

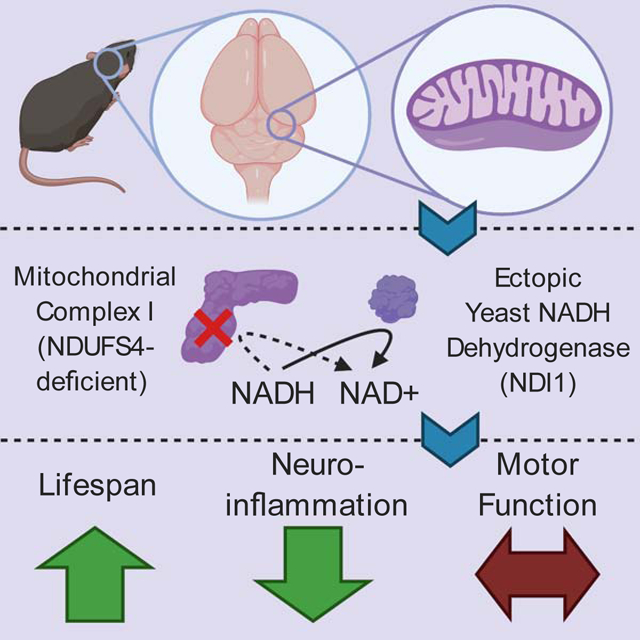
Summary:
Mitochondrial complex I regenerates NAD+ and proton pumps for TCA cycle function and ATP production, respectively. Mitochondrial complex I dysfunction has been implicated in many brain pathologies including Leigh Syndrome and Parkinson’s disease. We sought to determine whether NAD+ regeneration or proton pumping i.e. bioenergetic is the dominant function of mitochondrial complex I in protection from brain pathology. We generated a mouse that conditionally expresses the yeast NADH dehydrogenase (NDI1), a single enzyme that can replace the NAD+ regeneration capability of the 45-subunit mammalian mitochondrial complex I without proton pumping. NDI1 expression was sufficient to dramatically prolong lifespan without significantly improving motor function in a mouse model of Leigh Syndrome driven by the loss of NDUFS4, a subunit of mitochondrial complex I. Therefore, mitochondrial complex I activity in the brain supports organismal survival through its NAD+ regeneration capacity, while optimal motor control requires the bioenergetic function of mitochondrial complex I.
Keywords: Mitochondria, Leigh syndrome, NAD, neurometabolism
Graphical Abstract
Introduction:
The mitochondrial electron transport chain provides the proton-motive force that drives oxidative phosphorylation as well as the oxidation of NADH to NAD+ to allow the tricarboxylic acid cycle (TCA) to proceed (Chandel 2015). The TCA provides the building blocks for amino acids, nucleic acids, carbohydrates, and lipids. In mammals, a 45-subunit complex, mitochondrial complex I (MC1), couples the oxidation of NADH to proton pumping for the generation of ATP (Stroud et al., 2016). Dysfunction of MC1 has been implicated in neurodegenerative diseases such as Parkinson’s Disease and Leigh Syndrome (Area-Gomez et al., 2019). Given the essential role of MC1 in both NAD+ regeneration and ATP production, it is difficult to parse out which of these functions controls the effects of MC1 dysfunction in complex tissues like the brain.
Normally the bioenergetic and biosynthetic roles of MC1 are inseparable, because the oxidation of NADH and ubiquinone reduction are linked to the translocation of protons across the inner mitochondrial membrane to generate the protonmotive force. To dissect these two key functions of MC1 in vivo, we made use of the single subunit yeast enzyme NADH dehydrogenase (NDI1). In yeast, NDI1 serves as the major entry point into the electron transport chain, catalyzing the oxidation of NADH in the matrix like MC1. However, NDI1 does not proton pump to contribute to the production of ATP (Fig. 1a). Previously, NDI1 has been expressed in mammalian cells, both in vitro and through viral delivery (Seo et al., 1998, Wheaton et al., 2014), and it has been shown to increase the lifespan of Drosophila when ectopically expressed (Sanz et al., 2010).
Figure 1.
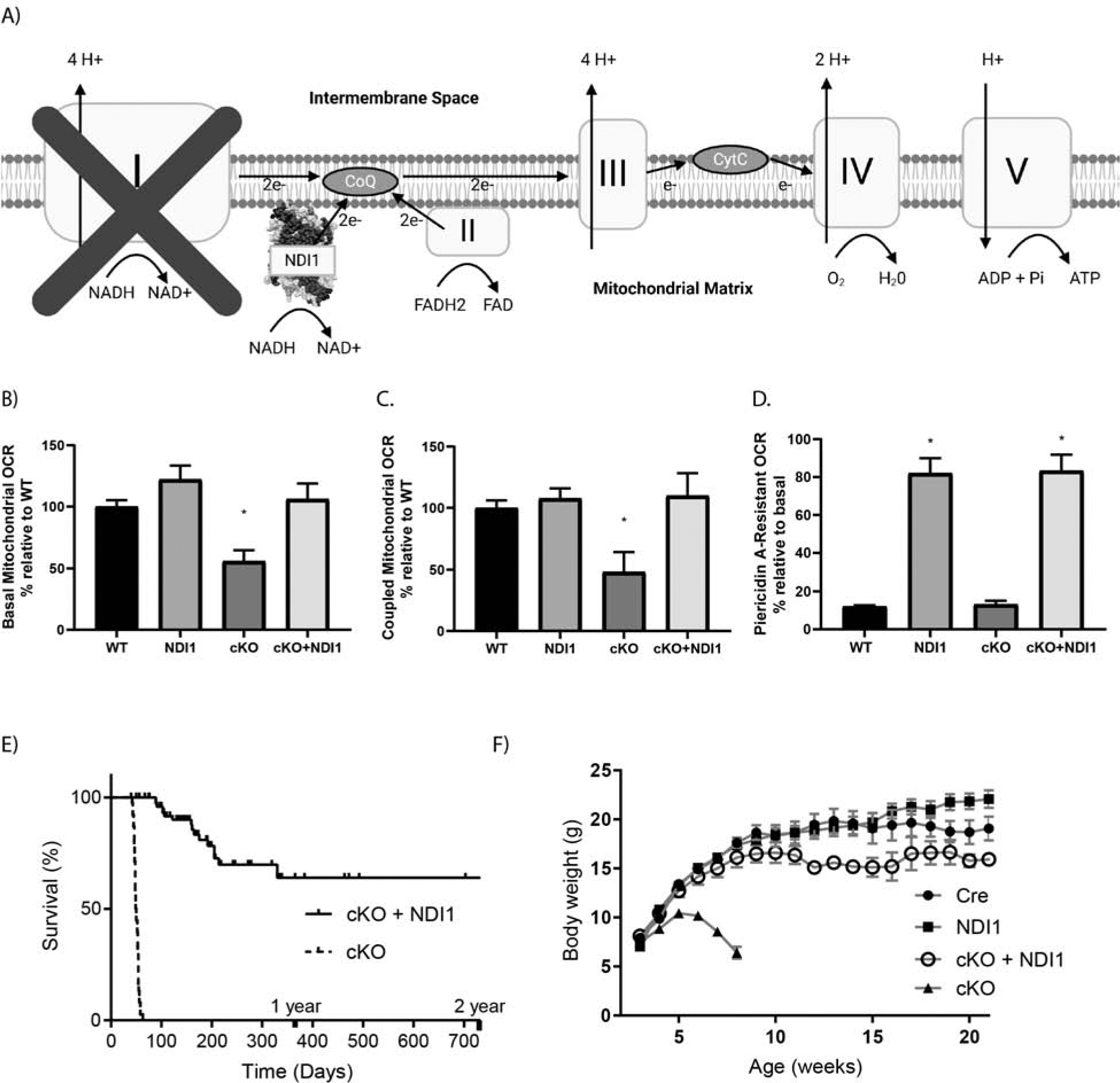
The yeast NDI1 protein is sufficient to rescue lifespan in a mouse model of Leigh Syndrome due to loss of mitochondrial complex I subunit NDUFS4. (A) Schematic of the mitochondrial electron transport chain with ectopic NDI1. (B) Basal oxygen consumption rate (OCR) of cerebellar granular neurons (CGNs) from postnatal day 2–7 mice. Data pooled from 6 independent experiments (N=7–14, ANOVA P=0.0008, Dunnett’s test to WT *P<0.05). (C) ATP-coupled OCR in CGNs with the ATPase inhibitor Oligomycin A. Data pooled from 4 independent experiments (N=4–11, ANOVA P=0.0008, Dunnett’s test to WT *P<0.05). (D) Percentage of basal OCR that is resistant to MC1 inhibitor Piericidin A at 500nM. (N=3–5, ANOVA P<0.0001, * P<0.05 Dunnett’s test to WT). (E) Kaplan-Meier curves of cKO vs cKO + NDI1. (N=40–59, Log-rank test P<0.0001). (F) Body weight in grams, cKO + NDI1 indicates 2 alleles of NDI1. (N=21–38, ANOVA P<0.0001, Dunnett’s test to Cre control P<0.0001). Data represent mean ± SEM.
We used a Cre/Lox approach to cause conditional loss of NDUFS4 expression of the yeast NDI1 in mice. NDUFS4 is an 18 kDa accessory subunit of MC1 involved in complex assembly and stability. Mice with NDUFS4 deficiency, globally or in the brain with Nestin-Cre recombination, recapitulates many features of human Leigh Syndrome, including the development of bilateral hyperintense MRI lesions, glial activation, seizures, ataxia, growth regression, and early mortality (Kruse et al., 2008, Quintana et al., 2010). Notably, deletion of NDUFS4 does not result in a complete loss of MC1 function, and the reduction in the enzymatic activity of MC1 varies by tissue (Kayser et al., 2016, Calvaruso et al., 2012). Interestingly, previous studies have shown that interventions specifically targeting NADH elevation such as NAD-precursor supplementation (Lee et al., 2019), mTOR inhibition (Johnson et al., 2014) and inhibition of mitochondrial serine catabolism (Yang et al., 2020). as well as hypoxia treatment (Jain et al. 2016) have demonstrated benefits in lifespan and function. A recent study demonstrated that the conditional loss of NDUFS4 in either GABAergic neurons (Gad2-Cre) or glutamatergic neurons (Vglut2-Cre) causes seizures or ataxia and breathing abnormalities, respectively (Bolea et al., 2019).
Results and Discussion
Expression of yeast NDI1 protein is sufficient to prolong lifespan in a Leigh Syndrome model
We designed a targeting construct containing Rosa26 homology arms and a Lox-STOP-Lox (LSL) cassette upstream of the NDI1 gene (Fig. S1A). Thus, yeast NDI1 is expressed only following Cre recombination and removal of the LSL cassette. We generated Ndufs4-floxed Nestin-Cre (cKO) mice and NDI1-LSL Ndufs4-floxed Nestin-Cre (cKO + NDI1) mice, where yeast NDI1 is expressed and NDUFS4 is lost specifically in the brain. Nestin cre causes LoxP recombination in neural lineage cells including astrocytes, oligodendrocytes, and neurons. Notably, microglia do not arise from a Nestin-positive precursor. To assess recombination, we measured NDI1 and NDUFS4 protein abundance in the brain (Fig. S1B, C). Additionally, we measured mRNA levels of NDI1 and Ndufs4 in sorted adult mouse brain cells. As expected, NDI1 expression and Ndufs4 deletion was observed in astrocytes, but not in microglia (Fig. S1D–F).
Recent single cell RNA-sequencing of the mouse brain found that in the cerebellum the Ndufs4 transcript is most highly expressed in granule neurons (CGNs) (Fig. S1G). We isolated CGNs and analyzed the basal oxygen consumption rate (OCR) (Fig. 1B) and OCR after treatment with the ATP synthase inhibitor oligomycin A (Fig. 1C) and the MC1 inhibitor piericidin A (Fig. 1D). CGNs expressing NDI1 demonstrated a mild increase in basal OCR (Fig. 1B). Loss of NDUFS4 in CGNs caused a significant decrease in both basal and coupled OCR that was rescued by NDI1 expression (Fig. 1B, C). Moreover, the CGNs from mice expressing NDI1 demonstrated resistance to OCR reduction upon treatment with the MC1 inhibitor piericidin A (Fig. 1B). Therefore, yeast NDI1 is expressed and functional in the brains of these mice.
NDUFS4 loss specifically in the brain causes pathologic changes resembling Leigh Syndrome, including reactive gliosis in the brainstem, abnormal breathing patterns, seizures, and ataxia (Kruse et al., 2008, Quintana et al., 2010). cKO mice die between the ages of day 45 and day 60 (Fig. 1E). Interestingly, when mice express yeast NDI1 in the cKO background (cKO + NDI1) their lifespan is drastically increased by a full order of magnitude in some mice, with a median survival greater than 1 year of age (Fig. 1E). Thus, a single yeast enzyme is sufficient to prolong the lifespan of a mouse in which the 45-subunit MC1 is dysfunctional. cKO mice reached an average weight of approximately 11g by 30–40 days of age and subsequently exhibited growth regression (Fig. 1F). In contrast, the cKO + NDI1 mice did not exhibit growth regression, but rather attained a body weight closer to their littermate controls (Fig. 1F).
Expression of yeast NDI1 is not sufficient to restore motor function in a Leigh Syndrome model.
To determine whether the NAD+ regeneration capability of NDI1 is sufficient to improve the progressive ataxia observed in NDUFS4-deficient mice, we measured coordination, balance, and ambulation by rotarod and open field behavioral testing. cKO + NDI1 mice after 2 months of age are almost completely incapable of performing the rotarod task due to severely impaired balance and coordination (Fig. 2A). NDI1 expression in the brain, in the absence of NDUFS4 deletion (NDI1 mice), does not impair the balance or coordination of the mice (Fig. 2A). Moreover, the cKO + NDI1 mice, like late stage cKO mice, have a diminished distance traveled compared to control mice (Fig. 2B). Due to ataxia and visual impairment (Quintana et al., 2010,), cKO mice exhibit increased variability in location preference with some mice spending significant proportions of time near the center, while others remain in periphery (Fig. 2C, D). This contrasts with control mice, which spend the majority of their time at the periphery (Fig. 2C, D). The cKO + NDI1 mice display a high variance of location preference similar to cKO mice (Fig. 2C, D).
Figure 2.
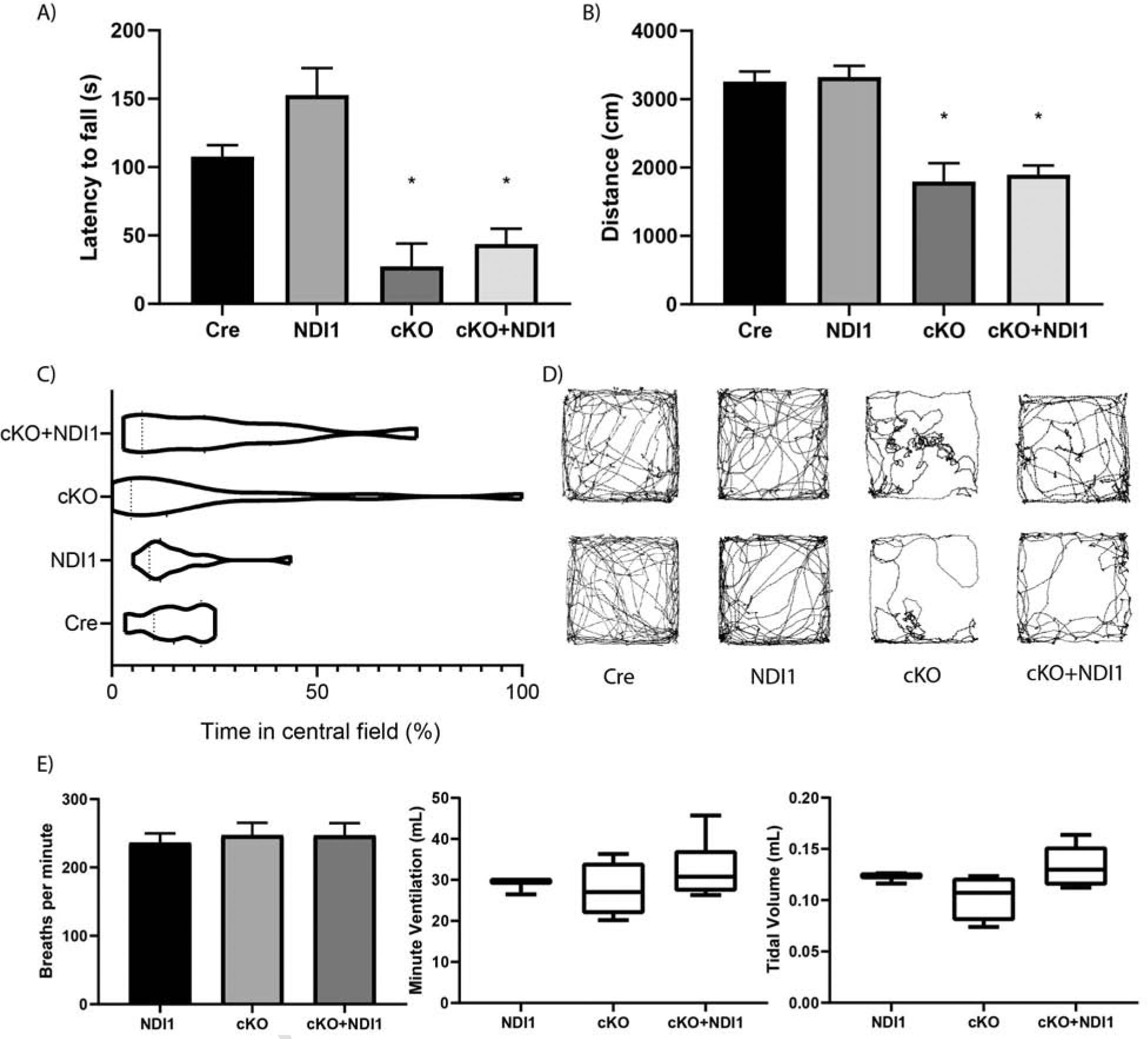
NDI1 expression in the brain does not rescue progressive ataxia or alter breathing parameters in a mouse model of Leigh Syndrome. (A) Latency to fall in an accelerating rotarod challenge in seconds (s)(N=8–22, ANOVA P<0.0001, Dunnett’s test to Cre control *P<0.01). (B) Distance traveled in 300 seconds in a novel open field environment (N=12–22, ANOVA P<0.0001, Dunnett’s test to Cre control *P<0.0001). (C) Percent time spent in the central zone of a novel open field environment. (Bartlett’s test P<0.0001). (D) Two representative path images of the open field test for each mouse genotype. (E) Whole body plethysmography breathing parameters: frequency, minute ventilation, and tidal volume. (N=3–6, ANOVA not significant). Data represent mean ± SEM.
Nestin Cre can also cause to recombination in skeletal muscle satellite cells, thus we profiled the quadriceps from ataxic cKO + NDI1 mice and NDI1 control mice over 12 months of age using H&E and trichrome stain (Fig. S2A–B). The average cross-sectional fiber diameter was not changed, and we saw no evidence of central nuclei (a marker of muscle regeneration after damage) or ragged red fibers (a pathognomonic finding in mitochondrial disease) (Fig. S2A–C). There was no change in the level of NDUFS4 protein in quadriceps (Fig. S2D). Also, there was no significant changes in soluble metabolites extracted from quadriceps of cKO + NDI1 and NDI1 mice (Fig. S2E–G). These data do not suggest that muscle dysfunction contributes to persistent ataxia.
NDUFS4-deficient mice are thought to develop brainstem dysfunction induced respiratory failure (Quintana et al., 2012). A recent report described an alternate cause of mortality due to seizures in GABAergic neuron-specific NDUFS4-deficient mice (Bolea et al., 2019). We observed tonic-clonic seizures in late stage cKO mice, but not the cKO + NDI1 mice. The average breathing frequency, tidal volume, and minute ventilation of both cKO mice and cKO + NDI1 mice were not significantly different compared to control mice but did display increased variability (Fig. 2E). Furthermore, there were no prolonged apneas in either cKO + NDI1 or cKO mice, despite some evidence of abnormal breathing patterns (Fig. S3A). Therefore, we speculate that seizures are the dominant cause of death in cKO mice.
Expression of yeast NDI1 is sufficient to prevent MRI lesions and microglial activation in a Leigh Syndrome model.
A key diagnostic feature of human mitochondrial disease is the presence of symmetric bilateral hyperintense T2-weighted MRI lesions, and MRI lesions develop in NDUFS4 deficient mice (Ferrari et al., 2017). MRI imaging of late stage cKO mice (8 weeks of age) revealed bilateral hyperintensities in the olfactory bulb and cerebellum near the fourth ventricle (Fig. 3A). cKO + NDI1 mice did not develop detectable MRI lesions in the cerebellum or brainstem, and only developed hyperintensities in the olfactory bulb at 6 months of age (Fig. 3A). Pathological analysis at 7 weeks of age showed microglial activation in the olfactory bulb, cerebellum, and brainstem of cKO mice, but not in age-matched cKO + NDI1 and control mice (Fig. 3B, S3C). We also observed activated astrocyte morphology in the brainstem, olfactory bulb, and cerebellum of cKO mice, but without a significant increase in GFAP+ cell numbers (Fig. S3B, Fig. 3C).
Figure 3.
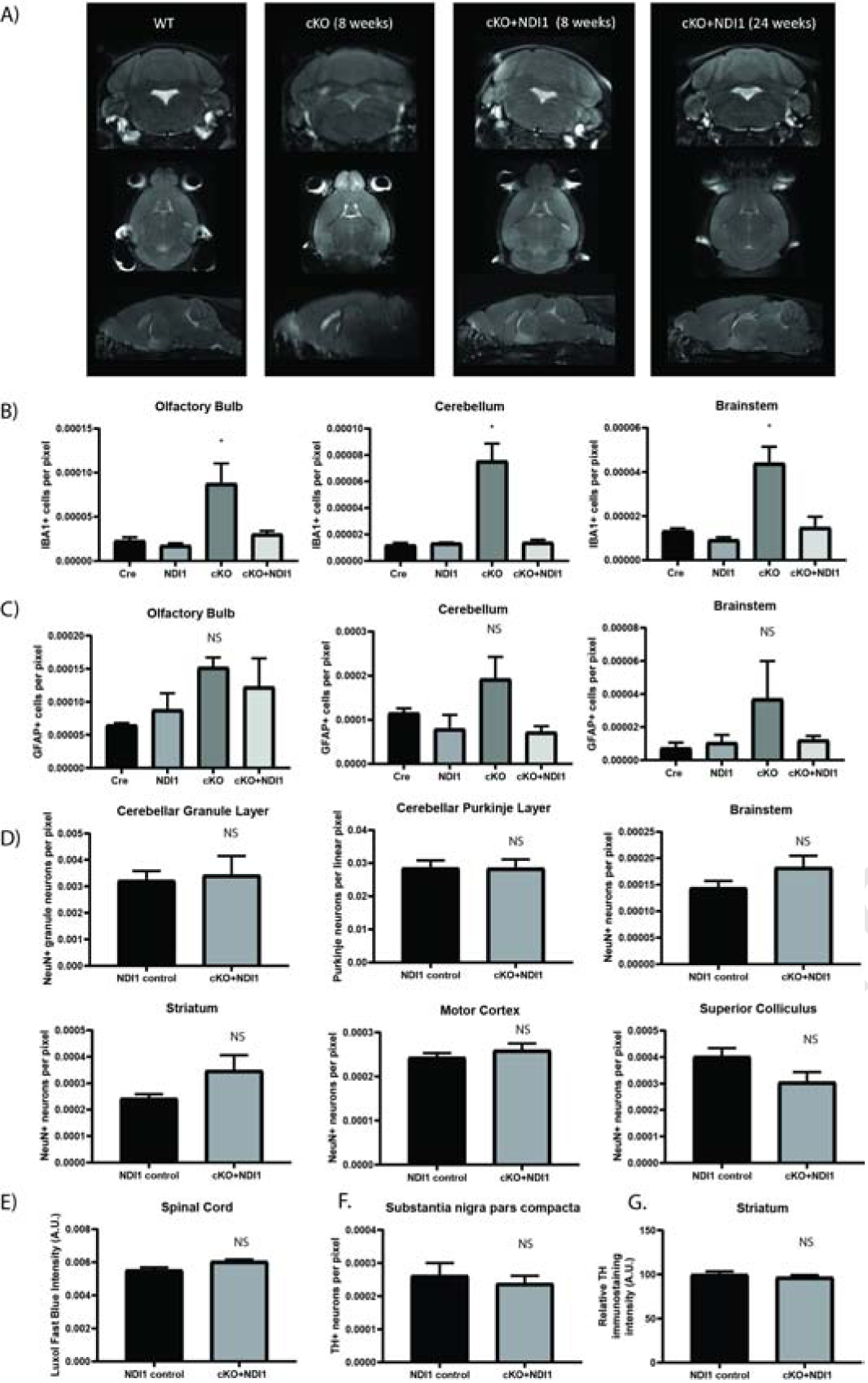
NDI1 expression prevents the formation of inflammatory glial activation and MRI lesions and does not cause neurodegeneration in a mouse model of Leigh Sydrome. (A) Representative MRI T2-weighted turbo spin echo sequence images. Coronal, horizontal, and sagittal planes are shown at the approximate level where cKO mice exhibit bilateral hyperintense lesions in the olfactory bulb, brainstem, and cerebellum. (B) Quantification of IBA1+ microglia in brain regions demonstrating cKO inflammation at 7–8 weeks of age (N=3–4, ANOVA with Dunnet’s test vs WT P<0.05*). See also Fig. S3. (C) Quantification of GFAP+ astrocytes at 7–8 weeks of age (N=3). (D) Quantification of neurons from motor control brain regions at over 1 year of age (N=6). (E) Quantification of spinal cord Luxol fast blue staining (N=3). (F) Quantification of tyrosine hydroxylase+ dopaminergic neurons (N=6) (G) Tyrosine hydroxylase+ immunoreactivity in striatum (N=5–6). (C-G) ANOVA and/or T-test not significant. Data represent mean ± SEM. See also Fig. S3.
To evaluate the persistent ataxia in cKO + NDI1 mice, we quantified neuron abundance in regions of motor control after one year of age (Fig. 3D). There were no significant changes in cell number and no evidence of inflammation or neurodegeneration between cKO + NDI1 and NDI1 controls. We also evaluated the spinal cord by H&E and Luxol fast blue myelin staining and observed no evidence of demyelination or pathology (Fig. 3E). Tyrosine hydroxylase (TH) quantification of dopaminergic neurons in the substantia nigra pars compacta displayed no significant changes between cKO + NDI1 mice and controls (Fig. 3F). There was also similar TH immunoreactivity in the striatum (Fig. 3G). There was no significant neurodegeneration or neuroinflammation in the brain regions involved in motor function in the cKO + NDI1 mice. The motor impairment in these mice is likely due to functional (rather than structural) deficits in cerebellar or brainstem circuits containing glutamatergic neurons, due to the lack of proton pumping by NDI1 thus its inability to contribute towards ATP production.
Expression of yeast NDI1 normalizes cerebellum metabolites and gene expression in a Leigh Syndrome model.
Next, we performed whole tissue metabolomic and transcriptomic analyses on cerebella (Fig. 4). We compared cKO and Cre control mice and identified the top 50 metabolites with altered relative abundance (Fig. 4A). We observed that cKO + NDI1 mice trended towards metabolite level normalization. Of note, the cerebella of cKO mice have increased lactate levels compared to the cerebella of control mice (Fig. 4B), consistent with previous models of mitochondrial dysfunction. To further characterize the ability of NDI1 expression to alter central carbon metabolism, we used U13C-glucose tracing in isolated astrocytes (Fig. S4A). Comparison of WT astrocytes to cKO revealed small but statistically significant decreases in the labeling of glutamate and citrate without significant changes in the labeling of lactate or TCA cycle intermediates (Fig. S4A). The relative lack of changes between WT and cKO is consistent with recent in vivo metabolic tracing experiments in global NDUFS4 deficient mice (Yang et al., 2020). Expression of NDI1, however, did cause significant changes in flux to the TCA cycle. NDI1-expressing astrocytes showed increased labeling of citrate, aspartate, malate, succinate, and glutamate, and decreased labeling of pyruvate and alanine (Fig. S4A). Therefore, NDI1 expression is sufficient to alter carbon flux from glucose into the TCA cycle. Next we used mitochondrial complex I (pyruvate plus malate) versus complex II (succinate) substrates to drive OCR in permeabilized astrocyte cultures. MC1-dependent OCR demonstrated a comparable pattern of OCR among the genotypes as that observed for CGNs in Figure 1B, with trends toward increased oxygen consumption in NDI1-expressing cells and decreased oxygen consumption in cKO cells, however, these changes were not statistically significant (Fig. S4B). Complex II-dependent OCR was similar in the astrocytes of all four genotypes (Fig. S4C). The NADH/NAD+ ratio was significantly increased following mitochondrial complex I inhibition in WT astrocytes while NDI1-expressing astrocytes were resistant to NADH/NAD+ ratio elevation following MC1 inhibition (Fig. S4D).
Figure 4.
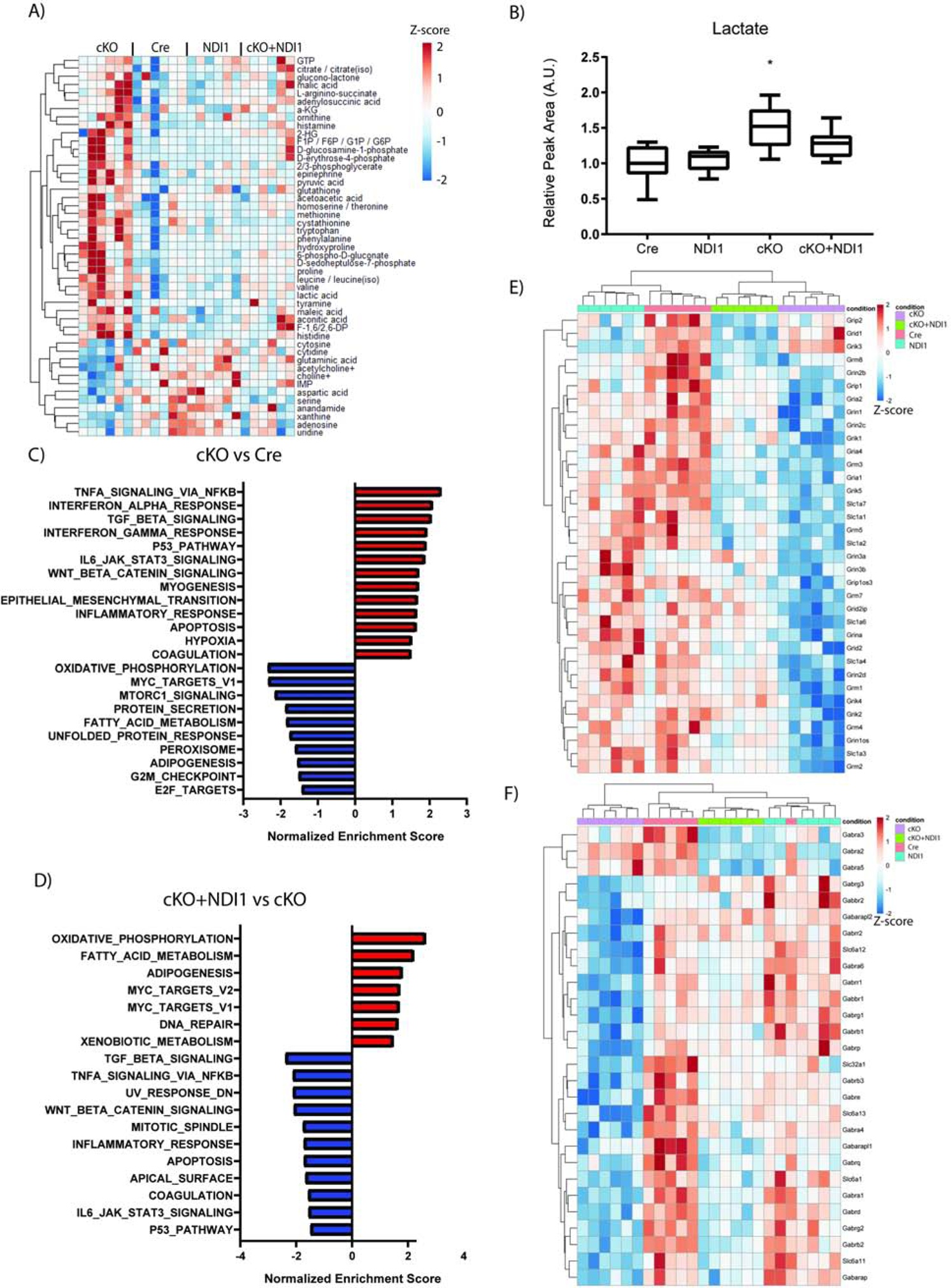
NDI1 expression in the brain partially restores cerebellum metabolites and gene expression in a mouse model of Leigh Syndrome. (A) Relative cerebellum metabolite levels. See also Fig. S4. (B) Relative abundance of cerebellum lactate (N=6 per genotype, Data represent mean ± SEM. ANOVA P=0.0003, Dunnett’s multiple comparisons test Cre compared to cKO *P=0.002). (C-F) Cerebellum differential gene expression analysis comparing Cre to cKO (C) and cKO to cKO + NDI1 (D) for enrichment of MSigDB Hallmark gene sets. (E) Heatmap of glutamate receptor and transporter gene expression. (F) Heatmap of GABA receptor and transporter gene expression. For A and C-F, N=6 per genotype: 3 male and 3 female mice at approximately 7 weeks of age.
RNA-sequencing and differential gene expression analysis on cerebellum tissue revealed numerous significant changes in gene expression among the four groups (Fig. S4E). Complete-linkage clustering dendrograms of samples (columns) and genes (rows) was carried out and revealed independent clustering of cKO samples, while cKO + NDI1 clustered with Cre and NDI1 controls (Fig. S4E). Pairwise differential gene expression analysis, followed by Gene Set Enrichment Analysis (GSEA), suggested that expression of a number of key pathways that are dysregulated in the cerebella of cKO mice are significantly enriched in the cerebella of cKO + NDI1 mice compared with cKO mice (Fig. 4C, D). This implies that the function of these pathways is fully or partially restored in cKO + NDI1 mice compared with cKO mice. For example, the gene sets for Oxidative Phosphorylation, MYC Targets, and Fatty Acid Metabolism are significantly depleted in cKO versus Cre (Fig. 4E), and these same pathways are significantly enriched in cKO + NDI1 versus cKO (Fig. 4F). Several of the most dysregulated genes among the groups were GABA receptor genes. Given the recent publication of conditional NDUFS4 deficiency in GABAergic and glutamatergic neurons (Bolea et al., 2019), we decided to specifically interrogate the transcriptomic data for GABA and glutamate receptors and channels. Glutamate receptor and transporter genes were downregulated in cKO mice compared to cKO+NDI1 (Figure 4E). GABA receptor and transporter genes were also downregulated in cKO mice compared to cKO + NDI1 mice and clustered separately (Fig. 4F). We surmise this provides intriguing preliminary data about the relative degree of rescue in different neuron types due to NDI1 expression. Since Gad2-Cre conditional NDUFS4 deficient mice exhibit seizure-induced-mortality at a time point consistent with Nestin-Cre mortality, a reduction in seizure burden due to NDI1 expression in GABAergic neurons in the midbrain may be a key mechanism by which lifespan is prolonged in our model. Furthermore, a population of glutamatergic neurons in the brainstem and/or cerebellum that remain impaired in our model may cause the persistent ataxia despite the increased survival.
Overall, our results indicate that a single yeast enzyme capable of regenerating mitochondrial NAD+ from NADH is sufficient to increase the lifespan, but not maintain the motor function, of mice with impairment of the 45-subunit MC1 in the brain. Perhaps restoration of NADH oxidation combined with a compensatory increase in glycolysis is sufficient to prevent reactive glial inflammation, but fails to completely restore neuronal function, as the latter may require ATP from MC1. Further genetic dissection of the cell-type specificity of the observed lifespan extension with NDI1 will be of interest in future studies, particularly with the use of Gad2-Cre and Vglut2-Cre to more directly determine the relative impact of these two key neuron populations in the prevention of seizures versus breathing abnormalities and ataxia, respectively.
Limitations of the present study:
A limitation of this study is our inability to measure the in situ mitochondrial NADH/NAD+ ratio or ATP production rates in this Leigh Syndrome model. Technical limitations, including the rapid changes that occur post-mortem, and the inability to rapidly isolate mitochondria in this model have contributed to our inability to detect them in tissue. Recent work in the literature has also demonstrated that the changes in whole tissue NADH/NAD+ ratio are quite subtle in global NDUFS4 deficient mice (Yang et al., 2020). Another limitation is that we have not definitively identified the mechanism by which cKO + NDI1 mice remain ataxic. Importantly, we do not observe significant neuroinflammation or neurodegeneration in our mouse model, and do not see any evidence of muscle or spinal cord pathology.
RESOURCE AVAILABILITY
Lead Contact:
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact: Navdeep Chandel (nav@northwestern.edu)
Materials Availability:
Mouse line generated in this study is freely available for distribution with an MTA with Northwestern University.
Data and Code Availability:
Datasets are available at Mendeley Data doi:10.17632/yp9tk59ngx.1 and GEO GSE149616.
Experimental Model and Subject Details:
Mouse models
NDUFS4 floxed and Nestin Cre mice were obtained from The Jackson Laboratory (B6.129S4-Ndufs4tm1Rpa/J JAX stock #026963 and B6.Cg-Tg(Nes-cre)1Kln/J JAX stock #003771, respectively). NDI1 knock-in mice were generated at the Northwestern Transgenic and Targeted Mutagenesis Laboratory. Briefly, we designed a targeting construct containing 5’ and 3’ Rosa26 homology arms as well as a Lox-STOP-Lox cassette upstream of the NDI1 gene from Saccharomyces cerevisiae (Fig. S1A). The targeting construct, which contains a neomycin resistance gene cassette, was electroporated into C57BL/6 embryonic stem cells. Neomycin-resistant ES clones were picked, and PCR was used to confirm proper incorporation of the targeting construct into the Rosa26 locus. Two independent ES clones containing the NDI1-LSL allele were injected into blastocysts to produce germline-transformed heterozygous NDI1-LSL mice. For generation of Kaplan-Meier survival plots, mice that were determined to have severe ataxia, were moribund, or lost greater than 10% body weight were euthanized and included as events (deaths) while data points were censored when non-terminal animals were euthanized for experimentation and tissue analysis. Animals were housed in Northwestern University’s Center for Comparative Medicine in ventilated microisolator cages with automatic water access, ad libitum fed standard rodent chow (Envigo/Teklad LM-485), housed in rooms with standard 12 hour light/dark cycles, and an ambient temperature of 23 °C with every other week cage changes in accordance with Northwestern University’s Center for Comparative Medicine and IACUC policies. Animal welfare was evaluated daily. The genetic background is C57BL/6J. Several distinct age ranges were used as noted in the text and figure legends: 0–1 weeks of age for ex vivo cellular analyses, the “late stage” of cKO disease corresponding to 7–8 weeks of age, and a later time point for persistent ataxic cKO + NDI1 and controls at 12–14 months of age. Males and females used for all studies. Subgroup analysis revealed no significant differences based on sex in the experimental mice, control mice had different body weight based on sex, but were averaged for the purpose of clarity. All animal procedures were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) at Northwestern University.
NDI1 Genotyping Primers:
Confirmation of Cre recombination (LSL Excision/NDI1 Expression) PCR:
- NDI1 LSL allele 180bp, NDI1 expressing allele 290bp
- CAG forward primer 5’ – CAACGTGCTGGTTATTGTGC
- Neo reverse primer 5’ – TCGCCTTCTTGACGAGTTCT
- NDI1 reverse primer 5’ – AACCCAGTATCAGCACGTTTG
NDI1-LSL allele PCR:
- Rosa26 wt allele (no NDI1) 224bp, NDI1-LSL knock-in allele 509bp
- Rosa B forward 5’ – GAGTTCTCTGCTGCCTCCTG
- Rosa B reverse 5’ – CCGACAAAACCGAAAATCTG
- WPRE B forward 5’ – GACGAGTCGGATCTCCCTTT
Method Details:
Oxygen consumption and mitochondrial complex I sensitivity in cerebellar granule neurons
Oxygen consumption of cerebellar granule neurons was carried out using a Seahorse XFe96 Analyzer. Cultured neurons basal oxygen consumption was determined by 3 baseline measurements of oxygen consumption followed by the injection of 500nM Piericidin A (an inhibitor of mammalian mitochondrial complex I. 3 measurements were made after injection 1 and then a second combined injection of 1uM Antimycin A and Piericidin A to completely inhibit electron transport through combined mitochondrial complex I and III blockade. The residual oxygen consumption after Piericidin A and Antimycin A combined injection (injection 2) was measured and subtracted from baseline and injection 1 measurements to determine the basal oxygen consumption and the complex I inhibitor-resistant oxygen consumption, respectively. Protocol for extraction and culture of early post-natal cerebellar granule neurons was adapted from previously published methods (Lee et al., 2009). Prior to experimentation, mouse genotype was determined, and tissue culture plates were coated with poly-D-lysine (Fisher) at two different concentrations: 100μg/mL for pre-plating/glial dishes and 500μg/mL for cerebellar granule neuron plates. The following day the developing cerebellum of individual early postnatal mice was removed, and meninges were removed under a dissecting microscope. Next, tissue was incubated at 37°C for 15 minutes in Papain from the Papain Dissociation System Kit (Worthington) combined with 1mg/mL of DNAse I. Tissue was then triturated using P1000 pipette tips pre-coated with serum and the suspension was allowed to settle for 30 seconds to 1 minute to allow large undissociated pieces to settle. Using serum-coated pipette tips, cells remaining in suspension were transferred to a new centrifuge vial and centrifuged at 200×g for 5 minutes. Cell pellets were resuspended in Minimum Essential Media + albumin-ovomucoid inhibitor. Resuspended cells were carefully layered over an additional 1mL of albumin-ovomucoid inhibitor solution in a new centrifuge vial and centrifuged at 70×g for 6 minutes, pelleting dissociated cells and leaving membrane fragments and lipid at the surface. Next, the cells were resuspended, passed through a 70-micron filter, and plated on pre-plating plates in Neurobasal A medium supplemented with 10% serum, glutamax, antibiotic/antimycotic, and KCl. Cells were allowed to settle for 20 minutes. After incubation, granule neurons and neuron progenitors were dislodged with gentle tapping while larger glia remained adhered. The pre-plating step for glial removal was repeated. Finally, an enriched population for cerebellar granule neurons was resuspended in serum-free media containing Neurobasal A Medium supplemented with 2% B-27, glutamax, antibiotic/antimycotic, and KCl and plated directly onto poly-D-lysine-coated Seahorse plates and cultured for 5 days prior to oxygen consumption analysis.
Open Field Test and Accelerating Rotarod Challenge
Spontaneous activity in an open field chamber (56 cm × 56 cm) was recorded with LimeLight4 animal tracing software to assess activity and exploratory behavior for 300 seconds. Distance travelled during the test as well as the percentage time spent in the perimeter of the chamber as compared with the center (based on the central 9 boxes of a 5×5 grid of the open field) were recorded. The rotarod uses an accelerating rotating cylindrical rod upon which mice balance and walk forward to prevent themselves from falling. Mice were acclimated to the rotarod equipment for 2 days at a constant rate of 12 RPM for 60 seconds for 4 trials per day with at least 5 minutes between trials. Next, the mice were tested for 3 days with an accelerating program from 4 to 40 RPM over 300 seconds for 4 trials per day with at least 5 minutes between trials. Rotarod operation and automatic detection of falls was used with Rod software. Open Field Testing and Accelerating Rotarod Challenge were carried out using equipment at the Northwestern Behavioral Phenotyping Core Facility.
Magnetic Resonance Imaging
MRI imaging was performed at the Small Animal Molecular Imaging Lab of CTI (Feinberg School of Medicine, Northwestern University) on a ClinScan 7 T Scanner (Bruker, Germany) using a mouse head 4-channel phase array coil as receiver and a volume quadrature coil as transmitter. The imaging protocol included a 3D Flash multi-echo sequence (TR = 40 ms, TEs = 2.53, 6.09, 9.65, 13.21 ms, flip angle = 15, voxel size = 0.15×0.15×0.15 mm3) and a high resolution 2D T2-RARE (TR = 5940 ms, TE = 41 ms; voxel size = 0.09×0.09×0.5 mm3).
Histology
For histological analyses mice at approximately 7 weeks of age (late stage of disease for cKO mice) or over 12 months of age were selected. Mice were euthanized and perfused with cold PBS supplemented with heparin and either 4% paraformaldehyde or 10% neutral buffered formalin and tissues were post-fixed for >24 hours in paraformaldehyde or formalin. Spinal cords were then decalcified using 0.5M EDTA pH 8.0. Fixed brains, quadriceps muscles, and spinal cords were trimmed and embedded in Paraffin blocks and sectioned at the Northwestern Mouse Histology and Phenotyping Core facility. Tissues were stained with hematoxylin and eosin (H&E), luxol fast blue (spinal cord only), and/or Gomori’s Trichrome stain (muscle only) using 4-micron sections. Immunohistochemistry (IHC) was carried out on 4-micron sections using tyramide signal amplification and avidin-biotin complex methods (automated) and using 3,3’-Diaminobenzidine (DAB) chromogen. 3–4 individual mice were analyzed per group for astrocyte IHC, microglia IHC, and spinal cord staining. 6 mice were analyzed per group for muscle and neuron quantification. Images were taken using a Nuance spectral camera on a Zeiss Axioskop upright microscope at the Northwestern Center for Advanced Microscopy. DAB staining quantification was carried out blinded in ImageJ using portions of 20x images with the number of cells determined using the Multi-point tool normalized to the analyzed area of pixels. For Purkinje neurons, the cell count was instead normalized to the analyzed linear pixel length rather than area. For quantification of Luxol intensity and tyrosine hydroxylase immunoreactivity in the striatum, the mean pixel intensities on a greyscale of uniformly DAB or Luxol stained portions of 20x images of striatum or spinal cord were measured in ImageJ, then the inverse of this value was used as a proxy for immunoreactivity and each individual value was normalized as a percentage of the average of controls. Histology was reviewed by a trained muscle and neuropathologist.
Metabolite measurement
Mice were euthanized and cerebellum was rapidly isolated, and flash frozen in liquid nitrogen. Samples were stored at −80°C until extraction. Soluble metabolites were extracted directly from tissue using cold methanol/water (80/20, v/v) at approximately 1μL per 50μg of tissue. Tissue was disrupted for 15 seconds by ultrasonication for brain (Branson Sonifier 250), or by rotor-stator homogenization for muscle (QIAGEN TissueRuptor II). Protein was precipitated by incubation at −80°C. Debris were pelleted by centrifugation at 18,000×g for 15 min at 4°C. The supernatant was transferred to a new tube and evaporated to dryness using a SpeedVac concentrator (Thermo Savant). Metabolites were reconstituted in 50% acetonitrile in analytical-grade water, vortex-mixed, and centrifuged to remove debris. Samples were analyzed by Ultra-High-Performance Liquid Chromatography and High-Resolution Mass Spectrometry and Tandem Mass Spectrometry (UHPLC-MS/MS). Specifically, the system consisted of a Thermo Q-Exactive in line with an electrospray source and an Ultimate3000 (Thermo) series HPLC consisting of a binary pump, degasser, and auto-sampler outfitted with an Xbridge Amide column (Waters; dimensions of 4.6mm × 100mm and a 3.5μm particle size). Mobile phase A contained 95% (vol/vol) water, 5% (vol/vol) acetonitrile, 10mM ammonium hydroxide, 10mM ammonium acetate, pH = 9.0; and mobile phase B was 100% Acetonitrile. The gradient was as follows: 0 min, 15% A; 2.5 min, 30% A; 7 min, 43% A; 16 min, 62% A; 16.1–18 min, 75% A; 18–25 min, 15% A with a flow rate of 400μL/min. The capillary of the ESI source was set to 275°C, with sheath gas at 45 arbitrary units, auxiliary gas at 5 arbitrary units, and the spray voltage at 4.0kV. In positive/negative polarity switching mode, an m/z scan range from 70 to 850 was chosen and MS1 data was collected at a resolution of 70,000. The automatic gain control (AGC) target was set at 1×106 and the maximum injection time was 200 ms. The top 5 precursor ions were subsequently fragmented, in a data-dependent manner, using the higher energy collisional dissociation (HCD) cell set to 30% normalized collision energy in MS2 at a resolution power of 17,500. Data acquisition and analysis were carried out by Xcalibur 4.1 software and Tracefinder 4.1 software, respectively (both from Thermo Fisher Scientific). The peak area for each detected metabolite was normalized by the total ion current which was determined by integration of all of the recorded peaks within the acquisition window. Downstream analysis was carried out using Microsoft Excel and R. The list of the top 50 candidate metabolites for differential abundance between Cre and cKO cerebella was created by sorting metabolites by p value of a t-test comparing 6 Cre and 6 cKO mice (3 male and 3 female mice of each genotype) at approximately 7 weeks of age which is the late stage of cKO disease. Heatmaps generated using R package Pheatmaps after transformation of peak areas for each metabolite to Z scores (for brain) and using MetaboAnalyst 4.0 for muscle. Pathway analysis of KEGG compound IDs, principal components analysis score plots, and multiple comparisons adjusted T-tests for muscle were performed using MetaboAnalyst 4.0 (Chong et al., 2018).
U13C-Glucose Flux
Cultured astrocyte-enriched primary cultures from the cerebellum and brainstem from mice less than 7 days of age were extracted and cultured for 7 days. Astrocyte-enriched primary cultures were then trypsinized and plated at 500,000 cells per mouse in 6-well plates. The following day, normal growth media was replaced with glucose flux media (Glucose-free DMEM powder supplemented with sodium bicarbonate, HEPES, antibiotics and 10% serum with 5mM U13C-Glucose and 2mM glutamine, pH 7.4) for 6 hours. After incubation, cells were rinsed with ice cold 0.9% NaCl, then 80% methanol was added to plates at a ratio of 700 cells per μL. Plates were then incubated at −80°C for 20 minutes. Cells were then extracted and homogenized using cell scrapers, vortexed for 1 minute, and centrifuged in a refrigerated table-top microcentrifuge at maximum speed for 10 minutes. Metabolite-containing supernatants were then processed and measured by mass-spectrometry as described above.
RNA Sequencing
6 mice per group (3 male and 3 female) were euthanized at approximately 7 weeks of age and the cerebellum was rapidly dissected and flash frozen in liquid nitrogen. Whole cerebellum RNA was extracted using a QIAGEN RNeasy Plus Mini Kit. Tissues were homogenized in RLT Plus Buffer + BME with a QIAGEN TissueRuptor II handheld rotor-stator homogenizer. After tissue homogenization, QIAshredder spin columns were used followed by gDNA elimination columns and an on-column DNase digestion with Ambion RNase-free DNase during the RNeasy RNA preparation manufacturer protocol. RNA was quantified and QC was checked using an Agilent 4200 TapeStation RNA ScreenTape. mRNA libraries were prepared using NEBNext Ultra Kit with polyA selection (New England BioLabs). Libraries were sequenced using a Next-Seq 500 High Output for 75 cycles (Illumina). Raw BCL read files were demultiplexed and FASTQ files were generated using bcl2fastq and trimmed using Trimmomatic (Bolger et al., 2014). Next, the reads were aligned to the mouse mm10 reference genome using STAR to generate BAM files (Dobin et al., 2013). HTSeq was used to count reads in the exons of genes (Anders et al., 2015). Likelihood ratio tests for all samples and all detected transcripts and pairwise differential gene expression analyses were carried out using the R package DESeq2 (Love et al., 2014). Gene set enrichment analysis was carried out using the Broad Institute GSEA software after conversion to human homologue gene symbols (Subramanian et al., 2005). The pairwise differential gene expression output generated from DESeq2 was submitted as a pre-ranked list based on the DESeq2 Wald Statistic (stat column) output sorting genes from significantly upregulated to significantly downregulated. GSEA was performed on Hallmark Gene Sets from the GSEA Molecular Signatures Database (Liberzon et al., 2015). Publicly available Drop-seq data from the mouse brain was accessed and a graph of NDUFS4 gene expression by cell type cluster in the cerebellum was generated through the DropViz platform (Saunders et al., 2018).
Protein and mRNA Quantification
Protein quantification was carried out on protein extracts from whole brain tissue from approximately 7-week-old mice and from mice >6 months of age and on muscle of mice >12 months of age. Mice were euthanized and tissue was flash frozen in liquid nitrogen. Protein was extracted using lysis buffer (Cell Signaling 9803S) and ultrasonication (Branson Sonifier 250) for brain and rotor-stator homogenization (QIAGEN TissueRuptor II) for muscle. Protein was quantified using the Pierce BCA assay (Thermo Fisher Scientific) and NDI1 and NDUFS4 were measured using a ProteinSimple Wes Simple Western System, an automated capillary based immunoassay. Approximately 30μg of total protein was loaded for each sample and antibodies were loaded at 1:100 (NDUFS4), 1:500 (NDI1), 1:200 (Vinculin), and 1:1000 (Tubulin). There is not a commercially available Anti-NDI1 antibody; however, we received a small amount of an Anti-NDI1 antibody from Dr. Eric Dufour from the University of Tampere, Finland to confirm the detection of NDI1 in brain tissue. Furthermore, we used the Anti-NDUFS4 antibody (Abcam 137064), the Anti-Vinculin antibody (Cell Signaling 12901), and the Anti-Tubulin antibody (Cell Signaling 2144). Virtual blot images were created within ProteinSimple Compass software. Peak area was normalized to loading control peak area and plotted in GraphPad Prism. mRNA was measured in cell types sorted from adult mouse brain to confirm the specific and appropriate Cre recombination in brain cell types. Nestin Cre should cause recombination in all adult brain cell types except for microglia. Therefore, we measured NDUFS4 and NDI1 mRNA in microglia and astrocytes from mice expressing NDI1 and conditional NDUFS4 knockout due to Nestin Cre expression. Brains from adult mice >7 weeks of age were dissociated for 30 minutes using the Papain Dissociation System Kit (Worthington) and Octomacs C Tube (Milltenyi Biotec) tissue homogenization. Cells were separated using a 27% Percoll centrifugation step to remove myelin debris. Cells were sorted directly into cell lysis buffer RLT+BME using a BD FACS Aria II Flow Cytometer at the Northwestern RHLCCC Flow Cytometry Core Facility. Antibody panel for cell selection consisted of CD11b and CD45.2 for microglia and ACSA2 for astrocytes. Specifically, we used an Anti-CD11b antibody (eBioscience 48-0112-80), an Anti-CD45.2 antibody (eBioscience 56-0454-82), and an Anti-ACSA2 antibody (Milltenyi Biotec 130-102-365). RNA was extracted using a QIAGEN RNeasy Plus Micro Kit following manufacturer protocol and qPCR was carried out using a CYBRFast 1-step RT-qPCR Lo-Rox Kit (Tonbo Biosciences) following manufacturer protocol with a BioRad CFX384 Real Time System. qPCR Primers:
NDUFS4 forward 5’-GAGCACATCCACTTGGAAGC
NDUFS4 reverse 5’-GATGTGCTCTTCTGGAACACC
NDI1 forward 5’-GCCGAAGAAGTCCAAATTCAC
NDI1 reverse 5’-CGACAGCCGTTCTCAGAT
Actin forward 5’-CTAAGGCCAACCGTGAAAAG
Actin reverse 5’-ACCAGAGGCATACAGGGACA
NADH/NAD+ Ratio
NADH/NAD+ ratio was determined on enriched astrocyte primary cultures from post-natal mouse cerebellum using the NAD/NADH Glo Assay (Promega) following the manufacturer-supplied protocol. Primary cultures enriched for cerebellar astrocytes were derived using a protocol adapted from the Cerebellar Granule Neuron culture protocol detailed in the section on oxygen consumption and mitochondrial complex I sensitivity in cerebellar granule neurons and Schildge et al. (2013). Pre-plating plates after cerebellar granule neuron culture were maintained as mixed cultures and enriched for astrocytes by providing astrocyte-selective media (DMEM supplemented with 10% serum) for 7 days followed by trypsinization and replating of the confluent astrocyte layer.
Whole Body Plethysmography
A Buxco Finepointe 4-site whole body plethysmography apparatus was used to record the breathing pattern and ventilation dynamics of awake, unrestrained mice. Frequency, tidal volume, and minute ventilation were recorded during a 20-minute period after 120 minutes of acclimation in the calibrated cylindrical chambers. Values were not adjusted for body weight.
Electron transport chain diagram generation and graphical abstract
The diagrams were generated using BioRender software, and NDI1 crystal structure representation was derived from Iwata et al. 2012.
Quantification and statistical analyses
Statistical analyses and graph generation were carried out using GraphPad Prism 8 and R. Figure alignment was carried out using Adobe Illustrator. Statistical tests are referenced in the figure legends. All T-tests were two-tailed and F tests were carried out to assess the assumption of equal group variances all other statistical assumptions were not directly assessed. ANOVA refers to an ordinary one-way ANOVA carried out with Dunnett’s multiple comparisons test comparing all groups to control unless otherwise indicated in the figure legend, additionally Brown-Forsythe tests were carried out to assess the assumption of equal group variances all other statistical assumptions were not directly assessed. Error bars indicate SEM. N refers to biological replicates (individual animals) per genotype group.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| IBA1 | Wako | NC9288364 |
| NDUFS4 | Abcam | 137064 |
| NDI1 | Eric Dufour – University of Tampere Finland | N/A |
| Vinculin | Cell Signaling | 12901 |
| Tubulin | Cell Signaling | 2144 |
| CD11b | eBioscience | 48-0112-80 |
| ACSA2 | Milltenyi Biotec | 130-102-365 |
| CD45.2 | eBioscience | 56-0454-82 |
| GFAP | Biocare | CP040A |
| Tyrosine Hydroxylase | Millipore Sigma | AB152 |
| NeuN | Abcam | Ab104225 |
| Bacterial and Virus Strains | ||
| N/A | ||
| Biological Samples | ||
| N/A | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Piericidin A | Sigma | A8674 |
| Antimycin A | Sigma | P4368 |
| Oligomycin A | Sigma | 75351 |
| U13C-Glucose | Cambridge Isotopes | CLM-1396–1 |
| Critical Commercial Assays | ||
| Seahorse XFe96 Flux Pack | Agilent | 102416–100 |
| Wes Simple Western System | Proteinsimple | SM-W004 |
| RNeasy plust mini kit | QIAGEN | 74134 |
| CYBRFast 1-step RT-qPCR Lo-Rox Kit | Tonbo Biosciences | 31–5201 |
| Ambion DNAse I | Invitrogen | AM2222 |
| Papain Dissociation System | Worthington | LK003150 |
| NAD/NADH Glo Assay | Promega | G9071 |
| NEBNext Ultra II Kit | NEB | E7645 |
| Octomacs C Tube | Milltenyi Biotec | 130-093-237 |
| Deposited Data | ||
| RNAseq data | This manuscript – GEO | GSE149616 |
| DropViz Single Cell RNA-seq data mouse brain | Dropviz.org | N/A |
| Raw data | This manuscript – Mendeley Data | doi:10.17632/yp9tk59ngx.1 |
| Experimental Models: Cell Lines | ||
| N/A | ||
| Experimental Models: Organisms/Strains | ||
| Mus musculus – NDUFS4 flox | The Jackson Laboratory | 026963 |
| Mus musculus – Nestin Cre | The Jackson Laboratory | 003771 |
| Mus musculus – NDI1 | This manuscript | N/A |
| Oligonucleotides | ||
| CAG forward primer 5’ – CAACGTGCTGGTTATTGTGC | Integrated DNA Technologies | N/A |
| Neo reverse primer 5’ – TCGCCTTCTTGACGAGTTCT | Integrated DNA Technologies | N/A |
| NDI1 reverse primer 5’ – AACCCAGTATCAGCACGTTTG | Integrated DNA Technologies | N/A |
| Rosa B forward primer 5’ – GAGTTCTCTGCTGCCTCCTG | Integrated DNA Technologies | N/A |
| Rosa B reverse primer 5’ – CCGACAAAACCGAAAATCTG | Integrated DNA Technologies | N/A |
| WPRE B forward primer 5’ – GACGAGTCGGATCTCCCTTT | Integrated DNA Technologies | N/A |
| qPCR NDUFS4 forward 5’ GAGCACATCCACTTGGAAGC | Integrated DNA Technologies | N/A |
| qPCR NDUFS4 reverse 5’ GATGTGCTCTTCTGGAACACC | Integrated DNA Technologies | N/A |
| qPCR NDI1 forward 5’ GCCGAAGAAGTCCAAATTCAC | Integrated DNA Technologies | N/A |
| qPCR NDI1 reverse 5’ CGACAGCCGTTCTCAGAT | Integrated DNA Technologies | N/A |
| qPCR Actin forward 5’ CTAAGGCCAACCGTGAAAAG | Integrated DNA Technologies | N/A |
| qPCR Actin reverse 5’ ACCAGAGGCATACAGGGACA | Integrated DNA Technologies | N/A |
| Recombinant DNA | ||
| Rosa 26 NDI1 targeting construct | This paper | N/A |
| Software and Algorithms | ||
| Trimmomatic | Bolger et al., 2014 | http://www.usadellab.org/cms/?page=trimmomatic |
| STAR | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| HTSeq | Anders et al., 2015 | https://htseq.readthedocs.io/en/master/ |
| DESeq2 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| GSEA | Subramanian et al., 2005 | https://www.gsea-msigdb.org/gsea/index.jsp |
| DropViz | Saunders et al., 2018 | http://dropviz.org/ |
| MetaboAnalyst | Chong et al., 2018 | https://www.metaboanalyst.ca/MetaboAnalyst/home.xhtml |
| Tracefinder | Thermo Scientific | https://www.thermofisher.com/us/en/home/industrial/mass-spectrometry/liquid-chromatography-mass-spectrometry-lc-ms/lc-ms-software/lc-ms-data-acquisition-software/tracefinder-software.html |
| Xcalibur | Thermo Scientific | https://www.thermofisher.com/order/catalog/product/OPTON-30965 |
| RStudio | RStudio, PBC | https://github.com/rstudio/rstudio |
| Mango | Research Imaging Institute, UTHSC | http://ric.uthscsa.edu/mango/ |
| Limelight | ActiMetrics | https://www.actimetrics.com/products/limelight/ |
| Prism | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Image J | NIH | https://imagej.nih.gov/ij/ |
| FlowJo | FlowJo | https://www.flowjo.com/ |
| FinePointe | Buxco | https://www.datasci.com/products/software/finepointe-software |
| Other | ||
Acknowledgments:
We thank the following core facilities at Northwestern: Transgenic and Targeted Mutagenesis Laboratory, CTI Small Animal Imaging Core, Behavioral Phenotyping Core, Pulmonary NextGen Sequencing Core, RHLCCC Metabolomics Core, Center for Advanced Microscopy, RHLCCC Flow Cytometry Core Facility. Histology services were provided by the Northwestern University Mouse Histology and Phenotyping Laboratory which is supported by NCI P30-CA060553 awarded to the RHLCC. We thank Michele Hadhazy and Elizabeth McNally for the use of Whole-Body Plethysmography. We thank the Center for Comparative Medicine staff. We thank Elizabeth Bartom for the use of the Ceto pipeline. We thank Eric Dufour from the University of Tampere for NDI1 antibody. Funding for this project was provided by the following NIH grants NSC: NIH2PO1HL071643-11A1, NIH1R35CA197532-01, NIH1PO1AG049665-01. GSM: NIH/NCI T32CA09560. CRR: NHLBI T32 HL076139-11. PAR: NHLBI F32HL136111. DSM: NINDS R25 NS070695. CMH: K08CA155764 and R01NS102669. PAR was supported by an American Thoracic Society/Boehringer Ingelheim Partner grant.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests: NSC is on SAB of Raphael Pharmaceuticals.
References:
- Anders S, Pyl PT, Huber W. (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Area-Gomez E, Guardia-Laguarta C, Schon EA, Przedborski S. (2019). Mitochondria, OxPhos, and neurodegeneration: cells are not just running out of gas. J Clin Invest 129, 34–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolea I, Gella A, Sanz E, Prada-Dacasa P, Menardy F, Bard AM, Machuca-Marquez P, Eraso-Pichot A, Modol-Caballero G, Navarro X, Kalume F, Quintana A. (2019). Defined neuronal populations drive fatal phenotype in a mouse model of Leigh Syndrome. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30,2114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvaruso MA, Willems P, van den Brand M, Valsecchi F, Kruse S, Palmiter R, Smeitink J, Nijtmans L. (2012). Mitochondrial complex III stabilizes complex I in the absence of NDUFS4 to provide partial activity. Hum Mol Genet 21, 115–20. [DOI] [PubMed] [Google Scholar]
- Chandel NS. (2015). Navigating metabolism. (Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press; ) [Google Scholar]
- Chong J, Soufan O, Li C, Caraus I, Li S, Bourque G, Wishart DS, Xia J. (2018). MetaboAnalyst 4.0: towards more transparent and integrative metabolomics analysis. Nucleic Acids Res 46, W486–W94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari M, Jain IH, Goldberger O, Rezoagli E, Thoonen R, Cheng KH, Sosnovik DE, Scherrer-Crosbie M, Mootha VK, Zapol WM. (2017). Hypoxia treatment reverses neurodegenerative disease in a mouse model of Leigh syndrome. Proc Natl Acad Sci USA 114, E4241–E50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata M, Lee Y, Yamashita T, Yagi T, Iwata S, Cameron AD, Maher MJ. (2012). The structure of the yeast NADH dehydrogenase (Ndi1) reveals overlapping binding sites for waterand lipid-soluble substrates. Proc Natl Acad Sci USA 109, 15247–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain IH, Zazzeron L, Goli R, Alexa K, Schatzman-Bone S, Dhillon H, Goldberger O, Peng J, Shalem O, Sanjana NE, Zhang F, Goessling W, Zapol WM, Mootha VK. (2016). Hypoxia as a therapy for mitochondrial disease. Science 352, 54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SC, Yanos ME, Kayser EB, Quintana A, Sangesland M, Castanza A, Uhde L, Hui J, Wall VZ, Gagnidze A, Oh K, Wasko BM, Ramos FJ, Palmiter RD, Rabinovitch PS, Morgan PG, Sedensky MM, Kaeberlein M. (2013). mTOR inhibition alleviates mitochondrial disease in a mouse model of Leigh syndrome. Science 342, 1524–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayser EB, Sedensky MM, Morgan PG. (2016). Region-Specific Defects of Respiratory Capacities in the Ndufs4(KO) Mouse Brain. PLoS One 11, e0148219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse SE, Watt WC, Marcinek DJ, Kapur RP, Schenkman KA, Palmiter RD. (2008). Mice with mitochondrial complex I deficiency develop a fatal encephalomyopathy. Cell Metab 7, 312–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CF, Caudal A, Abell L, Nagana Gowda GA, Tian R. (2019). Targeting NAD(+) Metabolism as Interventions for Mitochondrial Disease. Sci Rep 9, 3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HY, Greene LA, Mason CA, Manzini MC. (2009). Isolation and culture of post-natal mouse cerebellar granule neuron progenitor cells and neurons. J Vis Exp 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. (2015). The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 1, 417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana A, Kruse SE, Kapur RP, Sanz E, Palmiter RD. (2010). Complex I deficiency due to loss of Ndufs4 in the brain results in progressive encephalopathy resembling Leigh syndrome. Proc Natl Acad Sci USA 107, 10996–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana A, Zanella S, Koch H, Kruse SE, Lee D, Ramirez JM, Palmiter RD. (2012). Fatal breathing dysfunction in a mouse model of Leigh syndrome. J Clin Invest 122, 2359–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz A, Soikkeli M, Portero-Otin M, Wilson A, Kemppainen E, McIlroy G, Ellila S, Kemppainen KK, Tuomela T, Lakanmaa M, Kiviranta E, Stefanatos R, Dufour E, Hutz B, Naudi A, Jove M, Zeb A, Vartiainen S, Matsuno-Yagi A, Yagi T, Rustin P, Pamplona R, Jacobs HT. (2010). Expression of the yeast NADH dehydrogenase Ndi1 in Drosophila confers increased lifespan independently of dietary restriction. Proc Natl Acad Sci USA 107, 9105–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders A, Macosko EZ, Wysoker A, Goldman M, Krienen FM, de Rivera H, Bien E, Baum M, Bortolin L, Wang S, Goeva A, Nemesh J, Kamitaki N, Brumbaugh S, Kulp D, McCarroll SA. (2018). Molecular Diversity and Specializations among the Cells of the Adult Mouse Brain. Cell 174, 1015–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schildge S, Bohrer C, Beck K, Schachtrup C. (2013). Isolation and culture of mouse cortical astrocytes. J Vis Exp 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo BB, Kitajima-Ihara T, Chan EK, Scheffler IE, Matsuno-Yagi A, Yagi T. (1998). Molecular remedy of complex I defects: rotenone-insensitive internal NADH-quinone oxidoreductase of Saccharomyces cerevisiae mitochondria restores the NADH oxidase activity of complex I-deficient mammalian cells. Proc Natl Acad Sci USA 95, 9167–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroud DA, Surgenor EE, Formosa LE, Reljic B, Frazier AE, Dibley MG, Osellame LD, Stait T, Beilharz TH, Thorburn DR, Salim A, Ryan MT. (2016). Accessory subunits are integral for assembly and function of human mitochondrial complex I. Nature 538, 123–6. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 102, 15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheaton WW, Weinberg SE, Hamanaka RB, Soberanes S, Sullivan LB, Anso E, Glasauer A, Dufour E, Mutlu GM, Budigner GS, Chandel NS. (2014). Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. Elife 3, e02242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Canaveras JCG, Chen Z, Wang L, Liang L, Jang C, Mayr JA, Zhang Z, Ghergurovich JM, Zhan L, Joshi S, Zhixian H, McReynolds MR, Su X, White E, Morscher RJ, Rabinowitz JD. (2020). Serine Catabolism Feeds NADH when Respiration Is Impaired. Cell metab 10.1016/j.cmet.2020.02.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Datasets are available at Mendeley Data doi:10.17632/yp9tk59ngx.1 and GEO GSE149616.