

Abstract
Inactivation of the tumor suppressor lipid phosphatase INPP4B is common in triple negative breast cancer (TNBC). We generated a genetically-engineered TNBC mouse model deficient in INPP4B. We found a dose-dependent increase in tumor incidence in INPP4B homozygous and heterozygous knockout mice compared to wild-type, supporting a role for INPP4B as a tumor suppressor in TNBC. Tumors derived from INPP4B knockout mice are enriched for AKT and MEK gene signatures. Consequently, mice with INPP4B deficiency are more sensitive to PI3K or MEK inhibitors, compared to wild-type mice. Mechanistically, we found that INPP4B deficiency increases PI(3,4)P2 levels in endocytic vesicles but not at the plasma membrane. Moreover, INPP4B loss delays degradation of EGFR and MET, while promoting recycling of RTKs, thus enhancing the duration and amplitude of signaling output upon growth factor stimulation. Therefore, INPP4B inactivation in TNBC promotes tumorigenesis by modulating RTK recycling and signaling duration.
Keywords: INPP4B, triple negative breast cancer, EGFR, PI 3-kinase, phosphoinositide
Introduction
The phosphoinositide 3-kinase (PI3K) pathway is one of the most frequently altered signaling pathways in human cancer, and genetic gain oncogenes or loss of tumor suppressors that regulate or transduce the PI3K signal leads to tumorigenesis (1). In growth factor signaling, activation of class I PI3K results in production of the second messenger PI(3,4,5)P3 (PIP3), which recruits effector proteins such as the protein kinase AKT to promote cell growth, survival, migration and metabolic reprogramming (2). Termination of PI3K signaling is achieved by dephosphorylation of PIP3 by phosphatase and tensin homolog (PTEN). Alternatively, PIP3 can be removed by the sequential action of the SH2-domain containing inositol phosphatases 1 and 2 (SHIP1/2), to generate PI(3,4)P2, followed by inositol polyphosphate 4-phosphatases A and B (INPP4A/B) and PTEN, ultimately generating the precursor phosphoinositides PI(3)P and PI(4)P (3–5).
Gain-of-function, oncogenic mutations in PIK3CA, the gene that encodes the p110α catalytic subunit of class Ia PI3K, occur with high frequency in estrogen receptor positive (ER+ve) breast cancers (6). TNBC is a subtype of breast cancer that lacks targeted therapy options due to lack of expression of ER or HER2, and exhibits a high degree of molecular heterogeneity (7). In contrast to ER+ve breast cancers, PIC3CA is not frequently altered in TNBC, instead inactivating mutations or deletion of PTEN and heterozygous deletion of INPP4B are frequent (8–10). While PTEN has been established as a bona fide tumor suppressor in many cancer types and loss of PTEN sensitizes tumors to PI3K inhibitors (11), the function and mechanistic basis of INPP4B as a tumor suppressor is less clear. INPP4B was originally identified as a tumor suppressor in a genetic RNAi screen (12), and in cell-based and xenograft experiments, INPP4B inactivation leads to elevated PI(3,4)P2 levels, AKT activation, and increased tumor growth (13,14). By contrast, in ER+ve cells and tumors, INPP4B can actually function as an oncogene through activation of the serum and glucocorticoid-regulated kinase 3 (SGK3) pathway (15,16). Although loss of INPP4B protein expression is observed in 70%−80% of TNBC patient samples (13,17), to the extent that INPP4B-negativity is identified as the most specific biomarker in basal-like breast cancer (18,19), whether INPP4B loss functionally mediates TNBC development has not been evaluated in vivo.
The substrate of INPP4B, PI(3,4)P2, belongs to the family of phosphoinositides where the head group of the inositol ring can be reversibly phosphorylated and de-phosphorylated to generate seven distinct species, with preferential localization on distinct subcellular membrane compartments (20,21). By binding and recruiting effector proteins, phosphoinositides regulate a multitude of cellular functions including endocytosis and intracellular vesicle trafficking, cytoskeletal remodeling and signal transduction (20–22). Importantly, the mechanisms that regulate endocytosis, degradation or recycling of receptor tyrosine kinases (RTKs) including epidermal growth factor receptor (EGFR), MET and fibroblast growth factor receptor (FGFR), profoundly affect the amplitude and duration of signaling output and tumorigenesis (23–28). PI(3,4)P2 recruits specific effector proteins such as lamellipodin (29) and Tapp1/2 (30), as well as effectors with more promiscuous phosphoinositide binding such as AKT (31), Bam32 (32) and sorting nexins (33–35). In addition to promoting AKT signaling, regulating actin cytoskeletal rearrangements and facilitating clathrin-mediated endocytosis (36,37), localized production of PI(3,4)P2 at late endosome/lysosomes can suppress mTORC1 activation under specific nutrient-deprived conditions (38,39). However, the precise mechanism(s) by which INPP4B-loss contributing to TNBCs has not been determined. Here, we have generated a mouse model of INPP4B deletion in the context of TNBC, and have used it to decipher the role of PI(3,4)P2 and RTK trafficking in tumorigenesis.
Results
Tumor Penetrance Increases Upon Genetic Ablation of INPP4B in a TNBC Mouse Model
To determine whether genetic loss of INPP4B plays a functional role in the etiology of TNBC, we generated an INPP4B-deficient model by crossing INPP4B-phosphatase knockout mice (40) with a TNBC model in which Tp53 and Brca1 are deleted upon K14-driven Cre-expression in mammary epithelial cells (41) (Supplementary Figure S1A for breeding schematics). We confirmed deletion of exon 22 by genotyping (Supplementary Figure S1B) as described (42). As previously reported (40,43), INPP4B phosphatase deletion alone did not result in any appreciable phenotype, although we did observe age- and sex-dependent weight gain resulting in increased body weight in female mice over 8 months of age with regular chow (Supplementary Figure S1C). Similarly, modest alterations in glucose clearance when challenged with glucose, but not with insulin, were observed in INPP4B heterozygous (HET) and knockout (KO) mice compared to wild-type (WT) littermates (Supplementary Figure S1D). When crossed into the TNBC mouse model, INPP4B phosphatase loss resulted in a dose-dependent increase in mammary tumor penetrance. While mammary tumors developed in 17.2% of WT mice, 38% of mice developed mammary tumors in INPP4B HET mice, which increased to 53.7% in INPP4B in KO mice (Figure 1A). This significant increase in mammary tumor penetrance was also manifested as increased mammary tumor-related death (Figure 1B). In addition, we observed a slightly shortened lifespan for mammary tumor-bearing mice. The mean life span due to mammary tumor development for INPP4B WT mice was 290.8 days, while that for INPP4B HET mice was 232.9 days (p=0.006, one-way ANOVA), and similarly 239 days for INPP4B KO mice (p=0.01, one-way ANOVA) (Figure 1C).
Figure 1:

Genetic ablation of INPP4B promotes TNBC formation in a K14cre; Tp53flox/flox; Brca1flox/flox mouse model. A) Mammary tumor incidence in INPP4B WT, HET or KO mice. B) Survival in INPP4B WT (n=28), HET (n=53) or KO (n=43) mice. C) Life span of mammary tumor-bearing mice in INPP4B WT, HET or KO mice. D) Representative images of different histologies from K14cre; Tp53flox/flox; Brca1flox/flox; Inpp4B KO background, including adenocarcinomas, ductal carcinomas, mixed adenocarcinomas with focal squamous cell carcinomas, and cystic. E) AIMS analysis was performed using RNAseq data generated from tumors developed in K14cre; Tp53flox/flox; Brca1flox/flox; Inpp4B WT, HET and KO mice.
To understand the nature of mammary tumors developed from distinct INPP4B genetic backgrounds, we analyzed tumor histology by immunohistochemistry (IHC). The majority (76.19%) of tumors scored as mammary adenocarcinomas (Figure 1D:a), although ductular carcinomas, mammary adenocarcinoma mixed with focal squamous cell carcinoma and mammary cystic differentiated adenocarcinomas were also noted (Figure 1D:b-d). The triple negative nature of these mammary tumors was confirmed by estrogen receptor (ER), progesterone receptor (PR) and human epidermal growth factor receptor 2 (HER2) IHC as previously reported using this TNBC model (42), as well as RNAseq analyses using AIMS classifiers (Figure 1E), PAM50 classifiers (Supplementary Figures S1E), and unsupervised hierarchical clustering (Supplementary Figures S1F).
Gross Genome Instability is not Significantly Affected in Murine Tumors Upon INPP4B Deletion
In addition to its role in PI3K pathway signaling, loss of INPP4B elicits DNA repair defects in ovarian cancer which can result in chromosomal instability and increased tumor incidence (44). Therefore, we performed whole exome sequencing and evaluated markers for genome instability, including the number of chromosome breaks (Figure 2A), chromosomal translocations (Figure 2B), and small insertions and deletions (INDELs) (Figure 2C), but did not find any statistically significant differences in these parameters. By contrast, an increase in the number of point mutations was observed in INPP4B KO tumors compared to WT (Figure 2D). Among the different point mutations, we observed a significant increase in C to T mutations when comparing INPP4B HET or KO to WT tumors (Supplementary Figures S2A and S2B), but no differences in T to other nucleotide mutations (Supplementary Figure S2C). Overall, our data demonstrate that INPP4B loss does not affect gross chromosomal instability during tumor development in this model, although the number of single nucleotide point mutations is increased.
Figure 2.
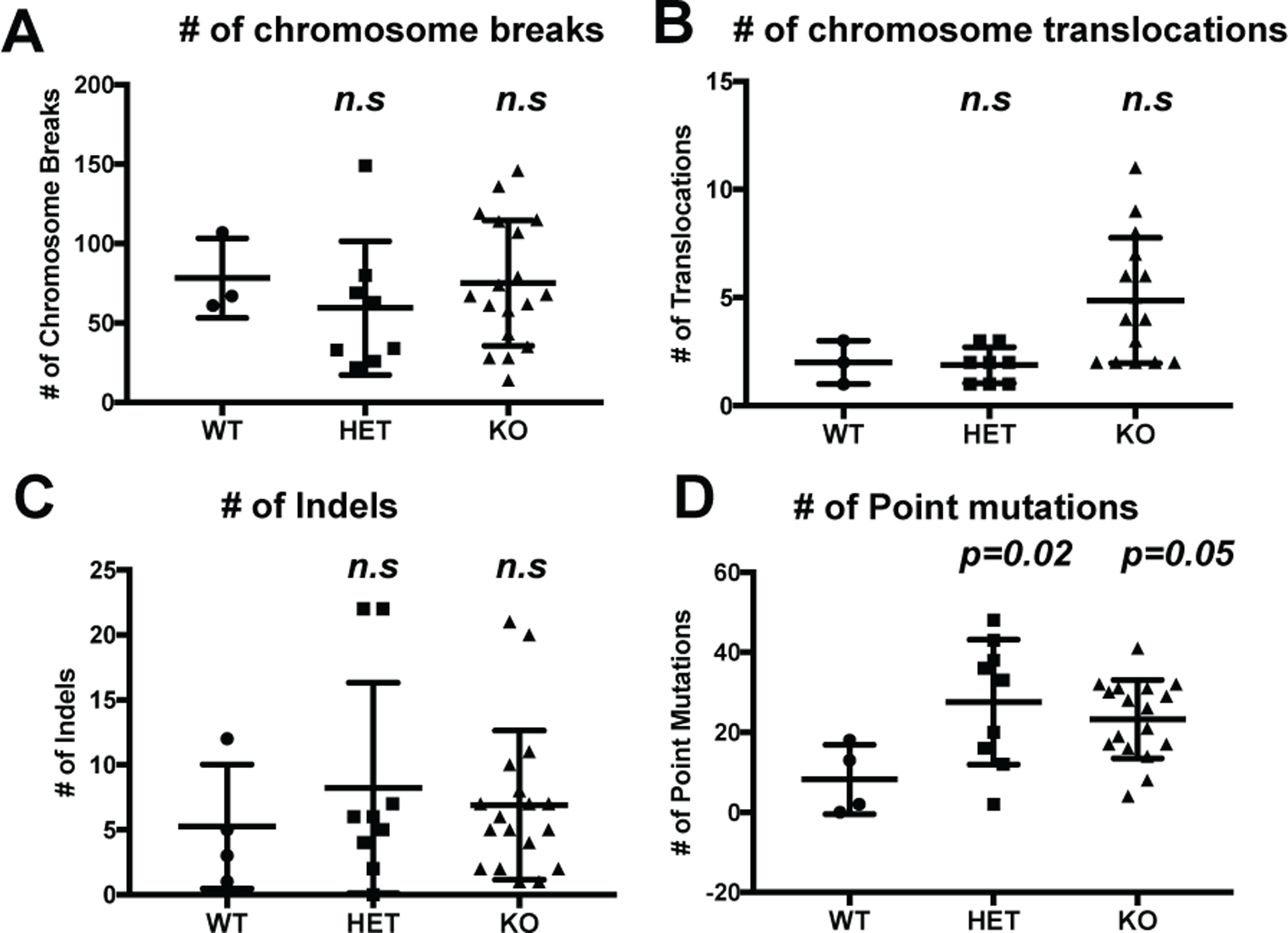
INPP4B loss does not significantly affect genomic instability. A) Number of chromosome breaks in INPP4B WT, HET and KO in the K14cre; Tp53flox/flox; Brca1flox/flox background. B) Number of chromosome translocations in same cohort as (A). C) number of INDELS in same cohort as (A). D) Number of point mutations in same cohort as (A).
INPP4B Loss Enhances PI3K and ERK Pathway Activation
We next compared the transcriptional profile of tumors developed from the INPP4B mouse models. Using gene set enrichment analysis (GSEA), we found an enhanced AKT pathway gene signature in INPP4B HET or KO mice compared to WT mice (Figure 3A). Surprisingly, tumors developed from INPP4B HET or KO mice also showed an increased mitogen-activated protein kinase kinase (MEK) pathway gene signature (Figure 3B). To confirm this observation in vitro, we knocked down INPP4B in breast epithelial MCF10A cells using shRNA, and this also resulted in enhanced PI3K pathway activation as reported by increased pAKT1, pAKT2 and pPRAS40 as well as increased extracellular-signal-regulated kinase (ERK) phosphorylation and pS6 (pS235/p236) (Figure 3C). Moreover, both the duration and magnitude of pAKT and pERK were enhanced in EGF-stimulated cells in which INPP4B was transiently downregulated using siRNA (Figure 3D and Supplementary Figure S3A and S3B). This was also observed in primary mammary epithelial cells stimulated with EGF over a time course (Supplementary Figure S3C). Phenotypically, INPP4B reduction promoted MCF10A cell proliferation under serum-deprived conditions when supplemented with EGF (Figure 3E), but not in cells grown with complete media (Supplementary Figure S3D). Finally, over-expression of INPP4B in the TNBC cell line MDA-MB-231 resulted in significantly reduced spheroid growth in 3D (Supplementary Figure S3E). In summary, the in vivo and in vitro results above show INPP4B loss promotes both PI3K and ERK pathway activation, which may contribute to mammary tumor development in TNBC.
Figure 3.

INPP4B loss enhances both PI3K and ERK signaling pathway activation in vivo and in vitro. A) GSEA analysis of tumors developed from INPP4B HET (left) or KO (right) backgrounds for AKT pathway activation compared to INPP4B WT background. B) GSEA analysis of tumors developed from INPP4B HET (left) or KO (right) backgrounds for MEK pathway activation compared to INPP4B WT background. C) MCF10A cells infected with virus harboring either pLK0.1 vector or pLK01-INPP4B-shRNA were washed, trypsinized, and pelleted. Total cell lysates were immunoblotted with the indicated antibodies. D) MCF10A cells transfected with non-target control siRNA (control-siRNA), or INPP4B smart-pool siRNA pool (si-pool-1, Dharmacon) or siRNA (si-2 from CST), serum starved and stimulated 50ng/ml EGF and immunoblotted with the indicated antibodies (n=5, representative images are shown). E) Proliferation of MCF10A-pLK0.1 cells or MCF10A-pLK-shINPP4B cells in serum-free medium containing 50ng/ml of EGF, figure is representative of 3 independent experiments, error bars represent SEM, statistics analysis was performed using 2-way ANOVA.
Endogenous Tumors with INPP4B Ablation are More Sensitive to PI3K and MEK Inhibition
We reasoned that if TNBC cells with reduced INPP4B levels become more dependent on PI3K and ERK signaling for tumor initiation and/or maintenance, then pathway inhibition may show a more pronounced effect compared to cells that retain INPP4B. We first tested this hypothesis in vitro, and generated INPP4B knockdown (using si/shRNA and CRISPR-Cas9) (Supplementary Figure S4A) and treated cells with PI3K pathway inhibitors, including the pan class I PI3K inhibitor BKM-120 and the catalytic AKT inhibitor GDC0068. We found that in both cases, INPP4B loss increased sensitivity to inhibitor treatment, resulting in statistically significant decreases in IC50 to both BKM120 (Figure 4A, Supplementary Figures S4B and S4C, n=3) and GDC0068 (Figure 4B, Supplementary Figure S4D, n=3). We also observed a trend showing decreased IC50 for the MEK inhibitor trametinib, however this decrease was not statistically significant (Supplementary Figure S4E). To test this hypothesis in vivo, we implanted tumors developed from INPP4B WT, HET, or KO backgrounds into the mammary glands of recipient nude mice(45), and carried out in vivo drug treatment using pre-determined doses once tumors reached 7–8 mm in diameter (42) (Figure 4C). We confirmed the efficacy of BKM120, the p110α-specific PI3K inhibitor BYL719 and the MEK inhibitor trametinib in pathway inhibition by immunoblotting tumor lysates for pAKT and pERK, respectively (Supplementary Figures S4F and S4G). We found that both BKM120 and BYL719 improved overall survival (Figure 4D), while trametinib delayed tumor growth (Figure 4E) without affecting overall survival (Supplementary Figure S4H). However, tumors developed from the INPP4B loss (HET and KO) background were more sensitive to either BKM120 or BYL719 (Figure 4F) or trametinib (Figure 4G), when compared to tumors developed from the INPP4B WT background. Therefore, INPP4B loss confers sensitivity to both PI3K and ERK pathway inhibition.
Figure 4.

INPP4B loss increases sensitivity to PI3K and MEK inhibitors. A) MCF10A cells harboring control-gRNA or INPP4B-gRNA1 treated with increasing concentrations of GDC0068 for 72 hours in serum-free medium containing 50ng/ml EGF. Live cells measured using CellTiter Glow and IC50 was calculated (n=4, student’s t-test, error bars represent SEM). B) MCF10A cells transiently transfected with control siRNA or INPP4B smart-pool siRNA and treated with increasing concentrations of BKM120 for 72 hours in serum-free medium containing 50ng/ml EGF. Live cells were measured as in (A) (n=3, student’s t-test, error bars represent SEM). C) Dose and frequency of in vivo drug treatment regimen. D) Nude mice implanted with GEMM tumors treated with BKM120 or BYL719. Statistical analysis of overall survival was performed using log-rank (Mantel-Cox) test. E) Nude mice bearing GEMM tumors were treated with trametinib, and tumor volume measured. Tumor sizes on day 9 were divided to those on day 1, statistical analysis was performed using student’s t-test. F) Overall survival of nude mice implanted with GEMM tumors and treated with BKM120 or BYL719 were analyzed based on INPP4B genotypes, WT and INPP4B loss (Loss=HET + KO). Statistical analysis was performed using log-rank (Mantel-Cox) test. G) Mice bearing GEMM tumors were treated with trametinib and percent change in tumor volume on day 9 of Trametinib was normalized to control treatment. Statistics was performed using student’s t-test.
INPP4B Loss Results in Increased PI(3,4)P2 in Intracellular Vesicles
INPP4B is a lipid phosphatase that dephosphorylates the 4’ position of PI(3,4)P2 to generate PI(3)P, and therefore loss or inactivation of INPP4B leads to increased pools of PI(3,4)P2. Consistent with this model, CRISPR-Cas9-mediated INPP4B reduction in MCF10A cells resulted in increased EGF-stimulated PI(3,4)P2 as measured by 3H-inositol labelling, whilst the levels of other phosphoinositides were unaffected (Figure 5A and Supplementary Figure S5A). PI(3,4)P2 has been shown to be localized to the plasma membrane where it regulates clathrin-mediated endocytosis, as well as at intracellular vesicles where it stimulates AKT signaling (46,47). Consistent with previous studies, immunofluorescence staining using anti-PI(3,4)P2 antibodies in INPP4B-downregulated cells revealed an increase in PI(3,4)P2 in intracellular vesicles following EGF stimulation (Figure 5B and 5C). By contrast, the PI(3,4)P2 plasma membrane pool only transiently increased at 3min and remained largely unaffected at other time points upon INPP4B downregulation (Supplementary Figures S5B and S5C). These data are consistent with the model that INPP4B contributes to the intracellular endomembrane pool of PI(3,4)P2 biosynthesis upon growth factor stimulation, leading to downstream pathway activation.
Figure 5.

INPP4B reduction results in increased PI(3,4)P2 in intracellular vesicles. A) MCF10A cells with control gRNA or INPP4B-gRNA1, labeled with 3H-inositol, serum-starved and stimulated with 50ng/ml of EGF for 0, 5min and 30min and phosphoinositide levels were measured. N=4, Statistics was performed using student’s t-test. B) MCF10A cells transiently transfected with non-target control siRNA or smart-pool INPP4B-siRNA were serum starved and stimulated with 50ng/ml of EGF, and stained for intracellular PI(3,4)P2, n=3. C) Quantification of intracellular PI(3,4)P2 levels upon EGF stimulation. ** p=0.003; *** p<=0.0001, student t test.
INPP4B Depletion Results in Delayed EGFR Degradation
In initial INPP4B knockdown experiments we noticed that in addition to enhanced pAKT and pERK, total EGFR protein levels were consistently elevated compared to control cells (Figure 3D and Figure 6A). Increased EGFR expression was confirmed by siRNA-mediated downregulation of INPP4B, with no alterations in EGFR and slight change in MET mRNA transcript levels (Figure 6B and Supplementary Figure S6A, S6B and S6C). Given the established functions of phosphoinositides, including PI(3,4)P2 in RTK trafficking (36), we investigated whether INPP4B loss affects RTK trafficking and signaling. We found that whilst total EGFR protein levels decreased in control siRNA-treated cells upon a time course of EGF stimulation, degradation of EGFR was delayed in INPP4B-siRNA treated cells (Figure 6C and 6D). This was confirmed using INPP4B CRISPR-Cas9 cells stimulated with EGF (Figure 6E and Supplementary Figures S6D and S6E). However, INPP4B reduction did not alter sensitivity to the EGFR inhibitor Erlotinib (Supplementary Figure S6F). In TNBC, EGFR overexpression has been shown to play an important role during tumor development, although other RTKs including MET also contribute (48). Consistent with this, total MET protein levels are increased in response to HGF stimulation in INPP4B knockdown cells compared to control (Figure 6F). In addition, pAKT and pERK were enhanced and prolonged in response to HGF (Figure 6F), similar to that observed in EGF-stimulated cells.
Figure 6.

INPP4B downregulation results in RTK degradation defects. A) MCF10A cell transiently transfected with control- or INPP4B-siRNA and immunoblotted with the indicated antibodies (left panel) and quantitated (right panel). B) qRT-PCR using mRNA from the same samples as in (A) (n=4, student’s t-test). C) MCF10A cells transfected with control or INPP4B-siRNA, serum-starved and treated with cycloheximide for 1-hour prior stimulation with 50ng/ml of EGF for the indicated times. D) Quantification of (C), n=3, 2-way ANOVA. E) MCF10A cells infected with Cas9/control gRNA or distinct INPP4B-gRNAs, serum starved, and treated with cycloheximide for 1 hour and stimulated with 50ng/ml EGF for the indicated times. F) MCF10A cells transfected with control or INPP4B siRNA, serum-starved and stimulated with 50ng/ml of EGF for the indicated times and immunoblotted with the indicated antibodies. G) INPP4B siRNA transfected MCF10A cells were serum-starved and stimulated with EGF for the indicated times. Immunofluorescence was performed with primary anti-EGFR antibody followed with Alexa Fluor-488 conjugated secondary antibody. H) Tumors developed from K14cre; Tp53flox/flox; BRCA1flox/flox ;INPP4B WT/HET/KO mice were sectioned and IHC was carried out using anti-mouse EGFR antibody. Quantitation is shown on the right (2-way ANOVA).
Next, we tracked EGFR subcellular localization dynamics upon EGF stimulation using immunofluorescence (IF). At early time points, accumulation of EGFR in intracellular vesicles was not affected by INPP4B reduction in response to EGF (Supplementary Figure S6G). By contrast, by 60 and 90 min EGFR trafficked to a perinuclear region in control siRNA-treated cells, but remained scattered in INPP4B siRNA-treated cells. By 180 min, EGFR staining was significantly diminished in control cells, whereas in INPP4B knockdown cells EGFR was still detectable with a perinuclear staining pattern (Figure 6G and Supplementary Figure S6H).
The prediction from the in vitro INPP4B knockdown experiments would be that in the setting of INPP4B loss in vivo, EGFR protein levels would be elevated. Immunohistochemistry staining of EGFR on the INPP4B mouse tumors revealed that while both tumors derived from the INPP4B WT cohort showed low levels of EGFR expression, 5/6 of tumors from the INPP4B HET or 7/10 tumors from the KO backgrounds showed medium to high level expression of total EGFR (Figure 6H).
INPP4B Affects Trafficking of EGFR to Late Endosome/Lysosomes and RTK Recycling
To further investigate trafficking of RTKs in the context of INPP4B loss, we performed IF to measure co-localization of EGFR with endosomal markers. At all time points tested, the intensity of staining and vesicle size of the early endosome antigen 1 (EEA1) marker was not affected upon INPPB reduction with siRNA (Supplementary Figure S7A and S7B). Similarly, the co-localization of EEA1 with EGFR in INPP4B siRNA-treated cells was unaffected at early time points, but persisted at later time points (60, 90 and 180min) (Figure 7A and Supplementary Figure 7C). At early time points (10 and 30 min), we also did not observe significant differences in the staining intensity of the late endosome/lysosome marker CD63 or CD63-positive vesicle size (Figure 7B). By contrast, at later time points when EGFR traffics to the late endosome/lysosome for degradation, CD63 intensity decreased with a concomitant increase in vesicle size in control cells, however, there was no increase in INPP4B knockdown cells (Figure 7B). Consistent with this observation, the dynamics of EGFR-CD63 co-localization was significantly altered: In control cells internalized EGFR progressively accumulated in CD63-positive late endosomes before being degraded, while a much less pronounced late endosomal EGFR accumulation was observed in INPP4B knockdown cells (Figure 7C and Supplementary Figure S7D). These data suggest a defect in EGFR trafficking from early endosomes to late endosomes/lysosomes. Furthermore, we found increased recycling of EGFR to the plasma membrane in INPP4B-siRNA treated cells (Figure 7D), explaining its increased surface levels (Figure 7E). Our data indicate that loss of INPP4B delays trafficking of the EGFR from early endosomes to the late endosomes/lysosomes, thus delaying receptor degradation, and thereby sustaining downstream signaling.
Figure 7.

INPP4B knockdown increases EGFR recycling and delayed trafficking to the lysosome. A) INPP4B siRNA-transfected MCF10A cells were serum starved, treated with cycloheximide, and stimulated with 50ng/ml EGF for the indicated times and stained with anti-EGFR and anti-EEA1. Images were analyzed and quantitated using Volocity. For each siRNA condition, percent co-localization at 3min was set as 100% and was normalized against other time points to evaluate the dynamic change in co-localization. Error bars represent SEM, and statistical analysis was carried out using 2-way ANOVA. B) Similar to (A), cells were stained with anti-EGFR and anti-CD63 and images analyzed using Volocity (Error Bars: SEM; Statistics: 2-way ANOVA). C) Using the same conditions described in (B), percent co-localization of CD63-EGFR was analyzed using Volocity. For each siRNA condition, percent co-localization at time 0 was set as 100% and normalized against other time points to evaluate the dynamics of co-localization over time (Error Bars: SEM; Statistics: 2-way ANOVA). D) After overnight starvation, cells were chilled to 4°C, loaded with 50ng/ml EGF for 30min, shifted to 37°C for 15min to allow internalization, acid washed and chased for the indicated times. Cells were fixed without permeabilization and stained for surface EGFR. Images were analyzed using Volocity. (Error Bars: SEM; Statistics: 2-way ANOVA). E) After overnight starvation, cells were trypsinized, fixed and surface expression of EGFR were stained for FACS analysis. (Error Bars: SEM; Statistics: student t test, n=5).
Discussion
In this study, we developed a genetically-engineered mouse model to provide evidence that INPP4B inactivation drives TNBC. Mechanistically, we uncovered a function for INPP4B in regulating the trafficking and degradation of EGFR and MET. INPP4B reduction results in increased PI(3,4)P2 accumulation in intracellular vesicles, delaying trafficking of EGFR from early endosomes to late endosomes/lysosomes upon EGF stimulation, whilst promoting recycling of EGFR to the cell surface. As a result, INPP4B loss delays EGFR degradation with a concomitant prolonged duration and amplitude of both AKT and ERK signaling, thereby promoting tumorigenesis. Consequently, INPP4B inactivation sensitizes TNBC cells to both PI3K and MEK inhibitors in vitro and in vivo.
Transcriptional profiling has revealed the extensive genetic heterogeneous nature of TNBC, with multiple distinct subgroups classified according to unique expression profiles with important clinical implications (49). Similarly, large-scale sequencing studies have detected diverse but low-frequency oncogenic mutations in numerous genes in TNBC, many of which contribute to PI3K and ERK pathway (8,50–52). Approximately 40% of ER+ve breast cancer patients harbor activating PIK3CA mutations, and the p110α-specific inhibitor Piqray (Alpelisib) was recently approved for the treatment of PIK3CA-mutant, ER+ve breast cancer in combination with fulvestrant in post-menopausal women with advanced or metastatic disease (53). By contrast, less than 10% of TNBC patients possess oncogenic PIK3CA mutations and to date there is no approved molecular targeted therapy for TNBC. Given the frequency of PI3K pathway hyperactivation in TNBC, targeting aberrant PI3K pathway activation remains a promising option. In our GEMM model, mammary tumors resulting from INPP4B heterozygous or homozygous deletion mice are more sensitive to PI3K pathway inhibition (Figure 4), suggesting that loss of INPP4B may be a predictive marker for sensitivity to PI3K inhibition, consistent with previous studies in cell lines (54).
Since AKT functions to modulate phenotypes associated with malignancy including proliferation, survival, migration, and metabolism, and is frequently hyperactivated in many cancers, numerous small molecule inhibitors have been developed for clinical use (2). As single agents, AKT inhibitors have shown minimal efficacy in clinical trials (55). In our GEMM model, INPP4B loss increases sensitivity to the AKT inhibitor GDC0068 (Ipatasertib) in vitro. In clinical trials, the AKT inhibitor AZD5363 did not significantly improve progression-free survival compared to paclitaxel alone in ER+ve breast cancer harboring PIK3CA mutations (NCT01625286, (56)). By contrast, addition of GDC0068 to paclitaxel as neoadjuvant therapy in early-stage TNBC patients harboring PIK3CA/AKT/PTEN alternations showed a favorable response (complete response 39% for GDC0068+Paclitaxel vs 9% Paclitaxel alone; NCT02301988 (57)). Given that INPP4B loss is common in TNBC (13,14,18,19) and promotes hyperactivation of AKT, it would therefore be interesting to evaluate the response of INPP4B-deficient TNBC to AKT inhibitors in vivo and in the clinic.
Compared to ER+ve or ERBB2- (HER2)-amplified breast cancer, EGFR overexpression is a frequent event in TNBC. Depending on the patient population and IHC test used, 13%−76% of TNBC tumors overexpress EGFR at the protein level (58). Yet, analyses of METABRIC and TCGA reveal that only approximately 5% of patients harbor ERBB1 (EGFR) gene amplification (59). This indicates that additional mechanisms must exist to account for increased EGFR protein in TNBC. Several mechanisms have been shown, including loss of PTEN (60), loss of BRCA1 (61) and increased expression of tissue transglutaminase (62). Our data are indicative of an additional mechanism whereby loss of INPP4B increases EGFR stability contributing to enhanced EGFR downstream signaling.
EGFR overexpression in TNBC indicates that it could serve as a potential therapeutic vulnerability for this subtype of breast cancer. However, numerous anti-EGFR therapeutics used as single agents or in combination with chemotherapy in TNBC have not shown durable therapeutic responses (63–66). One can speculate that more accurate patient stratification could improve treatment outcomes, for example by accruing patients expressing elevated levels of EGFR. In our studies, drug sensitivity assays with Erlotinib administered in cells with INPP4B reduction did not appreciably shift the IC50. These data are consistent with previous observations which showed that EGFR activation is required for initial endocytosis (27,67). Since Erlotinib inhibits EGFR phosphorylation, and stabilization of EGFR upon INPP4B reduction occurs after ligand-induced receptor endocytosis, EGFR inhibition would be predicted to be ineffective in INPP4B-null tumors.
The mechanism(s) by which loss of INPP4B and increased vesicular PI(3,4)P2 affect EGFR degradation and recycling, and as a result total EGFR expression, is presently unknown. Upon ligand-induced clathrin-mediated endocytosis, ubiquitinated EGFR and endosomal PI(3)P recruit ESCRT (endosomal sorting complexes required for transport) complexes for sorting into intraluminal vesicles for degradation (68–70). The INPP4B interactome has not been deciphered and it is not yet clear whether INPP4B can directly affect the ubiquitin ligase activity of c-Cbl docked onto the EGFR, thereby affecting EGFR/Hrs interaction, as has been shown for SHIP2 (71). Alternatively, INPP4B could directly or indirectly interact with ESCRT complexes, since INPP4A/B also localizes to endosomes (43,72,73). Our results show that INPP4B depletion results in increased PI(3,4)P2 in intracellular vesicles, consistent with previous studies (46,74). Although a subset of PI(3,4)P2-positive vesicles colocalize with EEA1, our preliminary studies show that only a small percentage (1.7%−6.1% depending on the time point) colocalize with internalized EGFR. By contrast, up to 50% of EGFR-positive vesicles are also positive for EEA1. Within this small subset of EGFR-positive vesicles, PI(3,4)P2 intensities were comparable between control siRNA- and INPP4B siRNA-treated cells. Several potential mechanisms may contribute to defects in RTK degradation and enhanced recycling. First, a block in PI(3,4)P2 degradation due to INPP4B loss may result in the reduction of a local endosomal pool of PI(3)P derived from PI(3,4)P2 (i.e. a minor pool not separable from the total cellular PI(3)P pool in our HPLC analysis), upstream of PI(3,5)P2 production via PIKFYVE and EGFR degradation. In addition, failure to dephosphorylate PI(3,4)P2 via INPP4B may promote funneling of PI(3,4)P2 into an alternative PTEN-mediated degradation pathway towards PI(4)P (3), a lipid that promotes recycling to the cell surface (75,76). Alternatively, PI(3,4)P2-rich endomembranes may recruit effectors to promote recycling. One possibility is that SNX18, a PX-BAR-containing sorting nexin that belongs to the SNX9/18/30 family may contribute to this mechanism. SNX18 binds to PI(3,4)P2 as well as other phosphoinositides (33,47) and colocalizes with Rab11 but not EEA1, and has been shown to promote tubulated recycling endomembranes (77–79).
Traditionally viewed as a breakdown product of PI(3,4,5)P3, recent studies have shown that PI(3,4)P2 has important signaling roles in its own right (36,80). Different enzymes contribute to the localized production of this signaling lipid, exerting seemingly different biological activities. For example, class II PI3K C2α at clathrin-coated pits is important for the synthesis of PI(3,4)P2 to mediate constriction of late-stage clathrin-coated pits (47,81,82). Class II PI3K C2β at late endosomes/lysosomes during growth factor starvation suppresses mTORC1 activity (39). Alternatively, class I PI3K activation by RTKs such as EGFR generates PI(3,4,5)P3, which is dephosphorylated by 5’ phosphatases to give rise to PI(3,4)P2, contributing to endosomal PI(3,4)P2 (5). Although the relative quantitative contribution from each of the routes during EGFR intracellular trafficking is not precisely understood, it is generally accepted that degradation from PI(3,4,5)P3 contributes to a significant fraction of PI(3,4)P2 in intracelllular vesicles upon RTK activation (36,37). Additional studies are required to determine whether class II PI3Ks can function in an analogous manner to INPP4B in TNBC etiology, especially given their importance in physiology and disease (83).
Recent studies have found that PTEN can also degrade PI(3,4)P2, and that combined depletion of INPP4B and PTEN results in synergistic accumulation of PI(3,4)P2 and AKT activation (3). Since PTEN inactivation is also a frequent event in TNBC and combined PTEN and INPP4B loss leads to AKT hyperactivation, this provides additional rationale for preclinical and clinical studies for AKT inhibitors in this setting. At the same time, it is important to note that while INPP4B functions as a bona fide tumor suppressor in TNBC and other cancers, studies in cell lines and mice have shown that in ER+ve breast cancer and colorectal cancer, INPP4B actually functions as an oncogene, potentially due to copy number gain or overexpression in these tumor types (15,84).
In summary, we have developed a mouse model of TNBC in which INPP4B functions as a tumor suppressor and regulates receptor tyrosine kinase trafficking and degradation. We propose a model whereby INPP4B inactivation results in PI(3,4)P2 accumulation in intracellular vesicles, delaying degradation of RTKs, prolonging both PI3K and ERK signaling and tumorigenesis and leading to sensitization to pathway inhibitors.
Materials and Methods
Mice and histology:
Animal experiments were conducted in accordance with Institutional Animal Care and Use Committee (IACUC) approved protocols (# 099–2015) at Beth Israel Deaconess Medical Center. K14cre; Brca1flox/flox; Trp53flox/flox mice were obtained from the Dr. Jos Jonkers (Netherlands Cancer Institute), and INPP4B del/del mice were obtained from Dr. Takehiko Sasaki (Tokyo Medical and Dental University). Mouse breeding was carried out as shown in Figure S1A, and genotyping was carried out as described (40,41)
Orthotopic tumor implantation:
Tumor pieces were cut into 2 mm in diameter and inserted into the 4th mammary fat pad of 8–10-week-old recipient mice via a 0.5 cm2 incision in the skin. The skin was closed with VetBond.
Tumor treatment and tumor measurement:
Once tumors reached approximately 8 mm in diameter as measured by electrical caliper (Fisher Scientific), mice were treated with indicated drugs obtained from MedChemExpress, LLC (BKM120, BYL719 and Tremetinib). For oral gavage, 100μl of drug suspension was administrated daily for six consecutive days, followed by one drug holiday. Tumor sizes were measured twice a week (length and width), and tumor volume was calculated as (3.14*length*width*width/6).
Genomic DNA and library preparation:
Genomic DNA from tumor or liver samples was prepared following the protocol for Promega ReliaPrep Tissue DNA Miniprep System (A2051). SureSelect or NimbleGen Mouse exome capture kits were used to generate DNA library according to manufacturer’s instructions. Sequencing was carried out using HiSeq4000 (Illumina) using paired end clustering and 51×2 cycles sequencing.
Mutation and copy number analysis:
See the supplementary methods for details. Somatic mutations were identified upon removing any mutations found in any tail, liver or normal mammary control samples, in mouse dbSNP, or with insufficient coverage in the control samples. Mutations were annotated with SnpEff. Copy number variants were called using CNVkit after removing low-quality reads. Sample-specific thresholds were computed to call amplifications and deletions.
RNA preparation and library preparation:
Total RNA was prepared following the protocol for Promega ReliaPrep RNA Tissue Miniprep System (Z6111), and RNA integrity and concentration were measured using the Agilent 2100 Bioanalyzer (Agilent Technologies). cDNA libraries were prepared from 15–35 ng RNA starting material (RIN values >6.0), using the TruSeq RNA Sample Preparation Kit (Illumina) according to the manufacturer’s instructions, and quality was checked on an Agilent 2100 Bioanalyzer (Agilent Technologies). Sequencing was carried out on the HiSeq 2500 (Illumina) using paired end clustering and 51×2 cycles sequencing.
Immunoblotting:
Tumors and cells were lysed in RIPA lysis buffer with protease inhibitors (Roche) and phosphatase inhibitors (Sigma). Equal amount of total protein lysates were used for immunoblotting. The following antibodies were from Cell Signaling Technology (Beverly, MA) and were used at 1:1000 dilution: pAKT (#3787), AKT (#4691), pERK (#4370), ERK (#4695), pMET (#3077), MET (#3127), pEGFR (#3777), EGFR (#4267), pPRAS40 (#2997), PRAS40 (#2691), Vinculin (#13901). Other antibodies used in this paper are as follows: INPP4B (Abcam, Ab81269), beta-actin (Sigma, A2228).
Cell lines:
MCF10A and MDA-MB-231cells were obtained from the American Type Culture Collection (ATCC) and authenticated using short tandem repeat (STR) profiling. Primary human mammary epithelial cells (HMECs) were obtained as described (42). MCF10A cells and HMECs were maintained in DMEM/Ham’s F12 (CellGro) supplemented with 5% equine serum (CellGro), 10 mg/mL insulin (Life Technologies), 500 ng/mL hydrocortisone (Sigma-Aldrich), 20 ng/mL EGF (R&D Systems), and 100 ng/mL cholera toxin (Sigma-Aldrich). MDA-MB-231 cells were maintained in DMEM (CellGro) supplemented with 10% fetal bovine serum (FBS) (Gemini). Cells were passaged for no more than 2 months and routinely assayed for mycoplasma contamination (MycoAlert, Lonza).
Lentivirus Production and Cell Infection:
Lentivirus preparation and infection were carried out as described using Lipofectamine 2000 (ThermoFisher) (42).
Cell Proliferation:
1500 cells were plated in 96-well plates in 200μl of cell culture medium and measured with CellTiter-Glo (Promega, G7572). For 3D culture, each well of the 8 well chamber slides were coated with 50μl growth-factor reduced Matrigel (Corning), followed by seeding 3000 cells in growth medium containing 2% Matrigel.
IC50 Measurement:
5000 cells were plated in 96 well plates and changed to medium (growth media or serum-free media/50ng/ml EGF) containing inhibitors at different concentrations. The following inhibitors were used: BKM-120 and BYL719 (MedChemExpress, LLC), GDC0068 (Selleck), Trametinib (Selleck), Erlotinib (Selleck). After 72 hours, the relative numbers of remaining cells were measured with CellTiter-Glo (Promega, G7572).
Immunofluorescence:
Cells were seeded on serum pre-coated glass cover slips overnight, serum-starved for 16–18 hours, and stimulated with 50ng/ml EGF. At indicated time points, cells were fixed in 4% paraformaldehyde, followed by 0.1% Triton X-100 in PBS (5min, RT), blocked at 37°C with 10% normal goat serum in PBS for 30min, and incubated with the following primary antibodies at 4°C overnight. After washing, cells were incubated with Alexa Fluor 488 or 568 conjugated secondary antibodies (Molecular Probes). Primary antibodies are as following: EGFR (CST #4267 for intracellular staining), EGFR-AF488 (Biolegend #352908 for surface staining); EEA1 (BD bioscience#610457), CD63 (BioLegend #353013). Images were acquired with Zeiss LSM 880 upright confocal system and analyzed with Volocity imaging software (Improvision, Perkin Elmer).
For PI(3,4)P2 staining, after fixation, cells were permeabilized and blocked with PBS containing 0.5% Saponin, 1% BSA and 10% normal goat serum for 30min for plasma membrane (PM) staining, or with 20μM Digitonin in buffer A (20mM Pipes,PH6.8, 137mM NaCl and 2.7mM KCl) followed by 30min in buffer A containing 5% normal goat serum and 50mM NH4Cl for intracellular vesicle (IV) staining. Anti-PI(3,4)P2 antibody (Echelon Biosciences Z-P034b) was diluted 1:150 in PBS or Buffer A (for PM or IV, respectively) containing 5% normal goat serum, incubated for 1 hour at room temperature, washed before incubating with Alexa Fluor 568 conjugated anti-mouse secondary antibody. Images were acquired using a spinning disk confocal microscope (Ultraview ERS, Perkin Elmer), analyzed with Volocity imaging software (Improvision, Perkin Elmer). PI(3,4)P2 levels were quantified using custom written ImageJ macros as described (47).
Immunohistochemistry (IHC):
Slides were deparaffinized, rehydrated and antigen retrieval was performed using SignalStain® Citrate Unmasking Solution (CST #14746). Slides were incubated with freshly prepared 3% H2O2 for 10min, washed twice with ddH2O, once with TBST, blocked in TBST/5% normal goat serum at R.T. for 1 hour. After incubating with anti-mouse-EGFR antibody (CST #71655) diluted in TBST/5% goat serum overnight at 4°C, the slides were washed 3 x TBST and incubated with goat-anti-rabbit-biotin secondary antibody at R.T. for 30min. Color development was carried out following instruction (Vectastain Elite ABC HRP Kit). Slides were counterstained hematoxylin (#14166), dehydrated and mounted.
Phosphoinositide measurements:
as previously reported (73). Briefly, cells were labeled for 48 hours in inositol-free DMEM with glutamine, 10% dialyzed FBS, and 20 mCi/mL 3H myo-inositol. At the indicated times, cells were washed and harvested by scraping in 1.5 mL ice-cold aqueous solution (1M HCl, 5 mM Tetrabutylammonium bisulfate, 25 mM EDTA) before adding 2 mL of MeOH and 4mL of CHCl3, vortexed and centrifuged. The aqueous layer was extracted 3 times using theoretical lower reagent (CHCl3:MeOH:aqueous solution in 8:4:3 v/v), and organic phase was collected and dried, deacylated at 55 degrees for 1 hour and dried. To the dried vials, 1 mL of theoretical upper and 1.5 mL of theoretical lower were added, vortexed, centrifuged and the aqueous phase was collected and dried. Samples were resuspended in 150 uL Buffer A (1mM EDTA), injected in anion-exchange HPLC using Partisphere SAX column and eluted with Buffer B (1mM EDTA, 1M NaH2PO4), detected using an on-line continuous flow scintillation detector (detailed gradient is provided in the supplementary methods).
Supplementary Material
Significance.
Inactivation of the lipid phosphatase INPP4B is frequent in triple negative breast cancer. Using a genetically engineered mouse model, we show that INPP4B functions as a tumor suppressor in TNBC. INPP4B regulates receptor tyrosine kinase trafficking and degradation, such that loss of INPP4B prolongs both PI3K and ERK activation.
Acknowledgements
We thank Drs. Jos Jonkers and Takehiko Sasaki for providing mouse strains, Junyan Zhang and Kangkang Yang for technical support, members of the Toker and Cantley laboratories for advice and discussion, Roderick Bronson at the DH/FCC Rodent Histopathology Core, Lay-Hong Ang and Aniket Gad at BIDMC Confocal Image Core, Suzanne L. White and Lena Liu for histology work and Eva Csizmadia for immunohistochemistry, and Luke Dow for plasmid constructs. This work was supported by: Susan G. Komen postdoctoral fellowship (H.L.); the Ludwig Center at Harvard (A.T.); the Breast Cancer Alliance (A.T); the Deutsche Forschungsgemeinschaft TRR186/A08 (V.H.); NIH R35 CA197588 (L.C.C.), NIH R01 CA226776 (G.M.W.), Breast Cancer Research Foundation (L.C.C. and G.M.W); a gift from the Jon and Mindy Gray Foundation (L.C.C.); NIH U54CA210184 (L.C.C.) and NCI F31 CA213460 (R.C.G.).
Footnotes
Conflict of Interest Statement:
A.T. is a consultant for Oncologie, Inc. and Bertis, Inc. L.C.C. is a founder and member of the SAB of Agios Pharmaceuticals and is a founder and member of SAB of Petra Pharmaceuticals and receives research support from Petra Pharmaceuticals. These companies are developing novel therapies for cancer.
References
- 1.Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, Abraham RT. The PI3K Pathway in Human Disease. Cell 2017;170(4):605–35 doi 10.1016/j.cell.2017.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Manning BD, Toker A. AKT/PKB Signaling: Navigating the Network. Cell 2017;169(3):381–405 doi 10.1016/j.cell.2017.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Malek M, Kielkowska A, Chessa T, Anderson KE, Barneda D, Pir P, et al. PTEN Regulates PI(3,4)P2 Signaling Downstream of Class I PI3K. Mol Cell 2017;68(3):566–80 e10 doi 10.1016/j.molcel.2017.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Norris FA, Majerus PW. Hydrolysis of phosphatidylinositol 3,4-bisphosphate by inositol polyphosphate 4-phosphatase isolated by affinity elution chromatography. J Biol Chem 1994;269(12):8716–20. [PubMed] [Google Scholar]
- 5.Rodgers SJ, Ferguson DT, Mitchell CA, Ooms LM. Regulation of PI3K effector signalling in cancer by the phosphoinositide phosphatases. Biosci Rep 2017;37(1) doi 10.1042/BSR20160432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004;304(5670):554 doi 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- 7.Garrido-Castro AC, Lin NU, Polyak K. Insights into Molecular Classifications of Triple-Negative Breast Cancer: Improving Patient Selection for Treatment. Cancer Discov 2019;9(2):176–98 doi 10.1158/2159-8290.CD-18-1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nik-Zainal S, Davies H, Staaf J, Ramakrishna M, Glodzik D, Zou X, et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016;534(7605):47–54 10.1038/nature17676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pereira B, Chin SF, Rueda OM, Vollan HK, Provenzano E, Bardwell HA, et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat Commun 2016;7:11479 doi 10.1038/ncomms11479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Testa U, Castelli G, Pelosi E. Breast Cancer: A Molecularly Heterogenous Disease Needing Subtype-Specific Treatments. Med Sci (Basel) 2020;8(1) doi 10.3390/medsci8010018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Juric D, Castel P, Griffith M, Griffith OL, Won HH, Ellis H, et al. Convergent loss of PTEN leads to clinical resistance to a PI(3)Kalpha inhibitor. Nature 2015;518(7538):240–4 doi 10.1038/nature13948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Westbrook TF, Martin ES, Schlabach MR, Leng Y, Liang AC, Feng B, et al. A genetic screen for candidate tumor suppressors identifies REST. Cell 2005;121(6):837–48 doi S0092–8674(05)00350–8 [pii] 10.1016/j.cell.2005.03.033. [DOI] [PubMed] [Google Scholar]
- 13.Fedele CG, Ooms LM, Ho M, Vieusseux J, O’Toole SA, Millar EK, et al. Inositol polyphosphate 4-phosphatase II regulates PI3K/Akt signaling and is lost in human basal-like breast cancers. Proc Natl Acad Sci U S A 2010;107(51):22231–6 doi 1015245107 [pii] 10.1073/pnas.1015245107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gewinner C, Wang ZC, Richardson A, Teruya-Feldstein J, Etemadmoghadam D, Bowtell D, et al. Evidence that inositol polyphosphate 4-phosphatase type II is a tumor suppressor that inhibits PI3K signaling. Cancer Cell 2009;16(2):115–25 doi S1535–6108(09)00180–9 [pii] 10.1016/j.ccr.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gasser JA, Inuzuka H, Lau AW, Wei W, Beroukhim R, Toker A. SGK3 mediates INPP4B-dependent PI3K signaling in breast cancer. Mol Cell 2014;56(4):595–607 doi S1097–2765(14)00780–1 [pii] 10.1016/j.molcel.2014.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Malik N, Macartney T, Hornberger A, Anderson KE, Tovell H, Prescott AR, et al. Mechanism of activation of SGK3 by growth factors via the Class 1 and Class 3 PI3Ks. Biochem J 2018;475(1):117–35 doi 10.1042/BCJ20170650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ooms LM, Binge LC, Davies EM, Rahman P, Conway JR, Gurung R, et al. The Inositol Polyphosphate 5-Phosphatase PIPP Regulates AKT1-Dependent Breast Cancer Growth and Metastasis. Cancer Cell 2015;28(2):155–69 doi 10.1016/j.ccell.2015.07.003. [DOI] [PubMed] [Google Scholar]
- 18.Asleh-Aburaya K, Sheffield BS, Kos Z, Won JR, Wang XQ, Gao D, et al. Basal biomarkers nestin and INPP4b identify intrinsic subtypes accurately in breast cancers that are weakly positive for oestrogen receptor. Histopathology 2017;70(2):185–94 doi 10.1111/his.13038. [DOI] [PubMed] [Google Scholar]
- 19.Bouchal P, Schubert OT, Faktor J, Capkova L, Imrichova H, Zoufalova K, et al. Breast Cancer Classification Based on Proteotypes Obtained by SWATH Mass Spectrometry. Cell Rep 2019;28(3):832–43 e7 doi 10.1016/j.celrep.2019.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Balla T Phosphoinositides: tiny lipids with giant impact on cell regulation. Physiol Rev 2013;93(3):1019–137 doi 10.1152/physrev.00028.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Di Paolo G, De Camilli P. Phosphoinositides in cell regulation and membrane dynamics. Nature 2006;443(7112):651–7 doi nature05185 [pii] 10.1038/nature05185. [DOI] [PubMed] [Google Scholar]
- 22.Posor Y, Eichhorn-Grunig M, Haucke V. Phosphoinositides in endocytosis. Biochim Biophys Acta 2015;1851(6):794–804 doi 10.1016/j.bbalip.2014.09.014. [DOI] [PubMed] [Google Scholar]
- 23.Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell 2010;141(7):1117–34 doi S0092–8674(10)00665–3 [pii] 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miaczynska M Effects of membrane trafficking on signaling by receptor tyrosine kinases. Cold Spring Harb Perspect Biol 2013;5(11):a009035 doi 10.1101/cshperspect.a009035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Palfy M, Remenyi A, Korcsmaros T. Endosomal crosstalk: meeting points for signaling pathways. Trends Cell Biol 2012;22(9):447–56 doi 10.1016/j.tcb.2012.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Platta HW, Stenmark H. Endocytosis and signaling. Curr Opin Cell Biol 2011;23(4):393–403 doi 10.1016/j.ceb.2011.03.008. [DOI] [PubMed] [Google Scholar]
- 27.Sorkin A, von Zastrow M. Endocytosis and signalling: intertwining molecular networks. Nat Rev Mol Cell Biol 2009;10(9):609–22 doi 10.1038/nrm2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stasyk T, Huber LA. Spatio-Temporal Parameters of Endosomal Signaling in Cancer: Implications for New Treatment Options. J Cell Biochem 2016;117(4):836–43 doi 10.1002/jcb.25418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krause M, Leslie JD, Stewart M, Lafuente EM, Valderrama F, Jagannathan R, et al. Lamellipodin, an Ena/VASP ligand, is implicated in the regulation of lamellipodial dynamics. Dev Cell 2004;7(4):571–83 doi S1534580704003302 [pii] 10.1016/j.devcel.2004.07.024. [DOI] [PubMed] [Google Scholar]
- 30.Dowler S, Currie RA, Campbell DG, Deak M, Kular G, Downes CP, et al. Identification of pleckstrin-homology-domain-containing proteins with novel phosphoinositide-binding specificities. Biochem J 2000;351(Pt 1):19–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Franke TF, Kaplan DR, Cantley LC, Toker A. Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate. Science 1997;275(5300):665–8. [DOI] [PubMed] [Google Scholar]
- 32.Ferguson KM, Kavran JM, Sankaran VG, Fournier E, Isakoff SJ, Skolnik EY, et al. Structural basis for discrimination of 3-phosphoinositides by pleckstrin homology domains. Mol Cell 2000;6(2):373–84 doi 10.1016/s1097-2765(00)00037-x. [DOI] [PubMed] [Google Scholar]
- 33.Chandra M, Chin YK, Mas C, Feathers JR, Paul B, Datta S, et al. Classification of the human phox homology (PX) domains based on their phosphoinositide binding specificities. Nat Commun 2019;10(1):1528 doi 10.1038/s41467-019-09355-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cullen PJ, Korswagen HC. Sorting nexins provide diversity for retromer-dependent trafficking events. Nat Cell Biol 2011;14(1):29–37 doi 10.1038/ncb2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cullen PJ, Steinberg F. To degrade or not to degrade: mechanisms and significance of endocytic recycling. Nat Rev Mol Cell Biol 2018;19(11):679–96 doi 10.1038/s41580-018-0053-7. [DOI] [PubMed] [Google Scholar]
- 36.Hawkins PT, Stephens LR. Emerging evidence of signalling roles for PI(3,4)P2 in Class I and II PI3K-regulated pathways. Biochem Soc Trans 2016;44(1):307–14 doi 10.1042/BST20150248. [DOI] [PubMed] [Google Scholar]
- 37.Li H, Marshall AJ. Phosphatidylinositol (3,4) bisphosphate-specific phosphatases and effector proteins: A distinct branch of PI3K signaling. Cell Signal 2015;27(9):1789–98 doi 10.1016/j.cellsig.2015.05.013. [DOI] [PubMed] [Google Scholar]
- 38.Ebner M, Koch PA, Haucke V. Phosphoinositides in the control of lysosome function and homeostasis. Biochem Soc Trans 2019;47(4):1173–85 doi 10.1042/BST20190158. [DOI] [PubMed] [Google Scholar]
- 39.Marat AL, Wallroth A, Lo WT, Muller R, Norata GD, Falasca M, et al. mTORC1 activity repression by late endosomal phosphatidylinositol 3,4-bisphosphate. Science 2017;356(6341):968–72 doi 10.1126/science.aaf8310. [DOI] [PubMed] [Google Scholar]
- 40.Kofuji S, Kimura H, Nakanishi H, Nanjo H, Takasuga S, Liu H, et al. INPP4B Is a PtdIns(3,4,5)P3 Phosphatase That Can Act as a Tumor Suppressor. Cancer Discov 2015;5(7):730–9 doi 2159–8290.CD-14–1329 [pii] 10.1158/2159-8290.CD-14-1329. [DOI] [PubMed] [Google Scholar]
- 41.Liu X, Holstege H, van der Gulden H, Treur-Mulder M, Zevenhoven J, Velds A, et al. Somatic loss of BRCA1 and p53 in mice induces mammary tumors with features of human BRCA1-mutated basal-like breast cancer. Proc Natl Acad Sci U S A 2007;104(29):12111–6 doi 0702969104 [pii] 10.1073/pnas.0702969104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu H, Murphy CJ, Karreth FA, Emdal KB, White FM, Elemento O, et al. Identifying and Targeting Sporadic Oncogenic Genetic Aberrations in Mouse Models of Triple-Negative Breast Cancer. Cancer Discov 2018;8(3):354–69 doi 10.1158/2159-8290.CD-17-0679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li Chew C, Lunardi A, Gulluni F, Ruan DT, Chen M, Salmena L, et al. In Vivo Role of INPP4B in Tumor and Metastasis Suppression through Regulation of PI3K-AKT Signaling at Endosomes. Cancer Discov 2015;5(7):740–51 doi 2159–8290.CD-14–1347 [pii] 10.1158/2159-8290.CD-14-1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ip LR, Poulogiannis G, Viciano FC, Sasaki J, Kofuji S, Spanswick VJ, et al. Loss of INPP4B causes a DNA repair defect through loss of BRCA1, ATM and ATR and can be targeted with PARP inhibitor treatment. Oncotarget 2015;6(12):10548–62 doi 10.18632/oncotarget.3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rottenberg S, Jaspers JE, Kersbergen A, van der Burg E, Nygren AO, Zander SA, et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci U S A 2008;105(44):17079–84 doi 10.1073/pnas.0806092105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Braccini L, Ciraolo E, Campa CC, Perino A, Longo DL, Tibolla G, et al. PI3K-C2gamma is a Rab5 effector selectively controlling endosomal Akt2 activation downstream of insulin signalling. Nat Commun 2015;6:7400 doi 10.1038/ncomms8400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Posor Y, Eichhorn-Gruenig M, Puchkov D, Schoneberg J, Ullrich A, Lampe A, et al. Spatiotemporal control of endocytosis by phosphatidylinositol-3,4-bisphosphate. Nature 2013;499(7457):233–7 doi 10.1038/nature12360. [DOI] [PubMed] [Google Scholar]
- 48.Masuda H, Zhang D, Bartholomeusz C, Doihara H, Hortobagyi GN, Ueno NT. Role of epidermal growth factor receptor in breast cancer. Breast Cancer Res Treat 2012;136(2):331–45 doi 10.1007/s10549-012-2289-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest 2011;121(7):2750–67 doi 45014 [pii] 10.1172/JCI45014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ciriello G, Gatza ML, Beck AH, Wilkerson MD, Rhie SK, Pastore A, et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 2015;163(2):506–19 doi S0092–8674(15)01195–2 [pii] 10.1016/j.cell.2015.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012;486(7403):346–52 doi nature10983 [pii] 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 2012;486(7403):395–9 doi nature10933 [pii] 10.1038/nature10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Andre F, Ciruelos E, Rubovszky G, Campone M, Loibl S, Rugo HS, et al. Alpelisib for PIK3CA-Mutated, Hormone Receptor-Positive Advanced Breast Cancer. N Engl J Med 2019;380(20):1929–40 doi 10.1056/NEJMoa1813904. [DOI] [PubMed] [Google Scholar]
- 54.Reed DE, Shokat KM. INPP4B and PTEN Loss Leads to PI-3,4-P2 Accumulation and Inhibition of PI3K in TNBC. Mol Cancer Res 2017;15(6):765–75 doi 10.1158/1541–7786.MCR-16–0183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jansen VM, Mayer IA, Arteaga CL. Is There a Future for AKT Inhibitors in the Treatment of Cancer? Clin Cancer Res 2016;22(11):2599–601 doi 10.1158/1078-0432.CCR-16-0100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Turner NC, Alarcon E, Armstrong AC, Philco M, Lopez Chuken YA, Sablin MP, et al. BEECH: a dose-finding run-in followed by a randomised phase II study assessing the efficacy of AKT inhibitor capivasertib (AZD5363) combined with paclitaxel in patients with estrogen receptor-positive advanced or metastatic breast cancer, and in a PIK3CA mutant sub-population. Ann Oncol 2019;30(5):774–80 doi 10.1093/annonc/mdz086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Oliveira M, Saura C, Nuciforo P, Calvo I, Andersen J, Passos-Coelho JL, et al. FAIRLANE, a double-blind placebo-controlled randomized phase II trial of neoadjuvant ipatasertib plus paclitaxel for early triple-negative breast cancer. Ann Oncol 2019;30(8):1289–97 doi 10.1093/annonc/mdz177. [DOI] [PubMed] [Google Scholar]
- 58.Nakai K, Hung MC, Yamaguchi H. A perspective on anti-EGFR therapies targeting triple-negative breast cancer. Am J Cancer Res 2016;6(8):1609–23. [PMC free article] [PubMed] [Google Scholar]
- 59.Levva S, Kotoula V, Kostopoulos I, Manousou K, Papadimitriou C, Papadopoulou K, et al. Prognostic Evaluation of Epidermal Growth Factor Receptor (EGFR) Genotype and Phenotype Parameters in Triple-negative Breast Cancers. Cancer Genomics Proteomics 2017;14(3):181–95 doi 10.21873/cgp.20030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vivanco I, Rohle D, Versele M, Iwanami A, Kuga D, Oldrini B, et al. The phosphatase and tensin homolog regulates epidermal growth factor receptor (EGFR) inhibitor response by targeting EGFR for degradation. Proc Natl Acad Sci U S A 2010;107(14):6459–64 doi 10.1073/pnas.0911188107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Burga LN, Hu H, Juvekar A, Tung NM, Troyan SL, Hofstatter EW, et al. Loss of BRCA1 leads to an increase in epidermal growth factor receptor expression in mammary epithelial cells, and epidermal growth factor receptor inhibition prevents estrogen receptor-negative cancers in BRCA1-mutant mice. Breast Cancer Res 2011;13(2):R30 doi 10.1186/bcr2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang J, Antonyak MA, Singh G, Cerione RA. A mechanism for the upregulation of EGF receptor levels in glioblastomas. Cell Rep 2013;3(6):2008–20 doi 10.1016/j.celrep.2013.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cowherd S, Miller LD, Melin SA, Akman S, Isom S, Cole J, et al. A phase II clinical trial of weekly paclitaxel and carboplatin in combination with panitumumab in metastatic triple negative breast cancer. Cancer Biol Ther 2015;16(5):678–83 doi 10.1080/15384047.2015.1026481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Damaskos C, Garmpi A, Nikolettos K, Vavourakis M, Diamantis E, Patsouras A, et al. Triple-Negative Breast Cancer: The Progress of Targeted Therapies and Future Tendencies. Anticancer Res 2019;39(10):5285–96 doi 10.21873/anticanres.13722. [DOI] [PubMed] [Google Scholar]
- 65.Gelmon K, Dent R, Mackey JR, Laing K, McLeod D, Verma S. Targeting triple-negative breast cancer: optimising therapeutic outcomes. Ann Oncol 2012;23(9):2223–34 doi 10.1093/annonc/mds067. [DOI] [PubMed] [Google Scholar]
- 66.Yardley DA, Ward PJ, Daniel BR, Eakle JF, Lamar RE, Lane CM, et al. Panitumumab, Gemcitabine, and Carboplatin as Treatment for Women With Metastatic Triple-Negative Breast Cancer: A Sarah Cannon Research Institute Phase II Trial. Clin Breast Cancer 2016;16(5):349–55 doi 10.1016/j.clbc.2016.05.006. [DOI] [PubMed] [Google Scholar]
- 67.Madshus IH, Stang E. Internalization and intracellular sorting of the EGF receptor: a model for understanding the mechanisms of receptor trafficking. J Cell Sci 2009;122(Pt 19):3433–9 doi 10.1242/jcs.050260. [DOI] [PubMed] [Google Scholar]
- 68.Bakker J, Spits M, Neefjes J, Berlin I. The EGFR odyssey - from activation to destruction in space and time. J Cell Sci 2017;130(24):4087–96 doi 10.1242/jcs.209197. [DOI] [PubMed] [Google Scholar]
- 69.Bergeron JJ, Di Guglielmo GM, Dahan S, Dominguez M, Posner BI. Spatial and Temporal Regulation of Receptor Tyrosine Kinase Activation and Intracellular Signal Transduction. Annu Rev Biochem 2016;85:573–97 doi 10.1146/annurev-biochem-060815-014659. [DOI] [PubMed] [Google Scholar]
- 70.Raiborg C, Stenmark H. The ESCRT machinery in endosomal sorting of ubiquitylated membrane proteins. Nature 2009;458(7237):445–52 doi 10.1038/nature07961. [DOI] [PubMed] [Google Scholar]
- 71.Prasad NK, Decker SJ. SH2-containing 5’-inositol phosphatase, SHIP2, regulates cytoskeleton organization and ligand-dependent down-regulation of the epidermal growth factor receptor. J Biol Chem 2005;280(13):13129–36 doi 10.1074/jbc.M410289200. [DOI] [PubMed] [Google Scholar]
- 72.Ivetac I, Munday AD, Kisseleva MV, Zhang XM, Luff S, Tiganis T, et al. The type Ialpha inositol polyphosphate 4-phosphatase generates and terminates phosphoinositide 3-kinase signals on endosomes and the plasma membrane. Mol Biol Cell 2005;16(5):2218–33 doi E04–09-0799 [pii] 10.1091/mbc.E04-09-0799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang H, Loerke D, Bruns C, Muller R, Koch PA, Puchkov D, et al. Phosphatidylinositol 3,4-bisphosphate synthesis and turnover are spatially segregated in the endocytic pathway. J Biol Chem 2020;295(4):1091–104 doi 10.1074/jbc.RA119.011774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liu SL, Wang ZG, Hu Y, Xin Y, Singaram I, Gorai S, et al. Quantitative Lipid Imaging Reveals a New Signaling Function of Phosphatidylinositol-3,4-Bisphophate: Isoform- and Site-Specific Activation of Akt. Mol Cell 2018;71(6):1092–104 e5 doi 10.1016/j.molcel.2018.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Henmi Y, Morikawa Y, Oe N, Ikeda N, Fujita A, Takei K, et al. PtdIns4KIIalpha generates endosomal PtdIns(4)P and is required for receptor sorting at early endosomes. Mol Biol Cell 2016;27(6):990–1001 doi 10.1091/mbc.E15-08-0564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ketel K, Krauss M, Nicot AS, Puchkov D, Wieffer M, Muller R, et al. A phosphoinositide conversion mechanism for exit from endosomes. Nature 2016;529(7586):408–12 doi 10.1038/nature16516. [DOI] [PubMed] [Google Scholar]
- 77.Haberg K, Lundmark R, Carlsson SR. SNX18 is an SNX9 paralog that acts as a membrane tubulator in AP-1-positive endosomal trafficking. J Cell Sci 2008;121(Pt 9):1495–505 doi 10.1242/jcs.028530. [DOI] [PubMed] [Google Scholar]
- 78.Knaevelsrud H, Soreng K, Raiborg C, Haberg K, Rasmuson F, Brech A, et al. Membrane remodeling by the PX-BAR protein SNX18 promotes autophagosome formation. J Cell Biol 2013;202(2):331–49 doi 10.1083/jcb.201205129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Park J, Kim Y, Lee S, Park JJ, Park ZY, Sun W, et al. SNX18 shares a redundant role with SNX9 and modulates endocytic trafficking at the plasma membrane. J Cell Sci 2010;123(Pt 10):1742–50 doi 10.1242/jcs.064170. [DOI] [PubMed] [Google Scholar]
- 80.Wang H, Lo WT, Haucke V. Phosphoinositide switches in endocytosis and in the endolysosomal system. Curr Opin Cell Biol 2019;59:50–7 doi 10.1016/j.ceb.2019.03.011. [DOI] [PubMed] [Google Scholar]
- 81.Domin J, Gaidarov I, Smith ME, Keen JH, Waterfield MD. The class II phosphoinositide 3-kinase PI3K-C2alpha is concentrated in the trans-Golgi network and present in clathrin-coated vesicles. J Biol Chem 2000;275(16):11943–50 doi 10.1074/jbc.275.16.11943. [DOI] [PubMed] [Google Scholar]
- 82.Goulden BD, Pacheco J, Dull A, Zewe JP, Deiters A, Hammond GRV. A high-avidity biosensor reveals plasma membrane PI(3,4)P2 is predominantly a class I PI3K signaling product. J Cell Biol 2019;218(3):1066–79 doi 10.1083/jcb.201809026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gulluni F, De Santis MC, Margaria JP, Martini M, Hirsch E. Class II PI3K Functions in Cell Biology and Disease. Trends Cell Biol 2019;29(4):339–59 doi 10.1016/j.tcb.2019.01.001. [DOI] [PubMed] [Google Scholar]
- 84.Guo ST, Chi MN, Yang RH, Guo XY, Zan LK, Wang CY, et al. INPP4B is an oncogenic regulator in human colon cancer. Oncogene 2016;35(23):3049–61 doi 10.1038/onc.2015.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.