

Abstract
Due to the rapid proliferation of antibiotic-resistant pathogenic bacteria, known as Carbapenem-Resistant Enterobacteriaceae (CRE), the efficacy of β-lactam antibiotics is threatened. β-Lactam antibiotics constitute over 50% of the available antibiotic arsenal. Recent efforts have been focused on developing inhibitors to these enzymes. In an effort to understand the mechanism of inhibition(s) of four FDA-approved thiol-containing drugs that were previously reported to be inhibitors of New Delhi Metallo-β-lactamase (NDM-1), various biochemical and spectroscopic techniques were used. Isothermal titration calorimetry demonstrated the binding affinity to NDM-1 corresponds to the reported IC50 values of the inhibitors. Equilibrium dialyses and metal analyses demonstrated that all of these inhibitors formed ternary complexes with ZnZn-NDM-1. Spectroscopic studies on CoCo-NDM-1, revealed two distinct binding modes for the thiol containing compounds. Our findings validate the need to further investigate the mechanism of inhibition of MBL inhibitors. Further research to identify inhibition capabilities beyond reported IC50 values is necessary for understanding the binding modes of these identified compounds and to provide the necessary foundation for developing clinically-relevant MBL inhibitors.
Keywords: Metallo-β-lactamase, thiorphan, tiopronin, spectroscopy, Co(II)-substituted enzyme
Introduction
Multidrug-resistant pathogenic bacteria, such as carbapenem-resistant Enterobacteriaceae (CRE), have been classified as an “urgent threat,” by the Centers for Disease Control and Prevention [1]. Gram-negative bacteria often contain several genes encoding β-lactamases, which hydrolyze β-lactam antibiotics rendering them ineffective. The significance of this problem lies in the fact that β-lactam antibiotics constitute over 50% of the available antibiotic arsenal. The three most clinically-relevant metallocarbapenemases are New Delhi metallo-β-lactamase (NDM), Verona integrin encoded metallo-β-lactamase (VIM), and imipenemase (IMP) [2]. These metallo-β-lactamases (MBLs) use a dinuclear Zn(II)-containing active site to hydrolyze nearly all β-lactam containing antibiotics. The substrate-binding abilities of these enzymes reside in the positioning of the two Zn(II) ions in the active site [3]. Of the three MBLs, NDM is the most widespread in U.S. patients, and NDM continues to evolve with twenty-eight variants identified at the time of authoring this manuscript [2,4]. Given the need to address MBLs in clinical strains, there have been numerous recent reports on new MBL inhibitors. There is also over 30 years of prior investigations into β-lactamase inhibition [5]. Unfortunately, there are no clinically-relevant MBL inhibitors yet reported [6]. The differences among the active sites, and the catalytic mechanisms, of these clinically-important enzymes have severely limited the development of common, selective inhibitors that do not strip metal from the active site in vivo [7,8].
Recently, Klinger et al. screened FDA-approved drugs and reported eleven potential MBL inhibitors containing thiol moieties [9]. Of the original eleven identified in their study: L-captopril, DL-thiorphan, tiopronin, and 2,3-dimercaprol exhibited inhibition of the three clinically-relevant MBLs (NDM, VIM and IMP). Dimercaprol is an approved drug against toxic metal poisoning. DL-thiorphan is the active metabolite in the antidiarrheal drug Racecadotril [9]. L-captopril is an approved angiotensin-converting enzyme (ACE) inhibitor for the treatment of hypertension, and the crystal structure of L-captopril complexed with NDM-1 was reported in 2012 [10]. Tiopronin is used as a treatment for metal poisoning and cystinuria [9]. Klinger et al. previously reported a crystal structure of NDM-1 complexed with tiopronin and proposed that the mode of inhibition is similar to that of L-captopril [9]. However, it was not possible for them to unambiguously discriminate all possible conformations of the ligand in the NDM-1 active site allowing for several proposed orientations (PDB ID 5A5Z).
Previous crystallographic studies suggest that there are two distinct binding modes for these thiol-containing compounds. Either the thiol sulfur bridges the two Zn(II) ions (as it does in the structure of the NDM-1/L-captopril complex) or the thiol sulfur coordinates terminally to only one Zn(II) ion (as in the structure of the NDM-1/tiopronin complex). It is not clear how thiorphan or dimercaprol bind to the metal center in the MBLs, and in the case of tiopronin, conflicting results exist. Raczynska et al. reported a reanalysis of the reported crystal structure of the tiopronin complex [11]. They successfully modelled and refined two water molecules in the active site replacing the tiopronin ligand from the original coordinates, after several failed attempts to model any ligand there, either tiopronin or such buffer components. They concluded that the observed electron density reported by Klinger et al. did not support modeling any ligand in the active site (PDB ID 5NBK) [12]. As we show below, spectroscopy provides valuable information to address this difference. We have been developing a “toolbox” approach to biophysical characterization to ascertain the mechanism of inhibition, through a series of solution-based analyses [13], and the discrepancy in interpretation of X-ray diffraction data is a perfect example to illustrate its utility. The studies presented below were designed to determine how tiopronin, thiorphan, and dimercaprol (Figure 1) inhibit NDM-1, and whether they are indeed good lead candidates for further elaboration and study. The work flow used herein can readily be used to examine any MBL inhibitor and is easily transferrable to other types of systems.
Figure. 1.
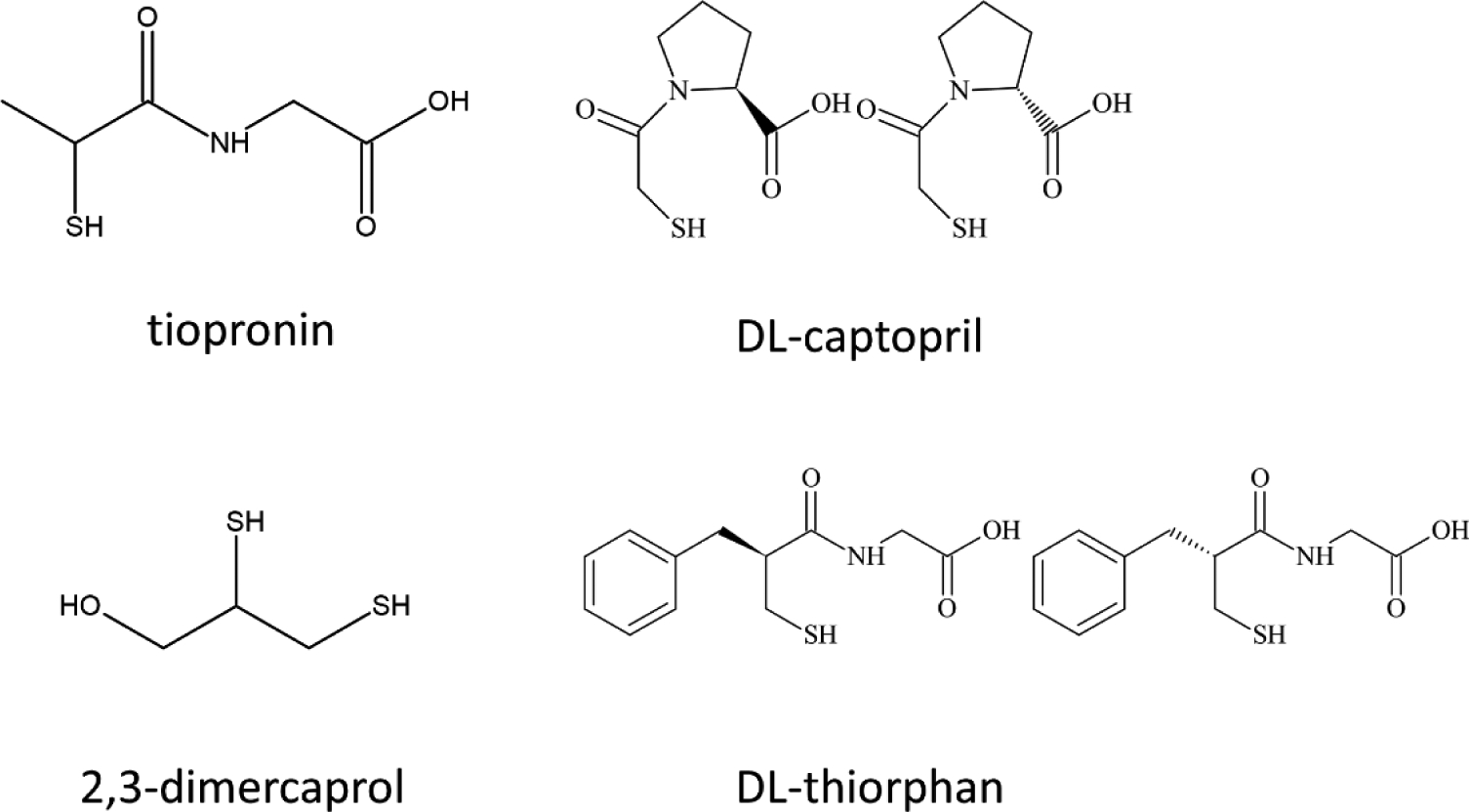
Structures of the three NDM-1 inhibitors under investigation, and the control compound DL-captopril.
Materials and Methods
Over-expression, purification, and characterization of NDM-1
Tag-free ZnZn-NDM-1 was prepared as described previously [15]. Briefly, over-expressed NDM-1 was purified with a single-step HisTrap column, using TEV protease to remove the His6-tag, as previously described [14]. The resulting NDM-1 exhibited Km and kcat values (5 ± 1 μM and 4.2 s−1), using chromacef as substrate, which are similar to previous reports [2]. ICP-AES confirmed that the resulting NDM-1 binds 1.9 ± 0.1 eq of Zn(II), also consistent with previous reports [2] [15] [16]. CoCo-NDM-1 was prepared by direct addition of 2 eq of Co(II) to apo-NDM-1, as we have reported previously [2].
Equilibrium dialysis
NDM-1 (final concentration 8 μM and final volume of 5 mL) in 50 mM HEPES, pH 7.5, was mixed with the compound of interest at concentrations of 0–128 μM. After incubation for 30 min, the solutions were dialyzed versus 500 mL of metal-free 50 mM HEPES, pH 7.5, overnight using dialysis tubing with MWCO of 6000–8000, Fisherbrand. The Zn(II) content in the resulting NDM-1 samples was determined ICP-AES.
Isothermal titration calorimetry
Isothermal titration calorimetry (ITC) experiments were carried out using a Nano ITC System (TA Instruments-Waters LLC., USA) with a 500 μL cell. All experiments were performed at 25 °C. Inhibitors were prepared by diluting a stock solution (in 100 mM DMSO) with 20 mM HEPES, pH 7.5, containing 150 mM NaCl. Tag-free NDM-1 was also diluted in the same buffer. The ITC cell was filled with 50 μM NDM-1 (300 μL), and the enzyme solutions were titrated with 500 μM L-captopril, DL-thiorphan, tiopronin, or 2,3-dimercaprol. The injection volume was 50 μL, and the time between injections was 210 s, allowing the inhibitor to bind. Kd values were determined by fitting using NanoAnalyze (TA Instruments-Waters LLC, USA).
UV-visible spectroscopy
Aliquots from stock solutions (20 mM) of the thiol compounds were added to 300 μM CoCo-NDM-1 samples, and the samples were then incubated on ice for 30 min. The UV-vis spectra of resulting samples between 250 and 800 nm were collected on a PerkinElmer Lambda 750 UV/vis/NIR spectrometer. The blank consisted of apo-NDM-1 (300 μM) in 50 mM HEPES, pH 6.8, containing 150 mM NaCl and 1 mM TCEP. Control spectra of an equimolar (600 μM) mixture of each inhibitor and aqueous Co(II) were recorded. The inhibitors were also examined, in the absence of protein, as a function of pH. To obtain pKa values, the absorptivity at λmax was fitted to Equation 1, a modified version of the Henderson-Hasselbach equation, where c1 and c2 represent the limiting extinction coefficients of the high- and low-pH forms, respectively [17].
| (1) |
NanoESI-mass spectrometry
ZnZn-NDM-1 (50 μM) was buffer-exchanged, using three Bio-Rad spin column wash tubes, into 100 mM ammonium acetate, pH 7.5. The eluted dizinc NDM-1 (50 μM) was mixed with 500 μM inhibitor and incubated for 10 minutes at room temperature. A Thermo Scientific LTQ Orbitrap XL™ hybrid ion trap-orbitrap mass spectrometer equipped with a nano-electrospray ionization (n-ESI) probe (Thermo Fisher Scientific, San Jose, CA, USA) with positive mode protein detection was used to analyze the samples. The major parameters were capillary temperature, 180 °C; sheath gas, 0; auxiliary gas, 0; sweep gas, 0; and spray voltage, 1.1–1.9 kV; tube-lens, 150 V; capillary voltage, 35 V. A full scan ranging from 1000 to 4000 (m/z) was used. The resolution was set at 30,000. Automated gain control was set at 3 × 104 in full scan, 1 × 104 in SIM, 1 × 104 in MSn, and 3000 in zoom for ion trap, 3 × 106 in full scan, 1 × 105 in SIM and 1 × 105 in MSn for Fourier transform. The nESI source was equipped with an offline unit (Cat # ES260), and the source was constructed based on published work with modification. Briefly, a platinum wire (0.25 mm diameter) was inserted in the center of the offline unit and in a pulled glass capillary with a tip i.d. of about 1 μm, produced in-house from a glass capillary (i.d. 0.8 mm, o.d. 1.5 mm) using a micropipette puller (model P-87 Flaming/Brown Micropipette Puller, Sutter Instrument Inc., USA). The sample solution (5 μL) was loaded into the pulled glass capillary by an infusion syringe (Thermo Scientific, USA). The platinum wire was inserted in the center of the capillary. The position of the capillary tip was then adjusted to around 3 mm away from the MS inlet.
NMR spectroscopy
Each NMR sample was buffered with 50 mM HEPES, pH 6.8, containing 150 mM NaCl and 10% D2O. Thiol inhibitors were dissolved in DMSO at high concentration (50–100 mM) so they could be titrated in small (~5 μL) aliquots. The inhibitor complex spectra presented here were obtained from samples containing 2 molar equivalents of inhibitor per molar equivalent of protein. Spectra were collected at 292 K on a Bruker ASX300 NMR spectrometer operating at a frequency of 300.16 MHz. Spectra were collected using a frequency switching method, applying a long, low power (270 ms) pulse centered at the water frequency, followed by a high power 3 μs pulse centered at 90 ppm [18]. This method allows for suppression of the water signal with enhancement of severely hyperfine-shifted resonances. Spectra consisted of 30,000 transients of 16k data points over a 333 ppm spectral window (100 kHz, λaq ≈ 51 ms); signal averaging took ~3 h per spectrum. The 1H NMR spectra of the four inhibitors with Co(II), in 2.5-fold molar excess and in the absence of protein, were recorded as controls.
EPR spectroscopy
EPR samples contained 1 mM CoCo-NDM-1, buffered with 50 mM HEPES, pH 7.5, containing 5 mM NaCl and ~10 % (v/v) glycerol, which served as a glassing agent. Inhibitor complexes were prepared containing 2 molar equivalents of inhibitor per molar equivalent of protein. Samples were loaded into EPR tubes and degassed by repeated evacuation/purgation with N2 prior to data collection. Spectra were collected on a Bruker EMX EPR spectrometer, equipped with an ER4116-DM dual mode resonator (9.37 GHz, parallel; 9.62 GHz perpendicular). The data were scaled so that the x-axes matched (perpendicular mode field values were scaled by 9.37/9.62). Temperature control was accomplished using an Oxford ESR900 cryostat and temperature controller. Other spectral conditions: microwave power = 0.2 mW; field modulation = 10 G (100 kHz); receiver gain = 104; time constant/conversion time = 41 ms.
Computational Modelling
An initial model of NDM-1 was prepared from PDB ID 4exs [19] using the “prepack” module within the Rosetta software suite. Initial three-dimensional coordinates for thiorphan were generated using OpenBabel with the simplified molecular-input line-entry system (SMILES) notation for thiorphan as input to generate a three-dimensional structure-data file (SDF). A family of 512 conformations of thiorphan were generated using OpenEye Omega version 3.1.2.2 [20] with the thiorphan SDF generated by OpenBabel [21]used as input. Rosetta ligand parameter files were prepared from the Omega-generated thiorphan conformations using the automatic RosettaLigand setup scripts packaged with Rosetta. Thiorphan was manually placed in a starting pose near the NDM-1 active site prior to ligand docking. Docking of thiorphan into NDM-1 was performed using Rosetta LigandDock [22,23]. Docking runs permitted side-chain and backbone flexibility with extra Chi1 and aromatic-Chi2 rotamers and ligand translations up to ±5 Å along the x, y, and z directions. A total of 10,260 decoys were generated and scored. A representative pose, selected as the lowest energy model of a cluster of 10 models based on the overall Rosetta energy score, was selected using the Rosetta clustering app.
Results and Discussion
Isothermal Titration Calorimetry (ITC)
IC50 values for L-captopril, D,L-thiorphan, tiopronin, and 2,3-dimercaprol binding to NDM-1, previously reported by Klinger et al., [3] ranged from 1.3 to 84 μM (Table 1; Figure 1). In an effort to expand on their findings, we examined the binding affinities of the set of thiol-containing compounds by ITC. All four appeared to form 1:1 complexes, and in general, the ITC-determined KD values (see Table 1 and Supporting Information, Figure S1) were consistent with the corresponding IC50, where tiopronin’s was 5 to 10-fold larger than the others. The data for thiorphan and captopril were sufficiently well-fitted to extract thermodynamic parameters, and both gave enthalpies of binding on the order of −30 kJ/mol (~ 7 kcal/mol). However, while the data for captopril indicate a positive entropy of binding, presumably from dehydration of the active site [24], the data for thiorphan clearly indicate a negative entropy of binding, possibly due to ordering of waters to solvate the thiorphan phenyl ring offsetting active site dehydration [24]. This difference may be due to the need to accommodate the hydrophobic pendant benzyl of thiorphan, which is not present in captopril, and may represent a position to be explored for further optimization.
Table 1.
Inhibition of ZnZn-NDM-1 by the current set of thiol-containing compounds.
Equilibrium Dialysis
To identify metal-stripping contributions, we incubated 8 μM ZnZn-NDM-1 with inhibitor concentrations ranging from 0–128 μM. After incubation for approximately 30 minutes, the mixture was dialyzed for four hours, to remove unbound Zn(II) and inhibitor. The resulting enzymes were analyzed with ICP-AES to determine their remaining metal content (Figure 2) [14]. Inhibitors that exhibit a metal-stripping mechanism are expected to show reduced metal content, while inhibitors that form a stable ternary complex should show little or no decrease in metal content [8]. EDTA was used as a control example of a metal-stripping inhibitor, while L-captopril served as an example of a ternary complex-forming inhibitor [25].As can be seen in Figure 2, exposure to increasing concentrations of EDTA led to proportionate reductions in NDM-1 metal content, while none of the thiol-containing inhibitors (L-captopril, DL-thiorphan, tiopronin, or 2,3-dimercaprol) appreciably altered the enzyme’s zinc content, even at 16-fold excess. These data, along with preceding studies, [14] [26] suggests that all four compounds form ternary complexes with NDM-1 in this concentration range (< 10 μM).
Figure 2.
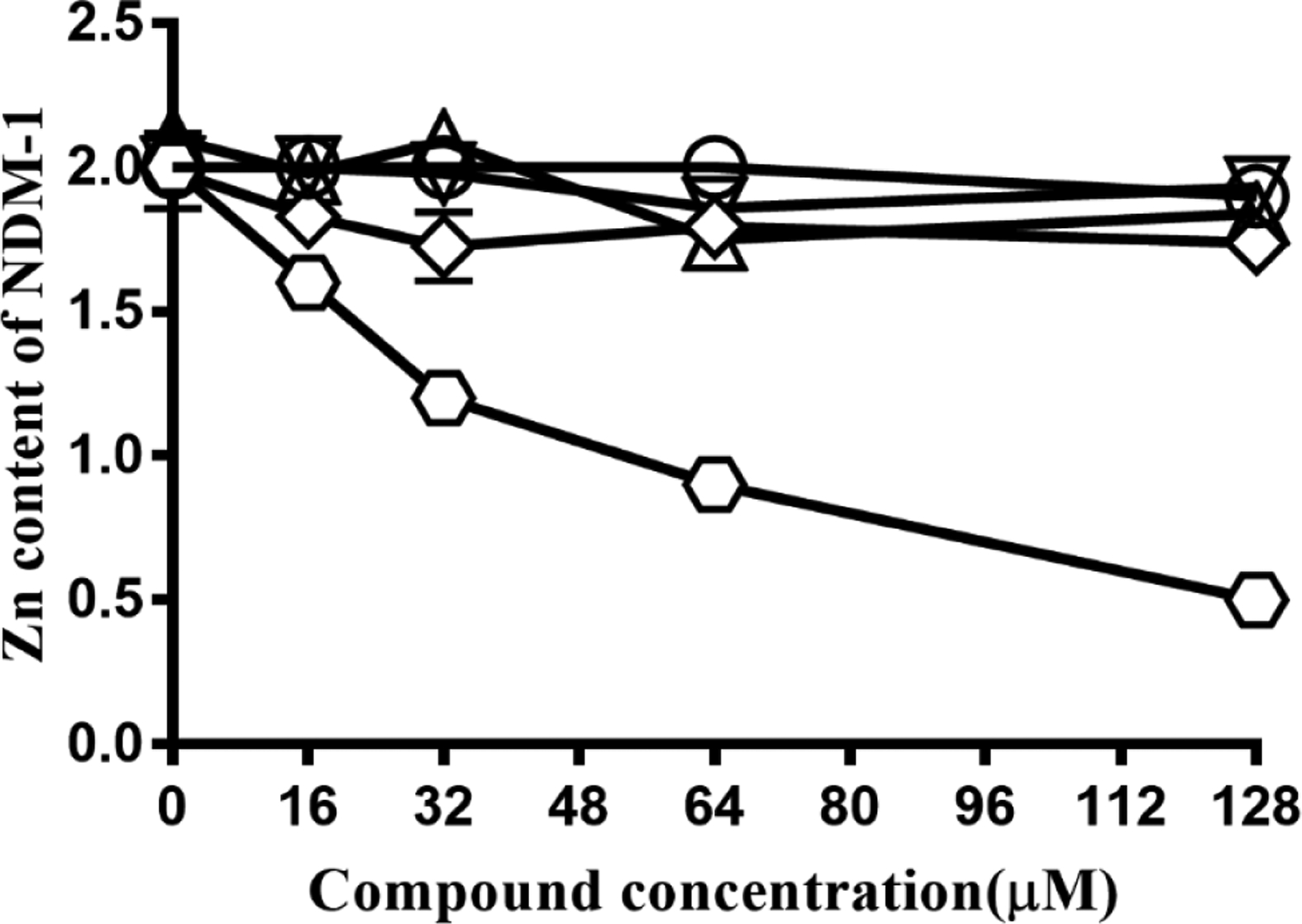
Equilibrium dialysis of 8 μM NDM-1 with increasing concentrations of EDTA (▢), captopril (◯), tiopronin (△), thiorphan (◇), and 2,3-dimercaprol (▽).
UV-visible Spectroscopy
To gain insight into the nature of the ternary complexes, we first turned to UV-visible spectroscopy. The presence of the thiol in the inhibitor structures provides a potential internal spectroscopic handle, but also a potential complication due to background absorption. Therefore, we first measured the optical spectrum of each inhibitor as a function of pH, shown in Supporting Information, Figure S2. In each case, at low-pH, where the protonated thiol is expected to be the dominant form, little (thiorphan) or no (tiopronin, dimercaprol, and captopril) absorption can be observed. However, again in each case, the spectra showed the onset of absorption with increasing pH. As perhaps could expected, the alkyl thiolates give rise to very low intensity features (λmax = 436 nm, ε < 2 M−1cm−1, for both), while thiorphan, with a pendant phenyl group, gives rise to a more intense absorption. Both thiorphan (ε > 240 M−1cm−1) and dimercaprol (ε > 2,400 M−1cm−1) show a higher energy λmax, in the UV, below 250 nm. Dimercaprol appears to show a second feature at longer wavelength (~ 260 nm), that levels off at ε ~ 2400 M−1cm−1, but the data are insufficient to claim the capture of multiple ionizations. Fitting the data shows that the pKa values range from 8.4 (tiopronin) to 8.5 (captopril) to 9.6 for thiorphan. The ionization(s) observed for dimercaprol indicate a minimum pKa of 8.5, with the longer wavelength better fitted at 9.4. For even the lowest of these values (8.4), the inhibitor thiols must be nearly fully protonated at pH 7.5. The relatively high pKa of the thiorphan thiol may help explain the negative entropy of binding, as it has a tenfold higher affinity for its thiol proton. Most importantly, the background spectra show that the enzyme-bound spectra presented below are unaffected by any contribution from unbound inhibitor.
We next examined the inhibitors’ effect on the optical spectra of Co(II)-substituted NDM-1. As previously reported, CoCo-NDM-1 exhibits an intense peak at 330 nm, attributed to a Cys-S to Co(II) LMCT, and a group of four ligand field transitions at 510, 549, 615, and 640 nm, with comparable contributions from both metal ions (Figure 3).[27,2,28] The addition of 2 eq of EDTA to CoCo-NDM-1 leads to disappearance of both the LMCT and ligand field transitions, consistent with the loss of both Co(II) ions, while captopril, which has been shown to bridge the metal ions via the thiol sulfur, leads to increased LMCT intensity and a significant shift the ligand field bands.[27] As shown in Figure 3, the addition of 2 eq of thiorphan resulted in increased LMCT intensity, with a shift of λmax to 345 nm (ε = 1,800 M−1 cm−1). Consistent with the relatively small KD from ITC (Table 1), the LMCT is nearly at its maximum with addition of the first eq, increasing only 20% on addition of the second eq. Meanwhile, the ligand field bands at 510 and 640 nm are reduced or eliminated when thiorphan binds, while the central pair shift closer together (to 567 and 607 nm, ε = 385 and 390 M−1 cm−1, respectively) and a new band appears at 675 nm. Based on the similarity to the effect of captopril on the spectrum of CoCo-NDM-1,[29] we must infer that thiorphan binds in a similar fashion, with the thiol sulfur occupying a bridging position between the two metal ions. In contrast, the addition of tiopronin to CoCo-NDM-1 resulted in a more intense LMCT band, without significantly affecting the ligand field transitions. Also consistent with the ITC (Table 1), it is clear that tiopronin does not saturate the active site as readily as thiorphan, as the LMCT intensity increases more than 50% on addition of the second eq (from ~1,100 to 1,800 M−1cm−1, Fig. 3A). Meanwhile, the lack of any significant effect on the ligand field region suggests it is terminally-bound through the thiol sulfur, possibly replacing the terminal water molecule at the Zn2 site.
Figure 3.
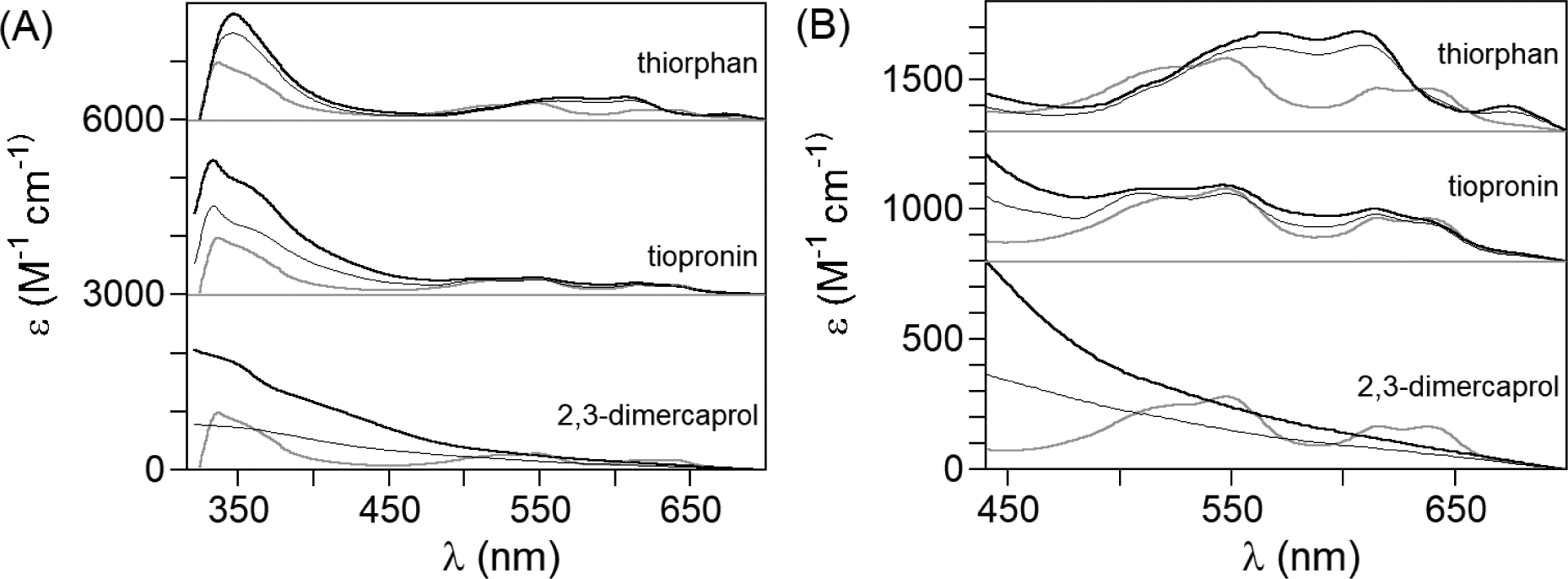
(A)UV-visible spectra of CoCo-NDM-1 with 1 eq (thin black line) and 2 eq thiol inhibitors (thick black line); the spectrum of CoCo-NDM-1 in the absence of inhibitor is presented in gray for reference. (B) Expansion of the ligand field region from ~ 450 to 700 nm.
Meanwhile, the addition of 2,3-dimercaprol to CoCo-NDM-1 results in wholesale changes in the UV-vis spectra with strong absorption spanning 300 to 500 nm that largely obscures the ligand field transitions. However, closer inspection of the ligand field region (see Figure 3B) shows that the longer wavelength ligand field transitions disappear with the first addition of 2,3-dimercaprol, indicative of a metal-stripping inhibitor at these concentrations. To gain further insight into the interaction with 2,3-dimercaprol, we performed native mass spectrometry (Figure S3). These experiments, performed at similar concentrations (500 μM vs. 300 μM for the optical spectra), also showed evidence of metal stripping. On exposure to 2,3-dimercaprol, the native-MS showed ZnZn-NDM-1 disappearing, with the population shifting to the metal-free form. Importantly, we note that we also observed a small portion of a binary complex between the metal-free enzyme and dimercaprol but did not observe a ternary complex between the Zn-loaded enzyme and the inhibitor. As 2,3-dimercaprol functions as a metal-stripping agent at the necessarily high concentrations of the spectroscopic studies, we will not present NMR and EPR data on dimercaprol-treated CoCo-NDM-1, which largely showed reductions in signal intensity, without the observation of new features.
Electron Paramagnetic Resonance Spectroscopy
EPR spectra for the CoCo-NDM-1/thiol complexes are shown in Figure 4. All of the spectra appear quasi-axial in perpendicular mode (Figure 4A, B1⊥B0), with g⊥ ~ 6.5 and g|| ~ 2.1. The width of the low-field, perpendicular feature shows some modulation on addition of inhibitor. Tiopronin causes the smallest perturbation, consistent with the UV-vis-derived suggestion that it binds terminally to one metal ion. Somewhat surprisingly, given the expectation that both thiorphan and captopril bind the NDM-1 active site in a bridging fashion, captopril broadens the EPR signal, while thiorphan leads to its sharpening. We believe this likely reflects differences in electron relaxation rates, and may be a result of secondary interactions. All three spectra are sufficiently altered to be considered indicative of binding to one or both of the Co(II) ions, but a deeper analysis of the perpendicular mode spectra is, at best, difficult [30]. However, previous studies on the di-Co(II) form of the related B1 metallo-α-lactamase, BcII, showed the spin coupling between the Co(II) ions could readily be probed by parallel mode EPR (B1||B0) [31].The parallel mode spectra of the NDM-1-inhibitor complexes are compared to the resting enzyme in Figure 4B. All of the inhibitor complexes show the same features as the resting enzyme, yet all show reduced signal intensity, suggesting reduced spin coupling. This result is particularly apparent in the negative feature near 800 G, and this feature has been correlated with coupling strength in previous work [31]. While all three inhibitors lead to reduced signal intensities, the level of attenuation at 800 G appears greater for thiorphan and captopril, consistent with the inhibitor replacing the bridging hydroxyl with the thiol sulfur. These most likely are facilitated by the thiol being protonated, which allows for protonation of the bridging hydroxyl, allowing it to then leave as water. The lesser attenuation seen in the tiopronin complex can be attributed to the addition of a second electron-rich sulfur donor, which should, in turn, weaken the bonds to the bridging group.
Figure 4.
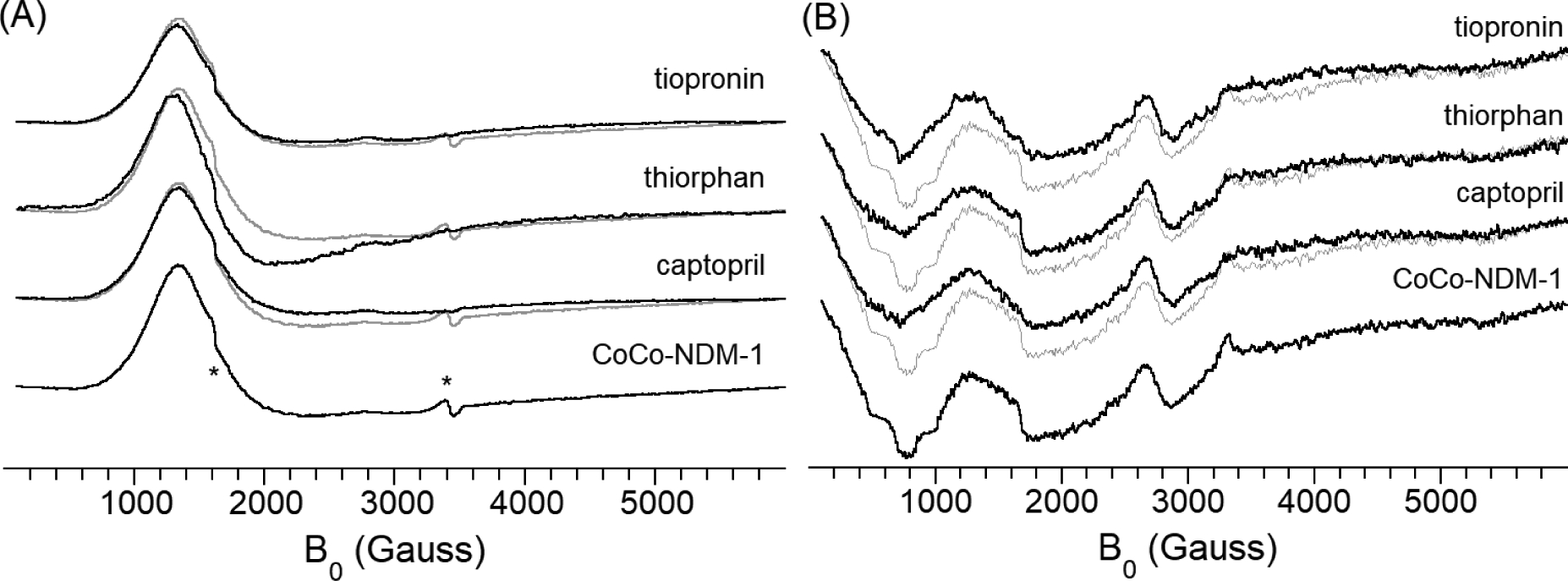
Perpendicular- (A) and parallel-mode (B) EPR of CoCo-NDM-1 thiol-inhibitor complexes. The features at ~1600 and ~3400 G in part A (marked with asterisks) are attributed to minor contamination from iron and copper, respectively. In both panels, the spectrum of the uncomplexed enzyme is overlaid as a gray line for comparison.
NMR Spectroscopy
To gain more atomic level detail, we examined the 1H NMR spectra of the thiol inhibitor complexes. The data, presented in Figure 5, are shown in three sections to allow them to be scaled differentially; the as-collected spectra are presented in Figure S4. None of the inhibitors alone form complexes with Co(II) free in solution (see Figure S5). The spectrum for CoCo-NDM-1 (bottom trace in Figure 5) has been previously reported and assigned, with solvent-exchangeable resonances at 78, 73, and 65 ppm, assigned to the exchangeable NH protons of three Co(II)-bound His residues at the Zn1 site. Meanwhile, peaks at 47 (aspartate β-CH2) and 107 (histidine NH) ppm along with the doublet near 170 ppm (cysteine β-CH2) were attributed to the Zn2 site [27].
Figure 5.
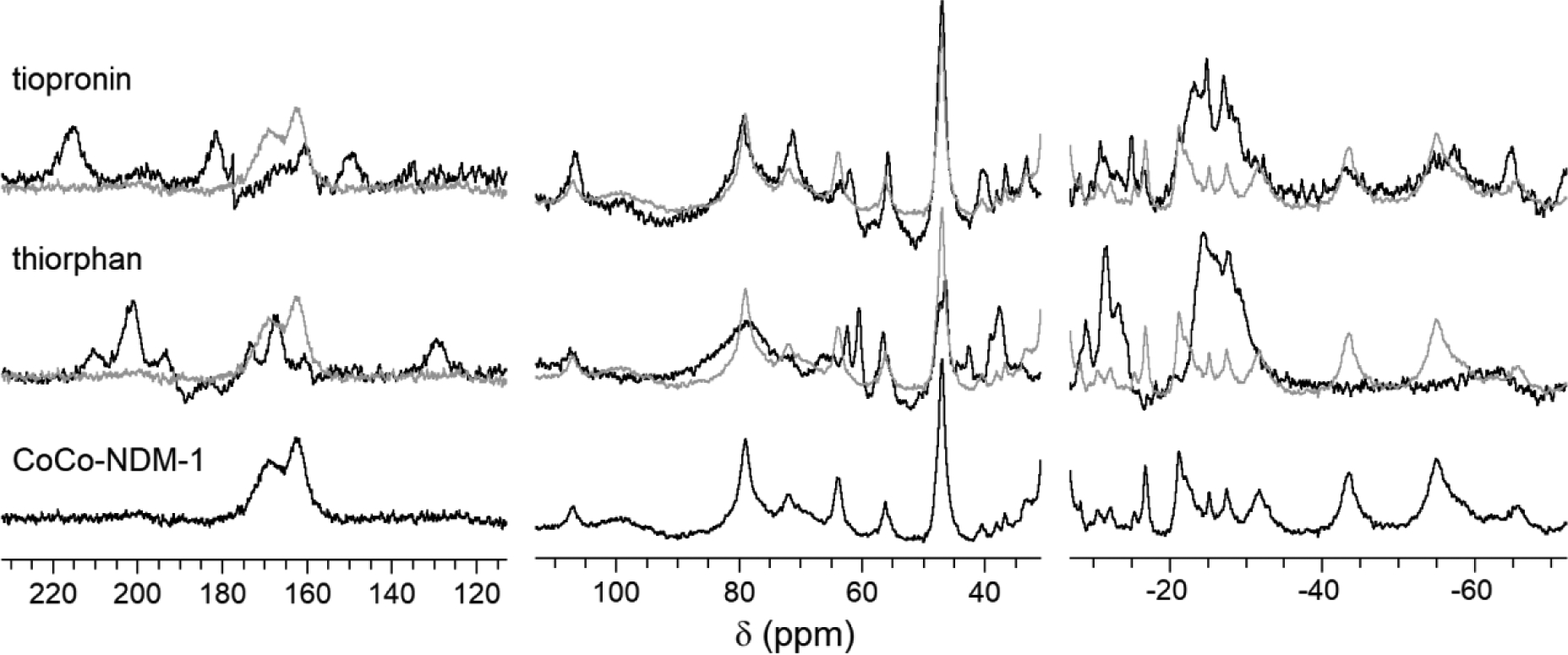
1H NMR spectra for CoCo-NDM-1 and thiol inhibitor complexes. The inhibitor-bound spectra are overlaid with the spectrum of the uncomplexed enzyme (gray lines) for reference.
The addition of tiopronin to CoCo-NDM-1 leads to the appearance of two new resonances to low field (> 140 ppm), consistent with coordination of a second thiolate, and one other new resonance at 60 ppm, which may arise from the terminal methyl of tiopronin. Aside from what appears to be a shift in the cysteine dihedral, based on a change in the separation of their NMR lines, and one fairly large change in the upfield region (~ 30 ppm, seen in all three inhibitor complexes, discussed below), tiopronin causes remarkably little else to change relative to the uncomplexed enzyme. This, again, is consistent with terminal coordination of tiopronin through its thiol sulfur.
We have previously reported the 1H NMR spectrum of CoCo-NDM-1 in complex with captopril, though the data were not discussed [14]. Addition of captopril to CoCo-NDM-1 has profound effects on signals attributed to both metal ions. These effects are evidenced by ~ 40 ppm chemical shift attenuation for two of the three Zn1 His ligands, as well as new resonances above 170 ppm, a splitting of the symmetry-equivalent aspartate β-CH2 (~ 47 ppm) and a sizable rearrangement of the resonances upfield of the diamagnetic window (δ < 0), which are commonly attributed to second sphere interactions with the Zn2 ion (see Figure S6) [32]. As shown in Figures 5 and S6, thiorphan produces many similar changes, including a set of new resonances above 170 ppm, indicative of coordination of the thiol, and similar changes to the aspartate and histidine resonances. Also present is one new broad resonance near 80 ppm which may come from the benzylic carbon of thiorphan, or perhaps a subset of the phenyl protons.
Computational Docking Models/Results of Thiorphan with NDM-1
To further explore the potential binding mechanism of this set of thiol-containing compounds as effective NDM-1 inhibitors, the computational modelling of DL-thiorphan, tiopronin, and 2,3-dimercaprol were attempted. The representative docking pose for DL-thiorphan (Figure 6B) features the sulfur positioned in a bridging position between the two active site zinc ions and the thiorphan phenyl ring positioned to enable π-stacking with the side chain imidazole ring of His122. Positioning of the acetyl-glycine substituent squarely within the active site channel opens up opportunities for solvent-mediated hydrogen bonds to surrounding residues; however, since the docking runs did not include explicit solvent, these potential interactions were not observed. Consistent with the EPR, NMR, and UV/visible spectroscopic results, the docking pose for tiopronin (Figure 6C) features binding to a single zinc ion, Zn2, through the tiopronin sulfur. Additional interactions between NDM-1 and tiopronin include packing of the tiopronin methyl group against the methylene groups of His189 and Cys208, and hydrogen bonds to the side chain ε-amino of Lys211 and the backbone amide proton of Asn220, each mediated by carbonyl oxygen atoms from tiopronin. Interestingly, the NDM-1 active site loops proximal to the pocket between His189 and Cys208 provide a rich source of hydrogen bond donors that could interact with tiopronin. In contrast with the binding modes observed for thiorphan and tiopronin, the docking pose for dimercaprol (Figure 6D) indicates interaction of two sulfur atoms with the active site zinc ions. One sulfur occupies a bridging position between the two zinc ions while the second sulfur coordinates to Zn2. Partly owing to the small size of dimercaprol, additional interactions with NDM-1 are limited to a potential hydrogen bond between the dimercaprol hydroxyl oxygen and the side chain amide of Asn220.
Figure 6.
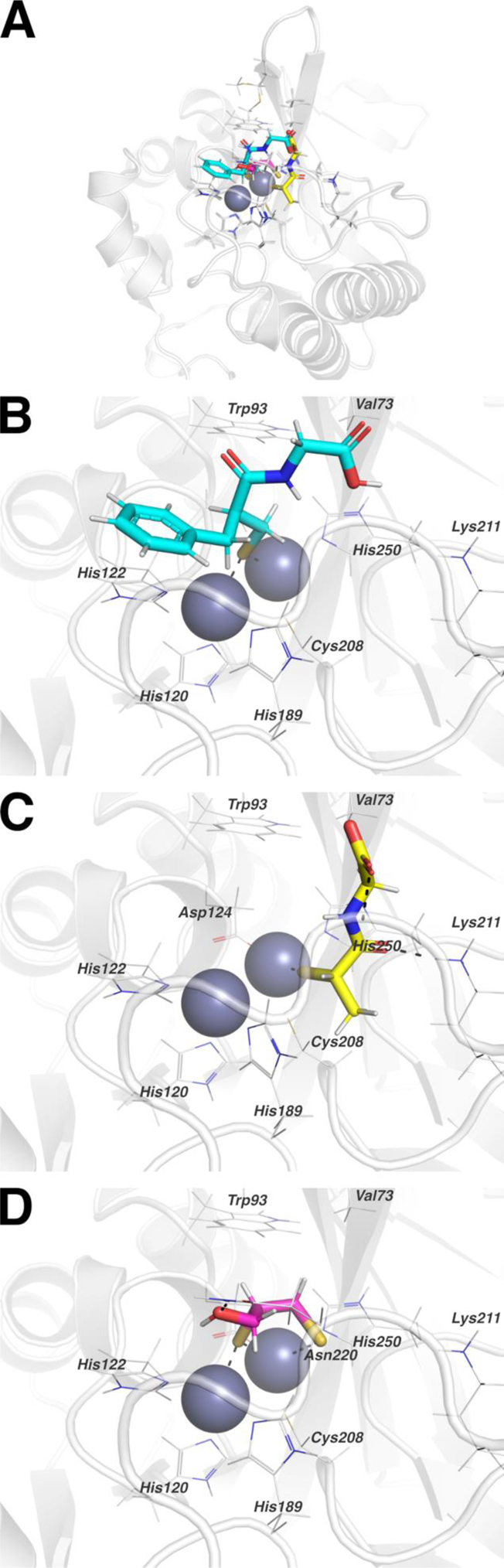
(A) Representative docking poses for thiorphan, tiopronin, and dimercaprol are shown with NDM-1 (grey). (B) The representative thiorphan (cyan) pose binds to the NDM-1 active site with the sulfur in a bridging position between the two zinc ions. (C) The representative tiopronin (yellow) pose binds to one zinc ion through the sulfur and interacts with neighboring atoms from the NDM-1 backbone and side chains. (D) The representative dimercaprol (magenta) pose interacts with both zinc ions using both sulfur atoms and interacts with a nearby NDM-1 side chain through the dimercaprol hydroxyl. Side chains in B-C interacting with ligands or zinc ions are labeled.
Summary and Conclusion
The sum of the methods utilized here have been developed by our labs and others over a number of years, specifically for the study of MBLs. The rationale for using such a wide array of approaches is summarized in Table 2. Each technique contributes through a core strength, and each provides a unique piece of the puzzle. However, each technique has limitations, and the complete understanding of the mechanism of an inhibitor requires multiple probes. We feel this progression is important. Equilibrium dialysis can quickly identify inhibitors that function via metal stripping, and, as was illustrated here, because it is performed at a substantially lower concentration than the spectroscopic studies, can validate potential hits that might otherwise be overlooked. A potential limitation is that multiple rounds of dialysis may lead to metal removal, generating a false positive with regards to metal stripping. In addition, this technique, by itself, cannot determine whether a ternary complex forms or not. Similarly, native mass spectrometry offers an avenue to determine the presence of a ternary complex, although, in most cases, it is not quantitative. ITC provides binding affinities and thermodynamic parameters associated with binding, using moderate amounts of enzyme and inhibitor. However as shown with dimercaprol in this study, dissociation constants can be obtained, even for inhibitors that strip metal from the MBL. UV/vis spectroscopic studies provide a rapid, albeit low resolution method for identifying interactions between the inhibitor and the active site metal ions. This technique, along with EPR and NMR discussed below, requires the use of large quantities of non-native, Co(II)-substituted MBL. EPR and NMR provide higher resolution data, with EPR directly interrogating the active site metal ions and NMR probing their interactions with the protons of functional groups that coordinate to the active site metal ions (both constitutive and inhibitor-derived). Finally, computational docking efforts offer an opportunity to propose specific binding modes which can be evaluated for agreement with the biophysical data from UV/Vis spectroscopy, EPR, and NMR, and potential positions that can benefit from further elaboration.
Table 2.
Comparison of the methods used here to examine inhibition mechanism.
| Technique | Enzyme Cone. | Information Content | Limitations |
|---|---|---|---|
| Equilibrium Dialysis | 8 μM | Does inhibitor strip metal from the enzyme? | No direct evidence for ternary complex. Long dialyses may lead to false positive of metal stripping. |
| ITC | 50 μM | What is the binding affinity (and associated thermodynamic parameters)? | Can yield binding parameters even for compounds that strip metal ions. |
| UV-visible | 300 μM | Does MBL inhibitor binding affect the coordination number of the Co(II) ion(s)? Does MBL inhibitor binding change the electronic properties of the Co(II) ion(s). Does MBL inhibitor strip metal ions from MBL? | Requires non-native, Co(II)- substituted protein; requires large amounts of protein; relatively low-resolution information. |
| EPR | 1 mM | Does MBL inhibitor binding affect the spin coupling between the Co(II) ions? Does MBL inhibitor strip metal ions from MBL? | Requires non-native, Co(II)- substituted protein; requires large amounts of protein; requires a non-standard piece of equipment in most research programs. |
| NMR | 1 mM | Does MBL inhibitor binding change the positioning of metal binding ligands? Does MBL inhibitor strip metal ions from MBL? | Requires non-native, Co(II)- substituted protein; requires large amounts of protein; |
| Nano-ESI-MS | 5 μM | Is a ternary (enzyme-metal-inhibitor) complex present in the sample? | Under most cases, technique is not quantitative; can only yield information on the presence of a ternary complex but not its concentration. |
| Computational | N/A | Does the MBL inhibitor bind? Technique can provide information about binding modes, potential active site interactions, and be used to interpret other data. | Technique requires a model of the enzyme, usually a crystal structure |
The approach we employed was needed to investigate with accuracy the mechanisms utilized by these thiol-containing compounds to inhibit MBLs. Our structuring of this approach, beginning with simpler experiments utilizing low enzyme and inhibitor concentrations prior to conducting more complex spectroscopic experiments requiring higher quantities of protein and inhibitor, offers an efficient route to identify those inhibitors that have the potential for clinically relevancy. In contrast, conventional studies examining enzymatic activities of MBLs, alone or in combination with cell-based minimum inhibitory concentration assays, identify the capability of compounds to inhibit MBL activities yet provide little understanding of their inhibition mechanisms. Using our integrated biophysical approach, we determined that DL-thiorphan exhibits a number of similarities to L-captopril, including the mode of ternary complex formation through use of a bridging sulfur between the two active site zinc ions, and thus likely uses a similar inhibition mechanism. In contrast, the binding modes for tiopronin and 2,3-dimercaprol are dissimilar to both thiorphan and captopril. Our studies with tiopronin also match up well with a recent reanalysis of the X-ray diffraction data for NDM-1 in complex with tiopronin [11] that refutes the originally proposed binding mode [26]. In particular, this result shows that with two conflicting views of the previously-reported tiopronin crystal structure, our toolbox of biochemical and spectroscopic methods allowed us to confirm the most recent set of findings [11]. We have suggested an innovative approach that allows for a more thorough determination of the mechanism of inhibition of MBL inhibitors using multiple techniques in combination [13]. Although the four inhibitors tested here were previously identified as potential clinically-relevant inhibitors of MBLs, our data suggest additional research with a broad toolbox of approaches is needed to identify novel MBL inhibitors with potential for clinically-relevant MBL inhibition.
Supplementary Material
Funding Sources
The authors acknowledge funding from National Institutes of Health awards R01 GM111926 to RCP, DLT and MWC; R35 GM128595 to RCP; R01AI063517 to RCP; and R15 GM134454 to MWC. MWC and DLT acknowledge support from National Science Foundation award CHE-1509285.
Abbreviations
- EPR
Electron paramagnetic resonance
- IPTG
Isopropyl β-D-1-thiogalactopyranoside
- ITC
Isothermal titration calorimetry
- MβL
metallo-β-lactamase
- NDM
New Delhi MβL
- NMR
Nuclear magnetic resonance
- TCEP
Tris(2-carboxyethyl)phosphine hydrochloride
- TEV
Tobacco Etch Virus
- EDTA
ethylenediaminetetraacetic acid
- ICP-AES
Inductively Coupled Plasma-Atomic Emissions Spectroscopy
- LMCT
ligand-metal charge transfer
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Declarations of conflict of interest: none
References
- 1.CDC (2018) Biggest Threats and Data. https://www.cdc.gov/drugresistance/biggest_threats.html.
- 2.Cheng Z, Thomas PW, Ju L, Bergstrom A, Mason K, Clayton D, Miller C, Bethel CR, VanPelt J, Tierney DL, Page RC, Bonomo RA, Fast W, Crowder MW (2018) Evolution of New Delhi metallo-beta-lactamase (NDM) in the clinic: Effects of NDM mutations on stability, zinc affinity, and mono-zinc activity. J Biol Chem 293 (32):12606–12618. doi: 10.1074/jbc.RA118.003835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Crowder MW, Spencer J, Vila AJ (2006) Metallo-beta-lactamases: Novel weaponry for antibiotic resistance in bacteria. Accounts Chem Res 39 (10):721–728. doi: 10.1021/ar0400241 [DOI] [PubMed] [Google Scholar]
- 4.Naas T, Oueslati S, Bonnin RA, Dabos ML, Zavala A, Dortet L, Retailleau P, Iorga BI (2017) Beta-lactamase database (BLDB) – structure and function. Journal of Enzyme Inhibition and Medicinal Chemistry 32 (1):917–919. doi: 10.1080/14756366.2017.1344235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Drawz SM, Bonomo RA (2010) Three decades of beta-lactamase inhibitors. Clin Microbiol Rev 23 (1):160–201. doi: 10.1128/CMR.00037-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Palzkill T (2013) Metallo-beta-lactamase structure and function. Ann N Y Acad Sci 1277:91–104. doi: 10.1111/j.1749-6632.2012.06796.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mojica MF, Bonomo RA, Fast W (2016) B1-Metallo-beta-Lactamases: Where Do We Stand? Curr Drug Targets 17 (9):1029–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.King AM, Reid-Yu SA, Wang W, King DT, De Pascale G, Strynadka NC, Walsh TR, Coombes BK, Wright GD (2014) Aspergillomarasmine A overcomes metallo-beta-lactamase antibiotic resistance. Nature 510 (7506):503–506. doi: 10.1038/nature13445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klingler FM, Wichelhaus TA, Frank D, Cuesta-Bernal J, El-Delik J, Muller HF, Sjuts H, Gottig S, Koenigs A, Pos KM, Pogoryelov D, Proschak E (2015) Approved Drugs Containing Thiols as Inhibitors of Metallo-beta-lactamases: Strategy To Combat Multidrug-Resistant Bacteria. J Med Chem 58 (8):3626–3630. doi: 10.1021/jm501844d [DOI] [PubMed] [Google Scholar]
- 10.King DT, Worrall LJ, Gruninger R, Strynadka NC (2012) New Delhi metallo-beta-lactamase: structural insights into beta-lactam recognition and inhibition. J Am Chem Soc 134 (28):11362–11365. doi: 10.1021/ja303579d [DOI] [PubMed] [Google Scholar]
- 11.Raczynska JE, Shabalin IG, Minor W, Wlodawer A, Jaskolski M (2018) A close look onto structural models and primary ligands of metallo-β-lactamases. Drug Resistance Updates 40:1–12. doi: 10.1016/j.drup.2018.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Raczynska JE, Shabalin IG, Minor W, Wlodawer A, Jaskolski M (2018) A close look onto structural models and primary ligands of metallo-beta-lactamases. Drug Resist Updat 40:1–12. doi: 10.1016/j.drup.2018.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ju LC, Cheng Z, Fast W, Bonomo RA, Crowder MW (2018) The Continuing Challenge of Metallo-beta-Lactamase Inhibition: Mechanism Matters. Trends Pharmacol Sci 39 (7):635–647. doi: 10.1016/j.tips.2018.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen ALY, Thomas PW, Stewart AC, Bergstrom A, Cheng ZS, Miller C, Bethel CR, Marshal SH, Credille CV, Riley CL, Page RC, Bonomo RA, Crowder MW, Tierney DL, Fast W, Cohen SM (2017) Dipicolinic Acid Derivatives as Inhibitors of New Delhi Metallo-beta-lactamase-1. Journal of Medicinal Chemistry 60 (17):7267–7283. doi: 10.1021/acs.jmedchem.7b00407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li T, Wang Q, Chen F, Li X, Luo S, Fang H, Wang D, Li Z, Hou X, Wang H (2013) Biochemical characteristics of New Delhi metallo-beta-lactamase-1 show unexpected difference to other MBLs. PLoS One 8 (4):e61914. doi: 10.1371/journal.pone.0061914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aitha M, Marts AR, Bergstrom A, Moller AJ, Moritz L, Turner L, Nix JC, Bonomo RA, Page RC, Tierney DL, Crowder MW (2014) Biochemical, mechanistic, and spectroscopic characterization of metallo-beta-lactamase VIM-2. Biochemistry 53 (46):7321–7331. doi: 10.1021/bi500916y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tierney DL, Fee JA, Ludwig ML, Penner-Hahn JE (1995) X-ray absorption spectroscopy of the iron site in Escherichia coli Fe(III) superoxide dismutase. Biochemistry 34 (5):1661–1668. doi: 10.1021/bi00005a022 [DOI] [PubMed] [Google Scholar]
- 18.Cheng ZS, VanPelt J, Bergstrom A, Bethel C, Katko A, Miller C, Mason K, Cumming E, Zhang H, Kimble RL, Fullington S, Bretz SL, Nix JC, Bonomo RA, Tierney DL, Page RC, Crowder MW (2018) A Noncanonical Metal Center Drives the Activity of the Sediminispirochaeta smaragdinae Metallo-beta-lactamase SPS-1. Biochemistry 57 (35):5218–5229. doi: 10.1021/acs.biochem.8b00728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.King DT, Worrall LJ, Gruninger R, Strynadka NCJ (2012) New Delhi Metallo-β-Lactamase: Structural Insights into β-Lactam Recognition and Inhibition. Journal of the American Chemical Society 134 (28):11362–11365. doi: 10.1021/ja303579d [DOI] [PubMed] [Google Scholar]
- 20.Hawkins PCD, Skillman AG, Warren GL, Ellingson BA, Stahl MT (2010) Conformer Generation with OMEGA: Algorithm and Validation Using High Quality Structures from the Protein Databank and Cambridge Structural Database. J Chem Inf Model 50 (4):572–584. doi: 10.1021/ci100031x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR (2011) Open Babel: An open chemical toolbox. Journal of Cheminformatics 3 (1):33. doi: 10.1186/1758-2946-3-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meiler J, Baker D (2006) ROSETTALIGAND: Protein–small molecule docking with full side-chain flexibility. Proteins: Structure, Function, and Bioinformatics 65 (3):538–548. doi: 10.1002/prot.21086 [DOI] [PubMed] [Google Scholar]
- 23.Davis IW, Baker D (2009) RosettaLigand Docking with Full Ligand and Receptor Flexibility. Journal of Molecular Biology 385 (2):381–392. doi: 10.1016/j.jmb.2008.11.010 [DOI] [PubMed] [Google Scholar]
- 24.Ichihara O, Shimada Y, Yoshidome D (2014) The Importance of Hydration Thermodynamics in Fragment-to-Lead Optimization. ChemMedChem 9 (12):2708–2717. doi: 10.1002/cmdc.201402207 [DOI] [PubMed] [Google Scholar]
- 25.Bergstrom A, Katko A, Adkins Z, Hill J, Cheng Z, Burnett M, Yang H, Aitha M, Mehaffey MR, Brodbelt JS, Tehrani KHME, Martin NI, Bonomo RA, Page RC, Tierney DL, Fast W, Wright GD, Crowder MW (2018) Probing the Interaction of Aspergillomarasmine A with Metallo-β-lactamases NDM-1, VIM-2, and IMP-7. Acs Infect Dis 4 (2):135–145. doi: 10.1021/acsinfecdis.7b00106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klingler FM, Wichelhaus TA, Frank D, Cuesta-Bernal J, El-Delik J, Muller HF, Sjuts H, Gottig S, Koenigs A, Pos KM, Pogoryelov D, Proschak E (2015) Approved Drugs Containing Thiols as Inhibitors of Metallo-beta-lactamases: Strategy To Combat Multidrug-Resistant Bacteria. Journal of Medicinal Chemistry 58 (8):3626–3630. doi: 10.1021/jm501844d [DOI] [PubMed] [Google Scholar]
- 27.Yang H, Aitha M, Marts AR, Hetrick A, Bennett B, Crowder MW, Tierney DL (2014) Spectroscopic and mechanistic studies of heterodimetallic forms of metallo-beta-lactamase NDM-1. J Am Chem Soc 136 (20):7273–7285. doi: 10.1021/ja410376s [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Periyannan GR, Costello AL, Tierney DL, Yang KW, Bennett B, Crowder MW (2006) Sequential binding of cobalt(II) to metallo-beta-lactamase CcrA. Biochemistry 45 (4):1313–1320. doi: 10.1021/bi051105n [DOI] [PubMed] [Google Scholar]
- 29.Bebrone C (2007) Metallo-beta-lactamases (classification, activity, genetic organization, structure, zinc coordination) and their superfamily. Biochem Pharmacol 74 (12):1686–1701. doi: 10.1016/j.bcp.2007.05.021 [DOI] [PubMed] [Google Scholar]
- 30.Baum RR, James CD, Tierney DL (2017) Paramagnetic Resonance of High-Spin Co(II) in Biologically-Relevant Environments: Models to Metalloproteins In: Hanson G, Berliner L (eds) Future Directions in Metalloprotein and Metalloenzyme Research. Springer International Publishing, Cham, pp 33–54. doi: 10.1007/978-3-319-59100-1_3 [DOI] [Google Scholar]
- 31.Llarrull LI, Tioni MF, Kowalski J, Bennett B, Vila AJ (2007) Evidence for a dinuclear active site in the metallo-beta-lactamase BcII with substoichiometric Co(II). A new model for metal uptake. J Biol Chem 282 (42):30586–30595. doi: 10.1074/jbc.M704613200 [DOI] [PubMed] [Google Scholar]
- 32.Orellano EG, Girardini JE, Cricco JA, Ceccarelli EA, Vila AJ (1998) Spectroscopic Characterization of a Binuclear Metal Site in Bacillus cereus β-Lactamase II. Biochemistry 37 (28):10173–10180. doi: 10.1021/bi980309j [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.