

Abstract
A genetic etiology is identified for one third of congenital heart disease (CHD) patients, including 8% attributable to coding de novo variants (DNVs). To assess the contribution of noncoding DNVs to CHD, we compared genome sequences from 749 CHD probands and their parents with 1,611 unaffected trios. Neural network prediction of noncoding DNV transcriptional impact identified a burden of DNVs in CHD (n = 2,238 DNVs) compared to controls (n = 4,177; P = 8.7 × 10−4). Independent analyses of enhancers showed excess DNVs in associated genes (27 genes vs. 3.7 expected, P = 1 × 10−5). We observed significant overlap between these transcription-based approaches (OR = 2.5, 95% CI 1.1–5.0, P = 5.4 × 10−3). CHD DNVs altered transcription levels in five of 31 enhancers assayed. Finally, we observed DNV burden in RNA-binding protein regulatory sites (OR = 1.13, 95% CI 1.1–1.2, P = 8.8 × 10−5). Our findings demonstrate an enrichment of potentially disruptive regulatory noncoding DNVs in a fraction of CHD at least as high as observed for damaging coding DNVs.
Congenital heart disease (CHD), which occurs in 1% of live births, has seen marked improvements in survival with modern surgical and medical management1. The decrease in infant mortality has increased CHD prevalence in older individuals and exposed co-morbidities impairing quality of life and life expectancy. Elucidation of CHD etiologies may improve outcomes, so the NHLBI-funded Pediatric Cardiac Genomics Consortium (PCGC) recruited >13,000 patients and utilized whole exome sequencing (WES) and chromosome microarrays to study CHD genetic architecture. Our analyses identified damaging rare transmitted and de novo variants (DNVs) in 8% of sporadic CHD (including 28% of syndromic and 3% of isolated CHD)2–5. Many DNVs identified in CHD patients alter proteins functioning in chromatin modification, regulation of transcription, and RNA processing4.
Based on these findings, we hypothesized that additional CHD causes may reside in noncoding elements functional during cardiac development. To explore that, we performed whole genome sequencing (WGS) to identify single nucleotide variants (SNVs) and small insertions/deletions (indels) in 763 CHD trios comprised of affected probands and unaffected parents and in 1,611 child-parent trios without CHD. First, 14 CHD probands with previously undetected likely causal genetic variants were identified; then, we compared noncoding DNVs using three approaches in the remaining cohort. Two strategies focused on cardiac gene regulatory elements, using a neural network model predicting variant-level resolution functional impact and by analyzing multiple DNVs in genes with human fetal heart enhancers overlapping cardiomyocyte differentiation open chromatin. We identified significant overlap between results from these complementary approaches and confirmed differences on transcription activity for 5 of 31 variants tested. Our third strategy, which interrogated RNA processing, found significant enrichment of noncoding DNVs in cases. Finally, we observed potentially contributory noncoding DNVs in isolated CHD probands as well as those with neurodevelopmental delays or extracardiac anomalies, suggesting varying degrees of cardiac specificity. Taken together, these results demonstrate a noncoding DNV contribution to CHD mediated through transcriptional and post-transcriptional regulatory effects on cardiac development.
RESULTS
Trio cohort characteristics and sequencing.
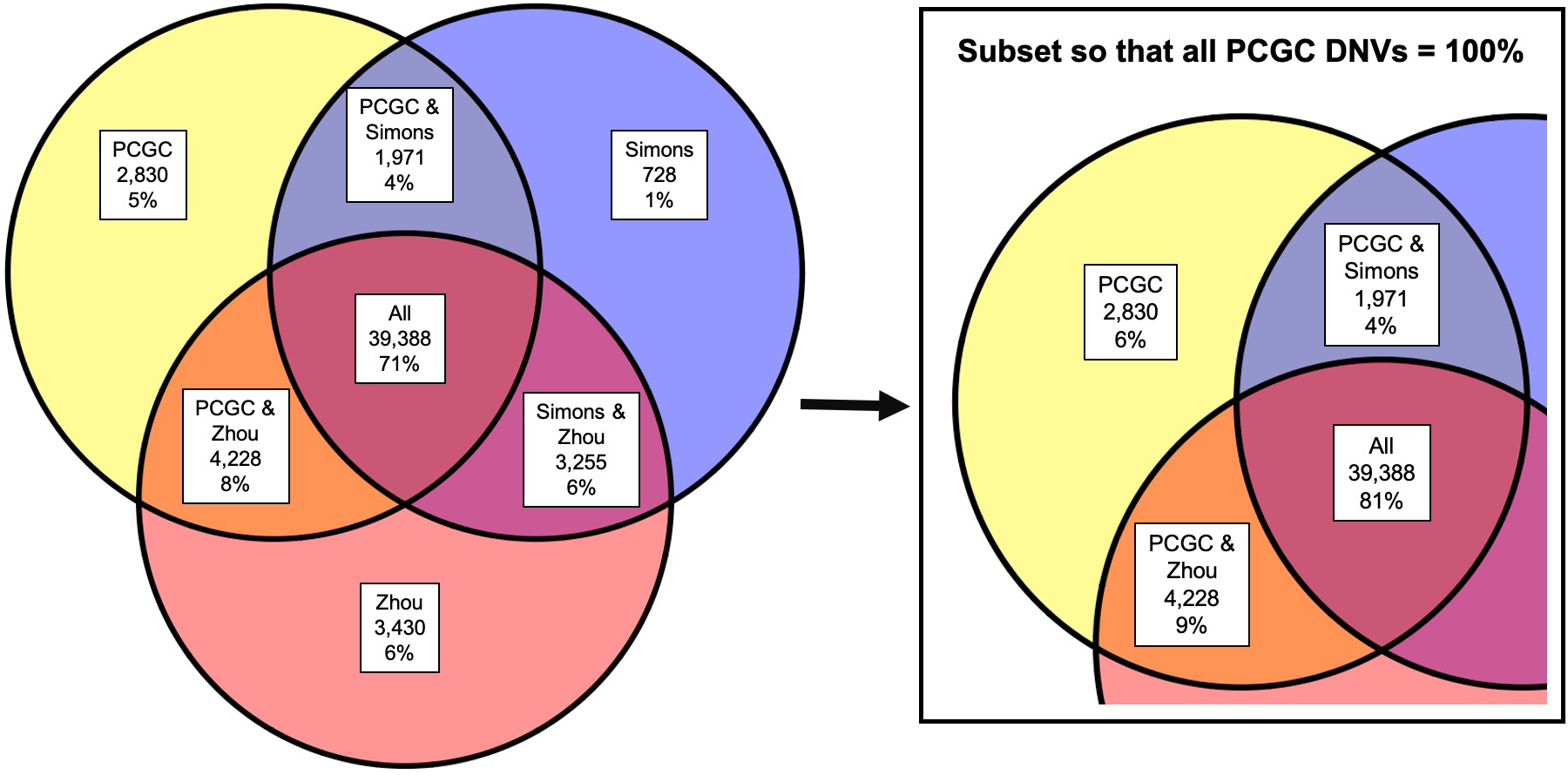
We performed WGS (coverage = 30x) on 763 CHD probands (311 with extra-cardiac anomalies; 452 with isolated heart malformations) and unaffected parents enrolled by the PCGC (Supplementary Table 1a, phenotype summary in Supplementary Table 1b)2. Subjects underwent WGS if prior WES studies5 failed to identify rare damaging missense or loss-of-function coding variants in CHD genes (Supplementary Table 5). We also studied DNVs in 1,611 individuals without CHD or autism, who had siblings with autism, and their parents from the Simons Simplex Collection6. To ensure accurate variant detection, de novo variants (DNVs) were identified using GATK and further evaluated with FreeBayes7 local realignment, classification by a neural network trained on Integrated Genomics Viewer (IGV) plots8, and manual curation of ambiguous variants (see Methods; Supplementary Tables 2 and 3). PCR-based Sanger sequencing validated 98% of 266 de novo SNVs and 94% of 83 de novo indels in cases. In controls, 94% of de novo SNVs were present in at least one published analysis (Extended Data Fig. 1)9,10. We identified a mean of 71 de novo SNVs and five de novo indels per CHD proband (58,090 DNVs) and 68 de novo SNVs and five de novo indels per control subject (117,344 DNVs), similar to WGS data obtained on similar platforms and coverage11.
As expected, DNVs per subject correlated with paternal (βCHD = 1.4, PCHD = 5 × 10−54, βcontrol = 1.4, Pcontrol = 6 × 10−86) and maternal (βCHD = 0.5, PCHD = 2 × 10−5, βcontrol = 0.4, Pcontrol = 3 × 10−8) ages (multiple variable linear regression; Extended Data Fig. 2)11,12. SNVs drove this association, but there was also a de novo indel association with paternal (βCHD = 0.07, PCHD = 2 × 10−4, βcontrol = 0.05, Pcontrol = 3 × 10−4) but not maternal age (βCHD = 0.01, PCHD = 0.6, βcontrol = 0.03, Pcontrol = 0.1)13. Without parental age adjustment, cases had more DNVs per subject than controls (P = 2 × 10−9, two-sided t-test), but not after adjustment (P = 0.1). To account for this difference, comparisons were made with respect to the total number of DNVs in CHD probands and controls.
Coding DNVs identified by WES and WGS.
WES data were available for 612 of 763 CHD probands4,5. Among 628 coding DNVs including 582 within WES capture regions (lifted over14 to hg38), WES and WGS both identified 509 (81%), while 38 of 69 DNVs called only by WES were confirmed by WGS IGV visualization (see Supplementary Note). Fifty coding DNVs identified solely by WGS (8%; 0.08/proband) included four within and 46 outside WES capture regions. One initially called by WES was removed for low read depth; three were not called by WES but confirmed by WES IGV visualization.
These analyses defined damaging DNVs in established CHD genes (PTPN11, NOTCH1 (n = 2), FBN1, FLT4, NR2F2, GATA4), and identified six subjects with 22q11 copy number variants and one with trisomy 21. The proband with a previously reported pathogenic FBN1 DNV in exon 42 (1–00761) had mitral stenosis, brachycephaly, short stature and other features consistent with geleophysic dysplasia (MIM 614185), 50% of which is caused by damaging DNVs in FBN1 exon 41 or 42. Damaging DNVs in known CHD genes were confirmed with reference-free DNV calling (see Methods) and IGV visualization. Six potentially damaging DNVs were identified in candidate CHD genes, including one insertion detected only with reference-free calling (Supplementary Table 4), but these individuals were retained for noncoding analyses. Excluding probands with likely causal genetics, 749 CHD probands were analyzed for noncoding DNVs.
Quantitative burden with categorical DNV classifications.
We observed no noncoding DNV enrichment in 749 CHD trios for DNVs associated with human (n = 210) or mouse (n = 614) CHD genes or genes highly expressed in heart development (n = 4,420) (Supplementary Tables 5 and 6). Similarly, we observed no enrichment in noncoding cardiac regulatory features comprising transcription factor binding sites (nhuman = 8, nmouse = 45), histone marks (nhuman = 45, nmouse = 60), and DNase hypersensitivity sites (nhuman = 23, nmouse = 3) assayed on cardiac cells (nhuman = 15), prenatal/fetal heart tissue (nhuman = 26, nmouse = 34), and postnatal heart tissue (nhuman = 35, nmouse = 74) (see Methods, Extended Data Fig. 3, and Supplementary Table 7)15–32.
Qualitative burden with HeartENN.
Finding no genome-wide significant DNV burden in global regions of cardiac transcriptional regulation among CHD probands, we predicted impact with variant-level resolution. We developed HeartENN (Heart Effect Neural Network, Fig. 1), an extension of DeepSEA33, which predicts molecular effect differences between any two alleles for every regulatory feature using convolutional neural networks. Another DeepSEA extension successfully identified noncoding DNV enrichment in autism34. HeartENN was trained on 1,000-bp genomic sequence context with the same 184 cardiac noncoding regulatory features used for previous region-based burden tests, but not those used for subsequent multiple-hit analysis (see Methods; Supplementary Table 7). Aside from using cardiac epigenomic training data and extending to mouse features, HeartENN is similar to DeepSEA. The HeartENN mean receiver operator characteristic area-under-the-curves (ROC AUCs) for mouse and human features were 0.9 and 0.85, respectively, similar to DeepSEA ROC AUCs; the area under the precision-recall curves were also comparable (Extended Data Fig. 4). Beyond restricting to heart-related features, we defined no other CHD relevance hypotheses. The maximum functional difference score observed in any feature was assigned to each DNV (Supplementary Table 8).
Figure 1 |. Analysis schematic.
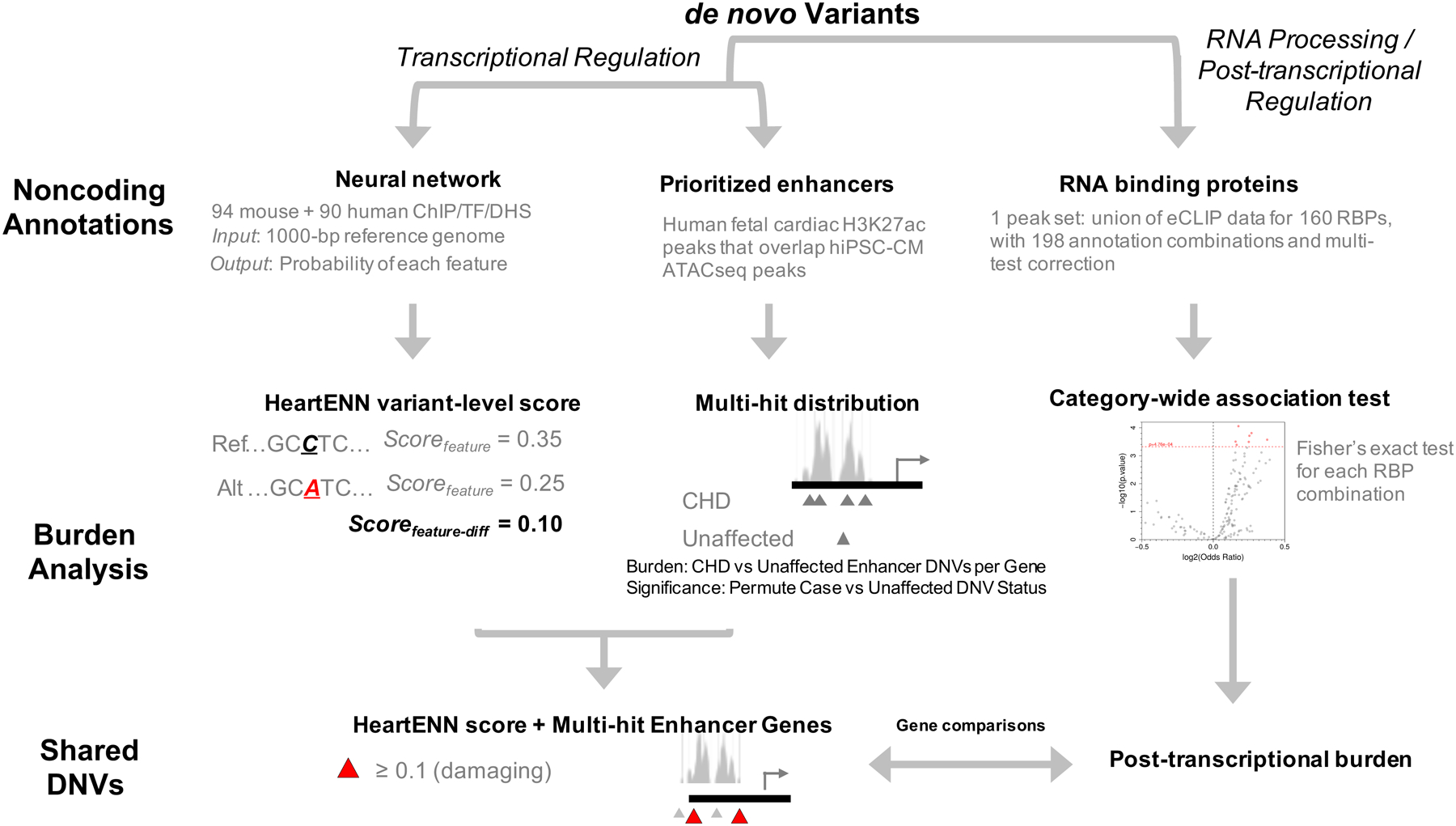
Overview of approach to identifying a noncoding de novo variant burden in congenital heart disease (CHD). ATAC, Assay for Transposase-Accessible Chromatin;, TF, transcription factor; DHS, DNase hypersensitivity sites; RBP, RNA-binding protein; NS, not significant; HeartENN, Heart Effect Neural Network.
We defined a range of scores relevant to congenital defects by contrasting maximum functional difference scores between Human Gene Mutation Database regulatory mutations (n = 1,564), inclusive of congenital defect pathogenic variants, and regulatory polymorphisms (n = 642). As these variants are from diverse malformations, we evaluated signals using DeepSEA33,34, which is generalizable to multiple organ systems. Pathogenic variants, but not polymorphisms, had an excess of DeepSEA scores ≥ 0.1 (Extended Data Fig. 5a). Lacking an equivalent dataset of CHD noncoding variants to evaluate HeartENN, we compared the DeepSEA and HeartENN null distributions. After randomly down-sampling DeepSEA to match the number of HeartENN annotations and applying HGMD polymorphism scores, we observed similar null distributions for HeartENN and DeepSEA (Extended Data Fig. 5b). We therefore set HeartENN scores ≥ 0.1 as potentially biologically meaningful for CHD.
The majority (>96%) of DNVs had HeartENN scores < 0.1, suggesting little functional impact from most variants. CHD cases were enriched for HeartENN scores ≥ 0.1 (n = 2,238 DNVs in CHD, n = 4,177 DNVs in controls, Fisher’s exact test P = 8.7 × 10−4, OR = 1.09, 95% CI 1.04–1.15, attributable risk = 183/2,283 DNVs). We tested enrichment across multiple cut-offs, observing (i) no marginal (P < 0.05) significance in controls at any cut-off, (ii) higher odds ratios with stricter thresholds (Fig. 2a and Supplementary Table 9), and (iii) significance when accounting for all thresholds (Fig. 2b, permutation P = 1.7 × 10−3, 10,000 permutations). Above 0.25, we observed consistent positive effect sizes despite decreased sample sizes, suggesting lack of power with more stringent thresholds. To test if the signal was consistent across functionally significant HeartENN scores, we placed every DNV into 0.02-HeartENN-score bins. We calculated the difference in fraction of DNVs in every 0.02 bin (Fig. 2c) and observed a strong propensity towards cases across bins.
Figure 2 |. Enrichment of noncoding de novo variants (DNVs) with functionally relevant HeartENN scores.
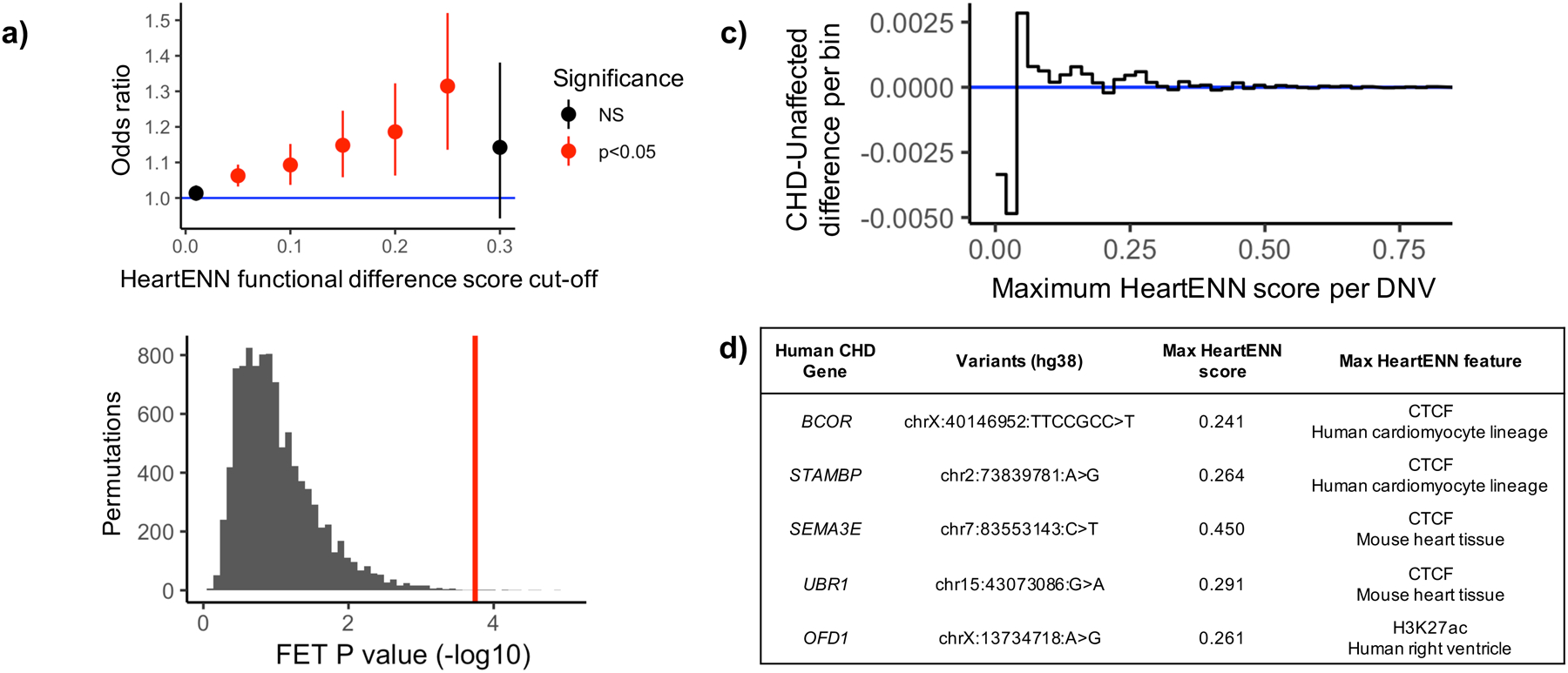
a, The number of noncoding DNVs above varying HeartENN thresholds (x-axis) was counted in CHD (n = 749) and unaffected (n = 1,611) individuals and compared to the total number of scored DNVs in CHD (n = 56,164) and unaffected (n = 114,065) individuals, plotted as odds ratios with 95% confidence intervals (counts, odds ratios, and Fisher’s exact test 2-sided unadjusted P-values are listed in Supplementary Table 9). b, Permutations of case-control status (n = 10,000, grey) found a significant P-value when accounting for all cut-offs (P = 1.7 × 10−3, 1-sided) by comparing the most significant observed P-value (red) to the most significant P-value per permutation (grey). c, The fraction of DNVs in 0.02-HeartENN-score bins demonstrated consistent propensity towards cases for functionally relevant HeartENN bins. d, Known human CHD genes with HeartENN-damaging (≥0.1) DNVs were enriched in CHD (nCHD = 18, nunaffected = 10, OR = 3.2, hypergeometric 1-sided P = 6 × 10−4), with the top five (shown here) predicted to disrupt CTCF and H3K27ac features. FET, Fisher’s exact test.
We tested whether other noncoding variant prioritization methods ranked HeartENN-damaging (score ≥ 0.1) variants as pathogenic. There was statistically significant support from all tested algorithms (LINSIGHT35, CADD36,37, DeepSEA33,34, GERP++38, and GWAVA TSS39) (Extended Data Fig. 6). We observed a case-control burden with CADD ≥ 15 (PBonferroni = 0.019), albeit without a dose-response relationship or cardiac-relevant interpretation.
Gene set enrichment of DNVs with HeartENN ≥ 0.1 upstream or downstream (<1 kb) or within 5’-UTR, intronic, or 3’-UTR sequences showed enrichment of known human CHD genes in cases (Fig. 2d and Supplementary Table 10; n = 18/959 genes in cases, n = 10/1,704 genes in controls, OR = 3.2, 95% CI 1.4–7.9, hypergeometric 1-sided P = 5.7 × 10−4). Notably, one proband with isolated CHD had a DNV (maximum HeartENN score 0.15, ID 1–07589) within a previously validated GATA4 enhancer with heart-constrained activity (Vista ID hs2205, heart-specific in 6/7 E11.5 embryos)27.
Burden of genes with multiple DNVs in human fetal cardiac enhancers.
A second approach interrogating noncoding DNVs focused on regions experimentally implicated in human cardiac developmental gene expression regulation. This strategy harnessed 31,555 human fetal heart enhancers identified by H3K27ac chromatin immunoprecipitation (ChIP) of human fetal cardiac tissues (8–17 weeks post-conception, see Methods). None was included in HeartENN. We intersected these fetal cardiac enhancers with open chromatin sequences from ATAC-seq during human induced pluripotent stem cells differentiation into cardiomyocytes (hiPSC-CMs). Based on prototypic gene expression, ATAC-seq was performed at two differentiation states: cardiac mesoderm (day 8; 155,989 ATAC peaks) and primordial cardiomyocytes (day 17; 62,326 ATAC peaks). The subset of ATAC peaks overlapping cardiac enhancers defined 21,618 prioritized human fetal heart enhancers (Supplementary Table 11). We assessed these sequences for DNVs.
Within prioritized human fetal heart enhancers, we identified 2,427 DNVs in CHD cases and 5,160 DNVs in controls (Fisher’s exact P = 1, Supplementary Table 12). Assignment of nearest genes defined 1,793 genes in CHD cases and 3,195 genes in controls. Among CHD cases, 27 genes were marginally enriched for DNVs. No gene was enriched for DNVs in controls (Fig. 3a and Supplementary Table 13). In 105 permutations of randomly assigned case or control status, fewer genes exhibited DNV enrichment than observed (P < 1 × 10−5, Fig. 3b).
Figure 3 |. Genes with multiple DNVs in prioritized human fetal heart enhancers.
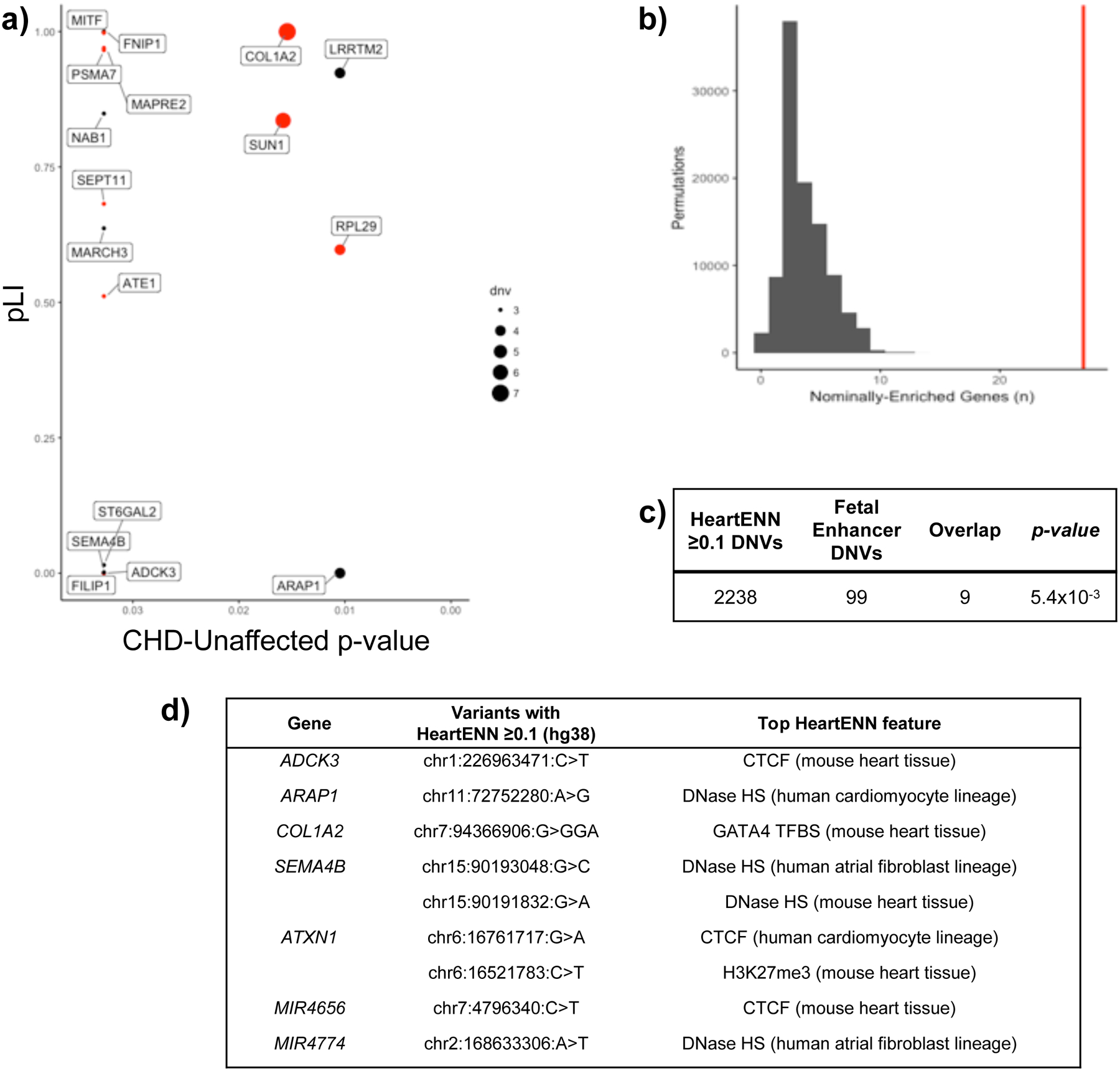
a, Genes with burden of DNVs in associated fetal cardiac enhancers with P < 0.05 for CHD (n = 749) compared to unaffected (n = 1,611). For each gene, the DNV burden P-value (x-axis) was determined with a two-sided Fisher’s exact test comparing DNVs in CHD (n = 56,164) versus unaffected individuals (n = 114,065) and is plotted against the pLI (y-axis). Dot size reflects the number of DNVs in the CHD cohort, and red denotes genes in the upper quartile of gene expression during heart development. Five genes without pLI values are not shown. b, Distribution of the number of nominally enriched genes by 100,000 random permutations of DNVs within prioritized human fetal heart enhancers demonstrates significant enrichment of genes with burden of CHD DNVs. As there were never 21 genes observed in permutation test, the most extreme P-value would be 10−5 (one-sided). c, Overlap between DNVs with HeartENN score ≥ 0.1 (n = 2,238) and those within prioritized human fetal heart enhancers (n = 99) is significantly enriched in CHD (1-sided hypergeometric distribution, no overlapping DNVs in controls). d, Top features representing a diverse spectrum of transcriptional regulation.
These 27 genes were associated with 99 case DNVs and 13 control DNVs (Supplementary Table 14). Nine case DNVs but none in controls had HeartENN scores ≥ 0.1 (Supplementary Table 14), significantly more than < 4% expected by chance (1-sided hypergeometric P = 5.4 × 10−3, Fig. 3c). Significance was assessed using the null hypothesis of proportional overlap, which was appropriate as HeartENN used different cardiac epigenomic data from the prioritized human fetal heart enhancers. Ten of the 27 genes enriched for DNVs in prioritized human fetal heart enhancers were highly expressed in E14.5 mouse hearts (P = 0.06): COL1A2, MAPRE2, SEPT11, PSMA7, SORBS1, RPL25, FILIP1, MITF, SUN1, and ATE1 (Supplementary Table 13). Twelve genes (FNIP1, COL1A2, MITF, MAPRE2, PSMA7, LRRTM2, NAB1, SUN1, SEPT11, MARCH3, RPL29, and ATE1) have a pLI > 0.5 (P = 0.03), reflecting a modest probability of LoF variant intolerance based on their prevalence in the Exome Aggregation Consortium (ExAC)40. One gene (COL1A2) was observed at the intersection of these findings: it includes a HeartENN ≥ 0.1 DNV, has a high pLI, and is highly expressed during mouse heart development. COL1A2 encodes a collagen that is highly expressed in developing cardiac valves37. Among the seven individuals with COL1A2-associated human fetal heart enhancer DNVs, all had pulmonary and/or aortic valve abnormalities, an enrichment trend compared to the 742 participants without such DNVs (486/742, P = 0.05).
Functional effects of de novo variation on transcriptional activity.
We assessed potential transcriptional effects of 31 DNVs (Fig. 4a) identified by HeartENN and/or prioritized human fetal heart enhancers using massively parallel reporter assays (MPRAs)41. Paired sequences (300–1,600 bp) containing reference or DNV sequences were synthesized and introduced into a pMPRA1 plasmid. At least three independent plasmid libraries were produced and transfected into multiple wells of iPSC-CMs at differentiation day 17 or 37. Transcriptional activity was assessed by comparing RNA/DNA test sequence reads per well. We observed no significant differences in transcriptional activity by construct length (Extended Data Fig. 7). Five of 31 construct pairs showed significant mean differences between reference and DNV sequences for at least three replicates (Fig. 4; t-test 2-sided BH-adjusted P < 0.05), including two DNVs that increased transcriptional activity. Two additional pairs showed DNV-reference transcriptional differences for two replicates but no overall statistical significance (Extended Data Fig. 8). These seven MPRA-positive variants were among 18 identified by both HeartENN (score ≥ 0.1) and prioritized human fetal heart enhancers or HeartENN (score ≥ 0.1) and ATAC-seq peaks. Among 13 variants selected with a single bioinformatic approach, none reproducibly yielded significant MPRA differences.
Figure 4 |. Massively parallel reporter assays for selected DNVs.
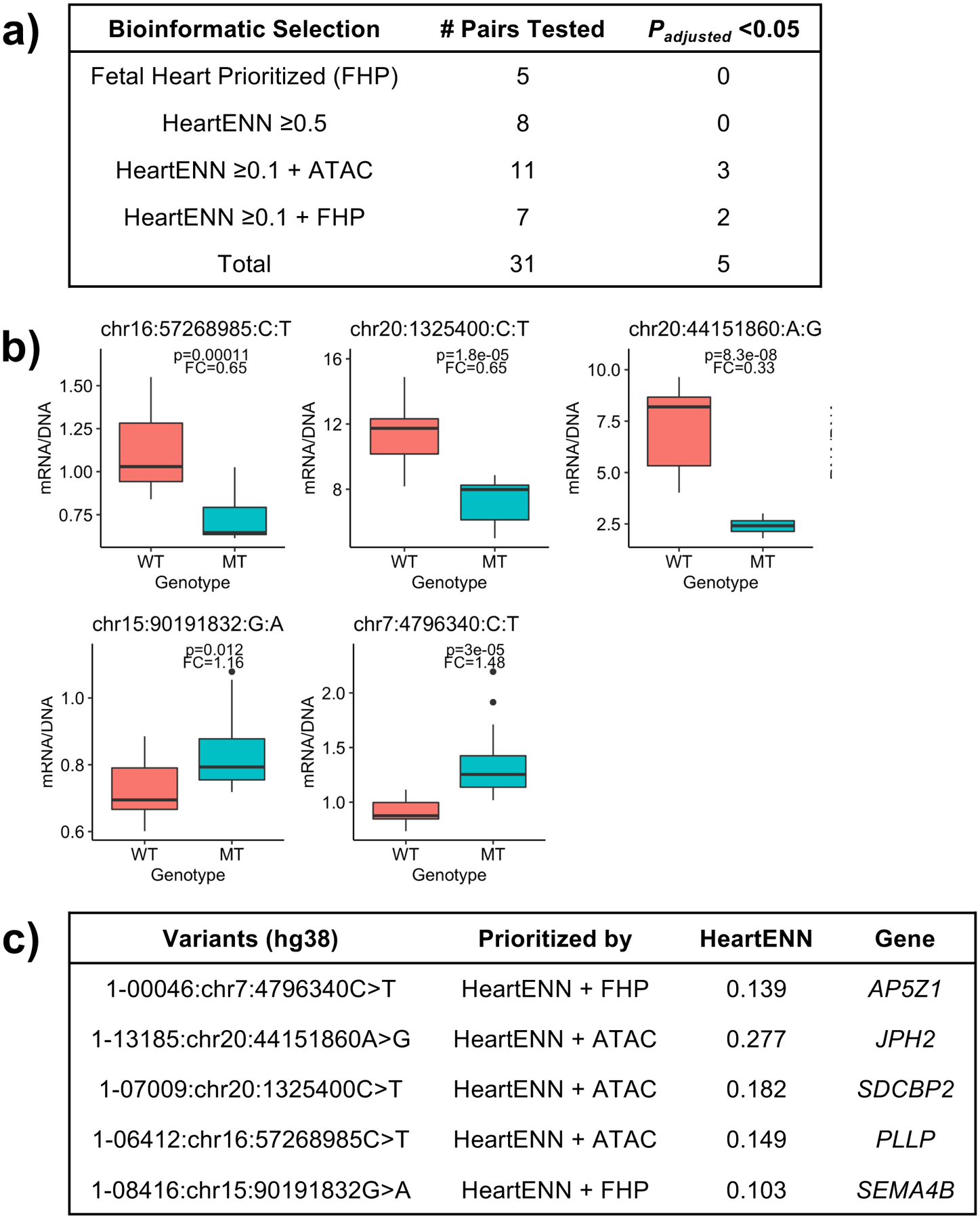
a, Pairs of reference and DNV sequences were selected based on bioinformatic analyses for the following classes: prioritized human fetal heart enhancer only, high HeartENN score (≥0.5) only, HeartENN score ≥ 0.1 at an ATAC-seq peak, and HeartENN score ≥ 0.1 in a prioritized human fetal heart enhancer. The numbers of pairs tested and the numbers for which the DNV sequence resulted in significantly different levels of transcription are indicated. b, Boxplots for the five pairs for which the transcription level from the DNV (MT) was significantly different from the reference (WT) sequence. Boxplots show the median fold change (FC), first and third quartiles (lower and upper hinges), and range of values (whiskers and outlying points). Both a and b show 2-sided t-test Benjamini-Hochberg P-values; each boxplot has at least 3 independent experiments with 4 technical replicates each, and the HeartENN ≥ 0.1 + FHP group was repeated 4 times. c, The genomic positions of the five DNVs for which transcription was significantly altered are indicated along with their bioinformatic classes, HeartENN functional difference scores, and associated genes.
Post-transcriptional regulatory enrichment.
In addition to transcriptional regulatory disruption, we tested noncoding DNV enrichment affecting post-transcriptional regulation. RNA-binding proteins (RBPs) mediate post-transcriptional regulation through pre-mRNA splicing, transport, localization, degradation, and translational control. We obtained 160 RBP eCLIP datasets from two ENCODE cell lines15. Because there are no cardiac eCLIP data, we inferred transcriptionally active cardiac binding sites by overlapping the human fetal heart H3K36me3 active transcription mark (used in HeartENN) and human embryonic stem cells (not used in HeartENN or prioritized human fetal heart enhancers). Using this narrower RBP binding site definition, we tested noncoding burden for all 162 annotation combinations (Fig. 5a): H3K36me3 histone mark, SNV and/or indel, constrained/haploinsufficient gene proximity, and transcription start site (TSS) or 3’-UTR anchor. The number of independent tests, determined with eigenvalue decomposition, was used to determine the Bonferroni P-value multiple testing adjustment42. This yielded 105 independent tests with significance threshold P = 4.76 × 10−4.
Figure 5 |. Enrichment of variants in RNA-binding protein category annotations.

a, Five groups of annotations were defined to investigate post-transcriptional regulation through disruption of RNA-binding protein binding, resulting in n = 162 combinations of (i) variant type; (ii) region type; (iii) RBP category; (iv) gene sets, specifically pLI constraint on nearest gene; and (v) histone mark annotations for actively transcribed regions in relevant proxy tissues*. These annotation categories were considered in the category-wide association test and yielded 105 independent tests, giving 4.76 × 10−4 as the strict Bonferroni threshold. b, Variant enrichment and significance for each test category, determined with a two-sided Fisher’s exact test: SNV-only tests used a total of n1 = 52,004 case SNVs and n0 = 106,438 control SNVs; SNV + indel tests used a total n1 = 55,827 case variants and n0 = 113,467 control variants. The association tests passing Bonferroni significance have been highlighted in red. c, Detailed tabulation of the seven Bonferroni-significant Fisher’s exact tests (two-sided).
We observed a significant enrichment of RBP DNVs overlapping H3K36me3 marks (Fig. 5b,c). The most significant result is RBP variants overlapping UCSF4 stem cell H3K36me3 (OR = 1.13, 95% CI 1.1–1.2, Fisher’s exact test 2-sided P = 8.77 × 10−5, 1,672 case DNVs, Supplementary Table 15). The signal was statistically significant for multiple embryonic stem cell types and when limited to constrained genes or TSS proximity. Intersecting these biologically relevant features yielded the largest statistically significant effect size (OR = 1.3, 95% CI 1.1–1.5, P = 2.68 × 10−4, 327 case DNVs).
We tested variant-level intersections between these post-transcriptional and transcriptional regulatory results. For the most significant RBP-implicated DNVs, there was a statistically significant overlap with DNVs in prioritized human fetal heart enhancers in cases (n = 10, OR = 3.6, 95% CI 1.9−∞, hypergeometric 1-sided P = 2.1 × 10−4) but not controls (n = 0). There was no significant overlap between RBP-implicated and HeartENN-damaging (score ≥ 0.1) DNVs in cases (n = 78, OR = 1.21, 95% CI 1.0−∞, P = 0.05) or controls (n = 122, OR = 1.12, 95% CI 0.9−∞, P = 0.10). In contrast, for RBP-implicated DNVs in constrained regions, we observed only one case DNV intersecting with prioritized fetal human heart enhancers and a statistically significant overlap with HeartENN-DNVs in cases (n = 19, OR = 1.52, 95% CI 1.0−∞, P = 0.033) but not controls (n = 16, OR = 0.86, 95% CI 0.5−∞, P = 0.7). Thus, in addition to transcriptional regulatory disruption, we find evidence that disturbed post-transcriptional regulation machinery may contribute to CHD.
Distribution of noncoding DNVs in canonical variant classes.
We characterized DNVs in canonical variant classes (intronic, promoter, UTR, etc.) for HeartENN-damaging (score ≥ 0.1) DNVs, prioritized human fetal heart enhancer multiple-hit DNVs, and post-transcriptional regulatory-disrupting DNVs (Extended Data Fig. 9). The majority of DNVs not identified by any bioinformatic method were intergenic (52% in cases, 52% in controls). In contrast, variants prioritized by the three methods were more likely intronic, with over-representation among other canonical categories depending on the method. This provides additional evidence that CHD-associated noncoding DNVs may have functional impacts.
Recurrently implicated genes with noncoding DNVs.
Among the union of implicated noncoding DNVs (HeartENN-damaging DNVs (2,238 cases, 4,177 controls), prioritized human fetal heart enhancer multiple-hit DNVs (99 cases, 13 controls), and post-transcriptional regulatory-disrupting DNVs from all seven Bonferroni-significant enrichments (2,149 cases, 3,963 controls)), 25 genes were recurrently implicated (unadjusted two-sided binomial P < 0.05) (Supplementary Table 16). High-interest genes were identified with haploinsufficiency constraint (pLI > 0.5 or missense Z-score >3), high mouse E14.5 heart expression rank, human or mouse CHD gene membership, and CHD-associated KEGG pathway membership. Results included two human CHD genes, but corresponding probands did not have the characteristic CHD phenotype, pulmonic stenosis. Candidate genes included SHOC2 (human CHD gene, constrained), ZNRF3, CPSF3 (CHD-associated KEGG pathway, constrained), and MAP4K4 (96th percentile embryonic heart expression, constrained).
Association between candidate noncoding DNVs and neurodevelopmental disorders or extracardiac anomalies.
We tested whether implicated noncoding DNVs were associated with phenotypic subgroups: isolated CHD (n=298), CHD with neurodevelopmental disorders (NDD) (n = 267), or CHD with extracardiac anomalies (ECA) (n = 305). Compared to probands with WES-identified damaging DNVs in highly expressed cardiac genes, CHD probands with DNVs in the 27 genes associated with prioritized human fetal heart enhancers had a lower frequency of NDD (odds 20/53 vs. 113/119; OR = 0.40, 95% CI 0.2–0.7, P = 0.002) but a similar prevalence of ECA (34/39 vs. 173/184; OR = 0.92, 95% CI 0.5–1.6, P = 0.87).
In contrast to probands with prioritized human fetal heart enhancer DNVs, most probands had at least one HeartENN-damaging (score ≥ 0.1) DNV, and presumably only a minority would be associated with CHD. Therefore, we tested phenotype associations by comparing HeartENN-damaging DNV enrichment within subgroups to controls (Extended Data Fig. 10a). A consistent enrichment was observed across all subgroups. We then tested the hypothesis that the parent algorithm, DeepSEA, which previously implicated noncoding DNVs in autism34, would also identify a burden in CHD cases with NDDs. No significant association was observed, but the highest effect size was observed for CHD with NDDs (OR = 1.05, 95% C.I. 1.0–1.1, two-sided Fisher’s exact test P = 0.18). A similarly consistent enrichment within sub-groups was observed for RBP-implicated DNVs (Extended Data Fig. 10b).
Contribution to CHD.
We estimated the mean attributable risk to CHD in the WES-negative cohort across all three methods (see Methods), assuming at most one causal, functional DNV per proband. HeartENN-damaging (score ≥ 0.1) DNVs contribute to a maximum of 24% of CHD in this cohort, and enrichment decreased with increasing HeartENN cut-offs (11% attributed to HeartENN ≥ 0.2, 2.9% attributed to HeartENN ≥ 0.3). This resulted in a final HeartENN contribution range of 3–24%. DNVs in prioritized human fetal heart enhancers contributed to 12.1% of CHD, including 1.1% attributable to shared HeartENN ≥ 0.1 DNVs. Lower percentages for DNVs associated with genes having pLI > 0.5 (5.4%) or high embryonic mouse heart expression (3.8%) resulted in a contribution range of 4–12%. Finally, DNVs implicated in post-transcriptional disruption contributed to 10.0% of CHD in this cohort. Although the cumulative percentage mean attributed risk (17–45%) suggests a substantial contribution in WES-negative CHD, these estimates must be refined in future studies. In summary, the fraction of CHD with contributory noncoding predicted functional DNVs in this WES-negative cohort is at least as high as the fraction of damaging coding DNVs identified with WES.
DISCUSSION
Noncoding variants remain potential contributors to disorders with unexplained genetics. Using WGS, we tested this hypothesis through systematic examination of noncoding regulatory elements in a mutation-negative CHD cohort. We, like others42–45, observed a lack of significant findings across broad noncoding annotation categories. By contrast, our alternative interrogation of noncoding variants implicated noncoding DNVs in CHD pathogenesis.
HeartENN, which provided variant-level scores, similar to the multifaceted DeepSEA algorithm that uncovered noncoding DNVs in autism34, defined significantly more DNVs in CHD probands. Separate analyses of prioritized human fetal heart enhancers identified distinct and some overlapping DNVs in CHD cases. Notably, functional assays were positive when these two strategies were combined. Although there was no transcriptional regulatory category-wide burden, we observed Bonferroni-significant category-wide burden among heart-transcribed RBP binding sites. These data implicate noncoding DNVs in CHD at both transcriptional and post-transcriptional regulatory levels. Our ability to detect signals was strongly influenced by availability of cardiovascular development noncoding genomic data, allowing us a narrow search space for DNV interrogation.
Through the two cardiac regulatory element strategies and their significant overlapping results, we identified known and potential human CHD genes. HeartENN-damaging variants were enriched for known human CHD genes (e.g., GATA4, OFD1), but there was little concordance between observed and reported cardiac/extracardiac phenotype constellations. Only one of seven genes identified with both approaches is implicated in heart development: COL1A2 encodes a collagen highly expressed in developing cardiac valves46, and all seven probands with noncoding COL1A2 DNVs had pulmonary and/or aortic valve abnormalities. Whether the other overlapping genes represent novel CHD genes or poor understanding of noncoding DNVs’ genic regulation remains uncertain. Among 20 non-overlapping genes with multi-DNV enrichment in prioritized human fetal heart enhancers, four are implicated in heart development: ATE1 depletion causes CHD in mice47; LRRTM2 resides within a CHD-associated region48; MITF regulates GATA4 expression49,50; and RPL29 encodes a target of LSD, a demethylase whose depletion causes CHD in mice51,52. Other gene associations include cardiomyopathy (FNIP1)53, striated muscle disorders (SUN1)54,55, and mouse embryonic lethality (SEPT11)56. When considering the union of transcriptional and post-transcriptional variants, SHOC2, CPSF3, ZNRF3, and MAP4K4 regulatory regions were consistently identified.
Among 31 DNV-containing sequences functionally tested in iPSC-CMs, five (16%) significantly altered transcription. While that rate is consistent with bioinformatic enrichment analyses, there are reasons to consider this a lower bound. The sequences were only tested in fetal cardiomyocytes at two time points using minimal promoters not in their native genomic context. Oligogenetic effects were not modeled in this functional assay. Genes associated with the five positive DNVs provide clues regarding CHD causality. JPH2 encodes junctophlilin-2, a membrane protein necessary for T-tubule formation, for which an N-terminal cleavage fragment modulates MEF2-mediated gene transcription, altering ERK and TGF-β signaling57. SEMA4B is in the top quartile for developing heart gene expression and encodes a semaphorin that signals through plexin receptors. Perturbations in semaphorin-plexin signaling can lead to CHD58,59. Future studies of additional DNVs incorporating more complex models will be needed to elaborate CHD pathogenesis precisely.
Our cohort was selected for WES-negative trios and higher paternal age to increase statistical power to identify a noncoding and de novo signal, respectively. Among this CHD cohort, we estimated that the fraction of subjects harboring noncoding predicted functional DNVs that contribute to CHD is at least as high as the fraction of CHD cases with contributory coding DNVs. We observed consistent results in isolated CHD and those with NDDs or ECAs, which is distinctly different than the NDD and ECA enrichment among CHD probands with damaging coding DNVs. For the prioritized human fetal heart enhancer DNVs, this manifested as depletion of patients with CHD and NDD compared to WES-implicated coding DNVs. These results could be explained by cardiac-specific effects in at least a subset of DNVs, suggesting future work could build on the cardiac relevance described here with a focus on cardiac specificity. The implicated GATA4 enhancer in a proband with isolated CHD illustrates the potential to uncouple frequently associated phenotypes through cardiac-selective regulatory effects.
While our findings established that noncoding DNVs contribute to CHD pathogenesis, the relevant genetic mechanisms remain to be explored. Previous studies of rare coding variants suggested some are sufficient to engender CHD (i.e., Mendelian genetic model), while many others are associated with incomplete penetrance, suggesting greater genetic complexity (e.g., oligogenic model) and/or environment effects. While noncoding DNVs contributing to CHD could act in a simple Mendelian manner (for instance, substantially reducing allelic expression), more modest gene expression effects would be congruous with an oligogenic mechanism. Future studies of noncoding variants observed in CHD are needed to establish transcriptional effect sizes and their ability to perturb heart development individually and in concert with other relevant factors.
These data are the first to systematically associate human CHD with cardiac regulatory DNVs. Our findings highlight the potential of WGS to more fully elucidate the genetic architecture of CHD. Extension of the statistical framework used is likely to define additional noncoding variants in CHD. When applied to larger cohorts, we expect to refine the magnitude of noncoding effects and to investigate complex CHD genetics, such as epistatic and pleiotropic effects of noncoding and coding variants.
METHODS
Participants.
Pediatric Cardiac Genomics Consortium (PCGC).
Patients with structural CHD and their parents (n = 763 trios) were enrolled in the PCGC’s Congenital Heart Disease Network Study (CHD GENES: ClinicalTrials.gov identifier NCT01196182)2. The protocols were approved by the Institutional Review Boards of Boston’s Children’s Hospital, Brigham and Women’s Hospital, Children’s Hospital of Los Angeles, Children’s Hospital of Philadelphia, Columbia University Medical Center, Great Ormond Street Hospital, Icahn School of Medicine at Mount Sinai, Rochester School of Medicine and Dentistry, Steven and Alexandra Cohen Children’s Medical Center of New York, and Yale School of Medicine. All participants or their parents provided informed consent. Individuals with a chromosomal aneuploidy, copy number variation associated with CHD, or likely causal variant identified with WES were excluded. The echocardiogram, catheterization, and operative reports were reviewed to determine cardiac phenotypes. Extracardiac structural anomalies were obtained from the medical records. Patients were classified as having neurodevelopmental disorders (NDDs) if parents reported the presence of developmental delay, learning disability, mental retardation, or autism for individuals at least 12 months old.
Controls.
Controls comprised 1,611 CHD- and autism-unaffected sibling-parent trios derived from sporadic autism quartets that consisted of one offspring with autism, one unaffected sibling, and their unaffected parents6. Controls were ascertained from 1,627 siblings after excluding 16 with a past medical history including CHD. The Simons Foundation kindly provided the phenotypic and genomic data for these unaffected trios.
Whole genome sequencing and variant identification.
DNA of the PCGC samples were sequenced at the Baylor College of Medicine Genomic and RNA Profiling Core (n = 900), the New York Genome Center (NYGC) Genomic Research Services (n = 75), and the Broad Institute for Genomic Services (n = 1,314) following the same protocol. Genomic DNAs from venous blood or saliva were prepared for sequencing using a PCR-free library preparation (n = 2,289) or SK2-IES library preparation (n = 75, Broad). All samples were sequenced on an Illumina Hi-Seq × Ten with 150-bp paired reads to a median depth >30x per individual. The controls were prepared similarly to cases. Specifically, the controls were sequenced at NYGC (n = 4,833) with 150-bp paired reads and median depth >30x per individual, using either a PCR-based library preparation on an Illumina Hi-Seq 2000 (n = 114) or a PCR-free library preparation on an Illumina Hi-Seq × Ten (n = 4,719). Previous Simons Simplex Collection sequencing of controls was performed at NYGC on the Illumina Hi-Seq 2500 (n = 120) or Illumina Hi-Seq × Ten (n = 4,761) to >30x coverage with 150-bp paired reads.
For both cases and controls, reads were aligned to GRCh37 or GRCh38 with the Burrows-Wheeler Aligner (BWA-MEM)60. GATK Best Practices recommendations were implemented for base quality score recalibration (QSR), indel realignment, and duplicate removal61. Standard hard filtering parameters were used for SNV and indel discovery across all 763 PCGC and 1,611 control trios, followed by N+1 joint genotyping and variant QSR62,63.
Identification and confirmation of de novo variants (DNVs).
DNV identification was performed for both cases and controls by pooling three pipelines from PCGC members at Mount Sinai, Columbia and Harvard. Mount Sinai used two tiers, a high stringency tier and a low stringency tier. High stringency tier parameters were GATK PASS (i.e., variants classified as true with an adaptive error model based on known true sites and artifacts), heterozygous ratio (AB) set to 0.3–0.7 in the proband, homozygous ratio (AB) less than 0.01 in both parents, depth (DP) ≥ 10, Joint Genotyping allele count (AC) = 1 across all trios, Genotype Quality (GQ) > 60 (proband and parents), Alternate Allele Depth (AAD) > 7 in the proband, and AAD < 3 in each parent. The lower tier consisted of de novo calls falling outside of the higher tier that did not fail the following filters: GATK PASS, heterozygous AB set to 0.2–0.8, DP 7–120, AC = 1 in all trios, GQ > 60 (proband), GQ > 30 (parents), AAD > 7. At Columbia, parameters for DNV identification were heterozygous or homozygous for the alternate allele in the proband, homozygous for the reference allele in the parents, not in a multiallelic site (3 or more), AC ≤ 2 in the cohort, Fisher’s exact test strand bias (FS) < 25, variant quality by depth (QD) > 2 for SNVs and QD > 1 for indels, ReadPosRankSum > −3 for indels, proband genotype Phred-scaled likelihood (PL) ≥ 70, proband AAD ≥ 6, proband heterozygous AB ≥ 0.28 if AAD ≥ 10 or heterozygous AB ≥ 0.20 if AAD < 10, parental GQ ≥ 30, parental DP ≥ 10, parental AB < 0.035, and population frequency < 0.1% (1KG, ESP, ExAC). For the third pipeline at Harvard, the parameters were AC = 1, DP 7–64 inclusive, ADD ≥ 5, heterozygous AB 0.2–0.8 inclusive, homozygous AB ≤ 0.1. Putative de novo calls near indels, in a homopolymer indel, or in a dinucleotide repeat were subsequently visually filtered with IGV. After consolidating de novo calls, all variants were force called with FreeBayes7. GATK and FreeBayes both perform local realignment. GATK uses a combination of known common variants, indels, and entropy calculations to generate log of the odds ratio (LOD) scores for alternative consensus sequences, replacing original alignments if LOD scores are higher. FreeBayes generalizes this Bayesian caller approach to allow for multiallelic loci and non-uniform copy number across samples, and the combination of GATK and FreeBayes variant calling was previously reported to improve the positive predictive value of indel identification to >97%64. Therefore, FreeBayes variant calling was performed on GATK-identified de novo variants to reduce false-positive variants. DNVs in Sinai’s high evidence tier but false with FreeBayes were manually reviewed. Finally, IGV plots of all the putative DNVs were passed through an 8-layer convolutional neural network trained on curated IGV plots, and classified into 6 categories (de novo SNVs, de novo insertions, de novo deletions, complex, uncertain, and false positives)8. Predicted false positives were excluded. Predicted de novo insertion, deletion, complex and uncertain events were subject to further manual inspection to remove additional false positives. DNVs with ExAC allele frequency > 0.1% as well as DNVs in nonstandard chromosomes, segmental duplications (score ≥ 0.99), low complexity regions, low mappability (300 bp, score < 1) regions, mucin or HLA genes, and ENCODE blacklisted sites were removed15,65–67. Finally, all DNVs within 50 bp in the same proband were considered a single event (i.e., a mutation cluster) for region-based and multiple-hit enrichment tests. DNVs identified using GRCh37 were lifted over to GRCh38.14. Sanger sequencing validation was performed for 266 de novo SNVs and 83 de novo indels.
Reference-free calling to identify candidate coding DNVs.
An alternative, reference-free DNV calling algorithm, RUFUS (https://github.com/jandrewrfarrell/RUFUS)68, was also used to call de novo variants in PCGC probands. Briefly, RUFUS compares the k-mer sequences directly from the raw Illumina reads of the proband-parent trio to identify unique DNA sequences present in the child that represent de novo genetic variation. Sequencing reads that contain these unique k-mer sequences are assembled using an in-built sequence assembler. Assembled contigs, containing the de novo allele, are mapped back to the human reference sequence for localization, using the BWA algorithm. RUFUS then interprets the aligned contigs to produce a VCF formatted variant report. All types of de novo variation (SNVs, short INDELs, and SVs of all types) are identified in a single run of the program.
Gene sets.
The three gene sets used in this study were genes in which coding mutations cause isolated or syndromic CHD in humans (human CHD genes), genes for which mouse knock-downs or knock-outs are associated with CHD (mouse CHD genes), and the top quarter of expressed genes during heart development (high heart expression, HHE genes)3,4. To generate the mouse CHD gene set, mammalian phenotype ontology (MPO) terms potentially relevant to CHD were identified. These were reviewed to remove cardiovascular terms not specific to CHD, such as cardiac dilation/hypertrophy, arrhythmias, and coronary artery disease69. Data on the mouse strains associated with these MPO terms were downloaded (http://www.mousemine.org/mousemine/). Only single-gene transgenic mutant mouse strains were kept, and these mouse genes were converted to their human orthologs (ftp://ftp.informatics.jax.org/pub/reports/HOM_MouseHumanSequence.rpt).
Multiple hypothesis testing correction for region-based test.
The P-value threshold was determined by correcting for the number of independently tested hypotheses. Because the 184 noncoding features were highly correlated (Supplementary Fig. 1), the number of independent hypothesis tests was set as the number of eigenvectors that explain ≥99% of the variance in the correlations between the features42. A P-value was simulated for all pair-wise correlations between features. The P-value is equal to the fraction of 10,000 permutations with a more extreme correlation than observed. Observed was calculated based on the overlap between DNVs and features. For each permutation, a random feature overlap matrix was generated by treating the observed overlaps as random variables and sampling from a binomial distribution. Eigenvalue decomposition of these P-values was used to estimate the number of effective tests that explain ≥99% of the variance in the 184 features. For the 184 noncoding cardiac gene regulatory features, this corresponded to 47 independent, effective tests and a Bonferroni P-value of 1.1×10−3 (0.05/47). These 184 features (i.e., 47 effective features) were tested in the context of six gene sets and genome-wide, so we corrected for these additional hypotheses. In order to account for testing six gene sets and genome-wide for 47 effective noncoding features, a final P-value cut-off of 1.3 × 10−4 = 0.05/(47×7) was used as a significance threshold for all comparisons.
HeartENN.
HeartENN encompasses two neural network-based epigenomic effects models: one for human heart chromatin data and one for mouse heart chromatin data. Both models use the same convolutional neural network architecture but predict different genome-wide features (90 for human, 94 for mouse) based on the heart-specific chromatin profiles available for each organism. The models were trained with PyTorch using the Selene library70.
Training and evaluation data for the genome-wide features (e.g., histone marks, transcription factors, and DNase I accessibility) included data processed from the Cistrome, ENCODE, and Roadmap Epigenomics projects, as well as a published dataset of 36 genome-wide p300/CBP and H3K27ac ChIP-seq profiles from ex vivo cardiac tissue samples in mouse and human across many conditions and developmental stages (Supplementary Table 7)11–28.
The architecture of the HeartENN models is extended from the DeepSEA33,34 architecture. In addition to HeartENN models predicting different regulatory features, the main changes are that (i) the HeartENN architecture contains double the number of convolution layers, (ii) the models predict the epigenomic features of the center 50-bp region and use the remaining 950-bp as the surrounding context sequence, and (iii) the number of kernels used in each convolution has been reduced (see Supplementary Note for details).
DNVs within RefSeq protein-coding exons were not scored with HeartENN (CHD probands, 792 DNVs; CHD-unaffected subjects, 1,749 DNVs); DNVs in noncoding exons were scored.
Accounting for varying HeartENN thresholds.
We compared the number of DNVs in CHD probands to unaffected subjects with HeartENN scores above varying thresholds. In this context, optimal power for rejecting the null hypothesis that cases and controls have similar rates of relevant HeartENN scores is achieved with the variable threshold test71. This was performed by DNV case-control label swapping across all HeartENN cut-offs in 0.05 intervals. For every resample, we randomly assigned case-control status to DNVs with replacement and identified the most significant P-value at any cut-off. Comparing this null distribution to the most extreme observed P-value resulted in a resampling P-value.
iPSC-derived cardiomyocyte differentiation and ATAC-seq.
Accessible chromatin regions during cardiomyocyte differentiation were identified via Assay for Transposase-Accessible Chromatin using sequencing (ATAC-seq) of isogenic human iPSC-CMs during several points during states of differentiation.
Cells were differentiated according to previously described methods with small modifications72. One million iPSCs were plated in 6-well plates and maintained for three days. The differentiation process was performed when cells were ~95% confluent. Differentiation was performed using the GSK inhibitor (ChIR 18 μM) and Wnt inhibitor protocol (IWP4 5 μM) referenced above. Selection was performed at days 12–15 using glucose-starved media. Cells were harvested at days 8, 17, and 30. Cell viability had to be >80% for cells collected. Cells were observed under a microscope, and for days 17 and 30, cells were only collected if the whole well was beating (wells that only had beating clusters were discarded).
ATAC-seq was performed as previously described73,74. Briefly, 50,000 cells were harvested and lysed to isolate nuclei. Nuclei were treated with Tn5 transposase (Nextera DNA Sample Prep Kit, Illumina) and DNA was isolated. Fragmented DNA was then amplified using bar-coded PCR primers and libraries were pooled. ATAC libraries were visualized on the tape station for characteristic nucleosome patterning before sequencing. Pooled libraries were then sequenced (Illumina Next-seq) to a depth of 100 million reads per sample. Reads were aligned to the hg19 reference genome using BWA-MEM and peaks were called using HOMERv4.975. Functional analysis of ATAC-seq peaks was performed using ChIPseeker (v.1.14.1)76. De novo motif enrichment was performed using HOMERv4.9. Differential peaks were identified using HOMERv4.9. Libraries that contained an excess of mitochondrial DNA (>15% for iPSC-CMs) were removed. Each replicate was analyzed individually (n = 3–4 per time point) and compared to other replicates at the same time point, and data were also visualized in IGV/UCSC Genome Browser. Comparison of any two replicates results in ~85–95% peak overlap between replicates.
Enrichment for genes with burden of DNVs in associated fetal cardiac enhancers.
Cardiac enhancer elements were identified by H3K27ac peaks from human cardiac tissue77. Enhancer peaks were assigned to the closest RefSeq transcription start site and intersected with ATAC-seq peaks from days 8 or 17 (see “iPSC-derived cardiomyocyte differentiation and ATAC-seq”). The likelihood of multiple genes having DNV enrichment was assessed by randomly permuting the 7,378 total DNVs associated with the prioritized human fetal heart enhancers to case or control status with the same 2,218:5,160 ratio. The number of genes with enrichment P < 0.05 in either cohort was calculated using a two-sided Fisher exact test.
Massively parallel reporter assays (MPRAs).
The effect of CHD noncoding DNVs on enhancer activity was assessed by MPRAs41, using constructs with longer sequences so as to assess those residing in broad ATAC peaks identified in D17 iPSC-CM peaks. DNVs were selected for study using the following criteria: HeartENN score ≥ 0.1 and with a prioritized human fetal heart enhancer (8 of 9 tested); HeartENN score ≥ 0.5 (11 of 22 tested); prioritized human fetal heart enhancer for which the associated gene was highly expressed in the developing heart (mouse E14.5 expression rank >75th percentile) and highly constrained (pLi > 0.8) (9 of 24 tested); and HeartENN score ≥ 0.1 and within a strong iPSC-CM D8 or D17 ATAC-seq peak as well as an overlapping human fetal H3K27ac peak (11 of 24 tested). Of note, most of the DNVs meeting those criteria that were not tested either contained a restriction site that would have prevented cloning of the full-length sequence or had repetitive sequences that were problematic for synthesis.
Gene fragments with 300–1,600 bp in length harboring reference and variant alleles were synthesized by Twist. Each fragment was separately PCR amplified, and SfiI restriction enzyme sites were incorporated. After cleaning with AmpureXP beads, equimolar amount pooled constructs were combined. To minimize occurrence of the restriction enzyme site in the enhancer sequences, SalI was substituted for XbaI when cloning the inserts and to accommodate this change, the SalI site downstream of the polyA signal in the pMPRA1 (Addgene 49349) was mutated, using MfeI and BbsI sites in proximity. Modified MPRA plasmid sequences were verified using Sanger sequencing.
Gene fragments were cloned using the published MPRA protocol41. In short, the pooled enhancer fragments were digested with SfiI and ligated to the modified and digested pMPRA1 backbone with T4 DNA ligase. Plasmids were transformed into 5’alpha electrocompetent E. coli and harvested with Qiagen Maxiprep. Isolated plasmid was digested with SalI and KpnI with Shrimp Alkaline Phosphatase. Promoter and luciferase isolated from pMPRAdonor2 (Addgene 49353) were than cloned into the intermediate plasmid. Final plasmid library was cleaned and concentrated with ethanol.
iPSCs were cultured under standard condition using mTesr. iPSCs were differentiated into CMs using the standard protocol78, and iPSC-CMs were selectively enriched using glucose starvation for 4 days. iPSC-CMs were replated into monolayers with 10x TrypLE. After replating, healthy cells that were vigorously beating were used for library transfection using Lipofectamine 3000 according to the manufacturer’s instruction. Total RNA was harvested with Trizol 48 h after transfection, and genomic DNA was removed with DNase1. cDNAs were synthesized using the SuperScript III First Strand Synthesis kit with oligo dT according to the manufacturer’s instruction. MPRA barcodes were amplified from cDNAs and plasmids using the Tagseq primers.
Sequencing reads containing correct plasmid sequences were selected from raw reads. Barcode sequences were then matched, counted, and normalized to the total number of barcode reads in the sequencing run.
Every variant in the “HeartENN ≥ 0.1 + FHP” group was replicated using four independent plasmid libraries; variants in the remaining three groups were replicated using three independent plasmid libraries. Libraries one, two, and four were transfected on differentiation day 17, while the third was transfected on day 37. Each plasmid library experiment was repeated in four or five wells. Together, this resulted in 12–20 expression measurements per mutant and wild-type variant with an extremely robust set of replicates incorporating different wells, plasmid libraries, and time points.
RNA binding protein eCLIP binding data.
Raw eCLIP binding data for the 160 available RNA binding proteins were obtained from ENCODE15. Peaks were called from replicates using CLIP Took Kit (CTK)79 and further processed80 into a narrower, higher confidence set of binding regions for each RBP. All peaks were then given 50 base pair padding on both sides to expand the genomic coverage and increase the number of variants associated with each RBP.
Analysis of disruption of post-transcriptional regulation.
Five groups of annotations were defined to investigate post-transcriptional regulation through disruption of RNA-binding protein binding: (i) 3 variant types (SNV, indel, all); (ii) 3 region types (TSS ± 20 kb region anchor where TSS is gene transcription start sites, 3’UTR region anchor defined as (TES – 5 kb, TES + 20 kb) where TES is gene transcription end sites, no region restriction); (iii) 1 RBP category (union of eCLIP peaks from 160 RBPs, padded on both sides with 50 bp); (iv) 2 gene sets (unconstrained or pLI > 0.5 constraint on nearest gene); and (v) histone mark annotations for actively transcribed regions in relevant proxy tissues, specifically H3K36me3 in eight human embryonic stem cell tissues: ES-I3 stem cells (E001), ES-WA7 stem cells (E002), H1 stem cells (E003), H9 stem cells (E008), HUES48 stem cells (E014), HUES6 stem cells (E015), HUES64 stem cells (E016), ES-UCSF4 stem cells (E024); plus human fetal heart tissue (E083).
Histone modification peaks were downloaded as broadPeak files, originally determined from Roadmap Epigenomics ChIP-Seq25. Raw broadPeaks were preprocessed as follows to include the majority of the area between the 5’ and 3’ UTRs for transcribed genes and smoothen noise in identifying actively transcribed regions in proxy tissues: gaps under 1 kb between histone peaks within this region were filled in, resulting in slightly improved signal for genes with many nearby peaks throughout.
Picking one annotation from each group resulted in 162 possible combinations. These annotation categories were considered in the combination-wide association test and yielded 105 independent tests, giving 4.76 × 10−4 as the strict Bonferroni threshold. Two-sided Fisher’s exact tests were used to obtain odds ratios and associated P-values for all test combinations. DNVs within RefSeq protein-coding exons were excluded.
Attributable risk calculation.
The fraction of CHD attributable to noncoding DNVs was calculated by determining the excess fraction of DNVs in cases compared to controls, and we then assumed at most one contributory DNV per proband to calculate the attributable fraction (Equation 1). This AR was calculated for HeartENN-damaging DNVs at successively stringent thresholds, DNVs within prioritized human fetal heart enhancers in multiple gene sets, DNVs shared between these results, and DNVs implicated in the top RBP enrichment. The AR is cumulative across methods (after subtracting out the contribution of shared DNVs) and represent estimates that should be refined in future studies.
Statistics.
All burden tests were calculated using 2-sided Fisher’s exact tests with base values set to the total number of DNVs in cases or controls. Using total number of DNVs as baseline, instead of number of trios, accounts for parental age. The significance threshold was P < 0.05, adjusted for multiple testing within each hypothesis space as specified in the preceding Methods.
Extended Data
Extended Data Fig. 1. Other pipelines identified 94% of DNVs in control trios.
Overlaps with DNVs identified in 1,470 control trios with two other pipelines9,10. Of note, a third analysis of these trios did not include de novo calls43. For consistency with other pipelines, only SNVs were included and variants in LCRs, blacklists, segmental duplications, and repeats were excluded. Together, 94% of de novo SNVs were called by at least one other pipeline.
Extended Data Fig. 2. Correlation between parental age at proband birth and DNVs/trio.
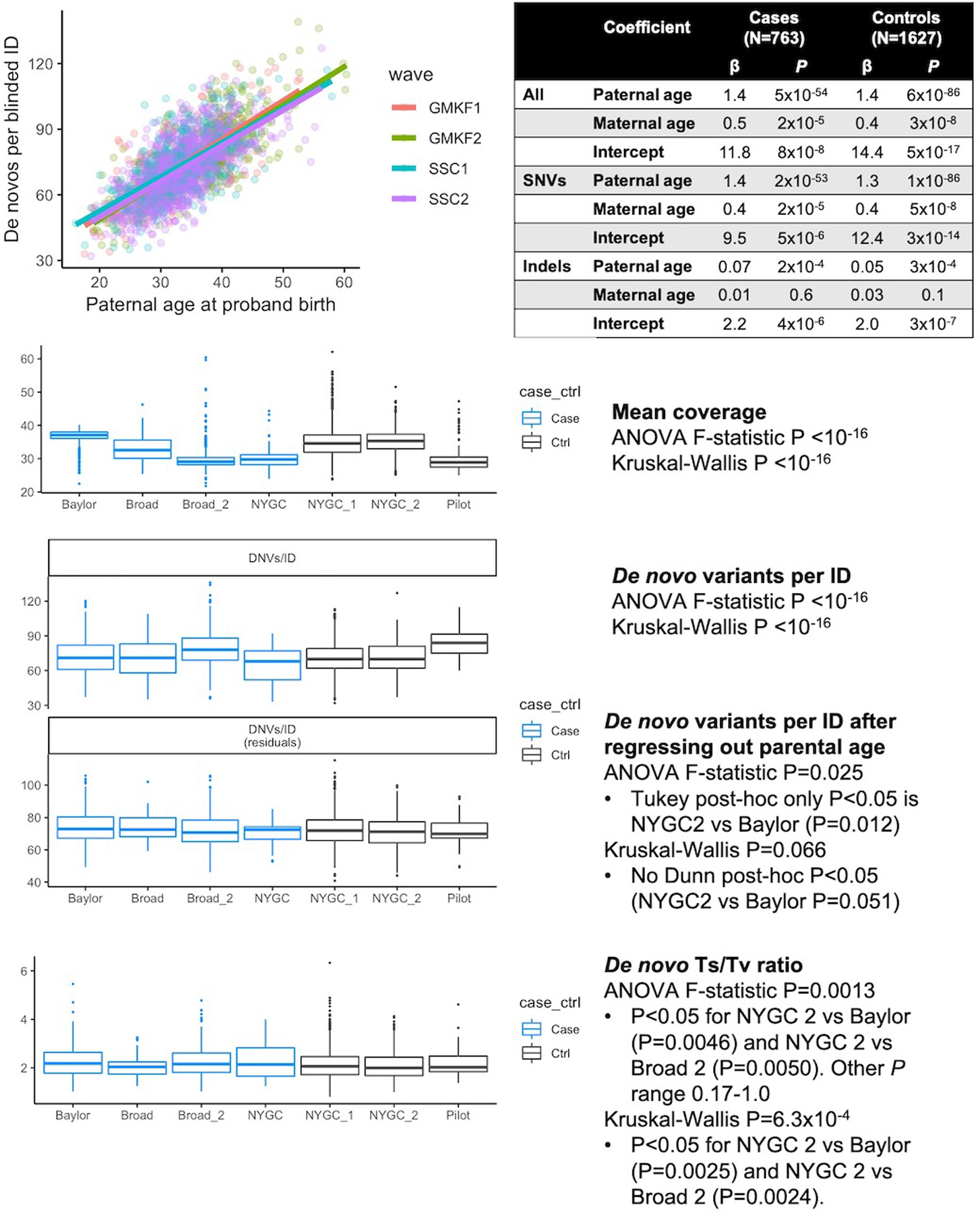
Multiple linear regression (βpaternal_agex + βmaternal_agex + βintercept + ε) was fitted on 763 CHD and 1,611 unaffected individuals to calculate the associations between paternal and maternal age for SNVs, indels, and combined. Regression coefficients and P-values are shown, uncorrected for multiple hypotheses. Sequencing metric comparisons between the centers, colored by cases (n = 763) and controls (n = 1,611), found moderate bias in DNV quantity, so the background statistical parameter throughout the manuscript is total number of DNVs. Box plots show medians and interquartile ranges.
Extended Data Fig. 3. De novo variant (DNV) CHD-unaffected burden.
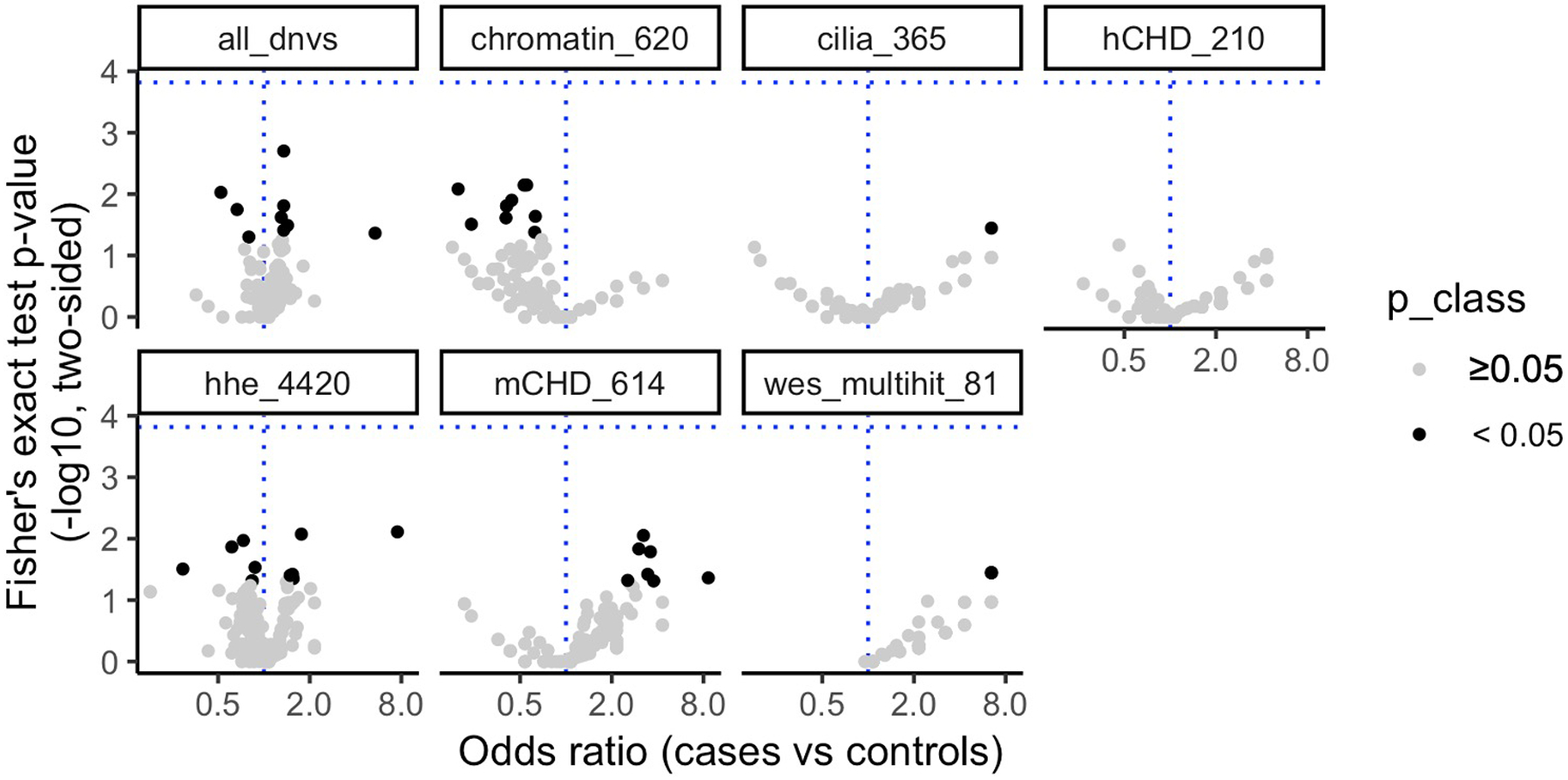
The number of DNVs in 184 noncoding annotations (points) genome-wide and within 10 kb of TSSs for 6 gene sets (facets) was counted in CHD (n = 749) and Simons unaffected (n = 1,611) individuals. The P value threshold (1.5 × 10−4, horizontal blue line) is 0.05 divided by the product of the number of effective annotations (n = 47) and number of gene sets (n = 7). The P value (y-axis) was calculated with a 2-sided Fisher’s exact test, the odds ratio (x-axis) was DNVsannotation,CHD/DNVstotal,CHD vs. DNVsannotation,unaffected/DNVstotal, unaffected. No annotations surpassed the P value threshold. CHD, congenital heart disease; HHE, high heart expression.
Extended Data Fig. 4. HeartENN performance was comparable to DeepSEA.
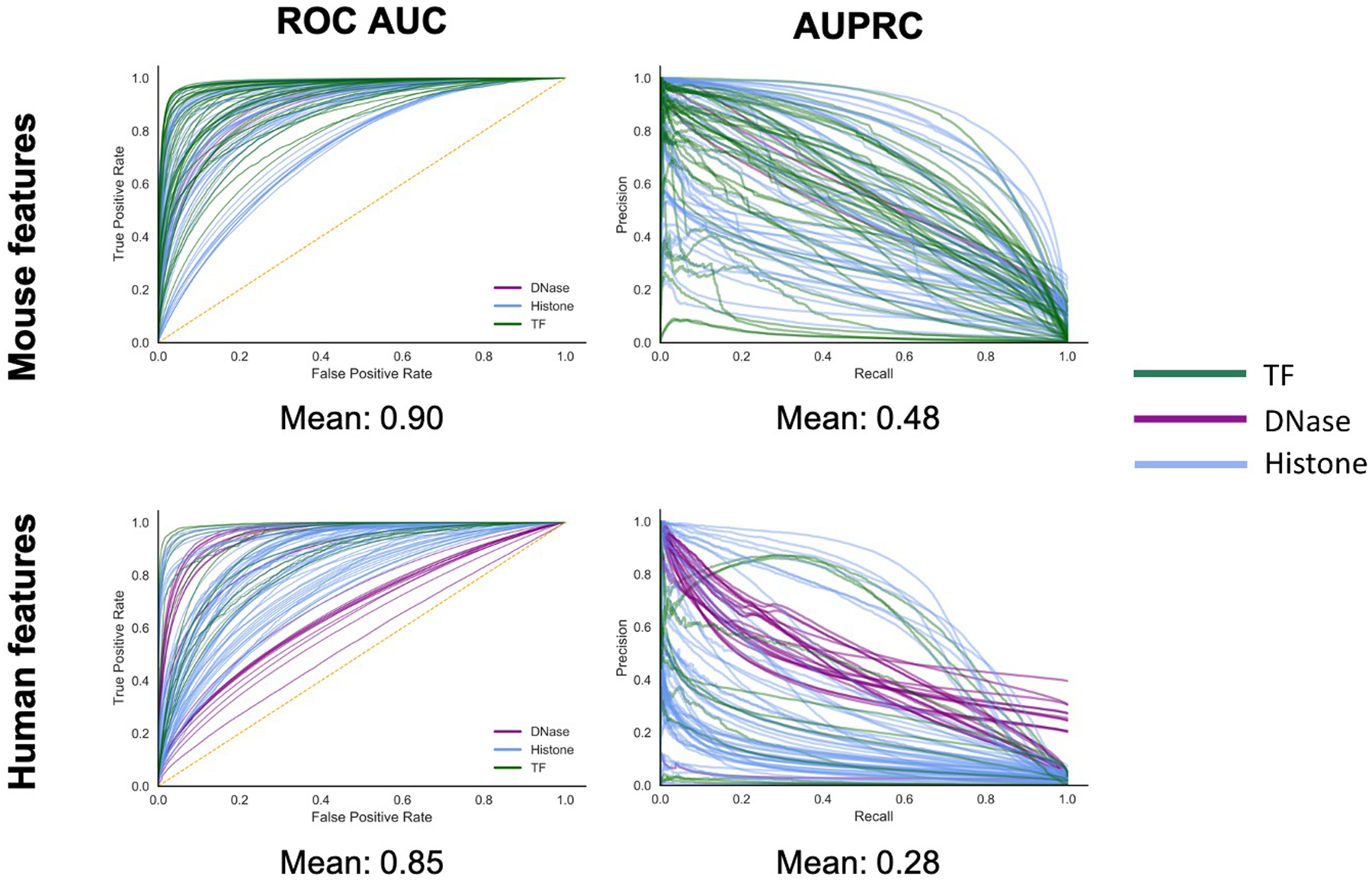
HearENN ROC AUC mean = 0.93 and AUPRC mean = 0.34. ROC AUC, receiver operator characteristics area under the curve; AUPRC, area under the precision recall curve.
Extended Data Fig. 5. Determining an absolute functional difference score range.
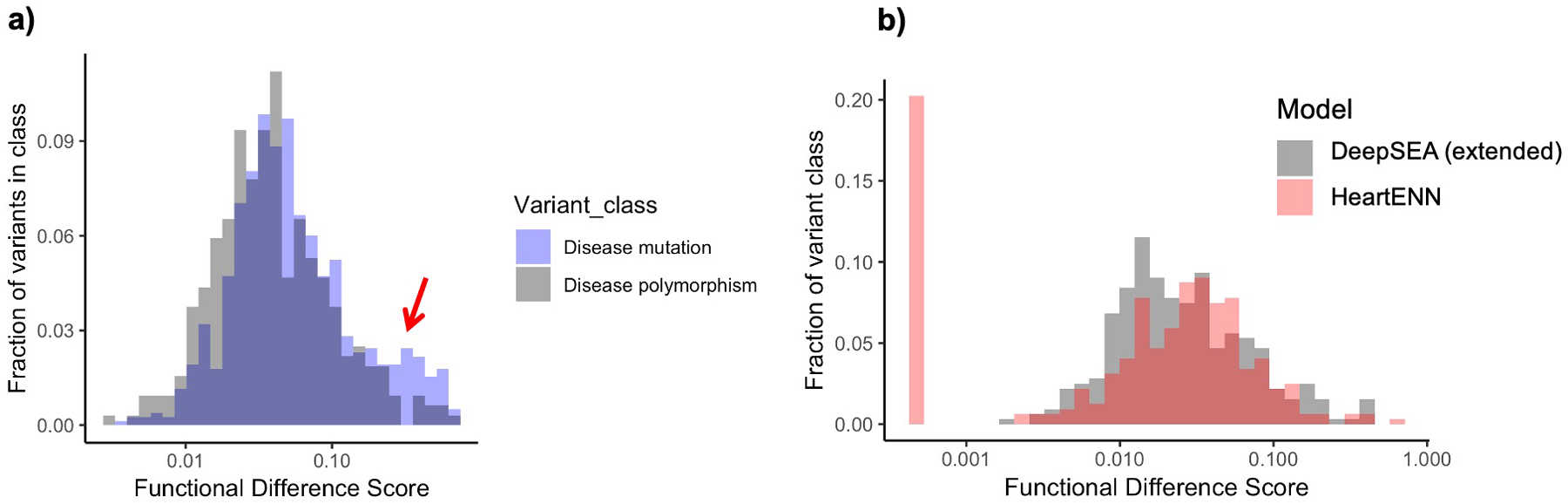
a, Comparison of HGMD disease mutations (blue, n = 1,564) and polymorphism (gray, n = 642) DeepSEA absolute functional difference scores at varying functional cut-offs illustrates a similar distribution and functionally impactful range ≥0.1 (arrow) for disease mutations. No statistical significance testing was performed. b, The similarity of null distributions for DeepSEA (gray, downsampled to 184 features) and HeartENN (heart) HGMD polymorphism scores suggested that the DeepSEA functional score range was also applicable to HeartENN (gray and red n = 642). Scores of 0 set off to left (as 10−4).
Extended Data Fig. 6. Support for HeartENN ≥ 0.1 functional ranking.
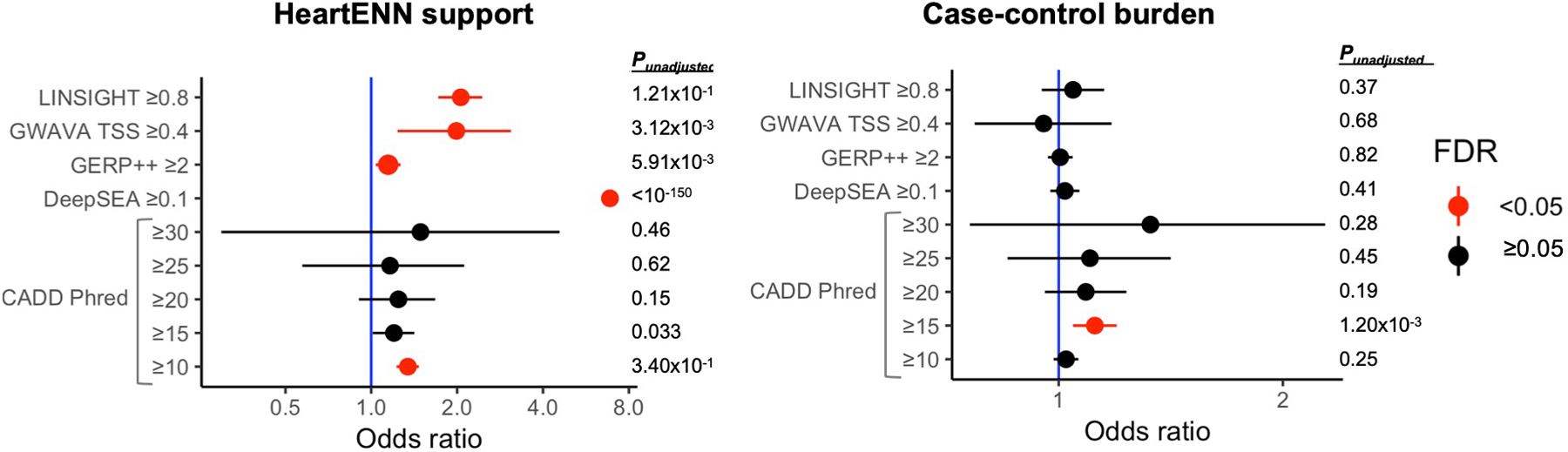
For all DNVs (n = 170,171), overlap between HeartENN ≥0.1 (n = 6,415) and other noncoding scores was assessed with a 2-sided Fisher’s exact test (left panel). Case-control burden for these other noncoding scores (right panel) was statistically significant for CADD ≥15 (PBonferroni = 0.019) with a 2-sided Fisher’s exact test (cases n = 56,164 and controls n = 114,065). For both panels, unadjusted P-values are tabulated, and red indicates a Benjamini-Hochberg-adjusted P-value false discovery rate (FDR) < 0.05.
Extended Data Fig. 7. Relationship between sequence length inserted into the pMRPA1 plasmid and the transcript reads/plasmid copies in MPRAs.
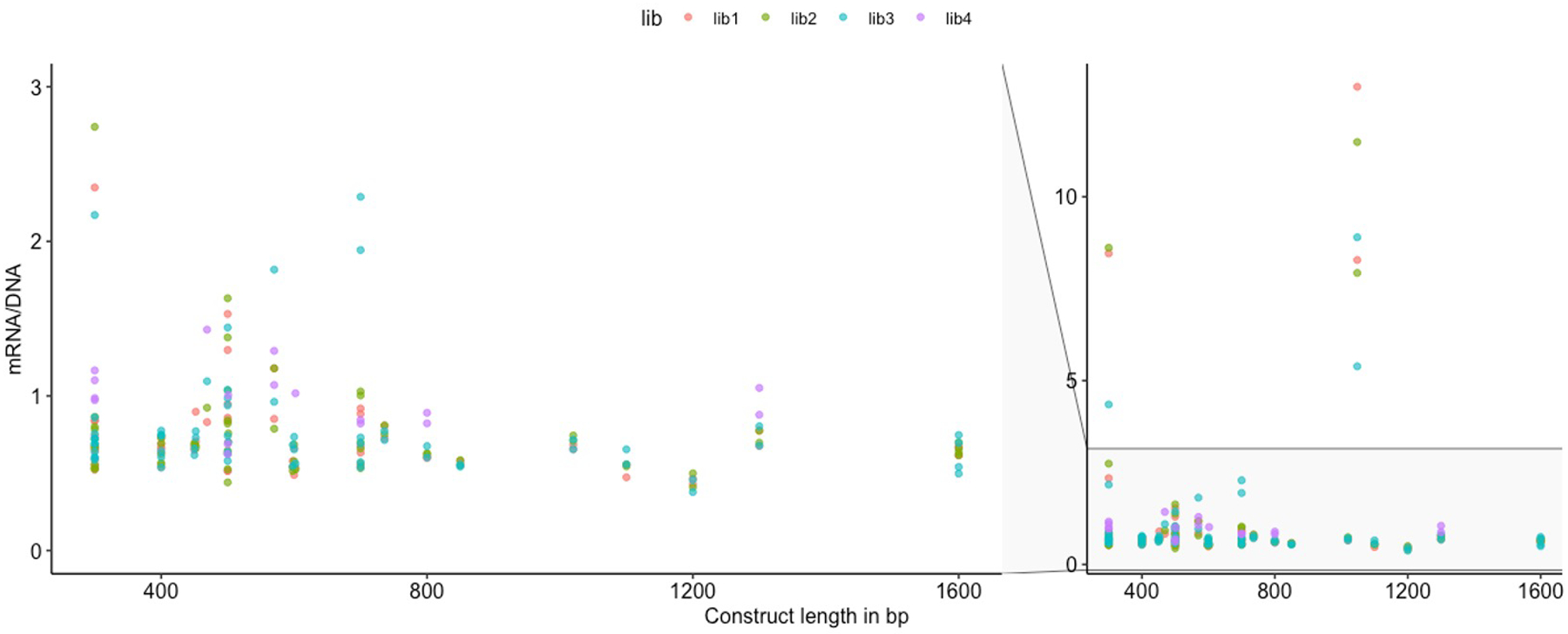
The length of the sequences inserted into the pMPRA1 plasmid (x-axis) ranged from 300 to 1,600 bp. After transfection of four libraries (color coded as per key) into the iPSC-CMs, the resulting ratios of transcript reads (mRNA) per plasmid copies (DNA) are graphed on the y-axis, showing no systematic relationship between insert length and transcriptional level.
Extended Data Fig. 8. DNVs with a trend towards decreased expression by MPRA assay.
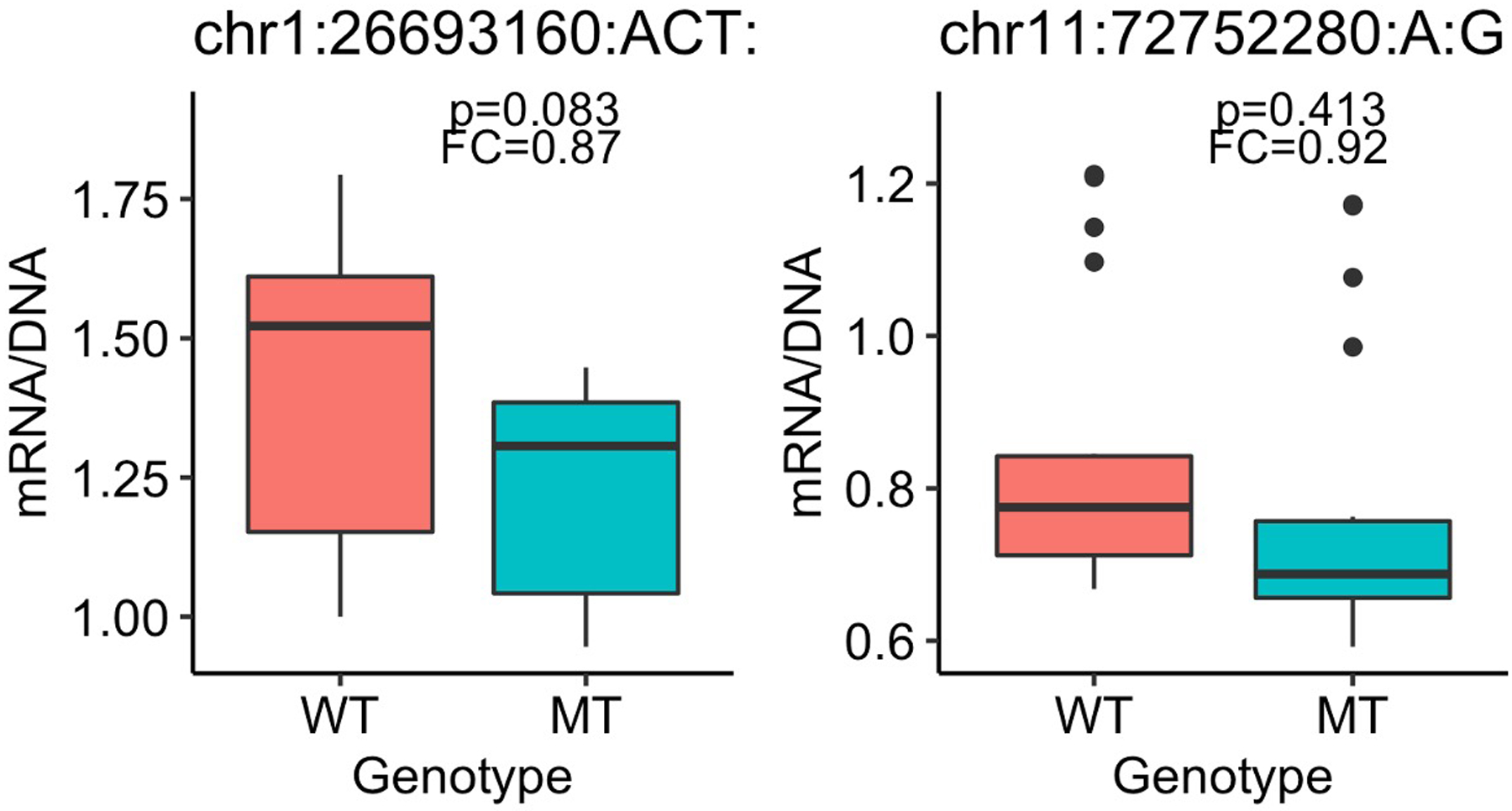
Box plots for two DNVs for which two MPRA replicates were significantly different but overall statistical significance across all replicates was not attained. Boxplots show the median fold change (FC), first and third quartiles (lower and upper hinges), and range of values (whiskers and outlying points). Statistical significance was assessed with 2-sided t-test Benjamini-Hochberg-adjusted P-values. Each boxplot has at least 3 independent experiments with 4 technical replicates each.
Extended Data Fig. 9. Fraction of DNVs in each of the canonical variant classes.
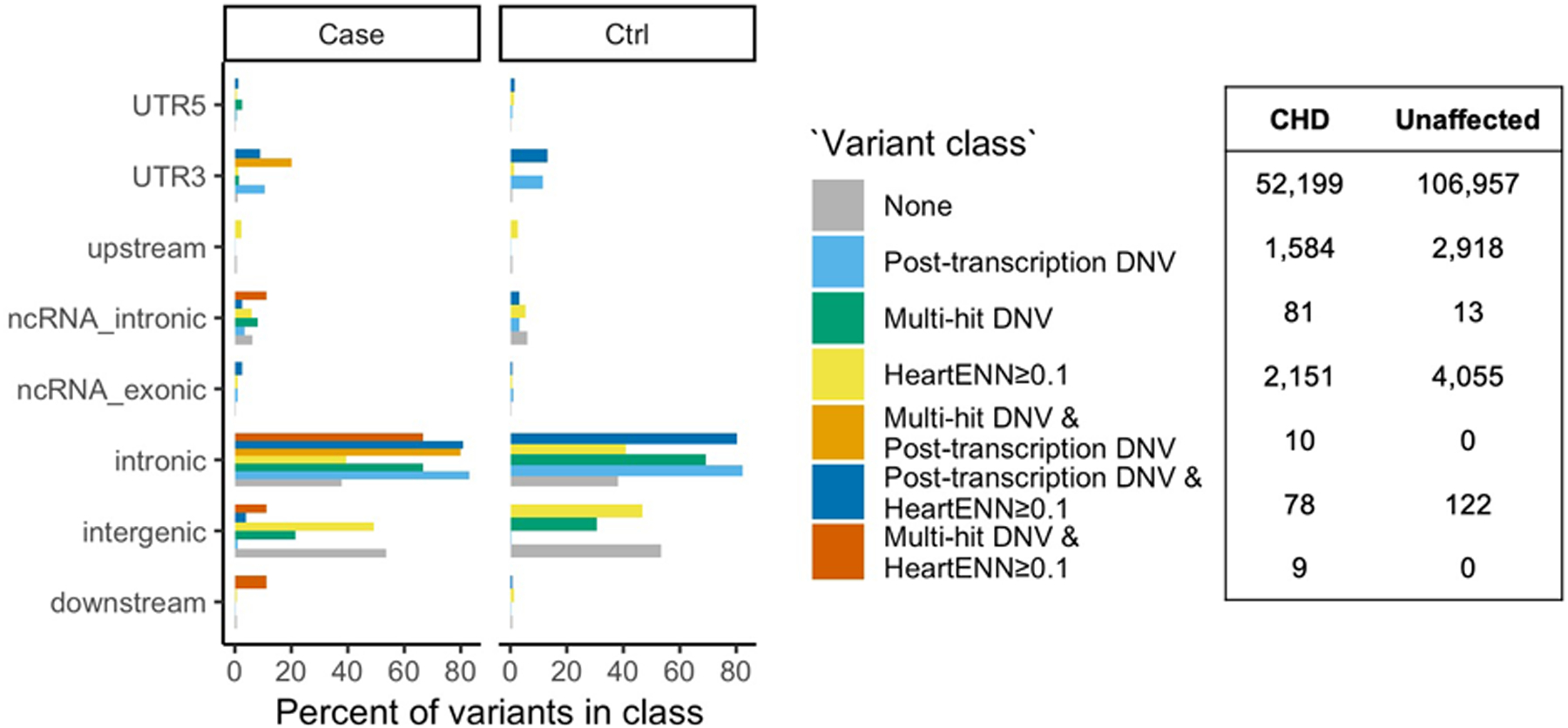
The fraction was calculated separately within CHD and unaffected subjects for each of the three methods (including overlaps) and the total number of variants in each group (right table).
Extended Data Fig. 10. DNV enrichment in phenotype subgroups.
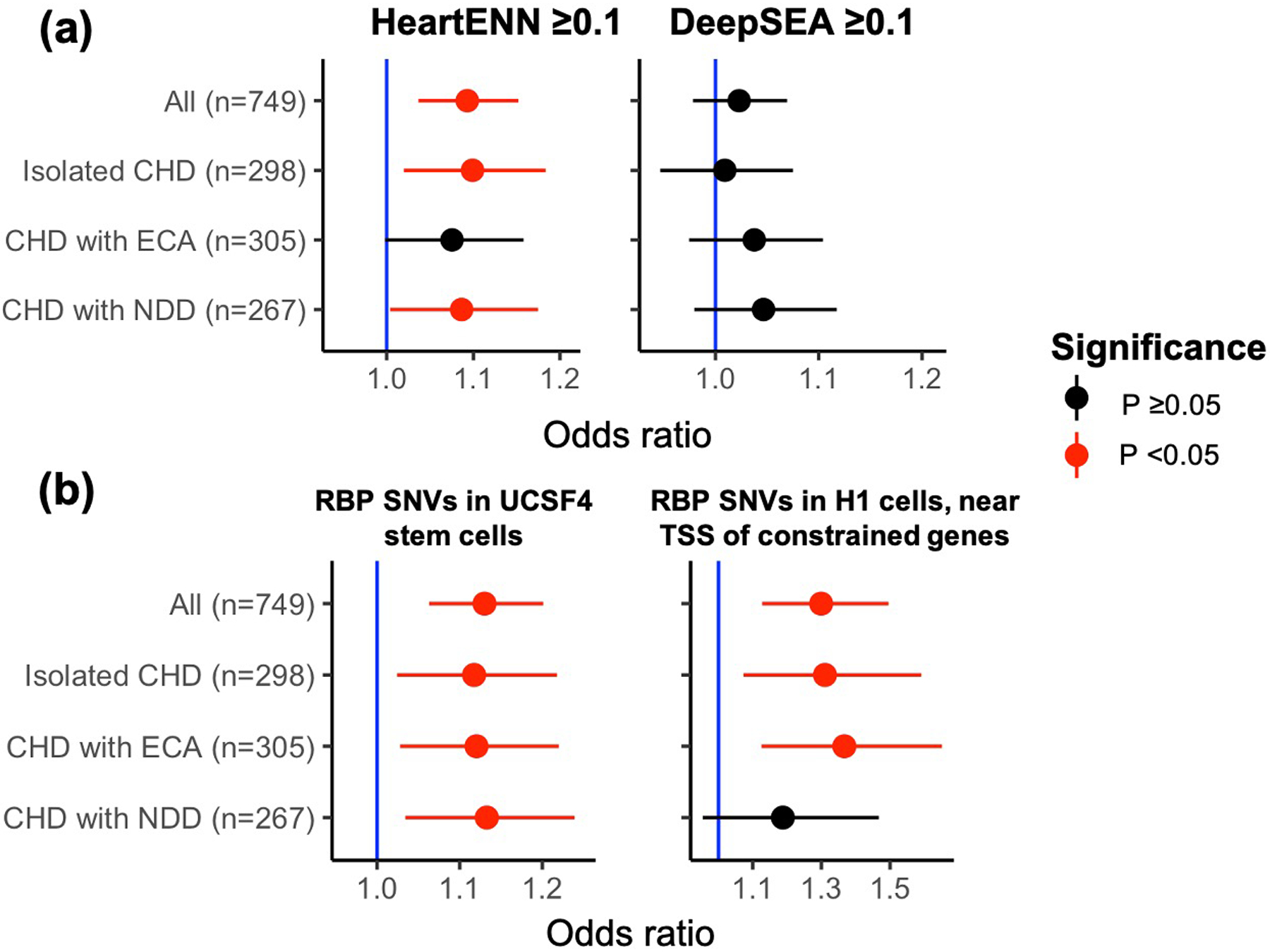
a, Enrichment of DNVs with predicted functional impacts (score ≥0.1) for HeartENN (left) and DeepSEA (right) within phenotype subgroups. b, Enrichment of de novo SNVs with H3K36me3 marks implicated in RNA-binding protein disruption in different subgroups for the most significant (left) and highest effect size (right) hits. Both a and b were performed with a 2-sided Fisher’s exact test (unadjusted P-values and 95% C.I.s shown) comparing the fraction of DNVs in each subgroup (HeartENN ≥ 0.1, DeepSEA ≥ 0.1, etc.) to the same control cohort. For HeartENN, there were n = 4,177 control DNVs with HeartENN ≥ 0.1 and n = 109,888 control DNVs with HeartENN < 0.1. NDD, neurodevelopmental disorder; ECA, extracardiac anomaly.
Supplementary Material
ACKNOWLEDGMENTS
We are enormously grateful to the patients and families who participated in this research. We thank the following for patient recruitment: A. Julian, M. Mac Neal, Y. Mendez, T. Mendiz-Ramdeen, and C. Mintz (Icahn School of Medicine at Mount Sinai); N. Cross (Yale School of Medicine); J. Ellashek and N. Tran (Children’s Hospital of Los Angeles); B. McDonough, J. Geva, and M. Borensztein (Harvard Medical School); K. Flack, L. Panesar, and N. Taylor (University College London); E. Taillie (University of Rochester School of Medicine and Dentistry); S. Edman, J. Garbarini, J. Tusi and S. Woyciechowski (Children’s Hospital of Philadelphia); D. Awad, C. Breton, K. Celia, C. Duarte, D. Etwaru, N. Fishman, E. Griffin, M. Kaspakoval, J. Kline, R. Korsin, A. Lanz, E. Marquez, D. Queen, A. Rodriguez, J. Rose, J.K. Sond, D. Warburton, A. Wilpers and R. Yee (Columbia Medical School); D. Gruber (Cohen Children’s Medical Center, Northwell Health). These data were generated by the Pediatric Cardiac Genomics Consortium (PCGC), under the auspices of the National Heart, Lung, and Blood Institute’s Bench to Bassinet Program (https://benchtobassinet.com). The results analyzed and published here are based in part on data generated by Gabriella Miller Kids First Pediatric Research Program projects phs001138.v1.p2/phs001194.v1.p2, and were accessed from the Kids First Data Resource Portal (https://kidsfirstdrc.org/) and/or dbGaP (www.ncbi.nlm.nih.gov/gap). This manuscript was prepared in collaboration with investigators of the PCGC and has been reviewed and/or approved by the PCGC. PCGC investigators are listed at https://benchtobassinet.com/Centers/PCGCCenters.aspx. This work was supported in part through the computational resources and staff expertise provided by Scientific Computing at the Icahn School of Medicine at Mount Sinai. We are grateful to all of the families at the participating Simons Simplex Collection (SSC) sites, as well as the principal investigators (A. Beaudet, R. Bernier, J. Constantino, E. Cook, E. Fombonne, D. Geschwind, R. Goin-Kochel, E. Hanson, D. Grice, A. Klin, D. Ledbetter, C. Lord, C. Martin, D. Martin, R. Maxim, J. Miles, O. Ousley, K. Pelphrey, B. Peterson, J. Piggot, C. Saulnier, M. State, W. Stone, J. Sutcliffe, C. Walsh, Z. Warren, E. Wijsman). We appreciate obtaining access to phenotypic and/or genetic data on SFARI Base. Approved researchers can obtain the SSC population dataset described in this study (https://www.sfari.org/resource/simons-simplex-collection/) by applying at https://base.sfari.org. This work was supported by the Mount Sinai Medical Scientist Training Program (5T32GM007280 to F.R.), National Institute of Dental and Craniofacial Research Interdisciplinary Training in Systems and Developmental Biology and Birth Defects (T32HD075735 to F.R.), Harvard Medical School Epigenetic and Gene Dynamics Award (S.U.M., C.E.S.), American Heart Association Post-Doctoral Fellowship (S.U.M.), and Howard Hughes Medical Institute (C.E.S.). Research conducted at the E.O. Lawrence Berkeley National Laboratory was supported by National Institutes of Health (NIH) grants (UM1HL098166 and R24HL123879) and performed under Department of Energy Contract DE-AC02-05CH11231, University of California. O.T. is a CIFAR fellow and this work was partially supported by NIH grant R01GM071966. The Pediatric Cardiac Genomics Consortium (PCGC) program is funded by the National Heart, Lung, and Blood Institute, National Institutes of Health, U.S. Department of Health and Human Services through grants UM1HL128711, UM1HL098162, UM1HL098147, UM1HL098123, UM1HL128761, and U01HL131003. The PCGC Kids First study includes data sequenced by the Broad Institute (U24 HD090743-01).
Footnotes
COMPETING INTERESTS
The authors have no competing interests as defined by Nature Research, or other interests that might be perceived to influence the results and/or discussion reported in this paper.
DATA AVAILABILITY
Whole genome sequencing data are deposited in the database of Genotypes and Phenotypes (dbGaP) under accession numbers phs001194.v2.p2 and phs001138.v2.p2.
CODE AVAILABILITY
Documentation, links, and availability of source code and select supplementary data are detailed at https://github.com/frichter/wgs_chd_analysis. The DNV identification pipeline is available at https://github.com/ShenLab/igv-classifier and https://github.com/frichter/dnv_pipeline. The HeartENN algorithmic framework is available at https://github.com/FunctionLab/selene/archive/0.4.8.tar.gz. HeartENN model weights and scripts for burden tests are available at https://github.com/frichter/wgs_chd_analysis. All source code is distributed under the MIT license.
REFERENCES
- 1.van der Linde D et al. Birth prevalence of congenital heart disease worldwide. J. Am. Coll. Cardiol 58, 2241–2247 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Gelb B et al. The Congenital Heart Disease Genetic Network Study: rationale, design, and early results. Circ. Res 112, 698–706 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zaidi S et al. De novo mutations in histone-modifying genes in congenital heart disease. Nature 498, 220–223 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Homsy J et al. De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science 350, 1262–1266 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jin SC et al. Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat. Genet 49, 1593–1601 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fischbach GD & Lord C The Simons Simplex Collection: a resource for identification of autism genetic risk factors. Neuron 68, 192–195 (2010). [DOI] [PubMed] [Google Scholar]
- 7.Garrison E & Marth G Haplotype-based variant detection from short-read sequencing. (2012). doi:arXiv:1207.3907v2
- 8.Richter F et al. Whole genome de novo variant identification with FreeBayes and neural network approaches. bioRxiv Genomics 2020.03.24.994160 (2020). doi: 10.1101/2020.03.24.994160 [DOI] [Google Scholar]
- 9.Zhou J et al. Whole-genome deep-learning analysis identifies contribution of noncoding mutations to autism risk. Nat. Genet 51, 973–980 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.An J-Y et al. Genome-wide de novo risk score implicates promoter variation in autism spectrum disorder. Science 362, eaat6576 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jónsson H et al. Parental influence on human germline de novo mutations in 1,548 trios from Iceland. Nature 549, 519–522 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Goldmann JM et al. Parent-of-origin-specific signatures of de novo mutations. Nat. Genet 48, 935–939 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Seiden AH et al. Elucidation of de novo small insertion/deletion biology with parent-of-origin phasing. Hum. Mutat 41, 800–806 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kent WJ et al. The human genome browser at UCSC. Genome Res. 12, 996–1006 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bernstein BE et al. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mei S et al. Cistrome Data Browser: a data portal for ChIP-Seq and chromatin accessibility data in human and mouse. Nucleic Acids Res. 45, D658–D662 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.He A et al. Dynamic GATA4 enhancers shape the chromatin landscape central to heart development and disease. Nat. Commun 5, 4907 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sayed D, Yang Z, He M, Pfleger JM & Abdellatif M Acute targeting of general transcription factor IIB restricts cardiac hypertrophy via selective inhibition of gene transcription. Circ. Heart Fail 8, 138–148 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stefanovic S et al. GATA-dependent regulatory switches establish atrioventricular canal specificity during heart development. Nat. Commun 5, 3680 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sayed D, He M, Yang Z, Lin L & Abdellatif M Transcriptional regulation patterns revealed by high resolution chromatin immunoprecipitation during cardiac hypertrophy. J. Biol. Chem 288, 2546–2558 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang L et al. KLF15 establishes the landscape of diurnal expression in the heart. Cell Rep. 13, 2368–2375 (2015). [DOI] [PubMed] [Google Scholar]
- 22.Anand P et al. BET bromodomains mediate transcriptional pause release in heart failure. Cell 154, 569–582 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Attanasio C et al. Tissue-specific SMARCA4 binding at active and repressed regulatory elements during embryogenesis. Genome Res. 24, 920–929 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sakabe NJ et al. Dual transcriptional activator and repressor roles of TBX20 regulate adult cardiac structure and function. Hum. Mol. Genet 21, 2194–2204 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Consortium RE et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.May D et al. Large-scale discovery of enhancers from human heart tissue. Nat. Genet 44, 89–93 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dickel DE et al. Genome-wide compendium and functional assessment of in vivo heart enhancers. Nat. Commun 7, 12923 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nord AS et al. Rapid and pervasive changes in genome-wide enhancer usage during mammalian development. Cell 155, 1521–1531 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blow MJ et al. ChIP-Seq identification of weakly conserved heart enhancers. Nat. Genet 42, 806–810 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yue F et al. A comparative encyclopedia of DNA elements in the mouse genome. Nature 515, 355–364 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shen Y et al. A map of the cis-regulatory sequences in the mouse genome. Nature 488, 116–120 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van den Boogaard M et al. Genetic variation in T-box binding element functionally affects SCN5A/SCN10A enhancer. J. Clin. Invest 122, 2519–2530 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou J & Troyanskaya OG Predicting effects of noncoding variants with deep learning–based sequence model. Nat. Methods 12, 931–934 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou J et al. Whole-genome deep-learning analysis identifies contribution of noncoding mutations to autism risk. Nat. Genet 51, 973–980 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang Y-F, Gulko B & Siepel A Fast, scalable prediction of deleterious noncoding variants from functional and population genomic data. Nat. Genet 49, 618–624 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kircher M et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet 46, 310–315 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rentzsch P, Witten D, Cooper GM, Shendure J & Kircher M CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 47, D886–D894 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Davydov EV et al. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput. Biol 6, e1001025 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ritchie GRS, Dunham I, Zeggini E & Flicek P Functional annotation of noncoding sequence variants. Nat. Methods 11, 294–296 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lek M et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Melnikov A, Zhang X, Rogov P, Wang L & Mikkelsen TS Massively parallel reporter assays in cultured mammalian cells. J. Vis. Exp (2014). doi: 10.3791/51719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Werling DM et al. An analytical framework for whole-genome sequence association studies and its implications for autism spectrum disorder. Nat. Genet 50, 727–736 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Turner TN et al. Genomic patterns of de novo mutation in simplex autism. Cell 171, 710–722.e12 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.C Yuen RK et al. Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat. Neurosci 20, 602–611 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hamdan FF et al. High rate of recurrent de novo mutations in developmental and epileptic encephalopathies. Am. J. Hum. Genet 101, 664–685 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Peacock JD, Lu Y, Koch M, Kadler KE & Lincoln J Temporal and spatial expression of collagens during murine atrioventricular heart valve development and maintenance. Dev. Dyn 237, 3051–3058 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kurosaka S et al. Arginylation regulates myofibrils to maintain heart function and prevent dilated cardiomyopathy. J. Mol. Cell. Cardiol 53, 333–341 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kleffmann W et al. 5q31 microdeletions: definition of a critical region and analysis of LRRTM2, a candidate gene for intellectual disability. Mol. Syndromol 3, 68–75 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mehta G et al. MITF interacts with the SWI/SNF subunit, BRG1, to promote GATA4 expression in cardiac hypertrophy. J. Mol. Cell. Cardiol 88, 101–110 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tshori S et al. Transcription factor MITF regulates cardiac growth and hypertrophy. J. Clin. Invest 116, 2673–2681 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nicholson TB et al. A hypomorphic lsd1 allele results in heart development defects in mice. PLoS One 8, e60913 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hamidi T et al. Identification of Rpl29 as a major substrate of the lysine methyltransferase Set7/9. J. Biol. Chem 293, 12770–12780 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Siggs OM et al. Mutation of Fnip1 is associated with B-cell deficiency, cardiomyopathy, and elevated AMPK activity. Proc. Natl. Acad. Sci. USA 113, E3706–E3715 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen C-Y et al. Accumulation of the inner nuclear envelope protein Sun1 is pathogenic in progeric and dystrophic laminopathies. Cell 149, 565–577 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Meinke P et al. Muscular dystrophy-associated SUN1 and SUN2 variants disrupt nuclear-cytoskeletal connections and myonuclear organization. PLoS Genet. 10, e1004605 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Röseler S et al. Lethal phenotype of mice carrying a Sept11 null mutation. Biol. Chem 392, 779–781 (2011). [DOI] [PubMed] [Google Scholar]
- 57.Guo A et al. E-C coupling structural protein junctophilin-2 encodes a stress-adaptive transcription regulator. Science 362, eaan3303 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yamagishi H et al. A History and Interaction of Outflow Progenitor Cells Implicated in “Takao Syndrome”. Etiology and Morphogenesis of Congenital Heart Disease: From Gene Function and Cellular Interaction to Morphology (2016). [Google Scholar]
- 59.Masuda T & Taniguchi M Congenital diseases and semaphorin signaling: Overview to date of the evidence linking them. Congenit. Anom. (Kyoto) 55, 26–30 (2015). [DOI] [PubMed] [Google Scholar]
- 60.Li H & Durbin R Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McKenna A et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.DePristo MA et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet 43, 491–498 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Van der Auwera GA et al. Current Protocols in Bioinformatics Current protocols in bioinformatics / editoral board, Baxevanis Andreas D. … [et al.] 11, (John Wiley & Sons, Inc., 2002). [Google Scholar]
- 64.Kim B-Y, Park JH, Jo H-Y, Koo SK & Park M-H Optimized detection of insertions/deletions (INDELs) in whole-exome sequencing data. PLoS One 12, e0182272 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bailey JA, Yavor AM, Massa HF, Trask BJ & Eichler EE Segmental duplications: organization and impact within the current human genome project assembly. Genome Res. 11, 1005–1017 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Derrien T et al. Fast computation and applications of genome mappability. PLoS One 7, e30377 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li H Towards better understanding of artifacts in variant calling from high-coverage samples. (2014). doi: 10.1093/bioinformatics/btu356 [DOI] [PMC free article] [PubMed]
- 68.Ostrander BEP et al. Whole-genome analysis for effective clinical diagnosis and gene discovery in early infantile epileptic encephalopathy. npj Genomic Med. 3, 22 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Blake JA et al. Mouse Genome Database (MGD)-2017: community knowledge resource for the laboratory mouse. Nucleic Acids Res. 45, D723–D729 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen KM, Cofer EM, Zhou J & Troyanskaya OG Selene: a PyTorch-based deep learning library for biological sequence-level data. bioRxiv 438291 (2018). doi: 10.1101/438291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Price AL et al. Pooled association tests for rare variants in exon-resequencing studies. Am. J. Hum. Genet 86, 832–838 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lian X et al. Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/β-catenin signaling under fully defined conditions. Nat. Protoc 8, 162–175 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Buenrostro JD, Wu B, Chang HY & Greenleaf WJ ATAC-seq: a method for assaying chromatin accessibility genome-wide in Current Protocols in Molecular Biology 109, 21.29.1–21.29.9 (John Wiley & Sons, Inc., 2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Corces MR et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Heinz S et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yu G, Wang L-G & He Q-Y ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31, 2382–2383 (2015). [DOI] [PubMed] [Google Scholar]
- 77.Spurrell CH et al. Genome-wide fetalization of enhancer architecture in heart disease. bioRxiv 591362 (2019). doi: 10.1101/591362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sharma A, Toepfer CN, Schmid M, Garfinkel AC & Seidman CE Differentiation and contractile analysis of GFP-sarcomere reporter hiPSC-cardiomyocytes. Curr. Protoc. Hum. Genet 96, 21.12.1–21.12.12 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shah A, Qian Y, Weyn-Vanhentenryck SM & Zhang C CLIP Tool Kit (CTK): a flexible and robust pipeline to analyze CLIP sequencing data. Bioinformatics 33, btw653 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Feng H et al. Modeling RNA-binding protein specificity in vivo by precisely registering protein-RNA crosslink sites. bioRxiv 428615 (2018). doi: 10.1101/428615 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.