

Abstract
Azole antifungals are an important class of antifungal drugs due to their low cost, ability to be administered orally, and broad-spectrum activity. However, their widespread and long-term use have given rise to adaptation mechanisms that render these compounds less effective against common fungal pathogens, including Candida albicans. New antifungals are desperately needed as drug resistant strains become more prevalent. We recently showed that copper supplementation potentiates the activity of the azole antifungal fluconazole against the opportunistic fungal pathogen C. albicans. Here, we report eight new azole analogues derived from fluconazole in which one triazole group has been replaced with a metal-binding group, a strategy designed to enhance potentiation of azole antifungal activity by copper. The bioactivity of all eight compounds was tested and compared to that of fluconazole. Three of the analogues showed activity against C. albicans and two had lower levels of trailing growth. One compound, Flu-TSCZ, was found to impact the levels, speciation, and bioavailability of cellular metals.
Keywords: Antifungal, azole, copper, metals in medicine, Candida albicans, homeostasis
Graphical Abstract
Introduction
More than 1.5 million deaths globally in 2017 were attributed to fungal infections, with another 1 billion individuals carrying a fungal infection in that year alone [1]. These infections are particularly dangerous to individuals with immune systems compromised from a variety of conditions ranging from chemotherapy to organ transplants to asthma. The azole antifungal fluconazole has been used in the clinic for nearly three decades to treat life-threatening fungal infections, including those caused by Candida albicans and Cryptococcus neoformans. Azoles bind and inhibit cytochrome P450 lanosterol 14⍺-demethylase (Cyp51) thereby blocking the biosynthesis of ergosterol, an important cell membrane component. Unfortunately, widespread and prolonged use of these drugs has allowed pathogens to develop several modes of resistance. These mechanisms include increased production of Cyp51, changes in the ergosterol biosynthetic pathway, modifications to Cyp51 that hinder substrate binding, and upregulation of efflux pumps that reduce the intracellular concentration of the drug [2–5]. It is becoming apparent that the use of fluconazole to treat serious infections may not be a viable long-term option, yet development of entirely new classes of antifungal compounds is a significant challenge owing to the similarity between eukaryotic pathogens and their hosts [6]. Repurposing existing drugs is one strategy to extend the utility of the currently limited antifungal arsenal [7–10]. Another approach is to create analogues of antifungal compounds to take advantage of a known fungal target while hopefully circumventing adaptation mechanisms associated with the original drug. There have been several reports of fluconazole analogues in which one of the triazole side arms is modified to accommodate features suggested from molecular docking and structure-activity relationship studies [11–21].
Other approaches to extend the biological activity and broaden the spectrum of azole drugs include taking advantage of metal coordination; metal complexes of econazole [22], ketoconazole [23–25], clotrimazole [23, 25–28], miconazole [25, 28], tioconazole [28], and fluconazole [29] have been reported. Of particular interest was the approximate 30% growth reduction reported for a binuclear Cu(II)–fluconazole complex as compared to the copper-free drug against several strains of C. albicans [29]. We have observed Cu-potentiated fluconazole activity with no evidence the Cu(II)–fluconazole complex forms under the conditions of our growth assays, leading us to conclude that complex formation is not required for Cu-potentiated biological activity [30]. Indeed, fluconazole binds Cu in aqueous buffer but not in the growth medium [30], illustrating that the presence of metal ions can impact a compound’s biological activity even if the complex dissociates. Although complex formation is not required, we reasoned that synthetically modifying azoles to encourage Cu binding may enhance the Cu-potentiated activity previously observed.
In the present study, we synthesized eight new analogues of fluconazole by replacing one of the 1,2,4-triazole rings with side arms that in principle should facilitate Cu binding. We hypothesized that such analogues may enhance the Cu-potentiated activity we have reported for fluconazole [30] or lead to new Cu-dependent modes of action. Although still fungistatic, three analogues were active against C. albicans at 25 μM or below and two exhibited lower trailing growth at 48 h compared to fluconazole. A thiosemicarbazone-based analogue, Flu-TSCZ, was capable of overriding a defect in Cu transport, distinguishing it from fluconazole and other analogues in its ability to supply cells with bioavailable Cu. Overall, we identify Flu-TSCZ as a promising compound for future development.
Results
Synthesis of fluconazole analogues with copper-binding side arms
As shown in Scheme 1, eight analogues of fluconazole were synthesized from a single commercially available building block in a way that retains the difluorophenyl and hydroxyl group pharmacophores of fluconazole, as well as one of its two triazole rings, as only one is needed for binding the heme Fe of Cyp51 [13]. The other triazole was replaced with side arms rich in nitrogen, sulfur, and oxygen donors that were selected due to their predilection for binding Cu(II) and/or Cu(I). Synthetic intermediates in Scheme 1 are labeled with Arabic numerals, whereas final compounds are given a classification I, II, or III depending on which of three fluconazole-based scaffolds were used to prepare them: either an azide, alkyne, or amine handle for conjugating with an alkyne-, azide-, or carboxylic acid-bearing side arm, respectively. The commercial availability or ease of synthesis was also considered, and some side arms were selected based on known biological activity. Specifically, thiosemicarbazones (TSCZs) have known antimicrobial properties that are often enhanced by metal-binding [31], making them an attractive class of compounds for our studies, and 8-hydroxyquinoline was investigated on the basis of the Cu-dependent fungicidal activity observed previously in C. neoformans [32, 33]. In addition to alpha-numerical labeling, each compound was assigned an abbreviated nickname that reflects both its fluconazole origin and its new metal-binding motif: Flu-Imidazole (Flu-Im, I-a), Flu-Phenol (I-b), Flu-Dimethylester (Flu-DME, II-a), Flu-Pyridyl (Flu-Pyr, II-b), Flu-Dipicolylamine (Flu-DPA, II-c), Flu-Aminopropanoic acid (Flu-APA, II-d), Flu-Thiosemicarbazone (Flu-TSCZ, III-a), and Flu-8-Hydroxyquinoline (Flu-8HQ, III-b).
Scheme 1. Synthesis of fluconazole analogues.

Commercially available ketone 1 was converted to alkyne-bearing fluconazole structure 2, which was then reacted with azide-bearing side arm 6 via CuAAC (route A) to obtain compounds with structure I. Alternatively, the ketone was converted to epoxide 3 via a Corey-Chaykovsky reaction. Epoxide 3 then underwent a ring-opening reaction with sodium azide to give azide-bearing fluconazole structure 4. Azide 4 then underwent CuAAC (route B) with alkyne-bearing side arm 7 to afford analogues with structure II or was reduced to the corresponding amine (5) via Pd/C catalyzed hydrogenation. Amine 5 was coupled to carboxylic acid-bearing side arm 8 via HBTU- or EEDQ-mediated coupling (route C) to yield analogues with structure III.
Beginning from commercially available ketone 1, the alkyne or azide versions of fluconazole (compounds 2 and 4, respectively) were synthesized using previously reported methods [12, 13]. Reduction of 4 via Pd/C-catalyzed hydrogenation with triethylsilane [34] yielded compound 5, the amine version of fluconazole. Analogues were synthesized from one of these three scaffolds via Cu-catalyzed azide-alkyne cycloaddition (CuAAC) starting from 2 and 4 or HBTU- or EEDQ-mediated coupling starting from 5. These conjugation strategies are favorable because they result in the formation of groups that could in principle participate in metal binding: a 1,2,3-triazole from CuAAC or an amide group from HBTU/EEDQ-mediated coupling.
Flu-Pyr, Flu-Phenol, and Flu-TSCZ have Cu-potentiated antifungal activity against C. albicans
Racemic mixtures of the eight fluconazole derivatives were tested for the ability to inhibit growth of C. albicans in liquid culture. As shown in Fig. 1, three of these compounds—Flu-Pyr, Flu-Phenol, and Flu-TSCZ—inhibited growth at a concentration of 25 μM or below either with or without supplemental Cu. Flu-8HQ and Flu-Im did not show inhibitory activity below 100 μM and were therefore considered inactive (Fig. S1, ESI). Flu-DME, Flu-APA, and Flu-DPA were also found to be inactive, exhibiting no activity up to 100 μM (Fig. S1, ESI).
Figure 1. Flu-Pyr, Flu-Phenol, and Flu-TSCZ exhibit Cu-potentiated growth inhibition against C. albicans.

Fluconazole is shown for comparison. Conditions: Growth at 30 °C in YPD (lot 2005064, metal analysis presented in Table S1, ESI) monitored at OD600 after 48 h. Data are reported as means with error bars representing standard deviation of three replicate conditions.
Fluconazole is a fungistatic drug to which C. albicans can develop tolerance that leads to trailing growth, defined as residual growth at a concentration above the minimal inhibitory concentration (MIC) [35]. We have found that Cu supplementation during treatment with fluconazole delays the onset of trailing growth in C. albicans [30, 36]. Similar to fluconazole (Fig. 1a), Flu-Pyr demonstrated trailing growth even at a concentration of 100 μM, but trailing was abrogated by addition of 10 μM Cu (Fig. 1b). Notably, Flu-Phenol and Flu-TSCZ gave rise to lower levels of trailing growth than fluconazole in the absence of added Cu (Fig. 1c–d). Potentiation by Cu was still evident for these analogues, though it was less dramatic due to the reduction in trailing growth observed for the compound alone.
Of note, there were two batches of YPD medium (lots 2044606 and 2101669) in which trailing growth was not evident at 48 h following exposure to the three active analogues or fluconazole (Fig. S2, ESI). We have previously observed minor fluctuations in the degree of 48-h trailing growth between batches of YPD medium received from a commercial supplier [30]. Trailing growth may not be evident at 48 h if the onset of tolerance is delayed due to environmental conditions (e.g. different nutrients in different batches of medium). Indeed, trailing growth does eventually occur when the growth assay is carried out to 72 h (Fig. S3a, ESI). We hypothesized that limiting Cu availability may restore 48-h trailing growth in this batch of YPD. To test this idea, the growth medium was supplemented with the membrane-impermeable extracellular Cu(I) chelator bathocuproinedisulfonic acid (BCS). Indeed, supplementing analogue-treated cells with BCS partially rescued growth for all three compounds (Fig. S2, ESI). The return of 48-h trailing growth upon supplementation with BCS bolsters the conclusion that Cu availability influences tolerance to the azole analogues.
Compounds that are fungicidal rather than fungistatic are desirable because they reduce the risk of the development of resistance [37]. To determine whether Flu-Phenol, Flu-Pyr, or Flu-TSCZ are fungicidal against C. albicans, colony forming units (CFUs) were determined following exposure to these agents with and without supplemental Cu in liquid culture. Fluconazole was also tested as a control. Colony growth, quantified as CFUs per mL, was observed for all treatment conditions, illustrating that the analogues are not fungicidal (Fig. 2a). For most conditions, the number of CFUs increased by approximately two orders of magnitude above that of the original inoculum, indicating that some trailing growth had occurred. Cu supplementation did not impact the number of CFUs for cells treated with fluconazole, Flu-Phenol, or Flu-Pyr. Treatment with Flu-TSCZ in the presence of supplemental Cu was the only condition in which no trailing growth was evident. This result makes sense, given that the experiment was performed in a batch of YPD medium that exhibited low levels of trailing growth at 48 h. The OD600 at 48 h is low for all of these conditions, but cells treated with Flu-TSCZ and Cu had the lowest level of growth by OD600 at 48 h (Fig. S3b, ESI). To investigate whether Cu concentration impacts the growth of cells treated with Flu-TSCZ, CFUs were determined for cultures exposed to Flu-TSCZ in the presence of a range of Cu concentrations. As shown in Fig. 2b, supplementation with very low (1 μM) or very high (1000 μM) Cu did not reduce CFUs relative to cells treated with Flu-TSCZ alone (0 μM supplemental Cu). However, supplementing with 10 μM Cu did reduce CFUs and supplementing with 100 μM Cu reduced CFUs even further, relative to cells treated with Flu-TSCZ alone. The number of CFUs per mL did not drop below the original inoculum, indicating the combination treatment is still fungistatic and not fungicidal. However, it is notable that there is a Cu-dependent decrease in the number of CFUs per mL for cells treated with 50 μM Flu-TSCZ. Cells treated with the inhibitory but lower concentration of 25 μM Flu-TSCZ did not exhibit a Cu-dependent decrease in CFUs (Fig. S4, ESI).
Figure 2. Colony forming units (CFUs) after exposure to analogues.

(a) CFUs/mL were determined following 48 h of exposure to 100 μM fluconazole (purple bars), Flu-Pyr (magenta bars), Flu-Phenol (green bars), or Flu-TSCZ (dark blue bars) in the absence (solid bars) or presence (striped bars) of 10 μM supplemental CuSO4 in YPD medium (lot 2044606). The number of CFUs/mL in the original inoculum is shown in light blue. (b) A Cu-dependent reduction in CFUs was observed for cells treated with 50 μM Flu-TSCZ. CFUs/mL were determined following 48 h of exposure to 50 μM Flu-TSCZ and 0–1000 μM supplemental CuSO4. Data are reported as means with individual data points indicating replicates within a single experiment.
Flu-TSCZ provides bioavailable Cu to C. albicans and C. neoformans
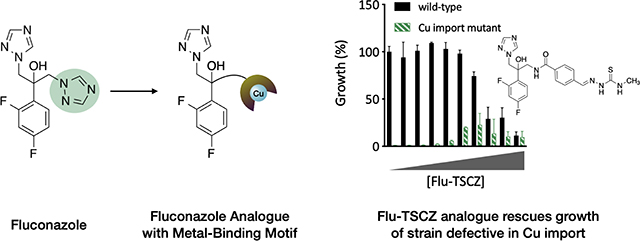
To determine whether the active analogues are capable of shuttling Cu into cells, they were tested for their ability to rescue growth of C. albicans and C. neoformans strains in which the Cu importers had been deleted. When grown in yeast peptone ethanol glycerol (YPEG) medium, cells are forced to respire, therefore requiring Cu for use in cytochrome c oxidase. C. albicans relies on a single high affinity Cu(I) importer Ctr1 and C. neoformans has two Cu importers, Ctr1 and Ctr4. As shown in Fig. 3, wild-type cells grow in YPEG while C. albicans ctr1Δ/Δ and C. neoformans ctr1Δ ctr4Δ cells do not. Flu-TSCZ was the only compound tested that rescued growth of both deletion strains (Fig. 3), indicating that Flu-TSCZ can deliver bioavailable Cu to C. albicans and C. neoformans while other compounds cannot (Fig. S5, ESI). This result distinguishes Flu-TSCZ from fluconazole, which does not rescue growth of either deletion strain [30]. Because Flu-TSCZ displayed antifungal activity against C. albicans and rescued growth of the strains lacking Cu import machinery, we chose to focus our attention on this compound for further characterization.
Figure 3. Flu-TSCZ recovers growth of cells lacking Cu import machinery.

48-h growth of C. albicans (a) or C. neoformans (b) WT (black bars) and Cu import mutants (green bars) treated with 0–100 μM Flu-TSCZ in YPEG medium. Dose dependent growth rescue was observed for cells lacking Cu import genes under these conditions. Data are reported as means with error bars representing standard deviation of three replicate conditions.
Flu-TSCZ binds Cu(II)
Analogues were endowed with side arms containing multiple donor atoms in order to promote Cu binding and, in turn, enhance potentiation of azole activity by Cu. As shown in Fig. 4a, Flu-TSCZ exhibits a characteristic absorption band at 321 nm in HEPES buffer that decreases in intensity upon addition of Cu(II), indicating formation of a Cu(II)–Flu-TSCZ complex. Similar spectral shifts were observed for other analogues, confirming that they also bind Cu(II) in HEPES buffer (Fig. S6, ESI).
Figure 4. Flu-TSCZ binds Cu(II) in HEPES buffer but not in YPD medium.

(a) UV-Vis spectra of Flu-TSCZ +/− Cu in 50 mM HEPES buffer, pH 7.4. Addition of Cu (blue trace) to Flu-TSCZ (purple trace) decreased the intensity of the absorption band at 321 nm and gave rise to a shoulder, indicating Cu complex formation. Addition of glycine (Gly, green trace) to the Cu(II)–Flu-TSCZ complex gave rise to a new feature at 413 nm, suggesting formation of a ternary complex, but addition of histidine (His, red trace) resulted in disappearance of the shoulder, indicating histidine competes with Flu-TSCZ for Cu(II). Conditions: [Flu-TSCZ] = 50 μM, [CuSO4] = 25 μM, [Gly] = 1 mM, [His] = 1 mM. (b) EPR spectra of Flu-TSCZ +/− Cu in YPD medium. Addition of Cu to YPD gives rise to an EPR spectrum (solid blue trace) that is unchanged by addition of Flu-TSCZ (dotted red trace). Conditions: [Flu-TSCZ] = 300 μM, [CuSO4] = 150 μM in YPD with 20% glycerol. Data collected at 77 K.
The ability of Flu-TSCZ to supply bioavailable Cu to the Cu import mutants raised the question of whether this compound may bind Cu in the growth medium. In YPD medium there is approximately 3 mM free glycine [38], which forms a 2:1 complex with Cu(II) (log β2 = 15.6 for Cu(Gly)2) [39, 40]. Thus, the ability of a compound to bind Cu in the presence of glycine indicates the likelihood that the complex will form in the growth medium during biological assays. We have previously shown that glycine outcompetes fluconazole for Cu(II) binding [30]. To determine whether glycine also competes with Flu-TSCZ for Cu(II), glycine was added to the preformed Cu(II)–Flu-TSCZ complex in HEPES buffer. As shown in Fig. 4a, glycine addition partially restored the intensity of the absorption band at 321 nm and gave rise to a new feature at 413 nm, suggesting the formation of a ternary complex. To determine whether another amino acid would have a different effect on the stability of Cu(II)–Flu-TSCZ in solution, a separate competition experiment was performed with L-histidine, which has a higher affinity than glycine for Cu(II) (log β2 = 18.6) [41, 42]. Like glycine, free histidine is present in YPD but at a lower concentration (approximately 0.5 mM) [38]. Upon addition of histidine to the Cu(II)–Flu-TSCZ complex, the ligand signal at 321 nm was almost fully restored and the shoulder unique to the Cu(II)–Flu-TSCZ complex disappeared (Fig. 4a), suggesting that histidine pulls Cu(II) away from Flu-TSCZ.
To test directly whether Flu-TSCZ binds Cu(II) under the conditions of our growth assays, we compared the EPR spectrum of Cu alone in YPD to that of Flu-TSCZ and Cu in YPD. There was no change to the EPR spectrum of Cu(II) in YPD upon addition of Flu-TSCZ, suggesting that the Cu(II)–Flu-TSCZ complex does not form under these conditions in YPD medium (Fig. 4b). Considering the data from the histidine competition, it is reasonable that amino acids present in YPD prevent formation of a stable Cu(II)–Flu-TSCZ complex.
Flu-TSCZ binds Cu(I)
Given that Cu(I) is the primary oxidation state of intracellular Cu [43, 44], a competitive binding assay was performed with the Cu(I) chelator ferrozine (log β2 = 15.1) [45, 46] to predict the ability of Cu(I)–Flu-TSCZ to form intracellularly. As shown in Fig. 5a, the absorption band for the pre-formed [CuI(Fz)2]3− complex disappeared upon addition of Flu-TSCZ, suggesting that Flu-TSCZ pulls Cu(I) away from this competing ligand. Furthermore, the [CuI(Fz)2]3− complex does not form upon addition of ferrozine to the pre-formed Cu(I)–Flu-TSCZ complex. As shown in Fig. 5b, addition of the reducing agent ascorbate to Cu(II)-Flu-TSCZ caused a slight shift in the absorption signal, indicating reduction of the complex to Cu(I). Addition of ferrozine to this solution did not change the absorption spectrum of this species and the [CuI(Fz)2]3− signal was not observed. To further probe the affinity of Flu-TSCZ for Cu(I), the higher affinity Cu(I) chelator bicinchoninate anion (BCA, log β2 = 17.2) [47, 48] was tested for its ability to bind Cu(I) in the presence of Flu-TSCZ. Addition of BCA gave rise to the characteristic signal at 562 nm, indicating formation of the [CuI(BCA)2]3− complex (Fig. 5b). Taken together, the competition data with these two Cu(I) probes [45] benchmark the affinity of Flu-TSCZ for Cu(I) in the picomolar range and suggest Flu-TSCZ may be a better ligand for Cu(I) than Cu(II). It is therefore possible that Flu-TSCZ rescues growth of cells lacking their Cu importers through redistribution of intracellular Cu(I) as opposed to shuttling in extracellular Cu(II).
Figure 5. Flu-TSCZ competes with the chelator ferrozine (Fz), but not bicinchoninate anion (BCA), for Cu(I) as determined by UV-Vis spectroscopy.

(a) Flu-TSCZ pulls Cu(I) away from the pre-formed [CuI(Fz)2]3− complex. (b) Presence of Flu-TSCZ in solution prevents formation of the [CuI(Fz)2]3− complex. CuCl2 was used as the source of Cu(II), and ascorbate (Asc) was added to generate Cu(I) in situ. Conditions: [Fz] = 100 μM, [CuCl2] = 40 μM, [Ascorbate] = 1 mM, [Flu-TSCZ] = 100 μM, [BCA] = 100 μM in 50 mM HEPES buffer, pH 7.4.
Potentiation of Flu-TSCZ activity is Cu specific
Although analogues were designed with the intention of strengthening the Cu potentiation observed for fluconazole, their efficacy could be affected by the availability of other metal ions in the growth medium. To determine the impact of varying the availability of individual biometals, growth assays were performed in Tris-buffered synthetic defined (Tris:SD) drop-out media rigorously controlled for metal content. Each medium was prepared from Chelex-treated water to remove trace metals and then nutrients were replenished to levels found in commercial SD media formulations [49]. Individual drop-out media were prepared by omitting Cu, Fe, Mn, or Zn, and assays were performed by titrating each omitted metal back to obtain a stepwise gradient of that metal. As shown in Fig. 6, growth of untreated control cells improved slightly as the dropped-out metal was returned, most clearly for Fe and Mn. Supplemental levels of Fe, Mn, and Zn were not inhibitory up to 50 μM, but 50 μM Cu inhibited growth by approximately 50%. Notably, increasing the levels of Cu (Fig. 6a), but not Fe, Mn, or Zn (Fig. 6b–d) potentiated the activity of Flu-TSCZ. In the Cu drop-out medium, 25–50 μM Flu-TSCZ was required to fully inhibit growth without Cu supplementation. By contrast, only 13 μM or 6 μM Flu-TSCZ was required for full inhibition when the growth medium was supplemented with 3 μM or 12.5 μM Cu, respectively.
Figure 6. Cu availability specifically has a significant impact on growth of Flu-TSCZ-treated cells.

48-h growth of untreated (dark purple lines) or Flu-TSCZ-treated C. albicans cells as a function of total Cu (a), Fe (b), Mn (c), and Zn (d) in the respective metal drop-out Tris:SD media. The lowest concentration of each metal represents actual trace levels of the dropped-out metal, analyzed by ICP-MS. The remaining concentration gradients represent theoretical metal concentrations based on what was added on top of trace levels. Shown are representative plots from n = 2 biologically independent experiments. Full metal analyses for each media formulation are reported in Tables S4–7, ESI.
C. albicans strains defective in Cu transport have increased tolerance to Flu-TSCZ
The observation that levels of Cu uniquely impact the efficacy of Flu-TSCZ led us to test how deletion of Cu transport genes impacts C. albicans’ tolerance to Flu-TSCZ. As shown in Fig. 7a, deletion of Cu exporter CRP1 increased the tolerance of C. albicans to Flu-TSCZ, demonstrating that cells are better equipped to adapt to Flu-TSCZ stress when they lack the Cu exporter. Interestingly, deletion of Cu importer CTR1 also increased tolerance, with even 100 μM Flu-TSCZ failing to fully inhibit growth (Fig. 7b). Of note, we have observed ctr1Δ/Δ cells grow in clumps in the overnight culture (Fig. S7a, ESI). The reason is unknown and there is no correlation between the size of the colony used to inoculate and whether cultures grew as planktonic cells or in clumps. Despite these morphological differences, similar results were obtained when Flu-TSCZ was tested against ctr1Δ/Δ cells growing in clumps (Fig. S7b, ESI) and planktonic ctr1Δ/Δ cells (Fig. 7b). Deletion of transcription factor MAC1, which controls the cellular response to Cu deficiency, including regulation of CTR1, did not affect tolerance (Fig. 7c). The antifungal activity of Flu-TSCZ against the CTR1, CRP1, and MAC1 deletion strains and their parent strains was not dramatically impacted by supplementation with 10 μM Cu (Fig. S8, ESI). Growth of ctr1Δ/Δ cells improved slightly upon Cu supplementation, consistent with these cells lacking high affinity Cu import capabilities. Growth inhibition of crp1Δ/Δ cells and parent strain KC2 was potentiated by Cu at high concentrations of Flu-TSCZ. In SC5314, Cu abrogated activity at 12.5 μM Flu-TSCZ, a concentration on the cusp of the MIC.
Figure 7. C. albicans strains lacking genes involved in Cu transport have reduced susceptibility to Flu-TSCZ.

C. albicans deletion strains were treated with 0–100 μM Flu-TSCZ in YPD medium at 30 °C for 48 h then growth was determined by measuring OD600. Deletion of Cu exporter CRP1 (a) or Cu importer CTR1 (b) rendered C. albicans more tolerant to Flu-TSCZ. Deletion of Cu-regulated transcription factor MAC1 (c) did not impact the tolerance of C. albicans to Flu-TSCZ. Data are normalized to the untreated control for each strain and reported as means with error bars representing standard deviation of three replicate conditions.
In contrast to the results obtained for Flu-TSCZ, we have previously found that both the ctr1Δ/Δ and mac1Δ/Δ strains are less tolerant than wild-type cells to fluconazole [50]. Thus, replacement of the triazole side arm of fluconazole with the thiosemicarbazone motif alleviates the sensitivity of the mac1Δ/Δ strain and fully reverses the sensitivity of the ctr1Δ/Δ strain. By intentionally incorporating metal-binding motifs, we have impacted the ability of strains defective in Cu homeostasis to tolerate azole stress.
Flu-TSCZ treatment alters cell-associated metal levels
The ability of Flu-TSCZ to recover growth of strains lacking Cu importers suggested this compound may increase levels of cell-associated Cu, despite the lack of conclusive spectroscopic evidence for Cu complex formation in the growth medium. To determine whether Flu-TSCZ alters cellular metal status, metal content was determined for cells treated with 10, 25, or 100 μM Flu-TSCZ with and without supplemental Cu over a 12 h period via inductively coupled plasma-mass spectrometry (ICP-MS). As we have observed previously for untreated C. albicans cells [36], Cu levels remained steady, Fe and Mn levels decreased, and Zn levels increased over time (Fig. 8). We found that treatment with 25 or 100 μM Flu-TSCZ did indeed increase cell-associated Cu above untreated levels, most notably at 6 h (Fig. 8a, Fig. S9, ESI). Supplementing these cells with 10 μM Cu increased these levels even further, such that cellular Cu concentrations in co-treated cells exceeded those of cells treated only with Cu. Levels of Fe and Mn were also elevated in cells treated with Flu-TSCZ, but Cu supplementation had no additional impact on these levels (Fig. 8b–c). Zn was the only metal tested that decreased in response to treatment with Flu-TSCZ (Fig. 8d), but these levels were also not impacted by Cu supplementation. Interestingly, in the absence of supplemental Cu, increasing the concentration of Flu-TSCZ from 25 to 100 μM did not further increase cell-associated levels of Cu, Fe, or Mn or further decrease levels of Zn. In the presence of supplemental Cu, however, increasing the concentration of Flu-TSCZ from 25 to 100 μM caused cells to accumulate additional Cu. This result suggests that Flu-TSCZ can increase cellular Cu in a dose-dependent manner, provided ample Cu is available. Without extra Cu in the growth environment, there seems to be a limit to the increase in cellular Cu Flu-TSCZ can provide. Treatment with 10 μM Flu-TSCZ did not impact metal levels, regardless of Cu supplementation (Fig. S10, ESI), indicating that the minimal concentration of Flu-TSCZ required to alter the metal status of C. albicans is between 10 and 25 μM.
Figure 8. Metal content of C. albicans cells reported as cellular concentration (μM).

Cu levels (a) increased during treatment with 25 or 100 μM Flu-TSCZ (red triangles and dark blue circles, respectively) relative to untreated control cells (green circles), with the most prominent difference at 6 h. Supplementing cells with Cu alone (light blue squares) caused Cu levels to increase steadily over time. Co-treatment with Flu-TSCZ and Cu (purple inverted triangles and orange squares) caused higher accumulation of Cu than Cu treatment alone. Treatment with Flu-TSCZ also increased levels of Fe (b) and Mn (c) with Cu supplementation having no additional effect. Zn levels (d) were lower in cells treated with Flu-TSCZ. Cells were grown in YPD medium for time indicated in figure legends. Cell-associated metal levels were analyzed by ICP-MS and cellular metal concentrations were calculated as described in Methods. Data are reported as mean ± SEM, n = 3 biologically independent samples per timepoint.
Flu-TSCZ depletes EPR-detectable Fe and increases EPR-detectable Mn
The subcellular speciation of metals detected by bulk ICP-MS analysis was probed by analyzing cells treated with Flu-TSCZ by electron paramagnetic resonance (EPR) spectroscopy. We have previously demonstrated that fluconazole treatment attenuates the signal at g = 4.3 [50], which represents the kinetically exchangeable, or labile, Fe pool [51]. Fluconazole treatment also modulated the multiline signal at g = 2 [50], taken to be from cellular Mn(II) [51]. As shown in Fig. 9a, treatment with Cu alone did not alter the EPR spectrum relative to the spectrum of untreated control cells. Treating with 10 μM Flu-TSCZ, which is below the minimal concentration tested that fully inhibited growth in YPD (12.5 μM), also did not impact the EPR spectrum, regardless of whether cells were also supplemented with Cu (Fig. 9b). Increasing the concentration of Flu-TSCZ to 25 μM attenuated the signal at g = 4.3 by approximately half and gave rise to a six-line pattern centered at g = 2 (Fig. 9c), which is characteristic of Mn(II) ions. Further increasing the Flu-TSCZ concentration to 100 μM did not result in any additional change to the g = 4.3 signal, but it did significantly increase the intensity of the signal centered at g = 2 (Fig. 9d).
Figure 9. Flu-TSCZ depletes EPR-detectable Fe and increases EPR-detectable Mn.

(a) Cu supplementation alone (red trace) did not impact the EPR signal at g = 4.3 relative to the untreated control cells (teal trace). (b) Treatment with 10 μM Flu-TSCZ with (red trace) or without (teal trace) Cu supplementation was not sufficient to impact the signal at g = 4.3. Increasing the concentration of Flu-TSCZ to 25 μM (c) or 100 μM (d) resulted in an attenuation of the g = 4.3 signal by approximately half and changed the signals in the g = 2 region, which are taken to be from Mn. Conditions: [Flu-TSCZ] is indicated in figure legends, [CuSO4] = 10 μM. Cells were treated for 6 h in YPD medium before analysis.
Discussion
The development of new strategies to combat invasive fungal infections is crucial as pathogens evolve to survive in the presence of existing frontline antifungal drugs. Herein, we report eight new derivatives of fluconazole created by installing Cu binding sites in place of one of the triazole arms, a strategy designed to capitalize on the Cu potentiation we have observed for fluconazole [30]. Although several analogues of fluconazole have been reported, this work provides a unique approach by focusing on structural motifs likely to interact with metal ions to impact metal homeostasis pathways. The idea of dual function azole compounds has also been explored by Papadopoulou and coworkers, who synthesized dual-function derivatives of fluconazole with the aim of inhibiting both Cyp51 and type I nitroreductase of protozoan parasite Trypanosoma cruzi [52].
Fluconazole is a fungistatic drug that inhibits cell growth but does not kill cells. C. albicans can therefore develop tolerance to fluconazole, a phenomenon in which a stable subpopulation of cells grow slowly during drug stress, despite the drug still interacting with the target [35]. Fungicidal drugs, on the other hand, result in cell death and do not give rise to trailing growth. Though none of our analogues exhibited fungicidal activity, cells treated with Flu-Phenol or Flu-TSCZ had a reduced level of trailing growth compared to those treated with fluconazole. It is possible that the chemical structures of these analogues have been sufficiently modified such that cells do not enact the same adaptative mechanisms associated with fluconazole. By contrast, cells treated with Flu-Pyr exhibited the same extent of trailing growth as those treated with fluconazole, suggesting this analogue is recognized in a way that causes a subpopulation of cells to adapt and tolerate this compound. Another possibility is that Flu-Pyr accumulates in cells at a lower concentration than Flu-Phenol and Flu-TSCZ. Trailing growth has been linked to a low intracellular concentration of fluconazole, although it does not result from lower uptake or higher efflux rates and the cause of the lower concentration is unknown [35]. Fluconazole is achiral, but modification of one of the side arms introduces a chiral center. All of the analogues were tested for antifungal activity as racemic mixtures (or a mixture of four diastereomers in the case of Flu-APA). It is possible that separation and testing of individual stereoisomers would reveal a preferable isomer and therefore a lower minimal inhibitory concentration, as has been previously reported for analogues of fluconazole [53].
Flu-TSCZ stood out among the active analogues due to its unique ability to partially rescue growth of the C. albicans ctr1Δ/Δ and C. neoformans ctr1Δ ctr4Δ strains, suggesting this compound is capable of providing bioavailable Cu for use in cytochrome c oxidase. This finding distinguishes Flu-TSCZ from fluconazole, which does not rescue growth of these strains [30]. Interestingly, both fluconazole [36] and Flu-TSCZ increase total levels of cell-associated Cu, yet only Flu-TSCZ rescues growth of the Cu import mutants, indicating that the speciation and/or location of cellular Cu pools differ when treated with Flu-TSCZ versus fluconazole. It was surprising that Flu-TSCZ did not show evidence of Cu(II) binding in YPD medium, given that ionophores that rescue growth of Cu import mutants are thought to do so by acquiring Cu(II) from the growth medium [54]. The ability of Flu-TSCZ to outcompete ferrozine for Cu(I) raises the possibility that Flu-TSCZ rescues growth of the Cu import mutant strains by mobilizing Cu(I). However, the mechanism by which cells accumulate higher levels of total Cu during azole treatment remains unexplained.
Attenuation of the EPR signal at g = 4.3 upon treatment with Flu-TSCZ is consistent with an increased need for cellular Fe to accommodate production of hemeprotein Cyp51 following inhibition by azoles [55]. We have reported a similar effect in the EPR spectrum of fluconazole-treated cells [36]. A notable difference is that 10 μM fluconazole is sufficient to deplete the signal but 10 μM Flu-TSCZ is not. This difference likely stems from the higher MIC of Flu-TSCZ and suggests that antifungal activity (i.e. sufficient inhibition of Cyp51 to cause cell stress) is necessary for cells to mobilize Fe from labile pools. Indeed, upon increasing the concentration of Flu-TSCZ from 10 to 25 μM, changes to bulk cellular metal levels and the whole cell EPR spectra were observed, further supporting the idea that there is a threshold Flu-TSCZ concentration required to impact metal homeostasis. Increasing the concentration of Flu-TSCZ from 25 to 100 μM did not further attenuate the g = 4.3 signal, revealing that mobilization of labile Fe pools is a set response, not one impacted by the amount of Flu-TSCZ. Interestingly, the signal at g = 2 does increase in magnitude with increasing Flu-TSCZ concentration, despite the fact that cell-associated levels of Cu, Fe, Mn, and Zn are the same whether cells were treated with 25 μM or 100 μM Flu-TSCZ. Together, these observations suggest that increasing the concentration of Flu-TSCZ causes metal redistribution without a change to total cellular metal levels. The number of paramagnetic metal species that are EPR detectable in the g = 2 region, including Fe-S clusters [56] and Mn species [51], make it difficult to definitively assign signals in this region for whole cell samples. However, given the six-line pattern of the signal, we hypothesize it arises primarily from Mn.
Conclusion
Innovative approaches are desperately needed to address the rise in incidences of invasive fungal infections and declining efficacy of currently available antifungal drugs. We designed analogues of the antifungal drug fluconazole with the aim of endowing them with Cu binding functionality that would enhance their antifungal activity against C. albicans. Two of these analogues resulted in lower trailing growth of C. albicans compared to the parent fluconazole, and one was shown to alter the levels and speciation of metals in cells. Incorporating metal binding moieties into known antifungal compounds is an avenue worth additional exploration.
Materials and Methods
General
Materials and General Methods
Chemicals and solvents were obtained from commercial suppliers and used as received unless otherwise noted. All reactions were performed under N2 and anhydrous solvent was used unless otherwise noted. All aqueous solutions were prepared using Milli-Q water. Stock solutions were prepared either in DMSO or Milli-Q water. Working stock solutions for each experiment were prepared by serial dilution into growth media. Stock solutions of Mg(II) (1.0 M), Ca(II) (1.0 M), Cu(II) (100 mM), Fe(III) (100 mM), Mn(II) (100 mM), Zn(II) (100 mM), Ni(II) (100 mM), and Co(II) (100 mM) were prepared from MgSO4·7H2O, CaCl2·H2O, CuSO4·5H2O or CuCl2, FeCl3·6H2O, MnCl2·4H2O, ZnSO4·7H2O, NiCl2, and CoCl2. Working solutions of metal salts were prepared for each experiment by serial dilution into growth media from concentrated stock solutions prepared in Milli-Q water.
Compound Characterization
Bulk purity was confirmed by NMR and analytical LC-MS and elemental composition by HRMS. NMR spectra (1H and 13C) were obtained on either a 400 MHz Varian INOVA or 500 MHz Varian spectrometer, as noted. High-resolution mass spectrometry was performed on an Agilent LCMS-TOF–DART at Duke University’s Department of Chemistry Instrumentation Facility.
Yeast Strains and Culture Conditions
Fungal stocks were maintained in 25% glycerol in YPD at −80 °C. Unless otherwise noted, experiments were performed with C. albicans clinical isolate SC5314, which was obtained from the American Type Culture Collection (ATCC). The SN152 strain and isogenic mac1Δ/Δ strain were obtained from the Fungal Genetics Stock Center [57, 58]. The KC2 strain and isogenic crp1Δ/Δ strain were generously provided by the Culotta Lab of Johns Hopkins University. A second SC5314 strain and isogenic ctr1Δ/Δ strain were kindly provided by the Brown Lab of the University of Exeter. The C. neoformans H99 and ctr1Δ ctr4Δ strains were generously provided by the Thiele Lab of Duke University. Cells were cultured at 30 °C in yeast peptone dextrose (YPD, Gibco, catalog number A1374501) medium, unless otherwise indicated. The SC5314 strain from the Brown lab was used as a control in experiments involving the ctr1Δ/Δ strain.
Growth Assays (General)
Prior to all experiments, cells were streaked onto YPD agar plates from frozen glycerol stocks and incubated at 30 °C for 24 h. A single colony was used to inoculate 10 mL of YPD medium, which was then incubated overnight (∼18 h) at 30 °C, 200 rpm. This overnight culture was diluted to an optical density at 600 nm (OD600) of 0.002 with fresh YPD medium and used as the working culture. Azole compounds to be tested were serially diluted 2-fold in YPD medium from DMSO stocks to final concentrations ranging from 0–100 μM, with < 1% DMSO and plated in a clear, flat-bottomed 96-well plate. For experiments in which supplemental Cu was used, fresh working solutions of Cu(II) were prepared by diluting 100 mM aqueous stocks of CuSO4 into YPD medium, and aliquots of these working solutions were added to appropriate wells at final concentrations indicated in figure legends. BCS working solutions were similarly prepared. Azole working solutions were serially diluted 2-fold to achieve final test concentrations indicated in figure legends. The working culture of C. albicans was then aliquoted to the 96-well plate to a final OD600 of 0.001 and a final volume of 200 μL per well. For each experiment, a compound-free positive growth control and a cell-free, negative control were included. Plates were incubated for 48 h at 30 °C, 200 rpm. Plates were covered with air-permeable AeraSeal film (Sigma) to minimize evaporation.
Fungal growth was evaluated by measuring OD600 using a PerkinElmer Victor3 V multilabel plate reader at 0, 24, and 48 h. For some experiments, additional timepoint data were collected. OD600 values were adjusted by subtracting the 0 h timepoint readings from other timepoint data to remove background signal from growth media. In cases where the OD was outside of the linear range (OD600 values approximately 0.0–0.8), cell suspensions were diluted 4-fold in fresh media and rescanned. To calculate actual OD values, the 0 h timepoint reading was subtracted from the diluted readings, and this value was multiplied by four. At least two biological replicates were performed with a minimum of three technical replicates per experiment. For a single experiment, each of the three replicate conditions were averaged and the error was calculated as standard deviation (SD), which is indicated by error bars in all figures. Final 48-h timepoint data is reported by plotting OD600 readings versus treatment conditions.
Metal Drop-Out Growth Assays (Defined Media)
All Tris-buffered Synthetic Defined (Tris:SD) media formulations were prepared from Chelex-treated Milli-Q water with individual addition of media components to allow for rigorous control of metal content. To deplete trace metals from water prior to media preparation, Milli-Q water was treated with Chelex 100 resin 100–200 mesh via batch method (50 g/L, Bio-Rad Laboratories). A concentrated stock of Synthetic Defined media omitted for Cu, Fe, Mn, and Zn (10X SD-) was prepared in the Chelex-treated Milli-Q water by adding glucose and yeast nitrogen base (YNB) ingredients at 10X concentrations. YNB components were added individually to avoid trace metals present in commercial YNB mixtures. To prepare working 1X Tris:SD medium, 10X SD- was diluted into Chelex-treated water (1:10), and Ultra-Pure Tris-HCl (VWR) was added to a final concentration of 50 mM. The pH of the media was adjusted to 7.4 using 1.0 M HCl or 1.0 M NaOH, and this media was filter-sterilized. Finally, CuSO4, FeCl3, MnCl2, and ZnSO4 were added, as appropriate, to create individual dropout medias for each metal. Final metal drop-out formulations were analyzed for metal content (Tables S4–S7, ESI).
ctr1Δ/Δ and ctr1Δ ctr4Δ Growth Recovery Assay
Overnight cultures of WT SC5314 and isogenic ctr1Δ/Δ (C. albicans) or WT H99 and isogenic ctr1Δ ctr4Δ (C. neoformans) in YPD were washed twice with PBS, pH 7.4 and resuspended in 10 mL YPEG medium with 2% EtOH and 3% glycerol. Cell suspensions were diluted to OD600 of 0.002 with fresh YPEG medium. WT was aliquoted to the top half of a 96-well plate and Cu importer deletion strain to the bottom half. Test compounds were added from DMSO stock solutions to final concentrations ranging from 0–100 μM. For each test run, a compound-free positive growth control and a cell-free, negative control were included. Plates were incubated at 30 °C and OD600 recorded at 0, 24, and 48 h. All tests were performed in triplicate for each condition in a single experiment, and two separate experiments were carried out. For a single experiment, each of the three replicate conditions were averaged and the error was calculated as standard deviation (SD).
Determination of CFUs
Fungicidal assays were initiated in 96-well plates by following the procedure described above for the growth assay for WT C. albicans in YPD medium. Aliquots of cell suspensions were removed after 48 h, diluted and plated on solid YPD medium. The number of colonies on each plate for each condition were counted after incubating the plates at 30 °C for 24 h. All tests were performed in duplicate for each condition in a single experiment, and two separate experiments were carried out.
UV-Vis and EPR Spectroscopy Measurements
UV-Visible absorption spectra were collected using a Varian Cary 50 UV-Visible spectrophotometer in quartz cuvettes with 1 cm pathlengths. UV-Vis studies probing Cu(I) binding were conducted in air using CuCl2 with excess ascorbate added as a reducing agent. X-band continuous wave (CW) EPR spectroscopy was conducted on a Bruker ESP 300 spectrometer equipped with an Oxford Instruments ESR 910 continuous helium flow cryostat. Typical experimental parameters were at 77 K, 9.37 GHz, 20 mW microwave power, and 5 G modulation amplitude. Spectra were baseline corrected using Bruker Xenon data processing software.
Preparation of Cells for EPR Spectroscopy
Overnight cultures of C. albicans in YPD medium were inoculated into fresh YPD medium at an OD600 of 0.3 and allowed to grow for 3 h at 30 °C with shaking at 200 rpm. Cultures were then either left untreated or treated with CuSO4 (10 μM), Flu-TSCZ (10, 25, or 100 μM), or both CuSO4 and Flu-TSCZ. After 6 h of treatment, 2 × 109 cells were harvested and washed with cold 5 mM disodium EDTA in PBS (1 mL) followed by cold 20 mM Tris-HCl, pH 7.4 (1 mL). The cell pellet was resuspended in 300 μL of 20 mM Tris-HCl, pH 7.4 containing 20% glycerol. The entire suspension was transferred to an EPR tube and cells were flash-frozen immediately and stored at –80 °C until EPR measurements were performed.
Preparation of Cells for ICP-MS
Overnight cultures of C. albicans in YPD medium were inoculated into fresh YPD medium at an OD600 of 0.3 and allowed to grow for 3 h at 30 °C with shaking at 200 rpm. Cultures were then either left untreated or treated with CuSO4 (10 μM), Flu-TSCZ (10, 25, or 100 μM), or both CuSO4 and Flu-TSCZ. At the timepoints indicated in figure legends, 1 × 108 cells were harvested and placed in 15-mL metal-free centrifuge tubes (VWR, cat. no. 89049–170). Cell suspensions were pelleted at 4,000 × g and washed with 1 mL of 1 mM disodium EDTA prepared in ultrapure water for trace metal analysis (VWR, cat. no. 87003–236), then with 1 mL of ultrapure water for trace metal analysis. Pellets were resuspended in 100 μL of concentrated nitric acid (Trace Metal Grade, Fisher) and heated at 90 °C for 1 h. After digestion, samples were diluted with 1.5 mL of 1% nitric acid.
ICP-MS Measurements
ICP-MS measurements were performed by Dr. Martina Ralle in the OHSU Elemental Analysis Core with partial support from the NIH instrumentation grant S10RR025512. Measurements were performed using an Agilent 7700x equipped with an ASX 500 autosampler. The system was operated at a radio frequency power of 1550 W, an argon plasma gas flow rate of 15 L/min, and Ar carrier gas flow rate of 0.9 L/min. Elements were measured in kinetic energy discrimination (KED) mode using He gas (4.3 ml/min). Data were quantified using an 11-point (0, 0.5, 1, 2, 5, 10, 20, 50, 100, 500, 1000 ppb (μg/kg)) for all elements except Mg and Ca (0, 0.05, 0.1, 0.2, 0.5, 1, 2, 5, 10, 50, 100 ppm (μg /kg)) using a multi-element standard (CEM 2, (VHG-SM70B-100)) and single element standards for Mg (Inorganic Ventures, CGMG-1) and Ca (Inorganic Ventures, CGCA-1). For each sample, data were acquired in triplicates and averaged. A coefficient of variance (CoV) was determined from frequent measurements of a sample containing approximately 10 ppb of all elements except Ca (1 ppm). An internal standard (Sc, Ge, Bi) continuously introduced with the sample was used to correct for detector fluctuations and to monitor plasma stability. Accuracy of the calibration curve was assessed by measuring NIST reference material (water, SRM 1643f) twice during the measurement and found to within ± 4% for all determined elements.
To obtain cellular metal concentrations (μM/cell), the analytical level of phosphorus (P) determined for each sample by ICP-MS was converted to cell count using a calculated constant for the mol P per cell. The constant, determined to be 1.3 ± 0.4 × 10−14 mol P/cell, was calculated by dividing the analytical mol P for each sample by the theoretical number of cells for each sample (1 × 108), then averaging these values. This constant represents the average P content per cell and corrects for the variation in the number of cells submitted for analysis. For μM/cell, a cell volume of 95 fL was used [59].
Synthetic Procedures
2-(2,4-difluorophenyl)-1-(1H-1,2,4-triazol-1-yl)pent-4-yn-2-ol (Flu-alkyne, 2) [14]
To a suspension of 1 (0.1986 g, 0.890 mmol) in DMF/THF (1:1, 2 mL), propargyl bromide (80 wt% in toluene, 0.30 mL, 3.37 mmol) was added. To this well-stirred mixture, activated zinc powder (0.1793 g, 2.74 mmol) was added. Prior to use, Zn powder was stirred in 2% HCl, washed with H2O, and vacuum dried. The exothermic reaction brought itself to reflux and was allowed to attain room temperature. The reaction mixture was then stirred under N2 for 6.5 hours at room temperature. HCl (5%) was then added to the reaction flask, and the product was extracted with DCM (3×40 mL), washed with H2O (2×20 mL) and brine (2×20 mL), dried over Na2SO4, and filtered. The solvent was removed by evaporation under reduced pressure, yielding a light orange sticky solid. In most cases, the product was used without further purification. The product can be recrystallized in MeOH to give a white solid as pure product, but with highly diminished yield. Yield (crude): 0.205 g (87%). 1H NMR (400 MHz, Chloroform-d) δ 7.97 (s, 1H), 7.84 (s, 1H), 7.53 (td, J = 9.0, 6.4 Hz, 1H), 6.85 – 6.75 (m, 2H), 4.82 – 4.66 (m, 2H), 4.50 (s, 1H), 2.85 (qd, J = 17.0, 2.7 Hz, 2H), 2.07 (t, J = 2.7 Hz, 1H). MS: m/z 264 (M + 1).
1-((2-(2,4-difluorophenyl)oxiran-2-yl)methyl)-1H-1,2,4-triazole (Flu-epoxide, 3) [60]
To a solution of 1 (0.5710 g, 2.56 mmol) in toluene (6 mL), trimethylsulfoxonium iodide (0.6851 g, 3.11 mmol) was added, followed by sodium hydroxide (20%, 0.6 mL). Trimethylsulfoxonium iodide is light-sensitive and was handled in the dark. The reaction mixture was heated to 60 °C and stirred under N2 in the dark for 4.5 hours. The product was extracted into toluene (3×50 mL), and the combined organic layers were washed with H2O (2×25 mL) and brine (2×25 mL), dried over Na2SO4, and filtered. The solvent was removed by evaporation under reduced pressure, yielding a sticky orange solid. Yield: 0.387 g (64%). 1H NMR (400 MHz, Chloroform-d) δ 7.99 (s, 1H), 7.76 (s, 1H), 7.08 (td, J = 8.4, 6.2 Hz, 1H), 6.72 (dddd, J = 12.6, 10.4, 8.2, 2.5 Hz, 2H), 4.58 (dd, J = 127.3, 14.9 Hz, 2H), 2.82 (dd, J = 28.0, 4.7 Hz, 2H). MS: m/z 238 (M + 1).
1-azido-2-(2,4-difluorophenyl)-3-(1H-1,2,4-triazol-1-yl)propan-2-ol (Flu-azide, 4) [12]
To a solution of 3 (0.6873 g, 2.90 mmol) in MeOH (15 mL), ammonium chloride (0.1906 g, 3.56 mmol) was added followed by sodium azide (0.5685 g, 8.74 mmol). The reaction mixture was heated to reflux under N2. After 6 hours, the mixture was diluted with 5% NaHCO3. The product was extracted into EtOAc (3×50 mL), and the combined organic layers were washed with H2O (1×25 mL) and brine (1×25 mL), dried over Na2SO4, and filtered. The compound was purified by column chromatography on silica gel using EtOAc/BuOH/AcOH/H2O (80:10:5:5) as eluent. The solvent was removed by evaporation under reduced pressure, yielding a light-yellow oil. Yield: 0.542 g (67%). 1H NMR (500 MHz, Chloroform-d) δ 7.94 (s, 1H), 7.76 (s, 1H), 7.49 (td, J = 9.0, 6.4 Hz, 1H), 6.82 – 6.71 (m, 2H), 5.80 (s, 1H), 4.74 – 4.61 (m, 2H), 3.59 (dd, J = 86.2, 12.9 Hz, 2H); 13C NMR (125 MHz, Chloroform-d) δ 163.03 (dd, J = 252.5, 13.9 Hz), 158.61 (dd, J = 245.9, 11.7 Hz), 151.62, 144.30, 130.19 (dd, J = 9.5, 5.5 Hz), 122.78 (dd, J = 13.0, 3.9 Hz), 111.98 (dd, J = 21.1, 3.4 Hz), 104.32 (t, J = 26.6 Hz), 76.26 (d, J = 5.1 Hz), 57.01 (d, J = 4.6 Hz), 54.74 (d, J = 5.9 Hz). MS: m/z 281 (M + 1).
1-amino-2-(2,4-difluorophenyl)-3-(1H-1,2,4-triazol-1-yl)propan-2-ol (Flu-amine, 5)
To a nitrogen-flushed solution of 4 (1.42 g, 5.07 mmol) in 10:1 MeOH/CHCl3 (17 mL), Pd/C (0.4468 g) was added. The reaction flask was fitted with a balloon to allow for gas expansion. To this suspension, triethylsilane (6.75 mL, 42.3 mmol) was slowly added dropwise, resulting in an exothermic reaction. Care must be taken not to add triethylsilane too quickly. The reaction mixture was left to stir at room temperature for 3 hours under N2, at which point the reaction mixture was filtered through Celite to remove Pd/C. The product was extracted into H2O (3×50 mL), the solvent was removed by evaporation under reduced pressure, and the product was recrystallized in CHCl3, yielding a white crystalline solid. Yield: 0.535 g (50%). 1H NMR (400 MHz, Methanol-d4) δ 8.41 (s, 1H), 7.90 (s, 1H), 7.56 (td, J = 9.1, 6.5 Hz, 1H), 7.12 – 6.94 (m, 2H), 4.77 (s, 2H), 3.49 (dd, J = 98.9, 13.3 Hz, 2H). MS: m/z 255 (M + 1).
4-(chloromethyl)-1H-imidazole (10) [61]
To a suspension of 4(5)-hydroxymethyl imidazole (0.5934 g, 6.04 mmol) in ACN (7 mL), a catalytic amount of DMF (0.3 mL) was added, followed by thionyl chloride (0.75 mL, 10.28 mmol). The exothermic reaction brought itself to reflux and was allowed to return to room temperature. After 2.5 hours, the solvent and remaining thionyl chloride were removed by evaporation under reduced pressure, yielding a brown solid. Yield: 0.617 g (88%). 1H NMR (400 MHz, Acetonitrile-d3) δ 13.76 (s, 1H), 8.56 (s, 1H), 7.45 (s, 1H), 4.78 (s, 2H). MS: m/z 117 (M + 1).
4-(azidomethyl)-1H-imidazole (6a) [61]
To a solution of 10 (0.3648 g, 3.13 mmol) in DMF (11 mL), sodium azide (0.4070 g, 6.26 mmol) was added. After stirring at room temperature for 19 hours, the reaction mixture was diluted with H2O, and the product was extracted into EtOAc (3×25mL). The combined organic layers were washed with H2O (2×25 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure, yielding a brown crystalline solid. Yield: 0.153 g (40%). 1H NMR (400 MHz, Acetonitrile-d3) δ 11.74 (s, 1H), 7.69 (s, 1H), 7.14 (s, 1H), 4.29 (s, 2H). MS: m/z 124 (M + 1).
2-azidophenol (6b) [62]
2-aminophenol (0.7642 g, 7.00 mmol) was dissolved in HCl (2.0 M, 10 mL) at −4 °C to give a light pink solution. A separate solution was prepared by dissolving sodium nitrite (0.5878 g, 8.52 mmol) in H2O (2 mL) at 0 °C, and this solution was added dropwise to the flask containing 2-aminophenol over a period of five minutes. After an additional five minutes, urea (0.0522 g, 0.869 mmol) was added to consume excess nitrous acid. In a separate flask, sodium azide (0.9136 g, 14.05 mmol) was added to a solution of sodium acetate (0.0028 g, 0.034 mmol) in H2O (10 mL) at −3 °C. To this solution, the previously prepared diazonium salt solution was added dropwise, and the reaction mixture was stirred at −3 °C for 2 hours. The product was extracted into diethyl ether (3×25 mL), and the combined organic layers were washed with H2O (1×25 mL), dried over Na2SO4, and filtered. Solvent was removed by evaporation under reduced pressure, giving a dark red oil. Yield: 0.934 g (99%). 1H NMR (400 MHz, DMSO- d6) δ 10.05 (s, 1H), 7.03 – 6.73 (m, 4H). MS: m/z 134 (M − 1).
N,N-bis(pyridin-2-ylmethyl)prop-2-yn-1-amine (DPA-alkyne, 7a) [63]
To a solution of bis(2-picolyl)amine (1.000 g, 5.02 mmol) in toluene (50 mL), propargyl bromide (80 wt% in toluene, 0.70 mL, 7.86 mmol) and triethylamine (0.75 mL, 5.38 mmol) were added. The reaction mixture was refluxed for 23 hours, during which time a brown solid precipitated. The solid was removed via vacuum filtration, and the filtrate was concentrated under reduced pressure. The compound was purified by column chromatography on silica gel using DCM/ACN/NH4OH (5:5:0.1) as eluent. The solvent was removed by evaporation under reduced pressure, yielding a dark brown oil. Yield: 0.766 g (64%). 1H NMR (400 MHz, Chloroform-d) δ 8.27 (d, J = 4.9 Hz, 2H), 7.40 – 7.31 (m, 2H), 7.24 (d, J = 7.8 Hz, 2H), 6.90 – 6.81 (m, 2H), 3.65 (s, 4H), 3.17 – 3.12 (m, 2H), 2.10 (q, J = 2.0 Hz, 1H). MS: m/z 238 (M + 1).
Methyl hydrazinecarbodithioate (11) [64]
To an ice-cold solution of potassium hydroxide (6.554 g, 117 mmol) in H2O (12.5 mL), 2-propanol (10 mL) was added, and this solution was maintained at 0 °C. Next, hydrazine hydrate (80% solution, 7.3 mL, 117 mmol) was added with stirring. Ice-cold carbon disulfide (7.0 mL, 117 mmol) was added dropwise to this solution over 1.5 h while maintaining 0 °C. The bright yellow mixture obtained was further stirred for 2.5 h, and then, ice-cold iodomethane (7.4 mL, 117 mmol) was added drop wise over a period of 2 h. Stirring was continued for an additional 1.5 h during which time a white precipitate formed. The precipitate was filtered, washed with cold H2O, and recrystallized from DCM to give a white crystalline solid. Yield: 4.155 g (29%). 1H NMR (400 MHz, Chloroform-d) δ 8.36 (s, 1H), 4.69 (s, 2H), 2.66 (s, 3H).
(E)-4-((2-(methylcarbamothioyl)hydrazineylidene)methyl)benzoic acid (8a) [65]
A solution of 4-formylbenzoic acid (0.4590 g, 3.06 mmol) and 11 (0.3566 g, 3.39 mmol) was refluxed in EtOH (6 mL) for 1.5 hours, during which time a solid precipitated. The precipitate was isolated via vacuum filtration, and triturated with EtOH (3×10 mL), yielding a white crystalline solid. Yield: 0.572 g (79%). 1H NMR (400 MHz, DMSO-d6) δ 13.07 (s, 1H), 11.64 (s, 1H), 8.65 – 8.60 (m, 1H), 8.09 (s, 1H), 7.98 – 7.89 (m, 4H), 3.02 (d, J = 4.5 Hz, 3H). MS: m/z 238 (M + 1).
1-(1-((1H-imidazol-4-yl)methyl)-1H-1,2,3-triazol-4-yl)-2-(2,4-difluorophenyl)-3-(1H-1,2,4-triazol-1-yl)propan-2-ol (Flu-Im, I-a)
to a mixture of 2 (0.6703 g, 2.55 mmol) and 6a (0.2980 g, 2.42 mmol) in tert-butanol and H2O (1:1, 35 mL), (+)-sodium L-ascorbate (7.203 g, 36.4 mmol) was added, followed by copper (II) sulfate pentahydrate (0.9095 g, 3.64 mmol). The reaction mixture stirred at room temperature for 20 hours before being diluted with a solution of ethanolamine and water (25 mL/100 mL). The product was extracted into EtOAc (5×25 mL), and the combined organic layers were washed with H2O (1×25 mL), dried over Na2SO4, and filtered. The solvent was removed by evaporation under reduced pressure, yielding an off-white solid. Yield: 0.083 g (9%). 1H NMR (400 MHz, DMSO-d6) δ 12.06 (s, 1H), 8.28 (s, 1H), 7.75 (s, 1H), 7.61 (s, 1H), 7.50 (s, 1H), 7.22 – 7.09 (m, 2H), 7.05 (s, 1H), 6.82 (td, J = 8.5, 2.6 Hz, 1H), 5.97 (s, 1H), 5.30 (s, 2H), 4.72 – 4.46 (m, 2H), 3.38 – 3.29 (d, 1H), 3.11 (d, J = 14.8 Hz, 1H). 13C NMR (125 MHz, DMSO-d6) δ 160.22 (ddd, J = 354.2, 246.3, 12.4 Hz), 150.56, 144.93, 141.53, 129.92 (dd, J = 9.6, 6.1 Hz), 123.26, 110.59 (dd, J = 20.5, 3.1 Hz), 103.84 (t, J = 26.8 Hz), 73.70 (d, J = 5.0 Hz), 56.88 (d, J = 4.6 Hz), 47.02, 34.56 (d, J = 4.5 Hz). ESI-HRMS calcd. for C17H16F2N8O [M+H]+ = 387.1488, found 387.1489.
2-(4-(2-(2,4-difluorophenyl)-2-hydroxy-3-(1H-1,2,4-triazol-1-yl)propyl)-1H-1,2,3-triazol-1-yl)phenol (Flu-Phenol, I-b)
To a solution of 2 (0.3127 g, 1.19 mmol) and 6b (0.2200 g, 1.63 mmol) in a mixture of tert-butanol and H2O (1:1, 8 mL), (+)-sodium L-ascorbate (0.0622 g, 0.314 mmol) was added, followed by copper (II) acetate monohydrate (0.0261, 0.131 mmol). The reaction mixture was allowed to stir for 5 days at room temperature. The product was extracted into EtOAc (3×25 mL), and the combined organic layers were washed with H2O (1×25 mL) and brine (1×25 mL), dried over Na2SO4, and filtered. The compound was purified by column chromatography on silica gel using 5% MeOH in DCM as eluent. The solvent was removed by evaporation under reduced pressure, yielding an off-white powder. Yield: 0.305 g (65%). 1H NMR (500 MHz, DMSO-d6) δ 10.49 (s, 1H), 8.31 (d, J = 1.1 Hz, 1H), 8.03 (s, 1H), 7.78 (d, J = 1.1 Hz, 1H), 7.55 (dt, J = 8.0, 1.5 Hz, 1H), 7.31 – 7.21 (m, 2H), 7.18 (ddd, J = 12.0, 9.1, 2.3 Hz, 1H), 7.06 (dt, J = 8.3, 1.3 Hz, 1H), 6.97 – 6.91 (m, 1H), 6.86 (td, J = 8.5, 2.7 Hz, 1H), 6.05 (s, 1H), 4.75 – 4.55 (m, 2H), 3.46 (d, J = 14.9 Hz, 1H), 3.25 (d, J = 14.8 Hz, 1H). 13C NMR (125 MHz, DMSO-d6) δ 160.32 (ddd, J = 351.8, 246.6, 12.7 Hz), 150.61, 149.02, 144.98, 141.44, 130.05 (t, J = 8.0 Hz), 129.71, 125.84 – 125.66 (m), 124.93, 124.55, 124.44, 119.60, 117.17, 110.72 (d, J = 20.3 Hz), 103.93 (t, J = 27.0 Hz), 73.79 (d, J = 5.0 Hz), 56.97, 34.62. ESI-HRMS calcd. for C19H16F2N6O2 [M+H]+ = 399.1376, found 399.1378.
Dimethyl 2-((1-(2-(2,4-difluorophenyl)-2-hydroxy-3-(1H-1,2,4-triazol-1-yl)propyl)-1H-1,2,3-triazol-4-yl)methyl)malonate (Flu-DME, II-a)
To a solution of 4 (0.305 g, 1.09 mmol) and dimethyl propargylmalonate (0.2 mL, 1.31 mmol) in tert-butanol and H2O (1:1, 20 mL), (+)-sodium L-ascorbate (0.0488 g, 0.246 mmol) was added, followed by copper (II) acetate monohydrate (0.0311 g, 0.156 mmol). The reaction mixture was stirred at room temperature for 21 hours. The product was extracted into EtOAc (3×25 mL), and the combined organic layers were washed with a saturated solution of disodium EDTA (1×25 mL) and H2O (1×25 mL), dried over Na2SO4, and filtered. The solvent was removed by evaporation under reduced pressure, yielding a yellow oil, which was purified by column chromatography on silica gel using 5% MeOH in DCM with 1% triethylamine as eluent. Fractions containing product were combined and further purified via HPLC on a C18 column using a linear gradient running from 95%–5% H2O/ACN over 40 min, giving a white shiny solid. Yield: 0.130 g (27%). 1H NMR (400 MHz, Chloroform-d) δ 7.98 (s, 1H), 7.79 (s, 1H), 7.45 (s, 1H), 7.37 (td, J = 8.6, 6.0 Hz, 1H), 6.82 – 6.71 (m, 2H), 5.43 (s, 1H), 4.83 (d, J = 14.3 Hz, 1H), 4.70 (q, J = 14.3 Hz, 2H), 4.25 (d, J = 14.3 Hz, 1H), 3.79 (td, J = 7.5, 1.6 Hz, 1H), 3.76 – 3.62 (m, 6H), 3.24 (d, J = 7.5 Hz, 2H). 13C NMR (125 MHz, Chloroform-d) δ 168.96, 165.39 – 1154.29 (m), 151.95, 144.63, 143.83, 130.10, 124.22, 121.95, 112.08 (d, J = 20.8 Hz), 104.39 (t, J = 26.9 Hz), 75.27 (d, J = 4.9 Hz), 55.87, 54.63, 52.72, 51.27, 30.96, 24.85. ESI-HRMS calcd. for C19H20F2N6O5 [M+H]+ = 451.1536, found 451.1539.
2-(2,4-difluorophenyl)-1-(4-(pyridin-2-yl)-1H-1,2,3-triazol-1-yl)-3-(1H-1,2,4-triazol-1-yl)propan-2-ol (Flu-Pyr, II-b)
To a solution of 4 (0.2850 g, 1.02 mmol) and 2-ethynylpyridine (0.1470 g, 1.43 mmol) in a mixture of tert-butanol and H2O (1:1, 8 mL), (+)-sodium L-ascorbate (0.0433 g, 0.219 mmol) was added, followed by copper (II) sulfate pentahydrate (0.0272, 0.109 mmol). The reaction mixture was allowed to stir for 21 hours at room temperature. The product was extracted into EtOAc (3×25 mL), and the combined organic layers were washed with H2O (1×25 mL) and brine (1×25 mL), dried over Na2SO4, and filtered. The compound was purified by column chromatography on silica gel using 5% MeOH in DCM as eluent. The solvent was removed by evaporation under reduced pressure, yielding a light pink crystalline solid. Yield: 0.178 g (46%). 1H NMR (500 MHz, Chloroform-d) δ 8.50 (ddd, J = 4.9, 1.8, 0.9 Hz, 1H), 8.25 (s, 1H), 8.05 (dt, J = 7.9, 1.1 Hz, 1H), 7.99 (s, 1H), 7.77 (s, 1H), 7.72 (td, J = 7.8, 1.8 Hz, 1H), 7.42 (td, J = 9.1, 6.3 Hz, 1H), 7.19 (ddd, J = 7.6, 4.9, 1.2 Hz, 1H), 6.79 (ddd, J = 12.0, 8.3, 2.5 Hz, 1H), 6.72 (tdd, J = 8.7, 2.5, 0.8 Hz, 1H), 5.68 (s, 1H), 4.91 – 4.77 (m, 3H), 4.37 (d, J = 14.3 Hz, 1H). 13C NMR (125 MHz, Acetonitrile-d3) δ 165.78 – 158.69 (m), 152.30, 151.01, 150.53, 148.48, 146.06, 137.95, 130.91, 125.21, 123.88, 123.59, 120.56, 112.07, 105.00, 75.87, 56.78, 55.87. ESI-HRMS calcd. for C18H15F2N7O [M+H]+ = 384.1379, found 384.1385.
1-(4-((bis(pyridin-2-ylmethyl)amino)methyl)-1H-1,2,3-triazol-1-yl)-2-(2,4-difluorophenyl)-3-(1H-1,2,4-triazol-1-yl)propan-2-ol (Flu-DPA, II-c)
To a solution of 4 (0.286 g, 1.02 mmol) and 7a (0.300, 1.26 mmol) in MeOH (7 mL), (+)-sodium L-ascorbate (0.0422 g, 0.213 mmol) was added, followed by copper (II) acetate monohydrate (0.0225 g, 0.113 mmol). The reaction mixture stirred at room temperature for 24 hours. The product was extracted into EtOAc (5×20 mL), and the combined organic layers were washed with disodium EDTA (3×30 mL), then H2O (1×20 mL) and brine (1×20 mL), dried over Na2SO4, and filtered. The solvent was removed by evaporation under reduced pressure to afford a metallic dark brown oil as crude product. Crude product was purified via HPLC on a C18 column using a linear gradient running from 95%–5% H2O/ACN over 40 min, giving a light brown oil. Yield: 0.098 g (19%). 1H NMR (400 MHz, Chloroform-d) δ 8.53 (dd, J = 4.9, 1.8 Hz, 2H), 8.02 (s, 1H), 7.83 (s, 1H), 7.67 (s, 1H), 7.64 (dd, J = 7.7, 1.9 Hz, 1H), 7.50 (d, J = 7.8 Hz, 2H), 7.38 (td, J = 9.0, 6.3 Hz, 1H), 7.18 – 7.12 (m, 2H), 6.71 (dtd, J = 16.3, 8.3, 4.2 Hz, 2H), 5.40 (s, 1H), 4.87 – 4.30 (m, 4H), 3.80 (s, 2H), 3.72 (s, 4H). 13C NMR (125 MHz, Chloroform-d) δ 164.77 – 156.89 (m), 159.15, 152.11, 149.23, 144.64 (d, J = 23.0 Hz), 136.63, 130.20 (dd, J = 9.6, 5.4 Hz), 125.35, 123.34, 122.23, 112.20 (dd, J = 20.9, 3.3 Hz), 105.18 – 103.90 (m), 75.52 (d, J = 4.7 Hz), 59.52, 55.98 (d, J = 4.8 Hz), 54.77 (d, J = 5.7 Hz), 48.49, 41.12. ESI-HRMS calcd. for C26H25F2N9O [M+H]+ = 518.2223, found 518.2224.
2-amino-3-(1-(2-(2,4-difluorophenyl)-2-hydroxy-3-(1H-1,2,4-triazol-1-yl)propyl)-1H-1,2,3-triazol-4-yl)propanoic acid (Flu-APA, II-d)
To a solution of 4 (0.2106 g, 0.752 mmol) and DL-propargylglycine (0.0908 g, 0.803 mmol) in a mixture of tert-butanol and H2O (1:1, 6 mL), (+)-sodium L-ascorbate (0.0382 g, 0.193 mmol) was added, followed by copper (II) acetate monohydrate (0.0207, 0.104 mmol). The reaction mixture was allowed to stir for 14 hours at room temperature. The reaction mixture was diluted with H2O (20 mL), and unreacted 4 was extracted into DCM (3×25 mL), while the product remained in the water layer. The solvent was removed by evaporation under reduced pressure, yielding a yellow sticky solid as crude product. The crude product was purified via HPLC on a C18 column using a linear gradient running from 95%–5% H2O/ACN over 40 min, giving a shiny cream-colored solid as clean product. Yield: 0.105 g (33%). 1H NMR (400 MHz, DMSO-d6) δ 8.79 (s, 1H), 8.27 (s, 1H), 8.24 (d, J = 11.2 Hz, 1H), 7.72 – 7.57 (m, 2H), 7.33 (td, J = 8.5, 8.1, 3.6 Hz, 1H), 7.06 (s, 1H), 5.40 (dd, J = 14.3, 5.0 Hz, 1H), 5.18 (dd, J = 27.3, 14.4 Hz, 2H), 5.01 (d, J = 14.4 Hz, 1H), 4.57 (dd, J = 10.0, 5.5 Hz, 1H), 3.64 (s, 2H), 3.57 (td, J = 15.6, 13.9, 6.4 Hz, 2H). 13C NMR (125 MHz, DMSO-d6) δ 169.99 (d, J = 7.1 Hz), 163.97 – 157.05 (m), 150.88, 145.22, 140.43, 129.91, 124.97 (d, J = 12.5 Hz), 123.14, 110.98 (d, J = 20.2 Hz), 104.00 (t, J = 26.8 Hz), 73.76 (d, J = 4.9 Hz), 55.38 (d, J = 5.9 Hz), 54.80, 52.02 (d, J = 9.6 Hz), 26.15 (d, J = 5.6 Hz). ESI-HRMS calcd. For C16H17F2N7O3 [M+H]+ = 394.1434, found 394.1440.
(E)-N-(2-(2,4-difluorophenyl)-2-hydroxy-3-(1H-1,2,4-triazol-1-yl)propyl)-4-((2-(methylcarbamothioyl)hydrazineylidene)methyl)benzamide (Flu-TSCZ, III-a)
To a solution of 8a (0.3058 g, 1.29 mmol) in dry DMF (6.5 mL), N-methylmorpholine (50% in DMF, 0.5 mL, 2.27 mmol) was added followed by HBTU (0.5337 g, 1.41 mmol). This mixture was allowed to stir under N2 for 15 min. Then, compound 5 (0.3573 g, 1.41 mmol) was added. The reaction mixture was allowed to stir at room temperature under N2 for 18 h. The product was extracted into EtOAc (3×50 mL), and the combined organic layers were washed with 5% LiCl (3×150 mL), NaHCO3 (1×200 mL), and H2O (2×200 mL), dried over Na2SO4, and filtered. Solvent was removed by evaporation under reduced pressure, and crude product was purified by column chromatography on silica gel using 100% EtOAc as eluent to give the pure compound as a light-yellow powder. Yield: 0.424 g (72%). 1H NMR (400 MHz, DMSO-d6) δ 11.61 (s, 1H), 8.59 (t, J = 6.1 Hz, 2H), 8.34 (s, 1H), 8.06 (s, 1H), 7.90 – 7.74 (m, 5H), 7.41 (td, J = 9.0, 6.7 Hz, 1H), 7.18 (ddd, J = 11.9, 9.1, 2.6 Hz, 1H), 6.93 (td, J = 8.5, 2.6 Hz, 1H), 6.27 (s, 1H, OH), 4.78 – 4.51 (m, 2H), 3.79 (d, J = 6.0 Hz, 2H), 3.02 (d, J = 4.5 Hz, 3H, CH3). 13C NMR (125 MHz, DMSO-d6) δ 177.83, 167.15, 163.71 – 156.77 (m), 150.53, 144.99, 140.46, 137.14, 134.42, 130.00 (d, J = 9.9 Hz), 127.62, 126.90, 124.77 (d, J = 12.6 Hz), 110.70 (d, J = 20.2 Hz), 103.94 (t, J = 27.1 Hz), 75.17 (d, J = 5.2 Hz), 55.11 (d, J = 5.8 Hz), 46.66, 30.86. ESI-HRMS calcd. for C21H21F2N7O2S [M+H]+ = 474.1518, found 474.1520.
N-(2-(2,4-difluorophenyl)-2-hydroxy-3-(1H-1,2,4-triazol-1-yl)propyl)-8-hydroxyquinoline-7-carboxamide (Flu-8HQ, III-b)
To a solution of 5 (0.43 g, 1.69 mmol) and 8-hydroxy-7-quinoline carboxylic acid (0.321 g, 1.697 mmol) in DMF (15 mL), 2-ethoxy-1-ethoxycarbonyl-1,2-dihydroquinoline (EEDQ, 0.466 g, 1.88 mmol) was added. The reaction mixture was stirred at room temperature for 18 hours, at which point still no reaction had occurred. The reaction mixture was heated to 60 °C for 3 days, and still no reaction occurred. Additional EEDQ was added (0.160 g, 0.647 mmol), and the reaction mixture was stirred for an additional 24 hours. Solvent was removed by evaporation under reduced pressure, and the crude products were reconstituted in EtOAc (50 mL). The organic layer was washed with a 10% solution of citric acid (4×40 mL), H2O (1×25 mL), and brine (1×25 mL), then dried over Na2SO4 and filtered. Crude product was purified by HPLC on a C18 column using a linear gradient running from 95%–5% H2O/ACN over 40 min, giving a bright yellow solid. Yield: 0.040 g (6%). 1H NMR (500 MHz, Chloroform-d) δ 8.81 (dd, J = 4.4, 1.5 Hz, 1H), 8.38 (t, J = 6.0 Hz, 1H), 8.19 – 8.13 (m, 2H), 8.07 (d, J = 8.9 Hz, 1H), 7.83 (s, 1H), 7.64 (td, J = 9.2, 6.5 Hz, 1H), 7.53 (ddd, J = 8.2, 4.2, 1.0 Hz, 1H), 7.35 (d, J = 8.8 Hz, 1H), 6.80 (dtd, J = 10.7, 8.3, 2.6 Hz, 2H), 6.41 (s, 2H), 4.71 – 4.62 (m, 2H), 4.13 – 3.92 (m, 2H), 2.17 (d, J = 1.1 Hz, 1H). 13C NMR (125 MHz, Chloroform-d) δ 168.12, 163.04 (d, J = 249.6 Hz), 151.77, 151.62, 148.69, 144.80, 138.26, 136.38, 130.83 – 130.50 (m), 130.36, 127.54, 124.07 (d, J = 4.0 Hz), 123.82, 118.07, 112.97, 111.87 (dd, J = 20.6, 3.5 Hz), 104.21 (t, J = 26.6 Hz), 76.75 (d, J = 5.1 Hz), 56.36 (d, J = 4.6 Hz), 48.06 (d, J = 4.7 Hz). ESI-HRMS calcd. for C21H17F2N5O3 [M+H]+ = 426.1372, found 426.1372.
Supplementary Material
Acknowledgments
We thank Prof. Val Culotta (Johns Hopkins University, Baltimore, MD) for providing the two strains of C. albicans (KC2 and crp1Δ/Δ), Prof. Alistair Brown (University of Exeter, Exeter, UK) for providing the SC5314 and ctr1Δ/Δ strains of C. albicans, and Prof. Dennis Thiele (Duke University, Durham, NC) for providing the two strains of C. neoformans (WT H99 and ctr1Δ ctr4Δ). This work was supported by the National Institutes of Health (Grant GM084176). E. W. H. acknowledges support from the United States Department of Education GAANN Fellowship (Award Number: P200A150114).
Footnotes
Conflict of Interest: The authors declare that they have no conflict of interest.
References
- 1.Bongomin F, Gago S, Oladele RO and Denning DW (2017) J Fungi 3:57 [Google Scholar]
- 2.Monk BC and Goffeau A (2008) Science 321:367. [DOI] [PubMed] [Google Scholar]
- 3.Lupetti A, Danesi R, Campa M, Del Tacca M and Kelly S (2002) Trends Mol Med 8:76–81 [DOI] [PubMed] [Google Scholar]
- 4.Cowen LE, Sanglard D, Howard SJ, Rogers PD and Perlin DS (2015) Cold Spring Harb Perspect Med 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Revie NM, Iyer KR, Robbins N and Cowen LE (2018) Curr Opin Microbiol 45:70–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roemer T and Krysan DJ (2014) Cold Spring Harb Perspect Med 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim K, Zilbermintz L and Martchenko M (2015) Ann Clin Microbiol Antimicrob 14:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cui J, Ren B, Tong Y, Dai H and Zhang L (2015) Virulence 6:362–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krysan DJ (2015) Fungal Genet Biol 78:93–98 [DOI] [PubMed] [Google Scholar]
- 10.Bisson WH (2012) Curr Top Med Chem 12:1883–1888 [DOI] [PubMed] [Google Scholar]
- 11.Motahari K, Badali H, Hashemi SM, Fakhim H, Mirzaei H, Vaezi A, Shokrzadeh M and Emami S (2018) Future Med Chem 10:987–1002 [DOI] [PubMed] [Google Scholar]
- 12.Liao J, Yang F, Zhang L, Chai X, Zhao Q, Yu S, Zou Y, Meng Q and Wu Q (2015) Arch Pharmacal Res 38:470–479 [DOI] [PubMed] [Google Scholar]
- 13.Pore VS, Agalave SG, Singh P, Shukla PK, Kumar V and Siddiqi MI (2015) Org Biomol Chem 13:6551–6561 [DOI] [PubMed] [Google Scholar]
- 14.Pore VS, Aher NG, Kumar M and Shukla PK (2006) Tetrahedron 62:11178–11186 [Google Scholar]
- 15.He X, Jiang Y, Zhang Y, Wu S, Dong G, Liu N, Liu Y, Yao J, Miao Z, Wang Y, Zhang W and Sheng C (2015) MedChemComm 6:653–664 [Google Scholar]
- 16.Zambrano-Huerta A, Cifuentes-Castañeda DD, Bautista-Renedo J, Mendieta-Zerón H, Melgar-Fernández RC, Pavón-Romero S, Morales-Rodríguez M, Frontana-Uribe BA, González-Rivas N and Cuevas-Yañez E (2019) Med Chem Res 28:571–579 [Google Scholar]
- 17.Ptaszyńska N, Olkiewicz K, Okońska J, Gucwa K, Łęgowska A, Gitlin-Domagalska A, Dębowski D, Lica J, Heldt M, Milewski S, Ng TB and Rolka K (2019) Peptides 117:170079. [DOI] [PubMed] [Google Scholar]
- 18.Thamban Chandrika N, Shrestha SK, Ngo HX, Howard KC and Garneau-Tsodikova S (2018) Biorg Med Chem 26:573–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Y, Xu K, Bai G, Huang L, Wu Q, Pan W and Yu S (2014) Molecules 19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zou Y, Yu S, Li R, Zhao Q, Li X, Wu M, Huang T, Chai X, Hu H and Wu Q (2014) Eur J Med Chem 74:366–374 [DOI] [PubMed] [Google Scholar]
- 21.Yu S, Wang L, Wang Y, Song Y, Cao Y, Jiang Y, Sun Q and Wu Q (2013) RSC Adv 3:13486–13490 [Google Scholar]
- 22.Karaoun N and Renfrew AK (2015) Chem Commun 51:14038–14041 [DOI] [PubMed] [Google Scholar]
- 23.Navarro M, Cisneros-Fajardo EJ, Lehmann T, Sánchez-Delgado RA, Atencio R, Silva P, Lira R and Urbina JA (2001) Inorg Chem 40:6879–6884 [DOI] [PubMed] [Google Scholar]
- 24.Iniguez E, Sanchez A, Vasquez MA, Martinez A, Olivas J, Sattler A, Sanchez-Delgado RA and Maldonado RA (2013) J Biol Inorg Chem 18:779–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Simpson PV, Nagel C, Bruhn H and Schatzschneider U (2015) Organometallics 34:3809–3815 [Google Scholar]
- 26.Betanzos-Lara S, Chmel NP, Zimmerman MT, Barron-Sosa LR, Garino C, Salassa L, Rodger A, Brumaghim JL, Gracia-Mora I and Barba-Behrens N (2015) Dalton Trans 44:3673–3685 [DOI] [PubMed] [Google Scholar]
- 27.Betanzos-Lara S, Gomez-Ruiz C, Barron-Sosa LR, Gracia-Mora I, Flores-Alamo M and Barba-Behrens N (2012) J Inorg Biochem 114:82–93 [DOI] [PubMed] [Google Scholar]
- 28.Kljun J, Scott AJ, Lanišnik Rižner T, Keiser J and Turel I (2014) Organometallics 33:1594–1601 [Google Scholar]
- 29.Ząbek A, Nagaj J, Grabowiecka A, Dworniczek E, Nawrot U, Młynarz P and Jeżowska-Bojczuk M (2015) Med Chem Res 24:2005–2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hunsaker EW and Franz KJ (2019) Dalton Trans 48:9654–9662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Paterson BM and Donnelly PS (2011) Chem Soc Rev 40:3005–3018 [DOI] [PubMed] [Google Scholar]
- 32.Helsel ME, White EJ, Razvi SZ, Alies B and Franz KJ (2017) Metallomics 9:69–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Festa RA, Helsel ME, Franz KJ and Thiele DJ (2014) Chem Biol 21:977–987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mandal PK and McMurray JS (2007) J Org Chem 72:6599–6601 [DOI] [PubMed] [Google Scholar]
- 35.Rosenberg A, Ene IV, Bibi M, Zakin S, Segal ES, Ziv N, Dahan AM, Colombo AL, Bennett RJ and Berman J (2018) Nat Commun 9:2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hunsaker EW and Franz KJ (2019) Metallomics 11:2020–2032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Robbins N, Caplan T and Cowen LE (2017) Annu Rev Microbiol 71:753–775 [DOI] [PubMed] [Google Scholar]
- 38.BDBiosciences (2006) Bd bionutrients technical manual: Advanced bioprocessing. https://www.bdbiosciences.com/documents/bionutrients_tech_manual.pdf.
- 39.Thompsett AR, Abdelraheim SR, Daniels M and Brown DR (2005) J Biol Chem 280:42750–42758 [DOI] [PubMed] [Google Scholar]
- 40.Xiao Z and Wedd AG (2010) Nat Prod Rep 27:768–789 [DOI] [PubMed] [Google Scholar]
- 41.Jones CE, Abdelraheim SR, Brown DR and Viles JH (2004) J Biol Chem 279:32018–32027 [DOI] [PubMed] [Google Scholar]
- 42.Sarkar B and Wigfield Y (1967) J Biol Chem 242:5572–5577 [PubMed] [Google Scholar]
- 43.Yang L, McRae R, Henary MM, Patel R, Lai B, Vogt S and Fahrni CJ (2005) Proc Natl Acad Sci USA 102:11179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ackerman CM, Lee S and Chang CJ (2017) Anal Chem 89:22–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xiao Z, Gottschlich L, van der Meulen R, Udagedara SR and Wedd AG (2013) Metallomics 5:501–513 [DOI] [PubMed] [Google Scholar]
- 46.Alies B, Badei B, Faller P and Hureau C (2012) Chem Eur J 18:1161–1167 [DOI] [PubMed] [Google Scholar]
- 47.Xiao Z, Brose J, Schimo S, Ackland SM, La Fontaine S and Wedd AG (2011) J Biol Chem 286:11047–11055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xiao Z, Donnelly PS, Zimmermann M and Wedd AG (2008) Inorg Chem 47:4338–4347 [DOI] [PubMed] [Google Scholar]
- 49.(2015) Cold Spring Harb Protoc, doi 10.1101/pdb.rec085639 [DOI] [Google Scholar]
- 50.Hunsaker EW and Franz KJ (2019) Metallomics, 11:2020–2032, doi 10.1039/C9MT00228F [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Holmes-Hampton GP, Jhurry ND, McCormick SP and Lindahl PA (2013) Biochemistry 52:105–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Papadopoulou MV, Bloomer WD, Lepesheva GI, Rosenzweig HS, Kaiser M, Aguilera-Venegas B, Wilkinson SR, Chatelain E and Ioset J-R (2015) J Med Chem 58:1307–1319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pore VS, Jagtap MA, Agalave SG, Pandey AK, Siddiqi MI, Kumar V and Shukla PK (2012) MedChemComm 3:484–488 [Google Scholar]
- 54.Helsel ME and Franz KJ (2015) Dalton Trans 44:8760–8770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Parker JE, Warrilow AGS, Price CL, Mullins JGL, Kelly DE and Kelly SL (2014) J Chem Biol 7:143–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hagen WR (2009) Metallomics 1:384–391 [DOI] [PubMed] [Google Scholar]
- 57.Noble SM and Johnson AD (2005) Eukaryot Cell 4:298–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McCluskey K, Wiest A and Plamann M (2010) J Biosci 35:119–126 [DOI] [PubMed] [Google Scholar]
- 59.Klis FM, de Koster CG and Brul S (2014) Eukaryot Cell 13:2–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Upadhayaya RS, Jain S, Sinha N, Kishore N, Chandra R and Arora SK (2004) Eur J Med Chem 39:579–592 [DOI] [PubMed] [Google Scholar]
- 61.Song YJ, Z. J.; Pandey A; Scarborough RM; Scarborough C (November 8, 2007). [Google Scholar]
- 62.Wu C-F, Zhao X, Lan W-X, Cao C, Liu J-T, Jiang X-K and Li Z-T (2012) J Org Chem 77:4261–4270 [DOI] [PubMed] [Google Scholar]
- 63.González Cabrera D, Koivisto BD and Leigh DA (2007) Chem Commun 4218–4220, doi 10.1039/B713501G:4218–4220 [DOI] [PubMed] [Google Scholar]
- 64.Kulandaivelu U, Shireesha B, Mahesh C, Vidyasagar JV, Rao TR, Jayaveera KN, Saiko P, Graser G, Szekeres T and Jayaprakash V (2013) Med Chem Res 22:2802–2808 [Google Scholar]
- 65.Liu K, Lu H, Hou L, Qi Z, Teixeira C, Barbault F, Fan BT, Liu S, Jiang S and Xie L (2008) J Med Chem 51:7843–7854 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.