

Abstract
Background:
In hemophilia bypass therapy, a platelet-dependent mechanism is believed to be primarily responsible for recombinant factor VIIa (rFVIIa)’s hemostatic effect. rFVIIa may also possibly interact with other cells through its binding to endothelial cell protein C receptor (EPCR) or cell surface phospholipids.
Objectives:
We aim to investigate the relative contribution of platelet-dependent and platelet-independent mechanisms in rFVIIa-mediated thrombin generation in hemophilic conditions at the injury site.
Methods:
Platelets were depleted in acquired and genetic hemophilia mice using anti-platelet antibodies. The mice were subjected to the saphenous vein injury, and the hemostatic effect of pharmacological concentrations of rFVIIa was evaluated by measuring thrombin generation at the injury site.
Results:
Administration of anti-mouse CD42 antibodies to mice depleted platelets by more than 95%. As expected, hemophilia mice, compared to wild-type mice, generated only a small fraction of thrombin at the injury site. The depletion of platelets in hemophilia mice further reduced thrombin generation. However, when pharmacological doses of rFVIIa were administered to hemophilia mice, substantial amounts of thrombin were generated even in the platelet-depleted hemophilia mice. No differences in thrombin generation were detected among FVIII−/−, EPCR-deficient FVIII−/−, and EPCR-overexpressing FVIII−/− mice depleted of platelets or not. Evaluation of platelets by flow cytometry as well as immunoblot analysis showed no detectable expression of EPCR.
Conclusions:
Our data suggest that pharmacological concentrations of rFVIIa generate thrombin in hemophilia in both platelet-dependent and platelet-independent mechanisms.
Keywords: Endothelial cell protein C receptor, factor VIIa, hemophilia, hemostasis, platelets, thrombin generation
1 ∣. INTRODUCTION
The recombinant factor VIIa (rFVIIa) is approved by the US Food and Drug Administration (FDA) as a bypassing therapeutic agent for the treatment of bleeding episodes in hemophilia patients with inhibitors and congenital factor VII-deficient patients. However, it is increasingly being used to treat other congenital and acquired bleeding disorders as well as an off-label emergency hemostatic agent in both pediatric and adult patients in trauma, surgery, and other indications [1, 2]. The supraphysiological doses of rFVIIa required for effective bleeding control not only increase thrombotic risk but is also cost ineffective [3-5]. Despite its successful use, the mechanism by which high doses of rFVIIa provide the hemostatic effect is not entirely clear [1, 4].
A few in vitro studies and mathematical modeling suggest a tissue factor (TF)-dependent mechanism [6-8]. In contrast, others suggest a phospholipid-dependent and TF-independent mechanism is responsible for the hemostatic effect of high doses of rFVIIa [9, 10]. Our earlier in vitro work with hemophilia patient’s plasma and recent in vivo studies utilizing murine hemophilia model suggest that pharmacological doses of rFVIIa restore hemostasis primarily in a TF-independent manner [11, 12]. In this mechanism, rFVIIa binds to anionic phospholipids exposed on activated platelets at the injury site and directly activates FX to FXa. FXa generated on platelets leads to prothrombinase assembly, and thrombin burst sufficient to achieve hemostasis [13].
However, the effective use of rFVIIa in treating bleeding episodes in patients with congenital and acquired platelet disorders such as thrombocytopenia [14, 15], uremia [16, 17], and severe inherited platelet dysfunctions such as Glanzmann thrombasthenia (GT) and Bernard-Soulier syndrome (BSS) pose a challenge to this hypothesis [18, 19]. A most plausible explanation for rFVIIa hemostatic effect under the above conditions is that rFVIIa potentiates activation of few platelets that are localized at the site of injury, thus resulting in efficient thrombin generation leading to increased fibrin deposition at the site of injury [20]. This hypothesis draws support from both in vitro static model utilizing washed platelets from subjects with GT and of normal platelets pre-treated to induce GT with inhibitors [21] and from ex vivo plasma-free flow models where recombinant FVIIa was shown to potentiate platelet activation [22, 23].
In conflict with the above hypothesis, an ex vivo flow model using whole blood and high concentrations of rFVIIa showed an increased fibrin deposition at the site of injury, but low platelet adhesion [24]. These experiments show that platelets isolated from patients with GT interacted with a more superficial layer of fibrin deposit [24]. A recent study by Ivanciu et al. suggests that the activated endothelium plays an unexpectedly important and major role in supporting prothrombinase assembly and thrombin generation at the site of injury [25]. Inhibition of platelet adhesion had no significant effect on fibrin formation at the site of injury. The above findings suggest the presence of platelet-independent mechanisms for thrombin generation at the site injury.
Our recent discovery that showed FVIIa also binds endothelial protein C receptor (EPCR) adds a new dimension to the potential mechanisms of rFVIIa action [26]. Our studies suggest that downregulation EPCR-mediated protein C activation by high doses of rFVIIa contributes to the hemostatic effect of rFVIIa in the treatment of hemophilia [27]. Recently, it has been reported that platelets express EPCR, and suggested that FVIIa binding to EPCR on platelets could synergistically contribute to the pharmacologic effect of high doses of rFVIIa [28]. The present study was conducted to evaluate the contribution of platelet-, EPCR-, and platelet-independent mechanisms, if any, to the hemostatic effect of rFVIIa in hemophilia treatment using murine hemophilia models.
2 ∣. MATERIALS AND METHODS
2.1 ∣. Reagents
Recombinant human FVIIa was provided the Late Walter Kisiel, University of New Mexico, Albuquerque (NM). rFVIIa was expressed and purified, as described earlier [29]. rFVIIa used in the present study was essentially similar to that of a non-clinical version of commercially available rFVIIa (NovoSeven). FVIIa protein concentration was determined from the absorbance of protein at 280 nm and using the extinction coefficient (E1%280) of FVIIa as 13.9 [30]. The generation of monoclonal (mAb) and polyclonal antibodies against mouse and human EPCR were described previously [31, 32]. Anti-human FVIII mAb that cross-reacts with murine FVIII and inhibits murine FVIII activity (GMA 8015) was obtained from Green Mountain Antibodies (Burlington, VT, USA). Rat anti-mouse GPIIb (CD41; clone MWReg30) was obtained from e-bioscience (San Diego, CA, USA). Rat monoclonal antibodies directed against mouse GPIbα(CD42b) was from Emfret Analytics (Eibelstadt, Germany). Rat isotype control IgG (IgG1k) and fluorophore-conjugated secondary antibodies were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Mouse TAT ELISA kit was from Assaypro (St Charles, MO, USA). The source of other reagents were: prostaglandin E1 from EMD Millipore-Sigma (St. Louis, MO, USA), apyrase from New England Biolabs Inc, (Boston, MA, USA), human alpha-thrombin from Enzyme Research Laboratories (South Bend, IN, USA), collagen type I from MP Biomedicals LLC (Irvine, CA, USA).
2.2 ∣. Mice
Wild-type mice (C57BL/6J) were obtained from Jackson Laboratories (Bar Harbor, ME) or bred in-house. Generation of hemophilia A (FVIII−/−) mice in C57BL/6J genetic background with normal EPCR expression levels and overexpression of EPCR (Tie2-EPCR/FVIII−/−) were described earlier [27]. The generation of EPCR deficient mice was described earlier [33]. To generate EPCR-deficient FVIII−/− mice, first Procrflox/flox/FVIII−/− and Procr+/floxMeox2+/cre/FVIII−/− mice were generated by back-crossing Procrflox/flox and Procr+/floxMeox2+/cre mice with FVIII−/− mice. EPCR-deficient FVIII−/− mice (Procr−/−/FVIII−/−) were generated by breeding female Procrflox/flox/FVIII−/− mice with male Procr+/floxMeox2+/cre/FVIII−/− mice. Ten to 14-week old mice, both sexes weighing between 24 to 30 g, were used in our experiments. All animal studies were reviewed and approved by the Institutional Animal Care and Use Committee and conducted according to the animal welfare guidelines outlined in the Guide for the Care and Use of Laboratory Animals.
2.3 ∣. Platelet depletion
Blood (~50 μl) was collected from mice a day before platelet depletion via submandibular vein puncture into EDTA (2.7 mM) anticoagulant. Platelet depletion antibodies (rat anti-CD41 or rat anti-CD42b) or control IgG (1 mg/kg) was administered to mice intravenously via the tail vein. Following the antibody administration, at varying times or a fixed interval, blood (~50 ul) was collected via submandibular vein puncture into EDTA anticoagulant. Blood samples were processed for platelet count using the HEMAVET-HV950FS analyzer (Drew Scientific, Inc., CT). Percent of platelet depletion was calculated by comparing the platelet counts before and after the administration of the antibodies.
2. 4 ∣. Saphenous vein bleeding
Mice were injected with a control IgG or anti-CD42b antibody via the tail vein 4 h prior to the saphenous vein bleeding. In the antibody-induced hemophilia model, anti-FVIII mAb (1 mg/kg) was injected along with control or the platelet-depleting antibody. After 4 h, rFVIIa (1 or 4 mg/kg) was given to mice via the tail vein in 100 μl volume. Five min following FVIIa administration, mice were subjected to the saphenous vein incision as described earlier [12] with a few modifications to fit with the objective of the current study. Briefly, the saphenous vein was punctured with a 23-G needle. After initial hemostasis, an approximately 1 mm longitudinal distal cut was made using a Student Vannas spring scissors by inserting one blade into the vessel using the needle hole as the entry point. Blood coming from the injury site was collected by a capillary action using a pipette tip and added to a microcentrifuge tube containing citrate anticoagulant. After each hemostasis incident, bleeding was reinitiated by disrupting the clot using a blunt 30G needle, and blood was collected throughout the bleeds. Since platelet depleted mice exhibited a high incidence of death after 15 min of the initiation of injury due to profuse bleeding, the collection of blood was limited to 15 min. Plasma was prepared by centrifuging the blood at 2, 000 x g for 10 min at 4°C, and immediately frozen on a dry ice bath to prevent further thrombin generation ex vivo and stored at −80°C until TAT levels were assayed.
2.5 ∣. Measurement of TAT levels.
Plasma isolated from blood collected at the injury site, via the sub-mandibular vein, or cardiac puncture was analyzed for TAT levels using AssayMax mouse ELISA kit (AssayPro, St. Charles, MO).
2.6 ∣. Isolation of human and murine platelets
Human venous blood was drawn from healthy volunteers after their consent. The human subject protocol was approved by the Institute Review Board at the University of Texas Health Science Center at Tyler, TX. The blood drawn was immediately mixed with an acid citrate-dextrose anticoagulant solution (1:6), and platelet-rich plasma (PRP) was obtained by centrifuging the blood at 200 x g for 20 min at room temperature. Similarly, blood was collected from 10-12 weeks old mice via sub-mandibular vein puncture into a one-sixth volume of the acid citrate-dextrose anticoagulant, and PRP was obtained by centrifugation as described above. PRP from 8 to 10 mice was pooled. Prostaglandin E1 was added to PRP (both human and murine) at 1 μM final concentration to prevent platelet activation. Platelets were pelleted by centrifuging the PRP at 900 x g for 10 min at room temperature. The platelet pellet was re-suspended in platelet resuspension buffer (Tyrode’s buffer with 12 mM NaHCO3, 5 mM dextrose, 2 mM MgCl2, and 0.3% BSA) containing 0.1 U/mL of apyrase. After resting the platelets for 30 min at room temperature, they were sedimented by centrifugation at 900 x g for 10 min at room temperature and resuspended in the resuspension buffer. Platelet numbers were counted using the HEMAVET-HV950FS analyzer, and the numbers were adjusted to 2x108 cells/ml before treating them with either collagen 1 (10 μg/ml) or thrombin (10 nM) or both for 30 min. The activated platelets were analyzed by flow cytometry or immune blot analysis with appropriate controls.
2.7 ∣. Flowcytometry
One mL of unactivated and activated platelets (both human as well as murine) were fixed with 1% paraformaldehyde for 30 min on ice. Specific primary antibodies against EPCR or non-specific isotype control IgG were added to the fixed platelets following blocking with 5% BSA in PBS for one hour. For analyzing human EPCR, goat anti-human EPCR polyclonal antibody or mouse monoclonal (JRK1500) antibody was used. For murine EPCR, the goat anti-mouse EPCR polyclonal antibody was used. Following the primary antibody incubation, Alexa Flour 488-tagged donkey anti-goat antibody or donkey anti-mouse antibody was used to detect the primary antibody. The labeled platelets were analyzed on the BD-FACS Caliber flow cytometer for EPCR expression. HUVEC and bEnd.3 cells were used as positive controls for human and mouse EPCR expression, respectively.
2.8 ∣. Immunoblot analysis
Two mL of unactivated and activated platelet suspension were pelleted down by centrifuging at 1,250 x g for 20 min at room temperature, supernatants were discarded, and the pellets were lysed in 1x SDS loading buffer. The samples were subjected to SDS-PAGE, followed by immunoblot analysis.
2.9 ∣. Data collection and statistical analysis
Three to 9 animals were used in each experimental group. The values of each parameter within a group were expressed as the mean ± SEM. Statistical significance between the two groups was assessed using the Mann-Whitney test. One-way analysis of variance (ANOVA) was used to determine statistically significant differences between more than two groups. Where ANOVA was used, Tukey’s post hoc multiple comparison test was used to obtain statistical significance between two treatment groups. GraphPad Prism version 8 was used for the statistical analysis and preparation of graphs.
3 ∣. RESULTS
3.1 ∣. Efficacy of platelet depletion in a murine model of hemophilia A
We tested two different platelet depleting antibodies - rat anti-mouse integrin alpha IIb (anti-mouse CD41) and rat anti-mouse GPIb (anti-mouse CD42b) - for their efficacy to deplete platelets. Intravenous administration of anti-mouse CD41 antibodies (1 mg/kg) to wild-type mice depleted 40-50% of platelets at 2 h following the antibody administration, and the platelet depletion reached about 80-85% at 24 h following the antibody administration (Figure 1A). Administration of anti-mouse CD42b antibodies (1 mg/kg) depleted 90-95% of platelets within 2 to 4 h after the intravenous administration of the antibody in both wild-type and FVIII−/− mice (Figure 1B). Injection of a similar concentration of control rat IgG did not result in any significant decrease in platelet number. Since rat anti-mouse CD42b antibodies were more effective than rat anti-mouse CD41 antibodies in depleting platelets, we used CD42b antibodies to deplete platelets in further experiments.
FIGURE 1.
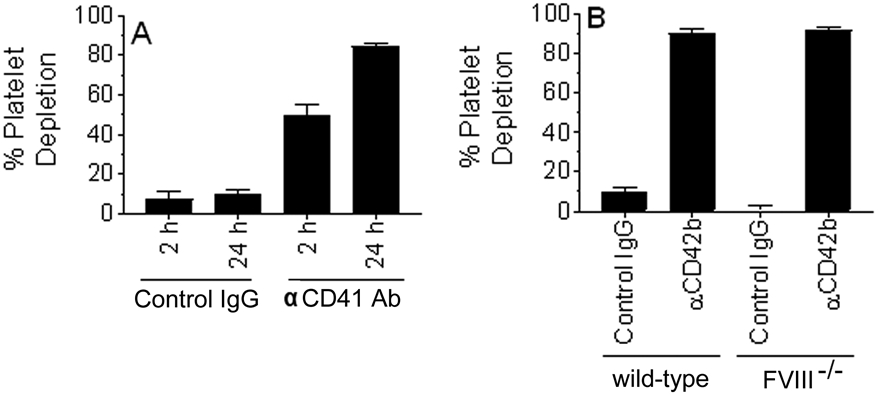
Depletion of platelets in wild-type and hemophilia A mice. (A) Wild-type mice were administered with control IgG or rat anti-murine CD41 antibody (1 mg/kg) intravenously via the tail-vein. A small aliquot of blood was drawn into EDTA anticoagulant at 2 and 24 h following the antibody administration. Platelets were counted using Hemavet. Percent of platelet depletion was calculated by comparing the platelet count after the antibody administration with the platelet count before the administration of the antibodies. (B) Wild-type or FVIII−/− mice were injected with control IgG or rat anti-murine CD42b antibodies. Blood was drawn 4 h after the antibody administration, and percent platelet depletion was calculated as described above (n = 4 to 10 mice/group).
3.2 ∣. Thrombin is generated in a localized manner at the injury site
Since platelets are essential for clot formation, the hemostatic effect of rFVIIa in platelet-depleted mice was assessed by measuring the amount of thrombin generated in the blood. TAT complex levels in the plasma were used as the indicator of the amount of thrombin generated in the blood. In initial experiments, wild-type mice were subjected to the saphenous vein injury, and the blood was collected directly from the injury site into citrate anticoagulant. Blood was also collected from the same mice via a submandibular vein or cardiac puncture at 15 min post-injury. Thrombin generated in the blood collected from different sites flowing the injury was compared with that of pre-injury collected blood. As shown in Figure 2, no significant increase in TAT levels was found in the plasma isolated from the blood collected from the submandibular vein or cardiac puncture, 8.03 ± 2.84 and 9.77 ± 4.82 ng/ml, respectively, compared to TAT levels in the plasma obtained from the blood collected before the injury (8.08 ±1.16 ng/ml).
FIGURE 2.
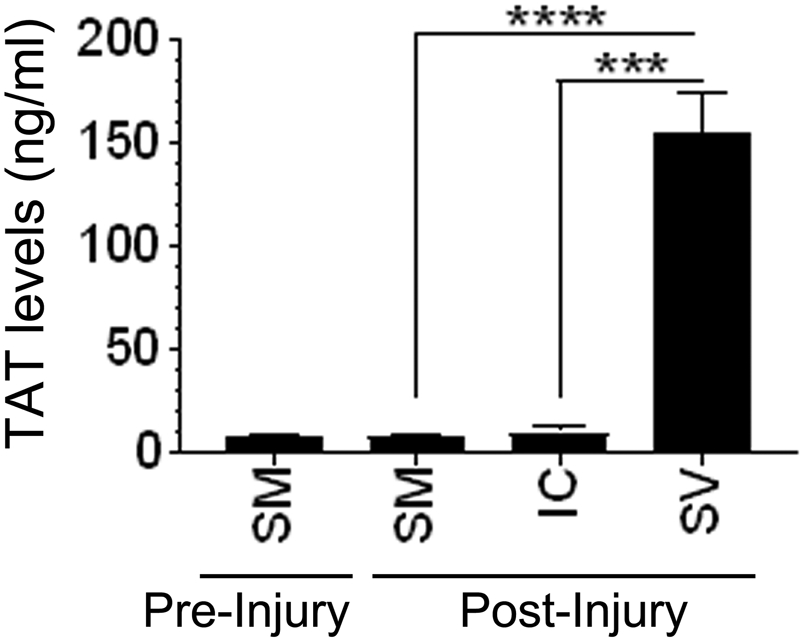
TAT levels in blood collected from different sites following the saphenous vein injury. Bleeding in wild-type mice was induced by the saphenous vein incision. Following the injury, blood from the injury site was collected directly into the citrate anticoagulant for 15 min (SV). At the end of 15 min, blood was also drawn into citrate anticoagulant via the sub-mandibular vein (SM) or intracardiac (IC) puncture. A small aliquot of blood (50 μl or less) was drawn from mice a day before the injury via the sub-mandibular vein (SM) puncture to serve as pre-injury control. TAT levels in the plasma were measured in ELISA. ***, p<0.001; ****, p<0.0001 (n = 3 to 7 mice/group).
In contrast, TAT levels were markedly higher in the plasma collected at the injury site (154 ± 35.2 ng/ml). We also found that the rate of TAT generation at the injury site was constant during the 30 min-bleeding period as we measured similar TAT levels in the blood collected over the different time courses (0-10 min; 10-20 min; and 20-30 min, data not shown). These results clearly indicate that injury was required for thrombin generation and that thrombin generation was limited strictly to the injury site and did not diffuse. These data suggest that the measurement of TAT levels in the systemic circulation does not reveal the extent of thrombin generated at the injury site.
3.3 ∣. Pharmacological doses of rFVIIa generate thrombin in platelet-dependent and platelet-independent mechanisms in the antibody-induced murine hemophilia models
Next, to investigate the contribution of platelet-dependent and -independent mechanisms to the restoration of hemostasis by pharmacological concentrations of rFVIIa in hemophilia with inhibitors, wild-type mice were either administered with anti-FVIII, anti-CD42b, or both. Then, rFVIIa was administered to mice, and mice were subjected to the saphenous vein injury. TAT levels in the blood collected at the injury site were measured. As reported earlier [27], the administration of anti-FVIII antibodies neutralized factor FVIII activity by about 95%. As shown in Figure 1, the administration of anti-CD42b antibodies depleted about 95% of platelets in the circulation. Wild-type mice that did not receive either anti-FVIII or anti-CD42b antibodies generated the highest TAT complex levels in the blood collected at the injury site (249.7 ± 40.65 ng/ml). Administration of isotype control IgG had no significant effect on thrombin generation (216 ± 30.8 ng/ml). Wild-type mice that received anti-CD42b antibodies to deplete platelets showed a marked decrease in TAT levels (50.9 ± 17.62 ng/ml) following the injury. Administration of FVIII antibodies also markedly decreased the generation of TAT (17.2 ± 4.09 ng/ml) at the injury site. The depletion of platelets in FVIII antibodies-administered mice further reduced the TAT levels to the lowest, 4.7 ± 0.58 ng/ml. These mice bled profusely, compared to mice administered with either anti-FVIII or anti-CD42b antibodies, and died within 15 to 20 min following the saphenous vein injury. Administration of rFVIIa (1 or 4 mg/kg) partially restored thrombin generation in all groups of mice to varying extents (Figure 3). For example, administration of 4 mg/kg rFVIIa to mice administered with anti-FVIII antibodies increased TAT levels 133.8 ± 26.06 ng/ml, approximately 60% of the TAT levels observed in wild-type mice. More importantly, the administration of rFVIIa (4 mg/kg) also increased TAT levels significantly in mice administered with FVIII antibodies and depleted of platelets (58.7 ± 5.39 ng/ml). These TAT levels constitute about 45% of the TAT levels observed in rFVIIa-treated acquired hemophilia A mice, in which platelets were not depleted. These data clearly illustrate that pharmacological concentrations of rFVIIa can generate significant amounts of thrombin in a platelet-independent mechanism in hemophilia with inhibitors.
FIGURE 3.

Pharmacological doses of rFVIIa generate thrombin in platelet-dependent and platelet-independent mechanisms in the antibody-induced murine hemophilia model. Wild-type mice were injected with anti-FVIII and/or platelet-depleting anti-CD42b antibodies or control IgG (1 mg/kg). After 4 h, rFVIIa was administered to mice at two different doses (1 mg/kg or 4 mg/kg) intravenously. Immediately, mice were subjected to the saphenous vein injury, and blood was collected from the injury site. TAT levels in the plasma were measured in ELISA. **, p<0.01 ***, p<0.001 (n = 6 to 9 mice/group).
3.4 ∣. FVIIa-EPCR interaction does not directly contribute to thrombin generation at the injury site
The relevance of FVIIa-EPCR interactions in hemostasis in the context of FVIIa therapy in disease is not completely understood. It has been suggested that FVIIa binding to EPCR on the vascular endothelium [34] or platelets [28] could enhance FVIIa activation of factor X, which could lead to enhanced thrombin generation in hemophilia patients treated with rFVIIa. To investigate the role of EPCR in platelet-dependent and platelet-independent rFVIIa-induced thrombin generation in hemophilia, FVIII−/−, Tie2-EPCR/FVIII−/−, and EPCR−/−/FVIII−/− mice were administered with anti-CD42b antibodies to deplete platelets or control IgG. These mice were administered with rFVIIa (4 mg/kg), subjected to the saphenous vein injury, and thrombin generation at the injury site was assessed by measuring TAT levels in the blood collected at the injury site. As shown in Figure 4, in the absence of any treatment, very low levels of TAT were generated in all three genotypes at injury site following the saphenous vein incision (FVIII−/−, 18.8 ± 4.8 ng/ml; EPCR−/−/FVIII−/−, 16.7 ± 2.6 ng/ml; and Tie2-EPCR/FVIII−/−, 17.5 ± 4.1 ng/ml). Administration of control isotype IgG had no significant effect on TAT generation in these mice. The depletion of platelets with CD42b antibodies further reduced TAT levels, but the reduction is not statistically significant compared to hemophilia mice administered with saline or control IgG (Figure 4). Administration of rFVIIa to FVIII−/−, Tie2-EPCR/FVIII−/−, or EPCR−/−/FVIII−/− mice that were administered with control IgG markedly increased TAT levels (FVIII−/−, 202 ± 24.0 ng/ml; EPCR−/−/FVIII−/−, 208 ± 32.8 ng/ml; and Tie2-EPCR/FVIII−/−, 129 ± 26.1 ng/ml). The differences in TAT levels among the genotypes were not statistically significant. Administration of rFVIIa to platelet-depleted FVIII−/−, Tie2-EPCR/FVIII−/−, and EPCR−/−/FVIII−/− mice also led to a significant increase in TAT levels, although to a lower extent compared to the corresponding genotype administered with control IgG (FVIII−/−, 54 ± 9.8 ng/ml; EPCR−/−/FVIII−/−, 53 ± 7.5 ng/ml; and Tie2-EPCR/FVIII−/−, 42 ± 3.5 ng/ml; Figure 4). These data indicate, as observed in acquired hemophilia model (Figure 3), rFVIIa generates about 30% of thrombin in a platelet-independent mechanism. The above data also suggest that FVIIa binding to EPCR does not directly contribute to thrombin generation.
FIGURE 4.

FVIIa-EPCR interaction does not directly contribute to thrombin generation. FVIII−/−, EPCR-deficient FVIII−/− mice, and Tie2-EPCR FVIII−/− mice were injected with platelet-depleting antibody (CD42 antibody) or control IgG (1 mg/kg). After 4 h, rFVIIa (4 mg/Kg) was injected via the tail vein, and immediately, bleeding was induced by the saphenous vein incision. Blood was collected from the injury site for 15 min following the injury, and TAT levels were measured. TAT levels in mice depleted of platelets ± rFVIIa treatment within the same genotype or TAT levels among various genotypes were compared. to determine statistical significance, ns, not statically significant difference; **, p<0.01 (n = 4 to 6 mice/group).
3.5 ∣. Neither murine not human platelets express EPCR
Recent studies reported that human platelets express EPCR, and FVIIa binding to platelet EPCR may contribute to the hemostatic effect of rFVIIa in hemophilia therapy [28]. There is no information at present on whether murine platelets express EPCR. Therefore, we next evaluated the status of EPCR expression in murine platelets. Washed platelets isolated from platelet-rich plasma of wild-type C57BL/6J mice were activated with thrombin (10 nM), collagen I (10 μg/ml), or both for 30 min, stained with murine EPCR-specific polyclonal antibodies, and subjected to flow cytometry. As shown in Figure 5A, we found no detectable staining of EPCR on either unactivated or activated platelets. Annexin V binding shows increased externalization of PS in thrombin- and collagen-stimulated platelets, indicating the activation of platelets by these treatments. In a positive control, bEnd.3 murine endothelial cells were stained positive for the EPCR. In additional studies, platelet lysates were subjected to immunoblot analysis. As shown in Figure 5B, we found no detectable EPCR band even when the lysate from 4 x 108 platelets was used for the immunoblot analysis. EPCR antibodies could detect EPCR from endothelial cell lysates, as low as from 1.5 x103 endothelial cells. To address whether the absence of EPCR in platelets is specific to murine EPCR, we next examined for the presence of EPCR in human platelets. As observed with murine platelets, we found no detectable signal to EPCR in human platelets too, either unactivated or activated (Figure 5C and 5D).
FIGURE 5.

Analysis of murine and human platelets for EPCR expression. Washed platelets were prepared from murine (C57BL/6J wild-type mice) and human (healthy volunteers) blood using standard procedures. The washed platelets were activated with either collagen 1 (10 μg/ml) or thrombin (10 nM) or both for 30 min. Murine unactivated and activated platelets were stained with polyclonal goat anti-mouse EPCR antibodies or annexin V and subjected to flow cytometry (A). Lysates of murine platelets (from 4 x108 platelets) were subjected to immunoblot analysis and probed with goat anti-mouse EPCR antibodies (B). As a control, cell lysates from 1.5 x 104, 0.75 x104, and 0.15 x104 murine endothelial cells (bEnd3) were used for immunoblot analysis. Human platelets were labeled with polyclonal goat anti-human EPCR antibodies or annexin V and subjected to flow cytometry (C). Lysates of human platelets (4 x 108 platelets) and HUVEC (from 4.0 to 0.1 x 104 cells) were subjected to immunoblot analysis and probed with goat anti-human EPCR antibodies. For probing ß-actin in platelets as a loading control, 10 times less protein was used in immunoblot analysis compared to probing for EPCR.
4 ∣. DISCUSSION
Data from experimental models and clinical observations strongly support the concept that high doses of rFVIIa provide the hemostatic effect in hemophilia and other bleeding disorders through platelet-dependent and TF-independent mechanism (see rev [35]). Binding of FVIIa to anionic phospholipids expressed on activated platelets and the anionic phospholipid-mediated FVIIa activation of FX to FXa on activated platelets were thought to be responsible to the generation of sufficient thrombin to correct the bleeding disorder in hemophilia patients with inhibitors [11, 36, 37]. Binding of FVIIa to glycoprotein 1b/IX/V complex on platelets was also shown to contribute to TF-independent thrombin generation by rFVIIa on the activated platelet surface [38]. In addition to activated platelets, other blood cells and the damaged vascular endothelium at the injury site may also provide anionic phospholipid procoagulant membrane surface [39]. However, at present, there is no information on whether high doses of rFVIIa can generate thrombin in the platelet-independent mechanism in hemophilia. Data presented in the manuscript provide strong evidence that pharmacological doses of rFVIIa generate substantial amounts of thrombin in platelet-independent mechanism in hemophilia animal model systems. Our data show that pharmacological doses of rFVIIa generate about 30% of thrombin in the platelet-independent mechanism. Our present data also show that rFVIIa-induced thrombin generation is strictly limited to the site of injury. In contrast to published data [28], in the present study, we found no evidence for detectable expression of EPCR in platelets.
It is unlikely that the platelet-independent generation of thrombin by rFVIIa at the saphenous vein injury site comes from the TF-dependent mechanism. Our earlier studies using the same injury model system showed that pharmacological concentrations of rFVIIa restore hemostasis independent of TF in an antibody-induced murine hemophilia model. This study documented that rFVIIa administration reduced the bleed times and blood loss to similar extents in hemophilic mice expressing very low levels (1%) or normal levels of human TF (HTF) [12]. In vivo studies, using a rFVIIa mutant that is completely devoid of TF binding also provides strong evidence that the TF-dependent mechanism does not play a role in rFVIIa-mediated hemostasis in hemophilia [10]. Consistent with the above data, we found that the administration of neutralizing TF mAb did not attenuate rFVIIa-induced thrombin generation in platelet-depleted FVIII−/− mice (data not shown).
rFVIIa binds very weakly to phospholipids, including anionic phospholipid, PS [40, 41]. Studies of Tavoosi et al. [42] showed that rFVIIa binds to phosphatic acid substantially better than to PS, but still with a low affinity (Kd = 1. 7 μM). Under these conditions, rFVIIa-mediated FX activation will linearly correlate with the amount of phospholipids available to interact with rFVIIa. Therefore, a tiny fraction of platelets left in the circulation (<5%) following the platelet depletion in our studies could not be responsible for the substantial amount of thrombin generated in hemophilia mice following rFVIIa administration.
In addition to platelets, other peripheral blood cells and the vascular endothelium interact with coagulation proteins, provide phospholipid membrane surface, and promote coagulation reactions leading to thrombin generation [43-45]. Therefore, it is possible that rFVIIa binding to membrane lipids on other cell surfaces may also lead to thrombin generation. This may account for a substantial amount of thrombin generated when high doses of rFVIIa were administered to hemophilia patients. Our present data do not identify the cellular source of platelet-independent thrombin generation. Given the large surface area provided by the vascular endothelium [46], the endothelium could be the major procoagulant source for the generation of thrombin by pharmacological concentrations of rFVIIa, independent of platelets. However, an injury to the endothelium appears to be necessary to trigger rFVIIa-mediated thrombin generation. It is possible that extracellular vesicles (EVs) originated from other cell types may also provide the phospholipid surface in platelet-depleted hemophilia mice to support the platelet-independent thrombin generation upon administration of pharmacological concentrations of rFVIIa. A recent study showed that about 50% of EVs were originated cells other than platelets, including a small fraction from activated endothelial cells, in healthy humans [47]. Although these EVs appear to lack procoagulant activity when analyzed in a fibrin generation assay in vitro [47], it is possible that these EVs could possess traces of procoagulant activity, and they could still support thrombin generation at the site of injury. It is possible that platelet-derived MVs, generated before depleting platelets, might contribute to rFVIIa-induced hemostasis in platelet-depleted hemophilia mice. However, it is unlikely as any such platelet-derived MVs would have been cleared from the circulation in a 4 h time period between the administration of platelet-depleting antibody and subjecting the mice to rFVIIa treatment and saphenous vein incision.
We found no evidence for systemic activation of coagulation with the administration of pharmacologic doses of rFVIIa. Thrombin generated at the injury site by rFVIIa is limited to the injury site and does not enter into the systemic circulation. Our data that suggest damaged endothelium and possibly other cells play an important role in providing a procoagulant membrane surface for FVIIa activation of FX, and the subsequent thrombin generation is consistent with the findings of Ivanciu et al. [25], who showed elegantly that prothrombinase bound in the vicinity of vascular damage is largely localized to activated endothelium, away from adherent activated platelets. In these studies, the reduction of adherent platelets to the site of injury did not substantially diminish prothrombinase assembly or thrombin formation. In this aspect, the importance of platelets in supporting rFVIIa activation of FX and prothrombinase assembly may differ. Although our studies suggest that cells other than platelets provide substantial amounts of rFVIIa-mediated thrombin generation at the injury site, platelets appear to play a major role in rFVIIa-induced thrombin generation.
Although EPCR on the endothelium specifically interacts with FVIIa, relatively with a high affinity [26], and localizes FVIIa to the endothelium [48], it does not play a direct role in rFVIIa-mediated thrombin generation. Our earlier studies showed that FVIIa binding to EPCR does not enhance FVIIa activation of FX [26]. Consistent with these data, in the current study, we found no differences in thrombin generation between hemophilia A mice expressing normal levels of EPCR and EPCR-deficient hemophilia A mice at the injury site following administration of high doses of rFVIIa. On the face value, the current data may appear contrary to the data reported in our recent article in which we showed that rFVIIa is more efficient in restoring the hemostasis in EPCR-deficient acquired hemophilia A mice due to the absence of EPCR-mediated activation of protein C [27]. However, the effect of EPCR deficiency on the efficacy of rFVIIa in restoring hemostasis in hemophilia is only discernible with the use of a lower dose of rFVIIa (0. 25 mg/kg). At higher doses of rFVIIa (1 and 4 mg/kg), there were no significant difference in rFVIIa’s effect in restoring hemostasis in EPCR-expressing and EPCR-deficient hemophilia mice [27]. The current study employed rFVIIa doses of 1 and 4 mg/kg. A slightly lower thrombin generation observed in EPCR-overexpressing hemophilia mice could be due to the enhanced anticoagulation in these mice and consistent with our earlier findings [27].
Recently, Fager et al. [28] demonstrated that human platelets express EPCR, and suggested that modulation of EPCR binding to platelets could be utilized to enhance the efficacy of rationally designed rFVIIa analogs. In the current study, we found no evidence for the presence of EPCR in murine platelets. It is possible that the murine platelets may differ from that of human platelets for the expression of EPCR. However, we also did not detect EPCR in human platelets too. At present, the reason for the differing results between these two studies is unknown. However, we speculate that trace contamination of leukocytes in platelet preparation may be responsible for detecting EPCR mRNA in platelets and EPCR protein in platelet lysates in the earlier study. Flow cytometry and microscopic studies that document the presence of EPCR in human platelets utilized commercially available EPCR antibodies that had no proven record of the specificity. In the current study, we employed both polyclonal and monoclonal EPCR antibodies generated in-house by one of the authors’ (Dr. Esmon) laboratory that were characterized extensively and used routinely in our laboratories over the past two decades [32, 48-52]. It is conceivable that human platelets express EPCR that is below the detection limit in our analyses. However, this seems unlikely as we could easily detect EPCR from as low as 1.0 x 103 endothelial cells in our immunoblot analysis. In contrast, no EPCR band was detected even when the lysate from 4 x 108 platelets was used for immunoblot analysis.
Our current observation that pharmacological concentrations of rFVIIa can generate thrombin independent of platelets provides an additional explanation on how rFVIIa treatment is effective in correcting bleeding disorders in patients with various platelets disorders, including GT, BSS, and severe thrombocytopenia, where thrombin generation is severely impaired [19, 53-56]. Although the mechanism by which rFVIIa corrects bleeding in these patients is not rigorously elucidated, it has been attributed that high concentrations of rFVIIa can generate sufficient thrombin via activation of FX by the platelet-bound rFVIIa and restoration of platelet aggregation by polymeric fibrin [19, 57]. Here, we suggest that thrombin generated by pharmacological doses of rFVIIa on the injured vascular endothelium promotes platelet activation and generates polymerized fibrin that facilitates adherence of activated platelets at the injured site.
In summary, our data indicate that, although the platelet-dependent mechanism plays a major role in achieving hemostasis by pharmacological concentrations of rFVIIa in hemophilia treatment, platelet-independent mechanisms may also contribute to rFVIIa-mediated hemostasis in hemophilia and other bleeding disorders. In addition to binding EPCR on the endothelium and down-regulating the EPCR-mediated anticoagulation pathway, rFVIIa, when administered in pharmacological concentrations, can also bind the membrane surface of damaged endothelium, in addition to activated platelets. The activated endothelium also supports the binding and assembly of other coagulation proteins [25, 43]. Thus, FVIIa activation of FX on the activated endothelium could lead to thrombin generation. Accumulating evidence indicates that pharmacological concentrations of rFVIIa generate thrombin by multiple mechanisms, and the combined effect may be responsible for the hemostatic effect of rFVIIa and correcting the bleeding disorder (Figure 6). Our current findings may have implications in designing and the development of new rFVIIa variants in treating bleeding disorders. Furthermore, a thorough understanding of the mechanism of rFVIIa action will allow us to reduce some of the risks associated with off-label use of rFVIIa.
FIGURE 6.

Mechanisms of thrombin generation by pharmacological doses of rFVIIa. When pharmacological doses of rFVIIa are administered, it generates traces of thrombin initially by binding to TF at the injury site and activating FX to FXa in the TF-dependent mechanism (1). This thrombin activates platelets. rFVIIa binds to activated platelets, and the platelet-bound rFVIIa generates the majority of thrombin by activating FX to FXa in a platelet phospholipid-dependent mechanism (2). rFVIIa also binds to the damaged endothelium at the injury site or other blood cells, and this leads to the generation of substantial amounts of thrombin in platelet-independent and TF-independent mechanism (3). rFVIIa also competes with protein C for EPCR binding, and displacement of protein C from the EPCR leads to down-regulation EPCR-mediated anticoagulation pathway, which indirectly contributes to thrombin generation (4).
Essentials.
rFVIIa is believed to provide hemostasis in hemophilia in a platelet-dependent mechanism.
We show now rFVIIa also induces thrombin generation independent of platelets in hemophilia mice.
rFVIIa-induced thrombin generation is localized strictly to the site of injury.
We found no evidence that either human or murine platelets express EPCR.
ACKNOWLEDGMENTS
This work is supported by the Investigator-initiated Research Grant H15-28004 from Baxalta, currently a part of Takeda and NHLBI grant HL107483
Footnotes
CONFLICT OF INTEREST
The authors state they have no conflict of interest. The study was partly funded by Investigator-initiated Research Grant from Takeda.
REFERENCES
- 1.Hedner U Factor VIIa and its potential therapeutic use in bleeding-associated pathologies. Thromb Haemost. 2008; 100: 557–562. 08100557 [pii]. [PubMed] [Google Scholar]
- 2.Logan AC, Yank V, Stafford RS. Off-label use of recombinant factor VIIa in U.S. hospitals: analysis of hospital records. Ann Intern Med. 2011; 154: 516–522. 154/8/516 [pii]; 10.7326/0003-4819-154-8-201104190-00002 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roberts HR, Monroe DM 3rd, Hoffman M. Safety profile of recombinant factor VIIa. Semin Hematol. 2004; 41: 101–108. 10.1053/j.seminhematol.2003.11.019. [DOI] [PubMed] [Google Scholar]
- 4.Abshire T, Kenet G. Recombinant factor VIIa: review of efficacy, dosing regimens and safety in patients with congenital and acquired factor VIII or IX inhibitors. J Thromb Haemost. 2004; 2: 899–909. 10.1111/j.1538-7836.2004.00759.x. [DOI] [PubMed] [Google Scholar]
- 5.Howes JL, Smith RS, Helmer SD, Taylor SM. Complications of recombinant activated human coagulation factor VII. Am J Surg. 2009; 198: 895–899. 10.1016/j.amjsurg.2009.05.026. [DOI] [PubMed] [Google Scholar]
- 6.van't V C., Golden NJ, Mann KG. Inhibition of thrombin generation by the zymogen factor VII: implications for the treatment of hemophilia A by factor VIIa. Blood. 2000; 95: 1330–1335. [PubMed] [Google Scholar]
- 7.Butenas S, Brummel KE, Branda RF, Paradis SG, Mann KG. Mechanism of factor VIIa-dependent coagulation in hemophilia blood. Blood. 2002; 99: 923–930. 10.1182/blood.v99.3.923. [DOI] [PubMed] [Google Scholar]
- 8.Shibeko AM, Woodle SA, Lee TK, Ovanesov MV. Unifying the mechanism of recombinant FVIIa action: dose dependence is regulated differently by tissue factor and phospholipids. Blood. 2012; 120: 891–899. blood-2011-11-393371 [pii]; 10.1182/blood-2011-11-393371 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Monroe DM, Hoffman M, Oliver JA, Roberts HR. Platelet activity of high-dose factor VIIa is independent of tissue factor. Br J Haematol. 1997; 99: 542–547. [DOI] [PubMed] [Google Scholar]
- 10.Feng D, Whinna H, Monroe D, Stafford DW. FVIIa as used pharmacologically is not TF dependent in hemophilia B mice. Blood. 2014; 123: 1764–1766. blood-2013-08-522987 [pii]; 10.1182/blood-2013-08-522987 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rao LV, Rapaport SI. Factor VIIa-catalyzed activation of factor x independent of tissue factor: its possible significance for control of hemophilic bleeding by infused factor VIIa. Blood. 1990; 75: 1069–1073. [PubMed] [Google Scholar]
- 12.Keshava S, Sundaram J, Rajulapati A, Pendurthi UR, Rao LV. Pharmacological concentrations of rFVIIa restore hemostasis independent of tissue factor in antibody-induced hemophilia mice. J Thromb Haemost. 2016. 10.1111/jth.13244 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoffman M, Monroe DM 3rd. The action of high-dose factor VIIa (FVIIa) in a cell-based model of hemostasis. Semin Hematol. 2001; 38: 6–9. 10.1016/s0037-1963(01)90140-4. [DOI] [PubMed] [Google Scholar]
- 14.Kristensen J, Killander A, Hippe E, Helleberg C, Ellegard J, Holm M, Kutti J, Mellqvist UH, Johansson JE, Glazer S, Hedner U. Clinical experience with recombinant factor VIIa in patients with thrombocytopenia. Haemostasis. 1996; 26 Suppl 1: 159–164. 10.1159/000217260. [DOI] [PubMed] [Google Scholar]
- 15.Barnes C, Blanchette V, Canning P, Carcao M. Recombinant FVIIa in the management of intracerebral haemorrhage in severe thrombocytopenia unresponsive to platelet-enhancing treatment. Transfus Med. 2005; 15: 145–150. 10.1111/j.0958-7578.2005.00564.x. [DOI] [PubMed] [Google Scholar]
- 16.Revesz T, Arets B, Bierings M, van den Bos C, Duval E. Recombinant factor VIIa in severe uremic bleeding. Thromb Haemost. 1998; 80: 353. [PubMed] [Google Scholar]
- 17.Moisescu E, Ardelean L, Simion I, Muresan A, Ciupan R. Recombinant factor VIIa treatment of bleeding associated with acute renal failure. Blood Coagul Fibrinolysis. 2000; 11: 575–577. 10.1097/00001721-200009000-00010. [DOI] [PubMed] [Google Scholar]
- 18.Poon MC, Demers C, Jobin F, Wu JW. Recombinant factor VIIa is effective for bleeding and surgery in patients with Glanzmann thrombasthenia. Blood. 1999; 94: 3951–3953. [PubMed] [Google Scholar]
- 19.Poon MC. Clinical use of recombinant human activated factor VII (rFVIIa) in the prevention and treatment of bleeding episodes in patients with Glanzmann's thrombasthenia. Vasc Health Risk Manag. 2007; 3: 655–664. [PMC free article] [PubMed] [Google Scholar]
- 20.Monroe DM, Hoffman M, Allen GA, Roberts HR. The factor VII-platelet interplay: effectiveness of recombinant factor VIIa in the treatment of bleeding in severe thrombocytopathia. Semin Thromb Hemost. 2000; 26: 373–377. 10.1055/s-2000-8455. [DOI] [PubMed] [Google Scholar]
- 21.Lisman T, Adelmeijer J, Heijnen HFG, deGroot PG. Recombinant factor VIIa restores aggregation of xIIbB3-deficient platelets via tissue factor-independent fibrin generation. Blood. 2004; 103: 1720–1727. [DOI] [PubMed] [Google Scholar]
- 22.Lisman T, Adelmeijer J, Cauwenberghs S, Van Pampus EC, Heemskerk JW, De Groot PG. Recombinant factor VIIa enhances platelet adhesion and activation under flow conditions at normal and reduced platelet count. J Thromb Haemost. 2005; 3: 742–751. 10.1111/j.1538-7836.2005.01227.x. [DOI] [PubMed] [Google Scholar]
- 23.Lisman T, Moschatsis S, Adelmeijer J, Nieuwenhuis HK, De Groot PG. Recombinant factor VIIa enhances deposition of platelets with congenital or acquired alpha IIb beta 3 deficiency to endothelial cell matrix and collagen under conditions of flow via tissue factor-independent thrombin generation. Blood. 2003; 101: 1864–1870. 10.1182/blood-2002-09-2761. [DOI] [PubMed] [Google Scholar]
- 24.Galan AM, Tonda R, Pino M, Reverter JC, Ordinas A, Escolar G. Increased local procoagulant action: a mechanism contributing to the favorable hemostatic effect of recombinant FVIIa in PLT disorders. Transfusion. 2003; 43: 885–892. 10.1046/j.1537-2995.2003.00427.x. [DOI] [PubMed] [Google Scholar]
- 25.Ivanciu L, Krishnaswamy S, Camire RM. New insights into the spatiotemporal localization of prothrombinase in vivo. Blood. 2014; 124: 1705–1714. blood-2014-03-565010 [pii]; 10.1182/blood-2014-03-565010 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ghosh S, Pendurthi UR, Steinoe A, Esmon CT, Rao LV. Endothelial cell protein C receptor acts as a cellular receptor for factor VIIa on endothelium. J Biol Chem. 2007; 282: 11849–11857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Keshava S, Sundaram J, Rajulapati A, Esmon C, Pendurthi U, Rao LVM. Factor VIIa interaction with EPCR modulates the hemostatic effect of rFVIIa in hemophilia therapy: Mode of its action. Blood Adv. 2017; 1: 1206–1214. 10.1182/bloodadvances.2016004143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fager AM, Machlus KR, Ezban M, Hoffman M. Human platelets express endothelial protein C receptor, which can be utilized to enhance localization of factor VIIa activity. J Thromb Haemost. 2018. 10.1111/jth.14165 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thim L, Bjoern S, Christensen M, Nicolaisen EM, Lund-Hansen T, Pedersen AH, Hedner U. Amino acid sequence and posttranslational modifications of human factor VIIa from plasma and transfected baby hamster kidney cells. Biochemistry. 1988; 27: 7785–7793. 10.1021/bi00420a030. [DOI] [PubMed] [Google Scholar]
- 30.Bajaj SP, Rapaport SI, Brown SF. Isolation and characterization of human factor VII. Activation of factor VII by factor Xa. J Biol Chem. 1981; 256: 253–259. [PubMed] [Google Scholar]
- 31.Li W, Zheng X, Gu J, Hunter J, Ferrell GL, Lupu F, Esmon NL, Esmon CT. Overexpressing endothelial cell protein C receptor alters the hemostatic balance and protects mice from endotoxin. J Thromb Haemost. 2005; 3: 1351–1359. 10.1111/j.1538-7836.2005.01385.x. [DOI] [PubMed] [Google Scholar]
- 32.Stearns-Kurosawa DJ, Kurosawa S, Mollica JS, Ferrell GL, Esmon CT. The endothelial cell protein C receptor augments protein C activation by the thrombin-thrombomodulin complex. Proc Natl Acad Sci U S A. 1996; 93: 10212–10216. 10.1073/pnas.93.19.10212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li W, Zheng X, Gu JM, Ferrell GL, Brady M, Esmon NL, Esmon CT. Extraembryonic expression of EPCR is essential for embryonic viability. Blood. 2005; 106: 2716–2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pavani G, Ivanciu L, Faella A, Marcos-Contreras OA, Margaritis P. The endothelial protein C receptor enhances hemostasis of FVIIa administration in hemophilic mice in vivo. Blood. 2014; 124: 1157–1165. 10.1182/blood-2014-04-567297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hoffman M Recombinant factor VIIa: Clinical uses, dosing, and adverse effects UpToDate: Wolters Kluwer, 2019. [Google Scholar]
- 36.Hoffman M, Monroe DM 3rd, Roberts HR. Activated factor VII activates factors IX and x on the surface of activated platelets: thoughts on the mechanism of action of high-dose activated factor VII. Blood Coagul Fibrinolysis. 1998; 9 Suppl 1: S61–65. [PubMed] [Google Scholar]
- 37.Hoffman M A cell-based model of coagulation and the role of factor VIIa. Blood Rev. 2003; 17 Suppl 1: S1–5. 10.1016/s0268-960x(03)90000-2. [DOI] [PubMed] [Google Scholar]
- 38.Weeterings C, de Groot PG, Adelmeijer J, Lisman T. The glycoprotein Ib-IX-V complex contributes to tissue factor-independent thrombin generation by recombinant factor VIIa on the activated platelet surface. Blood. 2008; 112: 3227–3233. 10.1182/blood-2008-02-139113. [DOI] [PubMed] [Google Scholar]
- 39.Zwaal RF, Comfurius P, Bevers EM. Surface exposure of phosphatidylserine in pathological cells. Cell Mol Life Sci. 2005; 62: 971–988. 10.1007/s00018-005-4527-3. [DOI] [PubMed] [Google Scholar]
- 40.McDonald JF, Shah AM, Schwalbe RA, Kisiel W, Dahlback B, Nelsestuen GL. Comparison of naturally occurring vitamin K-dependent proteins: correlation of amino acid sequences and membrane binding properties suggests a membrane contact site. Biochemistry. 1997; 36: 5120–5127. 10.1021/bi9626160. [DOI] [PubMed] [Google Scholar]
- 41.Nelsestuen GL, Kisiel W, Di Scipio RG. Interaction of vitamin K dependent proteins with membranes. Biochemistry. 1978; 17: 2134–2138. 10.1021/bi00604a017. [DOI] [PubMed] [Google Scholar]
- 42.Tavoosi N, Smith SA, Davis-Harrison RL, Morrissey JH. Factor VII and protein C are phosphatidic acid-binding proteins. Biochemistry. 2013; 52: 5545–5552. 10.1021/bi4006368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stern DM, Esposito C, Gerlach H, Gerlach M, Ryan J, Handley D, Nawroth P. Endothelium and regulation of coagulation. Diabetes Care. 1991; 14: 160–166. 10.2337/diacare.14.2.160. [DOI] [PubMed] [Google Scholar]
- 44.Gillis S, Furie BC, Furie B. Interactions of neutrophils and coagulation proteins. Semin Hematol. 1997; 34: 336–342. [PubMed] [Google Scholar]
- 45.Bouchard BA, Tracy PB. The participation of leukocytes in coagulant reactions. J Thromb Haemost. 2003; 1: 464–469. 10.1046/j.1538-7836.2003.00089.x. [DOI] [PubMed] [Google Scholar]
- 46.Aird WC. Spatial and temporal dynamics of the endothelium. J Thromb Haemost. 2005; 3: 1392–1406. 10.1111/j.1538-7836.2005.01328.x. [DOI] [PubMed] [Google Scholar]
- 47.Berckmans RJ, Lacroix R, Hau CM, Sturk A, Nieuwland R. Extracellular vesicles and coagulation in blood from healthy humans revisited. J Extracell Vesicles. 2019; 8: 1688936 10.1080/20013078.2019.1688936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sen P, Clark CA, Gopalakrishnan R, Hedner U, Esmon CT, Pendurthi UR, Rao LVM. Factor VIIa binding to endothelial cell protein C receptor: Differences between mouse and human systems. Thromb Haemost. 2012; 107: 951–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liaw PC, Mather T, Oganesyan N, Ferrell GL, Esmon CT. Identification of the protein C/activated protein C binding sites on the endothelial cell protein C receptor. Implications for a novel mode of ligand recognition by a major histocompatibility complex class 1-type receptor. J Biol Chem. 2001; 276: 8364–8370. 10.1074/jbc.M010572200. [DOI] [PubMed] [Google Scholar]
- 50.Nayak RC, Sen P, Ghosh S, Gopalakrishnan R, Esmon CT, Pendurthi UR, Rao LV. Endothelial cell protein C receptor cellular localization and trafficking: potential functional implications. Blood. 2009; 114: 1974–1986. 10.1182/blood-2009-03-208900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nayak RC, Keshava S, Esmon CT, Pendurthi UR, Rao LV. Rab GTPases regulate endothelial cell protein C receptor-mediated endocytosis and trafficking of factor VIIa. PLoS One. 2013; 8: e59304 10.1371/journal.pone.0059304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Clark CA, Vatsyayan R, Hedner U, Esmon CT, Pendurthi UR, Rao LV. Endothelial cell protein C receptor-mediated redistribution and tissue-level accumulation of factor VIIa. J Thromb Haemost. 2012; 10: 2383–2391. 10.1111/j.1538-7836.2012.04917.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ozelo MC, Svirin P, Larina L. Use of recombinant factor VIIa in the management of severe bleeding episodes in patients with Bernard-Soulier syndrome. Ann Hematol. 2005; 84: 816–822. 10.1007/s00277-005-1080-y. [DOI] [PubMed] [Google Scholar]
- 54.Gerotziafas GT, Zervas C, Gavrielidis G, Tokmaktsis A, Hatjiharissi E, Papaioannou M, Lazaridou A, Constantinou N, Samama MM, Christakis J. Effective hemostasis with rFVIIa treatment in two patients with severe thrombocytopenia and life-threatening hemorrhage. Am J Hematol. 2002; 69: 219–222. 10.1002/ajh.10056. [DOI] [PubMed] [Google Scholar]
- 55.Tengborn L, Petruson B. A patient with Glanzmann thrombasthenia and epistaxis successfully treated with recombinant factor VIIa. Thromb Haemost. 1996; 75: 981–982. [PubMed] [Google Scholar]
- 56.Franchini M The use of recombinant activated factor VII in platelet disorders: a critical review of the literature. Blood Transfus. 2009; 7: 24–28. 10.2450/2008.0015-08 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Seligsohn U Treatment of inherited platelet disorders. Haemophilia. 2012; 18 Suppl 4: 161–165. 10.1111/j.1365-2516.2012.02842.x. [DOI] [PubMed] [Google Scholar]