

Abstract
The application of gene therapy is widely expanding in research and continuously improving in preparation for clinical applications. The inner ear is an attractive target for gene therapy for treating environmental and genetic diseases in both the auditory and vestibular systems. With the lack of spontaneous cochlear hair cell replacement, hair cell regeneration in adult mammals is among the most important goals of gene therapy. In addition, correcting gene defects can open up a new era for treating inner ear diseases. The relative isolation and small size of the inner ear dictate local administration routes and carefully calculated small volumes of reagents. In the current review, we will cover effective timing, injection routes and types of vectors for successful gene delivery to specific target cells within the inner ear. Differences between research purposes and clinical applications are also discussed.
1. Introduction
The use of gene therapy in Otology may potentially address both genetic hearing loss and conditions that require regeneration of hair cells or neurons. To achieve these goals, we have to consider “where”, “when” and “how” to deliver specific gene(s) to the ear, and to select a therapeutic gene with the required efficacy. Several other chapters in this Special Issue of Hearing Research deal with specific diseases and targets for therapy. The main goal of this Review is to discuss practical aspect of HOW that can be accomplished. We first introduce target cells and tissues (WHERE), proceed to discussing timing for therapy (WHEN), and continue with a detailed consideration of the delivery routes, current outcomes with different types of vectors for each of these routes, side effect and complications and applicability to the human ear (HOW). Because our focus is on future clinical applications, we also attempt to summarize limitations and side effects, mostly covering work in mammals, primarily in mature ears, in vivo.
2. Where: Target cells or regions
Both hair cells and non-sensory cells are important targets for gene transfer (Figure 1). “Supporting cells” is a general term for non-sensory cells of the otocyst-derived epithelium that lines the scala media. Supporting cells in and around the organ of Corti are targeted because of their involvement in genetic deafness and their potential use for regenerative therapy. The most common gene related to non-syndromic hearing loss is GJB2, expressed in cochlear supporting cells (Del Castillo et al., 2017; Kelsell et al., 1997). In humans, the highest expression of GJB2 in normal ears is in sub-populations of supporting cells flanking the organ of Corti whereas pillar and Deiters cells appear devoid of immunoreactivity (Liu et al., 2017). Strong expression of CX26 was also found in the lateral wall area (Liu et al., 2009). However, in mice the level of CX26 in Deiters and pillar cells is higher than that found in humans, implicating a role in the active cochlea mechanism and making them a potential target for therapy (Zhu et al., 2015; Zong et al., 2017).
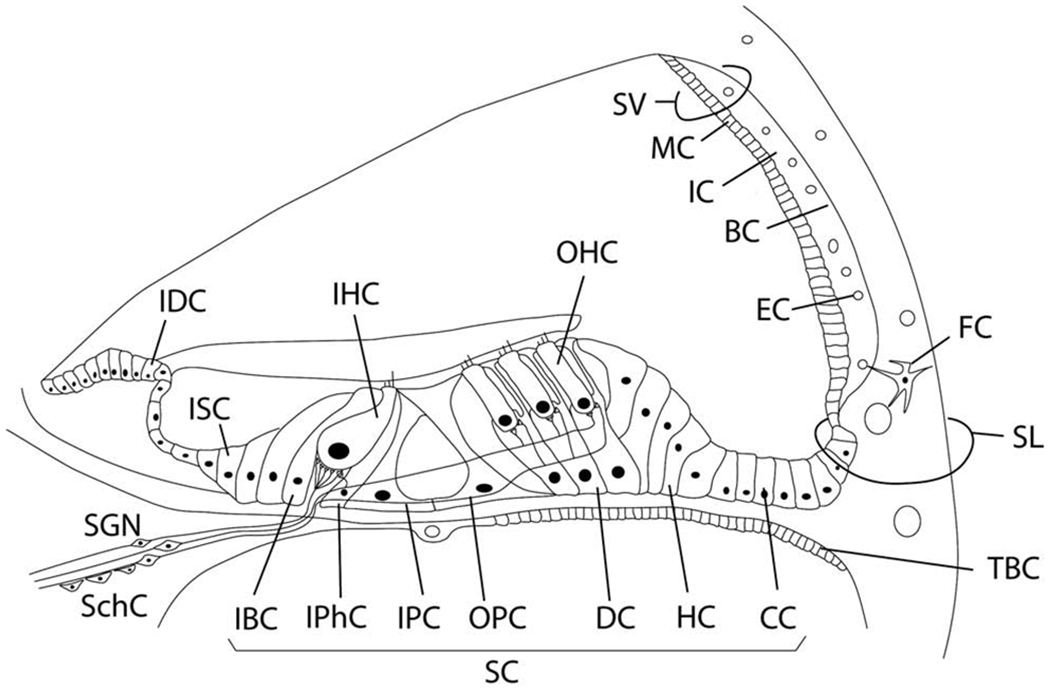
Figure 1. Target cells for the inner ear gene therapy.
IDC, Interdental cell; ISC, Inner sulcus cell; IHC, Inner hair cell; OHC, Outer hair cell; SV, Stria vacularis; MC, Marginal cell; IC, Intermediate cell; BC, Basal cell; EC, Endothelial cell; SL, Spiral ligament; FC, Fibrocyte; SGN, Spiral ganglion neuron; SchC, Schwann cell; IBC, Inner border cell; IPhC, Inner phalangeal cell; IPC, Inner pillar cell; OPC, Outer pillar cell; DC, Deiters cell; Hensen cell; CC, Claudius cell; SC, Supporting cell; TBC, tympanic border cells.
Supporting cells are also a target for hair cell regeneration via transdifferentiation, a phenotypic conversion that occurs spontaneously in the vestibular sensory epithelia but needs to be induced in the cochlea. Using only one gene, results can be inefficient (Kawamoto et al., 2003); but forced expression of combinatorial genes is being attempted for increasing the number of new hair cells (Minoda et al., 2007; Walters et al., 2017). As discussed below, adenovirus (AdV) vectors appear to be the most efficient gene transfer vehicles for transgene delivery into most types of supporting cells (Excoffon et al., 2006; Ishimoto et al., 2002; Venail et al., 2007).
Stria vascularis marginal cells are another important target. For instance, marginal cells are involved in cases of KCNQ1/KCNE1 mutations causing hearing loss (Knipper et al., 2006; Rivas et al., 2005). The presence of gap junctions in the stria vascularis, and the influence of mutations in these genes on endocochlear potential (EP) and hearing, make the strial cells important targets for gene therapy (Mei et al., 2017). Pannexin genes expressed in basal cells of the stria vascularis have also been linked to genetic deafness and may be a target for interventions via gene therapy (Zhao, 2016).
Hair cells are targets for gene transfer aimed at both genetic and environmental diseases. There are numerous genetic defects that affect hair cells, making them important targets for therapy (Table 1). Myosin VIIa is a motor protein in hair cells, essential for developing and arranging stereocilia bundles (Lefevre et al., 2008; Self et al., 1998) and related to Usher syndrome 1B. A mouse Myosin VIIa model showed disorganized stereocilia (Holme et al., 2002). Mutations in other myosins present in hair cells have been found in human families and modeled in mice, as reviewed by Rehman et al. (Rehman et al., 2016). The myosin genes are typically too large to load on AAV vectors, the choice vector for hair cell transduction, therefore gene therapy protocols will have to await future design of improved delivery vectors.
Table 1.
Target cells (or regions) for gene therapy related to human inner ear mutation. Genes that are not localized at present are excluded.
Several other hair cell genes involved in hearing loss are small enough to load into vectors for gene therapy approaches. The mutated transmembrane channel like 1 (TMC1) gene was identified in mammalian hereditary deafness. The TMC1 protein is a part of the mechanotransduction complex in inner hair cells and is essential for hearing (Corey et al., 2019). The feasibility of AAV mediated expression of exogenous Tmc genes for treating such mutations was shown by the Holt group (Askew et al., 2015) (Nist-Lund et al., 2019).
Mice lacking the vesicular glutamate transporter-3 (VGLUT3) are congenitally deaf due to loss of glutamate release at the inner hair cell afferent synapse. Cochlear delivery of VGLUT3 using adeno-associated virus type 1 (AAV1) leads to transgene expression in only inner hair cells (IHCs), despite broader viral uptake. Within 2 weeks of AAV1-VGLUT3 delivery, auditory brainstem response (ABR) thresholds normalize, along with partial rescue of the startle response. Lastly, partial reversal of the morphologic changes was seen within the afferent IHC ribbon synapse. These findings represent a successful restoration of hearing by gene replacement in mice, which is a significant advance toward gene therapy of human deafness (Akil et al., 2012). Other examples of gene transfer into the models for genetic deafness include mutations in whirlin (Chien et al., 2016; Isgrig et al., 2017), and harmonin (Pan et al., 2017).
As far as environmental hearing loss is concerned, hair cells can be targets for gene transfer for protection. For instance, patients who receive aminoglycosides therapy for cystic fibrosis were found, in some cases, to develop hearing loss (Zettner et al., 2018). Protective effects against hair cell loss due to aminoglycosides were demonstrated in animal models (Kawamoto et al., 2004) and may be advanced for use in cystic fibrosis patients. Similar approaches have been tried on animal models for cisplatinum ototoxicity (Chen et al., 2018a).
Other regions of the cochlea are also important., for instance, GJB2 is expressed not only in supporting cells in and around the organ of Corti, but also in fibrocytes and mesenchymal cells of the lateral wall, in basal and intermediate cells of the stria vascularis, and in spiral ganglion neurons (Iizuka et al., 2015; Lee et al., 2015; Liu et al., 2009; Liu et al., 2016; Mei et al., 2017; Yu et al., 2014). OtoGL is also expressed in multiple regions, such as interdental cell, tectorial membrane, hair cells, Claudius cells and spiral prominence (Yariz et al., 2012). Col4A3/4A4/4A5, which are genes related to Alport syndrome, are another example, being expressed at interdental cell, inner sulcus cell, basilar membrane and spiral ligament (Cosgrove et al., 1996).
As indicated by the data summarized above, several animal models are being employed for developing technology that would eventually be used in patient ears. The most commonly used animals are mice, because they serve as models for genetic deafness. However, mice ears are small and surgical procedures are complex. Some of the morphological analyses used for outcome analysis are also more challenging in the mouse ear compared to larger rodents. As such, guinea pigs (Komeda et al., 1999) rats (Venail et al., 2007) and other larger rodents serve as good models for gene therapy procedures for treating non-hereditary disease. Primates are also emerging as an important step prior to applying the therapy in the clinic (Maguire et al., in press, Hearing Research).
3. When: Time points for gene delivery
For hereditary hearing loss, the disease can be congenital or develop at a later time point, depending on the mutation. Early intervention will be best for most cases because the likelihood of rescue of cells and function is highest. Providing or preserving hearing is especially critical for early onset genetic hearing loss and genes involved in early development of the inner ear such as EYA1, CHD7, SIX1, ROR1 (Table 1), because development of language and other social and cognitive functions benefits from hearing. Progress in technology for in utero gene therapy may become applicable for treating early onset hereditary deafness. In mice, the cochlea is still developing and maturing after birth, facilitating rescue studies in immature ears (Akil et al., 2012; Akil et al., 2019; Askew et al., 2015; Iizuka et al., 2015; Nist-Lund et al., 2019)). Unlike mice, humans are born with mature cochleae and will likely require in utero gene therapy for treating developmental gene mutations (Gubbels et al., 2008; Wang et al., 2018a). Late onset hearing loss has longer time window for intervention. In general, diagnostic ability needs to improve to provide not only the identification of the mutated gene, but also an assessment of the condition and survival of the affected cells.
For acquired deafness due to environmental (non-genetic) factors, similar considerations apply as far as timing for the treatment. Because loss of hair cells due to overstimulation or aminoglycoside antibiotic treatment may lead to a flat epithelium, which is refractory to transdifferentiation (Izumikawa et al., 2008), early treatment would be preferable. However, the diagnostic tools currently available are insufficient for unequivocal determination of hair cell loss, its extent and location. In addition, some forms of hearing loss tend to improve over the first weeks after a trauma, and the expectation for this to occur is a contraindication for early gene transfer therapy.
4. How – Administration Routes
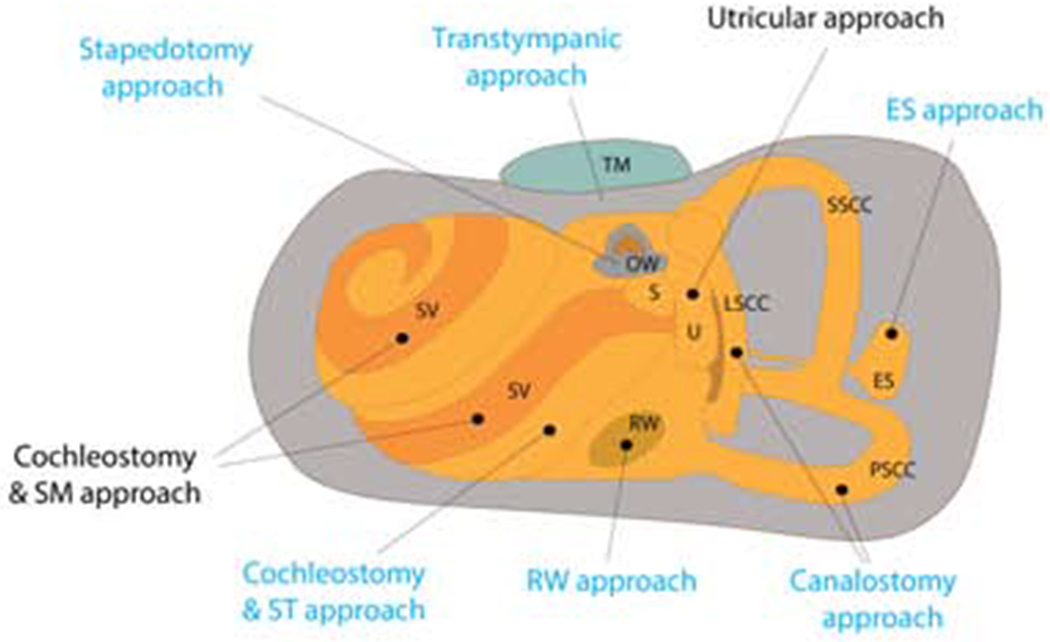
Choosing the right type of vector and right route of delivery will be important for both effective gene delivery and avoiding any off-target effects. Comparison between routes of gene delivery in human cochlea is summarized in table 2. Schematic representation of injection routes in rodent animal models and in humans is illustrated in figure 2.
Table. 2.
Routes of delivery to the human cochlea
| Route | Advantages | Disadvantages | Problems Mitigation |
|---|---|---|---|
| Systemic | Easy Procedure, Minimally Invasive, Procedure itself does not pose any risk to residual hearing | High risk for off-target spread, Difficult to preserve the therapeutic dose due to neutralizing antibodies, blood clearance of viral particles, and potential immune response of the recipient | Increase viral targeting specificity, reversibly modulating blood-labyrinth barrier permeability |
| Intra Cochlear (via Round or, Oval Windows, and via cochleostomy) | Delivery directly to the inner ear brings viruses to perilymph (scala tympani-via RW) or directly into endolymph (scala media – via OW). Viruses can transfect hair cells or supporting cells directed by their tropism. | Invasive procedures requiring full anesthesia. Risk of residual hearing loss. Risk of virus dissemination to the cerebellum and contralateral inner ear via the cochlear aqueduct or systematically through temporal bone marrow or via hematogenous spread. Viral concentration might be very high locally and not spread efficiently throughout the entire cochlea. | If the injection is made carefully residual hearing can be preserved, Virus spread via the cochlear aqueduct is less likely in adults, so the choice of the patient’s age should be considered. Viral injection in the cochlear fluids has to be very slow or used only if the target is located near the injection site. |
| Canalostomy via transmastoid surgery | Allows access to all three semicircular canals | A Very invasive procedure without established advantages over the intracochlear surgery | Demonstrate clear superiority of transmastoid viral delivery over the intracochlear injections. |
| Trans Tympanic | An easy and short procedure, minimally-invasive, with local anesthesia, already performed in humans | The concentration of the virus in the inner ear is poorly controlled due to the variable entry rate or imperviousness of the RW, to different viruses. | Increase the permeability of the RW using different compounds. Replace the viral particle with nanoparticle or liposome with stable RW permeation capability. |
Figure 2. Routes for gene delivery in rodent animal models and the human cochlea.
Sites in black font are currently only possible in rodents. OW, Oval window; ES, Endolymphatic sac; SM, Scala media; RW, Round window; ST, scala tympani; SV, Stria vascularis; S, Saccule; TM, tympanic membrane; U, Utricle; LSCC, Lateral semicircular canal; SSCC, Superior semicircular canal; PSCC, Posterior semicircular canal.
Systemic injection will be the least invasive delivery method possible. However, the blood-labyrinth barrier would reduce or restrict transfer into the cochlea (Nyberg et al., 2019). This barrier can be modulated somewhat by reagents such as diuretics, mannitol or by noise (Nyberg et al., 2019; Wu et al., 2014) but the safety of such manipulations and their utility for gene delivery are not yet established. In rats, the blood labyrinth barrier is not fully matured until postnatal day 14 (Suzuki et al., 1999), facilitating research on developing ears. rAAV2/9 injected intravenously to P1 neonatal mouse resulted in transgene expression in inner hair cells, spiral ganglion neurons and vestibular hair cells (Shibata et al., 2017). Shibata and colleagues (Shibata et al., 2017) experimented with systemic delivery of viruses to the wild type murine model and reported that the successful transduction of inner ear sensory epithelium depends on the dose, tropism of the viral serotype and the age of the recipient.
The major disadvantage of the systemically delivered virus-based genetic treatment is the potential for off-target effects. That said, a systemic route of administration for a viral-mediated genetic therapy has great promise for some types of disease processes. For example, a current clinical trial focus on the use of adeno-associated virus (AAV) serotypes and systemic intravenous injections to obtain global transduction of muscle tissue in an attempt to treat spinal muscle atrophy (SMA) (ClinicalTrials.gov Identifier: NCT02122952), and Duchenne muscular dystrophy (Duan, 2018) (ClinicalTrials.gov Identifiers: NCT03368742; NCT03375164; NCT03362502) as well as the AAV-mediated gene therapy for hemophilia A (Pasi et al., 2020) (ClinicalTrials.gov Identifier: NCT02576795). The success of the viral-based treatment depends among other factors on the optimal dose of the viral particles injected. The importance of this is recognized in clinical studies that use the determination of the viral dose as the primary outcome measure. So far, no serious adverse effects have been noted at various stages of these trials.
The systemic delivery of viral vectors to specifically target inner ear will require increased targeting specificity. This might be achieved by identification of specific serotypes whose natural, or genetically modified tropism will narrow the range of the target organs.
Improved viral targeting specificity for cochlear treatment will not address other limitations of systemic delivery. The blood-labyrinth barrier, neutralizing antibodies, blood clearance of viral particles, and the recipient’s immune-response will also have to be addressed, as reviewed in-depth by Duan (Duan, 2016).
Intracochlear delivery involves injecting materials into the cochlear fluids, endolymph or perilymph. The potential risks and negative effect of some of these procedures is summarized in Table 3 and explained in further detail below.
Table 3.
Negative clinical outcomes.
| Route | Potential negative outcomes | Disadvantages |
|---|---|---|
| Systemic | Viral dose and patient dependent Immune response. Other organs, besides the cochlea might also be targeted. | Difficult to preserve the therapeutic dose due to neutralizing antibodies, blood clearance of viral particles, and potential immune response of the recipient |
| Intra Cochlear (via Round or, Oval Windows, and via cochleostomy) | Loss of residual hearing, due to injection evoked mechanical damage to HCs. Full anesthesia related complications. Potential infection of the CNS. | Risk of virus dissemination to the cerebellum and contralateral inner ear via the cochlear aqueduct or systematically through temporal bone marrow or via hematogenous spread. Viral concentration might be very high locally and not spread efficiently throughout the entire cochlea. |
| Canalostomy via transmastoid surgery | Benign paroxysmal positional vertigo, deep vein thrombosis (Xie et al., 2017) | An invasive procedure used to treat severe Superior semicircular canal dehiscence. |
| Trans Tympanic | Post-injection pain, tongue numbness, transient dizziness, tinnitus, small persistent perforation of the tympanic membrane | The concentration of the virus in the inner ear is poorly controlled due to the variable entry rate or imperviousness of the RW, to different viruses. |
The oval window (OW) is readily visualized with a transcanal or transmastoid approach in humans; but in mice, the shape of the bulla and its position relative to the cochlea make the round window (RW) easier to visualize than the oval window. In addition, the RW is membraneous and thus is more easily penetrated than the bony stapes footplate in the oval window. Consequently, the RW approach is used to perform intracochlear surgery in many in vivo animal models. For instance, AAVs injected into perilymph via the RW transfected hair cells and effectively treated mutations of Vglut3 (Akil et al., 2012) and TMC1 (Nist-Lund et al., 2019). AAVs also can reach and transfect other cell types including supporting cells via RW injection (Isgrig et al., 2019), (Tan et al., 2019). Moreover, RW injection can preserve residual hearing with both AdV and AAV (Iizuka et al., 2008).
One consideration for RW injection is that virus can disseminate to the cerebellum and contralateral inner ear through the cochlear aqueduct which connects the perilymph with the CSF (Kho et al., 2000; Stover et al., 2000). This may be less of a problem in humans where the flow in the aqueduct is likely restricted or absent (Gopen et al., 1997; Rask-Andersen et al., 1977). Vector can also disseminate systematically through temporal bone marrow or via hematogenous spread (Kho et al., 2000).
Intracochlear gene delivery in humans can be achieved using an approach through the ear canal similar to that commonly used in stapedotomy for otosclerosis. The procedure is performed using a microscope with the patient under a general anesthetic, though potential use of a local and/or sedative type anesthetic could be considered. After the patient is anesthetized, the tympanic membrane is elevated allowing access to the middle ear space and lateral face of the otic capsule. A small hole can then be made in the footplate of the stapes using a drill or laser to enable access to the perilymph of the vestibule which is close to and in direct communication with the perilymph of the scala vestibuli. The only current gene therapy clinical trial in humans utilizes this approach to achieve intra-labyrinthine infusion of a virus carrying the basic helix-loop-helix transcription factor Atonal 1 (Atoh1), which speaks to the feasibility of this route for gene delivery. This trial aims to evaluate the safety, and tolerability of the tested agent, and its potential ability to improve hearing in humans (ClinicalTrials.gov Identifier: NCT02132130) however the results thereof have not yet been reported.
As an alternative to stapes footplate injection, the same transcanal surgical approach could be taken to allow access to the scala tympani through incision of the round window membrane. Occasionally overhanging round window niche bone and/or membranous duplications can present minor though manageable impediments to this approach. Alternatively, the scala tympani or scala vestibuli can be approached through a cochleostomy where precise drilling of otic capsule bone adjacent to the round window membrane would enable injection into these spaces. Surgical placement of a cochleostomy can be challenging and potentially traumatic, particularly for the scala vestibuli, therefore making a cochleostomy approach less ideal for intracochlear gene delivery.
Intracochlear delivery in humans, whether into the scala vestibuli or scala tympani, might be complicated by a number of factors. First, the fluids of the cochlea do not flow at any significant rate, although they may contribute to drug distribution if drug application can be extended to days (Salt et al., 2009). Therefore, a concentration gradient of viral solution during the single injection into the basal region might remain very high locally and may never reach the apical region at therapeutic concentrations. One solution to this would be to create a second fenestration to allow for displacement of perilymph upon injection so that the length of the cochlear duct could be perfused rather than simply injected. For example, if injecting virus into the stapes footplate one could incise the round window membrane to allow for efflux of displaced perilymph upon injection. While this solution might produce more rapid and uniform distribution of gene therapy agents along the length of the cochlear duct, trauma associated with the procedure may worsen any residual hearing that may be present.
Another factor that might complicate intracochlear delivery would be the potential for spread of an injected gene therapy agent through the cochlear aqueduct. This bony channel filled with cerebrospinal fluid enters scala tympani at the basal turn and connects to the subarachnoid space of the posterior cranial cavity at its opposite end. Anatomical analysis of the normal human cochlear aqueduct performed in the temporal bones of humans at ages from zero to one hundred years concluded that significant flow of fluid through the cochlear aqueduct would be unlikely (Gopen et al., 1997). That said, in children whose cochlear aqueducts are patent (Bachor et al., 1999), and also potentially in certain cases of adults, the injected gene therapy agent might migrate through the cochlear aqueduct away from the cochlea and transduce off-target cells in the brain and the contralateral ear, as seen in animal studies (Kho et al., 2000) (Stover et al., 2000). In addition to the dilution leading to reduced titer, flow to the CSF might result in off-target effects of the transgene on cells in the CNS.
One additional method that should be mentioned for intracochlear gene delivery would be delivery through a cochlear implant electrode array. Indeed cochlear implantation along with a drug for a one-time intracochlear delivery has been described clinically (Bento et al., 2016). Scala tympani is a preferred location for the cochlear implant insertion, and the electrode is generally inserted through the round window or via a cochleostomy (O’Connell et al., 2016). Gene delivery could be paired with cochlear implantation by injecting gene vectors through the electrode array port, or by insertion of an array wrapped in a porous medium impregnated with the vector. Both methods could provide access to the perilymph in the basal and middle turns of the cochlea. However apical access would be still limited as electrode arrays are not inserted that far along the cochlear duct. Cochlear implants procedures can be combined with delivery of plasmid or other gene vectors, with electrode-derived current driving electroporation of the genetic material into target cells (Pinyon et al., 2014; Pinyon et al., 2019).
Transtympanic approaches are widely used in human for drug delivery due to relative ease of its procedure. This route of administration relies on absorption of the injected agent from the middle ear to the inner ear through the RW. However, the RW is a three-layered membrane allowing only small molecules to diffuse, and gene vectors are too large. Attempts to increase the RW permeability were partly successful (Shibata et al., 2012).
The advantage of the transtympanic method of delivery stems from the relatively short, simple procedure and local, rather than general, anesthesia needed. The procedure can be performed during an office visit and with only topical anesthetic applied to the tympanic membrane. A small volume (0.5 – 1 milliliter) of the treatment solution is injected into the middle ear through a small needle that punctures the tympanic membrane. Ultimately, the round window membrane comes to contact with the injected solution, and the patient is positioned to minimize drug loss through the Eustachian tube for some time. The three-layer round window membrane permeability, however, might create variability between patients in the rate at which a drug solution enters the perilymph. Hahn and colleagues demonstrated variability between animals in the entry rate of applied compounds (Hahn et al., 2006).
Furthermore, the round window membrane may not be permeable to certain viruses (Jero et al., 2001). The inconsistency of the transtympanic delivery may be somewhat mitigated by increasing permeability of the round window membrane with different compounds such as by local anesthetics, histamine or healon (Chandrasekhar et al., 2000; Shibata et al., 2012). Perhaps in the future of gene therapy, viral vectors could be replaced by nanoparticles, and/or liposomes as vectors which may traverse the round window membrane more readily than viral vectors after transtympanic delivery (Maeda et al., 2007).
Canalostomy refers to the delivery of an agent into a semicircular canal and is a useful approach in rodent models and feasible in humans via transmastoid surgery. Various types of AAV infected inner hair cells and outer hair cells by posterior semicircular canal injection in mouse (Tao et al., 2018). Several serotypes of AAV could also preserve auditory functions by this way. Outcomes of combining RW and canalostomy showed enhanced gene expression (Yoshimura et al., 2018). While a mastoidectomy approach in humans allows for access to all three semicircular canals, the superiority of gene delivery via a canalostomy over round or oval window injections would need to be well established prior to this approach being realistically adopted in humans given the increased invasiveness of a transmastoid approach versus a transcanal approach and the risk of side effect such as those described following superior canal dehiscence repair (Xie et al., 2017).
Scala Media Cochleostomy:
In humans, the entire cochlea is embedded in the temporal bone and not visible to the surgeon, with the exception of the promontory, which is the most viable option for creation of a cochleostomy. In theory, access to the endolymph in the scala media could be achieved in humans through a precise cochleostomy on the promontory. That said, given the small size of the scala media, precise targeting of a cochleostomy to allow for injection of this space would likely be extremely challenging. In contrast, more of the rodent cochlea protrudes into the middle ear cavity facilitating access to perilymph as well as scala media endolymph. Thinner bone found in animal models, compared to humans, also allows visualization of the pigmented stria vascularis, providing a landmark for locating the scala media. The latter is significant for research purposes because for AdV to infect supporting cells in and around the organ of Corti, it needs to be injected into scala media (Excoffon et al., 2006; Ishimoto et al., 2002; Venail et al., 2007). Because the human ear does not provide such visible access to scala media injections, AdV cannot be easily delivered to supporting cells through the scala media, and therefore treatments focused on transdifferentiation of supporting cells to hair cells may need to await availability of appropriate vectors or sophisticated surgical technology.
5. How – Types of vectors to use
Adenovirus (AdV).
AdV can infect both dividing and non-dividing cells which makes its feasible for targeting terminally differentiated cells like HCs, supporting cells and inner ear neurons as well as epithelial progenitor cells. The most commonly used serotypes are 2 and 5 (Ahmed et al., 2017). Compared to other gene vectors, first and second generation AdV have the advantage of being able to carry larger genes (up to 14kb), but the disadvantage of the potential to provoke an immune reaction. Transgene expression peaks rapidly several days after injection and lasts for several months. Third generation AdV (gutted Ad) shows minimal immunogenicity and has an even larger cargo capacity of up to 36kb (Alba et al., 2005). Transgene expression was detected up to a year after injection in case of CNS (Soudais et al., 2004; Zou et al., 2000). AdV injection into scala media of mouse neonatal cochlea transduced mostly supporting cells however, cellular specificity depended on AdV vectors and the cochlear turn. Sometimes the inner and outer hair cells were transduced (Shu et al., 2016a) as well. Perilymphatic injection in adult cochleae showed transduction only of cells lining the perilymphatic space (mesothelial cells of scala tympani, scala vestibuli, basilar membrane and Reissner’s membrane) (Venail et al., 2007). Endolymphatic perfusion resulted in infection of Hensen, Deiters, pillar and phalangeal cells in the auditory epithelium and the satellite cells around spiral ganglion neurons. Venail et al. also showed that when forced injection into scala tympani caused membrane rupture, some inner and outer hair cells also were infected.
AAV has several advantages over AdV including longer gene expression and lower immune response and toxicity, and is becoming the vector of choice for gene therapy in several fields. After penetrating the host cell, AAV can either persist in a stable episome form or integrate into the host genome (Yang et al., 1999). One disadvantage of AAV is its small size: with a capsid diameter of 20-25 nm, it has a limited gene cargo capacity on only 4.8kb (Ahmed et al., 2017). For delivering larger genes, additional techniques such as a dual AAV strategy are needed (Akil et al., 2019). Inner ear transduction efficiency of several serotypes was evaluated in multiple studies, especially those targeting hair cells, as described above. AAV1, 2, 4, 5, 6, 7, and 8 can infect spiral limbus, spiral ligament and spiral ganglion cells. AAV1, 2, 3, 5, 6, and 8 are able to transduce genes into inner hair cells (Ahmed et al., 2017). AAV5 and 6.2 were able to infect supporting cells (Iizuka et al., 2015; Shu et al., 2016b). Recently, viral engineering techniques have been used to develop highly efficient AAVs: Exo-AAV, Anc80L65, AAV9-PHP.B, AAV2.7m8 and AAV-ie were reported to show high efficiency transduction into inner and outer hair cells. These vectors were also able to transfect some supporting cells, especially after injection to immature cochleae (Gyorgy et al., 2019; Isgrig et al., 2019; Landegger et al., 2017; Tan et al., 2019). So far, transfection restricted supporting cell has not been reported with any AAV, as recently reviewed (Martin et al., 2017). The ability to transfect supporting cells exclusively can only be accomplished using AdV vectors, but scala media delivery is needed.
Other viruses.
Lentivirus can also infect non-dividing cells and carry foreign DNA up to 8kb. After injection into scala media, it could infect marginal cells of stria vascularis and organ of Corti, and spiral ganglion neurons (Wei et al., 2013); however, its efficacy for transfecting hair cells was very limited (Pietola et al., 2008; Wang et al., 2013)). There were also trials with Sendai virus and Herpes virus vectors for inner ear gene delivery (Derby et al., 1999; Kanzaki et al., 2007). Safety of using these vectors remains to be addressed.
Other approaches for gene delivery.
Cationic lipid was used for Cas9-guide RNA-lipid complexes targeting the Tmc1 delivery by scala media injection. It successfully edited the genome and reduced progressive hearing loss (Gao et al., 2018). Electroporation can be used for gene transfer into cells of embryonic otocysts (Brigande et al., 2009). Plasmid electroporation driven by cochlear implant electrodes was used to deliver BDNF and GFP in guinea pig and stimulated survival and sprouting of spiral ganglion neurons (Pinyon et al., 2014).
6. Conclusions
The increasing interest in developing gene based therapy for hearing loss, combined with advances in genetic engineering, seems poised to deliver viable treatments in the future, with real potential for the pursuit of clinical trials. Our review of the delivery routes indicates the limits to which animal studies can improve surgical methods of delivery in a human patient. These limits are based on the anatomical differences between rodent and human ears, and the level of acceptable invasiveness of a surgical procedure. Nonetheless, animal models continue to be useful in the search for cell type specific genetic vectors, optimization of viruses to decrease their immunogenicity, increase transduction efficiency and regulate duration of gene expression. Better understanding of the differences between species will streamline the process of translation from lab to clinic, including customization of the chosen delivery vectors, specific cell types to be targeted, surgical route and the gene load to meet clinical standards for efficacy and safety.
Acknowledgements
We thank Donald Swiderski for assistance with the manuscript. This work was supported by the R. Jamison and Betty Williams Professorship and NIH-NIDCD Grants R01-DC01634, R01-DC015809 R01-DC014832, R01-DC013912, R01-DC009410 and R01-DC014456 and R01-DC013912.
Disclosures
SG is on the advisory boards for the Cystic Fibrosis Foundation, Applied Genetic Technologies Corporation and Roche. He also receives research support without financial remuneration from Med-El Corporation and is a consultant for Cochlear Corporation and Sirocco Therapeutics. YR is on the advisory board of Applied Genetic Technologies Corporation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Citations
- Abdelhak S, Kalatzis V, Heilig R, Compain S, Samson D, Vincent C, Weil D, Cruaud C, Sahly I, Leibovici M, Bitner-Glindzicz M, Francis M, Lacombe D, Vigneron J, Charachon R, Boven K, Bedbeder P, Van Regemorter N, Weissenbach J, Petit C 1997. A human homologue of the Drosophila eyes absent gene underlies branchio-oto-renal (BOR) syndrome and identifies a novel gene family. Nat Genet 15, 157–64. [DOI] [PubMed] [Google Scholar]
- Ahmed H, Shubina-Oleinik O, Holt JR 2017. Emerging Gene Therapies for Genetic Hearing Loss. J Assoc Res Otolaryngol 18, 649–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akil O, Seal RP, Burke K, Wang C, Alemi A, During M, Edwards RH, Lustig LR 2012. Restoration of hearing in the VGLUT3 knockout mouse using virally mediated gene therapy. Neuron 75, 283–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akil O, Dyka F, Calvet C, Emptoz A, Lahlou G, Nouaille S, Boutet de Monvel J, Hardelin JP, Hauswirth WW, Avan P, Petit C, Safieddine S, Lustig LR 2019. Dual AAV-mediated gene therapy restores hearing in a DFNB9 mouse model. Proc Natl Acad Sci U S A 116, 4496–4501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alba R, Bosch A, Chillon M 2005. Gutless adenovirus: last-generation adenovirus for gene therapy. Gene Ther 12 Suppl 1, S18–27. [DOI] [PubMed] [Google Scholar]
- Askew C, Rochat C, Pan B, Asai Y, Ahmed H, Child E, Schneider BL, Aebischer P, Holt JR 2015. Tmc gene therapy restores auditory function in deaf mice. Science translational medicine 7, 295ra108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avenarius MR, Jung JY, Askew C, Jones SM, Hunker KL, Azaiez H, Rehman AU, Schraders M, Najmabadi H, Kremer H, Smith RJH, Geleoc GSG, Dolan DF, Raphael Y, Kohrman DC 2018. Grxcr2 is required for stereocilia morphogenesis in the cochlea. PLoS One 13, e0201713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azaiez H, Decker AR, Booth KT, Simpson AC, Shearer AE, Huygen PL, Bu F, Hildebrand MS, Ranum PT, Shibata SB, Turner A, Zhang Y, Kimberling WJ, Cornell RA, Smith RJ 2015. HOMER2, a stereociliary scaffolding protein, is essential for normal hearing in humans and mice. PLoS Genet 11, e1005137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachor E, Byahatti S, Karmody CS 1999. New aspects in the histopathology of the cochlear aqueduct in children. Am J Otol 20, 612–20. [PubMed] [Google Scholar]
- Beisel KW, Nelson NC, Delimont DC, Fritzsch B 2000. Longitudinal gradients of KCNQ4 expression in spiral ganglion and cochlear hair cells correlate with progressive hearing loss in DFNA2. Brain Res Mol Brain Res 82, 137–49. [DOI] [PubMed] [Google Scholar]
- Ben Said M, Grati M, Ishimoto T, Zou B, Chakchouk I, Ma Q, Yao Q, Hammami B,Yan D, Mittal R, Nakamichi N, Ghorbel A, Neng L, Tekin M, Shi XR, Kato Y, Masmoudi S, Lu Z, Hmani M, Liu X 2016. A mutation in SLC22A4 encoding an organic cation transporter expressed in the cochlea strial endothelium causes human recessive non-syndromic hearing loss DFNB60. Hum Genet 135, 513–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bento RF, Danieli F, Magalhaes AT, Gnansia D, Hoen M 2016. Residual Hearing Preservation with the Evo(R) Cochlear Implant Electrode Array: Preliminary Results. Int Arch Otorhinolaryngol 20, 353–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brigande JV, Gubbels SP, Woessner DW, Jungwirth JJ, Bresee CS 2009. Electroporation-mediated gene transfer to the developing mouse inner ear. Methods Mol Biol 493, 125–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrasekhar SS, Rubinstein RY, Kwartler JA, Gatz M, Connelly PE, Huang E, Baredes S 2000. Dexamethasone pharmacokinetics in the inner ear: comparison of route of administration and use of facilitating agents. Otolaryngol Head Neck Surg 122, 521–8. [DOI] [PubMed] [Google Scholar]
- Chen DY, Liu XF, Lin XJ, Zhang D, Chai YC, Yu DH, Sun CL, Wang XL, Zhu WD, Chen Y, Sun LH, Wang XW, Shi FX, Huang ZW, Yang T, Wu H 2017. A dominant variant in DMXL2 is linked to nonsyndromic hearing loss. Genet Med 19, 553–558. [DOI] [PubMed] [Google Scholar]
- Chen H, Xing Y, Xia L, Chen Z, Yin S, Wang J 2018a. AAV-mediated NT-3 overexpression protects cochleae against noise-induced synaptopathy. Gene Ther 25, 251–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P, He L, Pang X, Wang X, Yang T, Wu H 2016. NLRP3 Is Expressed in the Spiral Ganglion Neurons and Associated with Both Syndromic and Nonsyndromic Sensorineural Deafness. Neural Plast 2016, 3018132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Xu K, Xie L, Cao HY, Wu X, Du AN, He ZH, Lin X, Sun Y, Kong WJ 2018b. The spatial distribution pattern of Connexin26 expression in supporting cells and its role in outer hair cell survival. Cell Death Dis 9, 1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien WW, Isgrig K, Roy S, Belyantseva IA, Drummond MC, May LA, Fitzgerald TS, Friedman TB, Cunningham LL 2016. Gene Therapy Restores Hair Cell Stereocilia Morphology in Inner Ears of Deaf Whirler Mice. Mol Ther 24, 17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corey DP, Akyuz N, Holt JR 2019. Function and Dysfunction of TMC Channels in Inner Ear Hair Cells. Cold Spring Harb Perspect Med 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosgrove D, Samuelson G, Pinnt J 1996. Immunohistochemical localization of basement membrane collagens and associated proteins in the murine cochlea. Hear Res 97, 54–65. [PubMed] [Google Scholar]
- Cryns K, Thys S, Van Laer L, Oka Y, Pfister M, Van Nassauw L, Smith RJ, Timmermans JP, Van Camp G 2003. The WFS1 gene, responsible for low frequency sensorineural hearing loss and Wolfram syndrome, is expressed in a variety of inner ear cells. Histochem Cell Biol 119, 247–56. [DOI] [PubMed] [Google Scholar]
- Del Castillo FJ, Del Castillo I 2017. DFNB1 Non-syndromic Hearing Impairment: Diversity of Mutations and Associated Phenotypes. Front Mol Neurosci 10, 428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derby ML, Sena-Esteves M, Breakefield XO, Corey DP 1999. Gene transfer into the mammalian inner ear using HSV-1 and vaccinia virus vectors. Hearing Research 134, 1–8. [DOI] [PubMed] [Google Scholar]
- Diaz-Horta O, Abad C, Sennaroglu L, Foster J 2nd, DeSmidt A, Bademci G, Tokgoz-Yilmaz S, Duman D, Cengiz FB, Grati M, Fitoz S, Liu XZ, Farooq A, Imtiaz F, Currall BB, Morton CC, Nishita M, Minami Y, Lu Z, Walz K, Tekin M. 2016. ROR1 is essential for proper innervation of auditory hair cells and hearing in humans and mice. Proc Natl Acad Sci U S A 113, 5993–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaudy F, Snoeckx R, Pfister M, Zenner HP, Blin N, Di Stazio M, Ferrara A, Lanzara C, Ficarella R, Declau F, Pusch CM, Nurnberg P, Melchionda S, Zelante L, Ballana E, Estivill X, Van Camp G, Gasparini P, Savoia A 2004. Nonmuscle myosin heavy-chain gene MYH14 is expressed in cochlea and mutated in patients affected by autosomal dominant hearing impairment (DFNA4). Am J Hum Genet 74, 770–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan D 2016. Systemic delivery of adeno-associated viral vectors. Curr Opin Virol 21, 16–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan D 2018. Systemic AAV Micro-dystrophin Gene Therapy for Duchenne Muscular Dystrophy. Mol Ther 26, 2337–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durruthy-Durruthy R, Sperry ED, Bowen ME, Attardi LD, Heller S, Martin DM 2018. Single Cell Transcriptomics Reveal Abnormalities in Neurosensory Patterning of the Chd7 Mutant Mouse Ear. Front Genet 9, 473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Excoffon KJ, Avenarius MR, Hansen MR, Kimberling WJ, Najmabadi H, Smith RJ, Zabner J 2006. The Coxsackievirus and Adenovirus Receptor: A new adhesion protein in cochlear development. Hear Res 215, 1–9. [DOI] [PubMed] [Google Scholar]
- Frenz S, Rak K, Volker J, Jurgens L, Scherzad A, Schendzielorz P, Radeloff A, Jablonka S, Hansen S, Mlynski R, Hagen R 2015. Mosaic pattern of Cre recombinase expression in cochlear outer hair cells of the Brn3.1 Cre mouse. Neuroreport 26, 309–13. [DOI] [PubMed] [Google Scholar]
- Gagnon LH, Longo-Guess CM, Berryman M, Shin JB, Saylor KW, Yu H, Gillespie PG, Johnson KR 2006. The chloride intracellular channel protein CLIC5 is expressed at high levels in hair cell stereocilia and is essential for normal inner ear function. J Neurosci 26, 10188–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Wang Q, Dong C, Chen S, Qi Y, Liu Y 2015. Whole Exome Sequencing Identified MCM2 as a Novel Causative Gene for Autosomal Dominant Nonsyndromic Deafness in a Chinese Family. PLoS One 10, e0133522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Tao Y, Lamas V, Huang M, Yeh WH, Pan B, Hu YJ, Hu JH, Thompson DB, Shu Y, Li Y, Wang H, Yang S, Xu Q, Polley DB, Liberman MC, Kong WJ, Holt JR, Chen ZY, Liu DR 2018. Treatment of autosomal dominant hearing loss by in vivo delivery of genome editing agents. Nature 553, 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giese APJ, Tang YQ, Sinha GP, Bowl MR, Goldring AC, Parker A, Freeman MJ, Brown SDM, Riazuddin S, Fettiplace R, Schafer WR, Frolenkov GI, Ahmed ZM 2017. CIB2 interacts with TMC1 and TMC2 and is essential for mechanotransduction in auditory hair cells. Nat Commun 8, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girotto G, Abdulhadi K, Buniello A, Vozzi D, Licastro D, d’Eustacchio A, Vuckovic D, Alkowari MK, Steel KP, Badii R, Gasparini P 2013. Linkage study and exome sequencing identify a BDP1 mutation associated with hereditary hearing loss. PLoS One 8, e80323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopen Q, Rosowski JJ, Merchant SN 1997. Anatomy of the normal human cochlear aqueduct with functional implications. Hear Res 107, 9–22. [DOI] [PubMed] [Google Scholar]
- Grati M, Chakchouk I, Ma Q, Bensaid M, Desmidt A, Turki N, Yan D, Baanannou A, Mittal R, Driss N, Blanton S, Farooq A, Lu Z, Liu XZ, Masmoudi S 2015. A missense mutation in DCDC2 causes human recessive deafness DFNB66, likely by interfering with sensory hair cell and supporting cell cilia length regulation. Hum Mol Genet 24, 2482–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillet N, Schwander M, Hildebrand MS, Sczaniecka A, Kolatkar A, Velasco J, Webster JA, Kahrizi K, Najmabadi H, Kimberling WJ, Stephan D, Bahlo M, Wiltshire T, Tarantino LM, Kuhn P, Smith RJ, Muller U 2009. Mutations in LOXHD1, an evolutionarily conserved stereociliary protein, disrupt hair cell function in mice and cause progressive hearing loss in humans. Am J Hum Genet 85, 328–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubbels SP, Woessner DW, Mitchell JC, Ricci AJ, Brigande JV 2008. Functional auditory hair cells produced in the mammalian cochlea by in utero gene transfer. Nature 455, 537–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyorgy B, Meijer EJ, Ivanchenko MV, Tenneson K, Emond F, Hanlon KS, Indzhykulian AA, Volak A, Karavitaki KD, Tamvakologos PI, Vezina M, Berezovskii VK, Born RT, O’Brien M, Lafond JF, Arsenijevic Y, Kenna MA, Maguire CA, Corey DP 2019. Gene Transfer with AAV9-PHP.B Rescues Hearing in a Mouse Model of Usher Syndrome 3A and Transduces Hair Cells in a Non-human Primate. Molecular therapy. Methods & clinical development 13, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn H, Kammerer B, DiMauro A, Salt AN, Plontke SK 2006. Cochlear microdialysis for quantification of dexamethasone and fluorescein entry into scala tympani during round window administration. Hear Res 212, 236–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holme RH, Steel KP 2002. Stereocilia defects in waltzer (Cdh23), shaker1 (Myo7a) and double waltzer/shaker1 mutant mice. Hear Res 169, 13–23. [DOI] [PubMed] [Google Scholar]
- Horn HF, Brownstein Z, Lenz DR, Shivatzki S, Dror AA, Dagan-Rosenfeld O, Friedman LM, Roux KJ, Kozlov S, Jeang KT, Frydman M, Burke B, Stewart CL, Avraham KB 2013. The LINC complex is essential for hearing. J Clin Invest 123, 740–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iizuka T, Kamiya K, Gotoh S, Sugitani Y, Suzuki M, Noda T, Minowa O, Ikeda K 2015. Perinatal Gjb2 gene transfer rescues hearing in a mouse model of hereditary deafness. Hum Mol Genet 24, 3651–61. [DOI] [PubMed] [Google Scholar]
- Iizuka T, Kanzaki S, Mochizuki H, Inoshita A, Narui Y, Furukawa M, Kusunoki T, Saji M, Ogawa K, Ikeda K 2008. Noninvasive in vivo delivery of transgene via adeno-associated virus into supporting cells of the neonatal mouse cochlea. Hum Gene Ther 19, 384–90. [DOI] [PubMed] [Google Scholar]
- Isgrig K, McDougald DS, Zhu J, Wang HJ, Bennett J, Chien WW 2019. AAV2.7m8 is a powerful viral vector for inner ear gene therapy. Nat Commun 10, 427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isgrig K, Shteamer JW, Belyantseva IA, Drummond MC, Fitzgerald TS, Vijayakumar S, Jones SM, Griffith AJ, Friedman TB, Cunningham LL, Chien WW 2017. Gene Therapy Restores Balance and Auditory Functions in a Mouse Model of Usher Syndrome. Mol Ther 25, 780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimoto S, Kawamoto K, Kanzaki S, Raphael Y 2002. Gene transfer into supporting cells of the organ of Corti. Hear Res 173, 187–97. [DOI] [PubMed] [Google Scholar]
- Izumikawa M, Batts SA, Miyazawa T, Swiderski DL, Raphael Y 2008. Response of the flat cochlear epithelium to forced expression of Atoh1. Hear Res 240, 52–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaworek TJ, Richard EM, Ivanova AA, Giese AP, Choo DI, Khan SN, Riazuddin S, Kahn RA, Riazuddin S 2013. An alteration in ELMOD3, an Arl2 GTPase-activating protein, is associated with hearing impairment in humans. PLoS Genet 9, e1003774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jero J, Mhatre AN, Tseng CJ, Stern RE, Coling DE, Goldstein JA, Hong K, Zheng WW, Hoque AT, Lalwani AK 2001. Cochlear gene delivery through an intact round window membrane in mouse. Hum Gene Ther 12, 539–48. [DOI] [PubMed] [Google Scholar]
- Kammerer R, Ruttiger L, Riesenberg R, Schauble C, Krupar R, Kamp A, Sunami K, Eisenried A, Hennenberg M, Grunert F, Bress A, Battaglia S, Schrewe H, Knipper M, Schneider MR, Zimmermann W 2012. Loss of mammal-specific tectorial membrane component carcinoembryonic antigen cell adhesion molecule 16 (CEACAM16) leads to hearing impairment at low and high frequencies. J Biol Chem 287, 21584–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanzaki S, Shiotani A, Inoue M, Hasegawa M, Ogawa K 2007. Sendai virus vector-mediated transgene expression in the cochlea in vivo. Audiol Neurootol 12, 119–26. [DOI] [PubMed] [Google Scholar]
- Kawamoto K, Ishimoto S, Minoda R, Brough DE, Raphael Y 2003. Math1 gene transfer generates new cochlear hair cells in mature guinea pigs in vivo. J Neurosci 23, 4395–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamoto K, Sha SH, Minoda R, Izumikawa M, Kuriyama H, Schacht J, Raphael Y 2004. Antioxidant gene therapy can protect hearing and hair cells from ototoxicity. Mol Ther 9, 173–81. [DOI] [PubMed] [Google Scholar]
- Kawashima Y, Geleoc GS, Kurima K, Labay V, Lelli A, Asai Y, Makishima T, Wu DK, Della Santina CC, Holt JR, Griffith AJ 2011. Mechanotransduction in mouse inner ear hair cells requires transmembrane channel-like genes. J Clin Invest 121,4796–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelsell DP, Dunlop J, Stevens HP, Lench NJ, Liang JN, Parry G, Mueller RF, Leigh IM 1997. Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature 387, 80–3. [DOI] [PubMed] [Google Scholar]
- Khan SY, Ahmed ZM, Shabbir MI, Kitajiri S, Kalsoom S, Tasneem S, Shayiq S, Ramesh A, Srisailpathy S, Khan SN, Smith RJ, Riazuddin S, Friedman TB, Riazuddin S 2007. Mutations of the RDX gene cause nonsyndromic hearing loss at the DFNB24 locus. Hum Mutat 28, 417–23. [DOI] [PubMed] [Google Scholar]
- Kharkovets T, Hardelin JP, Safieddine S, Schweizer M, El-Amraoui A, Petit C, Jentsch TJ 2000. KCNQ4, a K+ channel mutated in a form of dominant deafness, is expressed in the inner ear and the central auditory pathway. Proc Natl Acad Sci U S A 97, 4333–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kho ST, Pettis RM, Mhatre AN, Lalwani AK 2000. Safety of adeno-associated virus as cochlear gene transfer vector: analysis of distant spread beyond injected cochleae. Mol Ther 2, 368–73. [DOI] [PubMed] [Google Scholar]
- Kimura Y, Kubo S, Koda H, Shigemoto K, Sawabe M, Kitamura K 2013. RNA analysis of inner ear cells from formalin fixed paraffin embedded (FFPE) archival human temporal bone section using laser microdissection--a technical report. Hear Res 302, 26–31. [DOI] [PubMed] [Google Scholar]
- Knipper M, Claussen C, Ruttiger L, Zimmermann U, Lullmann-Rauch R, Eskelinen EL, Schroder J, Schwake M, Saftig P 2006. Deafness in LIMP2-deficient mice due to early loss of the potassium channel KCNQ1/KCNE1 in marginal cells of the stria vascularis. J Physiol 576, 73–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komeda M, Roessler BJ, Raphael Y 1999. The influence of interleukin-1 receptor antagonist transgene on spiral ganglion neurons. Hearing Research 131, 1–10. [DOI] [PubMed] [Google Scholar]
- Lalwani AK, Goldstein JA, Kelley MJ, Luxford W, Castelein CM, Mhatre AN 2000. Human nonsyndromic hereditary deafness DFNA17 is due to a mutation in nonmuscle myosin MYh9. Am J Hum Genet 67, 1121–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landegger LD, Pan B, Askew C, Wassmer SJ, Gluck SD, Galvin A, Taylor R, Forge A, Stankovic KM, Holt JR, Vandenberghe LH 2017. A synthetic AAV vector enables safe and efficient gene transfer to the mammalian inner ear. Nat Biotechnol 35, 280–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MY, Takada T, Takada Y, Kappy MD, Beyer LA, Swiderski DL, Godin AL, Brewer S, King WM, Raphael Y 2015. Mice with conditional deletion of Cx26 exhibit no vestibular phenotype despite secondary loss of Cx30 in the vestibular end organs. Hear Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefevre G, Michel V, Weil D, Lepelletier L, Bizard E, Wolfrum U, Hardelin JP, Petit C 2008. A core cochlear phenotype in USH1 mouse mutants implicates fibrous links of the hair bundle in its cohesion, orientation and differential growth. Development 135, 1427–37. [DOI] [PubMed] [Google Scholar]
- Li C, Bademci G, Subasioglu A, Diaz-Horta O, Zhu Y, Liu J, Mitchell TG, Abad C, Seyhan S, Duman D, Cengiz FB, Tokgoz-Yilmaz S, Blanton SH, Farooq A,Walz K, Zhai RG, Tekin M 2019. Dysfunction of GRAP, encoding the GRB2-related adaptor protein, is linked to sensorineural hearing loss. Proc Natl Acad Sci U S A 116, 1347–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Zhao X, Xin Q, Shan S, Jiang B, Jin Y, Yuan H, Dai P, Xiao R, Zhang Q, Xiao J, Shao C, Gong Y, Liu Q 2015. Whole-exome sequencing identifies a variant in TMEM132E causing autosomal-recessive nonsyndromic hearing loss DFNB99. Hum Mutat 36, 98–105. [DOI] [PubMed] [Google Scholar]
- Liu W, Bostrom M, Kinnefors A, Rask-Andersen H 2009. Unique expression of connexins in the human cochlea. Hear Res 250, 55–62. [DOI] [PubMed] [Google Scholar]
- Liu W, Lowenheim H, Santi PA, Glueckert R, Schrott-Fischer A, Rask-Andersen H 2018. Expression of trans-membrane serine protease 3 (TMPRSS3) in the human organ of Corti. Cell Tissue Res 372, 445–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Edin F, Blom H, Magnusson P, Schrott-Fischer A, Glueckert R, Santi PA, Li H, Laurell G, Rask-Andersen H 2016. Super-resolution structured illumination fluorescence microscopy of the lateral wall of the cochlea: the Connexin26/30 proteins are separately expressed in man. Cell Tissue Res 365, 13–27. [DOI] [PubMed] [Google Scholar]
- Liu W, Li H, Edin F, Brannstrom J, Glueckert R, Schrott-Fischer A, Molnar M, Pacholsky D, Pfaller K, Rask-Andersen H 2017. Molecular composition and distribution of gap junctions in the sensory epithelium of the human cochlea-a super-resolution structured illumination microscopy (SR-SIM) study. Ups J Med Sci 122, 160–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XZ, Walsh J, Tamagawa Y, Kitamura K, Nishizawa M, Steel KP, Brown SD 1997. Autosomal dominant non-syndromic deafness caused by a mutation in the myosin VIIA gene. Nat Genet 17, 268–9. [DOI] [PubMed] [Google Scholar]
- Locher H, de Groot JC, van Iperen L, Huisman MA, Frijns JH, Chuva de Sousa Lopes SM. 2015. Development of the stria vascularis and potassium regulation in the human fetal cochlea: Insights into hereditary sensorineural hearing loss. Dev Neurobiol 75, 1219–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Bigas N, Olive M, Rabionet R, Ben-David O, Martinez-Matos JA, Bravo O, Banchs I, Volpini V, Gasparini P, Avraham KB, Ferrer I, Arbones ML, Estivill X 2001. Connexin 31 (GJB3) is expressed in the peripheral and auditory nerves and causes neuropathy and hearing impairment. Hum Mol Genet 10, 947–52. [DOI] [PubMed] [Google Scholar]
- Maeda R, Kindt KS, Mo W, Morgan CP, Erickson T, Zhao H, Clemens-Grisham R, Barr-Gillespie PG, Nicolson T 2014. Tip-link protein protocadherin 15 interacts with transmembrane channel-like proteins TMC1 and TMC2. Proc Natl Acad Sci U S A 111, 12907–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda Y, Fukushima K, Kawasaki A, Nishizaki K, Smith RJ 2007. Cochlear expression of a dominant-negative GJB2R75W construct delivered through the round window membrane in mice. Neuroscience research 58, 250–4. [DOI] [PubMed] [Google Scholar]
- Martin DM, Raphael Y 2017. It’s All in the Delivery: Improving AAV Transfection Efficiency with Exosomes. Mol Ther 25, 309–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki S, Hosoya M, Okano H, Fujioka M, Ogawa K 2018. Expression pattern of EYA4 in the common marmoset (Callithrix jacchus) cochlea. Neurosci Lett 662, 185–188. [DOI] [PubMed] [Google Scholar]
- Mei L, Chen J, Zong L, Zhu Y, Liang C, Jones RO, Zhao HB 2017. A deafness mechanism of digenic Cx26 (GJB2) and Cx30 (GJB6) mutations: Reduction of endocochlear potential by impairment of heterogeneous gap junctional function in the cochlear lateral wall. Neurobiol Dis 108, 195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mhatre AN, Li J, Kim Y, Coling DE, Lalwani AK 2004. Cloning and developmental expression of nonmuscle myosin IIA (Myh9) in the mammalian inner ear. J Neurosci Res 76, 296–305. [DOI] [PubMed] [Google Scholar]
- Minoda R, Izumikawa M, Kawamoto K, Zhang H, Raphael Y 2007. Manipulating cell cycle regulation in the mature cochlea. Hear Res 232, 44–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki E, Okumura K, Ishikawa M, Yoshimoto S, Yamaguchi J, Seki Y, Wada K, Yokohama M, Ushiki T, Tokano H, Ishii R, Shitara H, Taya C, Kitamura K, Yonekawa H, Kikkawa Y 2010. Phenotypic and expression analysis of a novel spontaneous myosin VI null mutant mouse. Exp Anim 59, 57–71. [DOI] [PubMed] [Google Scholar]
- Molina L, Fasquelle L, Nouvian R, Salvetat N, Scott HS, Guipponi M, Molina F, Puel JL, Delprat B 2013. Tmprss3 loss of function impairs cochlear inner hair cell Kcnma1 channel membrane expression. Hum Mol Genet 22, 1289–99. [DOI] [PubMed] [Google Scholar]
- Nakano Y, Kelly MC, Rehman AU, Boger ET, Morell RJ, Kelley MW, Friedman TB, Banfi B 2018. Defects in the Alternative Splicing-Dependent Regulation of REST Cause Deafness. Cell 174, 536–548 e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neyroud N, Tesson F, Denjoy I, Leibovici M, Donger C, Barhanin J, Faure S, Gary F, Coumel P, Petit C, Schwartz K, Guicheney P 1997. A novel mutation in the potassium channel gene KVLQT1 causes the Jervell and Lange-Nielsen cardioauditory syndrome. Nat Genet 15, 186–9. [DOI] [PubMed] [Google Scholar]
- Nist-Lund CA, Pan B, Patterson A, Asai Y, Chen T, Zhou W, Zhu H, Romero S, Resnik J, Polley DB, Geleoc GS, Holt JR 2019. Improved TMC1 gene therapy restores hearing and balance in mice with genetic inner ear disorders. Nat Commun 10, 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyberg S, Abbott NJ, Shi X, Steyger PS, Dabdoub A 2019. Delivery of therapeutics to the inner ear: The challenge of the blood-labyrinth barrier. Science translational medicine 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell BP, Hunter JB, Wanna GB 2016. The importance of electrode location in cochlear implantation. Laryngoscope Investig Otolaryngol 1, 169–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odeh H, Hunker KL, Belyantseva IA, Azaiez H, Avenarius MR, Zheng L, Peters LM, Gagnon LH, Hagiwara N, Skynner MJ, Brilliant MH, Allen ND, Riazuddin S, Johnson KR, Raphael Y, Najmabadi H, Friedman TB, Bartles JR, Smith RJ, Kohrman DC 2010. Mutations in Grxcr1 are the basis for inner ear dysfunction in the pirouette mouse. Am J Hum Genet 86, 148–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olt J, Mburu P, Johnson SL, Parker A, Kuhn S, Bowl M, Marcotti W, Brown SD 2014. The actin-binding proteins eps8 and gelsolin have complementary roles in regulating the growth and stability of mechanosensory hair bundles of mammalian cochlear outer hair cells. PLoS One 9, e87331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan B, Askew C, Galvin A, Heman-Ackah S, Asai Y, Indzhykulian AA, Jodelka FM, Hastings ML, Lentz JJ, Vandenberghe LH, Holt JR, Geleoc GS 2017. Gene therapy restores auditory and vestibular function in a mouse model of Usher syndrome type 1c. Nat Biotechnol 35, 264–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasi KJ, Rangarajan S, Mitchell N, Lester W, Symington E, Madan B, Laffan M, Russell CB, Li M, Pierce GF, Wong WY 2020. Multiyear Follow-up of AAV5-hFVIII-SQ Gene Therapy for Hemophilia A. N Engl J Med 382, 29–40. [DOI] [PubMed] [Google Scholar]
- Pietola L, Aarnisalo AA, Joensuu J, Pellinen R, Wahlfors J, Jero J 2008. HOX-GFP and WOX-GFP lentivirus vectors for inner ear gene transfer. Acta Otolaryngol 128, 613–20. [DOI] [PubMed] [Google Scholar]
- Pinyon JL, Tadros SF, Froud KE, AC YW, Tompson IT, Crawford EN, Ko M, Morris R, Klugmann M, Housley GD 2014. Close-field electroporation gene delivery using the cochlear implant electrode array enhances the bionic ear. Science translational medicine 6, 233ra54. [DOI] [PubMed] [Google Scholar]
- Pinyon JL, von Jonquieres G, Crawford EN, Duxbury M, Al Abed A, Lovell NH, Klugmann M, Wise AK, Fallon JB, Shepherd RK, Birman CS, Lai W, McAlpine D, McMahon C, Carter PM, Enke YL, Patrick JF, Schilder AGM, Marie C, Scherman D, Housley GD 2019. Neurotrophin gene augmentation by electrotransfer to improve cochlear implant hearing outcomes. Hear Res 380, 137–149. [DOI] [PubMed] [Google Scholar]
- Qu Y, Tang W, Zhou B, Ahmad S, Chang Q, Li X, Lin X 2012. Early developmental expression of connexin26 in the cochlea contributes to its dominate functional role in the cochlear gap junctions. Biochem Biophys Res Commun 417, 245–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rask-Andersen H, Stahle J, Wilbrand H 1977. Human cochlear aqueduct and its accessory canals. Ann Otol Rhinol Laryngol Suppl 86, 1–16. [DOI] [PubMed] [Google Scholar]
- Rehman AU, Morell RJ, Belyantseva IA, Khan SY, Boger ET, Shahzad M, Ahmed ZM, Riazuddin S, Khan SN, Riazuddin S, Friedman TB 2010. Targeted capture and next-generation sequencing identifies C9orf75, encoding taperin, as the mutated gene in nonsyndromic deafness DFNB79. Am J Hum Genet 86, 378–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehman AU, Bird JE, Faridi R, Shahzad M, Shah S, Lee K, Khan SN, Imtiaz A, Ahmed ZM, Riazuddin S, Santos-Cortez RL, Ahmad W, Leal SM, Riazuddin S, Friedman TB 2016. Mutational Spectrum of MYO15A and the Molecular Mechanisms of DFNB3 Human Deafness. Hum Mutat 37, 991–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivas A, Francis HW 2005. Inner ear abnormalities in a Kcnq1 (Kvlqt1) knockout mouse: a model of Jervell and Lange-Nielsen syndrome. Otol Neurotol 26, 415–24. [DOI] [PubMed] [Google Scholar]
- Robertson NG, Resendes BL, Lin JS, Lee C, Aster JC, Adams JC, Morton CC 2001. Inner ear localization of mRNA and protein products of COCH, mutated in the sensorineural deafness and vestibular disorder, DFNA9. Hum Mol Genet 10, 2493–500. [DOI] [PubMed] [Google Scholar]
- Salt AN, Plontke SK 2009. Principles of local drug delivery to the inner ear. Audiology and Neuro-Otology 14, 350–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sang Q, Li W, Xu Y, Qu R, Xu Z, Feng R, Jin L, He L, Li H, Wang L 2015. ILDR1 deficiency causes degeneration of cochlear outer hair cells and disrupts the structure of the organ of Corti: a mouse model for human DFNB42. Biol Open 4, 411–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Cortez RL, Lee K, Azeem Z, Antonellis PJ, Pollock LM, Khan S, Irfanullah, Andrade-Elizondo PB, Chiu I, Adams MD, Basit S, Smith JD, University of Washington Center for Mendelian, G., Nickerson DA, McDermott BM Jr., Ahmad W, Leal SM. 2013. Mutations in KARS, encoding lysyl-tRNA synthetase, cause autosomal-recessive nonsyndromic hearing impairment DFNB89. Am J Hum Genet 93, 132–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider ME, Dose AC, Salles FT, Chang W, Erickson FL, Burnside B, Kachar B 2006. A new compartment at stereocilia tips defined by spatial and temporal patterns of myosin IIIa expression. J Neurosci 26, 10243–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze-Bahr E, Wang Q, Wedekind H, Haverkamp W, Chen Q, Sun Y, Rubie C, Hordt M, Towbin JA, Borggrefe M, Assmann G, Qu X, Somberg JC, Breithardt G, Oberti C, Funke H 1997. KCNE1 mutations cause jervell and Lange-Nielsen syndrome. Nat Genet 17, 267–8. [DOI] [PubMed] [Google Scholar]
- Self T, Mahony M, Fleming J, Walsh J, Brown SD, Steel KP 1998. Shaker-1 mutations reveal roles for myosin VIIA in both development and function of cochlear hair cells. Development 125, 557–66. [DOI] [PubMed] [Google Scholar]
- Shahin H, Walsh T, Sobe T, Abu Sa’ed J, Abu Rayan A, Lynch ED, Lee MK, Avraham KB, King MC, Kanaan M 2006. Mutations in a novel isoform of TRIOBP that encodes a filamentous-actin binding protein are responsible for DFNB28 recessive nonsyndromic hearing loss. Am J Hum Genet 78, 144–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata S, Miwa T, Wu HH, Levitt P, Ohyama T 2016. Hepatocyte Growth Factor-c-MET Signaling Mediates the Development of Nonsensory Structures of the Mammalian Cochlea and Hearing. J Neurosci 36, 8200–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata SB, Cortez SR, Wiler JA, Swiderski DL, Raphael Y 2012. Hyaluronic acid enhances gene delivery into the cochlea. Hum Gene Ther 23, 302–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata SB, Yoshimura H, Ranum PT, Goodwin AT, Smith RJH 2017. Intravenous rAAV2/9 injection for murine cochlear gene delivery. Scientific reports 7, 9609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin MJ, Lee JH, Yu DH, Kim HJ, Bae KB, Yuh HS, Kim MO, Hyun BH, Lee S, Park R, Ryoo ZY 2010. Spatiotemporal expression of tmie in the inner ear of rats during postnatal development. Comp Med 60, 288–94. [PMC free article] [PubMed] [Google Scholar]
- Shu Y, Tao Y, Li W, Shen J, Wang Z, Chen ZY 2016a. Adenovirus Vectors Target Several Cell Subtypes of Mammalian Inner Ear In Vivo. Neural Plast 2016, 9409846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu Y, Tao Y, Wang Z, Tang Y, Li H, Dai P, Gao G, Chen ZY 2016b. Identification of Adeno-Associated Viral Vectors That Target Neonatal and Adult Mammalian Inner Ear Cell Subtypes. Hum Gene Ther 27, 687–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirmaci A, Erbek S, Price J, Huang M, Duman D, Cengiz FB, Bademci G, Tokgoz-Yilmaz S, Hismi B, Ozdag H, Ozturk B, Kulaksizoglu S, Yildirim E, Kokotas H, Grigoriadou M, Petersen MB, Shahin H, Kanaan M, King MC, Chen ZY,Blanton SH, Liu XZ, Zuchner S, Akar N, Tekin M 2010. A truncating mutation in SERPINB6 is associated with autosomal-recessive nonsyndromic sensorineural hearing loss. Am J Hum Genet 86, 797–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soudais C, Skander N, Kremer EJ 2004. Long-term in vivo transduction of neurons throughout the rat CNS using novel helper-dependent CAV-2 vectors. FASEB J 18, 391–3. [DOI] [PubMed] [Google Scholar]
- Stover T, Yagi M, Raphael Y 2000. Transduction of the contralateral ear after adenovirus-mediated cochlear gene transfer. Gene Ther 7, 377–83. [DOI] [PubMed] [Google Scholar]
- Suzuki M, Kaga K 1999. Development of blood-labyrinth barrier in the semicircular canal ampulla of the rat. Hear Res 129, 27–34. [DOI] [PubMed] [Google Scholar]
- Tan F, Chu C, Qi J, Li W, You D, Li K, Chen X, Zhao W, Cheng C, Liu X, Qiao Y, Su B, He S, Zhong C, Li H, Chai R, Zhong G 2019. AAV-ie enables safe and efficient gene transfer to inner ear cells. Nat Commun 10, 3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Y, Huang M, Shu Y, Ruprecht A, Wang H, Tang Y, Vandenberghe LH, Wang Q, Gao G, Kong WJ, Chen ZY 2018. Delivery of Adeno-Associated Virus Vectors in Adult Mammalian Inner-Ear Cell Subtypes Without Auditory Dysfunction. Hum Gene Ther 29, 492–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoenes M, Zimmermann U, Ebermann I, Ptok M, Lewis MA, Thiele H, Morlot S, Hess MM, Gal A, Eisenberger T, Bergmann C, Nurnberg G, Nurnberg P, Steel KP, Knipper M, Bolz HJ 2015. OSBPL2 encodes a protein of inner and outer hair cell stereocilia and is mutated in autosomal dominant hearing loss (DFNA67). Orphanet J Rare Dis 10, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venail F, Wang J, Ruel J, Ballana E, Rebillard G, Eybalin M, Arbones M, Bosch A, Puel JL 2007. Coxsackie adenovirus receptor and alpha nu beta3/alpha nu beta5 integrins in adenovirus gene transfer of rat cochlea. Gene Ther 14, 30–7. [DOI] [PubMed] [Google Scholar]
- Walsh T, Shahin H, Elkan-Miller T, Lee MK, Thornton AM, Roeb W, Abu Rayyan A, Loulus S, Avraham KB, King MC, Kanaan M 2010. Whole exome sequencing and homozygosity mapping identify mutation in the cell polarity protein GPSM2 as the cause of nonsyndromic hearing loss DFNB82. Am J Hum Genet 87, 90–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters BJ, Coak E, Dearman J, Bailey G, Yamashita T, Kuo B, Zuo J 2017. In Vivo Interplay between p27(Kip1), GATA3, ATOH1, and POU4F3 Converts Non-sensory Cells to Hair Cells in Adult Mice. Cell Rep 19, 307–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Kempton JB, Brigande JV 2018a. Gene Therapy in Mouse Models of Deafness and Balance Dysfunction. Front Mol Neurosci 11, 300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Feng Y, Yan D, Qin L, Grati M, Mittal R, Li T, Sundhari AK, Liu Y,Chapagain P, Blanton SH, Liao S, Liu X 2018b. A dominant variant in the PDE1C gene is associated with nonsyndromic hearing loss. Hum Genet 137, 437–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Sun Y, Chang Q, Ahmad S, Zhou B, Kim Y, Li H, Lin X 2013. Early postnatal virus inoculation into the scala media achieved extensive expression of exogenous green fluorescent protein in the inner ear and preserved auditory brainstem response thresholds. J Gene Med 15, 123–33. [DOI] [PubMed] [Google Scholar]
- Webb SW, Grillet N, Andrade LR, Xiong W, Swarthout L, Della Santina CC, Kachar B, Muller U 2011. Regulation of PCDH15 function in mechanosensory hair cells by alternative splicing of the cytoplasmic domain. Development 138, 1607–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Fu Y, Liu S, Xia G, Pan S 2013. Effect of lentiviruses carrying enhanced green fluorescent protein injected into the scala media through a cochleostomy in rats. Am J Otolaryngol 34, 301–7. [DOI] [PubMed] [Google Scholar]
- Wu YX, Zhu GX, Liu XQ, Sun F, Zhou K, Wang S, Wang CM, Jia JW, Song JT, Lu LJ 2014. Noise alters guinea pig’s blood-labyrinth barrier ultrastructure and permeability along with a decrease of cochlear Claudin-5 and Occludin. BMC neuroscience 15, 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia AP, Ikeda K, Katori Y, Oshima T, Kikuchi T, Takasaka T 2000. Expression of connexin 31 in the developing mouse cochlea. Neuroreport 11,2449–53. [DOI] [PubMed] [Google Scholar]
- Xie Y, Sharon JD, Pross SE, Abt NB, Varma S, Della Santina CC, Minor LB, Carey JP 2017. Surgical Complications from Superior Canal Dehiscence Syndrome Repair: Two Decades of Experience. Otolaryngol Head Neck Surg 157, 273–280. [DOI] [PubMed] [Google Scholar]
- Yang J, Zhou W, Zhang Y, Zidon T, Ritchie T, Engelhardt JF 1999. Concatamerization of adeno-associated virus circular genomes occurs through intermolecular recombination. J Virol 73, 9468–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Scholl ES, Pan N, Fritzsch B, Haeseleer F, Lee A 2016. Expression and Localization of CaBP Ca2+ Binding Proteins in the Mouse Cochlea. PLoS One 11, e0147495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao H, Hill SF, Skidmore JM, Sperry ED, Swiderski DL, Sanchez GJ, Bartels CF, Raphael Y, Scacheri PC, Iwase S, Martin DM 2018. CHD7 represses the retinoic acid synthesis enzyme ALDH1A3 during inner ear development. JCI Insight 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yariz KO, Duman D, Zazo Seco C, Dallman J, Huang M, Peters TA, Sirmaci A, Lu N, Schraders M, Skromne I, Oostrik J, Diaz-Horta O, Young JI, Tokgoz-Yilmaz S, Konukseven O, Shahin H, Hetterschijt L, Kanaan M, Oonk AM, Edwards YJ, Li H, Atalay S, Blanton S, Desmidt AA, Liu XZ, Pennings RJ, Lu Z, Chen ZY, Kremer H, Tekin M 2012. Mutations in OTOGL, encoding the inner ear protein otogelin-like, cause moderate sensorineural hearing loss. Am J Hum Genet 91, 872–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasunaga S, Grati M, Cohen-Salmon M, El-Amraoui A, Mustapha M, Salem N, El-Zir E, Loiselet J, Petit C 1999. A mutation in OTOF, encoding otoferlin, a FER-1-like protein, causes DFNB9, a nonsyndromic form of deafness. Nat Genet 21, 363–9. [DOI] [PubMed] [Google Scholar]
- Yoshimura H, Shibata SB, Ranum PT, Smith RJH 2018. Enhanced viral-mediated cochlear gene delivery in adult mice by combining canal fenestration with round window membrane inoculation. Scientific reports 8, 2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Q, Wang Y, Chang Q, Wang J, Gong S, Li H, Lin X 2014. Virally expressed connexin26 restores gap junction function in the cochlea of conditional Gjb2 knockout mice. Gene Ther 21, 71–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zettner EM, Gleser MA 2018. Progressive Hearing Loss among Patients with Cystic Fibrosis and Parenteral Aminoglycoside Treatment. Otolaryngol Head Neck Surg 159, 887–894. [DOI] [PubMed] [Google Scholar]
- Zhang L, Hu L, Chai Y, Pang X, Yang T, Wu H 2014. A dominant mutation in the stereocilia-expressing gene TBC1D24 is a probable cause for nonsyndromic hearing impairment. Hum Mutat 35, 814–8. [DOI] [PubMed] [Google Scholar]
- Zhao HB 2016. Expression and function of pannexins in the inner ear and hearing. BMC Cell Biol 17 Suppl 1, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J, Shen W, He DZ, Long KB, Madison LD, Dallos P 2000a. Prestin is the motor protein of cochlear outer hair cells. Nature 405, 149–55. [DOI] [PubMed] [Google Scholar]
- Zheng L, Sekerkova G, Vranich K, Tilney LG, Mugnaini E, Bartles JR 2000b. The deaf jerker mouse has a mutation in the gene encoding the espin actin-bundling proteins of hair cell stereocilia and lacks espins. Cell 102, 377–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W, Huang L, Wei ZB, Silvius D, Tang B, Xu PX 2003. The role of Six1 in mammalian auditory system development. Development 130, 3989–4000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Chen J, Liang C, Zong L, Chen J, Jones RO, Zhao HB 2015. Connexin26 (GJB2) deficiency reduces active cochlear amplification leading to late-onset hearing loss. Neuroscience 284, 719–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong L, Chen J, Zhu Y, Zhao HB 2017. Progressive age-dependence and frequency difference in the effect of gap junctions on active cochlear amplification and hearing. Biochem Biophys Res Commun 489, 223–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L, Zhou H, Pastore L, Yang K 2000. Prolonged transgene expression mediated by a helper-dependent adenoviral vector (hdAd) in the central nervous system. Mol Ther 2, 105–13. [DOI] [PubMed] [Google Scholar]