

Abstract
G protein-coupled receptors (GPCR) are a large family of integral transmembrane proteins responsible for cellular signal transductions. Identification of therapeutic compounds to regulate the physiological processes is an important first step of drug discovery. We proposed MAGELLAN, a novel hierarchical virtual-screening (VS) pipeline, which starts with low-resolution protein structure prediction and structure-based binding-site identification, followed by homologous GPCR detections through structure and orthosteric binding-site comparisons. Ligand-profiles constructed from the homologous ligand-GPCR complexes are then used to thread through compound databases for virtual screening. The pipeline was first tested in a large-scale retrospective screening experiment against 224 human Class A GPCRs, where MAGELLAN achieved a median enrichment factor (EF) of 14.38, significantly higher than that using individual ligand profiles. Next, MAGELLAN was examined on 5 and 20 GPCRs from two public VS databases (DUD-E and GPCR-Bench) and resulted in an average EF of 9.75 and 13.70, respectively, which compare favorably with other state-of-the-art docking- and ligand-based methods, including AutoDock Vina (with EF=1.48/3.16 in DUD-E and GPCR-Bench), DOCK 6 (2.12/3.47 in DUD-E and GPCR-Bench), PoLi (2.2 in DUD-E), and FINDSITECcomb2.0 (2.90 in DUD-E). Detailed data analyses show that the major advantage of MAGELLAN is attributed to the power of ligand-profiling, which integrates complementary methods for ligand-GPCR interaction recognition and thus significantly improves the coverage and sensitivity of VS models. Finally, cases studies on opioid and motilin receptors show that new connections between functionally related GPCRs can be visualized in the minimum spanning tree built on the similarities of predicted ligand-binding ensembles, suggesting a novel use of MAGELLAN for GPCR deorphanization.
Introduction
G protein-coupled receptors (GPCR) are integral transmembrane proteins responsible for detecting, activating, and conducting cellular signal transductions. The malfunction of the receptors is the cause of a wide array of diseases, such as cancer and diabetes [1, 2]. Consequently, GPCRs are among the most clinically studied targets in drug discovery. A detailed analysis of the DrugBank shows that 23% of the 2,276 FDA-approved drugs on the market, including both small molecules and biologics, target GPCRs, while out of other 2,641 drugs that are under some form of clinical trial, 8% target GPCRs. Overall, GPCRs represent targets of 15% of all drugs that are either approved or investigational (Figure 1).
Figure 1.
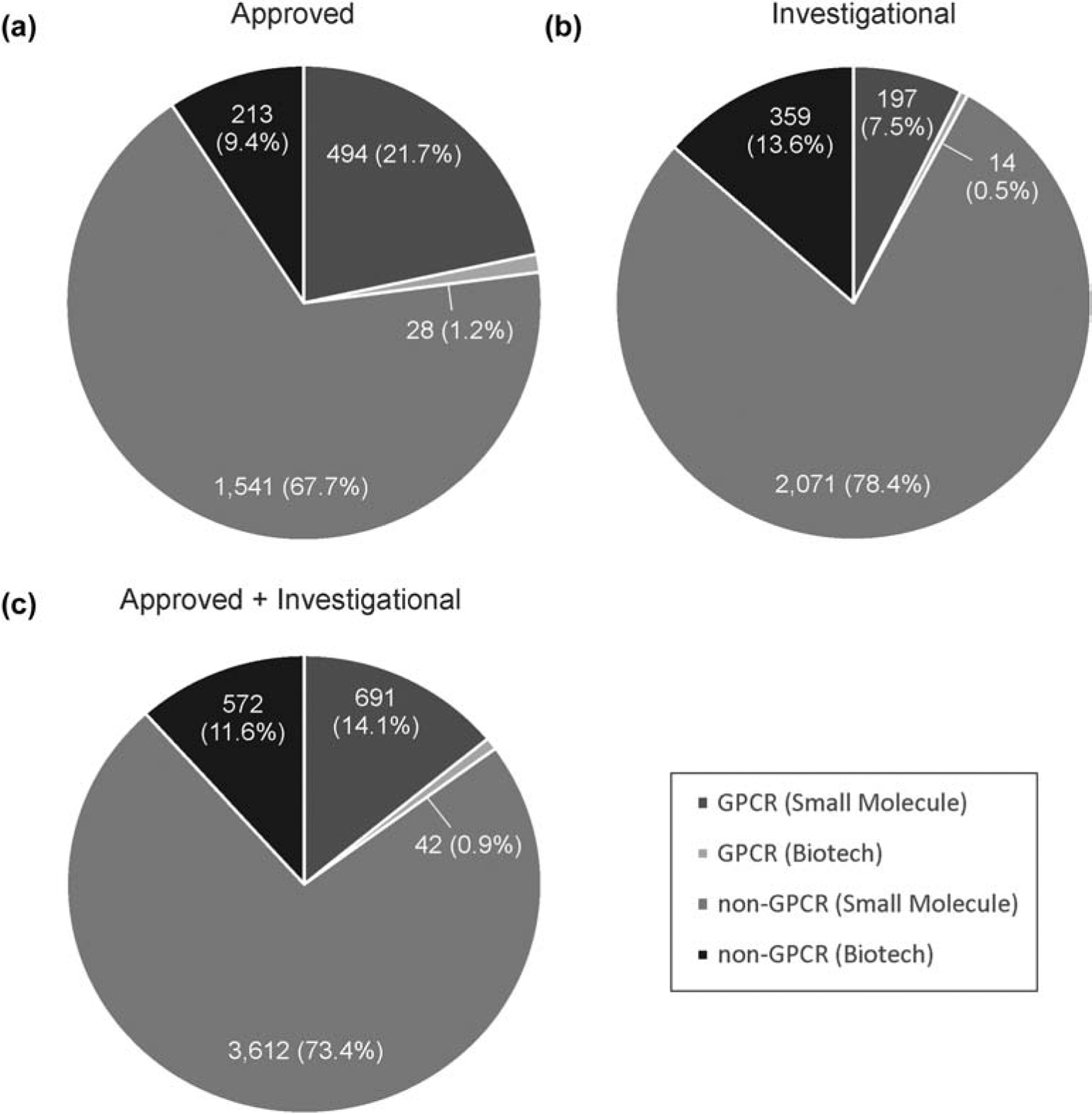
DrugBank statistics for GPCRs. Percentage of small molecule and biotech drugs shown for GPCR and non-GPCR targets under the groups of (A) approved, (B) investigational, and (C) total drugs.
The mainstay in drug development is high-throughput screening (HTS), a technique for biochemically assaying pharmacological targets against the candidate compounds on a large scale. However, HTS is usually costly and laborious, whereas various in silico approaches, which are much faster and less expensive, have been found useful in assisting and complementing HTS [3]. There are two general approaches that are commonly employed in the computer-aided drug screening: ligand-based and receptor-based virtual screening (VS). In the former, the knowledge of what ligands the receptor targets tend to bind with is used to develop a model for drug screening [4, 5], while the latter utilizes structural information of the receptors to predict ligand-binding affinity, normally through docking [6, 7]. Although the structure-based docking approaches are typically computationally expensive and take a long time to run through large compound libraries, they can be useful in providing physical configuration of ligand-receptor interactions and for producing results that are biochemically relevant; on the other hand, ligand-based approaches are usually very fast, but they tend to be biased towards ligands that are currently known [8].
Both receptor and ligand-based approaches require some sort of information, either known active ligands or a structure, which may not be available for a drug target of interest. The orphan GPCRs are one such example, many of which lack known endogenous ligands [9]. In this regard, chemical genomics approaches are often applied to infer ligand binding information, based on the assumption that similar receptors bind similar ligands [10]. One of the earliest applications of the idea was with the algorithm, FINDSITE, which uses ligand information from structurally-homologous receptors found through fold-recognition in a ligand-based VS [11, 12]. Another algorithm is PoLi, which looks for similar protein receptors by performing binding pocket structure comparison between the target and templates, followed by a ligand-based screening search [5]. More recently, the same group extended FINDSITE to FINDSITEcomb2.0, which utilizes threading and structure-based comparisons for template ligand selections [13].
Although structure is generally considered to be more conserved than sequence in evolution, relying solely on structural similarity can result in achieving a high rate of false positives in the selection of functionally relevant ligands, as receptors of similar structures often bind with different ligands. In particular, experimental structures are not always available for many medically relevant target proteins, where low-resolution models would have to be generated for the target receptors; this would further impact the accuracy and specificity of the structure-based ligand inferences. This is true especially for the case of GPCR families, which all have roughly similar global fold (7-TM helix bundle) but distinct helix packing and local structure at the binding sites [14]. Moreover, the majority of the structure-based approaches rely on selecting homologous proteins and their respective ligand sets from the Protein Data Bank (PDB) [15]; however, pharmacological data are often found in low quantities within the PDB. Currently, there are many more proteins with known pharmacological data, which are collected in various manually curated chemical databases, such as ChEMBL [16], BindingDB [17], and GLASS (for GPCRs) [18]. Using the wealth of information from such resources should help enhance the accuracy of the ligand-based chemical genomics approaches.
In this study, we present a novel ligand-profile based VS approach, MAGELLAN (standing for Michigan G protein-coupled receptor ligand-based virtual screen), specifically designed for G protein-coupled receptors. To enhance the reliability and robustness of a ligand-based screening approach, multiple modules are employed for the detection of a variety of receptor homologies from both structure- and sequence-based alignments, from which consensus ligand profiles, as represented by a 2D ligand fingerprint matrix, are created for the next step of VS. To carefully examine the strength and weakness of the pipeline, large-scale tests were performed on 224 representative Class A GPCRs, which were carefully controlled with various component. Additionally, stringent benchmarks were performed to test MAGELLAN with other state-of-the-art ligand and receptor based methods (including PoLi, FINDSITEcomb2.0 [13], AutoDock Vina [6], and DOCK 6 [7]). Here, Class A GPCRs were selected as the focus mainly because of the high popularity and diversities in structure and function and the clinical importance in drug discovery. Moreover, the conserved transmembrane domains of these receptors make it an ideal case for examining the sequence- and structure-based alignment modules, which helps increase the accuracy and specificity of the hybrid pipeline. The MAGELLAN webserver, together with the VS results for various human GPCRs and the filtered ligand sets, are available and downloadable at https://zhanglab.ccmb.med.umich.edu/MAGELLAN.
Methods
The process of MAGELLAN consists of three consecutive stages: 1) homologous GPCR detection, 2) ligand profile construction, and 3) profile-based virtual screening. The flowchart of the MAGELLAN pipeline is depicted in Figure 2, which starts with a single primary sequence of the target GPCR in FASTA format, where the output consists of a list of predicted ligands bound with the target.
Figure 2.
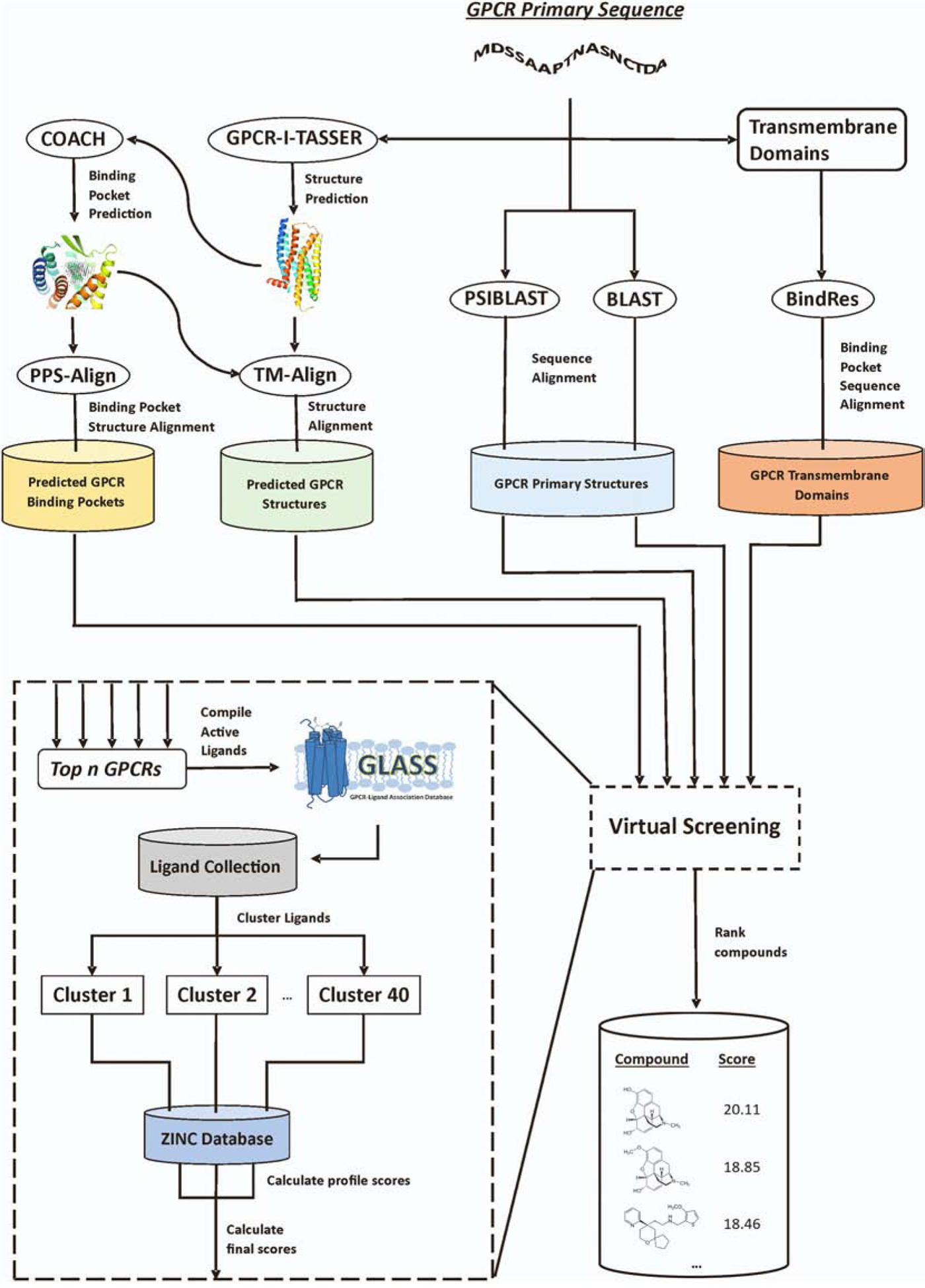
The MAGELLAN pipeline for GPCR virtual screening. It consists of three consecutive stages of homologous GPCR detection, ligand profile construction, and profile-based virtual screening.
Library Construction of Ligand-GPCR Association
As the ligand profile is a core concept in MAGELLAN, which is derived from known GPCR complexes, a comprehensive library of GPCR-ligand associations is first constructed from GLASS database [18]. Here, GLASS (GPCR-Ligand Association) database is a manually curated repository for experimentally validated GPCR-ligand interactions, with the data mined from literature and multiple primary chemical and biological databases, including ChEMBL [16], BindingDB [17], PDSP Ki [19], IUPHAR [20], and DrugBank [21]. In GLASS, only the entries with known experimental values of Ki, Kd, IC50, and EC50 are collected. In the cases that multiple experimental values exist for the same GPCR-ligand pair from different studies, the median was taken as the representative value to avoid outliers produced from incorrect or extreme environment setting. To filter out inactive ligands, a common threshold of 10 μM is used for both Ki and Kd values, while a threshold of 20 μM was set for IC50 and EC50 as previous studies found that a Ki-IC50 conversion factor of 2 is suitable. This relatively loose criterion could accommodate the variability in various conditions of assay experiments. After the filtration, the library contains 238,108 GPCR-ligand associations attached with 644 GPCRs.
Five Modules for Homologous GPCR Detection
Five complementary modules, built on TM-align [22], PPS-Align [23], BLAST [24], PSI-BLAST [25], and BindRes [26], are developed for detecting homologous and analogous GPCRs, where the first two are structure-based and other three are on sequence and sequence-profile comparisons (Figure 2). The idea of composite model approach by combining complementary pipelines is not new and has been previously used to improve modeling accuracy of fold recognition [27, 28] and ligand binding site predictions [29]. Here, we extend the idea to virtual screening and examine whether a combination of multiple sources of information could compensate for one another’s shortcomings and improve the overall quality of the final models. We have selected the five modules from TM-align, PPS-align, PSI/BLAST and BindRes, as these represent a collection of widely used algorithms that help extract quickly and robustly the structure, sequence and ligand-binding information associated with the targets GPCRs.
In the first GPCR detection module, TM-align [22] is used to structurally match (or align) the target to template GPCRs. To obtain a 3D structure model of the GPCR, the target sequence is submitted to GPCR-I-TASSER [30], which was designed to create full-length receptor structures by reassembling the structure fragments through replica-exchange Monte Carlo simulations [31], where the structure fragments are excited from the PDB template structures with the target-template alignments created by LOMETS [28, 32], a multi-threading approach for protein fold-recognitions. The resulting structure models are then compared by TM-align [22], a global structural alignment method, against the GPCRs in the pre-compiled GPCR-ligand library, in which the structures of all GPCRs are pre-generated with GPCR-I-TASSER. The TM-align based GPCR templates are scored by
| (1) |
Here, is the TM-score that measures the global structure similarity of the target and template models [33], where L is the length of the target sequence, Lali is the number of aligned residues by TM-align, di is the distance of ith aligned residue pair between target and template GPCRs, and is the scale factor. In Eq (1), f = m/n is the fraction of the aligned residues in the binding pocket (m) normalized by the total number of binding residues (n) on the GPCR template; accounts for the local structural similarity of the binding pockets between target and template; measures the evolutionary relation between the aligned binding residues, where is the BLOSUM62 mutation score; is the average Jensen-Shannon divergence over the binding pocket, where is the frequency of amino acid a at ith column of multiple sequence alignment (MSA) identified by PSI-Blast for the target GPCR and qa is the background frequency; and R is the residue chemical similarity of the binding site residues, where Figure S1 in Supporting Information (SI) provides an illustrative example for how the residue chemical similarity was calculated. The weight parameters for S, E, and J were taken from the qstr scoring function from TM-SITE [29], while the weight for T was reduced from 1 to 0.2 compared to that used in TM-SITE to de-emphasize the impact of the global structural similarity.
Second, PPS-Align is an algorithm recently designed for sequence-order independent structure alignments of binding pockets [23]. In this module, the GPCR-I-TASSER models of target and template GPCRs are first submitted to COACH [29] for ligand-binding site prediction through sequence and structure profile comparisons. Following COACH, the ligand binding pockets are determined by selecting the binding-site predictions with the highest confidence score by COACH for both target and template GPCRs, where the structures of binding pockets are finally aligned by PPS-Align for pocket comparisons. The GPCR templates by PPS-align from the library are scored by
| (2) |
where PPS in [0,1] is the pocket similarity score returned by PPS-Align, S and J are the same as defined in Eq. (1) with the weight parameters taken from the PPS-align program, and Ibs is the sequence identity of the binding-site residues in the PPS-align aligned regions between target and template GPCRs. In addition to normal compound molecules, COACH can also generate prediction on ion-binding sites, which typically consist of far fewer residues than that of a conventional binding pocket [29]. For example, the delta opioid receptor has a sodium ion bound that promotes negative allosteric effects (PDB: 4N6H) [34]. However, these sites typically have very low confidence scores and thus have never been selected by the clustering in our benchmark. Therefore, the small ion binding from COACH predictions does not have a detectable impact on the MAGELLAN procedure.
In the third BindRes module, we first parse the transmembrane (TM) domains of the target GPCR according to the UniProtKB/SwissProt annotation, which are then aligned with the TM domains of all template GPCRs in the library using Clustal Omega [35]. The template GPCRs are ranked by
| (3) |
where Ibs, R and J are defined similarly as in Eqs. (1–2). The calculations focus solely on the 44 orthosteric binding site residues on the TM-domains, as specified by Gloriam et al [26]. Since these orthosteric residues have been labelled in Ballesteros-Weinstein numbering system [36], the identities can be conveniently referred through the most conserved residue of each TM domain according to the Clustal Omega alignments. The weight for J was again taken from the qstr scoring function from TM-SITE [29]. Here, the binding was purely based on pharmacological data as extracted from the databases we used in the study. Although most of the databases do not differentiate between orthosteric ligands and allosteric modulators, GPCR allosteric modulators are still exceedingly uncommon and will most likely not get crowded out over the orthosteric ligands during the ligand clustering procedures in MAGELLAN.
Finally, the BLAST and PSI-BLAST modules use the programs from the NCBI BLAST+ software suite (version 2.2.29). For BLAST, the target sequence is matched against the GPCR templates, which are sorted by descending sequence identity to the target. The same is done for PSI-BLAST but with sequence-profile alignment, where the profiles were collected with 4 iterations from the non-redundant (NR) sequence database from NCBI under an E-value cutoff of 0.001; the parameters we used for number of iterations and E-value cutoff are standard values used in most sequence alignment and threading approaches with BLAST and PSI-BLAST [37]. The results are also ranked by descending sequence identity. Here, it is well-known that PSI-BLAST is more efficient than BLAST to detect distant-homology sequences due to the adopt of sequence-profile alignments. However, we found that BLAST can be useful to detect complementary templates due to the sensitivity in recognizing the similarity of local sequence motifs, which are important to homologous ligand detections since only the binding pocket residues are sensitive to relevant ligand binding in many GPCRs. Although MAGELLAN eventually ranks the templates based on their sequence identity to the target, the use of BLAST and PSI-BLAST in parallel can help increase the coverage and variation of the ligand profile as described below, due to the complementarity of the alignment algorithms.
It is worth noting that although MAGELLAN contains the BindRes module that focuses on the transmembrane orthosteric binding recognitions, the use of the MAGELLAN is not limited to screening compounds on the transmembrane orthosteric binding sites, because other four modules do not have such limit and a hybrid profile for all the modules could help detect ligands bound with other regions of GPCRs. Accordingly, the benchmark datasets used in this study were collected from multiple databases, which contain ligands from different binding locations, including those from both transmembrane and loop regions. In this regard, the benchmark results presented should reflect the overall performance of the pipelines on the ligands across all GPCR regions.
Ligand Profile Construction and Profile-Based VS
Associated active ligands from the ten top-ranked GPCRs are compiled for each of the five GPCR alignment modules. The resulting ligand collections are then clustered with the Taylor-Butina algorithm [38, 39] using the Chemfp Python library [40], where a Tanimoto coefficient (TC) cutoff of 0.8 was used. Here, all ligands are originally represented as InChI identifiers and keys, and converted into 1,024-bit Morgan fingerprints with a radius of 2 using RDKit [41]. The 40 largest GPCR clusters are selected for use in the next step of VS. If there are fewer than 40 clusters, all of them are used.
For a given cluster (k), a ligand profile is constructed for the target GPCR, which is represented by a n × Nk matrix, in which Nk is the number of non-redundant ligands in the cluster and each ligand has n-bit fingerprints taken from the ZINC12 database where n = 1,024 is the dimension of the Morgan fingerprints (Figure 3). The score for matching the profile with a compound in the library is defined by
| (4) |
where is the Tanimoto coefficient between the ith ligand and the zth compound in the database. Here, and are the bits in the ith and the zth compounds, respectively. is the weighting factor for the ith ligand, where Sm(i) is the scoring function of mth alignment module as defined in Eqs. (1–3) and Mi is the total number of modules that identified the ith ligand (Fig. 3).
Figure 3.
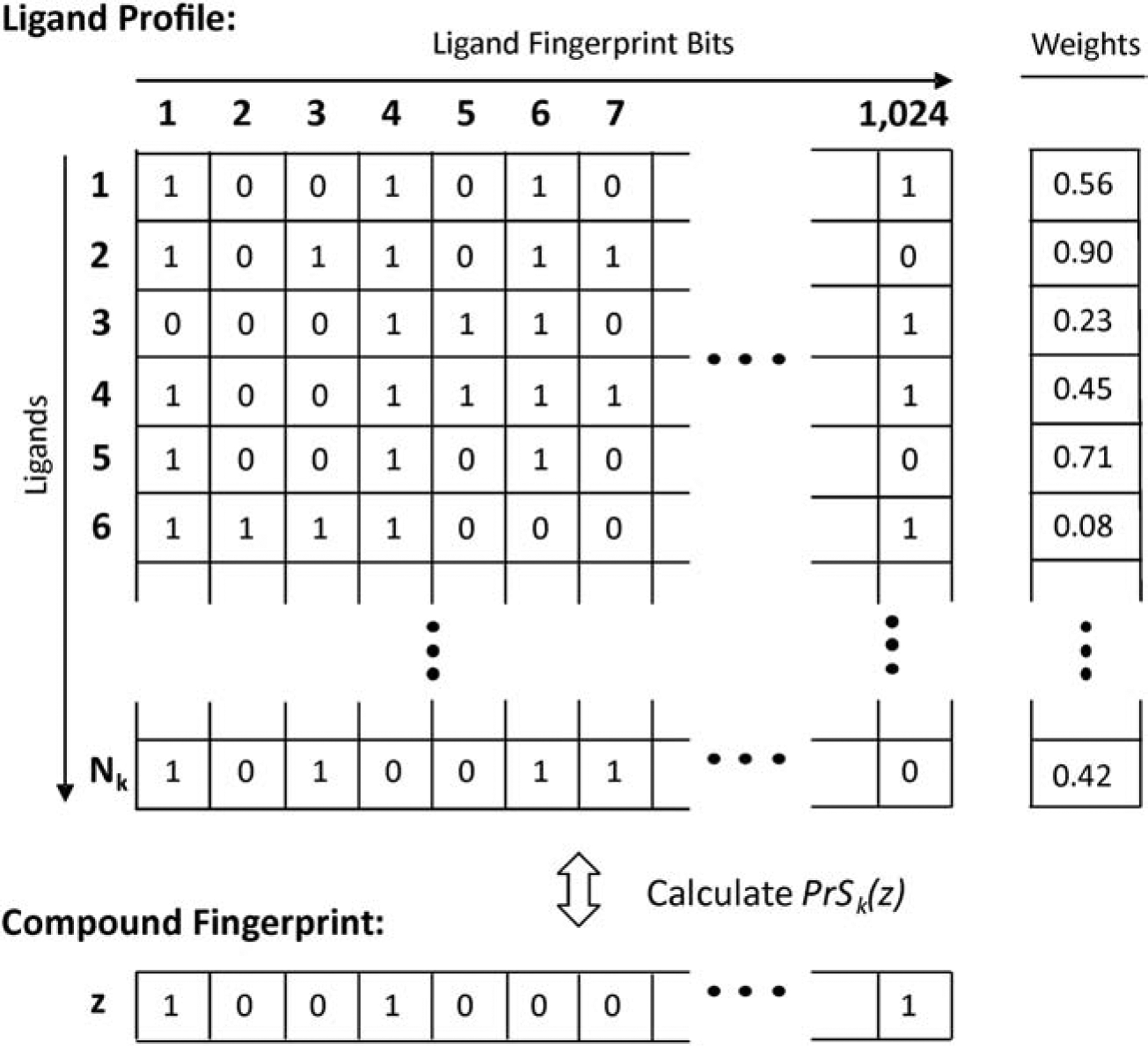
Illustration of ligand profile that summarizes feature information of all ligands from the kth cluster. In the profile, the horizontal index represents the ligand fingerprint bits, while the vertical index runs through all ligands. Each ligand is assigned a weight (wi) that equals to the average alignment score of different modules that have been used to identify the GPCR template associated with the ligand. The bottom panel indicates a compound fingerprint from the ZINC database, with which the profile is aligned.
The PrSk (z) can be converted into a renormalized Z-score by Zk(z) = (PrSk(z) – μk)/σk, where μk and σk are, respectively, the mean and standard deviation of PrSk(z) overall all compounds in ZINC for the kth cluster. This renormalization makes the matching score between clusters comparable despite of the different ligand populations in different clusters. A final score (Sz) for zth ZINC compound is calculated by taking the maximum Z-score among all clusters, i.e.,
| (5) |
Here, we note that there are overall 3 major free parameters involved in the binding-site clustering and ligand selection procedures, i.e., the number of GPCRs selected from each alignment module (=10), TC cutoff for clustering (=0.8), and the number of clusters used for VS (=40). These free parameters are open-ended variables in our algorithm, and we optimized their values using an independent dataset of 56 GPCRs that are non-redundant from the test proteins reported in this study. During the training process, the parameters were determined by maximizing the average enrichment factor of VS as defined in Eq. (7) below. There are also parameters involved in the individual modules where their values are mainly taken from the original sequence and structural alignment algorithms.
Construction of Minimum Spanning Tree by Similarity Ensemble Approach
To evaluate the similarity of GPCR proteins based on the similarity of their bound ligands, we construct a minimum spanning tree for the targets using the similarity ensemble approach (SEA) [42, 43]. To quantitatively assess the similarity between two ligand sets in a statistically stringent base, we collect multiple random ligand sets with sizes between 10 and 1,000 ligands from the GLASS database and calculate the TC-score between ligands of different sets. It was found that the mean (μ) and standard deviation (σ) of the TC-scores between two ligand sets is correlated with the product of the sizes of two ligand sets compared (s), which follows well with
| (6) |
where the parameters (k, m and r) can be obtained by linear regression fit with the data in Figure S2 in SI. Using Eq. (6), a raw TC-score of two ligand sets can be quantitatively converted into a size-independent Z-score by Z = (TC – μ)/σ.
Here, only the ligand pairs with a significant TC-score above a threshold are used in the statistical calculation, where a TC threshold of 0.84 is found to be optimal, which has the Z-score distribution follow the Gumbel extreme value distribution as shown in Figure S3. Using the Gumbel distribution data, a BLAST-like E-value can be calculated for each GPCR pairs. Finally, a minimum spanning tree (MST) based on the significance of E-value can be calculated using Kruskal’s algorithm [44], with the image produced with Cytoscape [45].
On-line Webserver Construction and Usage
Built on MAGELLAN, an online web server was constructed at https://zhanglab.ccmb.med.umich.edu/MAGELLAN, using Python CGI scripting, complemented with MySQL, Javascript and PHP. The only required input is the primary sequence for the GPCR of interest in FASTA format. An example is shown as follows:
>sp|P35372|OPRM_HUMAN Mu-type opioid receptor OS=Homo sapiens GN=OPRM1 PE=1 SV=2 MDSSAAPTNASNCTDALAYSSCSPAPSPGSWVNLSHLDGNLSDPCGPNRTDLGGRDSLCPPTGSPSMITAITIMALYSIVCVVGLFGNFLVMYVIVRYTKMKTATNIYIFNLALADALATSTLPFQSVNYLMGTWPFGTILCKIVISIDYYNMFTSIFTLCTMSVDRYIAVCHPVKALDFRTPRNAKIINVCNWILSSAIGLPVMFMATTKYRQGSIDCTLTFSHPTWYWENLLKICVFIFAFIMPVLIITVCYGLMILRLKSVRMLSGSKEKDRNLRRITRMVLVVVAVFIVCWTPIHIYVIIKALVTIPETTFQTVSWHFCIALGYTNSCLNPVLYAFLDENFKRCFREFCIPTSSNIEQQNSTRIRQNTRDHPSTANTVDRTNHQLENLEAETAPLP
The first line is a header and always starts with a greater-than sign (>) followed by descriptive text, while the other lines are comprised of the primary amino acid sequence (please refer to https://zhanglab.ccmb.med.umich.edu/FASTA/ for additional information). Users have also the option to supply other input information under the ‘Optional Input’ button, which MAGELLAN would have determined automatically if not provided. The first such parameter is the annotated transmembrane domains. An example is shown as follows:
| 1 | 64 | 98 | SPSMITAITIMALYSIVCVVGLFGNFLVMYVIVRY |
| 2 | 102 | 136 | KTATNIYIFNLALADALATSTLPFQSVNYLMGTWP |
| 3 | 138 | 170 | GTILCKIVISIDYYNMFTSIFTLCTMSVDRYIA |
| 4 | 181 | 212 | RTPRNAKIINVCNWILSSAIGLPVMFMATTKY |
| 5 | 226 | 260 | PTWYWENLLKICVFIFAFIMPVLIITVCYGLMILR |
| 6 | 275 | 311 | RNLRRITRMVL VVVA VFIVCWTPIHIYVIIKAL VTIP |
| 7 | 310 | 343 | IPETTFQTVSWHFCIALGYTNSCLNPVLYAFLDE |
Here, the columns are represented by (1) the domain number, (2) the starting residue number, (3) the ending residue number, and (4) the primary structure for each transmembrane domain separated by a tab character; each transmembrane domain goes on a separate line. The second parameter allows users to upload a query GPCR structure instead of running GPCR-I-TASSER for generating a structure model. The structure should be in PDB file format (http://www.wwpdb.org/documentation/file-format). Finally, the user can provide a list of residues corresponding to the orthosteric site of the GPCR in order to bypass running COACH. An example is shown as follows:
145,149,150,153,154,235,238,242,243,295,298,299,302,324,328
Thcby commas with no space in between. If a PDB structure was provided, the residues should match with the uploaded structure; otherwise, it should match with the input primary sequence. Using these optional parameters will help reduce the simulation time and improve the modeling accuracy if appropriately provided.
To facilitate relative GPCR studies, the datasets for all ligand and test sets filtered from the GLASS library are provided for download at the MAGELLAN homepage. Additionally, the top 1% of results from screening the full ZINC database for all human Class A GPCRs are pre-generated and made available publicly.
Algorithmic Distinctions MAGELLAN from Other Chemogenomic approaches
It is worth noting that the general principle of MAGELLAN, i.e., to perform ligand recognition and virtual screening through template-based ligand-protein association comparisons, is not new and similar to that used in several former studies [5, 12, 46, 47]. However, there are several critical differences between the approaches in library construction, template identifications, and ligand scoring. For example, instead of using the separate and raw PDB, DrugBank and ChEMBL libraries in these approaches, MAGELLAN exploits a uniformly-curated library GLASS [18], which constitutes the largest ligand-GPCR association library and has the binding affinity of all entries (including Ki, Kd, IC50, and EC50 values) carefully validated and filtered; these stringently validated ligand-receptor binding interactions should help improve the reliability of template-based binding interaction transferal. Second, MAGELLAN is designed specifically for GPCR screening. While it might be considered as a drawback compared to the pipelines performing general protein screening, it does allow for introduction of GPCR-specific interactions, including the transmembrane orthosteric binding-residue based recognition (Eq. 3), which has not been previously considered but proves helpful for increasing the specificity of GPCR screening. Finally, and probably most importantly, most of the existing approaches select ligands based on individual ligand-by-ligand Tanimoto coefficient comparison, which we found not always reliable for prioritizing ligands from a high number of compound decoys because an individual ligand from specific templates cannot appropriately represent the overall binding feature of the target protein and screening based on individual ligand comparison could often result in false positive results. To address this issue, MAGELLAN constructs a ligand profile from multiple ligand-receptor template clusters, and then uses the profile, which is represented by a 2D ligand fingerprint matrix (Fig 3), as a probe to detect from the library the ligands that match best with the ligand profile. As the ligand profiles are derived from multiple ligand-receptor templates that are appropriately weighted with confidence of different pipelines (Eq. 4), they should reflect better the overall binding feature of the target protein, which therefore help increase the sensitivity of virtual screening compared to the individual ligand-by-ligand comparisons. Conceptually, this is largely corresponding to the extension from sequence-sequence to profile-sequence alignments [25], which had achieved a fundamental advantage to the problems of protein homologous recognition and template-based protein structure prediction [48]; but here MAGELLAN employs the idea for ligand comparison and virtual screening.
Results and Discussion
Comparison of MAGELLAN with Component Modules
Five different alignment modules have been used in MAGELLAN for homologous GPCR collection. To justify the hybrid profile approach, we first examine the performance of MAGELLAN in comparison with that of the pipelines on individual alignment modules, in which the same procedure is performed as shown in Figure 2, but only with the homologous GPCRs detected by an individual module.
Dataset Construction and VS Experiment Design.
The test datasets are constructed from a comprehensive list of all 224 Class A GPCRs. For each GPCR, the active ligands are collected from the GLASS database [18], which are filtered with a stringent activity threshold of 1 μM for Ki, Kd, IC50, and EC50 values. In order to increase chemical diversity and to have a balanced sample size for each receptor, ligands are clustered for each GPCR by their Bemis-Murcko frameworks [49]. If there are >600 frameworks, the activity threshold will be decreased by a factor of 2 until there were fewer than 600 frameworks, where the highest activity ligands were selected from each framework. If the number of frameworks is between 100 and 600, the highest activity ligand is selected directly from each cluster. In case that fewer than 100 frameworks are formed, the highest activity ligands were chosen regardless of their framework until a total of 100 ligands was achieved. As a result, there are in total 54,438 active ligands collected, corresponding to 258 per GPCRs on average.
To test the VS methods, the active ligands from each GPCR are mixed with a set of 500,000 randomly selected compounds as decoys from the “Clean Drug-Like” subset of the ZINC database. The compounds downloaded are in SMILES string format, which are subsequently converted into Morgan fingerprints with RDKit [41], consisting of 1,024-bit fingerprints with a radius of 2. A retrospective virtual screen (RVS) experiment is implemented by different methods, where the goal of RVS is to prioritize the true active ligands using the proposed scoring functions. The performance of RVS can be qualitatively assessed by the enrichment factor (EF):
| (7) |
where Nact and Ntot are the total numbers of the active and all compounds in the ligand pool, respectively. and are, respectively, the numbers of true positive ligands and the number of all candidates in the top x% of the compounds selected by the RVS methods. A higher EFx indicates a better RVS performance, where EFx = 1 means a random selection without enrichment. While x% can be taken as different cutoff (1%, 2%, 5% etc), we focus mainly on 1% for concise data presentation.
Here we note that the randomly selected decoys from ZINC were not property matched or had a similarity threshold to known actives. In fact, we have considered using inactive compounds as the decoys. However, the number of active compounds greatly exceeds that of inactive compounds, as we have observed from our GPCR datasets. Thus, to have a balanced dataset to challenge our pipeline, we proceeded with the current experiment design. Given the sample size of our retrospective virtual screen (n=224) and the chemically-diverse compounds to which each independent GPCR binds, however, it should be highly unlikely that the randomly-chosen 500,000 compounds from ZINC (out of ~13M) would adversely bias our results.
To rule out the effect from using close homologous targets, a constraint was applied to the selection of GPCRs, where any homologous GPCR templates with >30% sequence identity to the target, based on the BLAST alignment, were excluded when inferring the ligand profiles. In case this constraint is turned off, only the target GPCR is excluded. Meanwhile, to challenge the pipelines, a homologous cutoff has been applied in the GPCR-I-TASSER structure modeling and COACH binding prediction, i.e., all structures with a sequence identity >30% to the target GPCR sequence are excluded from the threading template library, no matter if the constraint of binding GPCR is applied.
MAGELLAN Significantly Outperforms Component Modules in RVS Experiment.
In Figure 4A, we present a scatter plot of the enrichment factors (EF1%) acquired from MAGELLAN at the constraint of homologous GPCR filter, in comparison with that from the pipelines built on the five individual modules. It was shown that MAGELLAN achieves a higher enrichment factor than the individual modules for most of the GPCRs. For example, MAGELLAN outperforms the BindRes module in 130 cases, while BindRes does so in 72 cases. These numbers are 146/52, 115/77, 121/67, and 129/70 for BLAST, PSI-BLAST, PPS-align, and TM-align modules respectively.
Figure 4.
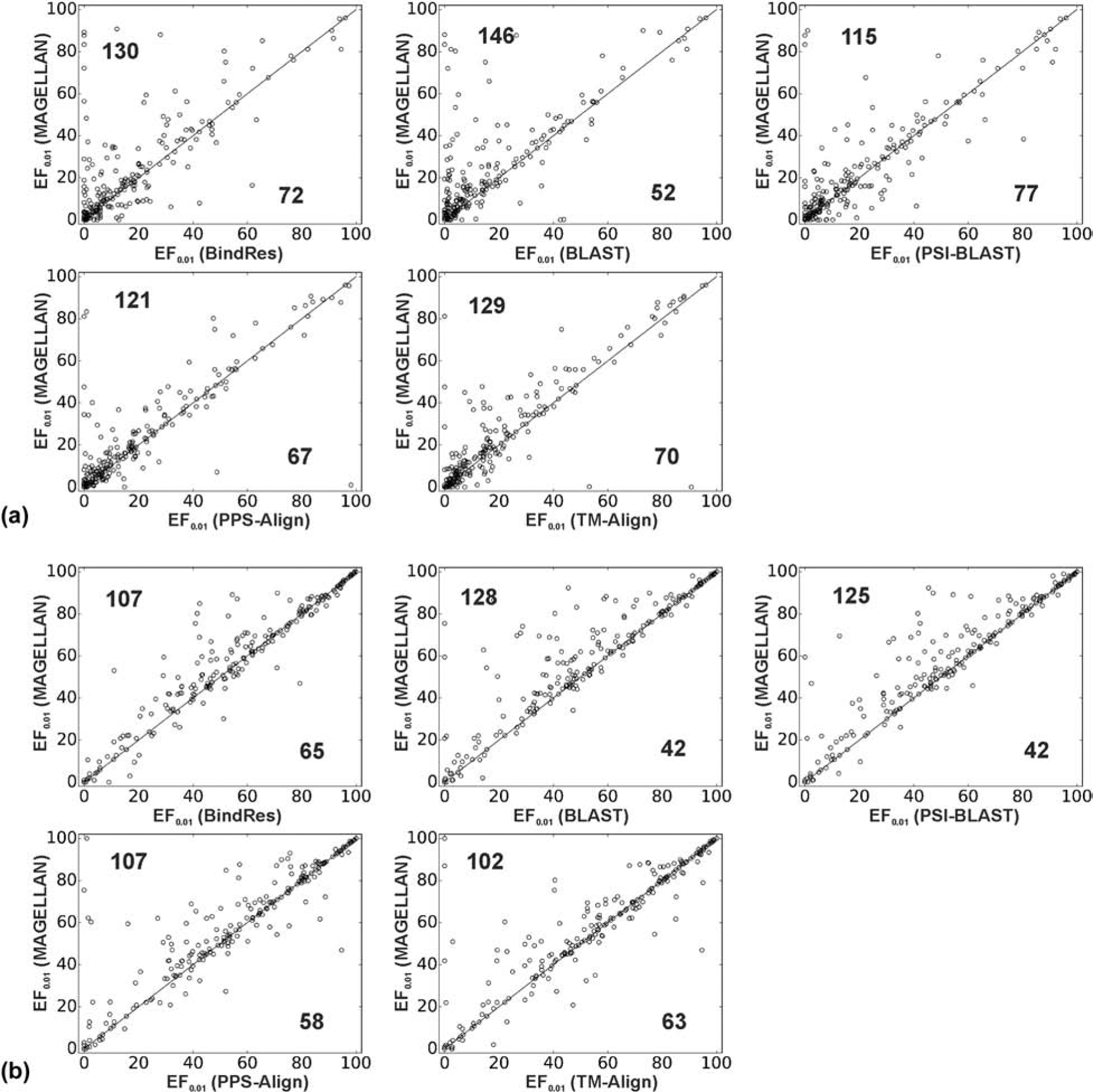
Comparison of MAGELLAN and the five component modules in the RVS experiment on 224 Class A GPCRs. Results are shown as EF1% either with (A) 30% sequence identity cutoff (constrained) or (B) no cutoff (not constrained but with target GPCR excluded). Each point represents one GPCR, where the numbers in each triangle represent the number of GPCRs for which the RVS pipeline outperforms the comparison ones.
In Table 1 (Columns 2), we also list the average and median EF1% values for each method, which shows again that MAGELLAN achieved a higher EF than all the individual modules. To examine the significance of the difference, a Wilcoxon signed-rank test is calculated for each pair of the comparison, where the two-tailed p-value is ≤0.005 in all the comparisons, indicating that the differences are statistically significant.
Table 1.
Summary of RVS results by MAGELLAN and that on the component modules. Data shown are median and average (in parentheses) EF1% values on 224 test Class A GPCRs. P-value is calculated in the Wilcoxon signed-rank test between MAGELLAN and the control modules. Alternative homology constraint cutoff was applied to remove all homologous templates with sequence identity >30%.
| Methods & Modules | With homology cutoff | Without homology cutoff | ||
|---|---|---|---|---|
| EF1% | p-value | EF1% | p-value | |
| MAGELLAN | 14.38 (23.03) | -- | 62.03 (59.13) | -- |
| BindRes | 10.19 (17.90) | 5 × 10−7 | 56.39 (56.35) | 4 × 10−6 |
| BLAST | 7.04 (16.49) | 5 × 10−14 | 53.02 (53.26) | 1 × 10−14 |
| PSI-BLAST | 11.83 (20.79) | 5 × 10−3 | 54.31 (54.18) | 2 × 10−14 |
| TM-align | 13.76 (20.15) | 8 × 10−9 | 56.92 (56.39) | 6 × 10−4 |
| PPS-Align | 10.99 (20.25) | 2 × 10−6 | 56.90 (55.55) | 1 × 10−6 |
Among the individual modules, the pipeline built on TM-align performed slightly better than other component modules, as evidenced by its higher median EF1% of 13.76. This may be understandable because structure is generally more conserved than sequence and thus comparisons on structure similarity detected more relevant GPCRs for ligand profiles.
In Figure 4B, we also present the results without using the constraint of a 30% sequence identity cutoff but with the target GPCRs excluded from the ligand profile detection process. As expected, the RVS performance becomes much better when homologous GPCRs are included in the ligand profile construction, where many of the points in the figure have been shifted to the right-upper quadrant in the plots, as compared to Figure 4A. Nevertheless, the full-version MAGELLAN still has a higher EF1% than that on the individual modules for the majority of the cases. As shown in Table 1 (Column 4), the average and median EF1% values of MAGELLAN are again significantly higher than that of the individual modules, with a Wilcoxon p-value ≤ 0.0006 for all the modules.
Here, with the homologous GPCRs included, the gap between the structure- and sequence-based methods disappears, which have similar median EF1%. This is not surprising because most of the homologous GPCRs with a strong structure similarity have also a high sequence identity, which could be detected by sequence-based modules as well.
Overall, the significant outperformance of MAGELLAN over the component modules demonstrated the necessity and advantage of the hybrid profile approach by collecting complementary homologous GPCR associations.
Both Sequence and Structural Alignments Are Essential to MAGELLAN Performance.
To further examine the impact of the individual GPCR alignment modules on the performance of MAGELLAN, we counted the highest-performing alignment method type for each GPCR under the sequence cutoff constraint, where the type was denoted as either sequence (BLAST, PSI-BLAST, BindRes) or structure based (TM-align, PPS-Align). Among the 224 Class A GPCRs, 89 have a structure-based module as their top-scoring module with the highest enrichment factors, while the sequence-based module does so in 135 cases. The overall number of GPCRs was relatively evenly distributed for each module, and no module stood out and outperform others.
We also examined the effect of running MAGELLAN without the homology cutoff constraint. 101 out of 224 targets resulted in a sequence-based module as their top-scoring module, while 123 were from structure-based modules. Among the 123 cases, the TM-align module produced 94 of the best cases, showing that the global structure comparison is more appropriate than local pocket comparison at the high homology level. Generally, in both scenarios with and without homology cutoff constraint, each module played a role in lending their predictive power to MAGELLAN. This is consistent with the observation that MAGELLAN outperforms all the component modules with a significant p-value, which signifies the synergistic effect of data fusion.
Taken together, these results appear to suggest that sequence-based methods helped compensate for where the structure-based methods failed and vice versa. Apparently, leaving any method types will result in reduction on overall performance, which highlights the advantages of MAGELLAN using a composite approach in the selection of template GPCRs for the construction of the ligand profile. It would be interesting and important to examine the effects of incorporating additional methods in a future study.
Benchmark of MAGELLAN with Other VS Approaches
To examine MAGELLAN with other state of the art approaches, we tested the performance in control with four widely-used VS programs, including AutoDock Vina [6], DOCK 6 [7], PoLi [5], and FINDSITEcomb2.0 [13]. The former two are receptor-docking based approaches, where the crystal structures of the target GPCRs were used as the input for molecular docking. Please refer to the Supplementary Text for details on how AutoDock Vina and DOCK 6 were set up and runs performed. PoLi is a ligand-based VS tool with the probe ligands detected by the binding-pocket structural comparisons between target and templates, while FINDSITEcomb2.0 is an extended approach from the same lab which utilizes threading and structure-based comparisons for template ligand selection. The benchmark tests of the methods were performed on two separate datasets from DUD-E [50] and GPCR-Bench [51], respectively.
As the software is not available for installation for PoLi and FINDSITEcomb2.0, the data for DUD-E was taken from the authors’ publication [5] and webserver (http://cssb2.biology.gatech.edu/FINDSITE-COMB-II/data/findsitecomb2data.zip) for these two methods, respectively. In both benchmarks against DUD-E, the authors applied a 30% sequence identity cutoff for template selection. Moreover, the method they employed for the calculation of EF1%was mathematically identical to that used in this study, making the data comparable with that of MAGELLAN.
Tests on DUD-E Dataset.
DUD-E [50] is a widely-used dataset specially designed for VS benchmarks. Benchmarks were run on GPCRs in DUD-E; this set contains five Class A GPCR proteins, where each protein has on average 224 active ligands from ChEMBL. Each active ligand is paired with 50 molecular decoys (with similar chemistry but of different topology) drawn from ZINC. While the turkey beta-1 adrenergic receptor (P07700) was included in DUD-E, there is no pharmacological data in any of the ligand databases. Thus, the ligand clusters from the human orthologue (P08588) were used in its place. To examine the performance, we run MAGELLAN and the control programs in an automated mode against the ligand dataset for each GPCR target, with the goal to pick up the true active ligands using their scoring functions.
In Table 2, we list the EF1% value of VS for the five GPCRs, calculated by MAGELLAN, AutoDock Vina, DOCK 6, FINDSITEcomb2.0, and PoLi, respectively. The data show a significantly better performance of MAGELLAN than the four control algorithms for three out of the five tested GPCRs, under the sequence identity cutoff of 30%. In particular, the human dopamine receptor DRD3 performed exceptionally well with MAGELLAN, with an EF1% 28.65, which is more than 5 times higher than that of the best control methods (5.2 by Poli). By manually checking the subtype name of the templates, a number of related GPCRs have been selected for the DRD3, including rat dopamine receptor 1B (P25115) and human dopamine receptor 1A (P21728), where the contribution of their ligands in the clusters was well established, accounting for 9 chemotypes (Figure 5A).
Table 2.
RVS results of EF1% on five Class A GPCRs in DUD-E Dataset. Values outside (and inside) parentheses are the results for MAGELLAN with (and without) homology constraint cutoffs. Data for PoLi was taken from Roy et al. [5] and data for FINDSITECcomb2.0 (FSc2.0) was downloaded from the authors’ webserver.
| Gene | UniProt ID | MAGELLAN | PoLi | FSc2.0 | AutoDock | AutoDock Refined | Dock6 |
|---|---|---|---|---|---|---|---|
| AA2AR | P29274 | 0.95 (39.03) | 1.2 | 0 | 1.42 | 4.03 | 2.86 |
| ADRB1 | P07700 | 5.47 (36.11) | 2.0 | 3.24 | 0.66 | 0.88 | 2.63 |
| ADRB2 | P07550 | 13.68 (34.75) | 2.6 | 5.20 | 2.69 | 0.90 | 1.35 |
| CXCR4 | P61073 | 0 (23.97) | 0 | 2.53 | 0 | 0 | 2.49 |
| DRD3 | P35462 | 28.65 (39.27) | 5.2 | 3.54 | 2.64 | 1.37 | 1.26 |
| Average | 9.75 (34.63) | 2.2 | 2.90 | 1.48 | 1.44 | 2.12 |
Figure 5.
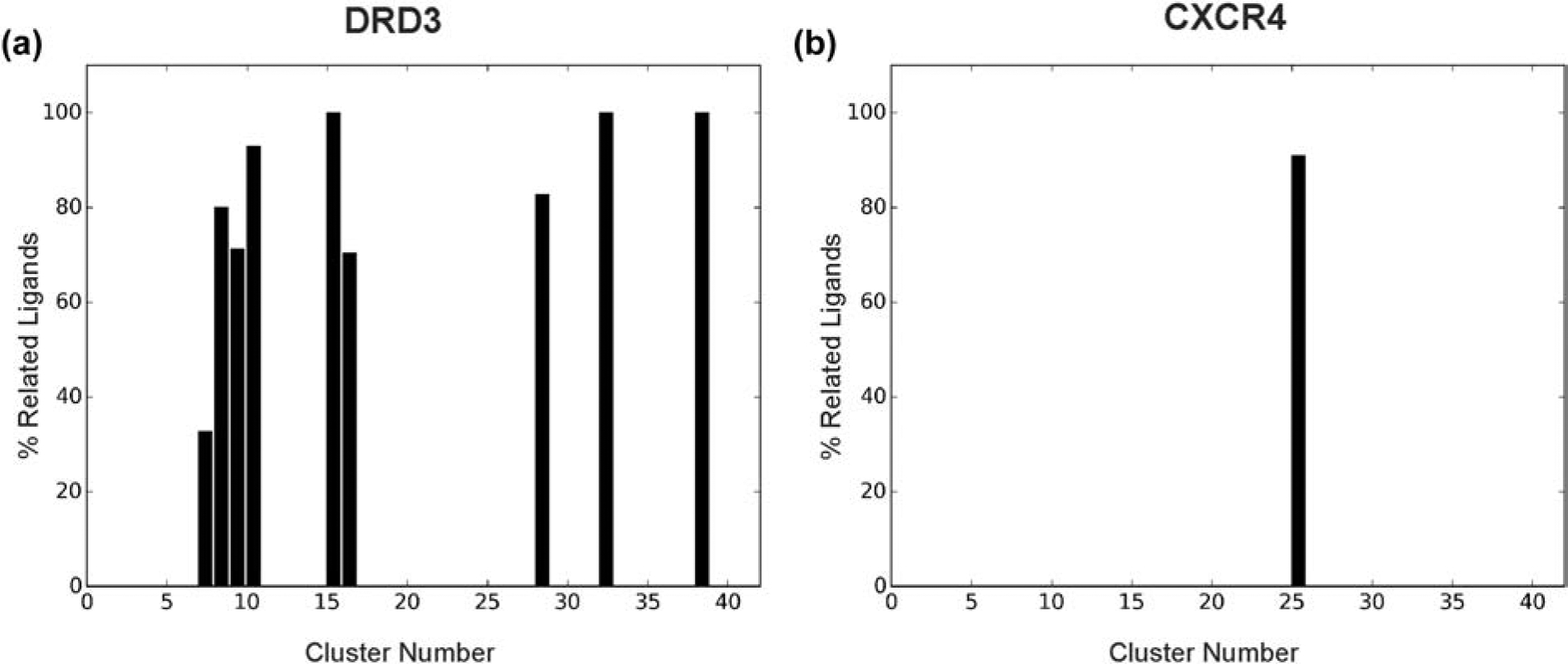
Proportion of ligands from related GPCRs in clusters from MAGELLAN with homology constraint cutoff. (A) Dopamine Receptor D3 (DRD3), (B) C-X-C chemokine receptor type 4 (CXCR4). The presence of chemotypes containing ligands from respective related GPCRs was examined, where one cluster represents one chemotype. The influence of related GPCRs resulted in 9 chemotypes for DRD3, while CXCR4 only had one.
Similarly, MAGELLAN outperforms the control methods on the beta-1 adrenergic receptor and beta-2 adrenergic receptor as well, with the EF1% being 1.7- and 2.6-folds higher than the best of the control methods, respectively. The main reason for the enhanced performance is due to complementary GPCR modules exploited in MAGELLAN which detected several closely-related GPCRs that bound with similar ligands, despite the low sequence identity; these include P25100, P18130 and O02824 for ADRB1, and Q01338 and P23944 for ADRB2. The ligand profiles constructed from the analogous ligands helped to prioritize the active compound hits. However, MAGELLAN yielded a low EF1% of 0.95 and 0 for the adenosine A2A receptor (AA2AR) and the C-X-C chemokine receptor type 4 (CXCR4). For AA2AR, this is mainly due to the lack of appropriate GPCR detection because no closely-related GPCRs exist in the GPCR-ligand library used by MAGELLAN after the 30% sequence identity cutoff is applied. For CXCR4, MAGELLAN detected a few relatives (including P51682, P51684 and O54814), but their associated ligands were only present in one out of the top 40 clusters used in MAGELLAN (Figure 5B), suggesting the need for chemotype diversity of the clusters. Moreover, the ligand set sizes of the clusters for the related GPCRs were very small (56, 65 and 34, respectively), lessening their influence overall. After turning off the homology cutoff constraint, however, MAGELLAN correctly detected all these homologous GPCRs, which resulted in a significantly higher EF1% value (39.03 and 23.97) in both cases.
The molecular docking algorithms in the current study utilized ligand flexibility with a rigid protein receptor. However, proteins are inherently flexible macromolecules, and the side chains of the orthosteric site could potentially adopt alternative rotameric states to accommodate an assortment of different chemical compounds. Therefore, using a rigid receptor in our docking simulations could be a drawback, potentially leading to erroneously decreased performance and/or unfair comparison results. To partially mitigate the effects of neglecting sidechain relaxation limitedly to the selected poses obtained with a rigid protein docking approach, we rescored the top-docked poses generated from AutoDock Vina using an induced fit protocol from Molecular Operating Environment (MOE), which allows for side chain flexibility on the receptor. As it was shown in Table 2, there was no clear improvement on average enrichment factor over ranking based solely on AutoDock Vina’s scoring function, although it did help in some cases such as AA2AR and ADRB1 (-but worsening two other cases of ADRB2 and DRD3).
There is, however, still a possibility that larger movement on the transmembrane domains or extracellular loops is required for the complete adoption of binding pockets amenable to chemotypes of the active compounds, though this would require expert curation per target on a case by case basis. In this regard, molecular dynamics simulations have been used in various studies to generate distinct conformers of the receptor for use in ensemble docking, and demonstrated some level of improvements on docking performance [52, 53]. Since the attention of the current study is to compare a newly-developed, automated VS pipeline (MAGELLAN) to easily automatable docking pipelines (AutoDock Vina, DOCK6), we have chosen to implement the docking programs with rigid-body receptor setting and limited the flexibility only on ligand conformations in our benchmark tests. Nevertheless, the involvements of more extensive flexible docking simulations, together with expert curation in the selection of key residues in the orthosteric site for flexibility and utilization of different receptor conformations, would likely considerably improve the docking performance results.
Altogether, these results highlight the importance of the inclusion of homologous templates for ligand profile constructions. This phenomenon was observed for many other receptors, in which EF1% was significantly increased by the inclusion of close homologous GPCRs in ligand profile construction. In particular, the number of chemotypes and size of ligand sets from related GPCRs appeared to play a role in performance, indicating that the success of RVS not only requires detection of the receptors but also with sufficient multiplicity of clustering ligands; this suggests again the importance of combination of multiple GPCR detection modules, as more of the consensus ligand-GPCR associations could be collected with an increasing number of complementary modules.
Overall, the average EF1% is 9.75 by MAGELLAN, which is 4.4 and 3.4 times higher than that by PoLi and FINDSITEcomb2.0, and 6.6 and 4.6 times higher than that by AutoDock and Dock6 respectively. The EF1% value will increase by 3.6 times if homologous GPCRs are allowed to be included in the profile construction process. Nevertheless, the two-tailed p-values of MAGELLAN compared to the control methods are not significant (i.e., 0.12, 0.15, 0.09, and 0.11 to PoLi, FINDSITEcomb2.0, AutoDock, and Dock6, respectively, with a Student’s t-test), probably due to the low number of proteins tested in this dataset.
Tests on GPCR-Bench Dataset.
The second benchmark dataset on which we conducted experiments is GPCR-Bench [51]; it contains 20 Class A GPCRs, each having between 100 to 600 active ligands accompanied by 50 decoys per active ligand. The RVS results on this benchmark are summarized in Table 3.
Table 3.
Summary of EF1% results on 20 Class A GPCRs in GPCR-Bench. Values outside (and inside) parentheses are the results for MAGELLAN with (and without) 30% sequence identity cutoff for template exclusion.
| Gene | UniProt ID | MAGELLAN | AutoDock | Dock6 |
|---|---|---|---|---|
| GPR40 | O14842 | 22.04 (48.92) | 24.28 | 21.84 |
| OX2R | O43614 | 7.92 (34.65) | 1.82 | 0 |
| ADRB2 | P07550 | 24.64 (60.87) | 0.20 | 9.40 |
| ADRB1 | P07700 | 1.03 (54.36) | 0 | 2.73 |
| ACM2 | P08172 | 23.00 (36.00) | 7.76 | 7.12 |
| ACM3 | P08483 | 24.88 (48.26) | 2.31 | 8.97 |
| S1PR1 | P21453 | 0.50 (51.24) | 0.47 | 0.16 |
| PAR1 | P25116 | 0.00 (0.00) | 13.39 | 2.00 |
| 5HT1B | P28222 | 16.83 (60.89) | 1.54 | 2.79 |
| AA2AR | P29274 | 0.48 (34.13) | 0 | 0.64 |
| OPRD | P32300 | 27.93 (65.77) | 3.77 | 0.75 |
| HRH1 | P35367 | 21.39 (50.75) | 3.28 | 0.22 |
| DRD3 | P35462 | 46.27 (60.70) | 1.24 | 1.03 |
| OPRK | P41145 | 12.94 (47.76) | 0.65 | 0.22 |
| OPRX | P41146 | 2.99 (24.38) | 0.25 | 2.85 |
| 5HT2B | P41595 | 18.41 (20.40) | 0.98 | 0.98 |
| OPRM | P42866 | 2.44 (58.54) | 0 | 1.08 |
| CCR5 | P51681 | 4.06 (23.86) | 0.53 | 4.36 |
| CXCR4 | P61073 | 4.26 (70.21) | 0 | 0.26 |
| P2Y12 | Q9H244 | 11.94 (13.43) | 0.76 | 2.03 |
| Average | 13.70 (43.26) | 3.16 | 3.47 | |
In total, MAGELLAN performed favorably (average EF1% = 13.70) in this benchmark, as compared with AutoDock Vina (3.16) and DOCK 6 (3.47). AutoDock Vina and DOCK 6 achieved the best enrichment for the free fatty acid receptor 1 (GPR40) with an EF1%=24.28 and 21.84 respectively. Since all of its active ligands belong to the same chemotype [51], the binding pocket of this target does not have as much variation compared with other targets. While active compounds from the other receptors may be chemically diverse, the snapshot of the binding pocket of GPR40 from the particular structure used (PDB: 4PHU) likely accommodates this single chemotype well (Figure S4). Also, since much less conformational variation is involved, the docking performance is less impacted by the rigid-body docking protocol employed in this experiment for such cases. MAGELLAN attained a comparable enrichment on this target with EF1%=22.04; if the homologous templates are included, however, the performance is significantly improved to EF1%=48.92. Additionally, AutoDock Vina achieved decent enrichment with the protease-activated receptor 1 (PAR1) at EF1% =13.39, while MAGELLAN has a zero EF1% with a homology constraint cutoff. In fact, MAGELLAN detected several protease-activated receptor subtypes (Q63645, P55085, Q96RI0), but their respective ligand sets were of a very small size (2, 59, 10, respectively). As a result, these relative ligands were not present in the top 40 clusters because of the minority of binding ligands, which resulted in the reduced performance. While most of the successful examples of GPCRs are found to have at least one related subtype that had a sizeable number of ligands, the data suggests that the number of ligands in the ligand sets of closely-related members is essential to the success of MAGELLAN, in addition to its ability to detect homologous GPCRs.
We also investigated whether there was any association between chemical diversity of the active compound sets for each GPCR and the performance of MAGELLAN. The results in Figure 5 did not show a visible overall correlation between these two variables. Nevertheless, MAGELLAN seems tending to perform better on the GPCRs for which there was a greater chemical diversity, especially in the cases where the ratio of the number of Bemis Murcko scaffolds to total active compounds was greater than 0.5. For some of the benchmark sets with lower ratios, such as with GPR40 (0.280), MAGELLAN performed well with and without the constraint, while for PAR1 which has a ratio=0.317 MAGELLAN generated a EF1%=0. Given that the benchmark sets for both of these receptors have a limited assortment of chemotypes, it is likely that MAGELLAN was not able to capture the corresponding chemotypes within the top 40 clusters. With respect to PAR1, numerous other active and chemically dissimilar compounds were overrepresented in its ligand profile, given the small amount of ligand sets of its homologues. On the other hand, the ratio for the benchmark set for AA2AR was 0.822, indicating a high chemical diversity. With the sequence identity cutoff constraint, MAGELLAN had a EF1%=0, while it was rescued with the removal of cutoff constraints and had a EF1% >30. Though this represents a special case in which having closely related homologues helps with the performance of MAGELLAN, it is interesting to observe the importance of homologue selection. Overall, our data seems to suggest a slight-to-no dependence of RVS performance on chemical diversity, while a better normalization of benchmark sets for virtual screening would be aided by more equal ratios of Bemis Murcko scaffolds to active compounds.
In Figure S6, we also present the log receiver operating characteristic curves (ROC) for the RVS results by MAGELLAN and the two control methods for all targets, where the corresponding Boltzmann-enhanced receiver operating characteristic (BEDROC, α=20) values are listed in Table S1. Here, as opposed to the conventional receiver operating characteristic (ROC), BEDROC has the advantage to better assess early enrichment [54], which is important as compounds selected for experimental validation are always chosen from top-ranked candidates. Overall, MAGELLAN exhibited a higher average early enrichment both with (BEDROC=0.32) and without (BEDROC = 0.68) the homology constraint cutoffs, as compared to AutoDock Vina (BEDROC=0.16) and DOCK 6 (BEDROC=0.14). This suggests again that MAGELLAN has a higher propensity to correctly select compounds that would potentially bind the GPCR of interest.
Case Studies on MAGELLAN-Based Deorphanization
Orphan GPCRs usually refer to those without identified endogenous ligands or known physiological functions. Recent survey has shown that 87 Class A, 8 Class C, and 26 adhesion GPCRs are orphan receptors in the human genome [20]. Despite the orphan status, these receptors likely have physiological roles intertwined with diseases and remain potential therapeutic targets in drug discovery. Here, we present case studies on two important GPCRs to illustrate the VS process of MAGELLAN and potential applications for GPCR deorphanization.
Mu Opioid Receptor.
Mu opioid receptor (UniProt ID: P35372) is a medically important target, which is closely involved in the reduction of pain. Common drugs in pain reduction include morphine and heroin, both of which are strong opioid agonists; but a major drawback is that their consumption often results in unwanted side effects, such as nausea, constipation, respiration depression, and addiction. Many current research efforts have been devoted to the search of new analgesic drugs with reduced or eliminated maladies [55].
The application of MAGELLAN on the human mu opioid receptor achieved an exceptionally high EF1% of 87.81. Figure 6 shows part of the minimum spanning tree of human GPCRs, involved with the mu opioid receptor. Here, the MST was constructed using SEA, described in Section 2.4 in Methods, where the GPCR similarity was assessed based on the ensemble of compounds recognized by the MAGELLAN pipeline for different receptors. It is shown that three other related GPCRs, including the kappa opioid, delta opioid and nociceptin receptors, are in close neighbor of the mu opioid receptor in the MST (blue nodes in Fig 6). This data first shows that MAGELLAN has ability to recognize compounds with a high accuracy for the important drug target receptor. Meanwhile, the SEA based analysis on the MAGELLAN products can detect untapped relations between GPCRs; which should help for GPCR deorphanization and function annotations.
Figure 6.
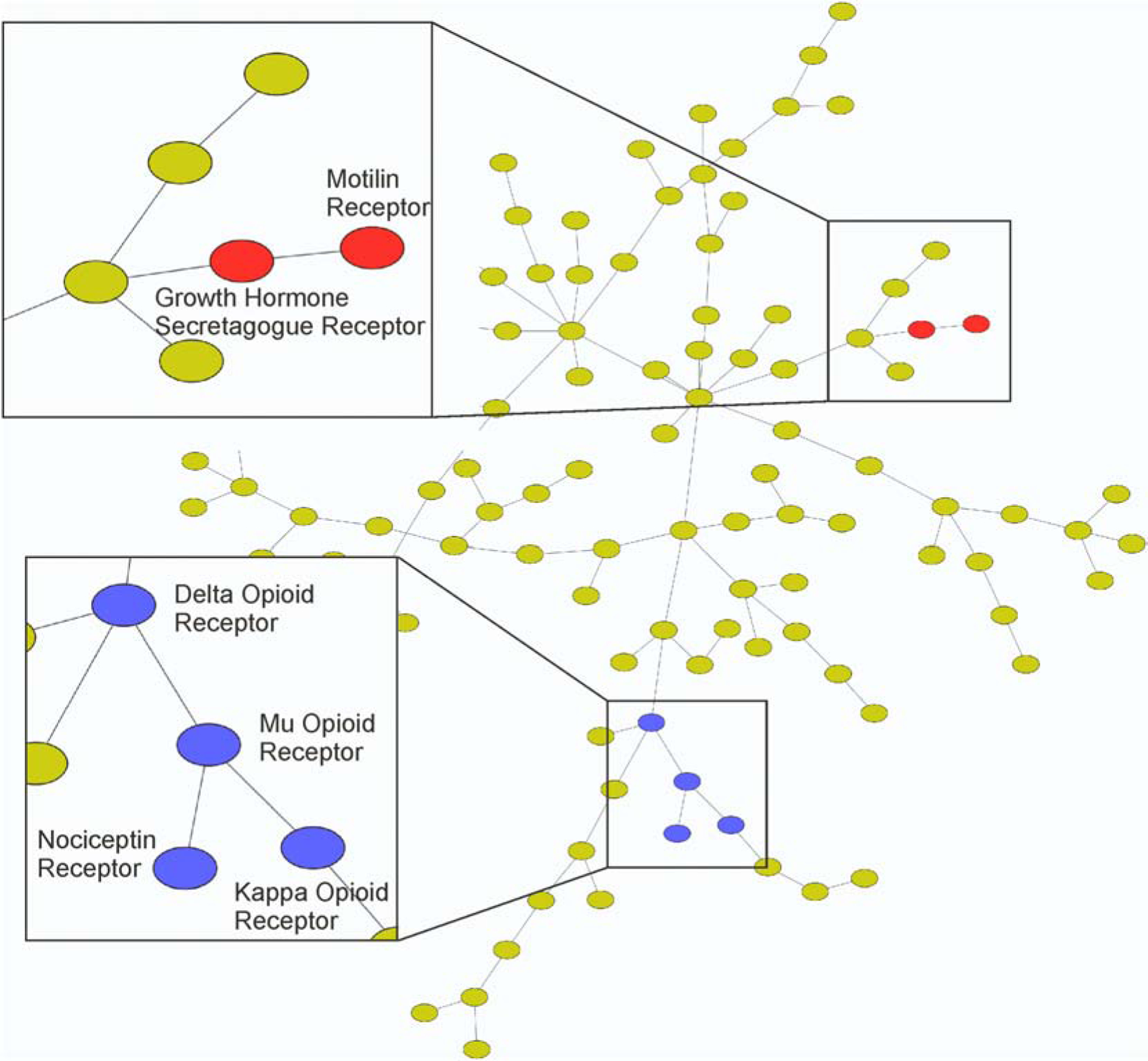
Minimum spanning tree of GPCRs constructed from MAGELLAN virtual screening results. The relation of GPCRs is assessed with the similarity ensemble approach (SEA), with the MST created using Kruskal’s algorithm on E-values of the ligand assembles associated with different GPCRs.
Motilin Receptor.
Human motilin receptor (UniProt ID: O43193) is a former orphan GPCR, whose closest homologue is the growth hormone secretagogue receptor type 1 (also known as ghrelin receptor). The MAGELLAN pipeline achieves a high EF1% of 33.33 on the human motilin receptor, where several alignment modules in MAGELLAN were able to select multiple orthologues of the growth hormone secretagogue receptor type 1. One example of the alignment detected by the BindRes module between the human motilin receptor and the pig growth hormone secretagogue receptor type 1 is shown in Figure S7, where a highly conserved local binding region was recognized, although the global sequence identity between the receptors is low.
Once again using the SEA and MST analyses, the motilin receptor and the growth hormone secretagogue receptor are shown to be pharmacologically similar in Figure 6 (red nodes). It should be noted that there are no ligand sets corresponding to any orthologues of the motilin receptor, except for the ligands of the human variant. Taking together, these results demonstrate again the feasibility of MAGELLAN for the application to deorphanize GPCRs. It should be noted that the BindRes score as defined in Eq. (3) relies on Ballesteros-Weinstein nomenclature, which is specific to Class A GPCRs. Thus, the current MAGELLAN pipeline and the case studies are focused on Class A GPCRs, to which most orphan GPCRs belong. Nevertheless, the extension of MAGELLAN to other GPCR classes should be feasible after necessary parameter reoptimizations. The work on this line is currently under progress.
Conclusions
Built on the assumption that similar receptors bind with similar ligands, we have developed a new hierarchical, ligand-profile based approach, MAGELLAN, for the virtual screening of human Class A GPCRs. Starting from amino acid sequence of the target proteins, MAGELLAN first utilizes GPCR-I-TASSER [30] to generate tertiary structure prediction for the target protein. Next, five modules of structure, sequence and orthosteric binding-site based alignment methods are extended to the detection of homologous and analogous proteins, where all known ligands bound with the proteins are clustered for the construction of chemical profiles; these profiles are finally used to thread through the compound libraries for screening putative ligands and drugs for the target receptor.
The pipeline was first tested on a comprehensive set of 224 Class A GPCRs and achieved a median enrichment factor EF1% of 15.31 after excluding all homologous templates in both structure prediction and GPCR template detection processes, which is significantly higher than the methods built on individual GPCR alignment modules. In addition, MAGELLAN was examined on two independent benchmark sets from DUD-E [50] and GPCR-Bench [51], consisting of 5 and 20 Class A GPCRs, with ligand selection results compared favorably with that of other state-of-the-art docking and ligand-based VS approaches, including AutoDock Vina [6], DOCK 6 [7], PoLi [5] and FINDSITEcomb2.0 [13]. Detailed data analysis shows that the major advantage of MAGELLAN lies in the utilization of both structure (including global and local) and orthosteric binding-site based comparisons for GPCR template detections, whereas the ligand profiles constructed from multiple resources of the data fusion help enhance the sensitivity and specificity of the VS through the compound databases. Here, we selected to focus MAGELLAN on Class A GPCRs, mainly due to the fact that the Class A GPCRs account for >85% of the GPCR superfamily and the overwhelming majority of GPCR drug targets are under Class A. This selection also provides a technical specificity on the approach, as one of the five modules by MAGELLEN (BindRes) considers conserved orthosteric binding profiles that are only available to the Class A GPCRs. Although the BindRes feature alone does not create the highest enrichment performance among all the modules (Table 1), our results showed that turning off the BindRes module reduced the MAGELLAN performance (with a P-value <0.05 in the Wilcoxon signed-rank test between results with and without BindRes), which partly highlights the specialization contribution of the selection.
Apart from the favorable benchmark performance, several advances may help future MAGELLAN developments. First, MAGELLAN is a ligand-profile based approach utilizing only ligand-GPCR associations. This is different from other ligand-oriented approaches, such as PoLi which relies on known ligand-protein complex structures from the BioLip [56]. Currently, CLASS contains 533,470 non-redundant ligand-GPCR associations, which is over 7,500 times higher than the number of known ligand-GPCR structural complexes in BioLiP; this is probably part of the reason for the significant improvement of MAGELLAN over PoLi. The gap between the ligand-receptor association and the protein binding structure databases are rapidly increasing [16–18, 57], which should give additional advantage and potential to the future development of the ligand-based methods such as MAGELLAN.
Compared to the docking-based approaches, MAGELLAN has the advantage in utilizing low-resolution predicted models, since high-resolution experimental structures are often unavailable to many important drug targets. Technically, it is also a benefit to exploit the global fold comparison for GPCR template detection because many experimentally solved structures are in unbound apo form, which can significantly impact the accuracy of the docking-based approaches that often have difficulty in modeling the ligand-induced conformational changes. In addition, docking target protein through a large library of compounds is very computationally expensive and time consuming. As experienced in this study, it typically took days to weeks for AutoDock Vina or DOCK 6 to complete a docking screen for a single GPCR, depending on the target; on the other hand, ligand-based screening using MAGELLAN only takes about an hour, after GPCR-I-TASSER that can take 10–20 hours for structural folding simulations. Nevertheless, docking based programs have the advantage to generate 3D model of binding structures that is often useful for function and drug-based analyses. Meanwhile, we also found that there are cases (such as PAR1) for which the docking-based approach achieve a much higher enrichment. Thus, structure-based docking methods have still their own value, where a combination of MAGELLAN with these approaches should further improve the functionality and accuracy of VS experiment; such development is currently under progress. Nevertheless, MAGELLAN has its own limit as its success relies on the detection of homologous GPCR templates. In principle, the approach could not work for the cases if no known GPCR-ligand associations exist or are detected. However, utilization of concept of ligand profile does allow the generation of reliable models from multiple weak template hits, which is the main reason that MAGELLAN outperformed the individual GPCR-detection modules and other chemogenomic- and docking-based approaches, even with the stringent homology-filtering conditions as shown in Table 1 and Figure 4A. In this regard, the development of new sensitive non-homologous ligand-GPCR associations should help to further improve the MAGELLAN modeling quality.
Finally, one advantage of MAGELLAN is on the ability to recognize ligands for orphan GPCRs, partly due to the fact that the pipeline does not rely on the availability of structure and function of known GPCRs. As illustrated by the case studies shown in Figure 6, reliable ligand compounds can be recognized for the orphan GPCRs, where new connections can be established between the orphan GPCRs and other receptors through the SEA analyses of ligand sets identified by MAGELLAN. While these and other examples can only be validated with pharmacological experiments, the data demonstrated a potential aspect of applications of MAGELLAN to the deorphanization of GPCRs.
Supplementary Material
Highlights.
Developed a new pipeline for Class-A GPCR virtual screening
Proposed a novel concept to improve virtual screening based on ligand profiling
A novel approach to GPCR deorphanization through ligand-binding ensembles
Integrating composite pipelines improves ligand-GPCR interaction recognition
Stringent tests of pipelines on 3 large-scale virtual screening experiments
Acknowledgements
This research was supported in part by National Institute of General Medical Sciences [GM136422], National Institute of Allergy and Infectious Diseases [AI134678], National Science Foundation [IIS1901191, DBI2030790], and Proteome Informatics of Cancer Training Program (T32 Training Grant). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].O’Hayre M, Vazquez-Prado J, Kufareva I, Stawiski EW, Handel TM, Seshagiri S, et al. The emerging mutational landscape of G proteins and G-protein-coupled receptors in cancer. Nat Rev Cancer. 2013;13:412–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Rompler H, Staubert C, Thor D, Schulz A, Hofreiter M, Schoneberg T. G protein-coupled time travel: evolutionary aspects of GPCR research. Molecular interventions. 2007;7:17–25. [DOI] [PubMed] [Google Scholar]
- [3].Tanrikulu Y, Kruger B, Proschak E. The holistic integration of virtual screening in drug discovery. Drug Discov Today. 2013;18:358–64. [DOI] [PubMed] [Google Scholar]
- [4].Geppert H, Vogt M, Bajorath J. Current trends in ligand-based virtual screening: molecular representations, data mining methods, new application areas, and performance evaluation. J Chem Inf Model. 2010;50:205–16. [DOI] [PubMed] [Google Scholar]
- [5].Roy A, Srinivasan B, Skolnick J. PoLi: A Virtual Screening Pipeline Based on Template Pocket and Ligand Similarity. J Chem Inf Model. 2015;55:1757–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Allen WJ, Balius TE, Mukherjee S, Brozell SR, Moustakas DT, Lang PT, et al. DOCK 6: Impact of new features and current docking performance. J Comput Chem. 2015;36:1132–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Drwal MN, Griffith R. Combination of ligand- and structure-based methods in virtual screening. Drug Discov Today Technol. 2013;10:e395–401. [DOI] [PubMed] [Google Scholar]
- [9].Civelli O, Saito Y, Wang ZW, Nothacker HP, Reinscheid RK. Orphan GPCRs and their ligands. Pharmacology & Therapeutics. 2006;110:525–32. [DOI] [PubMed] [Google Scholar]
- [10].Klabunde T Chemogenomic approaches to drug discovery: similar receptors bind similar ligands. Br J Pharmacol. 2007;152:5–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Brylinski M, Skolnick J. A threading-based method (FINDSITE) for ligand-binding site prediction and functional annotation. Proc Natl Acad Sci U S A. 2008;105:129–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zhou H, Skolnick J. FINDSITE(X): a structure-based, small molecule virtual screening approach with application to all identified human gpcrs. Mol Pharm. 2012;9:1775–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Zhou H, Cao H, Skolnick J. FINDSITE(comb2.0): A New Approach for Virtual Ligand Screening of Proteins and Virtual Target Screening of Biomolecules. Journal of chemical information and modeling. 2018;58:2343–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Shoichet BK, Kobilka BK. Structure-based drug screening for G-protein-coupled receptors. Trends Pharmacol Sci. 2012;33:268–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Rose PW, Prlic A, Altunkaya A, Bi C, Bradley AR, Christie CH, et al. The RCSB protein data bank: integrative view of protein, gene and 3D structural information. Nucleic acids research. 2017;45:D271–D81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Gaulton A, Bellis LJ, Bento AP, Chambers J, Davies M, Hersey A, et al. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic acids research. 2012;40:D1100–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Liu T, Lin Y, Wen X, Jorissen RN, Gilson MK. BindingDB: a web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic acids research. 2007;35:D198–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Chan WK, Zhang H, Yang J, Brender JR, Hur J, Ozgur A, et al. GLASS: a comprehensive database for experimentally validated GPCR-ligand associations. Bioinformatics. 2015;31:3035–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Roth BL, Lopez E, Patel S, Kroeze WK. The multiplicity of serotonin receptors: Uselessly diverse molecules or an embarrassment of riches? Neuroscientist. 2000;6:252–62. [Google Scholar]
- [20].Alexander SP, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE, et al. The Concise Guide to PHARMACOLOGY 2015/16: G protein-coupled receptors. Br J Pharmacol. 2015;172:5744–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Law V, Knox C, Djoumbou Y, Jewison T, Guo AC, Liu Y, et al. DrugBank 4.0: shedding new light on drug metabolism. Nucleic acids research. 2014;42:D1091–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhang Y, Skolnick J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005;33:2302–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Hu J, Li Y, Yu DJ, Zhang Y. PSS-align: Sequence-order independent comparison of protein binding pocket structures. 2019:submitted. [Google Scholar]
- [24].Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10. [DOI] [PubMed] [Google Scholar]
- [25].Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Gloriam DE, Foord SM, Blaney FE, Garland SL. Definition of the G protein-coupled receptor transmembrane bundle binding pocket and calculation of receptor similarities for drug design. J Med Chem. 2009;52:4429–42. [DOI] [PubMed] [Google Scholar]
- [27].Ginalski K, Elofsson A, Fischer D, Rychlewski L. 3D-Jury: a simple approach to improve protein structure predictions. Bioinformatics. 2003;19:1015–8. [DOI] [PubMed] [Google Scholar]
- [28].Zheng W, Zhang C, Wuyun Q, Pearce R, Li Y, Zhang Y. LOMETS2: improved meta-threading server for fold-recognition and structure-based function annotation for distant-homology proteins. Nucleic Acids Res. 2019;47:W429–W36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Yang J, Roy A, Zhang Y. Protein-ligand binding site recognition using complementary binding-specific substructure comparison and sequence profile alignment. Bioinformatics. 2013;29:2588–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Zhang J, Yang J, Jang R, Zhang Y. GPCR-I-TASSER: A hybrid approach to G protein-coupled receptor structure modeling and the application to the human genome. Structure. 2015;23:1538–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Swendsen RH, Wang JS. Replica Monte Carlo simulation of spin glasses. Physical Review Letters. 1986;57:2607–9. [DOI] [PubMed] [Google Scholar]
- [32].Wu S, Zhang Y. LOMETS: a local meta-threading-server for protein structure prediction. Nucleic Acids Res. 2007;35:3375–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zhang Y, Skolnick J. Scoring function for automated assessment of protein structure template quality. Proteins. 2004;57:702–10. [DOI] [PubMed] [Google Scholar]
- [34].Fenalti G, Giguere PM, Katritch V, Huang X-P, Thompson AA, Cherezov V, et al. Molecular control of δ-opioid receptor signalling. Nature. 2014;506:191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Sievers F, Higgins DG. Clustal Omega, Accurate Alignment of Very Large Numbers of Sequences. Multiple Sequence Alignment Methods. 2014;1079:105–16. [DOI] [PubMed] [Google Scholar]
- [36].Ballesteros JA, Weinstein H. [19] Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods in neurosciences. 1995;25:366–428. [Google Scholar]
- [37].Yan R, Xu D, Yang J, Walker S, Zhang Y. A comparative assessment and analysis of 20 representative sequence alignment methods for protein structure prediction. Sci Rep. 2013;3:2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Taylor R Simulation Analysis of Experimental-Design Strategies for Screening Random Compounds as Potential New Drugs and Agrochemicals. Journal of Chemical Information and Computer Sciences. 1995;35:59–67. [Google Scholar]
- [39].Butina D Unsupervised data base clustering based on Daylight’s fingerprint and Tanimoto similarity: A fast and automated way to cluster small and large data sets. Journal of Chemical Information and Computer Sciences. 1999;39:747–50. [Google Scholar]
- [40].Dalke A The FPS fingerprint format and chemfp toolkit. Journal of Cheminformatics. 2013;5:P36. [Google Scholar]
- [41].Landrum G RDKit: Open-source cheminformatics. Online) http://wwwrdkitorg Accessed. 2006;3:2012.
- [42].Keiser MJ, Roth BL, Armbruster BN, Ernsberger P, Irwin JJ, Shoichet BK. Relating protein pharmacology by ligand chemistry. Nat Biotechnol. 2007;25:197–206. [DOI] [PubMed] [Google Scholar]
- [43].Keiser MJ, Hert J. Off-target networks derived from ligand set similarity. Methods Mol Biol. 2009;575:195–205. [DOI] [PubMed] [Google Scholar]
- [44].Jnr JK. On the shortest spanning subtree and the traveling salesman problem. Proc Amer Math Soc 1956. p. 48–50. [Google Scholar]
- [45].Smoot ME, Ono K, Ruscheinski J, Wang P-L, Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2010;27:431–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Lee HS, Zhang Y. BSP-SLIM: a blind low-resolution ligand-protein docking approach using predicted protein structures. Proteins. 2012;80:93–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Roy A, Zhang Y. Recognizing protein-ligand binding sites by global structural alignment and local geometry refinement. Structure. 2012;20:987–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Progress Zhang Y. and challenges in protein structure prediction. Curr Opin Struct Biol. 2008;18:342–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Bemis GW, Murcko MA. The properties of known drugs. 1. Molecular frameworks. J Med Chem. 1996;39:2887–93. [DOI] [PubMed] [Google Scholar]
- [50].Mysinger MM, Carchia M, Irwin JJ, Shoichet BK. Directory of useful decoys, enhanced (DUD-E): better ligands and decoys for better benchmarking. J Med Chem. 2012;55:6582–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Weiss DR, Bortolato A, Tehan B, Mason JS. GPCR-Bench: A Benchmarking Set and Practitioners’ Guide for G Protein-Coupled Receptor Docking. J Chem Inf Model. 2016;56:642–51. [DOI] [PubMed] [Google Scholar]
- [52].Mangoni M, Roccatano D, Di Nola A. Docking of flexible ligands to flexible receptors in solution by molecular dynamics simulation. Proteins. 1999;35:153–62. [DOI] [PubMed] [Google Scholar]
- [53].Salmaso V, Moro S. Bridging Molecular Docking to Molecular Dynamics in Exploring Ligand-Protein Recognition Process: An Overview. Front Pharmacol. 2018;9:923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Truchon JF, Bayly CI. Evaluating virtual screening methods: good and bad metrics for the “early recognition” problem. J Chem Inf Model. 2007;47:488–508. [DOI] [PubMed] [Google Scholar]
- [55].Jordan BA, Cvejic S, Devi LA. Opioids and their complicated receptor complexes. Neuropsychopharmacology. 2000;23:S5–S18. [DOI] [PubMed] [Google Scholar]
- [56].Yang J, Roy A, Zhang Y. BioLiP: a semi-manually curated database for biologically relevant ligand-protein interactions. Nucleic Acids Res. 2013;41:D1096–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.