days. Half-media changes were performed weekly. NK cells were harvested after they reached maturity and co-cultured with irradiated K562-mbIL-21-4-1BB artificial antigen presenting cells (aAPCs) for expansion(Denman et al., 2012) with media containing RPMI 1640 and 10% FBS supplemented with 50 units/mL of hIL-2. aAPCs were kindly provided by Dr. Dean A. Lee (Nationwide Children’s Hospital).
Isolation and expansion of PB-NK cells
Mononuclear cells were isolated through density gradient centrifugation from an apheresis product (San Diego Blood Center), and NK cells were enriched by depleting CD3+ and CD19+ cells using EasySep™ Human NK Cell Enrichment Kit (Stemcell Technologies, 19055). Use of peripheral blood mononuclear cells from donors was approved by the Committee on the Use of Human Subjects in Research at the University of California, San Diego. PB-NK cells were expanded using the same method as used for expansion of iPSC-NK cells.
Immunoblotting
Immunoblotting was performed following standard methods from BioRad. Briefly, SDS-PAGE gels were used to resolve the cell lysates, and proteins were transferred to PVDF membranes using the wet transfer method. The detailed information of the antibodies is provided in Key Resource Table. All the antibody were used at 1:1000 dilution.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| APC-CD34 | BD Biosciences | Cat#345804 RRID: AB_2686894 |
| PE-CD45 | BD Biosciences | Cat#555483 RRID: AB_395875 |
| PE-CD43 | BD Biosciences | Cat#560199 RRID:AB_1645655 |
| PE-CD31 | BD Biosciences | Cat# 340297 RRID:AB_400016 |
| APC-CD56 | BD Biosciences | Cat#555518 RRID: AB_398601 |
| PE-CD56 | BD Biosciences | Cat#5555516 RRID:AB_395906 |
| FITC-CD94 | BD Biosciences | Cat#555888 RRID:AB_396200 |
| APC-CD117 | eBiosciences | Cat#17117842 RRID:AB_2016658 |
| PE-CD107a | BD Biosciences | Cat#555801 RRID:AB_396135 |
| PE-NKG2D | BD Biosciences | Cat#561815 RRID: AB_10896282 |
| PE-NKp46 | BD Biosciences | Cat#557991 RRID: AB_396974 |
| PE-NKp44 | BD Biosciences | Cat#558563 RRID: AB_647239 |
| PE-TRAIL | BD Biosciences | Cat#565499 RRID:AB_2732871 |
| PE-FAS Ligand | BD Biosciences | Cat#564261 RRID:AB_2738713 |
| PE-CD16 | BD Biosciences | Cat#560995 RRID:AB_10562387 |
| PacBlue-IFN-g | Biolegend | Cat#502522 RRID: AB_893525 |
| APC-CD2 | BD Biosciences | Cat#560642 RRID:AB_1727443 |
| PE-CD57 | BD Biosciences | Cat# 560844 RRID:AB_2033965 |
| PE-NKG2C | R&D Systems | Cat# FAB138P RRID:AB_2132983 |
| PE-NKG2A | Beckman Coulter | Cat# IM3291U RRID:AB_10643228 |
| PE-CD158a,h | Beckman Coulter | Cat#A09778 RRID:AB_ 2801261 |
| PE- CD158b1/b2,j | Beckman Coulter | Cat# IM2278U RRID:AB_ 2728104 |
| PE-CD158 e1/e2 | Beckman Coulter | Cat# IM3292 RRID:AB_ 131339 |
| PE-CD122 | BD Biosciences | Cat#554522 RRID:AB_395451 |
| PE-TRA-1–81 | BD Biosciences | Cat#560161 RRID:AB_1645540 |
| APC-SSEA4 | Biolegend | Cat#330417 RRID:AB_2616818 |
| Rabbit monoclonal anti-CIS (D4C10) | Cell Signaling Technology | Cat# 8431 RRID:AB_11179218 |
| Mouse monoclonal anti-JAK1 (73/JAK1) | BD Biosciences | Cat# 610231 RRID:AB_397626 |
| Rabbit monoclonal anti-phospho-JAK1 (Y1022 + Y1023) antibody [EPR1899(2)] | Abcam | Cat# ab138005 RRID:N/A |
| Rabbit monoclonal anti-STAT3 (D3Z2G) | Cell Signaling Technology | Cat# 12640 RRID:AB_2629499 |
| Rabbit polyclonal anti-phospho-STAT3 (Tyr705) | Cell Signaling Technology | Cat# 9131 RRID:AB_331586 |
| Mouse monoclonal anti-STAT5 (251619) | R&D System | Cat# MAB2174 RRID:AB_2239992 |
| Rabbit monoclonal anti-phospho-STAT5 (Tyr694) (D47E7) | Cell Signaling Technology | Cat# 4322 RRID:AB_10544692 |
| Rabbit monoclonal anti-phospho-p70 S6 Kinase (Thr389) (D5U1O) | Cell Signaling Technology | Cat# 97596 RRID:AB_2800283 |
| Rabbit polyclonal anti-p70 S6 Kinase | Cell Signaling Technology | Cat# 9202 RRID:AB_331676 |
| Rabbit monoclonal anti-phospho-S6 Ribosomal Protein (Ser235/236) (D57.2.2E) | Cell Signaling Technology | Cat# 4858 RRID:AB_916156 |
| Rabbit monoclonal anti-S6 Ribosomal Protein (5G10) | Cell Signaling Technology | Cat# 2217 RRID:AB_331355 |
| Rabbit monoclonal anti-GAPDH (D16H11) | Cell Signaling Technology | Cat# 5174 RRID:AB_10622025 |
| Rabbit monoclonal anti- Vinculin (E1E9V) | Cell Signaling Technology | Cat# 13901 RRID:AB_2728768 |
| Bacterial and Virus Strains | ||
| pKT2-mCAG-IRES-GFPZEO | Branden Moriarity lab | N/A |
| pCMV(CAT)T7-SB100 | Addgene | Cat#34879 |
| pSpCas9 | GenScript | PX165 |
| pGS-gRNA | GenScript | N/A |
| Biological Samples | ||
| Human Serum AB | Sigma-Aldrich | Cat#H4522 |
| Peripheral blood buffy coat | San Diego Blood Bank (https://www.sandiegobloodbank.org/) | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant human IL-2 | Proleukin | Cat#NDC-66483-116-07 |
| Recombinant human IL-3 | Pepro Technology | Cat#200–03 |
| Recombinant human IL-7 | R&D Systems | R&D Systems Cat#207-IL |
| Recombinant human IL-15 | R&D Systems | Cat#247-IL |
| Recombinant human FLT-3 Ligand | Pepro Technology | Cat#300–19 |
| Recombinant human SCF | R&D Systems | Cat#255-SC/CF |
| Recombinant human VEGF | R&D Systems | Cat#293-VE |
| Recombinant human BMP-4 | R&D Systems | Cat#314-BP |
| Recombinant human bFGF basic | R&D Systems | Cat#4114-TC |
| Recombinant human IL-12 | R&D Systems | Cat#219-IL-005 |
| Recombinant human IL-18 | R&D Systems | Cat#9124-IL-010 |
| a-MEM culture medium | Fisher Scientific | Cat#12634 |
| Horse serum | Fisher Scientific | Cat#16050130 |
| Fetal bovine serum | Fisher Scientific | Cat# 10437010 |
| CellEvent Caspase-3/7 Green Detection Reagent | Thermo fisher | Cat#C10423 |
| IncuCyte Caspase-3/7 Green Apoptosis Assay | Essenbioscience | Cat#4440 |
| GolgiStop | BD Biosciences | Cat#554724 |
| GolgiPlug | BD Biosciences | Cat#555029 |
| Rapmycine | Sigma | Cat#R8781 |
| STEMdiff™ APEL™2 Medium | Stemcell Technologies | Cat#05270 |
| CellTrace™ Violet Cell Proliferation Kit | Thermo fisher | Cat#C34571 |
| CellTrace™ Far Red Cell Proliferation Kit | Thermo fisher | Cat#C34564 |
| Critical Commercial Assays | ||
| EasySep Human NK Cell Enrichment Kit | Stemcell Technologies | Cat#19055 |
| Human Stem Cell Nucleofector™ Kit | Lonza | Cat# VPH-5012 |
| Seahorse XF Glycolytic Rate Assay Kit | Agilent | Cat# 103344–100 |
| Seahorse XF Cell Mito Stress Test Kit | Agilent | Cat# 103015–100 |
| Deposited Data | ||
| RNA sequencing | This paper | Accession number: GSE150155 |
| Experimental Models: Cell Lines | ||
| Human: iPS cells | Dan S. Kaufman lab | N/A |
| Human: K-562 cells | ATCC | Cat#CCL-243 |
| Human: SKOV-3 cells | ATCC | Cat#HTB-77 |
| Human: MOL-M13 cells | DSMZ | Cat#ACC 554 |
| aAPC | Dean A. Lee lab | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ | Jackson lab | Cat#005557 |
| Oligonucleotides | ||
| gRNAs targeting exon 3 of human CISH gene: crRNA-1#: CAAGGGCTGCATGACTGGCT | This paper | N/A |
| gRNAs targeting exon 3 of human CISH gene: crRNA-2#: TGCTGGGGCCTTCCTCGAGG | This paper | N/A |
| Software and Algorithms | ||
| IncuCyte real-time image system | Essenbioscience | N/A |
| Xenogen IVIS imaging system | Caliper Life Science | N/A |
| NovoExpress software | ACEA Biosciences | N/A |
| Cytobank | Cytobank | N/A |
| Seahorse Wave Desktop Software | Agilent | N/A |
| Prism 8 | Graphpad | N/A |
Flow cytometry
Flow cytometry was done on a NovoCyte (ACEA Biosciences) and data were analyzed using NovoExpress or FloJo software. The antibodies used for flow cytometry are listed in the Antibody section.
Mass cytometry
For viability assessment, cells were stained with Cell-ID Intercalator-103Rh (Fluidigm, San Francisco, CA, 201103B) in complete medium for 20 minutes at 37°C. Maxpar Cell Staining Buffer (Fluidigm, 201068) was used for all antibody staining and subsequent washing. Samples were incubated with Fc receptor binding inhibitor (Thermo Fisher Scientific, 14-9161-73) for 10 minutes at room temperature, before adding surface antibodies and incubating for 30 minutes at 4°C. Subsequently, cells were fixed in Maxpar PBS (Fluidigm, 201058) with 2% paraformaldehyde, transferred to methanol and stored at −20°C. The day after, cells were stained with an intracellular antibody cocktail for 40 minutes at 4°C and labeled with Cell-ID Intercalator-Ir (Fluidigm, 201192B). Samples were supplemented with EQ Four Element Calibration Beads (Fluidigm, 201078) and acquired on a CyTOF 2 (Fluidigm) equipped with a SuperSampler (Victorian Airship, Alamo, CA) at an event rate of <500/sec. Antibodies were either obtained pre-labeled from Fluidigm or conjugated with metal isotopes using Maxpar X8 antibody labeling kits (Fluidigm) (Supplemental Table 1). FCS files were normalized using Helios software (Fluidigm) and gated on CD45+ CD19- CD14- CD32- CD3- viable single cells using Cytobank (Cytobank Inc., Santa Clara, CA). For subsequent analysis, data was imported into R (R Core Team, 2019) using the flowCore package, and transformed using arcsinh(x/5). 20,000 events were randomly sampled from each file and concatenated. t-Distributed Stochastic Neighbor Embedding (t-SNE) was then performed using the Rtsne R package with default settings and the following parameters: 2B4, CD16, CD161, CD2, CD27, CD3, CD34, CD38, CD56, CD57, CD8, CD94, DNAM-1, Granzyme B, LILRB1, Ki-67, KSP37, NKG2A, NKG2C, NKG2D, NKp30, Perforin, Siglec-7, Syk, TIGIT and TIM-3. Results were visualized using the ggplot2 R package.
CD107a expression and IFN-γ staining:
NK cells were harvested, washed with their culture media and co-cultured with tumor targets at a 1:1 effector-to-target ratio. Different combinations of NK cells, tumors, stimulation molecules and antibodies were incubated with anti-CD107a PE (BD Biosciences, Franklin Lakes, NJ, 555801) for 1 hour. Following incubation, 1:1000 Golgi Stop and 1:1000 Golgi Plug solutions (BD Biosciences, 554724, 555029) were added and the cells incubated for an additional 2 hours. Cells were then counterstained with Live/Dead Fixable Aqua Dye (Thermo Fisher Scientific, L34957), IgG1 Isotype APC (BD Biosciences, 551442) and CD56-APC (BD Biosciences, 555518) for 30 min at 4°C. After staining, the cells were washed with buffer containing DPBS (Lonza, Basel, Switzerland, 17–512), 2% standard FBS (Life Technologies, 10100147), 0.1% NaN3 (Sigma Aldrich, 71289) and then fixed with BD Cytofix (BD Biosciences, 554655) for 20 min at 4C. Next, cells were washed and permeabilized using BD Perm/Wash (BD Biosciences, 554723) for 15 min at 4C. Afterwards, cells were stained for 30 min with IFN-γ Pacific Blue (Biolegend, San Diego, CA, 502521). After washing, cells were analyzed.
CellEvent™ Caspase-3/7 Green Flow Cytometry assay:
Target cells were pre-stained with CellTrace™ Violet (Thermo-Fisher Scientific, C34557) at a final concentration of 5μM in PBS for 15min at 37°C. After staining, the cells were washed in complete culture medium prior to being mixed with NK cell cultures at the indicated effector to target (E:T) ratios. After a brief centrifugation, co-cultures were incubated at 37°C for 3.5hrs. Afterwards, CellEvent® Caspase-3/7 Green Detection Reagent (Thermal Fisher Scientific, C10423) was added for an additional 30min of culture for a total incubation time of 4 hours. During the final 5 minutes of staining, SYTOX™ AADvanced™ dead cell stain solution (Thermal Fisher Scientific, S10349) was added and mixed gently. Cells were then analyzed by flow cytometry.
IncuCyte Caspase-3/7 Green Apoptosis assay:
Target cells were labeled with CellTrace™ Far Red (ThermoFisher, C34564). Adherent target cells were seeded in a 96-well plate at a density of 4000 cells/well 24hr before addition of IncuCyte Caspase-3/7 Green Apoptosis Assay Reagent (Essen Bioscience, 4440) to each well diluted by a factor of 1,000. Non-adherent target cells were seeded in fibronectin coated 96-well plates at a density of 30,000 to 50,000 cells/well and further incubated at room temperature for 30 min before the addition of IncuCyte Caspase-3/7 Green Apoptosis Assay Reagent. After incubation, NK cells were added at various E:T ratios and monitored on the IncuCyte ZOOM to acquire images every 1 h for adherent cells and every 30 min for non-adherent cells. Experiments were performed with 3 independent biological triplicates. The cytotoxicity of target cells was analyzed by quantifying red cell number and/or overlay of Caspase 3/7 (green) within the red cells.
RNA Sequencing
Total RNA was isolated from cells using the miRNeasy Kit (Qiagen) according to the manufacturer’s protocol. RNA quality control, library construction, sequencing and data analysis were performed by Novogene Inc. Briefly, RNA quantification and qualification was performed using Nanodrop for checking RNA purity (OD260/0D280), agarose gel electrophoresis and Agilent 2100 for checking RNA integrity. mRNA was purified from total RNA using poly-T oligo-attahched magnetic beads. cDNA library was synthesized using random hexamer primer and M-MuLV Reverse Transcriptase and sequencing using Illumina Hiseq 2000. The threshold of differential expression genes in Volcano diagram is padj<0.05. Color descending from red to blue in heatmap of differential expression genes indicated log10 (FPKM+1) from large to small. ClusterProfiler software was used for all enrichment analysis, including Gene Ontology (GO) enrichment, Disease Ontology (DO) enrichment and Kyoto Encyclopedia of Genes and Genomes (KEGG, http:www.kegg.jp/). padj<0.05 was considered as significant enrichment.
NK cell polyfunctionality evaluation by single-cell cytokine profiling and calculation of PSI
Single-cell cytokine profiling was analyzed using IsoPlexis 32-plex immune cytokine response panel as previous reported(Rossi et al., 2018; Xue et al., 2017). Briefly, cryopreserved NK cells were thawed and resuspended in complete media (RPMI+10%FBS) with 5 ng/ml IL-15 at a density of 1×106 cells/ml. Cells were recovered at 37 °C, 5% CO2 overnight. Next day, cells were stimulated with IL-12 (10 ng/ml, R&D System, 219-IL-005) and IL-18 (100 ng/ml, R&D System, 9124-IL-010) for 1 hour then cells were loaded into the single-cell barcode chip (SCBC) microchip for single-cell secretomics evaluation. A single cell functional profile was determined for each NK cell type. Profiles were categorized into effector (Granzyme B, IFN-γ, MIP-1α, Perforin, TNF-α, TNF-β), stimulatory (GM-CSF, IL-2, IL-5, IL-7, IL-8, IL-9, IL-12, IL-15, IL-21), regulatory (IL-4, IL-10, IL-13, IL-22, TGF-β1, sCD137, sCD40L), chemoattractive (CCL-11, IP-10, MIP-1β, RANTES), and inflammatory (IL-1b, IL-6, IL-17A, IL-17F, MCP-1, MCP-4) groups. Polyfunctional NK cells were defined as cells co-secreting at least 2 proteins from the prespecified panel per cell coupled with the amount of each protein produced (ie, combination of number of proteins secreted and at what intensity). Polyfunctional strength index (PSI) was defined as the percentage of polyfunctional cells, multiplied by mean fluorescence intensity (MFI) of the proteins secreted by those cells, calculated using a prespecified formula(Rossi et al., 2018).
Metabolic studies:
WT iPSC-NK cells and CISH−/− iPSC NK cells were incubated with low concentration of IL-15 (1 ng/ml) for 7 days with or without rapamycin (100 ng/ml) (Sigma, R8781). Cells were resuspended in Seahorse XF Assay Medium (Agilent Technologies) and seeded at 150,000 cells/well (96-well plate). Plate was pre-coated with Poly-D-Lysine (Sigma, P6407) over-night at 37 °C. The extracellular acidification rate and the oxygen consumption rate were measured (pmoles/min) in real time in an XFe96 analyzer using Seahorse XF Glycolytic Rate Assay Kit (Agilent, 103344–100) and Seahorse XF Cell Mito Stress Test Kit (Agilent, 103015–100). Basal glycolysis was measured before the addition of rotenone/antimycin A (0.5 μM) and glycolytic was elucidated by subtracting the rate of glycolysis before and after addition of 2-deoxy-D-glucose (2DG, 100 mM). Basal respiration was measured after addition of glucose, ATP-linked respiration was calculated from subtraction of basal respiration and respiration after addition of oligomycin (1 μM), and mitochondrial spare respiratory capacity (SRC) was measured after addition of carbonyl cyanide-4 (trifluoromethoxy)phenylhydrazone (FCCP, 1 μM).
In vivo xenograft studies:
Mice were sub-lethally irradiated (225 cGy) 1 day prior to tumor engraftment. Mice were given 1×106 luc-expressing MOLM-13 cells via Intravenous (IV). NK cells (107cells/mouse) were injected IV 1 day after tumor cells infusion. NK cells were supported by the injection of IL-2 and/or IL-15 as reported previously(Li et al., 2018). Tumor burden was determined by BLI using the Xenogen IVIS Imaging system. 7 days or 14 days after infusion of NK cells, 50 μl blood was collected from mice tail vein and red blood cells were removed by ACK lysing buffer (Gibco, A10492). The remaining cells were subjected to flow cytometric staining of human CD56-APC (BD Biosciences, 555518) or CD45-PE (BD Biosciences, 555483). Human NK cell persistence in PB was calculated as number of human CD56+ or CD45+ cells per μl blood.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data are presented as the mean ± standard error of the mean. Differences between groups were evaluated using the one-way ANOVA or Two-tailed T test (noted in figure legends). For the quantification of in vivo image, data are presented as the mean ± SEM and differences between groups were analyzed using Two-tailed T test. Survival curve was analyzed using Log-rank (Mantel-Cox) test. Statistical analysis is performed in the environment of GraphPad Prism Statistical. All tests were considered significant at p < 0.05.
Supplementary Material
Highlights:
Deletion of CISH in human NK cells leads to improved anti-tumor activity
CISH−/− NK cells demonstrate more efficient glycolytic and OxPhos activity
The improved metabolic profile is mediated by mTOR signaling
CISH−/− NK cells more effectively treat AML in vivo with longer NK cell persistence
Acknowledgements:
We thank Dr. Dean Lee (Nationwide Children’s Hospital) for providing aAPCs. We thank Jon Chen, Sean Mackay, Tom Cain, Jens Eberlein and Brianna Flynn from Isoplexis for carrying out the polyfunctionality studies. We appreciate helpful discussion and assistance from all Kaufman lab members. KJM was supported by grants from the Norwegian Cancer Society, the Norwegian Research Council, the South-Eastern Norway Regional Health Authority, the Norwegian, the KG Jebsen Center for Cancer Immunotherapy and Radium Hospitalets Legater. These studies supported by NIH R01CA203348 and U01CA217885 (DSK), and CIRM DISC2-09615 (DSK).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure of Conflict of Interest:
Dan S. Kaufman: Consultant for Fate Therapeutics, has equity and receives income. The terms of this arrangement have been reviewed and approved by the University of California, San Diego in accordance with its conflict of interest policies. He also has patents filed or issued related to this work.
Karl-Johan Malmberg: Consults and receives research support from Fate Therapeutics
Kun-Liang Guan: Co-founder and has an equity interest in Vivace Therapeutics, Inc, and OncoImmune, Inc.
Huang Zhu: Has patent filed related to this work.
References:
- Assmann N, O’Brien KL, Donnelly RP, Dyck L, Zaiatz-Bittencourt V, Loftus RM, Heinrich P, Oefner PJ, Lynch L, Gardiner CM, et al. (2017). Srebp-controlled glucose metabolism is essential for NK cell functional responses. Nat Immunol 18, 1197–1206. [DOI] [PubMed] [Google Scholar]
- Bremm M, Pfeffermann LM, Cappel C, Katzki V, Erben S, Betz S, Quaiser A, Merker M, Bonig H, Schmidt M, et al. (2019). Improving Clinical Manufacturing of IL-15 Activated Cytokine-Induced Killer (CIK) Cells. Front Immunol 10, 1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cichocki F, Wu CY, Zhang B, Felices M, Tesi B, Tuininga K, Dougherty P, Taras E, Hinderlie P, Blazar BR, et al. (2018). ARID5B regulates metabolic programming in human adaptive NK cells. J Exp Med 215, 2379–2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong JJ, Wang XW, Zheng XH, Wang D, Fu BQ, Sun R, Tian ZG, and Wei HM (2018). Dysfunction of Natural Killer Cells by FBP1-Induced Inhibition of Glycolysis during Lung Cancer Progression. Cell Metab 28, 243–255.e245. [DOI] [PubMed] [Google Scholar]
- Delconte RB, Kolesnik TB, Dagley LF, Rautela J, Shi W, Putz EM, Stannard K, Zhang JG, Teh C, Firth M, et al. (2016). CIS is a potent checkpoint in NK cell-mediated tumor immunity. Nat Immunol 17, 816–824. [DOI] [PubMed] [Google Scholar]
- Denman CJ, Senyukov VV, Somanchi SS, Phatarpekar PV, Kopp LM, Johnson JL, Singh H, Hurton L, Maiti SN, Huls MH, et al. (2012). Membrane-bound IL-21 promotes sustained ex vivo proliferation of human natural killer cells. PloS one 7, e30264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolstra H, Roeven MWH, Spanholtz J, Hangalapura BN, Tordoir M, Maas F, Leenders M, Bohme F, Kok N, Trilsbeek C, et al. (2017). Successful Transfer of Umbilical Cord Blood CD34(+) Hematopoietic Stem and Progenitor-derived NK Cells in Older Acute Myeloid Leukemia Patients. Clinical cancer research : an official journal of the American Association for Cancer Research 23, 4107–4118. [DOI] [PubMed] [Google Scholar]
- Donnelly RP, Loftus RM, Keating SE, Liou KT, Biron CA, Gardiner CM, and Finlay DK (2014). mTORC1-dependent metabolic reprogramming is a prerequisite for NK cell effector function. Journal of immunology 193, 4477–4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felices M, Lenvik AJ, McElmurry R, Chu S, Hinderlie P, Bendzick L, Geller MA, Tolar J, Blazar BR, and Miller JS (2018). Continuous treatment with IL-15 exhausts human NK cells via a metabolic defect. Jci Insight 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floros T, and Tarhini AA (2015). Anticancer Cytokines: Biology and Clinical Effects of Interferon-alpha 2, Interleukin (IL)-2, IL-15, IL-21, and IL-12. Semin Oncol 42, 539–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu L, Liu Y, An Q, Zhang J, Tong Y, Zhou F, Lu W, Liang X, and Gu Y (2019). Glycolysis metabolic changes in sperm cryopreservation based on a targeted metabolomic strategy. Int J Clin Exp Pathol 12, 1775–1781. [PMC free article] [PubMed] [Google Scholar]
- Gardiner CM (2019). NK cell metabolism. Journal of leukocyte biology. [DOI] [PubMed] [Google Scholar]
- Geller MA, Cooley S, Judson PL, Ghebre R, Carson LF, Argenta PA, Jonson AL, Panoskaltsis-Mortari A, Curtsinger J, McKenna D, et al. (2011). A phase II study of allogeneic natural killer cell therapy to treat patients with recurrent ovarian and breast cancer. Cytotherapy 13, 98–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilton DJ, Richardson RT, Alexander WS, Viney EM, Willson TA, Sprigg NS, Starr R, Nicholson SE, Metcalf D, and Nicola NA (1998). Twenty proteins containing a C-terminal SOCS box form five structural classes. P Natl Acad Sci USA 95, 114–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgins JJ, Khan ST, Park MM, Auer RC, and Ardolino M (2019). Killers 2.0: NK cell therapies at the forefront of cancer control. J Clin Invest 129, 3499–3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntington ND, Legrand N, Alves NL, Jaron B, Weijer K, Plet A, Corcuff E, Mortier E, Jacques Y, Spits H, et al. (2009). IL-15 trans-presentation promotes human NK cell development and differentiation in vivo. J Exp Med 206, 25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawalekar OU, O’Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD Jr., Patel PR, Guedan S, Scholler J, Keith B, et al. (2016). Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 44, 380–390. [DOI] [PubMed] [Google Scholar]
- Keppel MP, Saucier N, Mah AY, Vogel TP, and Cooper MA (2015). Activation-specific metabolic requirements for NK Cell IFN-gamma production. Journal of immunology 194, 1954–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kershaw NJ, Murphy JM, Liau NPD, Varghese LN, Laktyushin A, Whitlock EL, Lucet IS, Nicola NA, and Babon JJ (2013). SOCS3 binds specific receptor-JAK complexes to control cytokine signaling by direct kinase inhibition. Nat Struct Mol Biol 20, 469–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khor CC, Vannberg FO, Chapman SJ, Guo HY, Wong SH, Walley AJ, Vukcevic D, Rautanen A, Mills TC, Chang KC, et al. (2010). CISH and Susceptibility to Infectious Diseases. New Engl J Med 362, 2092–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knorr DA, Ni Z, Hermanson D, Hexum MK, Bendzick L, Cooper LJ, Lee DA, and Kaufman DS (2013). Clinical-scale derivation of natural killer cells from human pluripotent stem cells for cancer therapy. Stem cells translational medicine 2, 274–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, and Mattarollo SR (2017). Natural killer cell metabolism. Molecular immunology. [DOI] [PubMed] [Google Scholar]
- Li Y, Hermanson DL, Moriarity BS, and Kaufman DS (2018). Human iPSC-Derived Natural Killer Cells Engineered with Chimeric Antigen Receptors Enhance Anti-tumor Activity. Cell Stem Cell 23, 181–192 e185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mah AY, and Cooper MA (2016). Metabolic Regulation of Natural Killer Cell IFN-gamma Production. Crit Rev Immunol 36, 131–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Y, van Hoef V, Zhang X, Wennerberg E, Lorent J, Witt K, Masvidal L, Liang S, Murray S, Larsson O, et al. (2016). IL-15 activates mTOR and primes stress-activated gene expression leading to prolonged antitumor capacity of NK cells. Blood 128, 1475–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marais A, Cherfils-Vicini J, Viant C, Degouve S, Viel S, Fenis A, Rabilloud J, Mayol K, Tavares A, Bienvenu J, et al. (2014). The metabolic checkpoint kinase mTOR is essential for IL-15 signaling during the development and activation of NK cells. Nat Immunol 15, 749–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcais A, Marotel M, Degouve S, Koenig A, Fauteux-Daniel S, Drouillard A, Schlums H, Viel S, Besson L, Allatif O, et al. (2017). High mTOR activity is a hallmark of reactive natural killer cells and amplifies early signaling through activating receptors. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelet X, Dyck L, Hogan A, Loftus RM, Duquette D, Wei K, Beyaz S, Tavakkoli A, Foley C, Donnelly R, et al. (2018). Metabolic reprogramming of natural killer cells in obesity limits antitumor responses. Nat Immunol 19, 1330–1340. [DOI] [PubMed] [Google Scholar]
- Miller JS, and Lanier LL (2019). Natural Killer Cells in Cancer Immunotherapy. Annu Rev Canc Biol 3, 77–103. [Google Scholar]
- Miller JS, Morishima C, McNeel DG, Patel MR, Kohrt HEK, Thompson JA, Sondel PM, Wakelee HA, Disis ML, Kaiser JC, et al. (2018). A First-in-Human Phase I Study of Subcutaneous Outpatient Recombinant Human IL15 (rhIL15) in Adults with Advanced Solid Tumors. Clinical cancer research : an official journal of the American Association for Cancer Research 24, 1525–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JS, Soignier Y, Panoskaltsis-Mortari A, McNearney SA, Yun GH, Fautsch SK, McKenna D, Le C, Defor TE, Burns LJ, et al. (2005). Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood 105, 3051–3057. [DOI] [PubMed] [Google Scholar]
- Morvan MG, and Lanier LL (2016). NK cells and cancer: you can teach innate cells new tricks. Nat Rev Cancer 16, 7–19. [DOI] [PubMed] [Google Scholar]
- O’Brien KL, and Finlay DK (2019). Immunometabolism and natural killer cell responses. Nat Rev Immunol 19, 282–290. [DOI] [PubMed] [Google Scholar]
- Palmer DC, Guittard GC, Franco Z, Crompton JG, Eil RL, Patel SJ, Ji Y, Van Panhuys N, Klebanoff CA, Sukumar M, et al. (2015). Cish actively silences TCR signaling in CD8(+) T cells to maintain tumor tolerance. J Exp Med 212, 2095–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce EL, Poffenberger MC, Chang CH, and Jones RG (2013). Fueling immunity: insights into metabolism and lymphocyte function. Science 342, 1242454.24115444 [Google Scholar]
- Pomeroy EJ, Hunzeker JT, Kluesner MG, Lahr WS, Smeester BA, Crosby MR, Lonetree CL, Yamamoto K, Bendzick L, Miller JS, et al. (2019). A Genetically Engineered Primary Human Natural Killer Cell Platform for Cancer Immunotherapy. Mol Ther. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomeroy EJ, Hunzeker JT, Kluesner MT, Crosby MR, Lahr WS, Bendzick L, Miller JS, Webber BR, Geller MA, Walcheck B, et al. (2018). A Genetically Engineered Primary Human Natural Killer Cell Platform for Cancer Immunotherapy. bioRxiv, 430553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW, Bagg A, Marcucci KT, Shen A, Gonzalez V, et al. (2015). Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Science translational medicine 7, 303ra139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poznanski SM, and Ashkar AA (2019). What Defines NK Cell Functional Fate: Phenotype or Metabolism? Front Immunol 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putz EM, Guillerey C, Kos K, Stannard K, Miles K, Delconte RB, Takeda K, Nicholson SE, Huntington ND, and Smyth MJ (2017). Targeting cytokine signaling checkpoint CIS activates NK cells to protect from tumor initiation and metastasis. Oncoimmunology 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, Scott DA, Inoue A, Matoba S, Zhang Y, et al. (2013). Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154, 1380–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranson T, Vosshenrich CAJ, Corcuff E, Richard O, Muller W, and Di Santo JP (2003). IL-15 is an essential mediator of peripheral NK-cell homeostasis. Blood 101, 4887–4893. [DOI] [PubMed] [Google Scholar]
- Rautela J, Surgenor E, and Huntington ND (2018). Efficient genome editing of human natural killer cells by CRISPR RNP. bioRxiv, 406934. [DOI] [PubMed] [Google Scholar]
- Romee R, Cooley S, Berrien-Elliott MM, Westervelt P, Verneris MR, Wagner JE, Weisdorf DJ, Blazar BR, Ustun C, Defor TE, et al. (2018). First-in-human phase 1 clinical study of the IL-15 superagonist complex ALT-803 to treat relapse after transplantation. Blood 131, 2515–2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romee R, Rosario M, Berrien-Elliott MM, Wagner JA, Jewell BA, Schappe T, Leong JW, Abdel-Latif S, Schneider SE, Willey S, et al. (2016). Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Science translational medicine 8, 357ra123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi J, Paczkowski P, Shen YW, Morse K, Flynn B, Kaiser A, Ng C, Gallatin K, Cain T, Fan R, et al. (2018). Preinfusion polyfunctional anti-CD19 chimeric antigen receptor T cells are associated with clinical outcomes in NHL. Blood 132, 804–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Jin YQ, Shen C, Qi H, Chu P, Yin QQ, Li JQ, Tian JL, Jiao WW, Xiao J, et al. (2014). Genetic contribution of CISH promoter polymorphisms to susceptibility to tuberculosis in Chinese children. PloS one 9, e92020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner J, Pfannenstiel V, Waldmann A, Bergs JWJ, Brill B, Huenecke S, Klingebiel T, Rodel F, Buchholz CJ, Wels WS, et al. (2017). A Two-Phase Expansion Protocol Combining Interleukin (IL)-15 and IL-21 Improves Natural Killer Cell Proliferation and Cytotoxicity against Rhabdomyosarcoma. Front Immunol 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegiel B, Vuerich M, Daneshmandi S, and Seth P (2018). Metabolic Switch in the Tumor Microenvironment Determines Immune Responses to Anti-cancer Therapy. Frontiers in oncology 8, 284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrangle JM, Velcheti V, Patel MR, Garrett-Mayer E, Hill EG, Ravenel JG, Miller JS, Farhad M, Anderton K, Lindsey K, et al. (2018). ALT-803, an IL-15 superagonist, in combination with nivolumab in patients with metastatic non-small cell lung cancer: a non-randomised, open-label, phase 1b trial. Lancet Oncol 19, 694–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Q, Bettini E, Paczkowski P, Ng C, Kaiser A, McConnell T, Kodrasi O, Quigley MF, Heath J, Fan R, et al. (2017). Single-cell multiplexed cytokine profiling of CD19 CAR-T cells reveals a diverse landscape of polyfunctional antigen-specific response. Journal for immunotherapy of cancer 5, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JG, Farley A, Nicholson SE, Willson TA, Zugaro LM, Simpson RJ, Moritz RL, Cary D, Richardson R, Hausmann G, et al. (2015). The conserved SOCS box motif in suppressors of cytokine signaling binds to elongins B and C and may couple bound proteins to proteasomal degradation (vol 96, pg 2071, 1999). P Natl Acad Sci USA 112, E2979–E2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Blum RH, Bjordahl R, Gaidarova S, Rogers P, Lee TT, Abujarour R, Bonello GB, Wu J, Tsai PF, et al. (2020). Pluripotent stem cell-derived NK cells with high-affinity noncleavable CD16a mediate improved antitumor activity. Blood 135, 399–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, and Kaufman DS (2019). An Improved Method to Produce Clinical-Scale Natural Killer Cells from Human Pluripotent Stem Cells. Methods in molecular biology 2048, 107–119. [DOI] [PubMed] [Google Scholar]
- Zhu H, Lai YS, Li Y, Blum RH, and Kaufman DS (2018). Concise Review: Human Pluripotent Stem Cells to Produce Cell-Based Cancer Immunotherapy. Stem Cells 36, 134–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
• The published article includes all datasets generated or analyzed during this study
• RNA sequencing data is available in GEO via accession number: GSE150155