

Abstract
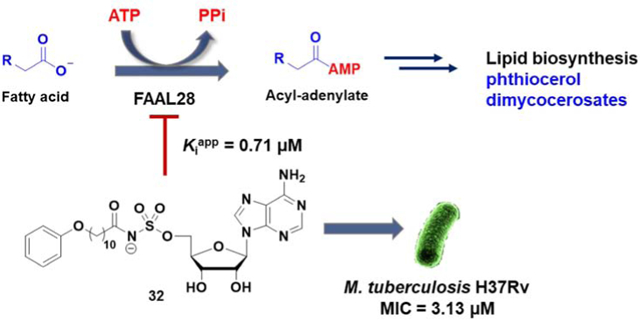
Lipid metabolism in Mycobacterium tuberculosis (Mtb) relies on 34 fatty acid adenylating enzymes (FadDs) that can be grouped into two classes: fatty acyl-CoA ligases (FACLs) involved in lipid and cholesterol catabolism and long chain fatty acyl-AMP ligases (FAALs) involved in biosynthesis of the numerous essential and virulence-conferring lipids found in Mtb. The precise biochemical roles of many FACLs remain poorly characterized while the functionally nonredundant FAALs are much better understood. Here we describe the systematic investigation of 5′-O-[N-(alkanoyl)sulfamoyl]adenosine (alkanoyl adenosine monosulfamate, alkanoyl-AMS) analogs as potential multitarget FadD inhibitors for their antitubercular activity and biochemical selectivity towards representative FAAL and FACL enzymes. We identified several potent compounds including 12-azidododecanoyl-AMS 28, 11-phenoxyundecanoyl-AMS 32, and nonyloxyacetyl-AMS 36 with minimum inhibitory concentrations (MICs) against M. tuberculosis ranging from 0.098–3.13 μM. Compound 32 was notable for its impressive biochemical selectivity against FAAL28 (apparent Ki = 0.7 μM) versus FACL19 (Ki >100 μM), and uniform activity against a panel of multidrug and extensively drug-resistant TB strains with MICs ranging from 3.13–12.5 μM in minimal (GAST) and rich (7H9) media. The SAR analysis provided valuable insights for further optimization of 32 and also identified limitations to overcome.
Keywords: Mycobacterium tuberculosis, fatty acyl-AMP ligases, FAAL28, fatty acyl-CoA ligases, FACL19, acyl-AMS analogs
GRAPHICAL ABSTRACT
1. Introduction
Tuberculosis (TB) has afflicted humans for thousands of years and is the leading cause of death from a single infectious agent.[1] One quarter of the world’s population is infected asymptomatically with the pathogenic bacteria Mycobacterium tuberculosis (Mtb), the causative agent of TB.[1] According to a World Health Organization (WHO) estimate, approximately 10.4 million individuals fell ill with TB in 2018 resulting in 1.5 million deaths.[1] Chemotherapy for the simplest drug-susceptible TB requires a four-drug regimen comprised of isoniazid, rifampicin, pyrazinamide and ethambutol for 2 months, followed by a continuation phase of isoniazid and rifampicin for another 4 months. Drug resistant TB (DR-TB) is considerably more challenging to treat and involves more complicated and longer drug regimens from 9–20 months with treatment success rates declining to 56% for multidrug resistant TB and 39% for extensively drug resistant TB.[2] To combat the growing crisis of DR-TB and achieve the WHO End TB Strategy, improved diagnostics, more effective vaccines and new drugs will be required to stop TB transmission.
Mycobacteria produce a tremendously diverse repertoire of lipophilic molecules. These molecules range from simple short chain fatty acids to the very complex mycolic acids.[3-5] Lipids in the cell envelope of Mtb include the essential mycolic acids as well as the virulence-conferring phthiocerol dimycocerosates (PDIMs), phenolic glycolipids (PGL), sulfolipids, and conditionally essential mycobactins (Fig. 1).[6-9] Lipid degradation is also critical and mycobacteria utilize host lipids (fatty acids and cholesterol) to fuel central metabolic pathways and as substrates for many of the aforementioned complex mycobacterial lipids. [10-13]
Fig. 1. Unique lipids found in cell envelope of Mtb.
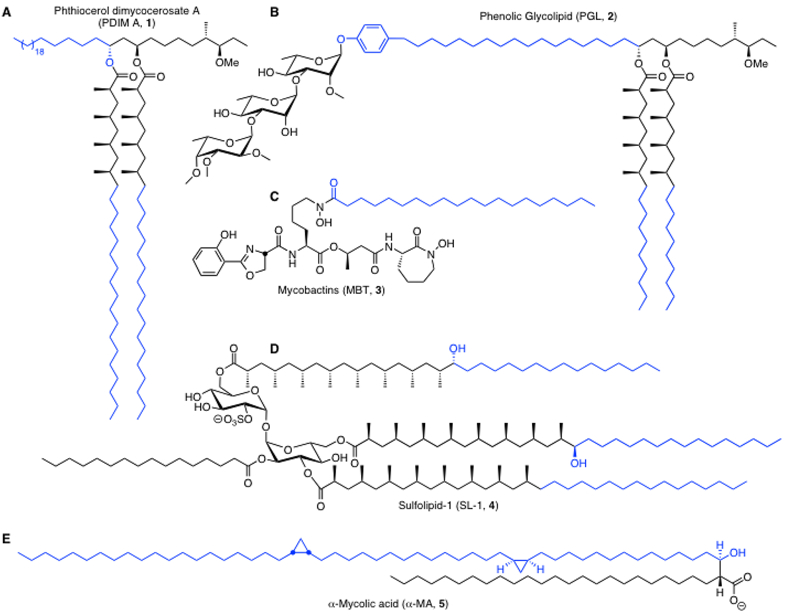
All of the molecules shown exist as a suite of related isomers that vary in the lipid chain length. If reported, the major isomer is shown otherwise a representative molecule is depicted. Specific FadDs are responsible for installation of the lipid chains highlighted in blue. A. PDIM A (1) biosynthesis requires FAAL26 and FAAL28 for synthesis of phthiocerol and mycocerosic acid moieties, respectively. B. PGLs (2) require FAAL22 and FAAL29 for assembly of the phenolphthiocerol lipid as well as FAAL28 for the two mycocerosic acids. C. MBTs (3) employ FAAL33 for installation of the C-20 lipid residue on the central lysine moiety. D. Sulfolipids represented by SL-1 (4) require FAAL23 for biosynthesis of the phthioceranic acid and two hydroxyphthioceranic acid groups. E. The mycolic acids represented by the most abundant α-MA (5) employ FAAL32 for introduction of the meromycolic acid subunit.
The fadD family of genes are involved in both lipid biosynthesis and catabolism. Highlighting the importance of lipid metabolism in mycobacteria the Mtb genome contains an astonishing 34 fadD genes, whereas E. coli encodes for a single fadD involved in fatty acid degradation. Based on their function, the mycobacterial fadD’s are grouped into two subclasses: fatty acyl-CoA ligases (FACLs) involved in lipid degradation and fatty acyl-AMP ligases (FAALs) dedicated to lipid biosynthesis.[11-19] Gokhale has proposed to rename the FadDs by their functional classification, for example FadD19, a fatty acyl-CoA ligase, is renamed FACL19 and FadD28, a fatty acyl-AMP ligase, is renamed FAAL28.[17] Both FACLs and FAALs catalyze the ATP-dependent activation of fatty acid substrates to an intermediate acyl-adenylate, but transfer the acyl group onto different substrates: coenzyme A (CoA) for FACLs and polyketide synthases (PKS) for FAALs (Fig. 2).
Fig. 2. FadD enzyme mechanism.
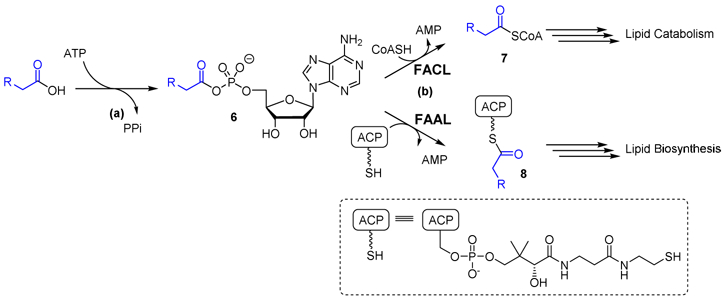
FadDs catalyze a two-step reaction. In the first step (a) FadDs catalyze the adenylation of a fatty acid to afford an intermediate acyladenylate 6. In the second reaction (b) FadDs catalyze the acylation of an acceptor molecule resulting in thioester products 7 or 8. FadDs that form CoA esters 7 are classified as fatty acyl CoA ligases (FACLs) whereas FadDs that load the ACP domain of polyketide synthase enzymes to provide 8 are known as fatty acyl AMP ligases (FAALs).
The precise biochemical roles of the 20 FACLs are largely unknown. Four acyl-CoA synthetases FACL3, FACL17, FACL18 and FACL19 were shown to be up-regulated during growth of Mtb on cholesterol.[20, 21] Subsequently, FACL3, FACL17 and FACL19 have been biochemically validated as CoA ligases involved in cholesterol degradation. FACL3 is required in catabolism of the steroid rings C and D metabolism whereas FACL17 and FACL19 are involved in degradation of the C17 side chain of cholesterol.[20, 22-25] The sequence of FACL18 is nearly identical to FACL19 and Sampson has hypothesized FACL18 may have arisen from gene duplication.[23] FACL13 is part of the mymA operon in remodeling the cell envelope of intracellular Mtb under acidic conditions and exhibits a distinct preference for C24 and C26 fatty acids.[26] Transposon mutagenesis studies suggest most FACLs are not essential, which may be due to their functional redundancy.[27] Indeed, FACL6, FACL15, and FACL19 were shown to possess broad substrate specificity.[14]
By contrast, the FAAL class of FadDs appears to be functionally nonredundant and serve to link fatty acid and polyketide synthesis in mycobacteria.[13, 16] FadD10, involved in the synthesis of a virulence-related lipopeptide, was misannotated as a FACL; however, it is in fact a FAAL that transfers fatty acids to an acyl carrier protein (Rv0100).[28] FAAL21 is the fatty acyl AMP ligase that provides the activated fatty acyl starter unit to Pks3/4.[29] FAAL22 is essential for synthesis of the phenolic glycolipids and is responsible for the activation and transfer of 4-hydroxybenzoic acid onto PKS15/1.[30-32] FAAL23 was found to be involved in sulfolipid production.[33] There are three acyl-AMP ligases: FAAL26, FAAL28 and FAAL29 that are required for the biosynthesis of PDIM, a major virulence lipid in the cell wall.[34-38] FAAL26 initiates phthiocerol synthesis by loading the polyketide synthase PpsA with long-chained fatty acids while FAAL28 initiates mycocerosic acid synthesis by loading the PKS protein mycocerosic acid synthase.[7, 17, 31, 39-43] Finally, FAAL32 is required for activation of the long meromycolic chain and is essential for mycobacterial growth. [44-47]
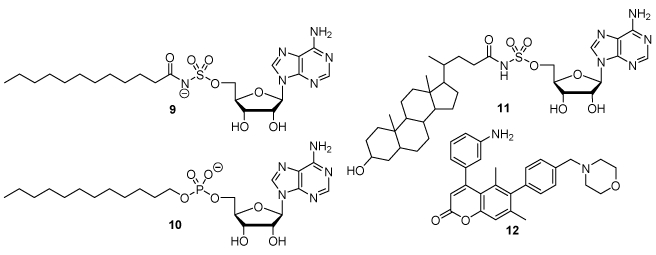
The identification of specific inhibitors as tool compounds against each class of FadDs (FACL or FAAL) or selective inhibitors of an individual FadD enzyme can help to decipher the functional role of FadDs in lipid metabolism. Inhibitors that target crucial nodes or simultaneously disrupt the lipid metabolic network through multitarget inhibition could lead to the development of new class of antitubercular agents.[11, 12] Since FadDs are a newly discovered family of adenylate-forming enzymes in Mtb there are few reported inhibitors.[48] Among them, 5′-O-[N-(dodecanoyl)sulfamoyl]adenosine 9 (Fig. 3) was shown to inhibit FAAL28 and FACL19 with apparent Ki values of 1.5 μM and 4.9 μM respectively and possessed very modest antitubercular activity with a minimum inhibitory concentration (MIC) of only 100 μM against Mtb.[17] The corresponding dodecylphosphate-AMP analogue 10 (Fig. 3) inhibited FAAL32 with an apparent Ki of 0.11 μM and was also a weak inhibitor of growth of M. smegmatis with an MIC of 20 μM.[45] Niu et al. hypothesized that non-hydrolyzable analogues of steroid metabolites (e.g., cholestenoic acyl-AMP or choloyl-AMP) could act as a class-specific inhibitor of the acyl-CoA synthetases responsible for steroid side chain degradation. [49] They designed 5′-O-[N-lithocholoyl)sulfamoyl]adenosine (LCA-AMS) 11 (Fig. 3) which exhibited highly selective inhibition toward mycobacteria. LCA-AMS 11 inhibited M. smegmatis FadD17 and FadD1 with apparent Ki values at 23 and 67 nM, respectively, but again displayed weak growth inhibition with an MIC of 50 μM against Mtb. The only non-substrate inhibitor of an FadD was described by Hung and co-workers, who discovered a coumarin analog as a FadD32 inhibitor from phenotypic high-throughput screening and identified the target through whole-genome sequencing.[50-52] The optimized inhibitor CCA34 12 (Fig. 3) did not block the adenylation activity of FadD32, but rather lipid transfer onto Pks13 with an IC50 of ~5 μM.[52]
Fig. 3. Previously described nucleoside-based FadD inhibitors.
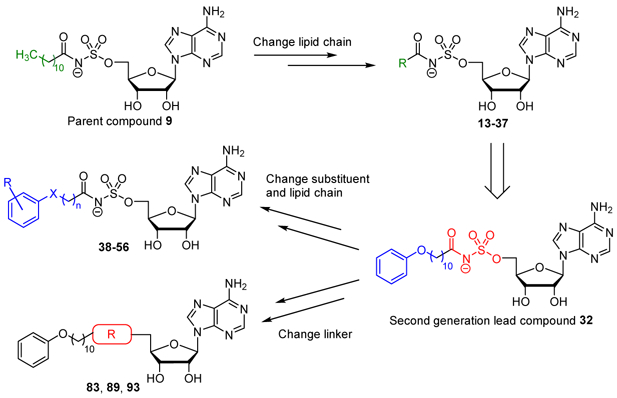
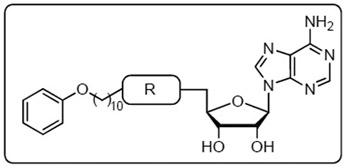
Herein we describe our efforts to prepare nucleoside inhibitors of FAAL enzymes using decanoyl-AMS 9 as a template. All compounds were initially evaluated against representative FACL and FAAL enzymes as well as for their whole-cell activity against Mtb H37Rv in minimal and rich media. We examined the importance of the acyl chain length, introduced conformational constraints into the acyl chain, and explored substituents in the vicinal and distal ends of the acyl side chain of 9. These efforts led to the identification of 11-phenoxyundecanoyl-AMS 32 with significantly improved anti-mycobacterial activity and selectivity (Fig. 4). We then performed two independent SAR campaigns of 32 to investigate both the placement and flexibility of the terminal phenoxy group as well as the importance of acyl-sulfamate linker. Finally, the most promising analogs were evaluated against a panel of MDR-TB and XDR-TB strains.
Fig. 4. Rational design of the new acyl-sulfamoyl adenosine-based inhibitors.
2. Results and discussion
2.1. Chemistry
All inhibitors were synthesized following the general approach described in the Scheme 1. Fatty acids were converted to the corresponding N-hydroxysuccinimide ester (9c, 13c-56c) and then coupled with 2′,3′-O-isopropylidene-5′-O-sulfamoyladenosine 57 in the presence of cesium carbonate to afford protected acyl-sulfamoyladenosine intermediates 9d and 13d–56d. Deprotection of the isopropylidene group with aqueous TFA afforded the desired bisubstrate inhibitors 9d and 13d–56d in low to moderate yields in greater than 95% purities (Scheme 1).
Scheme 1.
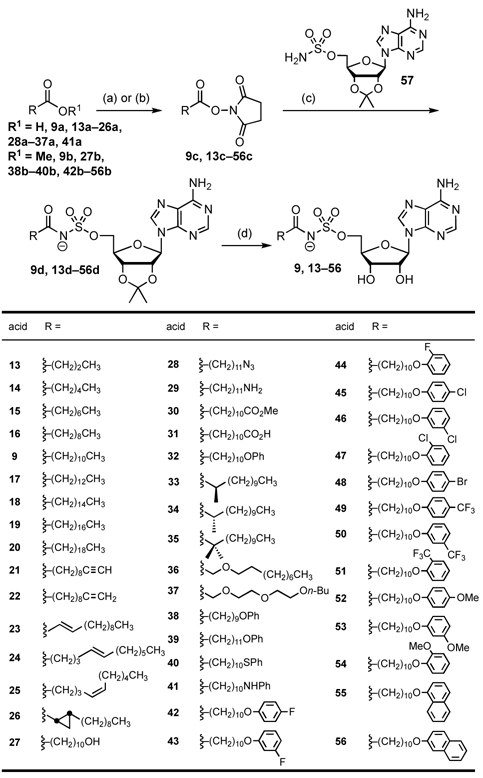
Synthesis of acyl sulfamate inhibitors 9 and 13–56. Reaction conditions: (a) N-hydroxysuccinimide, DCC, CH2Cl2; (b) 1 N aqueous NaOH/MeOH, 100 °C, then N-hydroxysuccinimide, EDC, CH2Cl2; (c) Cs2CO3, DMF, 78–93%; (d) 80% aqueous TFA, 18–89%.
Catalytic hydrogenation of the 12-azido analogue 28 afforded its 12-amino counterpart 29 in good yield while saponification of 12-methoxy carbonyl derivative 30 provided the corresponding acid 31. Attempts to prepare the (Z)-2-dodecanoyl-AMS analogue 23 revealed that the α,β-unsaturated carbonyl moiety of 23d readily isomerizes to the more stable trans-3-dodecanoyl-AMS intermediate providing 23d as a 2:1 E:Z mixture. Moreover, attempts to synthesize pure (E)-2-dodecenoyl-23d from the corresponding N-hydroxysuccinimide ester of cis-2-dodecenoic, provided the same 2:1 E:Z mixture of products (not shown), which was inseparable by flash chromatography conditions suggesting the 2:1 E:Z mixture represents the equilibrium ratio of products under the basic reaction conditions.
Fatty acids that were not available commercially were synthesized as described in Schemes 2-6. (Z)-2-Dodecanoic acid 23a was prepared using the improved method of Rappe by bromination of 2-dodecanone 58 to yield 1,3-dibromoketone 59, which was converted to 23a in good yield by Favorskii rearrangement mediated by sodium hydroxide (Scheme 2A).[53] The constitutional isomer (E)-5-dodecanoic acid 24a was synthesized from 1-octyne 60 by hydrozirconation with Schwarz’s reagent and Negishi coupling of the corresponding vinylzirconium intermediate with ethyl 4-bromobutyrate employing Pd2(dba)3. Saponification of the ethyl ester 61 provided (E)-5-dodecanoic acid (Scheme 2B).[54] (Z)-5-Undecanoic acid 25a was synthesized in one step by Wittig reaction between hexanal 62 and (4-carboxybutyl)triphenylphosphonium bromide 63 in the presence of sodium bis(trimethylsilyl)amide (Scheme 2C).[55] (1S,2R)-2-Nonyl-cyclopropanecarboxylic acid 26a was synthesized from 2-dodecen-1-ol 64 using the method of Charette for the enantioselective cyclopropanation of allylic alcohols.[56] The resulting alcohol 65 was then oxidized by Jones reagent to the desired carboxylic acid 26a (Scheme 2D). The (Z)-2-dodecenoic acid 23a is not stable at room temperature and readily converts to the more stable E isomer.
Scheme 2.
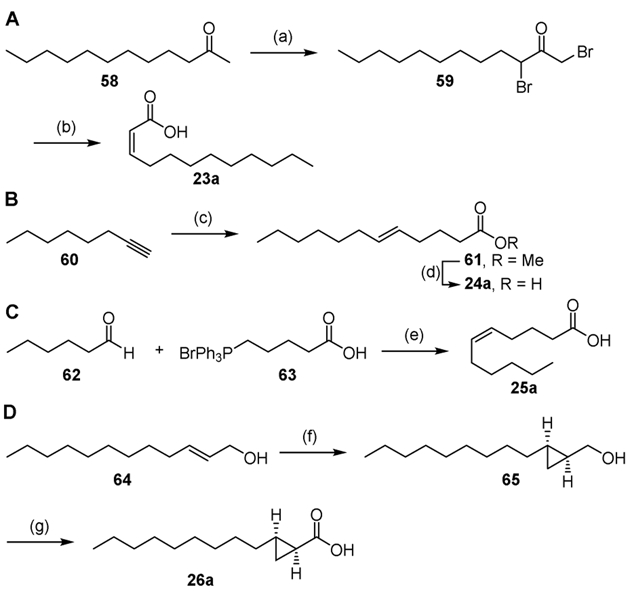
Synthesis of acids 23a–26a. Reaction conditions: (a) HBr (aq), Br2, 99%; (b) NaOH (aq), then HCl (aq), 87%; (c) Cp2ZrHCl, Pd2(dba)3, LiBr, NMP/THF, then ethyl 4-bromobutyrate, 79%; (d) LiOH, MeOH/H2O, 70%; (e) NaHMDS, THF, 40%; (f) (4R,5R)-2-butyl-N,N,N′N′-tetramethyl-1,3,2-dioxaborolane-4,5-dicarboxamide, Zn(CH2I)2, CH2Cl2, 66%; (g) CrO3, H2SO4, H2O/acetone, 75%.
Scheme 6.
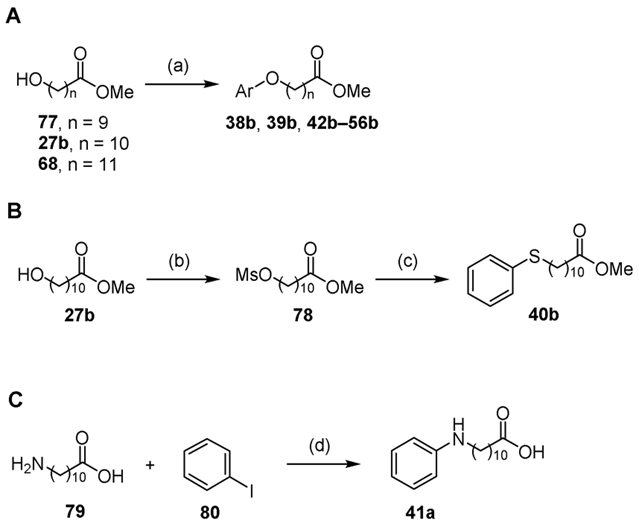
Synthesis of 41a, 38b–56b. Reaction conditions: (a) ArOH, DIAD, PPh3, THF, 65–95%; (b) MsCl, Et3N, THF, 97%; (c) NaH, THF, thiophenol, 98%; (d) 10 mol% CuI, K3PO4·H2O, H2O/decanol, 84%.
12-Azidododecanoic acid 28a was prepared from 12-bromododecanoic acid 66 by nucleophilic substitution with sodium azide (Scheme 3A).[57] 12-Methoxy-12-oxododecanoic acid 30a was synthesized by Baeyer-Villiger oxidation of cyclododecanone 67 with potassium peroxydisulfate to afford 12-hydroxy methyl ester 68 followed by Jones oxidation (Scheme 3B).[58, 59]
Scheme 3.
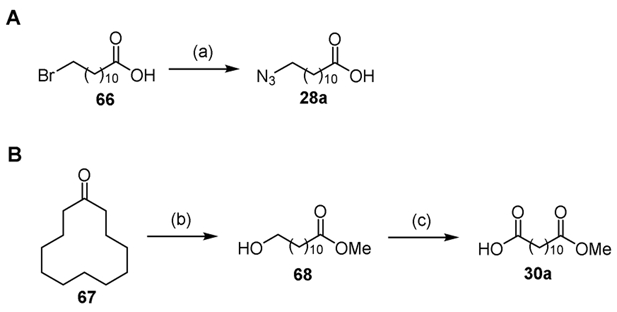
Synthesis of acids 28a and 30a. Reaction conditions: (a) NaN3, DMSO, 97%; (b) H2SO4, K2S2O8, MeOH, 83%; (c) CrO3, H2SO4, H2O/acetone, 57%.
The enantiopure 2-methyldodecanoic acids 33a and 34a (Scheme 4A-B) were prepared by Evan’s asymmetric alkylation methodology.[60] The requisite N-acyloxazolidinones 69 and 71 were prepared by lithiation of (S) and (R)-4-benzyloxazolidin-2-one and subsequent reaction with the mixed pivalic dodecanoic anhydride. Methylation of the Z-enolates of 69 and 71 derived by deprotonation with sodium bis(trimethylsilyl)amide furnished 70 and 72, respectively. The chiral auxiliary was removed with lithium hydroperoxide to afford the desired (R) and (S)-2-methyldodecanoic acids 33a and 34a.[61, 62] Two cycles of methylation of the lithiated dodecanoyl enolate of 9b afforded the desired 2,2-dimethyl ester 73, which was then saponified with lithium hydroxide to provide the desired 2,2-dimethyldodecanoic acid 35a (Scheme 4C).[63]
Scheme 4.
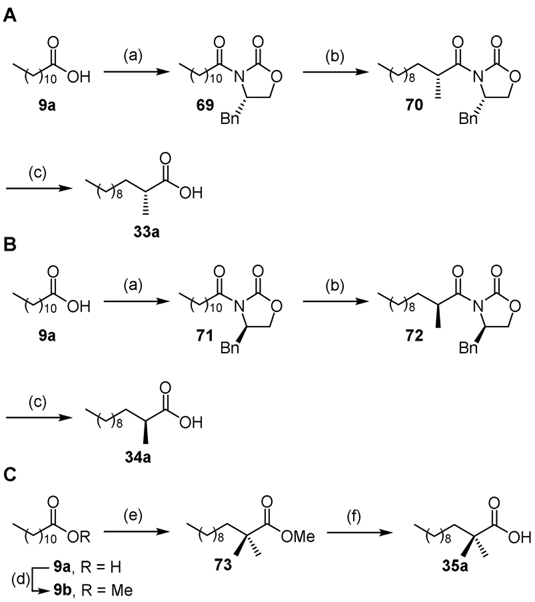
Synthesis of acids 33a–35a. Reaction conditions: (a) PivCl, DIPEA, THF, then n-BuLi, (S) or (R)-4-benzyl-2-oxazolidinone, THF, 97%; (b) NaHMDS, MeI, THF, 78%; (c) 30% H2O2, LiOH, THF/H2O, 99%; (d) H2SO4, MeOH, 99%; (e) LDA, THF, then MeI, repeated 2×, 53% (2 steps), (f) LiOH, THF/MeOH/H2O, 88%.
2-(Nonyloxy)acetic acid 36a and 2-[2-(2-butoxyethoxy)ethoxy]acetic acid 37a were synthesized in moderate yields by the coupling of nonanol 75 and 2-(2-butoxyethoxy)ethanol 76, respectively with chloroacetic acid 74 in the presence of NaH (Scheme 5A-B).[64]
Scheme 5.
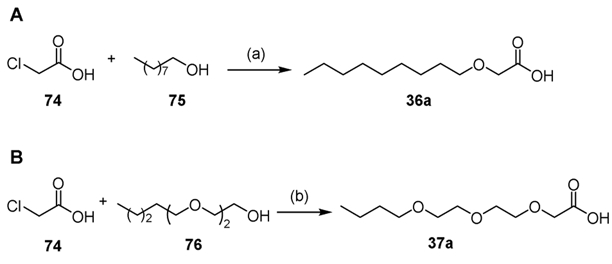
Synthesis of 36a and 37a. Reaction conditions: (a) NaH, THF: 30% (36a), 55% (37a).
Fatty acyl methyl esters 38b, 39b, and 42b–56b were prepared from 10, 12, or 11-hydroxy-undecanoic acid methyl ester 77, 68, or 27b respectively under Mitsunobu coupling conditions with the appropriate phenol (Scheme 6A). Mesylation of 11-hydroxyundecanoic acid methyl ester 27b followed by coupling with thiophenol in the presence of sodium hydride provided methyl 11-(phenylthio)undecanoate 40b (Scheme 6B). 11-(Phenylamino)undecanoic acid 41a was synthesized from 11-aminoundecanoic acid 79 via coupling with iodobenzene 80 in the presence of copper iodide and potassium phosphate to afford the desired acid in good yield (Scheme 6C).
Previous work in our group with other adenylating enzyme inhibitors showed that the acylsulfamide is also an excellent bioisostere of the acyl-adenylate and is more stable.[65] Consequently, we prepared acylsulfamide 83 by coupling 11-phenoxyundecanoate N-hydroxysuccinimide 32c with 2′,3′-O-isopropylidene-5′-N-sulfamoylamideadenosine 81 in the presence of cesium carbonate followed by the removal of the isopropylidene group with aqueous TFA (Scheme 7).
Scheme 7.
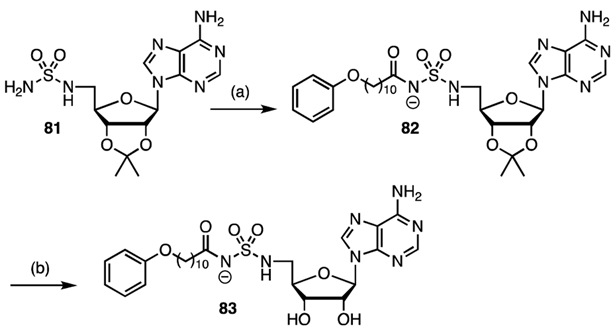
Synthesis of acylsulfamide 83. Reaction conditions: (a) Cs2CO3, DMF, 32c; (b) 80% aqueous TFA, 42% (2 steps).
Alkyl sulfamate 89 was created to explore the importance of the carbonyl group for activity. Reduction of 11-phenoxyundecanoic acid 32a via LiAlH4 provided the alcohol 84, which was subsequently tosylated to supply ether 86. Alkylation of N6,N6-bis(tert-butoxycarbonyl)-2′,3′-O-isopropylidene-5′-O-sulfamoyladenosine 87 with ether 86 employing Cs2CO3 gave sulfamate 88, which was deprotected with aqueous TFA to yield the desired alkyl sulfamate 89 (Scheme 8).
Scheme 8.
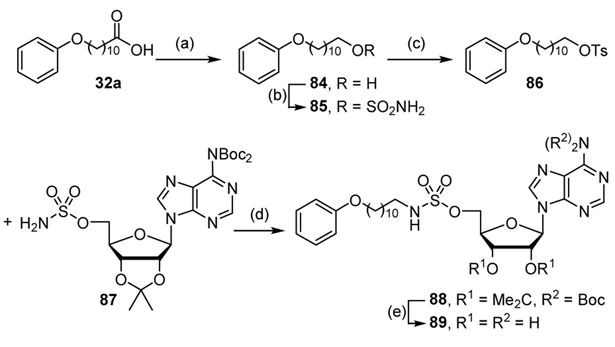
Synthesis of sulfamate 89. Reaction conditions: (a) LiAlH4, THF, 80%; (b) ClSO2NH2, NaH, THF, 100%; (c) TsCl, DMAP, CH2Cl2, 100%; (d) Cs2CO3, DMF, 22%; (e) 80% aqueous TFA, 97%.
We also prepared the reverse alkyl sulfamate by first tosylating N6,N6-bis(tert-butoxycarbonyl)-2′,3′-O-isopropylideneadenosine 90 to provide compound 91. Alkylation of 91 with sulfamate 85 utilizing Cs2CO3 followed by deprotection with aqueous TFA afforded the desired compound 93 (Scheme 9).
Scheme 9.
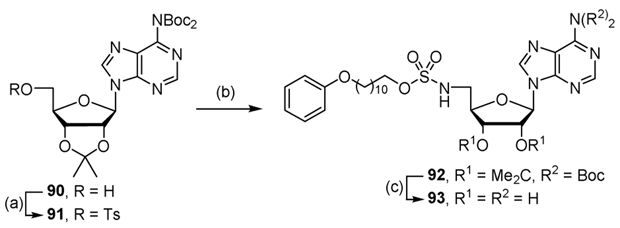
Synthesis of reverse sulfamate 93. Reaction conditions: (a) TsCl, DMAP, CH2Cl2, 98%; (b) Cs2CO3, DMF, 85; (c) 80% aqueous TFA, 11% (2 steps).
2.2. Antimicrobial Activity
Acyl-AMS analogues 9, 13–56, 83, 89 and 93 were evaluated for their whole-cell activity against Mycobacterium tuberculosis H37Rv in GAST and 7H9 media. GAST medium is a minimal medium where the sole carbon source is glycerol. We also evaluated compounds in 7H9 medium containing glycerol and glucose as the carbon sources supplemented with and without palmitate. The minimum inhibitory concentration (MIC) defined here is the concentration that results >99% inhibition of cell growth. The MICs for the acyl-AMS analogues exhibit a high media dependence with the best activity observed in GAST medium and poor activity in 7H9 medium either with and without palmitate (Table 1). Hence in the discussion of SAR trends below, the relative microbiological activity refers to the activity in GAST medium unless otherwise noted. The attenuated activity in 7H9 medium may be due to the high protein binding of these acidic lipophilic molecules since 7H9 contains bovine serum albumin. Alternatively, nutrient-rich media 7H9 contain lipids and other fatty acids that can potentially rescue activity of some FadD’s.
Table 1.
Biological activity and cytotoxicity of 5′-O-[N-(acyl)sulfamoyl]adenosine analogues 9, 13–37.
 |
|||||||
|---|---|---|---|---|---|---|---|
| MIC (μM) | Kiapp (μM) | Cytotox | |||||
| Cmpd | R= | GASTa | 7H9b | 7H9/palmitatec | FACL19d | FAAL28e | IC50 (μM)f |
| 13 | CH3(CH2)2- | >100 | >100 | >100 | >100 | >100 | 81.4 |
| 14 | CH3(CH2)4- | 100 | >100 | >100 | >100 | >100 | 48.6 |
| 15 | CH3(CH2)6- | 12.5–25 | 100 | >100 | >100 | >100 | 6.0 |
| 16 | CH3(CH2)8- | 1.56 | 100 | >100 | >100 | 88 | 10.1 |
| 9 | CH3(CH2)10- | 0.19–0.39 | 50 | 50 | 67.4 | 6.4 | 28.7 |
| 17 | CH3(CH2)12- | 0.39–0.78 | 50 | 100 | 10.0 | 1.4 | 0.9 |
| 18 | CH3(CH2)14- | 6.25 | 100 | >100 | 3.2 | 1.0 | 0.3 |
| 19 | CH3(CH2)16- | 100 | 100 | >100 | 2.4 | 0.5 | 0.1 |
| 20 | CH3(CH2)18- | >100 | >100 | >100 | ndg | nd | 0.1 |
| 21 | HCC(CH2)8- | 1.56 | 50 | 50 | >100 | >100 | 9.7 |
| 22 | H2CHC(CH2)8- | 0.39–0.78 | 25 | 25 | >100 | >100 | 11.6 |
| 23 | CH3(CH2)7CH2CHCH-trans- | 100 | >100 | >100 | >100 | >100 | 10.4 |
| 24 | CH3(CH2)5CHCH(CH2)3-trans- | 100 | >100 | >100 | >100 | >100 | 16.8 |
| 25 | CH3(CH2)4CHCH(CH2)3-cis- | 6.25 | 100 | 100 | >100 | >100 | 14.3 |
| 26 | CH3(CH2)8CH(CH2)CH-S,S- | 50 | >100 | >100 | >100 | 69.8 | 6.1 |
| 27 | HO(CH2)10- | 6.25 | 100 | 100 | >100 | 92.7 | 42.7 |
| 28 | N3(CH2)11- | 0.19 | 12.5–25 | 25 | >100 | 28.5 | 5.6 |
| 29 | NH2(CH2)11- | 3.13 | 50 | 12.5–25 | 93.3 | >100 | 27.1 |
| 30 | MeOOC(CH2)10- | 3.13 | 100 | >100 | >100 | >100 | 14.0 |
| 31 | HOOC(CH2)10- | 25–50 | >100 | >100 | >100 | >100 | >100 |
| 32 | C6H5O(CH2)10- | 3.13 | 50 | 50 | >100 | 0.71 | 2.0 |
| 33 | CH3(CH2)9CH(CH3)-R- | 12.5–25 | >100 | >100 | >100 | >100 | 39.4 |
| 34 | CH3(CH2)9CH(CH3)-S- | 50 | >100 | >100 | >100 | >100 | 14.5 |
| 35 | CH3(CH2)9C(CH3)2- | 12.5–25 | >100 | >100 | 54.7 | >100 | 6.7 |
| 36 | CH3(CH2)8OCH2- | 0.098–0.19 | 25 | 25–50 | >100 | >100 | 8.7 |
| 37 | CH3(CH2)3O(CH2)2O(CH2)2OCH2- | 6.25 | >100 | >100 | >100 | >100 | 16.0 |
Microbiological and biochemical data is from 3 replicates.
Whole cell inhibitory activity against Mtb H37Rv in GAST medium.
Whole cell inhibitory activity against Mtb H37Rv in 7H9 medium.
Whole cell inhibitory activity against Mtb H37Rv in 7H9/palmitate medium.
Inhibitory activity against FACL19. The standard deviation was ≤10% of the mean.
Inhibitory activity against FAAL28. The standard deviation was ≤10% of the mean.
Cytotoxicity against Vero monkey cells using the MTT assay.
nd = no data.
Compounds 9 and 13–20 consisting of lipid chains ranging from C4 to C20 were evaluated initially to determine the optimum chain length. The activity followed a parabolic relationship with activity monotonically increasing with length of the lipid chain from C4 to C12, peaking at C12, then decreasing from C14 to C20. Among them, the three analogs bearing C10-, C12- and C14-side alkyl chains, corresponded to the highest potency in the series, having a MIC in the range 0.19–1.56 μM in GAST medium. Altering the lipid chain by shortening to C8 or lengthening to C16 displayed 64– and 16–fold lower activity respectively when compared to C12 compound. Analogs containing the shortest C4 chain (13) and the longest C20 lipid (20) were completely inactive.
Based on the impressive whole cell activity of dodecanoyl-AMS 9, we conducted a systematic SAR analysis to further refine the acyl chain. Introduction of unsaturation with a terminal alkyne (21) or terminal alkene (22) was well tolerated resulting in a modest 2–4-fold loss in activity. However, incorporation of trans-unsaturation or trans-cyclopropanes within the chain in trans-Δ2-23, trans-Δ5-24 and trans-2-cyclopropyl 26 almost completely abolished activity with MICs increasing to 50–100 μM. By contrast, cis-Δ5-25 retained considerable activity with an MIC of 6.25 μM. Introduction of alcohol, amino, methyl ester, and phenoxy functional groups on the terminus of the lipid chain (27, 29, 30 and 32) led to 8–16-fold reduction in potency relative to 9, but these analogs still retained appreciable activity with MICs of 3.13–6.25 μM demonstrating some flexibility at this position. The lipophilic terminal azide 28 was equipotent while the polar carboxylic acid was 128-fold less active. Taken together, these results suggest only neutral or positively charged groups are permitted at the lipid terminus. Introduction of R or S-configured α-methyl substituents in 33 and 34 or an α-gem-dimethyl in 35 resulted in a greater than 64–fold loss in activity indicating steric bulk is poorly tolerated at this position. Lastly, introduction of oxygen atoms in the lipid chain was explored with mono-ether 36 and tri-ether 37. Amazingly, mono-ether showed a 2–fold increase in activity (MIC = 0.098–0.19 μM), but this gain in activity was lost by incorporation of additional oxygen atoms in the lipid chain.
Although not the most potent in terms of MIC, we discovered that 11-phenoxyundecanoyl-AMS 32 exhibited biochemical selectivity for FAAL28 over FACL19 (vide infra) and thus elected to perform additional SAR of this new lead molecule with the goal to further improve antimicrobial activity while retaining selectivity for the acyl-AMP ligase FAAL28. We first evaluated homologs 38–39 one carbon shorter and longer, respectively (Table 2) and observed the parent compound 32 possessed optimal activity. Replacement of the ether oxygen atom for a sulfur atom obliterated activity. However, exchanging the oxygen for nitrogen led to a modest 4-fold loss in activity, a result consistent with 12-amino derivative 29 affirming the tolerance for a positively charged group at the lipid terminus. We next explored modification of the aryl ring with a range of electron donating and withdrawing substituents at the ortho-, meta-, and para-positions (42–54) as well as two ring-fused analogs (55–56). In general, the SAR was remarkably flat with MICs ranging from 12.5–50 μM for most active compounds, but some trends were observed. In general, para-substituted analogs were more potent (MIC = 6.25–50 μM), meta-substituted analogs were less active (MIC = 25–50 μM) and ortho-substituted analogs were inactive (MIC = > 50 μM). Among the para-substituted analogs potency was found to increase as the size of the atom/functional group decreased (MIC trend: F = Cl < CF3 < Br = fused benzo = OMe).
Table 2.
The enzymatic and antimicrobial activity of 5′-O-[N-(phenoxyacyl)sulfamoyl]adenosine and related analogues 32, 38–56.
 |
||||||
|---|---|---|---|---|---|---|
| MIC (μM)a | Kiapp (μM)b | Kiapp (μM)c | ||||
| Cmpd | R = | X | n = | GAST | FACL19 | FAAL28 |
| 32 | H | O | 10 | 3.13 | >100 | 0.7 |
| 38 | H | O | 9 | 50 | >100 | 6.7 |
| 39 | H | O | 11 | 12.5–25 | >100 | 2.6 |
| 40 | H | S | 10 | >50 | 41.6 | 6.3 |
| 41 | H | NH | 10 | 12.5 | 63.4 | 7.3 |
| 42 | p-F | O | 10 | 12.5–25 | 11.4 | 0.3 |
| 43 | m-F | O | 10 | 25–50 | 7.3 | 1.6 |
| 44 | o-F | O | 10 | >50 | >100 | 1.5 |
| 45 | p-Cl | O | 10 | 6.25–12.5 | 13.7 | 2.2 |
| 46 | m-Cl | O | 10 | 25–50 | 26.4 | 0.5 |
| 47 | o-Cl | O | 10 | >50 | >100 | 3.6 |
| 48 | p-Br | O | 10 | >50 | 10.4 | 4.0 |
| 49 | p-CF3 | O | 10 | 25–50 | 34.4 | 15.6 |
| 50 | m-CF3 | O | 10 | 25–50 | 13.5 | 4.4 |
| 51 | o-CF3 | O | 10 | >50 | 28.5 | 1.3 |
| 52 | p-OMe | O | 10 | >50 | >100 | 6.6 |
| 53 | m-OMe | O | 10 | 25–50 | >100 | 1.7 |
| 54 | o-OMe | O | 10 | >50 | >100 | >100 |
| 55 | 2,3-benzo | O | 10 | >50 | 9.64 | 25.24 |
| 56 | 3,4-benzo | O | 10 | >50 | 20.39 | 1.88 |
Microbiological data is from 3 replicates.
Whole cell inhibitory activity against Mtb H37Rv in GAST medium.
Inhibitory activity against FACL19.
Inhibitory activity against FAAL28.
Substitution of the sulfamate 5′-oxygen in 32 provided acyl sulfamide 83, which has comparable antimicrobial activity with a MIC of 1.56–3.13 μM (Table 3). Removal of the carbonyl group to yield alkyl sulfamate 89 caused a greater than 16–fold loss in activity revealing the importance of the carbonyl group. Finally, the reverse alkyl sulfamate 93 was also inactive displaying no inhibition of growth up to 50 μM.
Table 3.
The enzymatic and antimicrobial activity of linker analogues 83, 89 and 93.
 |
||||
|---|---|---|---|---|
| MIC (μM)a |
Kiapp (μM)b |
Kiapp (μM)c |
||
| Cmpd | R = | GAST | FACL19 | FAAL28 |
| 32 | 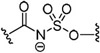 |
3.13 | >100 | 0.7 |
| 83 | 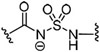 |
1.56–3.13 | >100 | 3.1 |
| 89 | >50 | >100 | >100 | |
| 93 | >50 | >100 | >100 | |
Microbiological and biochemical data is from 3 replicates.
Whole cell inhibitory activity against Mtb H37Rv in GAST medium.
Inhibitory activity against FACL19.
Inhibitory activity against FAAL28.
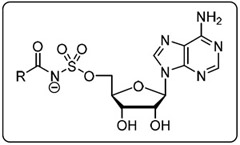
Acyl-AMS derivatives displaying the most potent antimicrobial activity against M. tuberculosis H37Rv were tested against a panel of multidrug resistant (MDR) and extensively drug resistant (XDR) TB strains in GAST and 7H9 medium (Table 4).[66] These drug-resistant pathogens displayed less differential activity in media than the reference drug susceptible strain. As a result, most of the selected analogues were more active against the MDR-TB and XDR-TB strains in 7H9 medium compared to the reference H37Rv strain, but less active in GAST medium relative to the reference H37Rv strain. For example, compounds 9 and 17 containing C12 and C14 saturated fatty acyl chains displayed MICs in 7H9 medium ranging from 3.13–12.5 μM against this MDR and XDR Mtb panel, whereas their activity against M. tuberculosis H37Rv in 7H9 medium was only 50 μM. By contrast, the activity of 9 and 17 against the MDR/XDR strains in GAST medium was diminished (MICs 3.13–25 μM) compared to their activity against M. tuberculosis H37Rv (MIC: 0.19–0.78 μM). The lead 11-phenoxyundecanoyl-AMS 23 was notable for its fairly uniform activity against the MDR-TB and XDR-TB panel in both media with MICs of 3.13–12.5 μM for 8 of the 9 tested conditions.
Table 4.
The activity against MDR-TB and XDR-TB strains of most potent compounds.
 |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| MIC (μM) | |||||||||||
| HREZSKPTha,b | HREZSKPTha,b | HREKOa,b | HREPa,c | HRESPOCTha,c | |||||||
| Cmpd | R= | 7H9d | GASTe | 7H9 | GAST | 7H9 | GAST | 7H9 | GAST | 7H9 | GAST |
| 9 | CH3(CH2)10- | 12.5 | ngf | 12.5 | 25 | 6.25 | 3.13 | 6.25 | 6.25 | 12.5 | 12.5 |
| 17 | CH3(CH2)12- | 12.5 | ng | 25 | 12.5 | 3.13 | 6.25 | 6.25 | 6.25 | 12.5 | 12.5 |
| 22 | H2CHC(CH2)8- | 6.25 | ng | 50 | 3.13 | 6.25 | 3.13 | 12.5 | 3.13 | 25 | 3.13 |
| 28 | N3(CH2)11- | 6.25 | ng | 50 | 3.13 | 6.25 | 3.13 | 12.5 | 3.13 | 25 | 6.25 |
| 32 | C6H6O(CH2)10- | 12.5 | ng | 12.5 | 6.25 | 12.5 | 3.13 | 12.5 | 3.13 | 50 | 6.25 |
| 36 | CH3(CH2)8OCH2- | 12.5 | ng | 12.5 | 1.56 | 6.25 | 0.39 | 6.25 | 0.78 | 50 | 6.25 |
| Isoniazidg | 2 | nt | 2 | >8 | 2 | 4 | 2 | 4 | 8 | 8 | |
Whole cell inhibitory activity against Mtb with resistance pattern listed as such: H, isoniazid; R, rifampicin; E, ethambutol; Z, pyrazinamide; S, streptomycin; K, kanamycin; P, para-aminosalicylic acid; Th, prothionamide; O, ofloxacin.
MDR-TB strains.
XDR-TB strains.
Whole cell inhibitory activity against Mtb in 7H9 medium/tyloxapol.
Whole cell inhibitory activity against Mtb in GAST medium.
ng = no growth.
The critical concentration for high-level resistance is 1 μM.
2.2. Enzyme Inhibition
All compounds were tested for their inhibitory activity against two FadD enzymes: fatty acyl CoA ligase (FACL19) and fatty AMP ligase (FAAL28). These enzymes were selected as representative members of the FACL and FAAL class of enzymes since they have been structurally and biochemically well characterized and can be readily overexpressed and purified from E. coli.[17, 67] Shorter chain (C4 to C10) analogues 13–16 were essentially inactive against both enzymes. However, once the alkyl chain length reached C12 in dodecanoyl-AMS 9, enzyme inhibition was observed with an apparent Ki of 67 μM for FACL19 and 6 μM for FAAL28 (Table 1). Potency monotonically increased as the alkyl chain increased reaching a Ki of 2.4 μM for FACL19 and 0.48 μM for FAAL28 with steroyl-AMS 19. These results are consistent with previous observations demonstrating FACL19 and FAAL28 favor medium to long chain fatty acids.[17]
Introduction of unsaturation within the alkyl chain at the terminal, internal or vicinal position in analogs 21–25 ablated all biochemical inhibition of FACL19 and FAAL28. The trans-cyclopropane derivative 26 displayed weak inhibition of FAAL28 indicating some tolerance to conformational constraints within the alkyl chain of these acyl-AMS inhibitors. Substitution on the terminus of the lipid chain with hydroxy, amino, azido, ester and carboxylic acid functional groups in 27–31 was poorly tolerated. However, 11-phenoxyundecanoyl-AMS 32 displayed potent inhibition of FAAL28 with an apparent Ki of 0.7 μM, but was completely inactive against FACL19 demonstrating the highest level of biochemical selectivity observed for any compound evaluated. Introduction of methyl groups at the α-carbon in 33–35 and oxygen atoms within the alky chain in 36–37 obliterated activity.
Based on the promising biochemical selectivity and potent antitubercular activity of 11-phenoxyundecanoyl-AMS 32, a series of related analogs was synthesized and evaluated. Decreasing the alkyl chain of 32 by one carbon in 38 led to a 10-fold decrease in potency for FAAL28 while increasing the chain length by one carbon resulted in a modest 4-fold decrease in potency for FAAL28. These biochemical results with FAAL28 were closely mirrored in the microbiological activity of 38 and 39, which were 16-fold and 4-fold less active than 32 against M. tuberculosis H37Rv (Table 2). Both compounds maintained excellent biochemical selectivity with no inhibition of FACL19. Exchanging the terminal oxygen atom of 32 for a sulfur atom or nitrogen atom resulted in a 10–fold decrease in activity for FAAL28 and loss of biochemical selectivity as both analogs exhibited modest inhibition of FACL19. Among the phenyl substituted analogs 42–56, ortho-substituted analogues tended to be more selective for FAAL28 in this series of compounds; however, biochemical selectivity for most compounds was severely degraded relative to 32 as the majority of analogs also showed good-to-moderate inhibition of FACL19 (Table 2).
The impact of the acyl sulfamate moiety of 32 on biochemical inhibition was examined with acyl sulfamide 83 and alkyl sulfamates 89 and 93 (Table 3). Acyl sulfamide 83 maintained biochemical selectivity with no inhibition of FACL19 and a modest 4-fold diminished inhibition of FAAL28 compared to 32. Removal of the carbonyl in alkyl sulfamate 89 and reverse alkyl sulfamate 93 derivatives ablated biochemical inhibition of FAAL28.
2.3. Cytotoxicity
The cytotoxicity of compounds 9 and 13–37 was evaluated against Vero cells (ATCC) using the MTT assay with DMSO as a positive control (Table 1). Every compound except 31 containing a terminal carboxylic acid in the lipid tail was cytotoxic with IC50 values for inhibition of 50% cell viability between 0.07–81 μM. Unlike the antitubercular activity, which exhibited a parabolic relationship peaking at C12, the cytotoxicity monotonically increases with chain length from 81 μM with 13 containing a C4 lipid to 0.07 μM for 20 containing a C20 lipid. The SAR of the remaining analogs 21–26, 28, 30, and 34–37 containing unsaturation, conformational constraints, α-methyl groups, and ether functional groups was relatively flat with IC50‘s between 5-17 μM. Compounds with polar and hydrogen-bond donating functional groups including alcohol 27 and amino 29 were slightly less cytotoxic with IC50 of 27 and 42 μM, respectively. Ether 36 exhibited moderate cytotoxicity (IC50 = 8.7 μM), but had the highest therapeutic index [MIC/IC50 = 46–92] of all analogues due to its exceptional potency (MIC = 0.098–0.19 μM). The lead compound 11-phenoxyundecanoyl-AMS 32 possesses notable cytotoxicity with an IC50 of 2 μM, a result consistent with the hydrophobic trend observed with the C4–C20 lipid analogues, resulting in a therapeutic index of less than one.
3. Conclusions
The goal of this study was to define the structure-activity relationships of 5′-O-[N-(alkanoyl)sulfamoyl]adenosine (alkanoyl adenosine monosulfamate, alkanoyl-AMS) inhibitors that govern antitubercular activity. A secondary goal was to identify a chemical probe selective for either FAAL or FACL enzymes using FAAL28 and FACL19 as proxies for each class, recognizing that such an analysis is intrinsically limited since these two enzymes cannot capture the substrate specificities of all 12 FAALs and 24 FACLs.[16] This was accomplished through the synthesis of a systematic series of 48 compounds. We initially explored the impact of the acyl chain length activity with a series of even-chained analogs from C4 to C20 and observed a parabolic relationship between chain length and antitubercular activity where antitubercular activity was optimal at a chain length of C12-C14. Our second round of SAR focused on the tolerance of the lipid chain of dodecanoyl-AMS 9 to modification through introduction of alkene and cyclopropane conformation constraints at terminal and internal positions throughout the lipid chain. We also explored the impact of a range of polar and nonpolar functional groups at the lipid terminus, the effect of sterics at the α-carbon, and the ability to tolerate oxygen atoms in the lipid chain. These efforts led to the identification of several promising analogs including 12-azidodecanoyl-AMS 28, 12-aminodecanoyl-AMS 29, 11-phenoxyundecanoyl-AMS 32, and nonyloxyacetyl-AMS 36 whose MICs in GAST medium are 0.19, 3.13, 3.13, and 0.19 μM, respectively. Concurrent biochemical evaluation against FACL19 and FAAL28 revealed 11-phenoxyundecanoyl-AMS 32 exhibited greater than 142-fold selectivity for FAAL28 over FACL19 providing the first compound with high biochemical selectivity. We had initially hypothesized that analogs selective for the FAAL class of enzymes would allow the separation of antitubercular activity from cytotoxicity since FAAL enzymes are functionally unique to mycobacteria whereas FACL enzymes are ubiquitous in mammals.[17] However, compound 32 retained appreciable cytotoxicity suggesting the alkanoyl-AMS scaffold is intrinsically cytotoxic. Indeed, all of the synthesized compounds 13–37 with the exception of the double ionized 11-carboxyundecanoyl-AMS 31 displayed some cytotoxicity. Our third series of compounds studied the SAR of the terminal phenoxy group of 11-phenoxyundecanoyl-AMS 32 on antitubercular activity and biochemical selectivity. Substitution on the terminal phenoxy group in all cases reduced antitubercular activity and lowered FAAL28 potency as well as decreased biochemical selectivity (KiFAAL28/KiFAC19). Finally, we investigated the role of the acylsulfamate linkage of 32 demonstrating the carbonyl moiety is absolutely critical for activity, but the 5′-oxygen atom can be replaced with a NH moiety, results consistent with previous studies of acyl-adenylate inhibitors.[68, 69]
4. Experimental section
4.1. Chemistry
All commercial reagents (Sigma-Aldrich, Acros, Fisher) were used as provided. Sulfamoyl chloride was prepared by the method of Heacock except that it was used directly without recrystallization.[70] 2′,3′-O-Isopropylidene-5′-O-sulfamoyladenosine 57 and 5’-deoxy-2’,3’-O-isopropylidene-5’-N-(sulfamoyl)aminoadenosine 81 were prepared as previously described.[69] N6,N6-Bis(tert-butoxycarbonyl)-5’-O-sulfamoyl-2’,3’-O-isopropylideneadenosine 87 and N6,N6-bis(tert-butoxycarbonyl)-2′,3′-O-isopropylideneadenosine 90 were prepared by the method of Ikeuchi et al.[68] An anhydrous solvent dispensing system (J. C. Meyer) using two packed columns of neutral alumina was used for drying tetrahydrofuran (THF), CH2Cl2, and N,N-dimethylformamide (DMF), and the solvents were dispensed under Argon. Anhydrous grade 1,2-dimethoxyethane (DME), methanol (MeOH), and acetonitrile (MeCN) were purchased from Sigma-Aldrich and used as provided. All reactions were performed under an inert atmosphere of dry Argon in oven-dried glassware (150 °C). Flash chromatography was performed with an ISCO Combiflash Companion® purification system with prepacked silica gel cartridges with the indicated solvent system. 1H and 13C NMR spectra were recorded on a Varian 600 MHz spectrometer. Proton chemical shifts are reported in ppm from an internal standard of residual dimethyl sulfoxide (2.50 ppm), methanol (3.31 ppm) or chloroform (7.21 ppm) and carbon chemical shifts are reported using an internal standard of residual dimethyl sulfoxide (39.52 ppm) or methanol (49.19 ppm). Proton chemical data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, m = multiplet, br = broad), coupling constant, integration. Low and high resolution mass spectra were acquired on an Agilent TOF II TOF/MS instrument equipped with either an ESI or APCI interface. The purity of the final compounds was greater than 95%.
4.1.1. General Procedure for Synthesis of NHS Esters (Method A)
To a solution of the appropriate fatty acid 9b, 13b–37b (1.0 equiv) in CH2Cl2 (0.1 M) at 0°C was added N-hydroxysuccinimide (NHS, 1.0 equiv) and N,N′-dicyclohexylcarbodiimide (1.0 equiv). The reaction mixture was warmed to 23 °C and stirred 16 h. The resulting mixture was filtered and the filtrate was concentrated under reduced pressure to provide NHS esters 9c, 13c–37c without further purification.
4.1.2. General Procedure for Synthesis of NHS Esters (Method B)
To the appropriate fatty acid methyl ester 38b–56b (1.0 equiv) was added NaOH/MeOH (aqueous 1N NaOH, 1:1). The resulting solution was refluxed at 100 °C for 2 h. Next, the mixture was acidified with aqueous 1N HCl until pH < 2 and then extracted with EtOAc (5×10 mL). The combined extracts were washed with aqueous NaCl (20 mL), dried (MgSO4), and concentrated. To the resulting crude mixture was added NHS (1.1 equiv), 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (1.1 equiv) and CH2Cl2 (8 mL). The reaction mixture was allowed to stir for 6 h and then quenched with aqueous 1N HCl (5 mL). The aqueous solution was extracted with CH2Cl2 (3×15 mL). The combined extracts were washed with aqueous NaCl (20 mL), dried (MgSO4), and concentrated. Purification by flash chromatography (4:1 Hexanes/EtOAc) afforded 38c–56c as white solids.
4.1.2.1. N-Hydroxysuccinimidyl 10-phenoxydecanoate (38c).
The title compound was prepared from 38b using the general procedure for the synthesis of NHS esters, method B (90%): 1H NMR (600 MHz, CDCl3) δ 1.31–1.37 (m, 6H), 1.40–1.46 (m, 4H), 1.72–1.79 (m, 4H), 2.60 (t, J = 7.2 Hz, 2H), 2.83 (br s, 4H), 3.95 (t, J = 7.2 Hz, 2H), 6.89–6.90 (m, 2H), 6.91–6.94 (m, 1H), 7.26–7.28 (m, 2H); HRMS (ESI+) calcd for C20H28NO5 [M+H]+ 362.1962, found 362.1950 (error 3.3 ppm).
4.1.2.2. N-Hydroxysuccinimdyl 12-phenoxydodecanoate (39c).
The title compound was prepared from 39b using the general procedure for the synthesis of NHS esters, method B (89%): 1H NMR (600 MHz, CDCl3) δ 1.30–1.36 (m, 10H), 1.39–1.46 (m, 4H), 1.73–1.79 (m, 4H), 2.60 (t, J = 7.2 Hz, 2H), 2.81 (br s, 4H), 3.95 (t, J = 7.2 Hz, 2H), 6.89–6.93 (m, 3H), 7.26–7.28 (m, 2H); HRMS (ESI+) calcd for C22H32NO5 [M+H]+ 390.2275, found 390.2257 (error 4.6 ppm).
4.1.2.3. N-Hydroxysuccinimdyl 11-(phenylthio)undecanoate (40c).
The title compound was prepared from 40b using the general procedure for the synthesis of NHS esters, method B (78%): 1H NMR (600 MHz, CDCl3) δ 1.20–1.23 (m, 8H), 1.32–1.38 (m, 4H), 1.57–1.62 (m, 2H), 1.66–1.71 (m, 2H), 2.54 (t, J = 7.2 Hz, 2H), 2.76 (br s, 4H), 2.86 (t, J = 7.2 Hz, 2H), 7.09–7.12 (m, 1H), 7.21–7.23 (m, 2H), 7.26–7.27 (m, 2H); HRMS (ESI+) calcd for C21H30NO4S [M+H]+ 392.1890, found 392.1893 (error 0.8 ppm).
4.1.2.4. N-Hydroxysuccinimdyl 11-(phenylamino)undecanoate (41c).
The title compound was prepared from 41a using the general procedure for the synthesis of NHS esters, method B (74%): 1H NMR (600 MHz, CDCl3) δ 1.26–1.31 (m, 8H), 1.37–1.42 (m, 4H), 1.59–1.64 (m, 2H), 1.72–1.77 (m, 2H), 2.60 (t, J = 7.2 Hz, 2H), 2.82 (br s, 4H), 3.10 (t, J = 7.2 Hz, 2H), 6.59–6.60 (m, 2H), 6.66–6.69 (m, 1H), 7.15–7.18 (m, 2H); HRMS (ESI+) calcd for C21H31N2O4 [M+H]+ 375.2278, found 375.2267 (error 2.9 ppm).
4.1.2.5. N-Hydroxysuccinimdyl 11-(4-fluorophenoxy)undecanoate (42c).
The title compound was prepared from 42b using the general procedure for the synthesis of NHS esters, method B (73%): 1H NMR (600 MHz, CDCl3) δ 1.30–1.36 (m, 8H), 1.37–1.46 (m, 4H), 1.71–1.77 (m, 4H), 2.59 (t, J = 7.2 Hz, 2H), 2.82 (br s, 4H), 3.89 (t, J = 7.2 Hz, 2H), 6.80–6.82 (m, 2H), 6.93–6.96 (m, 2H); HRMS (ESI+) calcd for C21H29FNO5 [M+H]+ 394.2024, found 394.2009 (error 3.8 ppm).
4.1.2.6. N-Hydroxysuccinimdyl 11-(3-fluorophenoxy)undecanoate (43c).
The title compound was prepared from 43b using the general procedure for the synthesis of NHS esters, method B (82%): 1H NMR (600 MHz, CDCl3) δ 1.31–1.33 (m, 8H), 1.39–1.45 (m, 4H), 1.71–1.78 (m, 4H), 2.59 (t, J = 7.2 Hz, 2H), 2.81–2.82 (br s, 4H), 3.92 (t, J = 7.2 Hz, 2H), 6.58–6.64 (m, 2H), 6.66–6.67 (m, 1H), 7.17–7.21 (m, 1H); MS HRMS (ESI+) calcd for C21H29FNO5 [M+H]+ 394.2024, found 394.2003 (error 5.3 ppm).
4.1.2.7. N-Hydroxysuccinimdyl 11-(2-fluorophenoxy)undecanoate (44c).
The title compound was prepared from 44b using the general procedure for the synthesis of NHS esters, method B (76%): 1H NMR (600 MHz, CDCl3) δ 1.31–1.35 (m, 8H), 1.37–1.41 (m, 2H), 1.43–1.48 (m, 2H), 1.71–1.76 (m, 2H), 1.78–1.83 (m, 2H), 2.59 (t, J = 7.2 Hz, 2H), 2.81–2.82 (br s, 4H), 4.01 (t, J = 7.2 Hz, 2H), 6.85–6.88 (m, 1H), 6.94–6.97 (m, 1H), 7.02–7.26 (m, 2H); HRMS (ESI+) calcd for C21H29FNO5 [M+H]+ 394.2024, found 394.2010 (error 3.6 ppm).
4.1.2.8. N-Hydroxysuccinimdyl 11-(4-chlorophenoxy)undecanoate (45c).
The title compound was prepared from 45b using the general procedure for the synthesis of NHS esters, method B (74%): 1H NMR (600 MHz, CDCl3) δ 1.30–1.33 (m, 8H), 1.38–1.44 (m, 4H), 1.71–1.78 (m, 4H), 2.60 (t, J = 7.2 Hz, 2H), 2.82–2.83 (br s, 4H), 3.90 (t, J = 7.2 Hz, 2H), 6.80–6.82 (m, 2H), 7.20–7.22 (m, 2H); HRMS (ESI+) calcd for C21H29C1NO5 [M+H]+ 410.1729, found 410.1710 (error 4.6 ppm).
4.1.2.9. N-Hydroxysuccinimdyl 11-(3-chlorophenoxy)undecanoate (46c).
The title compound was prepared from 46b using the general procedure for the synthesis of NHS esters, method B (70%): 1H NMR (600 MHz, CDCl3) δ 1.31–1.39 (m, 8H), 1.39–1.45 (m, 4H), 1.72–1.78 (m, 4H), 2.60 (t, J = 7.2 Hz, 2H), 2.82–2.83 (br s, 4H), 3.92 (t, J = 7.2 Hz, 2H), 6.76–6.78 (m, 1H), 6.88–6.91 (m, 2H), 7.16–7.19 (m, 1H); HRMS (ESI+) calcd for C21H29C1NO5 [M+H]+ 410.1729, found 410.1708 (error 5.1 ppm).
4.1.2.10. N-Hydroxysuccinimdyl 11-(2-chlorophenoxy)undecanoate (47c).
The title compound was prepared from 47b using the general procedure for the synthesis of NHS esters, method B (72%): 1H NMR (600 MHz, CDCl3) δ 1.31–1.41 (m, 10H), 1.46–1.51 (m, 2H), 1.71–1.76 (m, 2H), 1.80–1.85 (m, 2H), 2.60 (t, J = 7.2 Hz, 2H), 2.82–2.83 (br s, 4H), 4.02 (t, J = 7.2 Hz, 2H), 6.85–6.88 (m, 1H), 6.90–6.92 (m, 1H), 7.17–7.20 (m, 1H), 7.34–7.35 (m, 1H); HRMS (ESI+) calcd for C21H29C1NO5 [M+H]+ 410.1729, found 410.1717 (error 2.9 ppm).
4.1.2.11. N-Hydroxysuccinimdyl 11-(4-bromophenoxy)undecanoate (48c).
The title compound was prepared from 48b using the general procedure for the synthesis of NHS esters, method B (77%): 1H NMR (600 MHz, CDCl3) δ 1.30–1.32 (m, 8H), 1.37–1.44 (m, 4H), 1.71–1.78 (m, 4H), 2.59 (t, J = 7.2 Hz, 2H), 2.81–2.83 (br s, 4H), 3.90 (t, J = 7.2 Hz, 2H), 6.75–6.78 (m, 2H), 7.33–7.36 (m, 2H) calcd for C21H29BrNO5+ [M+H] 454.1229, 456.1209, found 454.1208, 456.1188 (error 4.6 ppm).
4.1.2.12. N-Hydroxysuccinimdyl 11-[(4-trifluoromethyl)phenoxy] undecanoate (49c).
The title compound was prepared from 49b using the general procedure for the synthesis of NHS esters, method B (83%): 1H NMR (600 MHz, CDCl3) δ 1.31–1.34 (m, 8H), 1.38–1.47 (m, 4H), 1.71–1.81 (m, 4H), 2.59 (t, J = 7.2 Hz, 2H), 2.82–2.83 (br s, 4H), 3.98 (t, J = 7.2 Hz, 2H), 6.93–6.94 (m, 2H), 7.51–7.53 (m, 2H).
4.1.2.13. N-Hydroxysuccinimdyl 11-[(3-trifluoromethyl)phenoxy] undecanoate (50c).
The title compound was prepared from 50b using the general procedure for the synthesis of NHS esters, method B (77%): 1H NMR (600 MHz, CDCl3) δ 1.32–1.35 (m, 8H), 1.40–1.47 (m, 4H), 1.73–1.80 (m, 4H), 2.60 (t, J = 7.2 Hz, 2H), 2.82–2.83 (br s, 4H), 3.98 (t, J = 7.2 Hz, 2H), 7.05–7.06 (m, 1H), 7.11 (s, 1H), 7.17–7.18 (m, 1H), 7.35–7.38 (m, 1H).
4.1.2.14. N-Hydroxysuccinimdyl 11-[(2-trifluoromethyl)phenoxy] undecanoate (51c).
The title compound was prepared from 51b using the general procedure for the synthesis of NHS esters, method B (97%): 1H NMR (600 MHz, CDCl3) δ 1.25–1.41 (10H, m), 1.44–1.49 (m, 2H), 1.71–1.76 (m, 2H), 1.78–1.82 (m, 2H), 2.59 (t, J = 7.2 Hz, 2H), 2.81 (br s, 4H), 4.03 (t, J = 7.2 Hz, 2H), 6.95–6.98 (m, 2H), 7.44–7.46 (m, 1H), 7.54–7.55 (m, 1H).
4.1.2.15. N-Hydroxysuccinimdyl 11-(4-methoxyphenoxy)undecanoate (52c).
The title compound was prepared from 52b using the general procedure for the synthesis of NHS esters, method B (75%): 1H NMR (600 MHz, CDCl3) δ 1.30–1.33 (m, 8H), 1.37–1.45 (m, 4H), 1.71–1.76 (m, 4H), 2.59 (t, J = 7.2 Hz, 2H), 2.81–2.83 (br s, 4H), 3.76 (s, 3H), 3.89 (t, J = 7.2 Hz, 2H), 6.81–6.83 (s, 4H); HRMS (ESI+) calcd for C22H32NO6 [M+H]+ 406.2224, found 406.2207 (error 4.2 ppm).
4.1.2.16. N-Hydroxysuccinimdyl 11-(3-methoxyphenoxy)undecanoate (53c).
The title compound was prepared from 53b using the general procedure for the synthesis of NHS esters, method B (77%): 1H NMR (600 MHz, CDCl3) δ 1.31–1.33 (m, 8H), 1.39–1.45 (m, 4H), 1.72–1.79 (m, 4H), 2.60 (t, J = 7.2 Hz, 2H), 2.81–2.82 (br s, 4H), 3.78 (s, 3H), 3.93 (t, J = 7.2 Hz, 2H), 6.45–6.46 (m, 1H), 6.48–6.50 (m, 2H), 7.15–7.17 (m, 1H); HRMS (ESI+) calcd for C22H32NO6 [M+H]+ 406.2224, found 406.2241 (error 4.2 ppm).
4.1.2.17. N-Hydroxysuccinimdyl 11-(2-methoxyphenoxy)undecanoate (54c).
The title compound was prepared from 54b using the general procedure for the synthesis of NHS esters, method B (74%): 1H NMR (600 MHz, CDCl3) δ 1.26–1.34 (m, 8H), 1.37–1.47 (m, 4H), 1.71–1.76 (m, 2H), 1.81–1.86 (m, 2H), 2.59 (t, J = 7.2 Hz, 2H), 2.82 (br s, 4H), 3.86 (s, 3H), 4.00 (t, J = 7.2 Hz, 2H), 6.89 (s, 4H); HRMS (ESI+) calcd for C22H32NO6 [M+H]+ 406.2224, found 406.2224 (error 0 ppm).
4.1.2.18. N-Hydroxysuccinimdyl 11-(1-naphthalenyloxy)undecanoate (55c).
The title compound was prepared from 55b using the general procedure for the synthesis of NHS esters, method B (77%): 1H NMR (600 MHz, CDCl3) δ 1.33–1.41 (m, 10H), 1.54–1.59 (m, 2H), 1.72–1.77 (m, 2H), 1.90–1.95 (m, 2H), 2.60 (t, J = 7.2 Hz, 2H), 2.80–2.83 (br s, 4H), 4.13 (t, J = 7.2 Hz, 2H), 6.80–6.81 (m, 1H), 7.35–7.38 (m, 1H), 7.40–7.42 (m, 1H), 7.45–7.50 (m, 2H), 7.78–7.80 (m, 1H), 8.28–8.30 (m, 1H); HRMS (ESI+) calcd for C25H32NO5 [M+H]+ 426.2275, found 426.2251 (error 5.6 ppm).
4.1.2.19. N-Hydroxysuccinimdyl 11-(2-naphthalenyloxy)undecanoate (56c).
The title compound was prepared from 56b using the general procedure for the synthesis of NHS esters, method B (75%): 1H NMR (600 MHz, CDCl3) δ 1.33–1.41 (m, 10H), 1.49–1.52 (m, 2H), 1.74–1.76 (m, 2H), 1.84–1.86 (m, 2H), 2.60 (t, J = 7.2 Hz, 2H), 2.78 (br s, 4H), 4.07 (t, J = 7.2 Hz, 2H), 7.13–7.16 (m, 2H), 7.31–7.34 (m, 1H), 7.42–7.44 (m, 1H), 7.72–7.77 (m, 3H); HRMS (ESI+) calcd for C25H32NO5 [M+H]+ 426.2275, found 426.2265 (error 2.3 ppm).
4.1.3. General Procedure for Acylation
To a solution of 5′-O-sulfamoyl-2′,3′-O-isopropylideneadenosine (1.0 equiv) in anhydrous DMF (0.1 M) at 0 °C were added cesium carbonate (3.0 equiv) and the appropriate N-hydroxysuccinimyl ester 9c, 13c–56c (1.5 equiv). The mixture was warmed to 23 °C and stirred for 16 h. The resulting mixture was filtered and the filtrate was concentrated under reduced pressure. Purification by flash chromatography (0–20% EtOAc/MeOH + 0.5% Et3N) afforded the title compounds 9d, 13d–56d as colorless oils.
4.1.3.1. 5′-O-[N-(Dodecanoyl)sulfamoyl]-2′,3′-O-isopropylideneadenosine Triethylammonium salt (9d).
NHS ester 9c was coupled with 5′-O-sulfamoyl-2′,3′-O-isopropylideneadenosine using the general procedure for acylation to afford the title compound (69%): Rf 0.6 (8:2 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 0.87 (t, J = 7.2 Hz, 3H), 1.21–1.30 (m, 25H), 1.37 (s, 3H), 1.57 (p, J = 7.2 Hz, 2H), 1.60 (s, 3H), 2.16 (t, J = 7.2 Hz, 2H), 3.15 (q, J = 7.2 Hz, 6H), 4.21–4.26 (m, 2H), 4.52–4.54 (m, 1H), 5.12 (dd, J = 6.0, 3.0 Hz, 1H), 5.36 (dd, J = 6.0, 3.0 Hz, 1H), 6.23 (d, J = 3.0 Hz, 1H), 8.20 (s, 1H), 8.45 (s, 1H); MS (ESI–) calcd for C25H39N6O7S [M − H]− 567.3, found 567.3.
4.1.3.2. 5′-O-[N-(Butanoyl)sulfamoyl]-2′,3′-O-isopropylideneadenosine Triethylammonium salt (13d).
NHS ester 13c was coupled with 5′-O-sulfamoyl-2′,3′-O-isopropylideneadenosine using the general procedure for acylation to afford the title compound (64%): Rf 0.4 (8:2 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 0.92 (t, J = 7.2 Hz, 3H), 1.29 (t, J = 7.2 Hz, 9H), 1.39 (s, 3H), 1.57–1.61 (m, 2H), 1.61 (s, 3H), 2.14 (t, J = 7.2 Hz, 2H), 3.18 (q, J = 7.2 Hz, 6H), 4.21–4.24 (m, 2H), 4.53 (br s, 1H), 5.13 (d, J = 6.0 Hz, 1H), 5.36 (d, J = 6.0 Hz, 1H), 6.24 (br s, 1H), 8.21 (s, 1H), 8.45 (s, 1H); HRMS (ESI−) calcd for C17H23N6O7S [M − H]− 455.1354, found 455.1340 (error 3.1 ppm).
4.1.3.3. 5′-O-[N-(Hexanoyl)sulfamoyl]-2′,3′-O-isopropylideneadenosine Triethylammonium salt (14d).
NHS ester 14c was coupled with 5′-O-sulfamoyl-2′,3′-O-isopropylideneadenosine using the general procedure for acylation to afford the title compound (68%): Rf 0.5 (8:2 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 0.88 (t, J = 7.2 Hz, 3H), 1.27–1.30 (m, 13H), 1.38 (s, 3H), 1.58 (p, J = 7.2 Hz, 2H), 1.61 (s, 3H), 2.16 (t, J = 7.2 Hz, 2H), 3.17 (q, J = 7.2 Hz, 6H), 4.21–4.24 (m, 2H), 4.54 (br s, 1H), 5.12 (d, J = 6.0 Hz, 1H), 5.35 (dd, J = 6.0, 3.0 Hz, 1H), 6.24 (d, J = 3.0 Hz, 1H), 8.21 (s, 1H), 8.45 (s, 1H); HRMS (ESI−) calcd for C19H27N6O7S [M − H]− 483.1667, found 483.1666 (error 0.2 ppm).
4.1.3.4. 5′-O-[N-(Octanoyl)sulfamoyl]-2′,3′-O-isopropylideneadenosine Triethylammonium salt (15d).
NHS ester 15c was coupled with 5′-O-sulfamoyl-2′,3′-O-isopropylideneadenosine using the general procedure for acylation to afford the title compound (74%): Rf 0.5 (8:2 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 0.85 (t, J = 7.2 Hz, 3H), 1.24–1.28 (m, 17H), 1.38 (s, 3H), 1.58 (p, J = 7.2 Hz, 2H), 1.60 (s, 3H), 2.16 (t, J = 7.2 Hz, 2H), 3.17 (q, J = 7.2 Hz, 6H), 4.22–4.28 (m, 2H), 4.53 (br s, 1H), 5.12 (dd, J = 6.0, 3.0 Hz, 1H), 5.36 (dd, J = 6.0, 3.0 Hz, 1H), 6.23 (d, J = 3.0 Hz, 1H), 8.21 (s, 1H), 8.43 (s, 1H); HRMS (ESI−) calcd for C21H31N6O7S [M − H]− 511.1980, found 511.1993 (error 2.5 ppm).
4.1.3.5. 5′-O-[N-(Decanoyl)sulfamoyl]-2′,3′-O-isopropylideneadenosine Triethylammonium salt (16d).
NHS ester 16c was coupled with 5′-O-sulfamoyl-2′,3′-O-isopropylideneadenosine using the general procedure for acylation to afford the title compound (60%): Rf 0.5 (8:2 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 0.87 (t, J = 7.2 Hz, 3H), 1.21–1.31 (m, 21H), 1.38 (s, 3H), 1.57 (p, J = 7.2 Hz, 2H), 1.60 (s, 3H), 2.16 (t, J = 7.2 Hz, 2H), 3.18 (q, J = 7.2 Hz, 6H), 4.20–4.25 (m, 2H), 4.52 (br s, 1H), 5.12 (dd, J = 6.0, 3.0Hz, 1H), 5.35 (dd, J = 6.0, 3.0 Hz, 1H), 6.23 (d, J = 3.0 Hz, 1H), 8.21 (s, 1H), 8.44 (s, 1H); MS (ESI−) calcd for C23H35N6O7S [M − H]− 539.2, found 539.3.
4.1.3.6. 5′-O-[N-(Tetradecanoyl)sulfamoyl]-2′,3′-O-isopropylideneadenosine Triethylammonium salt (17d).
NHS ester 17c was coupled with 5′-O-sulfamoyl-2′,3′-O-isopropylideneadenosine using the general procedure for acylation to afford the title compound (68%): Rf 0.6 (8:2 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 0.87 (t, J = 7.2 Hz, 3H), 1.24–1.29 (m, 29H), 1.37 (s, 3H), 1.58 (p, J = 7.2 Hz, 2H), 1.59 (s, 3H), 2.16 (t, J = 7.2 Hz, 2H), 3.16 (q, J = 7.2 Hz, 6H), 4.20–4.25 (m, 2H), 4.52 (br s, 1H), 5.12 (dd, J = 6.0, 3.0 Hz, 1H), 5.36 (dd, J = 6.0, 3.0 Hz, 1H), 6.23 (d, J = 3.0 Hz, 1H), 8.20 (s, 1H), 8.43 (s, 1H); MS (ESI−) calcd for C27H43N6O7S [M − H]− 595.3, found 595.3.
4.1.3.7. 5′-O-[N-(Palmitoyl)sulfamoyl]-2′,3′-O-isopropylideneadenosine Triethylammonium salt (18d).
NHS ester 18c was coupled with 5′-O-sulfamoyl-2′,3′-O-isopropylideneadenosine using the general procedure for acylation to afford the title compound (65%): Rf 0.6 (8:2 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 0.89 (t, J = 7.2 Hz, 3H), 1.24–1.32 (m, 33H), 1.38 (s, 3H), 1.57 (p, J = 7.2 Hz, 2H), 1.60 (s, 3H), 2.17 (t, J = 7.2 Hz, 2H), 3.17 (q, J = 7.2 Hz, 6H), 4.21–4.26 (m, 2H), 4.52–4.54 (m, 1H), 5.12 (dd, J = 6.0, 3.0 Hz, 1H), 5.36 (dd, J = 6.6, 3.0 Hz, 1H), 6.24 (d, J = 3.0 Hz, 1H), 8.21 (s, 1H), 8.44 (s, 1H); HRMS (ESI−) calcd for C29H47N6O7S [M − H]− 623.3232, found 623.3204 (error 4.5 ppm).
4.1.3.8. 5′-O-[N-(Stearoyl)sulfamoyl]-2′,3′-O-isopropylideneadenosine Triethylammonium salt (19d).
NHS ester 19c was coupled with 5′-O-sulfamoyl-2′,3′-O-isopropylideneadenosine using the general procedure for acylation to afford the title compound (59%): Rf 0.6 (8:2 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 0.89 (t, J = 7.2 Hz, 3H), 1.25–1.28 (m, 37H), 1.38 (s, 3H), 1.57 (p, J = 7.2 Hz, 2H), 1.60 (s, 3H), 2.17 (t, J = 7.2 Hz, 2H), 3.18 (q, J = 7.2 Hz, 6H), 4.22–4.27 (m, 2H), 4.53 (br s, 1H), 5.12 (d, J = 6.0 Hz, 1H), 5.36 (d, J = 6.0 Hz, 1H), 6.24 (d, J = 3.0 Hz, 1H), 8.21 (s, 1H), 8.44 (s, 1H); MS (ESI−) calcd for C31H51N6O7S [M − H]− 651.4, found 651.4.
4.1.3.9. 5′-O-[N-(Icosanoyl)sulfamoyl]-2′,3′-O-isopropylideneadenosine Triethylammonium salt (20d).
NHS ester 20c was coupled with 5′-O-sulfamoyl-2′,3′-O-isopropylideneadenosine using the general procedure for acylation to afford the title compound (48%): Rf 0.6 (8:2 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 0.88 (t, J = 7.2 Hz, 3H), 1.24–1.30 (m, 41H), 1.38 (s, 3H), 1.56 (p, J = 7.2 Hz, 2H), 1.60 (s, 3H), 2.16 (t, J = 7.2 Hz, 2H), 3.19 (q, J = 7.2 Hz, 6H), 4.21–4.27 (m, 2H), 4.52 (br s, 1H), 5.11 (d, J = 6.0 Hz, 1H), 5.34 (dd, J = 6.0, 3.0 Hz, 1H), (d, J = 3.0 Hz, 1H), 8.20 (s, 1H), 8.44 (s, 1H); MS (ESI−) calcd for C33H55N6O7S [M − H]− 679.4, found 679.4.
4.1.3.10. 5′-O-[N-(Undec-10-ynoyl)sulfamoyl]-2′,3′-O-isopropylideneadenosine Triethylammonium salt (21d).
NHS ester 21c was coupled with 5′-O-sulfamoyl-2′,3′-O-isopropylideneadenosine using the general procedure for acylation to afford the title compound (70%): Rf 0.5 (8:2 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.28 (t, J = 7.2 Hz, 9H), 1.30–1.36 (m, 8H), 1.38 (s, 3H), 1.43–1.50 (m, 2H), 1.56 (p, J = 7.2 Hz, 2H), 1.60 (s, 3H), 2.11–2.17 (m, 5H), 3.18 (q, J = 7.2 Hz, 6H), 4.20–4.26 (m, 2H), 4.52–4.54 (m, 1H), 5.12 (dd, J = 6.0, 3.0 Hz, 1H), 5.35 (dd, J = 6.0, 3.0 Hz, 1H), 6.23 (d, J = 3.0 Hz, 1H), 8.21 (s, 1H), 8.44 (s, 1H); MS (ESI−) calcd for C24H33N6O7S [M − H]− 549.2, found 549.2.
4.1.3.11. 5′-O-[N-(10-Undecenoyl)sulfamoyl]-2′,3′-O-isopropylideneadenosine Triethylammonium salt (22d).
NHS ester 22c was coupled with 5′-O-sulfamoyl-2′,3′-O-isopropylideneadenosine using the general procedure for acylation to afford the tile compound (58%): Rf 0.5 (8:2 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.30–1.36 (m, 19H), 1.39 (s, 3H), 1.59 (p, J = 7.2 Hz, 2H), 1.60 (s, 3H), 1.99–2.02 (m, 2H), 2.20 (t, J = 7.2 Hz, 2H), 3.19 (q, J = 7.2 Hz, 6H), 4.20–4.26 (m, 2H), 4.52–4.55 (m, 1H), 4.89 (d, J = 9.6 Hz, 1H), 4.96 (d, J = 17.4 Hz, 1H), 5.12 (dd, J = 6.0, 3.0 Hz, 1H), 5.35 (dd, J = 6.0, 3.0 Hz, 1H), 6.23 (d, J = 3.0 Hz, 1H), 5.74–5.82 (m, 1H), 6.08 (d, J = 6.0 Hz, 1H), 8.20 (s, 3H), 8.49 (s, 1H); MS (ESI−) calcd for C24H35N6O7S [M − H]− 551.2, found 551.3.
4.1.3.12. 5′-O-{N-[(E)-Dodec-2-enoyl]sulfamoyl}-2′,3′-O-isopropylideneadenosine Triethylammonium salt (23d).
NHS ester 23c was coupled with 5′-O-sulfamoyl-2′,3′-O-isopropylideneadenosine using the general procedure for acylation to afford the title compound (62%): Rf 0.5 (8:2 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 0.88 (t, J = 7.2 Hz, 3H), 1.24–1.30 (m, 23H), 1.38 (s, 3H), 1.60 (s, 3H), 1.96 (q, J = 7.2 Hz, 2H), 2.87 (d, J = 7.2 Hz, 2H), 3.17 (q, J = 7.2 Hz, 6H), 4.19–4.24 (m, 2H), 4.52–4.53 (m, 2H), 5.10 (dd, J = 6.0, 3.0 Hz, 1H), 5.33 (dd, J = 6.0, 3.0 Hz, 1H), 5.49 (dt, J=15.0, 7.2 Hz, 1H), 5.57 (dt, J = 15.0, 7.2 Hz, 1H) 6.23 (d, J = 3.0 Hz, 1H), 8.21 (s, 1H), 8.45 (s, 1H); MS (ESI−) calcd for C25H37N6O7S [M − H]− 565.3, found 565.3.
4.1.3.13. 5′-O-{N-[(E)-Dodec-5-enoyl]sulfamoyl}-2′,3′-O-isopropylideneadenosine Triethylammonium salt (24d).
NHS ester 24c was coupled with 5′-O-sulfamoyl-2′,3′-O-isopropylideneadenosine using the general procedure for acylation to afford the title compound: Rf 0.5 (8:2 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 0.88 (t, J = 7.2 Hz, 3H), 1.26–1.31 (m, 17H), 1.38 (s, 3H), 1.60 (s, 3H), 1.62 (p, J = 7.2 Hz, 2H), 1.94–1.99 (m, 4H), 2.17 (t, J = 7.2 Hz, 2H), 3.17 (q, J = 7.2 Hz, 6H), 4.22–4.24 (m, 2H), 4.53 (br s, 1H), 5.12 (d, J = 6.0 Hz, 1H), 5.35–5.37 (m, 3H), 6.24 (br s, 1H), 8.21 (s, 1H), 8.44 (s, 1H); MS (ESI−) calcd for C25H37N6O7S [M − H]− 565.2, found 565.3.
4.1.3.14. 5′-O-{N-[(Z)-Undec-5-enoyl]sulfamoyl}-2′,3′-O-isopropylideneadenosine Triethylammonium salt (25d).
NHS ester 25c was coupled with 5′-O-sulfamoyl-2′,3′-O-isopropylideneadenosine using the general procedure for acylation to afford the title compound (65%): Rf 0.5 (8:2 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 0.88 (t, J = 7.2 Hz, 3H), 1.25–1.33 (m, 15H), 1.38 (s, 3H), 1.60 (s, 3H), 1.62 (p, J = 7.2 Hz, 2H), 1.99–2.06 (m, 4H), 2.18 (t, J = 7.2 Hz, 2H), 3.17 (q, J = 7.2, 6H), 4.22–4.24 (m, 2H), 4.53 (br s, 1H), 5.12 (dd, J = 6.0, 3.0 Hz, 1H), 5.32–5.34 (m, 3H), 6.23 (d, J = 3.0 Hz, 1H), 8.20 (s, 1H), 8.47 (s, 1H); MS (ESI−) calcd for C24H35N6O7S [M − H]− 551.2, found 551.2.
4.1.3.15. 5′-O-{N-[(1S,2R)-2-Nonylcyclopropanecarbonyl]sulfamoyl}-2′,3′-O-isopropylideneadenosine Triethylammonium salt (26d).
NHS ester 26c was coupled with 5′-O-sulfamoyl-2′,3′-O-isopropylideneadenosine using the general procedure for acylation to afford the title compound (59%): Rf 0.7 (8:2 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 0.50–0.54 (m, 1H), 0.87 (t, J = 7.2 Hz, 3H), 0.99–1.04 (m, 1H), 1.24–1.35 (m, 27H), 1.38 (s, 3H), 1.60 (s, 3H), 3.17 (q, J = 7.2 Hz, 6H), 4.20–4.26 (m, 2H), 4.52 (br s, 1H), 5.09 (d, J = 6.0 Hz, 1H), 5.33 (dd, J = 6.0, 3.0 Hz, 1H), 6.23 (d, J = 3.0 Hz, 1H), 8.21 (s, 1H), 8.43 (s, 1H); MS (ESI−) calcd for C26H39N6O7S [M − H]− 579.3, found 579.3.
4.1.3.16. 5′-O-[N-(11-Hydroxyundecanoyl)sulfamoyl]-2′,3′-O-isopropylideneadenosine Triethylammonium salt (27d).
NHS ester 27c was coupled with 5′-O-sulfamoyl-2′,3′-O-isopropylideneadenosine using the general procedure for acylation to afford the title compound (69%): Rf 0.5 (8:2 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.26–1.30 (m, 21H), 1.38 (s, 3H), 1.51 (p, J = 7.2 Hz, 2H), 1.57 (p, J = 7.2 Hz, 2H), 1.61 (s, 3H), 2.16 (t, J = 7.2 Hz, 2H), 3.16 (q, J = 7.2 Hz, 6H), 3.53 (t, J = 7.2 Hz, 2H), 4.22–4.23 (m, 2H), 4.54 (br s, 1H), 5.12 (dd, J = 6.0, 3.0 Hz, 1H), 5.33 (dd, J = 6.0, 3.0 Hz, 1H), 6.23 (d, J = 3.0 Hz, 1H), 8.21 (s, 1H), 8.47 (s, 1H); MS (ESI−) calcd for C24H37N6O8S [M − H]− 569.2, found 569.2.
4.1.3.17. 5′-O-[N-(12-Azidododecanoyl)sulfamoyl]-2′,3′-O-isopropylideneadenosine Triethylammonium salt (28d).
NHS ester 28c was coupled with 5′-O-sulfamoyl-2′,3′-O-isopropylideneadenosine using the general procedure for acylation to afford the title compound (53%): Rf 0.6 (8:2 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.26–1.31 (m, 23H), 1.38 (s, 3H), 1.53–1.59 (m, 4H), 1.60 (s, 3H), 2.17 (t, J = 7.2 Hz, 2H), 3.16 (q, J = 7.2 Hz, 6H), 3.26 (t, J = 7.2 Hz, 2H), 4.19–4.25 (m, 2H), 4.53 (br s, 1H), 5.12 (dd, J = 6.0, 3.0 Hz, 1H), 5.36 (dd, J = 6.0, 3.0 Hz, 1H), 6.24 (d, J = 3.0 Hz, 1H), 8.22 (s, 1H), 8.44 (s, 1H); MS (ESI−) calcd for C25H38N9O7S [M − H]− 608.3, found 608.2.
4.1.3.18. 5′-O-[N-(12-Methoxy-12-oxododecanoyl)sulfamoyl]-2′,3′-O-isopropylideneadenosine Triethylammonium salt (30d).
NHS ester 30c was coupled with 5′-O-sulfamoyl-2′,3′-O-isopropylideneadenosine using the general procedure for acylation to afford the title compound (62%): Rf 0.6 (8:2 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.20–1.26 (m, 21H), 1.37 (s, 3H), 1.53–1.58 (m, 2H), 1.57 (p, J = 7.2 Hz, 2H), 1.59 (s, 3H), 2.16 (t, J = 7.2 Hz, 2H), 2.27 (t, J = 7.2 Hz, 2H), 3.16 (q, J = 7.2 Hz, 6H), 4.19–4.26 (m, 2H), 4.52 (br s, 1H), 5.12 (dd, J = 6.0, 3.0 Hz, 1H), 5.36 (dd, J = 6.0, 3.0 Hz, 1H), 6.23 (d, J = 3.0 Hz, 1H), 8.21 (s, 1H), 8.44 (s, 1H); MS (ESI−) calcd for C26H39N6O9S [M − H]− 611.3, found 611.2.
4.1.3.19. 5′-O-[N-(11-Phenoxyundecanoyl)sulfamoyl]-2′,3′-O-isopropylideneadenosine Triethylammonium salt (32d).
NHS ester 32c was coupled with 5′-O-sulfamoyl-2′,3′-O-isopropylideneadenosine using the general procedure for acylation to afford the title compound (66%): Rf 0.5 (8:2 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.24–1.31 (m, 21H), 1.38 (s, 3H), 1.43 (p, J = 7.2 Hz, 2H), 1.57 (p, J = 7.2 Hz, 2H), 1.60 (s, 3H), 2.16 (t, J = 7.2 Hz, 2H), 3.15 (q, J = 7.2 Hz, 6H), 4.19–4.25 (m, 2H), 4.52 (br s, 1H), 5.11 (dd, J = 6.0, 3.0 Hz, 1H), 5.34 (dd, J = 6.0, 3.0 Hz, 1H), 6.22 (d, J = 3.0 Hz, 1H), 6.86–6.88 (m, 3H), 7.22 (t, J = 7.2 Hz, 2H), 8.21 (s, 1H), 8.44 (s, 1H); MS (ESI−) calcd for C30H41N6O8S [M − H]− 645.3, found 645.3.
4.1.3.20. 5′-O-{N-[(R)-2-Methyldodecanoyl]sulfamoyl}-2′,3′-O-isopropylideneadenosine Triethylammonium salt (33d).
NHS ester 33c was coupled with 5′-O-sulfamoyl-2′,3′-O-isopropylideneadenosine (0.52 mmol, 200 mg) using the general procedure for acylation to afford the title compound (57%): Rf 0.6 (8:2 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 0.87 (t, J = 7.2 Hz, 3H), 1.06 (d, J = 7.2 Hz, 3H), 1.23–1.28 (m, 25H), 1.36 (s, 3H), 1.59 (p, J = 7.2 Hz, 2H), 1.60 (s, 3H), 2.25–2.29 (m, 1H), 3.15 (q, J = 7.2 Hz, 6H), 4.19–4.27 (m, 2H), 4.51 (br s, 1H), 5.12 (dd, J = 6.0, 3.0 Hz, 1H), 5.36 (dd, J = 6.0, 3.0 Hz, 1H), 6.24 (d, J = 3.0 Hz, 1H), 8.21 (s, 1H), 8.44 (s, 1H); MS (ESI−) calcd for C26H41N6O7S [M − H]− 581.3, found 581.3.
4.1.3.21. 5′-O-{N-[(S)-(+)-2-Methyldodecanoyl]sulfamoyl}-2′,3′-O-isopropylideneadenosine Triethylammonium salt (34d).
NHS ester 34c was coupled with 5′-O-sulfamoyl-2′,3′-O-isopropylideneadenosine using the general procedure for acylation to afford the title compound (62%): Rf 0.6 (8:2 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 0.87 (t, J = 7.2 Hz, 3H), 1.05 (d, J = 7.2 Hz, 3H), 1.21–1.27 (m, 25H), 1.36 (s, 3H), 1.59 (p, J = 7.2 Hz, 2H), 1.60 (s, 3H), 2.24–2.29 (m, 1H), 3.16 (q, J = 7.2 Hz, 6H), 4.20–4.26 (m, 2H), 4.52 (br s, 1H), 5.12 (dd, J = 6.0, 3.0 Hz, 1H), 5.34 (dd, J = 6.0, 3.0 Hz, 1H), 6.23 (d, J = 3.0 Hz, 1H), 8.21 (s, 1H), 8.45 (s, 1H); MS (ESI−) calcd for C26H41N6O7S [M − H]− 581.3, found 581.3.
4.1.3.22. 5′-O-[N-(2,2-Dimethyldodecanoyl)sulfamoyl]-2′,3′-O-isopropylideneadenosine Triethylammonium salt (35d).
NHS ester 35c was coupled with 5′-O-sulfamoyl-2′,3′-O-isopropylideneadenosine using the general procedure for acylation to afford the title compound (48%): Rf 0.7 (8:2 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 0.87 (t, J = 7.2 Hz, 3H), 1.08 (s, 6H), 1.21–1.29 (m, 25H), 1.37 (s, 3H), 1.47 (p, J = 7.2 Hz, 2H), 1.59 (s, 3H), 3.15 (q, J = 7.2 Hz, 6H), 4.18–4.24 (m, 2H), 4.51 (br s, 1H), 5.13 (dd, J = 6.0, 3.0 Hz, 1H), 5.34 (dd, J = 6.0, 3.0 Hz, 1H), 6.23 (d, J = 3.0 Hz, 1H), 8.21 (s, 1H), 8.45 (s, 1H); MS (ESI−) calcd for C27H43N6O7S [M − H]− 595.3, found 595.3.
4.1.3.23. 5′-O-[N-(Nonyloxy)acetylsulfamoyl]-2′,3′-O-isopropylideneadenosine Triethylammonium salt (36d).
NHS ester 36c was coupled with 5′-O-sulfamoyl-2′,3′-O-isopropylideneadenosine using the general procedure for acylation to afford the title compound (59%): Rf 0.4 (8:2 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 0.87 (t, J = 7.2 Hz, 3H), 1.20–1.32 (m, 21H), 1.36 (s, 3H), 1.55 (p, J = 7.2 Hz, 2H), 1.60 (s, 3H), 3.11 (q, J = 7.2 Hz, 6H), 3.45 (t, J = 7.2 Hz, 2H), 3.90 (s, 2H), 4.21–4.27 (m, 2H), 4.52 (br s, 1H), 5.12 (dd, J = 6.0, 3.0 Hz, 1H), 5.37 (dd, J = 6.0, 3.0 Hz, 1H), 6.24 (d, J = 3.0 Hz, 1H), 8.21 (s, 1H), 8.44 (s, 1H); MS (ESI−) calcd for C24H37N6O8S [M − H]− 569.2, found 569.3.
4.1.3.24. 5′-O-{N-2-[2-(2-Butoxyethoxy)ethoxy]acetylsulfamoyl}-2′,3′-O-isopropylideneadenosine Triethylammonium salt (37d).
NHS ester 37c was coupled with 5′-O-sulfamoyl-2′,3′-O-isopropylideneadenosine using the general procedure for acylation to afford the title compound (56%): Rf 0.3 (8:2 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 0.92 (t, J = 7.2 Hz, 3H), 1.28 (t, J = 7.2 Hz, 9H), 1.33–1.37 (m, 2H), 1.39 (s, 3H), 1.51–1.54 (m, 2H), 1.61 (s, 3H), 3.19 (q, J = 7.2 Hz, 6H), 3.46 (t, J = 7.2 Hz, 2H), 3.55–3.56 (m, 2H), 3.60–3.62 (m, 2H), 3.63–3.65 (m, 4H), 3.96 (s, 2H), 4.24–4.25 (m, 2H), 4.54–4.55 (m, 1H), 5.13 (dd, J = 6.0, 3.0 Hz, 1H), 5.36 (dd, J = 6.6, 3.0 Hz, 1H), 6.24 (d, J = 3.0 Hz, 1H), 8.21 (s, 1H), 8.46 (s, 1H); MS (ESI−) calcd for C23H35N6O10S [M − H]− 587.2, found 587.3.
4.1.3.25. 5′-O-[N-(10-Phenoxydecanoyl)sulfamoyl]-2′,3′-O-isopropylideneadenosine Triethylammonium Salt (38d).
NHS ester 38c was coupled with 5′-O-sulfamoyl-2′,3′-O-isopropylideneadenosine using the general procedure for acylation to afford the title compound (85%): Rf 0.3 (7:3 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.28 (t, J = 7.2 Hz, 9H), 1.26–1.32 (m, 8H), 1.38 (s, 3H), 1.40–1.44 (m, 2H), 1.60 (s, 3H), 1.56–1.61 (m, 2H), 1.70–1.75 (m, 2H), 2.17 (t, J = 7.2 Hz, 2H), 3.17 (q, J = 7.2 Hz, 6H), 3.92 (t, J = 7.2 Hz, 2H), 4.20-4.26 (m, 2H), 4.52–4.54 (m, 1H), 5.11–5.13 (m, 1H), 5.34–5.35 (m, 1H), 6.23 (d, J = 3.0 Hz, 1H), 6.86–6.88 (m, 3H), 7.22–7.24 (m, 2H), 8.21 (s, 1H), 8.44 (s, 1H); MS (ESI−) calcd for C29H39N6O8S [M − H]− 631.2556, found 631.2540 (error 2.5 ppm).
4.1.3.26. 5′-O-[N-(12-Phenoxydodecanoyl)sulfamoyl]-2′,3′-O-isopropylideneadenosine Triethylammonium Salt (39d).
NHS ester 39c was coupled with 5′-O-sulfamoyl-2′,3′-O-isopropylideneadenosine using the general procedure for acylation to afford the title compound (88%): Rf 0.3 (7:3 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.28 (t, J = 7.2 Hz, 9H), 1.26–1.33 (m, 12H), 1.38 (s, 3H), 1.42–1.46 (m, 2H), 1.55–1.59 (m, 2H), 1.60 (s, 3H), 1.71–1.76 (m, 2H), 2.17 (t, J = 7.2 Hz, 2H), 3.17 (q, J = 7.2 Hz, 6H), 3.94 (t, J = 7.2 Hz, 2H), 4.22–4.24 (m, 2H), 4.52–4.53 (m, 1H), 5.11–5.13 (m, 1H), 5.34–5.36 (m, 1H), 6.23 (d, J = 3.0 Hz, 1H), 6.86–6.88 (m, 3H), 7.22–7.24 (m, 2H), 8.21 (s, 1H), 8.44 (s, 1H); HRMS (ESI−) calcd for C31H43N6O8S [M − H]− 659.2869, found 659.2894 (error 3.7 ppm).
4.1.3.27. 5′-O-{N-[11-(Phenylthio)undecanoyl]sulfamoyl}-2′,3′-O-isopropylideneadenosine Triethylammonium Salt (40d).
NHS ester 40c was coupled with 5′-O-sulfamoyl-2′,3′-O-isopropylideneadenosine using the general procedure for acylation to afford the title compound (86%): Rf 0.3 (7:3 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.27 (t, J = 7.2 Hz, 9H), 1.24–1.28 (m, 10H), 1.37–1.38 (m, 2H), 1.38 (s, 3H), 1.54–1.60 (m, 4H), 1.60 (s, 3H), 2.16 (t, J = 7.2 Hz, 2H), 2.88–2.91 (m, 2H), 3.14–3.19 (m, 6H), 4.20–4.26 (m, 2H), 4.53–4.54 (m, 1H), 5.11–5.12 (m, 1H), 5.33–5.35 (m, 1H), 6.23 (d, J = 3.0 Hz, 1H), 7.14–7.16 (m, 1H), 7.24–7.27 (m, 2H), 7.29–7.30 (m, 2H), 8.21 (s, 1H), 8.44 (s, 1H); HRMS (ESI−) calcd for C30H41N6O7S2− [M − H]− 661.2484, found 661.2458 (error 3.9 ppm).
4.1.3.28. 5′-O-{N-[11-(Phenylamino)undecanoyl]sulfamoyl}-2′,3′-O-isopropylideneadenosine Triethylammonium Salt (41d).
NHS ester 41c was coupled with 5′-O-sulfamoyl-2′,3′-O-isopropylideneadenosine using the general procedure for acylation to afford the title compound (91%): Rf 0.3 (7:3 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.26 (t, J = 7.2 Hz, 9H), 1.25–1.29 (m, 10H), 1.34–1.38 (m, 2H), 1.38 (s, 3H), 1.55–1.60 (m, 4H), 1.60 (s, 3H), 2.16 (t, J = 7.2 Hz, 2H), 3.03 (t, J = 7.2 Hz, 2H), 3.14 (q, J = 7.2 Hz, 6H), 4.20–4.26 (m, 2H), 4.52–4.54 (m, 1H), 5.11–5.12 (m, 1H), 5.33–5.35 (m, 1H), 6.23 (d, J = 3.0 Hz, 1H), 6.57–6.62 (m, 3H), 7.06–7.09 (m, 2H), 8.21 (s, 1H), 8.43 (s, 1H); HRMS (ESI−) calcd for C30H42N7O7S [M − H]− 644.2872, found 644.2879 (error 1.1 ppm).
4.1.3.29. 5′-O-{N-[11-(4-Fluorophenoxy)undecanoyl]sulfamoyl}-2′,3′-O-isopropylideneadenosine Triethylammonium Salt (42d).
NHS ester 42c was coupled with 5′-O-sulfamoyl-2′,3′-O-isopropylideneadenosine using the general procedure for acylation to afford the title compound (90%): Rf 0.3 (7:3 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.29 (t, J = 7.2 Hz, 9H), 1.28–1.32 (m, 10H), 1.38 (s, 3H), 1.40–1.45 (m, 2H), 1.55–1.60 (m, 2H), 1.60 (s, 3H), 1.70–1.75 (m, 2H), 2.17 (t, J = 7.2 Hz, 2H), 3.18 (q, J = 7.2 Hz, 6H), 3.91 (t, J = 7.2 Hz, 2H), 4.21–4.26 (m, 2H), 4.53–4.54 (m, 1H), 5.11–5.12 (m, 1H), 5.34–5.35 (m, 1H), 6.23 (d, J = 3.0 Hz, 1H), 6.85–6.88 (m, 2H), 6.95–6.98 (m, 2H), 8.21 (s, 1H), 8.45 (s, 1H); HRMS (ESI−) calcd for C30H40FN6O8S [M − H]− 663.2618, found 663.2586 (error 4.8 ppm).
4.1.3.30. 5′-O-{N-[11-(3-Fluorophenoxy)undecanoyl]sulfamoyl}-2′,3′-O-isopropylideneadenosine Triethylammonium Salt (43d).
NHS ester 43c was coupled with 5′-O-sulfamoyl-2′,3′-O-isopropylideneadenosine using the general procedure for acylation to afford the title compound (95%): Rf 0.3 (7:3 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.28 (t, J = 7.2 Hz, 9H), 1.27–1.32 (m, 10H), 1.38 (s, 3H), 1.41–1.45 (m, 2H), 1.55–1.59 (m, 2H), 1.60 (s, 3H), 1.71–1.76 (m, 2H), 2.17 (t, J = 7.2 Hz, 2H), 3.17 (q, J = 7.2 Hz, 6H), 3.94 (t, J = 7.2 Hz, 2H), 4.20–4.26 (m, 2H), 4.53–4.54 (m, 1H), 5.11–5.13 (m, 1H), 5.33–5.35 (m, 1H), 6.23 (d, J = 3.0 Hz, 1H), 6.60–6.65 (m, 2H), 6.70–6.71 (m, 1H), 7.20–7.24 (m, 1H), 8.21 (s, 1H), 8.45 (s, 1H); HRMS (ESI−) calcd for C30H40FN6O8S [M − H]− 663.2618, found 663.2641 (error 3.4 ppm).
4.1.3.31. 5′-O-{N-[11-(2-Fluorophenoxy)undecanoyl]sulfamoyl}-2′,3′-O-isopropylideneadenosine Triethylammonium Salt (44d).
NHS ester 44c was coupled with 5′-O-sulfamoyl-2′,3′-O-isopropylideneadenosine using the general procedure for acylation to afford the title compound (86%): Rf 0.3 (7:3 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.28 (t, J = 7.2 Hz, 9H), 1.27–1.32 (m, 10H), 1.38 (s, 3H), 1.48–1.43 (m, 2H), 1.59–1.55 (m, 2H), 1.60 (s, 3H), 1.78–1.74 (m, 2H), 2.16 (t, J = 7.2 Hz, 2H), 3.18 (q, J = 7.2 Hz, 6H), 4.01 (t, J = 7.2 Hz, 2H), 4.25–4.20 (m, 2H), 4.54–4.53 (m, 1H), 5.12–5.11 (m, 1H), 5.35–5.34 (m, 1H), 6.23 (d, J = 3.0 Hz, 1H), 6.89–6.86 (m, 1H), 7.07–7.03 (m, 3H), 8.21 (s, 1H), 8.46 (s, 1H); HRMS (ESI−) calcd for C30H40FN6O8S [M − H]− 663.2618, found 663.2632 (error 2.1 ppm).
4.1.3.32. 5′-O-{N-[11-(4-Chlorophenoxy)undecanoyl]sulfamoyl}-2′,3′-O-isopropylideneadenosine Triethylammonium Salt (45d).
NHS ester 45c was coupled with 5′-O-sulfamoyl-2′,3′-O-isopropylideneadenosine using the general procedure for acylation to afford the title compound (80%): Rf 0.3 (7:3 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.28 (t, J = 7.2 Hz, 9H), 1.27–1.32 (m, 10H), 1.38 (s, 3H), 1.40–1.45 (m, 2H), 1.55–1.58 (m, 2H), 1.60 (s, 3H), 1.70–1.75 (m, 2H), 2.17 (t, J = 7.2 Hz, 2H), 3.18 (q, J = 7.2 Hz, 6H), 3.92 (t, J = 7.2 Hz, 2H), 4.21–4.26 (m, 2H), 4.53–4.54 (m, 1H), 5.11–5.13 (m, 1H), 5.34–5.35 (m, 1H), 6.23 (d, J = 3.0 Hz, 1H), 6.86–6.87 (m, 2H), 7.20–7.22 (m, 2H), 8.21 (s, 1H), 8.44 (s, 1H); HRMS (ESI−) calcd for C30H40ClN6O8S [M − H]− 679.2322, found 679.2337 (error 2.2 ppm).
4.1.3.33. 5′-O-{N-[11-(3-Chlorophenoxy)undecanoyl]sulfamoyl}-2′,3′-O-isopropylideneadenosine Triethylammonium Salt (46d).
NHS ester 46c was coupled with 5′-O-sulfamoyl-2′,3′-O-isopropylideneadenosine using the general procedure for acylation to afford the title compound (90%): Rf 0.3 (7:3 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.26 (t, J = 7.2 Hz, 9H), 1.25–1.33 (m, 10H), 1.38 (s, 3H), 1.40–1.45 (m, 2H), 1.55–1.58 (m, 2H), 1.60 (s, 3H), 1.70–1.75 (m, 2H), 2.17 (t, J = 7.2 Hz, 2H), 3.13 (q, J = 7.2 Hz, 6H), 3.93 (t, J = 7.2 Hz, 2H), 4.20–4.25 (m, 2H), 4.53–4.55 (m, 1H), 5.11–5.12 (m, 1H), 5.33–5.35 (m, 1H), 6.23 (d, J = 3.0 Hz, 1H), 6.82–6.83 (m, 1H), 6.88–6.89 (m, 2H), 7.20–7.22 (m, 1H), 8.21 (s, 1H), 8.46 (s, 1H); HRMS (ESI−) calcd for C30H40ClN6O8S [M − H]− 679.2322, found 679.2300 (error 3.2 ppm).
4.1.3.34. 5′-O-{N-[11-(2-Chlorophenoxy)undecanoyl]sulfamoyl}-2′,3′-O-isopropylideneadenosine Triethylammonium Salt (47d).
NHS ester 47c was coupled with 5′-O-sulfamoyl-2′,3′-O-isopropylideneadenosine using the general procedure for acylation to afford the title compound (93%): Rf 0.3 (7:3 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.27 (t, J = 7.2 Hz, 9H), 1.26–1.35 (m, 10H), 1.38 (s, 3H), 1.45–1.50 (m, 2H), 1.55–1.58 (m, 2H), 1.60 (s, 3H), 1.75–1.80 (m, 2H), 2.17 (t, J = 7.2 Hz, 2H), 3.15 (q, J = 7.2 Hz, 6H), 4.01 (t, J = 7.2 Hz, 2H), 4.20–4.25 (m, 2H), 4.52–4.54 (m, 1H), 5.11–5.13 (m, 1H), 5.33–5.35 (m, 1H), 6.23 (d, J = 3.0 Hz, 1H), 6.86–6.88 (m, 1H), 7.00–7.02 (m, 1H), 7.20–7.23 (m, 1H), 7.31–7.32 (m, 1H), 8.21 (s, 1H), 8.45 (s, 1H); HRMS (ESI−) calcd for C30H40ClN6O8S [M − H]− 679.2322, found 679.2340 (error 2.6 ppm).
4.1.3.35. 5′-O-{N-[11-(4-Bromophenoxy)undecanoyl]sulfamoyl}-2′,3′-O-isopropylideneadenosine Triethylammonium Salt (48d).
NHS ester 48c was coupled with 5′-O-sulfamoyl-2′,3′-O-isopropylideneadenosine using the general procedure for acylation to afford the title compound (97%): Rf 0.3 (7:3 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.28 (t, J = 7.2 Hz, 9H), 1.27–1.32 (m, 10H), 1.38 (s, 3H), 1.40–1.44 (m, 2H), 1.56–1.59 (m, 2H), 1.60 (s, 3H), 1.70–1.75 (m, 2H), 2.17 (t, J = 7.2 Hz, 2H), 3.17 (q, J = 7.2 Hz, 6H), 3.92 (t, J = 7.2 Hz, 2H), 4.22–4.24 (m, 2H), 4.53–4.54 (m, 1H), 5.11–5.13 (m, 1H), 5.34–5.35 (m, 1H), 6.23 (d, J = 3.0 Hz, 1H), 6.81–6.83 (m, 2H), 7.34–7.36 (m, 2H), 8.21 (s, 1H), 8.45 (s, 1H); HRMS (ESI−) calcd for C30H40BrN6O8S [M − H]− 723.1812, 725.1791, found 723.1813, 725.1797 (error 0.1 ppm).
4.1.3.36. 5′-O-{N-[11-(4-Trifluoromethylphenoxy)undecanoyl]sulfamoyl}-2′,3′-O-isopropylideneadenosine Triethylammonium Salt (49d).
NHS ester 49c was coupled with 5′-O-sulfamoyl-2′,3′-O-isopropylideneadenosine using the general procedure for acylation to afford the title compound (94%): Rf 0.3 (7:3 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.28 (t, J = 7.2 Hz, 9H), 1.27–1.33 (m, 10H), 1.38 (s, 3H), 1.42–1.48 (m, 2H), 1.54–1.58 (m, 2H), 1.60 (s, 3H), 1.74–1.78 (m, 2H), 2.17 (t, J = 7.2 Hz, 2H), 3.18 (q, J = 7.2 Hz, 6H), 4.01 (t, J = 7.2 Hz, 2H), 4.23–4.28 (m, 2H), 4.52–4.54 (m, 1H), 5.11–5.13 (m, 1H), 5.33–5.35 (m, 1H), 6.23 (d, J = 3.0 Hz, 1H), 7.02–7.03 (m, 2H), 7.54–7.55 (m, 2H), 8.21 (s, 1H), 8.43 (s, 1H); HRMS (ESI−) calcd for C31H40F3N6O8S [M − H]− 713.2586, found 713.2614 (error 3.9 ppm).
4.1.3.37. 5′-O-{N-[11-(3-Trifluoromethylphenoxy)undecanoyl]sulfamoyl}-2′,3′-O-isopropylideneadenosine Triethylammonium Salt (50d).
NHS ester 50c was coupled with 5′-O-sulfamoyl-2′,3′-O-isopropylideneadenosine using the general procedure for acylation to afford the title compound (78%): Rf 0.3 (7:3 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.28 (t, J = 7.2 Hz, 9H), 1.27–1.32 (m, 10H), 1.38 (s, 3H), 1.44–1.50 (m, 2H), 1.56–1.58 (m, 2H), 1.60 (s, 3H), 1.74–1.78 (m, 2H), 2.17 (t, J = 7.2 Hz, 2H), 3.18 (q, J = 7.2 Hz, 6H), 4.00 (t, J = 7.2 Hz, 2H), 4.21–4.26 (m, 2H), 4.53–4.54 (m, 1H), 5.11–5.13 (m, 1H), 5.34–5.35 (m, 1H), 6.23 (d, J = 3.0 Hz, 1H), 7.13–7.19 (m, 3H), 7.42–7.45 (m, 1H), 8.21 (s, 1H), 8.44 (s, 1H); HRMS (ESI−) calcd for C31H40F3N6O8S [M − H]− 713.2586, found 713.2608 (error 3.1 ppm).
4.1.3.38. 5′-O-{N-[11-(2-Trifluoromethylphenoxy)undecanoyl]sulfamoyl}-2′,3′-O-isopropylideneadenosine Triethylammonium Salt (51d).
NHS ester 51c was coupled with 5′-O-sulfamoyl-2′,3′-O-isopropylideneadenosine using the general procedure for acylation to afford the title compound (89%): Rf 0.3 (7:3 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.25 (t, J = 7.2 Hz, 9H), 1.24–1.32 (m, 10H), 1.38 (s, 3H), 1.45–1.51 (m, 2H), 1.55–1.59 (m, 2H), 1.60 (s, 3H), 1.75–1.80 (m, 2H), 2.17 (t, J = 7.2 Hz, 2H), 3.10 (q, J = 7.2 Hz, 6H), 4.05 (t, J = 7.2 Hz, 2H), 4.20–4.24 (m, 2H), 4.52–4.53 (m, 1H), 5.11–5.12 (m, 1H), 5.33–5.35 (m, 1H), 6.23 (d, J = 3.0 Hz, 1H), 6.99–7.02 (m, 1H), 7.12–7.13 (m, 1H), 7.51–7.54 (m, 2H), 8.21 (s, 1H), 8.46 (s, 1H); HRMS (ESI−) calcd for C31H40F3N6O8S [M − H]− 713.2586, found 713.2587 (error 0.1 ppm).
4.1.3.39. 5′-O-{N-[11-(4-Methoxyphenoxy)undecanoyl]sulfamoyl}-2′,3′-O-isopropylideneadenosine Triethylammonium Salt (52d).
NHS ester 52c was coupled with 5′-O-sulfamoyl-2′,3′-O-isopropylideneadenosine using the general procedure for acylation to afford the title compound (82%): Rf 0.3 (7:3 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.25 (t, J = 7.2 Hz, 9H), 1.24–1.36 (m, 10H), 1.38 (s, 3H), 1.40–1.44 (m, 2H), 1.55–1.59 (m, 2H), 1.60 (s, 3H), 1.68–1.73 (m, 2H), 2.17 (t, J = 7.2 Hz, 2H), 3.10 (q, J = 7.2 Hz, 6H), 3.72 (s, 3H), 3.88 (t, J = 7.2 Hz, 2H), 4.19–4.25 (m, 2H), 4.53–4.55 (m, 1H), 5.11–5.12 (m, 1H), 5.33–5.35 (m, 1H), 6.23 (d, J = 3.0 Hz, 1H), 6.81–6.82 (m, 4H), 8.21 (s, 1H), 8.46 (s, 1H); HRMS (ESI−) calcd for C31H43N6O9S [M − H]− 675.2818, found 675.2789 (error 4.2 ppm).
4.1.3.40. 5′-O-{N-[11-(3-Methoxyphenoxy)undecanoyl]sulfamoyl}-2′,3′-O-isopropylideneadenosine Triethylammonium Salt (53d).
NHS ester 53c was coupled with 5′-O-sulfamoyl-2′,3′-O-isopropylideneadenosine using the general procedure for acylation to afford the title compound (87%): Rf 0.3 (7:3 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.27 (t, J = 7.2 Hz, 9H), 1.26–1.36 (m, 10H), 1.38 (s, 3H), 1.39–1.44 (m, 2H), 1.55–1.58 (m, 2H), 1.60 (s, 3H), 1.69–1.74 (m, 2H), 2.17 (t, J = 7.2 Hz, 2H), 3.16 (q, J = 7.2 Hz, 6H), 3.74 (s, 3H), 3.90 (t, J = 7.0 Hz, 2H), 4.20–4.27 (m, 2H), 4.51–4.53 (m, 1H), 5.11–5.12 (m, 1H), 5.35–5.36 (m, 1H), 6.23 (d, J = 3.0 Hz, 1H), 6.42–6.43 (m, 1H), 6.45–6.47 (m, 2H), 7.10–7.13 (m, 1H), 8.22 (s, 1H), 8.42 (s, 1H); HRMS (ESI−) calcd for C31H43N6O9S [M − H]− 675.2818, found 675.2812 (error 0.9 ppm).
4.1.3.41. 5′-O-{N-[11-(2-Methoxyphenoxy)undecanoyl]sulfamoyl}-2′,3′-O-isopropylideneadenosine Triethylammonium Salt (54d).
NHS ester 54c was coupled with 5′-O-sulfamoyl-2′,3′-O-isopropylideneadenosine using the general procedure for acylation to afford the title compound (83%): Rf 0.3 (7:3 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.27 (t, J = 7.2 Hz, 9H), 1.25–1.34 (m, 10H), 1.38 (s, 3H), 1.41–1.45 (m, 2H), 1.56–1.58 (m, 2H), 1.60 (s, 3H), 1.73–1.76 (m, 2H), 2.17 (t, J = 7.2 Hz, 2H), 3.16 (q, J = 7.2 Hz, 6H), 3.81 (s, 3H), 3.96 (t, J = 7.2 Hz, 2H), 4.20–4.24 (m, 2H), 4.52–4.53 (m, 1H), 5.11–5.12 (m, 1H), 5.34–5.35 (m, 1H), 6.23 (d, J = 3.0 Hz, 1H), 6.86–6.88 (m, 2H), 6.90–6.93 (m, 2H), 8.21 (s, 1H), 8.44 (s, 1H); HRMS (ESI−) calcd for C31H43N6O9S [M − H]− 675.2818, found 675.2808 (error 1.5 ppm).
4.1.3.42. 5′-O-{N-[11-(Naphthalen-1-yloxy)undecanoyl]sulfamoyl}-2′,3′-O-isopropylideneadenosine Triethylammonium Salt Triethylammonium (55d).
NHS ester 55c was coupled with 5′-O-sulfamoyl-2′,3′-O-isopropylideneadenosine using the general procedure for acylation to afford the title compound (81%): Rf 0.3 (7:3 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.22 (t, J = 7.2 Hz, 9H), 1.29–1.32 (m, 10H), 1.37 (s, 3H), 1.36–1.40 (m, 2H), 1.59 (s, 3H), 1.54–1.60 (m, 2H), 1.88–1.91 (m, 2H), 2.17 (t, J = 7.2 Hz, 2H), 3.04 (q, J = 7.2 Hz, 6H), 4.12 (t, J = 7.2 Hz, 2H), 4.21–4.23 (m, 2H), 4.52–4.54 (m, 1H), 5.10–5.12 (m, 1H), 5.32–5.34 (m, 1H), 6.22 (d, J = 3.0 Hz, 1H), 6.84–6.86 (m, 1H), 7.33–7.38 (m, 2H), 7.42–7.45 (m, 2H), 7.76–7.77 (m, 1H), 8.19–8.20 (m, 1H), 8.21 (s, 1H), 8.45 (s, 1H); HRMS (ESI−) calcd for C34H43N6O8S [M − H]− 695.2869, found 695.2893 (error 3.4 ppm).
4.1.3.43. 5′-O-{N-[11-(Naphthalen-2-yloxy)undecanoyl]sulfamoyl}-2′,3′-O-isopropylideneadenosine Triethylammonium Salt (56d).
NHS ester 56c was coupled with 5′-O-sulfamoyl-2′,3′-O-isopropylideneadenosine using the general procedure for acylation to afford the title compound (93%): Rf 0.3 (7:3 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.26 (t, J = 7.2 Hz, 9H), 1.24–1.35 (m, 10H), 1.37 (s, 3H), 1.44–1.50 (m, 2H), 1.55–1.59 (m, 2H), 1.59 (s, 3H), 1.77–1.81 (m, 2H), 2.17 (t, J = 7.2 Hz, 2H), 3.14 (q, J = 7.2 Hz, 6H), 4.04 (t, J = 7.2 Hz, 2H), 4.20–4.25 (m, 2H), 4.52–4.53 (m, 1H), 5.10–5.12 (m, 1H), 5.33–5.34 (m, 1H), 6.22 (d, J = 3.0 Hz, 1H), 7.09–7.11 (m, 1H), 7.17–7.18 (m, 1H), 7.26–7.29 (m, 1H), 7.37–7.40 (m, 1H), 7.70–7.73 (m, 3H), 8.21 (s, 1H), 8.44 (s, 1H); HRMS (ESI−) calcd for C34H43N6O8S [M − H]− 695.2869, found 695.2893 (error 3.4 ppm).
4.1.4. General Procedure for TFA Deprotection
To 5′-O-[N-(acyl)sulfamoyl]-2′,3′-O-isopropylideneadenosine triethylammonium salt 9d, 13d–56d was added aqueous 80% TFA. The reaction was allowed to stir at 0 °C for 0.5 h, then warmed to 23 °C and stirred for an additional 1.5 h. The resulting mixture was concentrated under reduced pressure to remove all traces of TFA. Purification by flash chromatography (0–20% EtOAc/MeOH + 0.5% NEt3) afforded the title compounds 9 and 13–56.
4.1.4.1. 5′-O-[N-(Dodecanoyl)sulfamoyl]adenosine Triethylammonium Salt (9).
The title compound was prepared from 9d using the general procedure for TFA deprotection (32%): Rf 0.28 (9:1 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 0.89 (t, J = 7.2 Hz, 3H), 1.21–1.32 (m, 25H), 1.58–1.61 (m, 2H), 2.20 (t, J = 7.2 Hz, 2H), 3.19 (q, J = 7.2 Hz, 6H), 4.28–4.31 (m, 2H), 4.35–4.40 (m, 2H), 4.67 (t, J = 6.0 Hz, 1H), 6.08 (d, J = 6.0 Hz, 1H), 8.20 (s, 1H), 8.47 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 9.2, 14.5, 23.7, 27.3, 30.45, 30.52, 30.6, 30.68, 30.74, 30.76, 33.1, 39.9, 47.9, 69.6, 72.3, 76.0, 84.4, 89.3, 120.2, 141.1, 150.9, 153.9, 157.3, 182.2; HRMS (ESI−) calcd for C22H35N6O7S [M − H]− 527.2293, found 527.2316 (error 4.3 ppm).
4.1.4.2. 5′-O-[N-(Butanoyl)sulfamoyl]adenosine Triethylammonium Salt (13).
The title compound was prepared from 13d using the general procedure for TFA deprotection (20%): Rf 0.12 (8:2 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 0.93 (t, J = 7.2 Hz, 3H), 1.29 (t, J = 7.2 Hz, 9H), 1.62 (p, J = 7.2 Hz, 2H), 2.18 (t, J = 7.2 Hz, 2H), 3.19 (q, J = 7.2 Hz, 6H), 4.27–4.29 (m, 2H), 4.32–4.34 (m, 1H), 4.37–4.39 (m, 1H), 4.69 (t, J = 6.0 Hz, 1H), 6.08 (d, J = 6.0 Hz, 1H), 8.20 (s, 1H), 8.49 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 9.4, 14.4, 20.8, 42.3, 48.0, 72.5, 76.2, 84.7, 89.3, 120.3, 141.3, 151.0, 154.0, 157.4, 183.1; HRMS (ESI−) calcd for C14H19N6O7S [M − H]− 415.1041, found 415.1049 (error 1.9 ppm).
4.1.4.3. 5′-O-[N-(Hexanoyl)sulfamoyl]adenosine Triethylammonium Salt (14).
The title compound was prepared from 14d using the general procedure for TFA deprotection (34%): Rf 0.15 (8:2 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 0.88 (t, J = 7.2 Hz, 3H), 1.29–1.31 (m, 13H), 1.58–1.61 (m, 2H), 2.19 (t, J = 7.8 Hz, 2H), 3.19 (q, J = 7.2 Hz, 6H), 4.27–4.29 (m, 2H), 4.31–4.33 (m, 1H), 4.37–4.39 (m, 1H), 4.68 (t, J = 6.0 Hz, 1H), 6.09 (d, J = 6.0 Hz, 1H), 8.20 (s, 1H), 8.50 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 9.5, 14.4, 23.7, 27.3, 32.9, 40.4, 47.9, 69.3, 72.5, 76.2, 84.7, 89.2, 120.3, 141.3, 151.0, 154.0, 157.4, 183.4; HRMS (ESI−) calcd for C16H23N6O7S [M − H]− 443.1354, found 443.1354 (error 0 ppm).
4.1.4.4. 5′-O-[N-(Octanoyl)sulfamoyl]adenosine Triethylammonium Salt (15).
The title compound was prepared from 15d using the general procedure for TFA deprotection (61%): Rf 0.18 (8:2 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 0.86 (t, J = 7.2 Hz, 3H), 1.23–1.31 (m, 17H), 1.59 (p, J = 7.2 Hz, 2H), 2.19 (t, J = 7.2 Hz, 2H), 3.19 (q, J = 7.2 Hz, 6H), 4.28–4.30 (m, 2H), 4.33–4.35 (m, 1H), 4.37–4.39 (m, 1H), 4.68 (t, J = 6.0 Hz, 1H), 6.09 (d, J = 6.0 Hz, 1H), 8.20 (s, 1H), 8.49 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 9.3, 14.5, 23.8, 27.5, 30.4, 30.6, 33.0, 40.2, 48.0, 69.5, 72.4, 76.2, 84.6, 89.3, 120.3, 141.3, 151.0, 154.0, 157.4, 182.8; HRMS (ESI−) calcd for C18H27N6O7S [M − H]− 471.1667, found 471.1659 (error 1.7 ppm).
4.1.4.5. 5′-O-[N-(Decanoyl)sulfamoyl]adenosine Triethylammonium Salt (16).
The title compound was prepared from 16d using the general procedure for TFA deprotection (29%): Rf 0.25 (9:1 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 0.87 (t, J = 7.2 Hz, 3H), 1.24–1.30 (m, 21H), 1.59 (p, J = 7.2 Hz, 2H), 2.19 (t, J = 7.2 Hz, 2H), 3.19 (q, J = 7.2 Hz, 6H), 4.28–4.30 (m, 2H), 4.32–4.35 (m, 1H), 4.38–4.39 (m, 1H), 4.68 (t, J = 6.0 Hz, 1H), 6.09 (d, J = 6.0 Hz, 1H), 8.20 (s, 1H), 8.49 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 9.3, 14.5, 23.8, 27.5, 30.5, 30.6, 30.7, 30.8, 33.1, 40.3, 48.0, 69.4, 72.4, 76.2, 84.6, 89.3, 120.3, 141.2, 151.0, 154.0, 157.4, 183.2; HRMS (ESI−) calcd for C20H31N6O7S [M − H]− 499.1980, found 499.1971 (error 1.8 ppm).
4.1.4.6. 5′-O-[N-(Tetradecanoyl)sulfamoyl]adenosine Triethylammonium Salt (17).
The title compound was prepared from 17d using the general procedure for TFA deprotection (30%): Rf 0.18 (8:2 EtOAc/MeOH): 1H NMR (600 MHz, CD3OD) δ 0.89 (t, J = 7.2 Hz, 3H), 1.24–1.31 (m, 29H), 1.59 (p, J = 7.2 Hz, 2H), 2.19 (t, J = 7.2 Hz, 2H), 3.19 (q, J = 7.2 Hz, 6H), 4.28–4.30 (m, 2H), 4.32–4.35 (m, 1H), 4.37–4.39 (m, 1H), 4.67 (t, J = 6.0 Hz, 1H), 6.08 (d, J = 6.0 Hz, 1H), 8.20 (s, 1H), 8.50 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 9.4, 14.5, 23.8, 27.5, 30.5, 30.6, 30.7, 30.8, 30.86, 30.87, 30.88. 30.89, 33.2, 40.3, 48.0, 69.4, 72.5, 76.2, 84.7, 89.3, 120.3, 141.3, 151.0, 154.0, 157.4, 183.2; HRMS (ESI−) calcd for C24H39N6O7S [M − H]− 555.2606, found 555.2587 (error 3.4 ppm).
4.1.4.7. 5′-O-[N-(Palmitoyl)sulfamoyl]adenosine Triethylammonium Salt (18).
The title compound was prepared from 18d using the general procedure for TFA deprotection (50%); Rf 0.21 (8:2 EtOAc/MeOH): 1H NMR (600 MHz, CD3OD) δ 0.90 (t, J = 7.2 Hz, 3H), 1.24–1.32 (m, 33H), 1.57–1.59 (m, 2H), 2.20 (t, J = 7.2 Hz, 2H), 3.21 (q, J = 7.2 Hz, 6H), 4.28–4.33 (m, 2H), 4.35–4.39 (m, 2H), 4.67 (t, J = 6.0 Hz, 1H), 6.08 (d, J = 6.0 Hz, 1H), 8.20 (s, 1H), 8.50 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 9.4, 14.6, 23.9, 27.6, 30.4, 30.62, 30.67, 30.73, 30.77, 30.80, 30.85, 30.90, 30.92, 30.93, 33.2, 40.4, 48.0, 69.4, 72.5, 76.3, 84.7, 89.3, 120.3, 141.3, 151.0, 154.0, 157.4, 183.3; HRMS (ESI−) calcd for C24H39N6O7S [M − H]− 583.2919, found 583.2884 (error 6.0 ppm).
4.1.4.8. 5′-O-[N-(Stearoyl)sulfamoyl]adenosine Triethylammonium Salt (19).
The title compound was prepared from 19d using the general procedure for TFA deprotection (24%): Rf 0.24 (8:2 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 0.89 (t, J = 7.2 Hz, 3H), 1.24–1.30 (m, 37H), 1.59 (p, J = 7.8 Hz, 2H), 2.19 (t, J = 7.2 Hz, 2H), 3.19 (q, J = 7.2 Hz, 6H), 4.29–4.31 (m, 2H), 4.33–4.36 (m, 1H), 4.38–4.40 (m, 1H), 4.67 (t, J = 6.0 Hz, 1H), 6.09 (d, J = 6.0 Hz, 1H), 8.20 (s, 1H), 8.48 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 9.3, 14.5, 23.8, 27.47, 27.49, 30.4, 30.56, 30.62, 30.66, 30.72, 30.75, 30.79, 30.85 (2C), 30.88 (2C), 33.2, 40.2, 48.0, 69.4, 72.4, 76.2, 84.6, 89.3, 120.3, 141.2, 151.0, 154.0, 157.4, 183.0; HRMS (ESI−) calcd for C28H47N6O7S [M − H]− 611.3232, found 611.3202 (error 4.9 ppm).
4.1.4.9. 5′-O-[N-(Icosanoyl)sulfamoyl]adenosine Triethylammonium Salt (20).
The title compound was prepared from 20d using the general procedure for TFA deprotection (18%): Rf 0.27 (8:2 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 0.90 (t, J = 7.2 Hz, 3H), 1.24–1.32 (m, 41H), 1.55–1.60 (m, 2H), 2.20 (t, J = 7.2 Hz, 2H), 3.21 (q, J = 7.2 Hz, 6H), 4.29–4.30 (m, 1H), 4.32–4.34 (m, 1H), 4.37–4.39 (m, 2H), 4.66 (t, J = 6.0 Hz, 1H), 6.08 (d, J = 6.0 Hz, 1H), 8.20 (s, 1H), 8.46 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 9.6, 14.6, 23.8, 23.9, 27.7, 30.6, 30.74, 30.78, 30.86, 30.89, 30.92 (3C), 30.93 (3C), 32.9, 33.2, 40.5, 48.0, 69.3, 72.5, 76.3, 84.8, 89.3, 120.2, 141.3, 151.1, 154.0, 157.4, 183.4 (Missing 1 C in the alkyl chain); HRMS (ESI−) calcd for C30H51N6O7S [M − H]− 639.3545, found 639.3535 (error 1.6 ppm).
4.1.4.10. 5′-O-[N-(Undec-10-ynoyl)sulfamoyl]adenosine Triethylammonium Salt (21).
The title compound was prepared from 21d using the general procedure for TFA deprotection (51%): Rf 0.55 (8:2 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.22–1.32 (m, 17H), 1.42 (p, J = 7.2 Hz, 2H), 1.54 (p, J = 7.2 Hz, 2H), 2.08 (t, J = 7.2 Hz, 2H), 2.10–2.16 (m, 3H), 3.13 (q, J = 7.2 Hz, 6H), 4.23–4.30 (m, 3H), 4.35–4.37 (m, 1H), 4.66 (t, J = 6.0 Hz, 1H), 6.05 (d, J = 6.0 Hz, 1H), 8.16 (s, 1H), 8.45 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 9.2, 19.0, 27.4, 29.6, 29.7, 30.0, 30.39, 30.43, 40.2, 47.7, 69.2, 69.4, 72.3, 76.0, 84.4, 85.1, 89.2, 120.1, 141.1, 150.8, 153.9, 157.2, 183.1; HRMS (ESI−) calcd for C21H29N6O7S [M − H]− 509.1824, found 509.1830 (error 1.2 ppm).
4.1.4.11. 5′-O-[N-(Undec-10-enoyl)sulfamoyl]adenosine Triethylammonium Salt (22).
The title compound was prepared from 22d using the general procedure for TFA deprotection (39%): Rf 0.65 (7:3 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.26–1.30 (m, 19H), 1.59 (p, J = 7.2 Hz, 2H), 1.99–2.02 (m, 2H), 2.20 (t, J = 7.2 Hz, 2H), 3.19 (q, J = 7.2 Hz, 6H), 4.28–4.30 (m, 2H), 4.32–4.35 (m, 1H), 4.38–4.40 (m, 1H), 4.68 (t, J = 6.0 Hz, 1H), 4.89 (d, J = 9.6 Hz, 1H), 4.96 (d, J = 17.4 Hz, 1H), 5.74–5.82 (m, 1H), 6.08 (d, J = 6.0 Hz, 1H), 8.20 (s, 3H), 8.49 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 9.3, 27.5, 30.20, 30.27, 30.30, 30.60, 30.65, 35.0, 40.3, 48.0, 69.4, 72.4, 76.2, 84.7, 89.3, 114.7, 120.3, 140.3, 141.3, 151.0, 154.0, 157.4, 183.0; HRMS (ESI−) calcd for C21H31N6O7S [M − H]− 511.1980, found 511.1973 (error 1.3 ppm).
4.1.4.12. 5′-O-{N-[(E)-Dodec-2-enoyl]sulfamoyl}adenosine Triethylammonium Salt (23).
The title compound was prepared from 23d using the general procedure for TFA deprotection (25%); Rf 0.65 (7:3 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 0.89 (t, J = 7.2 Hz, 3H), 1.28 (t, J = 7.2 Hz, 9H), 1.23–1.30 (m, 14H), 1.98 (q, J = 7.2 Hz, 2H), 2.91 (d, J = 7.2 Hz, 2H), 3.19 (q, J = 7.2 Hz, 6H), 4.25–4.30 (m, 2H), 4.37–4.39 (m, 2H), 4.68 (t, J = 6.0 Hz, 1H), 5.49 (dt, J = 15.0, 7.2 Hz, 1H), 5.57 (dt, J = 15.0, 7.2 Hz, 1H), 6.08 (d, J = 6.0 Hz, 1H), 8.20 (s, 1H), 8.49 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 9.2, 14.4, 23.7, 30.2, 30.3, 30.4, 30.5, 30.6, 32.7, 33.1, 47.9, 69.4, 72.3, 76.1, 84.5, 89.2, 120.2, 125.5, 134.3, 141.1, 150.9, 153.9, 157.3, 182.0; HRMS (ESI−) calcd for C22H33N6O7S [M − H]− 525.2137, found 525.2146 (error 1.7 ppm).
4.1.4.13. 5′-O-{N-[(E)-Dodec-5-enoyl]sulfamoyl}adenosine Triethylammonium Salt (24).
The title compound was prepared from 24d using the general procedure for TFA deprotection (53%): Rf 0.25 (8:2 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 0.89 (t, J = 7.2 Hz, 3H), 1.23–1.32 (m, 17H), 1.63 (p, J = 7.2 Hz, 2H), 1.93–2.00 (m, 4H), 2.20 (t, J = 7.2 Hz, 2H), 3.20 (q, J = 7.2 Hz, 6H), 4.28–4.34 (m, 2H), 4.37–4.39 (m, 2H), 4.67 (t, J = 6.0 Hz, 1H), 5.33–5.41 (m, 2H), 6.08 (d, J = 6.0 Hz, 1H), 8.20 (s, 1H), 8.45 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 9.3, 14.6, 23.8, 25.1, 26.2, 26.4, 30.1, 30.8, 30.9, 33.2, 48.1, 69.9, 71.9, 75.7, 85.4, 89.9, 121.4, 130.6, 132.6, 140.7, 150.7, 153.9, 157.2, 182.3; HRMS (ESI−) calcd for C22H33N6O7S [M − H]− 525.2137, found 525.2137 (error 0 ppm).
4.1.4.14. 5′-O-{N-[(Z)-Undec-5-enoyl]sulfamoyl}adenosine Triethylammonium Salt (25).
The title compound was prepared from 25d using the general procedure for TFA deprotection (38%): Rf 0.26 (8:2 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 0.87 (t, J = 7.2 Hz, 3H), 1.23–1.32 (m, 15H), 1.63 (p, J = 7.8 Hz, 2H), 1.98–2.06 (m, 4H), 2.21 (t, J = 7.2 Hz, 2H), 3.19 (q, J = 7.2 Hz, 6H), 4.28–4.30 (m, 2H), 4.32–4.35 (m, 1H), 4.37–4.39 (m, 1H), 4.68 (t, J = 6.0 Hz, 1H), 5.32–5.34 (m, 2H), 6.08 (d, J = 6.0 Hz, 1H), 8.19 (s, 1H), 8.48 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 9.4, 14.6, 23.8, 27.6, 28.1, 28.3, 30.7, 32.8, 39.8, 48.1, 69.6, 72.5, 76.2, 84.7, 89.4, 120.4, 130.3, 131.6, 141.3, 151.0, 154.0, 157.4, 182.4; HRMS (ESI−) calcd for C21H31N6O7S [M − H]− 511.1980, found 511.1958 (error 4.3 ppm).
4.1.4.15. 5′-O-{N-[(1S,2R)-2-Nonylcyclopropanecarbonyl]sulfamoyl} adenosine Triethylammonium Salt (26).
The title compound was prepared from 26d using the general procedure for TFA deprotection (41%): Rf 0.38 (7:3 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 0.51–0.54 (m, 1H), 0.89 (t, J = 7.2 Hz, 3H), 1.00–1.03 (m, 1H), 1.21–1.33 (m, 23H), 1.34–1.41 (m, 4H), 3.16 (q, J = 7.2 Hz, 6H), 4.24–4.30 (m, 2H), 4.30–4.32 (m, 1H), 4.36–4.38 (m, 1H), 4.68 (t, J = 6.0 Hz, 1H), 6.08 (d, J = 6.0 Hz, 1H), 8.21 (s, 1H), 8.49 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 9.3, 14.5, 15.7, 23.3, 23.7, 25.8, 30.39, 30.41, 30.5, 30.67, 30.72, 33.0, 34.4, 47.7, 69.2, 72.5, 76.0, 84.6, 89.1, 120.2, 141.1, 150.9, 153.9, 157.3, 182.7; HRMS (ESI−) calcd for C23H35N6O7S [M − H]− 539.2293, found 539.2290 (error 0.6 ppm).
4.1.4.16. 5′-O-[N-(11-Hydroxyundecanoyl)sulfamoyl]adenosine Triethylammonium Salt (27).
The title compound was prepared from 27d using the general procedure for TFA deprotection (56%): Rf 0.28 (8:2 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.26–1.31 (m, 21H), 1.51 (p, J = 7.2 Hz, 2H), 1.57–1.61 (m, 2H), 2.19 (t, J = 7.2 Hz, 2H), 3.19 (q, J = 7.2 Hz, 6H), 3.52 (t, J = 7.2 Hz, 2H), 4.27–4.29 (m, 2H), 4.32–4.35 (m, 1H), 4.37–4.39 (m, 1H), 4.68 (t, J = 6.0 Hz, 1H), 6.09 (d, J = 6.0 Hz, 1H), 8.20 (s, 1H), 8.51 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 9.3, 27.0, 27.5, 30.67, 30.70, 30.75, 30.80, 30.84, 33.8, 40.3, 48.0, 63.1, 69.4, 72.4, 76.1, 84.6, 89.3, 120.3, 141.2, 150.9, 154.0, 157.4, 183.2; HRMS (ESI−) calcd for C21H33N6O8S [M − H]− 529.2086, found 529.2078 (error 1.5 ppm).
4.1.4.17. 5′-O-[N-(12-Azidododecanoyl)sulfamoyl]adenosine Triethylammonium Salt (28).
The title compound was prepared from 28d using the general procedure for TFA deprotection (33%): Rf 0.18 (8:2 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.25–1.35 (m, 23H), 1.54–1.61 (m, 4H), 2.19 (t, J = 7.2 Hz, 2H), 3.19 (q, J = 7.2 Hz, 6H), 3.26 (t, J = 7.2 Hz, 2H), 4.27–4.30 (m, 2H), 4.33–4.35 (m, 1H), 4.37–4.39 (m, 1H), 4.67 (t, J = 6.0 Hz, 1H), 6.09 (d, J = 6.0 Hz, 1H), 8.20 (s, 1H), 8.49 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 9.2, 27.4, 27.8, 29.9, 30.2, 30.53, 30.57, 30.60, 30.62 (2C), 40.2, 47.9, 52.5, 69.2, 72.3, 76.1, 84.5, 89.2, 120.2, 141.2, 150.9, 153.9, 157.3, 183.2; HRMS (ESI−) calcd for C22H34N9O7S [M − H]− 568.2307, found 568.2190 (error 20.6 ppm).
4.1.5. 5′-O-[N-(12-Aminododecanoyl)sulfamoyl]adenosine (29)
To compound 28 (0.13 mmol, 90 mg) in MeOH (20 mL) was added Pd/C (20 mg) at 23 °C. The mixture was shaken under H2 (g) (40 psi) for 3 h. The crude mixture was filtered through a short pad of Celite. The filtrate was concentrated under reduced pressure, dissolved in MeOH, and concentrated onto Celite. Purification by flash chromatography (0–50% MeOH/EtOAc + 0.5% NEt3) afforded the title compound (52 mg, 62%) as a colorless oil: Rf 0.11 (7:3 EtOAc/MeOH);1H NMR (600 MHz, CD3OD) δ 1.26–1.36 (m, 23H), 1.57–1.63 (m, 4H), 2.19 (t, J = 7.2 Hz, 2H), 2.90 (t, J = 7.2 Hz, 2H), 4.26–4.29 (m, 2H), 4.31–4.34 (m, 1H), 4.37–4.39 (m, 1H), 4.67 (t, J = 6.0 Hz, 1H), 6.08 (d, J = 6.0 Hz, 1H), 8.20 (s, 1H), 8.50 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 27.2, 27.4, 28.6, 29.85, 29.89, 30.10, 30.12, 40.3, 41.0, 60.3, 69.3, 72.5, 76.3, 84.7, 89.4, 120.4, 141.3, 151.0, 154.0, 157.5, 183.6 (missing 1C in alkyl chain); HRMS (ESI−) calcd for C22H36N7O7S [M − H]− 542.2402, found 542.2367 (error 6.4 ppm).
4.1.4.18. 5′-O-[N-(12-Methoxy-12-oxododecanoyl)sulfamoyl]adenosine Triethylammonium Salt (30).
The title compound was prepared from 30d using the general procedure for TFA deprotection (40%): Rf 0.37 (8:2 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.24–1.31 (m, 21H), 1.54–1.60 (m, 4H), 2.18 (t, J = 7.2 Hz, 2H), 2.29 (t, J = 7.2 Hz, 2H), 3.19 (q, J = 7.2 Hz, 6H), 3.64 (s, 3H), 4.26–4.29 (m, 2H), 4.31–3.34 (m, 1H), 4.37–4.39 (m, 1H), 4.67 (t, J = 6.0 Hz, 1H), 6.08 (d, J = 6.0 Hz, 1H), 8.19 (s, 1H), 8.50 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 9.4, 26.2, 27.5, 30.3, 30.5, 30.59, 30.67, 30.69, 30.7, 35.0, 40.3, 48.0, 52.1, 69.5, 72.4, 76.2, 84.6, 89.4, 120.3, 141.3, 150.9, 154.0, 157.4, 176.2, 183.1; HRMS (ESI−) calcd for C23H35N6O9S [M − H]− 571.2192, found 571.2180 (error 2.1 ppm).
4.1.6. 5′-O-[N-(11-Carboxyundecanoyl)sulfamoyl]adenosine Triethylammonium Salt (31)
To compound 30 (0.13 mmol, 80 mg) in MeOH/H2O (1:1, 4 mL) was added LiOH (0.39 mmol, 17mg). The mixture was stirred at 23 °C for 16 h. The resulting mixture was concentrated under reduced pressure. The title compound was prepared from the crude mix using the general procedure for TFA deprotection (41%): Rf 0.38 (7:3 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.25–1.28 (m, 21H), 1.56–1.59 (m, 4H), 2.15 (t, J = 7.2 Hz, 2H), 2.19 (t, J = 7.2 Hz, 2H), 3.14 (q, J = 7.2 Hz, 6H), 4.26–4.29 (m, 2H), 4.31–3.34 (m, 1H), 4.37–4.39 (m, 1H), 4.68 (t, J = 6.0 Hz, 1H), 6.09 (d, J = 6.0 Hz, 1H), 8.20 (s, 1H), 8.50 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 9.3, 27.5, 27.6, 30.60, 30.64, 30.70, 30.73, 30.79, 30.82, 38.8, 40.4, 47.6, 69.1, 72.4, 76.1, 84.6, 89.2, 120.2, 141.2, 150.9, 153.9, 157.3, 182.5, 183.5; HRMS (ESI−) calcd for C22H32N6O9S [M − H]− 557.2035, found 557.2064 (error 5.2 ppm).
4.1.1.19. 5′-O-[N-(11-Phenoxyundecanoyl)sulfamoyl]adenosine Triethylammonium Salt (32).
. The title compound was prepared from 32d using the general procedure for TFA deprotection (62%): Rf 0.29 (8:2 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.25–1.32 (m, 21H), 1.43 (p, J = 7.2 Hz, 2H), 1.59 (p, J = 7.2 Hz, 2H), 1.73 (p, J = 7.2 Hz, 2H), 2.19 (t, J = 7.2 Hz, 2H), 3.17 (q, J = 7.2 Hz, 6H), 3.93 (t, J = 7.2 Hz, 2H), 4.26–4.29 (m, 2H), 4.31–4.34 (m, 1H), 4.38–4.40 (m, 1H), 4.69 (t, J = 6.0 Hz, 1H), 6.08 (d, J = 6.0 Hz, 1H), 6.87 (d, J = 7.2 Hz, 3H), 7.23 (t, J = 7.2 Hz, 2H), 8.20 (s, 1H), 8.50 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 9.2, 27.1, 27.5, 30.4, 30.47, 30.53, 30.55, 30.58, 30.64, 40.3, 47.8, 68.9, 69.2, 72.4, 76.1, 84.5, 89.2, 115.5 (2C), 120.2, 121.4, 130.4 (2C), 141.1, 150.9, 153.9, 157.3, 160.6, 183.2; HRMS (ESI−) calcd for C27H37N6O8S [M − H]− 605.2399, found 605.2391 (error 1.3 ppm).
4.1.4.20. 5′-O-{N-[(R)-2-Methyldodecanoyl]sulfamoyl}adenosine Triethylammonium Salt (33).
The title compound was prepared from 33d using the general procedure for TFA deprotection (51%): Rf 0.31 (8:2 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 0.89 (t, J = 7.2 Hz, 3H), 1.08 (d, J = 6.6 Hz, 3H), 1.23–1.30 (m, 25H), 1.59–1.65 (m, 2H), 2.27–2.31 (m, 1H), 3.16 (q, J = 7.2 Hz, 6H), 4.26–4.28 (m, 2H), 4.31–4.34 (m, 1H), 4.38–4.40 (m, 1H), 4.69 (t, J = 6.0 Hz, 1H), 6.08 (d, J = 6.0 Hz, 1H), 8.19 (s, 1H), 8.48 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 9.4, 14.6, 18.8, 23.9, 28.9, 30.60, 30.61, 30.90, 30.94, 31.1, 33.2, 36.0, 44.7, 48.1, 69.5, 72.6, 76.2, 84.8, 89.3, 120.4, 141.3, 151.1, 154.0, 157.5, 183.0; HRMS (ESI−) calcd for C23H37N6O7S[M − H]− 541.2450, found 541.2459 (error 1.7 ppm).
4.1.4.21. 5′-O-{N-[(S)-2-Methylodecanoyl]sulfamoyl}adenosine Triethylammonium Salt (34).
The title compound was prepared from 34d using the general procedure for TFA deprotection (57%): Rf 0.28 (8:2 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 0.87 (t, J = 7.2 Hz, 3H), 1.06 (d, J = 7.2 Hz, 3H), 1.23–1.28 (m, 25H), 1.59–1.63 (m, 4H), 2.27–2.31 (m, 1H), 3.16 (q, J = 7.2 Hz, 6H), 4.26–4.28 (m, 2H), 4.31–4.34 (m, 1H), 4.38–4.40 (m, 1H), 4.69 (t, J = 6.0 Hz, 1H), 6.08 (d, J = 6.0 Hz, 1H), 8.19 (s, 1H), 8.48 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 9.3, 14.6, 18.8, 23.9, 28.6, 30.6, 30.62, 30.89, 30.93, 31.1, 33.2, 36.0, 44.7, 48.0, 69.4, 72.4, 76.2, 84.5, 89.3, 120.4, 141.3, 151.4, 154.0, 157.4, 183.2; HRMS (ESI−) calcd for C23H37N6O7S [M − H]− 541.2450, found 541.2435 (error 2.8 ppm).
4.1.4.22. 5′-O-[N-(2,2-Dimethyldodecanoyl)sulfamoyl]adenosine Triethylammonium Salt (35).
The title compound was prepared from 35d using the general procedure for TFA deprotection (40%): Rf 0.27 (8:2 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 0.88 (t, J = 7.2 Hz, 3H), 1.11 (s, 6H), 1.19–1.24 (m, 16H), 1.29 (t, J = 7.2 Hz, 9H), 1.47–1.51 (m, 2H), 3.19 (q, J = 7.2 Hz, 6H), 4.27–4.30 (m, 2H), 4.32–4.35 (m, 1H), 4.37–4.40 (m, 1H), 4.69 (t, J = 6.0 Hz, 1H), 6.08 (d, J = 6.0 Hz, 1H), 8.20 (s, 1H), 8.49 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 9.2, 14.4, 23.7, 26.2, 26.5 (2C), 30.4, 30.7 (2C), 30.8, 31.6, 33.1, 42.7, 45.1, 47.9, 69.6, 72.4, 76.1, 84.5, 89.2, 120.2, 141.2, 150.9, 153.9, 157.3, 182.9; HRMS (ESI−) calcd for C24H39N6O7S [M − H]− 555.2606, found 555.2579 (error 4.9 ppm).
4.1.4.23. 5′-O-[N-2-(Nonyloxy)acetylsulfamoyl]adenosine Triethylammonium Salt (36).
The title compound was prepared from 36d using the general procedure for TFA deprotection (54%): Rf 0.3 (8:2 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 0.89 (t, J = 7.2 Hz, 3H), 1.24–1.32 (m, 21H), 1.56 (p, J = 7.2 Hz, 2H), 3.19 (q, J = 7.2 Hz, 6H), 3.46 (t, J = 7.2 Hz, 2H), 3.93 (s, 2H), 4.27–4.29 (m, 2H), 4.31–4.34 (m, 1H), 4.37–4.39 (m, 1H), 4.68 (t, J = 6.0 Hz, 1H), 6.08 (d, J = 6.0 Hz, 1H), 8.20 (s, 1H), 8.48 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 9.2, 14.4, 23.7, 27.1, 30.4, 30.58, 30.61, 30.7, 33.0, 47.9, 69.4, 72.3, 72.5, 72.6, 76.0, 84.4, 89.3, 120.2, 141.1, 150.9, 153.8, 157.2, 178.3; HRMS (ESI−) calcd for C21H33N6O8S [M − H]− 529.2086, found 529.2083 (error 0.6 ppm).
4.1.4.24. 5′-O-{N-2-[2-(2-Butoxyethoxy)ethoxy]acetylsulfamoyl}adenosine Triethylammonium Salt (37).
The title compound was prepared from 37d using the general procedure for TFA deprotection (46%): Rf 0.10 (8:2 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 0.90 (t, J = 7.2 Hz, 3H), 1.29 (t, J = 7.2 Hz, 9H), 1.33–1.37 (m, 2H), 1.51–1.54 (m, 2H), 3.19 (q, J = 7.2 Hz, 6H), 3.45 (t, J = 7.2 Hz, 2H), 3.54–3.64 (m, 8H), 3.98 (s, 2H), 4.27–4.30 (m, 2H), 4.32–4.34 (m, 1H, 4.37–4.40 (m, 1H), 4.67 (t, J = 6.0 Hz, 1H), 6.08 (d, J = 6.0 Hz, 1H), 8.19 (s, 1H), 8.50 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 9.3, 14.3, 20.4, 32.9, 48.0, 69.3, 71.1, 71.4, 71.50, 71.55, 72.2, 72.4, 73.0, 76.2, 84.6, 89.3, 120.3, 141.3, 151.0, 154.0, 157.4, 178.6; HRMS (ESI−) calcd for C20H31N6O10S [M − H]− 547.1828, found 547.1806 (error 4.0 ppm).
4.1.4.25. 5′-O-[N-(10-Phenoxydecanoyl)sulfamoyl]adenosine Triethylammonium Salt (38).
The title compound was prepared from 38d using the general procedure for TFA deprotection (87%): Rf 0.1 (7:3 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.27 (t, J = 7.2 Hz, 9H), 1.26–1.34 (m, 8H), 1.39–1.43 (m, 2H), 1.57–1.61 (m, 2H), 1.69–1.74 (m, 2H), 2.19 (t, J = 7.2 Hz, 2H), 3.16 (q, J = 7.2 Hz, 6H), 3.92 (t, J = 7.2 Hz, 2H), 4.26–4.34 (m, 3H), 4.38–4.39 (m, 1H), 4.68 (t, J = 6.0 Hz, 1H), 6.09 (d, J = 6.0 Hz, 1H), 6.86–6.89 (m, 3H), 7.22–7.24 (m, 2H), 8.20 (s, 1H), 8.50 (s, 1H); 13C NMR (150 MHz, CD3OD): δ 9.1, 27.1, 27.4, 30.39, 30.46, 30.50, 30.58, 40.2, 47.7, 68.8, 69.2, 72.2, 76.0, 84.4, 89.2, 115.4, 120.1, 121.4, 130.3, 141.1, 150.7, 153.8, 157.2, 160.5, 183.2; HRMS (ESI−) calcd for C26H35N6O8S [M − H]− 591.2243, found 591.2224 (error 3.2 ppm).
4.1.4.26. 5′-O-[N-(12-Phenoxydodecanoyl)sulfamoyl]adenosine Triethylammonium Salt (39).
The title compound was prepared from 39d using the general procedure for TFA deprotection (52%): Rf 0.1 (7:3 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.28 (t, J = 7.2 Hz, 9H), 1.24–1.34 (m, 12H), 1.42–1.46 (m, 2H), 1.58–1.60 (m, 2H), 1.71–1.76 (m, 2H), 2.19 (t, J = 7.2 Hz, 2H), 3.18 (q, J = 7.2 Hz, 6H), 3.93 (t, J = 7.2 Hz, 2H), 4.25–4.34 (m, 3H), 4.39 (m, 1H), 4.68 (t, J = 6.0 Hz, 1H), 6.09 (d, J = 6.0 Hz, 1H), 6.87–6.88 (m, 3H), 7.22–7.24 (m, 2H), 8.20 (s, 1H), 8.50 (s, 1H); 13C NMR (150 MHz, CD3OD): δ 9.0, 27.0, 27.3, 30.28, 30.36, 30.40, 30.45, 30.51, 30.53, 30.55, 40.1, 47.6, 68.87, 69.1, 72.1, 75.9, 84.3, 89.1, 115.3, 120.0, 121.3, 130.2, 141.0, 150.7, 153.7, 157.1, 160.4, 183.1; HRMS (ESI−) calcd for C28H39N6O8S [M − H]− 619.2556, found 619.2565 (error 1.4 ppm).
4.1.4.27. 5′-O-{N-[11-(Phenylthio)undecanoyl]sulfamoyl}adenosine Triethylammonium Salt (40).
The title compound was prepared from 40d using the general procedure for TFA deprotection (74%): Rf 0.1 (7:3 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.28 (t, J = 7.2 Hz, 9H), 1.20–1.30 (m, 13H), 1.36–1.39 (m, 2H), 1.55–1.60 (m, 5H), 2.19 (t, J = 7.2 Hz, 2H), 2.89 (t, J = 7.2 Hz, 2H), 3.18 (q, J = 7.2 Hz, 6H), 4.27–4.35 (m, 3H), 4.38–4.40 (m, 1H), 4.68 (t, J = 6.0 Hz, 1H), 6.09 (d, J = 6.0 Hz, 1H), 7.13–7.15 (m, 1H), 7.25–7.30 (m, 2H), 7.30–7.31 (m, 2H), 8.19 (s, 1H), 8.49 (s, 1H); 13C NMR (150 MHz, CD3OD): δ 9.2, 23.1, 27.4, 29.7, 30.21, 30.26, 30.52, 30.54, 34.29, 40.3, 47.8, 69.3, 72.3, 76.1, 84.5, 89.3, 119.5, 120.2, 126.7 (2C), 129.9, 130.1, 138.3, 141.2, 150.9, 153.9, 157.3, 183.2 (missing 1C in alkyl chain); HRMS (ESI−) calcd for C27H37N6O7S2 [M − H]− 621.2171, found 621.2145 (error 4.2 ppm).
4.1.4.28. 5′-O-{N-[11-(Phenylamino)undecanoyl]sulfamoyl}adenosine Triethylammonium Salt (41).
The title compound was prepared from 41d using the general procedure for TFA deprotection (89%): Rf 0.1 (7:3 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.29 (t, J = 7.2 Hz, 9H), 1.27–1.31 (m, 12H), 1.35–1.38 (m, 2H), 1.57–1.60 (m, 2H), 2.19 (t, J = 7.2 Hz, 2H), 3.04 (t, J = 7.2 Hz, 2H), 3.18 (q, J = 7.2 Hz, 6H), 4.27–4.34 (m, 3H), 4.38–4.39 (m, 1H), 4.68 (t, J = 6.0 Hz, 1H), 6.08 (d, J = 6.0 Hz, 1H), 6.59–6.63 (m, 3H), 7.07–7.10 (m, 2H), 8.20 (s, 1H), 8.50 (s, 1H); 13C NMR (150 MHz, CD3OD): δ 9.2, 23.7, 27.4, 28.3, 30.36, 30.52, 30.57, 30.61, 30.68, 40.3, 45.1, 47.7, 69.2, 72.3, 76.1, 84.5, 89.3, 114.1 (2C), 117.9, 129.9 (2C), 141.15, 141.16, 150.4, 150.8, 153.9, 157.3, 183.5; HRMS (ESI−) calcd for C27H38N7O7S [M − H]− 604.2559, found: 604.2551 (error 1.3 ppm).
4.1.4.29. 5′-O-{N-[11-(4-Fluorophenoxy)undecanoyl]sulfamoyladenosine Triethylammonium Salt (42).
The title compound was prepared from 42d using the general procedure for TFA deprotection (77%): Rf 0.1 (7:3 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.27 (t, J = 7.2 Hz, 9H), 1.26–1.32 (m, 10H), 1.40–1.45 (m, 2H), 1.57–1.61 (m, 2H), 1.70–1.74 (m, 2H), 2.19 (t, J = 7.2 Hz, 2H), 3.12 (q, J = 7.2 Hz, 6H), 3.91 (t, J = 7.2 Hz, 2H), 4.26–4.33 (m, 3H), 4.38–4.39 (m, 1H), 4.68 (t, J = 6.0 Hz, 1H), 6.09 (d, J = 6.0 Hz, 1H), 6.86–6.88 (m, 2H), 6.95–6.98 (m, 2H), 8.19 (s, 1H), 8.50 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 9.2, 27.1, 27.4, 30.4, 30.47, 30.52, 30.56, 30.58, 30.7, 40.2, 47.9, 69.2, 69.6, 72.4, 76.1, 84.6, 89.2, 116.5, (d, J = 23.2 Hz, 2C), 116.6 (d, J = 8.0 Hz, 2C), 120.3, 141.2, 150.9, 153.9, 156.9, 157.3, 159.3 (d, J = 234.9 Hz), 183.0; HRMS (ESI−) calcd for C27H36FN6O8S [M − H]− 623.2305, found 623.2278 (error 4.3 ppm).
4.1.4.30. 5′-O-{N-[11-(3-Fluorophenoxy)undecanoyl]sulfamoyladenosine Triethylammonium Salt (43).
The title compound was prepared from 43d using the general procedure for TFA deprotection (81%): Rf 0.1 (7:3 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.28 (t, J = 7.2 Hz, 9H), 1.26–1.30 (m, 10H), 1.39–1.44 (m, 2H), 1.56–1.60 (m, 2H), 1.70–1.74 (m, 2H), 2.19 (t, J = 7.2 Hz, 2H), 3.18 (q, J = 7.2 Hz, 6H), 3.93 (t, J = 7.2 Hz, 2H), 4.27–4.34 (m, 3H), 4.36–4.40 (m, 1H), 4.66 (t, J = 6.0 Hz, 1H), 6.09 (d, J = 6.0 Hz, 1H), 6.59–6.64 (m, 2H), 6.68–6.70 (m, 1H), 7.20–7.24 (m, 1H), 8.19 (s, 1H), 8.49 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 9.1, 26.9, 27.3, 30.1, 30.3, 30.37, 30.40, 30.43, 30.5, 40.1, 47.7, 69.1 (d, J = 11.8 Hz), 72.1, 75.9, 84.3, 89.1, 102.7 (d, J = 25.0 Hz), 107.8 (d, J = 21.5 Hz), 111.3 (d, J = 2.9 Hz), 120.0, 131.2 (d, J = 10.4 Hz), 141.0, 150.7, 153.7, 157.1, 162.0, 163.1 (d, J = 314.0 Hz), 165.7 183.1; HRMS (ESI−) calcd for C27H36FN6O8S [M − H]− 623.2305, found 623.2312 (error 1.1 ppm).
4.1.4.31. 5′-O-{N-[11-(2-Fluorophenoxy)undecanoyl]sulfamoyladenosine Triethylammonium Salt (44).
The title compound was prepared from 44d using the general procedure for TFA deprotection (83%): Rf 0.1 (7:3 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.29 (t, J = 7.2 Hz, 9H), 1.28–1.32 (m, 10H), 1.42–1.46 (m, 2H), 1.58–1.60 (m, 2H), 1.73–1.78 (m, 2H), 2.19 (t, J = 7.2 Hz, 2H), 3.19 (q, J = 7.2 Hz, 6H), 4.01 (t, J = 7.2 Hz, 2H), 4.27–4.34 (m, 3H), 4.38–4.39 (m, 1H), 4.68 (t, J = 6.0 Hz, 1H), 6.08 (d, J = 6.0 Hz, 1H), 6.86–6.89 (m, 1H), 7.03–7.07 (m, 3H), 8.20 (s, 1H), 8.48 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 9.3, 27.1, 27.5, 30.46, 30.55, 30.65, 30.67, 30.69, 30.75, 40.4, 47.9, 69.4, 70.5, 72.4, 76.2, 84.6, 89.4, 116.3, 116.9 (d, J = 18.4 Hz), 120.3, 122.1 (d, J = 6.9 Hz), 125.6 (d, J = 3.9 Hz), 141.3, 148.6 (d, J = 10.4 Hz), 150.9, 154.0, 154.5 (d, J = 242.7 Hz), 157.4, 183.3; HRMS (ESI−) calcd for C27H36FN6O8S [M − H]− 623.2305, found 623.2320 (error 2.4 ppm).
4.1.4.32. 5′-O-{N-[11-(4-Chlorophenoxy)undecanoyl]sulfamoyl} adenosine Triethylammonium Salt (45).
The title compound was prepared from 45d using the general procedure for TFA deprotection (82%): Rf 0.1 (7:3 EtOAc/MeOH);1H NMR (600 MHz, CD3OD) δ 1.30 (t, J = 7.2 Hz, 9H), 1.27–1.33 (m, 10H), 1.40–1.45 (m, 2H), 1.58–1.60 (m, 2H), 1.70–1.75 (m, 2H), 2.19 (t, J = 7.2 Hz, 2H), 3.19 (q, J = 7.2 Hz, 6H), 3.92 (t, J = 7.2 Hz, 2H), 4.27–4.34 (m, 3H), 4.38–4.39 (m, 1H), 4.68 (t, J = 6.0 Hz, 1H), 6.08 (d, J = 6.0 Hz, 1H), 6.86–6.88 (m, 2H), 7.21–7.23 (m, 2H), 8.20 (s, 1H), 8.50 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 9.2, 27.1, 27.4, 30.2, 30.3, 30.46, 30.51, 30.55, 30.57, 30.63, 40.2, 47.9, 69.28, 69.34, 72.3, 76.1, 84.5, 89.2, 117.0 (2C), 120.2, 130.2 (2C), 141.2, 150.9, 153.9, 157.3, 159.4, 183.1; HRMS (ESI−) calcd for C27H36ClN6O8S [M − H]− 639.2009, found 639.2019 (error 1.6 ppm).
4.1.4.33. 5′-O-{N-[11-(3-Chlorophenoxy)undecanoyl]sulfamoyl} adenosine Triethylammonium Salt (46).
The title compound was prepared from 46d using the general procedure for TFA deprotection (88%): Rf 0.1 (7:3 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.28 (t, J = 7.2 Hz, 9H), 1.27–1.31 (m, 10H), 1.40–1.45 (m, 2H), 1.57–1.62 (m, 2H), 1.71–1.75 (m, 2H), 2.20 (t, J = 7.2 Hz, 2H), 3.18 (q, J = 7.2 Hz, 6H), 3.93 (t, J = 7.2 Hz, 2H), 4.26–4.34 (m, 3H), 4.38–4.39 (m, 1H), 4.68 (t, J = 6.0 Hz, 1H), 6.09 (d, J = 6.0 Hz, 1H), 6.82–6.83 (m, 1H), 6.88–6.90 (m, 2H), 7.20–7.22 (m, 1H), 8.20 (s, 1H), 8.50 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 9.3, 27.1, 27.4, 30.3, 30.46, 30.52, 30.56, 30.58, 30.64, 40.3, 47.9, 69.26, 69.33, 72.3, 76.1, 84.5, 89.3, 114.1, 115.8, 120.2, 121.5, 131.4, 135.8, 141.2, 150.9, 153.9, 157.3, 161.5, 183.3; HRMS (ESI−) calcd for C27H36ClN6O8S [M − H]− 639.2009, found 639.1982 (error 4.2 ppm).
4.1.4.34. 5′-O-{N-[11-(2-Chlorophenoxy)undecanoyl]sulfamoyl}adenosine Triethylammonium Salt (47).
The title compound was prepared from 47d using the general procedure for TFA deprotection (80%): Rf 0.1 (7:3 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.29 (t, J = 7.2 Hz, 9H), 1.25–1.32 (m, 10H), 1.45–1.50 (m, 2H), 1.56–1.60 (m, 2H), 1.75–1.80 (m, 2H), 2.19 (t, J = 7.2 Hz, 2H), 3.18 (q, J = 7.2 Hz, 6H), 4.01 (t, J = 7.2 Hz, 2H), 4.27–4.34 (m, 3H), 4.38–4.39 (m, 1H), 4.68 (t, J = 6.0 Hz, 1H), 6.09 (d, J = 6.0 Hz, 1H), 6.86–6.89 (m, 1H), 7.01–7.02 (m, 1H), 7.21–7.23 (m, 1H), 7.31–7.33 (m, 1H), 8.20 (s, 1H), 8.49 (s, 1H); 13C NMR (150 MHz, CD3OD): δ 9.2, 27.1, 27.4, 30.2, 30.38, 30.45, 30.56, 30.63, 40.3, 47.8, 69.2, 70.1, 72.3, 76.1, 84.5, 89.2, 114.7, 120.2, 122.2, 123.8, 128.9, 131.1, 141.2, 150.8, 153.9, 156.0, 157.3, 183.3 (missing 1C in alkyl chain); HRMS (ESI−) calcd for C27H36ClN6O8S [M − H]− 639.2009, found 639.2004 (error 0.8 ppm).
4.1.4.35. 5′-O-{N-[11-(4-Bromophenoxy)undecanoyl]sulfamoyl} adenosine Triethylammonium Salt (48).
The title compound was prepared from 48d using the general procedure for TFA deprotection (72%): Rf 0.1 (7:3 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.29 (t, J = 7.2 Hz, 9H), 1.27–1.31 (m, 10H), 1.39–1.43 (m, 2H), 1.57–1.60 (m, 2H), 1.70–1.75 (m, 2H), 2.19 (t, J = 7.2 Hz, 2H), 3.19 (q, J = 7.2 Hz, 6H), 3.92 (t, J = 7.2 Hz, 2H), 4.28–4.36 (m, 3H), 4.38–4.39 (m, 1H), 4.68 (t, J = 6.0 Hz, 1H), 6.08 (d, J = 6.0 Hz, 1H), 6.81–6.84 (m, 2H), 7.34–7.37 (m, 2H), 8.20 (s, 1H), 8.48 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 9.1, 27.1, 27.3, 30.3, 30.42, 30.48, 30.52, 30.55, 30.61, 40.1, 47.7, 69.3, 69.4, 72.2, 76.0, 84.4, 89.3, 113.3 (2C), 117.5, 120.2, 133.2 (2C), 141.1, 150.8, 153.9, 157.3, 159.8, 182.9; HRMS (ESI−) calcd for C27H36BrN6O8S [M − H]− 683.1499, 685.1478, found 683.1500, 685.1483 (error 0.1 ppm).
4.1.4.36. 5′-O-{N-[11-(4-Trifluoromethylphenoxy)undecanoyl] sulfamoyl}adenosine Triethylammonium Salt (49).
The title compound was prepared from 49d using the general procedure for TFA deprotection (68%): Rf 0.1 (7:3 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.30 (t, J = 7.2 Hz, 9H), 1.27–1.33 (m, 10H), 1.42–1.48 (m, 2H), 1.57–1.62 (m, 2H), 1.74–1.79 (m, 2H), 2.20 (t, J = 7.2 Hz, 2H), 3.20 (q, J = 7.2 Hz, 6H), 4.02 (t, J = 7.2 Hz, 2H), 4.26–4.36 (m, 3H), 4.38–4.40 (m, 1H), 4.67 (t, J = 5.4 Hz, 1H), 6.08 (d, J = 5.8 Hz, 1H), 7.03 (d, J = 8.4 Hz, 2H), 7.55 (d, J = 8.4 Hz, 2H), 8.20 (s, 1H), 8.49 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 9.2, 27.1, 27.3, 30.4, 30.50, 30.52, 30.54, 30.56, 30.62, 40.2, 47.9, 69.3, 69.4, 72.3, 76.1, 84.5, 89.2, 115.7, 120.2, 123.4 (q, J = 32.4 Hz), 126.1 (q, J = 269.4 Hz), 127.9 (q, J = 3.6 Hz), 140.6, 141.2, 150.3, 150.9, 153.9, 157.3, 163.3, 182.9; HRMS (ESI−) calcd for C28H36F3N6O8S [M − H]− 673.2273, found 673.2260 (error 1.9 ppm).
4.1.4.37. 5′-O-{N-[11-(3-Trifluoromethylphenoxy)undecanoyl]sulfamoyladenosine Triethylammonium Salt (50).
The title compound was prepared from 50d using the general procedure for TFA deprotection (70%): Rf 0.1 (7:3 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.29 (t, J = 7.2 Hz, 9H), 1.28–1.33 (m, 10H), 1.42–1.47 (m, 2H), 1.58–1.60 (m, 2H), 1.74–1.79 (m, 2H), 2.19 (t, J = 7.2 Hz, 2H), 3.19 (q, J = 7.2 Hz, 6H), 4.00 (t, J = 7.2 Hz, 2H), 4.27–4.34 (m, 3H), 4.38–4.39 (m, 1H), 4.68 (t, J = 6.0 Hz, 1H), 6.08 (d, J = 6.0 Hz, 1H), 7.13–7.19 (m, 3H), 7.43 (t, J = 7.9 Hz, 1H), 8.20 (s, 1H), 8.50 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 9.2, 27.1, 27.5, 30.2, 30.46, 30.53, 30.56, 30.59, 30.64, 40.3, 47.8, 69.2, 69.4, 72.3, 76.1, 84.5, 89.2, 112.2 (q, J = 3.4 Hz), 117.9 (q, J = 3.7 Hz), 119.17, 120.2, 124.62 (q, J = 270.3 Hz), 131.4, 132.9 (q, J = 32.1 Hz), 141.1, 150.8, 153.9, 157.3, 160.9, 183.3; HRMS (ESI−) calcd for C28H36F3N6O8S [M − H]− 673.2273, found 673.2295 (error 3.3 ppm).
4.1.4.38. 5′-O-{N-[11-(2-Trifluoromethylphenoxy)undecanoyl]sulfamoyladenosine Triethylammonium Salt (51).
The title compound was prepared from 51d using the general procedure for TFA deprotection (61%): Rf 0.1 (7:3 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.29 (t, J = 7.2 Hz, 9H), 1.28–1.32 (m, 10H), 1.44–1.49 (m, 2H), 1.57–1.61 (m, 2H), 1.75–1.79 (m, 2H), 2.19 (t, J = 7.2 Hz, 2H), 3.18 (q, J = 7.2 Hz, 6H), 4.05 (t, J = 7.2 Hz, 2H), 4.27–4.35 (m, 3H), 4.38–4.39 (m, 1H), 4.68 (t, J = 6.0 Hz, 1H), 6.09 (d, J = 6.0 Hz, 1H), 7.00 (t, J = 7.6 Hz, 1H), 7.12 (d, J = 8.3 Hz, 1H), 7.51–7.54 (m, 2H), 8.20 (s, 1H), 8.50 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 9.2, 26.9, 27.4, 30.1, 30.2, 30.3, 30.48, 30.53, 30.6, 40.2, 47.8, 69.2, 69.6, 72.3, 76.1, 84.5, 89.2, 114.1, 119.6 (q, J = 29.8 Hz), 120.2, 120.8, 125.3 (q, J = 270.3 Hz), 127.7 (q, J = 5.7 Hz), 134.7, 141.1, 150.8, 153.9, 157.3, 158.5, 183.2; HRMS (ESI−) calcd for C28H36F3N6O8S [M − H]− 673.2273, found 673.2263 (error 1.5 ppm).
4.1.4.39. 5′-O-{N-[11-(4-Methoxyphenoxy)undecanoyl]sulfamoyl} adenosine Triethylammonium Salt (52).
The title compound was prepared from 52d using the general procedure for TFA deprotection (86%): Rf 0.1 (7:3 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.29 (t, J = 7.2 Hz, 9H), 1.27–1.32 (m, 10H), 1.39–1.44 (m, 2H), 1.57–1.62 (m, 2H), 1.68–1.73 (m, 2H), 2.20 (t, J = 7.2 Hz, 2H), 3.18 (q, J = 7.2 Hz, 6H), 3.73 (s, 3H), 3.88 (t, J = 7.2 Hz, 2H), 4.29–4.36 (m, 3H), 4.38–4.39 (m, 1H), 4.68 (t, J = 6.0 Hz, 1H), 6.08 (d, J = 6.0 Hz, 1H), 6.81–6.82 (m, 4H), 8.20 (s, 1H), 8.48 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 9.2, 27.1, 27.4, 30.49, 30.51, 30.53, 30.56, 30.59, 30.7, 40.3, 47.8, 56.1, 69.2, 69.6, 72.3, 76.1, 84.5, 89.2, 115.6 (2C), 116.5 (2C), 120.2, 141.2, 150.9, 153.9, 154.7, 155.2, 157.3, 183.2; HRMS (ESI−) calcd for C28H39N6O9S [M − H]− 635.2505, found 635.2476 (error 4.6 ppm).
4.1.4.40. 5′-O-{N-[11-(3-Methoxyphenoxy)undecanoyl]sulfamoyl} adenosine Triethylammonium Salt (53).
The title compound was prepared from 53d using the general procedure for TFA deprotection (67%): Rf 0.1 (7:3 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.28 (t, J = 7.2 Hz, 9H), 1.27–1.32 (m, 10H), 1.40–1.45 (m, 2H), 1.57–1.61 (m, 2H), 1.70–1.74 (m, 2H), 2.19 (t, J = 7.2 Hz, 2H), 3.17 (q, J = 7.2 Hz, 6H), 3.75 (s, 3H), 3.91 (t, J = 7.2 Hz, 2H), 4.26–4.34 (m, 3H), 4.38–4.39 (m, 1H), 4.68 (t, J = 6.0 Hz, 1H), 6.09 (d, J = 6.0 Hz, 1H), 6.43–6.44 (m, 1H), 6.46–6.48 (m, 2H), 7.11–7.14 (m, 1H), 8.20 (s, 1H), 8.50 (s, 1H); 13C NMR (150 MHz, CD3OD): δ 9.2, 27.1, 27.4, 30.4, 30.48, 30.52, 30.56, 30.58, 30.65, 40.2, 47.8, 55.6, 68.9, 69.3, 72.3, 76.1, 84.5, 89.2, 101.9, 107.1, 107.8, 120.2, 130.8, 141.1, 150.8, 153.9, 157.3, 161.8, 162.3, 183.1; HRMS (ESI−) calcd for C28H39N6O9S [M − H]− 635.2505, found 635.2475 (error 4.7 ppm).
4.1.4.41. 5′-O-{N-[11-(2-Methoxyphenoxy)undecanoyl]sulfamoyl}adenosine Triethylammonium Salt (54).
The title compound was prepared from 54d using the general procedure for TFA deprotection (74%): Rf 0.1 (7:3 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.30 (t, J = 7.2 Hz, 9H), 1.27–1.31 (m, 10H), 1.42–1.46 (m, 2H), 1.56–1.60 (m, 2H), 1.73–1.77 (m, 2H), 2.20 (t, J = 7.2 Hz, 2H), 3.20 (q, J = 7.2 Hz, 6H), 3.82 (s, 3H), 3.96 (t, J = 7.2 Hz, 2H), 4.29–4.36 (m, 3H), 4.37–4.38 (m, 1H), 4.67 (t, J = 6.0 Hz, 1H), 6.08 (d, J = 6.0 Hz, 1H), 6.87–6.88 (m, 2H), 6.91–6.94 (m, 2H), 8.20 (s, 1H), 8.49 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 9.2, 27.1, 27.4, 30.4, 30.48, 30.54, 30.57, 30.59, 30.7, 40.3, 47.8, 56.6, 69.2, 70.3, 72.3, 76.1, 84.5, 89.3, 113.6, 115.0, 120.2, 122.2, 122.3, 141.2, 150.1, 150.9, 151.0, 153.9, 157.3, 183.2; HRMS (ESI−) calcd for C28H39N6O9S [M − H]− 635.2505, found 635.2477 (error 4.4 ppm).
4.1.4.42. 5′-O-{N-[11-(Naphthalen-1-yloxy)undecanoyl]sulfamoyl}adenosine Triethylammonium Salt (55).
The title compound was prepared from 55d using the general procedure for TFA deprotection (66%): Rf 0.1 (7:3 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.27 (t, J = 7.2 Hz, 9H), 1.26–1.31 (m, 8H), 1.36–1.40 (m, 2H), 1.53–1.60 (m, 4H), 1.88–1.92 (m, 2H), 2.19 (t, J = 7.2 Hz, 2H), 3.14 (q, J = 7.2 Hz, 6H), 4.13 (t, J = 7.2 Hz, 2H), 4.26–4.33 (m, 3H), 4.38–4.39 (m, 1H), 4.68 (t, J = 6.0 Hz, 1H), 6.08 (d, J = 6.0 Hz, 1H), 6.86–6.87 (m, 1H), 7.33–7.39 (m, 2H), 7.41–7.46 (m, 2H), 7.76–7.77 (m, 1H), 8.20 (s, 1H), 8.20–8.21 (m, 1H), 8.50 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 9.1, 27.3, 27.4, 30.3, 30.46, 30.52, 30.54, 30.56, 30.63, 40.2, 47.8, 69.2, 69.2, 72.2, 76.0, 84.4, 89.3, 105.7, 120.2, 120.9, 122.9, 125.9, 127.0, 127.1, 127.2, 128.4, 135.9, 141.1, 150.8, 153.8, 156.0, 157.2, 183.2; HRMS (ESI−) calcd for C31H39N6O8S [M − H]− 655.2556, found 655.2529 (error 4.1 ppm).
4.1.4.43. 5′-O-{N-[11-(Naphthalen-2-yloxy)undecanoyl]sulfamoyl}adenosine Triethylammonium Salt (56).
The title compound was prepared from 56d using the general procedure for TFA deprotection (60%): Rf 0.1 (7:3 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.28 (t, J = 7.2 Hz, 9H), 1.27–1.38 (m, 10H), 1.46–1.51 (m, 2H), 1.57–1.61 (m, 2H), 1.78–1.83 (m, 2H), 2.19 (t, J = 7.2 Hz, 2H), 3.17 (q, J = 7.2 Hz, 6H), 4.06 (t, J = 7.2 Hz, 2H), 4.27–4.34 (m, 3H), 4.38–4.39 (m, 1H), 4.68 (t, J = 6.0 Hz, 1H), 6.08 (d, J = 6.0 Hz, 1H), 7.10–7.12 (m, 1H), 7.19–7.19 (m, 1H), 7.27–7.30 (m, 1H), 7.38–7.41 (m, 1H), 7.71–7.74 (m, 3H), 8.20 (s, 1H), 8.50 (s, 1H); 13C NMR (150 MHz, CD3OD): δ 9.2, 27.2, 27. 5, 30.40, 30.52, 30.54, 30.57, 30.60, 30.64, 30.68, 40.3, 47.8, 69.0, 69.2, 72.4, 76.1, 84.5, 89.2, 107.6, 119.9, 124.4, 127.2, 127.7, 128.5, 130.2, 130.3, 136.2, 141.1, 150.8, 153.8, 157.2, 158.5, 183.2; HRMS (ESI−) calcd for C31H39N6O8S [M − H]− 655.2556, found 655.2526 (error 4.6 ppm).
4.1.7. 5′-N-(N-(11-Phenoxyundecanoyl)sulfamoyl)-2′,3′-O-isopropylideneadenosine (82)
To a solution of 5′-deoxy-2′,3′-O-isopropylidene-5′-[(sulfamoyl)amino]adenosine 81 (0.42 mmol, 1.0 equiv, 160 mg) in DMF (4 mL) was added 11-phenoxyundecanoate N-hydroxysuccinimide 32c (0.63 mmol, 1.5 equiv, 233 mg) and cesium carbonate (1.26 mmol, 3.0 equiv, 405 mg). The reaction mixture was stirred at 23 °C for 16 h. The resulting mixture was filtered and the filtrate was concentrated under reduced pressure. The crude product was used in next step without further purification.
4.1.4.44. 5′-deoxy-5′-{N-[(11-Phenoxyundecanoyl)sulfamoyl] amino} adenosine Triethylammonium Salt (83).
The title compound was prepared from 82 using the general procedure for TFA deprotection (42% over 2 steps): Rf 0.3 (9:1 CH2Cl2/MeOH + 1% NEt3); 1H NMR (400 MHz, CD3OD) 1.28 (t, J = 7.3 Hz, 9H), 1.24–1.36 (m, 10H), 1.44 (m, 2H), 1.60 (m, 2H), 1.70–1.77 (m, 2H), 2.20 (t, J = 7.4 Hz, 2H), 3.18 (q, J = 7.3 Hz, 6H), 3.93 (t, J = 6.4 Hz, 2H), 4.25–4.36 (m, 3H), 4.39 (m, 1H), 4.69 (t, J = 5.6 Hz, 1H), 6.09 (d, J = 5.8 Hz, 1H), 6.85–6.91 (m, 3H), 7.20–7.26 (m, 2H), 8.20 (s, 1H), 8.50 (s, 1H); 13C NMR (101 MHz, CD3OD): δ 9.2, 27.1, 27.4, 30.42, 30.49, 30.52, 30.56, 30.59, 30.66, 40.2, 47.9, 68.9, 69.3, 72.4, 76.1, 84.6, 89.2, 115.5, 121.4, 121.3, 130.4, 141.1, 150.9, 153.9, 157.3, 160.6, 182.9; HRMS (ESI+) calcd for C27H39N7O7S [M + H]+ 606.2704, found 606.2709 (error 0.8 ppm).
4.1.8. 1-(11-Phenoxy)undecanol (84)
Lithium aluminum hydride (2.0 mmol, 2.0 equiv, 76 mg) was suspended in THF (8 mL) and cooled to 0°C. 11-Phenoxyundecanoic acid 32a (1.0 mmol, 1.0 equiv, 278 mg) was added in THF (2 mL). The reaction mixture was allowed to warm to 23 °C and stirred for 16 h. The reaction mixture was cooled to 0 °C and water (3 mL) was added slowly. The resulting slurry was filtered through Celite, dried (MgSO4) and concentrated under reduced pressure to yield the title compound (211 mg, 80%) as a white solid: Rf 0.25 (2:8 EtOAc/Hexanes); 1H NMR (600 MHz, CDCl3) δ 1.2–1.4 (m, 12H), 1.45 (p, J = 7.2 Hz, 2H), 1.56 (p, J = 7.2 Hz, 2H), 1.78 (p, J = 7.2 Hz, 2H), 3.63 (t, J = 7.2 Hz, 2H), 3.95 (t, J = 7.2 Hz, 2H), 6.89 (d, J = 7.2 Hz, 2H), 6.93 (t, J = 7.2 Hz, 1H), 7.27 (t, J = 7.2 Hz, 2H).
4.1.9. 11-Phenoxyundecyl sulfamate (85)
11-Phenoxyundecan-1-ol 84 (0.35 mmol, 1.0 equiv, 100 mg) was dissolved in THF (5 mL) and cooled to 0°C. Sodium hydride (60% in mineral oil, 0.36 mmol, 1.03 equiv, 14 mg) was added and the reaction was stirred for 10 minutes. Sulfamoyl chloride (0.36 mmol, 1.03 equiv, 41 mg) was added and the reaction was allowed to warm 23 °C and stirred for 16 h. Water (30 mL) was added to the reaction mixture and the mixture was extracted with EtOAc (3 × 20 ml). The combined organic extracts were washed with saturated aqueous NaCl (20 mL), dried (MgSO4) and concentrated under reduced pressure to yield the title compound (96 mg, 80%) as a colorless oil: Rf; 0.18 (2:8 EtOAc/Hexanes); 1H NMR (600 MHz, CD3OD) δ 1.2–1.4 (m, 12H), 1.45 (p, J = 7.2 Hz, 2H), 1.51 (p, J = 7.2 Hz, 1H), 1.69 (p, J = 7.2 Hz, 1H), 1.74 (p, J = 7.2 Hz, 2H), 3.52 (t, J = 7.2 Hz, 1H), 3.92 (t, J = 7.2 Hz, 2H), 4.09 (t, J = 7.2 Hz, 1H), 6.8–6.9 (m, 3H), 7.22 (t, J = 7.2 Hz, 2H); MS (ESI +) cald for C17H29NO4S [M + H]+ 344.2, found 344.2.
4.1.10. 11-Phenoxyundecyl 4-methylbenzenesulfonate (86)
11-Phenoxyundecan-1-ol 84 (2.0 mmol, 1.0 equiv, 528 mg), 4-dimethylaminopyridine (6.0 mmol, 3.0 equiv, 733 mg), and 4-toluenesulfonyl chloride (2.4 mmol, 1.2 equiv, 0.458 g) were dissolved in CH2Cl2 (20 mL) and allowed to stir for 16 h at 23 °C. The reaction mixture was diluted with additional CH2Cl2 (20 mL) and washed with water (30 mL), aqueous 1M HCl (30 mL), and saturated aqueous NaHCO3 (30 mL). The organic extract was dried (MgSO4) and concentrated under reduced pressure to yield the title compound: Rf; 0.45 (2:8 EtOAc/Hexanes); 1H NMR (600 MHz, CDCl3) δ 1.2–1.4 (m, 12H), 1.45 (p, J = 7.2 Hz, 2H), 1.63 (p, J = 7.2 Hz, 2H), 1.78 (p, J = 7.2 Hz, 2H), 2.45 (s, 3H), 3.53 (t, J = 6.8 Hz, 1H), 3.95 (t, J = 6.8 Hz, 2H), 4.02 (t, J = 6.8 Hz, 1H), 6.88–6.95 (m, 3H), 7.28 (t, J = 7.2 Hz, 2H), 7.34 (d, J = 8.0 Hz, 2H), 7.79 (d, J = 8.2 Hz, 2H).
4.1.11. N6,N6-bis(tert-Butoxycarbonyl)-5′-O-[N-(11-phenoxyundecyl)sulfamoyl]-2′,3′-O-isopropylideneadenosine (88)
To a solution of N6,N6-bis(tert-butoxycarbonyl)-2′,3′-O-isopropylidene-5′-O-sulfamoyladenosine 87 (1.0 mmol, 1.0 equiv, 386 mg) in DMF (25 mL) was added 11-phenoxyundecyl 4-methylbenzenesulfonate 86 (1.5 mmol, 1.5 equiv, 628 mg) and cesium carbonate (2.5 mmol, 2.5 equiv, 341 mg). The reaction mixture was stirred at 23°C for 16 h and then concentrated under reduced pressure. Purification by flash chromatography (0–10% CH2Cl2/MeOH) afforded the title compound (106 mg, 21%) as a white solid: Rf 0.5 (9:1 CH2Cl2/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.24–1.35 (m, 14H), 1.38 (s, 3H), 1.45 (p, J = 7.2 Hz, 2H), 1.56–1.58 (m, 18H), 1.59 (s, 3H), 1.74 (p, J = 7.2 Hz, 2H), 2.79 (m, 2H), 3.93 (t, J = 7.2 Hz, 2H), 4.21 (m, 1H), 4.24 (m, 1H), 4.48 (q, J = 3.0 Hz, 1H), 5.13 (dd, J = 6.0, 3.0, 1H), 5.44 (dd, J = 6.0, 3.0 Hz, 1H), 6.24 (d, J = 3.0 Hz, 1H), 6.85–6.89 (m, 3H), 7.22 (t, J = 7.2 Hz, 2H), 8.21 (s, 1H), 8.25 (s, 1H); MS (ESI+) calcd for C40H60N6O11S [M + H]+ 833.4, found 833.4.
4.1.12. 5′-O-[N-(11-Phenoxyundecyl)sulfamoyl]adenosine (89)
The title compound was prepared from 88 using the general procedure for TFA deprotection (97%): Rf 0.26 (9:1 CH2Cl2/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.28–1.35 (m, 12H), 1.40–1.47 (m, 4H), 1.74 (p, J = 7.2 Hz, 2H), 2.92 (t, J = 7.2 Hz, 2H), 3.94 (t, J = 7.2 Hz, 2H), 4.28–4.32 (m, 2H), 4.34–4.36 (m, 1H), 4.43 (t, J = 6.0 Hz, 1H), 4.68 (t, J = 6.0 Hz, 1H), 6.07 (d, J = 6.0 Hz, 1H), 6.85–6.90 (m, 3H), 7.23 (t, J = 7.2 Hz, 2H), 8.21 (s, 1H), 8.28 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 27.2, 28.2, 30.2, 30.4, 30.51, 30.54, 30.67, 30.70, 30.8, 45.1, 59.7, 68.9, 71.7, 77.2, 85.2, 95.3, 115.5 (2C), 121.2, 121.5, 130.4 (2C), 140.6, 140.9, 150.3, 158.7, 160.6; HRMS (ESI+) calcd for C27H40N6O7S [M + H]+ 593.2757, found 593.2756 (error 0.2 ppm).
4.1.13. N6,N6-bis(tert-Butoxycarbonyl)-5′-O-4-methylbenzenesulfonate-2′,3′-O-isopropylideneadenosine (91)
N6,N6-bis(tert-Butoxycarbonyl)-2′,3′-O-isopropylideneadenosine 90 (0.25 mmol, 1.0 equiv, 129 mg), 4-toluenesulfonyl chloride (0.31 mmol, 1.2 equiv, 59 mg), and 4-dimethylaminopyridine (0.76 mmol, 3.0 equiv, 93 mg) were dissolved in CH2Cl2 (8 mL) and stirred at 23 °C for 16 h. The reaction mixture was diluted with additional CH2Cl2 (22 mL) and washed with water (30 mL), aqueous 1M HCl (30 mL), and saturated aqueous NaHCO3 (30 mL). The organic extract was dried (MgSO4) and concentrated under reduced pressure to give the title compound (162 mg, >98%) as a white solid: Rf 0.44 (1:1 Hexanes/EtOAc); 1H NMR (600 MHz, CDCl3) δ 1.38 (s, 3H), 1.49 (s, 18H), 1.61 (s, 3H), 2.41 (s, 3H), 4.23 (qd, J = 10.8, 5.0 Hz, 2H), 4.46–4.69 (m, 1H), 5.04 (dd, J = 6.3, 3.1 Hz, 1H), 5.33 (dd, J = 6.3, 2.5 Hz, 1H), 6.14 (d, J = 3.0 Hz, 1H), 7.25 (d, J = 7.2 Hz, 2H), 7.65 (d, J = 7.2 Hz, 2H), 8.11 (s, 1H), 8.77 (s, 1H); MS (ESI+) cald for C30H39N5O10S [M + H]+ 662.2490, found 662.2496 (error 0.9 ppm).
4.1.14. N6,N6-bis(tert-Butoxycarbonyl)-5′-deoxy-5′-[(11-phenoxyundecyloxy)sulfonyl]amino-2′,3′-O-isopropylideneadenosine (92)
To a solution of N6,N6-bis(tert-butoxycarbonyl)-5′-O-4-methylbenzenesulfonate-2′,3′-O-isopropylideneadenosine 91 (0.23 mmol, 1.0 equiv, 149 mg) in DMF (10 mL) was added 11-phenoxyundecyl sulfamate 85 (0.28 mmol, 1.2 equiv, 96 mg) and cesium carbonate (0.69 mmol, 3.0 equiv, 225 mg). The reaction was stirred at 23°C for 16 h. The resulting mixture was heated to 50 °C for an additional 24 h. The resulting mixture was filtered and the filtrate was concentrated under reduced pressure. The crude product was used in next step without further purification.
4.1.15. 5′-Deoxy-5′-{[(11-phenoxyundecyloxy)sulfonyl]amino}adenosine (93)
The title compound was prepared from 92 using the general procedure for TFA deprotection (11% over 2 steps): Rf 0.4 (9:1 CH2Cl2/MeOH); 1H NMR (600 MHz, CD3OD) δ 1.24–1.36 (m, 12H), 1.45 (p, J = 7.2 Hz, 2H), 1.68 (p, J = 7.2 Hz, 2H), 1.74 (p, J = 7.2 Hz, 2H), 3.42 (d, J = 3.3 Hz, 1H), 3.94 (t, J = 6.4 Hz, 2H), 4.04–4.11 (m, 2H), 4.28–4.30 (m, 1H), 4.32 (dd, J = 5.4, 2.6 Hz, 1H), 4.82–4.84 (m, 1H), 5.90 (d, J = 7.2 Hz, 1H), 6.86–6.90 (m, 3H), 7.23 (t, J = 7.2 Hz, 2H), 8.21 (s, 1H), 8.24 (s, 1H); 13C NMR (125 MHz, (CD3)2SO) δ 25.1, 25.7, 28.3, 28.6, 28.8, 28.9, 29.0, 29.0, 29.1, 45.2, 67.4, 69.7, 71.3, 72.6, 83.3, 88.4, 114.5 (2C), 119.7, 120.4, 129.6 (2C), 140.6, 148.9, 152.4, 156.4, 158.8; HRMS (ESI+) calcd for C27H39N6O7S [M + H]+ 593.2752, found 593.2759 (error 1.2 ppm).
4.2. Minimum Inhibitory Concentration Assay
Minimum inhibitory concentrations (MICs) were determined in quadruplicate in GAST[71] or 7H9 broth base supplemented with 5g/L BSA fraction V, 0.8g/L NaCl, 0.05% Tyloxapol and the appropriate carbon source according to the broth microdilution method[72] using compounds from DMSO stock solutions or with control wells treated with an equivalent amount of DMSO. The carbon sources were either 0.05 mM sodium palmitate or 20 mM glycerol/10 mM glucose. All MIC experiments were performed in triplicate and used an initial cell density of 104-105 cells/assay in a volume of 100 μL and growth was monitored at 7 and 14 days. The strains used were Mtb H37Rv ATCC 27294 or a subset of previously reported clinical strains.[73]
4.3. Enzyme Kinetic Assay
Apparent Ki values were determined using a coupled continuous assay employing FAAL28 or FACL19, pyrophosphatase, and nucleoside phosphorylase under initial velocity conditions as described.[74] Reactions contained 0.45 μM FAAL28 or 0.167 μM FACL19 in a buffer of 50 mM Trizma·HCl pH 8.0, 2.5 mM ATP, 5 mM MgCl2, 0.5 mM DTT, 150 mM hydroxylamine pH 7, 0.1 U nucleoside phosphorylase, 0.04 U pyrophosphatase, 0.2 mM 7-methylthioguanosine (MesG), and 33 μM tetradecanoic acid (KM = 5.3 μM FAAL28) or 250 μM dodecanoic acid. Reactions were run in triplicate in 96-well half-area UV Star plates (Greiner) and the cleavage of MesG was monitored at A360 on a Molecular Devices Spectramax M5e plate reader. Kiapp values were determined by fitting the concentration-response plots to the Morrison equation since the inhibitors exhibited tight-binding behavior (Kiapp ≤ 100 × [E]).[75]
4.4. Cytotoxicity Assay
African green monkey Cercopithecus aethiops kidney cells (Vero, ATCC) cells were plated in 96-well plates at (2.5–5.0) × 104 cells per well (200 μL). Vero cells were maintained in minimum essential medium (MEM) supplemented with 5% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 μg/mL streptomycin. Compounds were prepared as 20 mM stock solutions in DMSO, and 1 μL of the compound stock solution was added to each well in 200 μL of MEM, yielding a final compound concentration of 100 μM. Control wells contained either 50% DMSO (negative control) or 0.5% DMSO (positive control), and all reactions were done in triplicate. The plate was incubated for 72 h at 37 °C in a 5% CO2/95% air humidified atmosphere. Measurement of cell viability was carried out using a modified method of Mosmann based on 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT).[76] MTT was prepared fresh at 1 mg/mL in serum-free, phenol red free RPMI 1640 media. MTT solution (200 μL) was added to each well, and the plate was incubated as described above for 3 h. The MTT solution was removed, and the formazan crystals were solubilized with 200 μL of isopropanol. The plate was read on a M5e spectrophotometer (Molecular Devices) at 570 nm for formazan and 650 nm for background subtraction. Cell viability was estimated as the percentage absorbance of sample relative to the DMSO control.
Highlights.
5′-O-[N-(alkanoyl)sulfamoyl]adenosine (alkanoyl adenosine monosulfamate, alkanoyl-AMS) analogues were synthesized and evaluated for their antitubercular activity.
11-Phenoxyundecanoyl-AMS 32 inhibits Mycobacterium tuberculosis with a minimum inhibitory concentration of 3 μM.
Compound 32 selectively inhibits the fatty acyl AMP ligase (FAAL28) over the fatty acyl CoA ligase (FACL19).
Acknowledgement
This research was supported by a grant from the Great Lakes Regional Center of Excellence (GLRCE, 2U54AI057153), the National Institutes of Health (AI136445) and funded (in part) by the Intramural Research Program of the NIAID, NIH.
Abbreviations
- TB
tuberculosis
- Mtb
Mycobacterium tuberculosis
- FadD
fatty acid adenylating enzyme
- FAAL
fatty acyl-AMP ligase
- FACL
fatty acyl-CoA ligase
- PKS
polyketide synthase
- PDIM
phthiocerol dimycocerosate
- FAS
fatty acid synthase
- MDR
multi-drug resistant
- XDR
extensively drug resistant
- SAR
structure-activity relationship
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].WHO 2019 Global Tuberculosis Report, World Health Organization: France, 2019; Chapter 3, pp 27–70. https://apps.who.int/iris/handle/10665/329368 (accessed April 4, 2020). [Google Scholar]
- [2].WHO 2019 consolidated guidelines on drug-resistant tuberculosis treatment, World Health Organization: Switzerland, 2019; pp 19–41. https://www.who.int/tb/publications/2019/consolidated-guidelines-drug-resistant-TB-treatment/en/ (accessed April 4, 2020). [PubMed] [Google Scholar]
- [3].Gokhale RS, Saxena P, Chopra T, Mohanty D, Versatile polyketide enzymatic machinery for the biosynthesis of complex mycobacterial lipids, Nat Prod Rep, 24 (2007) 267–277. [DOI] [PubMed] [Google Scholar]
- [4].Quadri LE, Biosynthesis of mycobacterial lipids by polyketide synthases and beyond, Critical reviews in biochemistry and molecular biology, 49 (2014) 179–211. [DOI] [PubMed] [Google Scholar]
- [5].Takayama K, Wang C, Besra GS, Pathway to synthesis and processing of mycolic acids in Mycobacterium tuberculosis, Clinical microbiology reviews, 18 (2005) 81–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Duckworth BP, Nelson KM, Aldrich CC, Adenylating Enzymes in Mycobacterium tuberculosis as Drug Targets, Current Topics in Medicinal Chemistry, 12 (2012) 766–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Grimes KD, Aldrich CC, A high-throughput screening fluorescence polarization assay for fatty acid adenylating enzymes in Mycobacterium tuberculosis, Analytical Biochemistry, 417 (2011) 264–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Jackson M, Stadthagen G, Gicquel B, Long-chain multiple methyl-branched fatty acid-containing lipids of Mycobacterium tuberculosis: biosynthesis, transport, regulation and biological activities, Tuberculosis (Edinb), 87 (2007) 78–86. [DOI] [PubMed] [Google Scholar]
- [9].Miyazawa T, Takahashi S, Kawata A, Panthee S, Hayashi T, Shimizu T, Nogawa T, Osada H, Identification of Middle Chain Fatty Acyl-CoA Ligase Responsible for the Biosynthesis of 2-Alkylmalonyl-CoAs for Polyketide Extender Unit, J Biol Chem, 290 (2015) 26994–27011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wilburn KM, Fieweger RA, VanderVen BC, Cholesterol and fatty acids grease the wheels of Mycobacterium tuberculosis pathogenesis, Pathogens and disease, 76 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Le NH, Molle V, Eynard N, Miras M, Stella A, Bardou F, Galandrin S, Guillet V, Andre-Leroux G, Bellinzoni M, Alzari P, Mourey L, Burlet-Schiltz O, Daffe M, Marrakchi H, Ser/Thr Phosphorylation Regulates the Fatty Acyl-AMP Ligase Activity of FadD32, an Essential Enzyme in Mycolic Acid Biosynthesis, J Biol Chem, 291 (2016) 22793–22805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Mohanty D, Sankaranarayanan R, Gokhale RS, Fatty acyl-AMP ligases and polyketide synthases are unique enzymes of lipid biosynthetic machinery in Mycobacterium tuberculosis, Tuberculosis (Edinb), 91 (2011) 448–455. [DOI] [PubMed] [Google Scholar]
- [13].Gokhale RS, Saxena P, Chopra T, Mohanty D, Versatile polyketide enzymatic machinery for the biosynthesis of complex mycobacterial lipids, Natural Product Reports, 24 (2007) 267–277. [DOI] [PubMed] [Google Scholar]
- [14].Arora P, Vats A, Saxena P, Mohanty D, Gokhale RS, Promiscuous fatty acyl CoA ligases produce acyl-CoA and acyl-SNAC precursors for polyketide biosynthesis, J Am Chem Soc, 127 (2005) 9388–9389. [DOI] [PubMed] [Google Scholar]
- [15].Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE 3rd, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Krogh A, McLean J, Moule S, Murphy L, Oliver K, Osborne J, Quail MA, Rajandream MA, Rogers J, Rutter S, Seeger K, Skelton J, Squares R, Squares S, Sulston JE, Taylor K, Whitehead S, Barrell BG, Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence, Nature, 393 (1998) 537–544. [DOI] [PubMed] [Google Scholar]
- [16].Trivedi OA, Arora P, Sridharan V, Tickoo R, Mohanty D, Gokhale RS, Enzymic activation and transfer of fatty acids as acyl-adenylates in mycobacteria, Nature, 428 (2004) 441–445. [DOI] [PubMed] [Google Scholar]
- [17].Arora P, Goyal A, Natarajan VT, Rajakumara E, Verma P, Gupta R, Yousuf M, Trivedi OA, Mohanty D, Tyagi A, Sankaranarayanan R, Gokhale RS, Mechanistic and functional insights into fatty acid activation in Mycobacterium tuberculosis, Nat Chem Biol, 5 (2009) 166–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Goyal A, Verma P, Anandhakrishnan M, Gokhale RS, Sankaranarayanan R, Molecular basis of the functional divergence of fatty acyl-AMP ligase biosynthetic enzymes of Mycobacterium tuberculosis, J Mol Biol, 416 (2012) 221–238. [DOI] [PubMed] [Google Scholar]
- [19].Lux MC, Standke LC, Tan DS, Targeting adenylate-forming enzymes with designed sulfonyladenosine inhibitors, The Journal of Antibiotics, 72 (2019) 325–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Casabon I, Crowe AM, Liu J, Eltis LD, FadD3 is an acyl-CoA synthetase that initiates catabolism of cholesterol rings C and D in actinobacteria, Mol Microbiol, 87 (2013) 269–283. [DOI] [PubMed] [Google Scholar]
- [21].Casabon I, Zhu SH, Otani H, Liu J, Mohn WW, Eltis LD, Regulation of the KstR2 regulon of Mycobacterium tuberculosis by a cholesterol catabolite, Mol Microbiol, 89 (2013) 1201–1212. [DOI] [PubMed] [Google Scholar]
- [22].Casabon I, Swain K, Crowe AM, Eltis LD, Mohn WW, Actinobacterial acyl coenzyme A synthetases involved in steroid side-chain catabolism, Journal of bacteriology, 196 (2014) 579–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wipperman MF, Sampson NS, Thomas ST, Pathogen roid rage: cholesterol utilization by Mycobacterium tuberculosis, Critical reviews in biochemistry and molecular biology, 49 (2014) 269–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wilbrink MH, Petrusma M, Dijkhuizen L, van der Geize R, FadD19 of Rhodococcus rhodochrous DSM43269, a steroid-coenzyme A ligase essential for degradation of C-24 branched sterol side chains., Applied and Environmental Microbiology, 77 (2011) 4455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Bragin EY, Shtratnikova VY, Dovbnya DV, Schelkunov MI, Pekov YA, Malakho SG, Egorova OV, Ivashina TV, Sokolov SL, Ashapkin VV, Donova MV, Comparative analysis of genes encoding key steroid core oxidation enzymes in fast-growing Mycobacterium spp. strains, The Journal of steroid biochemistry and molecular biology, 138 (2013) 41–53. [DOI] [PubMed] [Google Scholar]
- [26].Khare G, Gupta V, Gupta RK, Gupta R, Bhat R, Tyagi AK, Dissecting the role of critical residues and substrate preference of a Fatty Acyl-CoA Synthetase (FadD13) of Mycobacterium tuberculosis, PLoS One, 4 (2009) e8387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Sassetti CM, Boyd DH, Rubin EJ, Genes required for mycobacterial growth defined by high density mutagenesis, Mol Microbiol, 48 (2003) 77–84. [DOI] [PubMed] [Google Scholar]
- [28].Liu Z, Ioerger TR, Wang F, Sacchettini JC, Structures of Mycobacterium tuberculosis FadD10 protein reveal a new type of adenylate-forming enzyme, J Biol Chem, 288 (2013) 18473–18483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Belardinelli JM, Larrouy-Maumus G, Jones V, Sorio de Carvalho LP, McNeil MR, Jackson M, Biosynthesis and translocation of unsulfated acyltrehaloses in Mycobacterium tuberculosis, J Biol Chem, 289 (2014) 27952–27965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Ferreras JA, Stirrett KL, Lu X, Ryu JS, Soll CE, Tan DS, Quadri LE, Mycobacterial phenolic glycolipid virulence factor biosynthesis: mechanism and small-molecule inhibition of polyketide chain initiation, Chem Biol, 15 (2008) 51–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Simeone R, Leger M, Constant P, Malaga W, Marrakchi H, Daffe M, Guilhot C, Chalut C, Delineation of the roles of FadD22, FadD26 and FadD29 in the biosynthesis of phthiocerol dimycocerosates and related compounds in Mycobacterium tuberculosis, The FEBS journal, 277 (2010) 2715–2725. [DOI] [PubMed] [Google Scholar]
- [32].Samanta S, Singh A, Biswas P, Bhatt A, Visweswariah SS, Mycobacterial phenolic glycolipid synthesis is regulated by cAMP-dependent lysine acylation of FadD22, Microbiology (Reading, England), 163 (2017) 373–382. [DOI] [PubMed] [Google Scholar]
- [33].Lynett J, Stokes RW, Selection of transposon mutants of Mycobacterium tuberculosis with increased macrophage infectivity identifies fadD23 to be involved in sulfolipid production and association with macrophages, Microbiology (Reading, England), 153 (2007) 3133–3140. [DOI] [PubMed] [Google Scholar]
- [34].Díaz C, Pérez del Palacio J, Valero-Guillén PL, Mena García P, Pérez I, Vicente F, Martín C, Genilloud O, Sánchez Pozo A, Gonzalo-Asensio J, Comparative Metabolomics between Mycobacterium tuberculosis and the MTBVAC Vaccine Candidate, ACS infectious diseases, (2019). [DOI] [PubMed] [Google Scholar]
- [35].Koster K, Largen A, Foster JT, Drees KP, Qian L, Desmond EP, Wan X, Hou S, Douglas JT, Whole genome SNP analysis suggests unique virulence factor differences of the Beijing and Manila families of Mycobacterium tuberculosis found in Hawaii, PLOS ONE, 13 (2018) e0201146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Broset E, Martín C, Gonzalo-Asensio J, Evolutionary Landscape of the Mycobacterium tuberculosis Complex from the Viewpoint of PhoPR: Implications for Virulence Regulation and Application to Vaccine Development, mBio, 6 (2015) 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Vergnolle O, Chavadi SS, Edupuganti UR, Mohandas P, Chan C, Zeng J, Kopylov M, Angelo NG, Warren JD, Soll CE, Quadri LEN, Biosynthesis of Cell Envelope-Associated Phenolic Glycolipids in Mycobacterium marinum, Journal of bacteriology, 197 (2015) 1040–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Brodin P, Poquet Y, Levillain F, Peguillet I, Larrouy-Maumus G, Gilleron M, Ewann F, Christophe T, Fenistein D, Jang J, Jang MS, Park SJ, Rauzier J, Carralot JP, Shrimpton R, Genovesio A, Gonzalo-Asensio JA, Puzo G, Martin C, Brosch R, Stewart GR, Gicquel B, Neyrolles O, High content phenotypic cell-based visual screen identifies Mycobacterium tuberculosis acyltrehalose-containing glycolipids involved in phagosome remodeling, PLoS Pathog, 6 (2010) e1001100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Chuang PC, Chen HY, Jou R, Single-nucleotide polymorphism in the fadD28 gene as a genetic marker for East Asia Lineage Mycobacterium tuberculosis, J Clin Microbiol, 48 (2010) 4245–4247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Trivedi OA, Arora P, Vats A, Ansari MZ, Tickoo R, Sridharan V, Mohanty D, Gokhale RS, Dissecting the mechanism and assembly of a complex virulence mycobacterial lipid, Mol Cell, 17 (2005) 631–643. [DOI] [PubMed] [Google Scholar]
- [41].Infante E, Aguilar LD, Gicquel B, Pando RH, Immunogenicity and protective efficacy of the Mycobacterium tuberculosis fadD26 mutant, Clinical and experimental immunology, 141 (2005) 21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Sirakova TD, Fitzmaurice AM, Kolattukudy P, Regulation of expression of mas and fadD28, two genes involved in production of dimycocerosyl phthiocerol, a virulence factor of Mycobacterium tuberculosis, Journal of bacteriology, 184 (2002) 6796–6802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Menendez-Bravo S, Comba S, Sabatini M, Arabolaza A, Gramajo H, Expanding the chemical diversity of natural esters by engineering a polyketide-derived pathway into Escherichia coli, Metabolic engineering, 24 (2014) 97–106. [DOI] [PubMed] [Google Scholar]
- [44].Gavalda S, Leger M, van der Rest B, Stella A, Bardou F, Montrozier H, Chalut C, Burlet-Schiltz O, Marrakchi H, Daffe M, Quemard A, The Pks13/FadD32 crosstalk for the biosynthesis of mycolic acids in Mycobacterium tuberculosis, J Biol Chem, 284 (2009) 19255–19264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Leger M, Gavalda S, Guillet V, van der Rest B, Slama N, Montrozier H, Mourey L, Quemard A, Daffe M, Marrakchi H, The dual function of the Mycobacterium tuberculosis FadD32 required for mycolic acid biosynthesis, Chem Biol, 16 (2009) 510–519. [DOI] [PubMed] [Google Scholar]
- [46].Kuhn ML, Alexander E, Minasov G, Page HJ, Warwrzak Z, Shuvalova L, Flores KJ Wilson DJ, Shi C, Aldrich CC, Anderson WF, Structure of the Essential Mtb FadD32 Enzyme: A Promising Drug Target for Treating Tuberculosis, ACS infectious diseases, 2 (2016. 579–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Guillet V, Galandrin S, Maveyraud L, Ladeveze S, Mariaule V, Bon C, Eynard N, Daffe M, Marrakchi H, Mourey L, Insight into Structure-Function Relationships and Inhibition of the Fatty Acyl-AMP Ligase (FadD32) Orthologs from Mycobacteria, J Biol Chem, 291 (2016) 7973–7989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Zhang Z, Zhou R, Sauder JM, Tonge PJ, Burley SK, Swaminathan S, Structural and functional studies of fatty acyl adenylate ligases from E. coli and L. pneumophila, J Mol Biol, 406 (2011) 313–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Niu Y, Ge F, Yang Y, Ren Y, Li W, Chen G, Wen D, Liu F, Xiong L, Biochemical characterization of acyl-coenzyme A synthetases involved in mycobacterial steroid side-chain catabolism and molecular design: synthesis of an anti-mycobacterial agent, 3 Biotech, 9 (2019) 169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Fang C, Lee KK, Nietupski R, Bates RH, Fernandez-Menendez R, Lopez-Roman EM, Guijarro-Lopez L, Yin Y, Peng Z, Gomez JE, Fisher S, Barros-Aguirre D, Hubbard BK, Serrano-Wu MH, Hung DT, Discovery of heterocyclic replacements for the coumarin core of anti-tubercular FadD32 inhibitors, Bioorganic & medicinal chemistry letters, 28 (2018) 3529–3533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Kawate T, Iwase N, Shimizu M, Stanley SA, Wellington S, Kazyanskaya E, Hun DT, Synthesis and structure-activity relationships of phenyl-substituted coumarins with anti-tubercular activity that target FadD32, Bioorganic & medicinal chemistry letters, 23 (2013) 6052–6059. [DOI] [PubMed] [Google Scholar]
- [52].Stanley SA, Kawate T, Iwase N, Shimizu M, Clatworthy AE, Kazyanskaya E, Sacchettini JC, Ioerger TR, Siddiqi NA, Minami S, Aquadro JA, Grant SS, Rubin EJ, Hung DT, Diarylcoumarins inhibit mycolic acid biosynthesis and kill <em>Mycobacterium tuberculosis</em> by targeting FadD32, Proceedings of the National Academy of Sciences, 110 (2013) 11565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Rappe C, Cis-α,β-unsaturated acids: isocrotonic acid, Org. Synth, 53 (1973) 123–127. [Google Scholar]
- [54].Wiskur SL, Korte A, Fu GC, Cross-Couplings of Alkyl Electrophiles under “Ligandless” Conditions: Negishi Reactions of Organozirconium Reagents, Journal of the American Chemical Society, 126 (2004) 82–83. [DOI] [PubMed] [Google Scholar]
- [55].Coxon GD, Douglas JD, Minnikin DE, Facile synthesis of (Z)-tetracos-5-enoic acid and racemic cis-4-(2-octadecylcyclopropane-1-yl)-butanoic acid, Chemistry and Physics of Lipids, 126 (2003) 49–53. [DOI] [PubMed] [Google Scholar]
- [56].Charette AB, Juteau H, Lebel H, Molinaro C, Enantioselective Cyclopropanation of Allylic Alcohols with Dioxaborolane Ligands: Scope and Synthetic Applications, Journal of the American Chemical Society, 120 (1998) 11943–11952. [Google Scholar]
- [57].O'Neil EJ, DiVittorio KM, Smith BD, Phosphatidylcholine-Derived Bolaamphiphiles via Click Chemistry, Organic Letters, 9 (2007) 199–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Strawn LM, Martell RE, Simpson RU, Leach KL, Counsell RE, Iodoaryl analogues of dioctanoylglycerol and 1-oleoyl-2-acetylglycerol as probes for protein kinase C, Journal of medicinal chemistry, 32 (1989) 2104–2110. [DOI] [PubMed] [Google Scholar]
- [59].Yan J, Travis BR, Borhan B, Direct Oxidative Cleavage of α- and β-Dicarbonyls and α-Hydroxyketones to Diesters with KHSO5, The Journal of Organic Chemistry, 69 (2004) 9299–9302. [DOI] [PubMed] [Google Scholar]
- [60].Evans DA, Ennis MD, Mathre DJ, Asymmetric alkylation reactions of chiral imide enolates. A practical approach to the enantioselective synthesis of .alpha.-substituted carboxylic acid derivatives, Journal of the American Chemical Society, 104 (1982) 1737–1739. [Google Scholar]
- [61].Hermann C, Giammasi C, Geyer A, Maier ME, Syntheses of hapalosin analogs by solid-phase assembly of acyclic precursors, Tetrahedron, 57 (2001) 8999–9010. [Google Scholar]
- [62].Ghosh AK, Gong G, Total Synthesis and Revision of C6 Stereochemistry of (+)- Amphidinolide W, The Journal of Organic Chemistry, 71 (2006) 1085–1093. [DOI] [PubMed] [Google Scholar]
- [63].Mitsumori S, Tsuri T, Honma T, Hiramatsu Y, Okada T, Hashizume H, Inagaki M, Arimura A, Yasui K, Asanuma F, Kishino J, Ohtani M, Synthesis and Biological Activity of Various Derivatives of a Novel Class of Potent, Selective, and Orally Active Prostaglandin D2 Receptor Antagonists. 1. Bicyclo[2.2.1]heptane Derivatives, Journal of medicinal chemistry, 46 (2003) 2436–2445. [DOI] [PubMed] [Google Scholar]
- [64].Denmark SE, Chung W.-j., Lewis Base Activation of Lewis Acids: Catalytic, Enantioselective Addition of Glycolate-Derived Silyl Ketene Acetals to Aldehydes, The Journal of Organic Chemistry, 73 (2008) 4582–4595. [DOI] [PubMed] [Google Scholar]
- [65].Duckworth BP, Geders TW, Tiwari D, Boshoff HI, Sibbald PA, Barry CE 3rd, Schnappinger D, Finzel BC, Aldrich CC, Bisubstrate adenylation inhibitors of biotin protein ligase from Mycobacterium tuberculosis, Chem Biol, 18 (2011) 1432–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Bockman MR, Engelhart CA, Cramer JD, Howe MD, Mishra NK, Zimmerman M, Larson P, Alvarez-Cabrera N, Park SW, Boshoff HIM, Bean JM, Young VG, Ferguson DM, Dartois V, Jarrett JT, Schnappinger D, Aldrich CC, Investigation of (S)-(−)-Acidomycin: A Selective Antimycobacterial Natural Product That Inhibits Biotin Synthase, ACS infectious diseases, 5 (2019) 598–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Goyal A, Yousuf M, Rajakumara E, Arora P, Gokhale RS, Sankaranarayanan R, Crystallization and preliminary X-ray crystallographic studies of the N-terminal domain of FadD28, a fatty-acyl AMP ligase from Mycobacterium tuberculosis, Acta crystallographica. Section F, Structural biology and crystallization communications, 62 (2006) 350–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Ikeuchi H, Meyer ME, Ding Y, Hiratake J, Richards NGJ, A critical electrostatic interaction mediates inhibitor recognition by human asparagine synthetase, Bioorganic & Medicinal Chemistry, 17 (2009) 6641–6650. [DOI] [PubMed] [Google Scholar]
- [69].Somu RV, Boshoff H, Qiao C, Bennett EM, Barry CE, Aldrich CC, Rationally Designed Nucleoside Antibiotics That Inhibit Siderophore Biosynthesis of Mycobacterium tuberculosis, Journal of medicinal chemistry, 49 (2006) 31–34. [DOI] [PubMed] [Google Scholar]
- [70].Heacock D, Forsyth CJ, Shiba K, Musier-Forsyth K, Synthesis and Aminoacyl-tRNA Synthetase Inhibitory Activity of Prolyl Adenylate Analogs, Bioorganic Chemistry, 24 (1996) 273–289. [Google Scholar]
- [71].De Voss JJ, Rutter K, Schroeder BG, Su H, Zhu Y, Barry CE 3rd, The salicylate-derived mycobactin siderophores of Mycobacterium tuberculosis are essential for growth in macrophages, Proc Natl Acad Sci U S A, 97 (2000) 1252–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Domenech P, Reed MB, Barry CE 3rd, Contribution of the Mycobacterium tuberculosis MmpL protein family to virulence and drug resistance, Infect Immun, 73 (2005) 3492–3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Jeon CY, Hwang SH, Min JH, Prevots DR, Goldfeder LC, Lee H, Eum SY, Jeon DS, Kang HS, Kim JH, Kim BJ, Kim DY, Holland SM, Park SK, Cho SN, Barry CE 3rd, Via LE, Extensively drug-resistant tuberculosis in South Korea: risk factors and treatment outcomes among patients at a tertiary referral hospital, Clin Infect Dis, 46 (2008) 42–49. [DOI] [PubMed] [Google Scholar]
- [74].Wilson DJ, Aldrich CC, A continuous kinetic assay for adenylation enzyme activity and inhibition, Anal Biochem, 404 (2010) 56–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].e. Copeland RA, Evaluation of enzyme inhibitors in drug discovery., John Wiley & Sons, Inc., New Jersey, 2005. [Google Scholar]
- [76].Mosmann T, Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays, Journal of immunological methods, 65 (1983) 55–63. [DOI] [PubMed] [Google Scholar]