

SUMMARY:
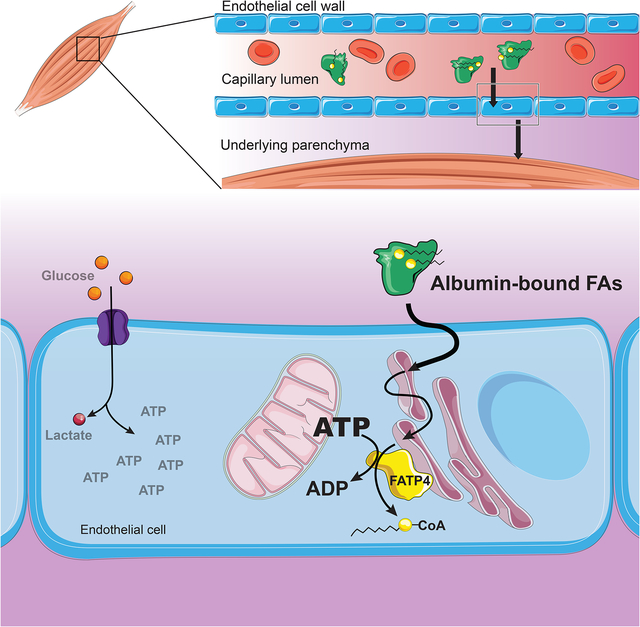
Most organs use fatty acids (FAs) as a key nutrient, but little is known of how blood-borne FAs traverse the endothelium to reach underlying tissues. We conducted a small-molecule screen and identified niclosamide as a suppressor of endothelial FA uptake and transport. Structure/activity relationship studies demonstrated that niclosamide acts through mitochondrial uncoupling. Inhibitors of oxidative phosphorylation and the ATP/ADP translocase also suppressed FA uptake, pointing principally to ATP production. Decreasing total cellular ATP by blocking glycolysis did not decrease uptake, indicating that specifically mitochondrial ATP is required. Endothelial FA uptake is promoted by fatty acid transport protein 4 (FATP4) via its ATP-dependent acyl-CoA synthetase activity. Confocal microscopy revealed that FATP4 resides in the endoplasmic reticulum (ER), and that endothelial ER is intimately juxtaposed with mitochondria. Together, these data indicate that mitochondrial ATP production, but not total ATP levels, drives endothelial FA uptake and transport via acyl-CoA formation in mitochondrial/ER microdomains.
Graphical Abstract
eTOC Blurb
Ibrahim et al. discover that mitochondrial (but not glycolytic) ATP regulates endothelial fatty acid uptake and transport. Endothelial mitochondria are closely juxtaposed to endoplasmic reticulum, and ATP locally produced by mitochondria is used by FATP4, which resides in the ER, to promote fatty acid uptake via its ATP-dependent acyl-CoA synthetase activity.
INTRODUCTION:
Endothelial cells (ECs) make up the innermost lining of all blood vessels (Aird, 2010). The capillary endothelium presents a barrier to the transport of solutes, particularly in those organs that harbor continuous, unfenestrated endothelium (Rose and Goresky, 1977; Sukriti et al., 2014). Fuels such as sugars and fats must therefore be actively transported across this tight barrier to the surrounding tissue. Organs like the skeletal muscle and the heart contain a continuous endothelium and rely heavily on fatty acids (FAs) as a source of fuel (Van der Vusse et al., 1998). FAs travel the blood stream largely in two forms: esterified as triglycerides in lipoprotein particles like chylomicrons and VLDL, or as free FAs non-covalently conjugated to albumin (Niot and Besnard, 2003; van der Vusse, 2009). If esterified, FAs are liberated from triglycerides by lipoprotein lipase, located on the luminal side of the endothelium (Frayn and Langin, 2003; Goldberg, 1996). Thus, no matter the mode of transport, FAs must cross the endothelium to reach the underlying parenchyma. Recent work has indicated that this process can be physiologically regulated in a paracrine fashion by factors secreted from the underlying tissue. For example, fat-consuming oxidative muscle fibers and cardiomyocytes secrete VEGFB, a member of the VEGF family, which then promotes FA uptake and transport in ECs (Hagberg et al., 2010). Likewise, muscle also secretes 3-hydroxy-isobutyrate (3-HIB), an intermediate catabolic product of valine, to promote trans-endothelial fatty acid transport (Jang et al., 2016a). These and other regulatory mechanisms coordinate muscle metabolism with FA delivery (Arany et al., 2008).
Little is known, however, of how FAs cross the endothelial barrier in response to these physiological signals. Various mechanisms have been proposed, including paracellular transport; flip-flop into, and diffusion within, endothelial membranes; and active intracellular transport (Iso et al., 2013; Kampf et al., 2006; Komarova and Malik, 2010; Mehrotra et al., 2014; Minshall and Malik, 2006). Animals lacking endothelial CD36, a multi-functional surface receptor that facilitates FA uptake via unclear mechanisms, have significant reductions in FA uptake in heart and other organs with continuous endothelium, demonstrating that ECs present a barrier to fatty acid transport (Pepino et al., 2014; Son et al., 2018). The identification of fatty acid transport proteins (FATPs) that can promote FA import in various cell types have strengthened the argument that FA uptake is an intracellular, protein-regulated process (Anderson and Stahl, 2013; Black et al., 2009; Glatz et al., 2010; Pohl et al., 2004). In ECs, FATP3 and FATP4 are required for VEGFB-induced and 3HIB-induced FA uptake (Hagberg et al., 2010; Jang et al., 2016a). However, how FATPs are regulated, and how they themselves mediate FA uptake, remains poorly understood.
Here, we undertook a high-throughput small-molecule screen to identify novel processes that mediate and control the uptake of FA in ECs. Surprisingly, we found that endothelial mitochondria play a key role in FA uptake and transport. Specifically, the ATP produced within mitochondria, but not from glycolysis, drives the mechanism by which the FATPs promote endothelial FA uptake.
RESULTS:
Niclosamide inhibits endothelial FA uptake and transport
For our small-molecule screen, we employed a luminescent FA uptake system (Henkin et al., 2012). Briefly, a compound comprised of luciferin covalently bound via a disulfide bond to palmitic acid (henceforth termed FFA-Luc) is non-covalently conjugated to bovine serum albumin (BSA) and administered to luciferase-expressing ECFCs (LUCEs). Once taken up by these endothelial cells, the FFA-Luc is cleaved by the reducing condition of the cytosol to liberate luciferin, which is then oxidized by luciferase to produce a photon. Detection of luminescence thus acts as a proxy for FA uptake (Figure 1A). Pilot experiments determined that 4 μM FFA-Luc conjugated to 3 μM BSA produced the highest Z-factor (Z’ > 0.5), using the stimulatory effects of 3-HIB as a positive control (Figure S1A). Using these concentrations, we found that uptake of FFA-Luc occurs within seconds and plateaus at approximately five minutes (Figure S1B). Additionally, calculating the area under these curves demonstrated that LUCEs increased FFA-Luc uptake over 3-fold in response to 3-HIB stimulation, indicating the robustness of the assay.
Figure 1. Chemical screening identifies niclosamide as a potent inhibitor of endothelial FA uptake and transport, mediated via mitochondrial uncoupling.
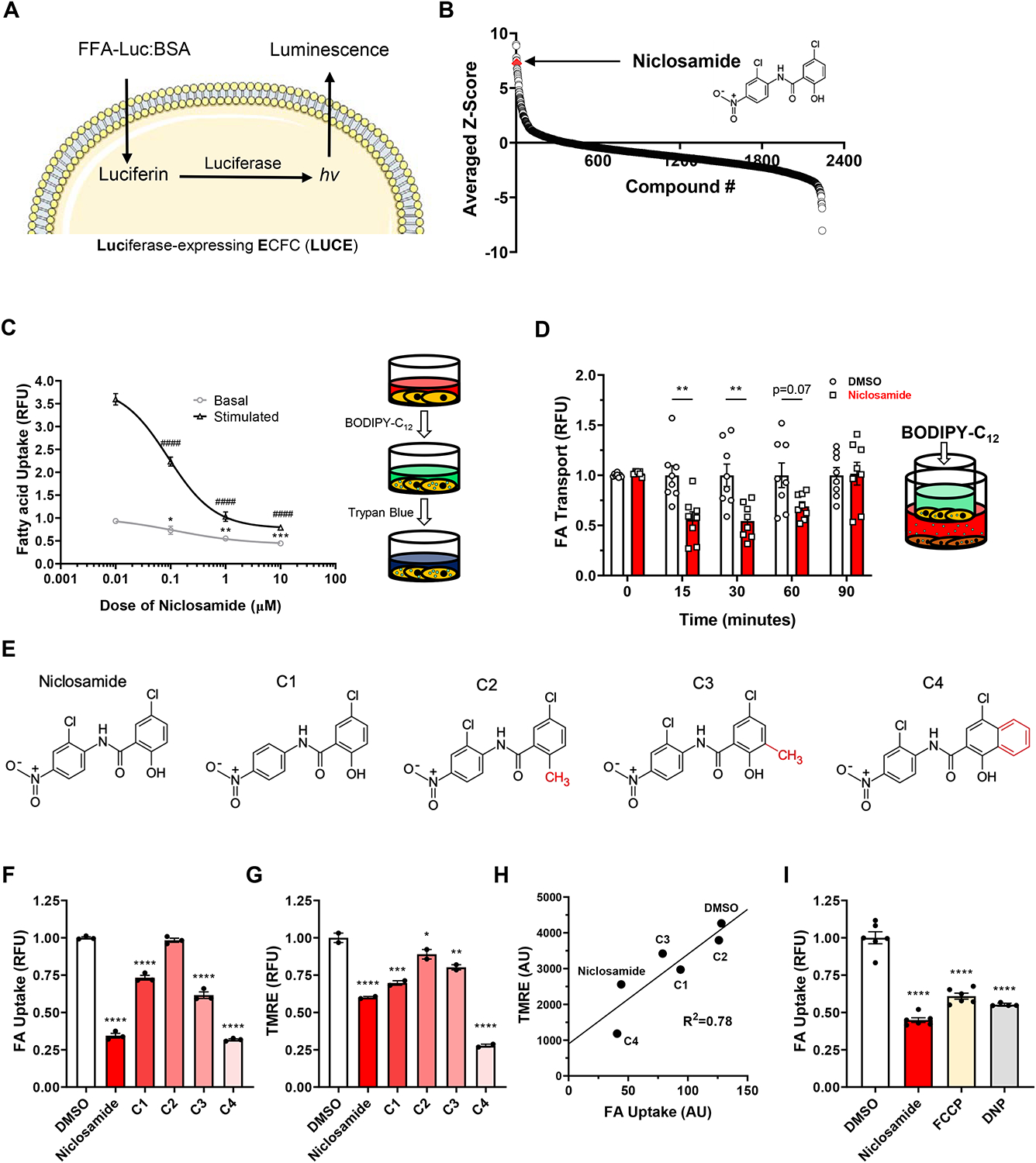
(A) Schematic for the luminescent FA uptake assay used for chemical screening. Palmitic acid bound via disulfide bond to luciferin is given to ECFCs expressing luciferase. The reducing environment of the cytosol frees the luciferin, which is then oxidized to produce light.
(B) Niclosamide (red diamond) ranks highly in terms of average z-score among the chemicals administered in the primary screening, indicating strong inhibition of FA uptake. The chemical structure is indicated on the right side of the graph.
(C) Dose-dependent decrease of basal and stimulated (25 mM 3-HIB) endothelial FA uptake (RFU, relative fluorescence units) in response to 1-hour treatment of niclosamide. Pictured on right is the schematic for the fluorescent BODIPY-C12 based assay used to measure FA uptake.
(D) Kinetics of FA transport across a confluent bEnd.3 cell monolayer treated with 1 μM niclosamide. Pictured on right is the schematic for this BODIPY-C12 based transport assay.
(E) Structure of niclosamide (leftmost) and chemical analogues (compounds 1 through 4, labeled C1-C4). The red-colored moiety in each of the analogues identifies their structural difference as compared to niclosamide. For C1, the difference is the absence of the chlorine that is meta to the nitro group in the structure of niclosamide.
(F) FA uptake in response to drugs in (E) administered for 1 hour at 1 μM.
(G) Mitochondrial membrane potential as determined by flow cytometric analysis of tetramethylrhodamine ethyl ester (TMRE) signal in response to drugs in (E) administered for 1 hour at 1 μM.
(H) Correlation of FA uptake and mitochondrial membrane potential in cells treated with drugs depicted in (E).
(I) FA uptake in response to 3 different mitochondrial uncoupling drugs for 1 hour: 1 μM niclosamide, 0.5 μM 2-[2-[4-(trifluoromethoxy)phenyl]hydrazinylidene]-propanedinitrile (FCCP) and 500 μM 2,4-dinitrophenol (DNP).
BODIPY-C12 and BSA were used at 2 and 1 μM, respectively, in all FA uptake assays unless otherwise specified. Data are means and error bars are ± SEM. All statistics were determined using one-way ANOVA with Dunnett’s test for multiple comparisons. *p<0.05, **p<0.01, **p<0.001, ****p<0.0001 (compared to DMSO treated control); ####p<0.0001 (compared to DMSO + 3-HIB treated control).
We next conducted the full chemical screen, using a library of >2200 diverse chemical perturbagens, including kinase inhibitors, epigenetic inhibitors, GPCR/Ion channel modifiers, metabolic inhibitors, microbiology agents, and FDA-approved marketed drugs with annotated biological activities, predictable activities, and proven scaffolds directed against a wide range of drug targets (Figure S1C). We delivered these compounds (and 3-HIB) in triplicate to LUCEs plated onto 384-well plates and measured uptake of FFA-Luc. We identified several compounds that strongly suppressed or induced endothelial FA uptake (Figures 1B, S1D). Eight-point serial dilution curves on the top and bottom 1% (FA uptake inhibitors and activators, respectively) and testing both basal (vehicle) and stimulated (3HIB) uptake confirmed many of these hits, with several of them in the low micromolar range. One compound in particular, niclosamide, potently inhibited both basal and stimulated FA uptake during this secondary screening (Figure S1E). We next subjected these compounds to an orthogonal assay that measured uptake of the fluorescent FA analog BODIPY-C12 (Figure S1F). Niclosamide again strongly suppressed FA uptake with a relative IC50 of 0.1 μM for both basal and stimulated uptake (Figure 1C). Niclosamide suppressed FA uptake across a wide range of concentrations of BODIPY-C12 and BSA (Figure S1G), and the suppression of FA uptake by niclosamide was rapid, achieving maximum effect within five minutes (Figure S1H). Finally, niclosamide also reduced endothelial FA transport in vitro, quantified by the transport of BODIPY-C12 across a tight monolayer of endothelial cells (Figure 1D).
Endothelial FA uptake and transport requires ATP production
Since its discovery as an anti-helminth medication (Thompson, 1967), niclosamide has been observed to have effects on several molecular pathways in various cell types, including suppression of stimulatory phosphorylation of the transcription factor STAT3 (Ren et al., 2010). However, these effects are typically observed after an hour or longer, whereas we found that niclosamide decreases FA uptake within minutes (Figure S1H), suggesting a signaling independent mechanism. More recently, Tao et al. observed that niclosamide caused rapid uncoupling of mitochondria in cell culture and in vivo (Tao et al., 2014). To ascertain whether mitochondrial uncoupling by niclosamide is responsible for suppression of FA uptake in ECs, we undertook structure/activity relationship (SAR) studies, using a number of mild modifications on niclosamide’s structure (Figure 1E). These compounds exhibited varying degrees in reduction of endothelial FA uptake, as well as varying extent of mitochondrial uncoupling, and the two functions were highly correlated (Figures 1F–H), strongly supporting the notion that niclosamide suppresses FA uptake via uncoupling mitochondria. Furthermore, we found that two other structurally unrelated mitochondrial uncouplers, FCCP and 2,4-dinitrophenol, also strongly decreased endothelial FA uptake (Figures 1I, S1I).
To determine if other perturbations of mitochondrial function also impact FA uptake, we next tested a number of known mitochondrial inhibitors (Figure 2A). Both rotenone and oligomycin, which inhibit complex I and ATP synthase, respectively, reduced FA uptake as much as did FCCP, despite their opposing effect on mitochondrial membrane potential (Figures 2B, S2A). Neither rotenone nor oligomycin affect endothelial migration or viability at these doses (Kim et al., 2017). These data indicated that suppression of FA uptake does not directly rely on effects on membrane potential. Treatment with β-etomoxir, which suppresses fatty acid oxidation (FAO) by inhibiting the Acyl-CoA transporter CPT1α, had no effect on FA uptake (Figure S2B), indicating that reduced capacity for FAO does not cause suppression of FA uptake. Another possibility was the NAD+/NADH ratio, which is important for proper endothelial cell function (Diebold et al., 2019). However, expression of LbNOX, a bacterial enzyme that oxidizes NADH to NAD+ and thus increases NAD+/NADH, had no effect on FA uptake (Figures S2C–D). Together, these data suggested that no aspect of electron transport, membrane potential, or redox state was likely to fully explain the suppression of FA uptake by these reagents.
Figure 2. ATP production is required for endothelial FA uptake in vitro and ex vivo.
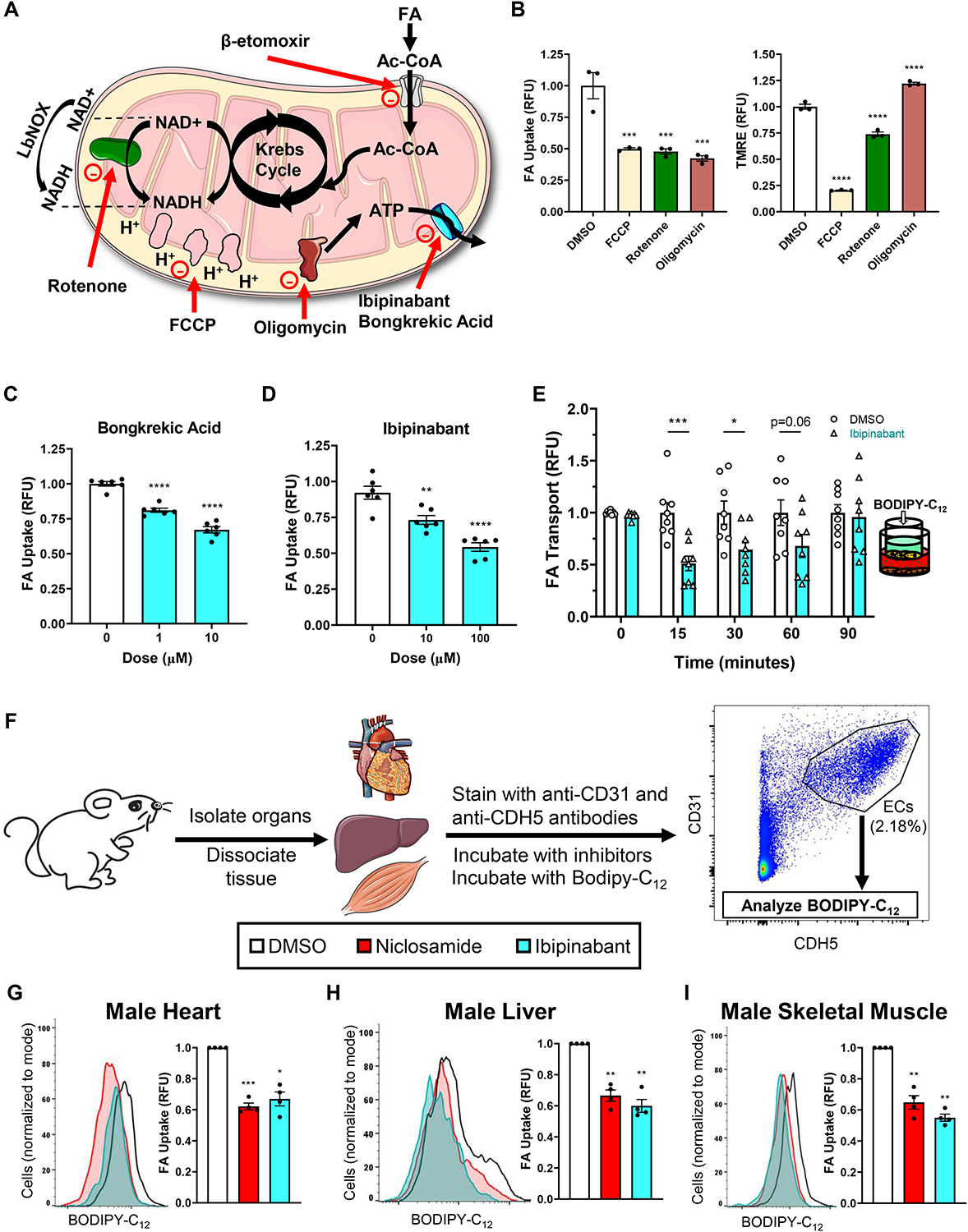
(A) Diagram depicting the target of various mitochondrial perturbagens used below (and related Supplementary Figure S2).
(B) FA uptake (left) and mitochondrial membrane potential (right) in response to 0.5 μM FCCP, 0.5 μM rotenone and 1 μM oligomycin.
(C-E) Dose-dependent FA uptake in response to ANT inhibitors Bongkrekic acid (C) and ibipinabant (D). The latter drug was then tested for its effects on FA transport (E).
(F) Schematic depicting the process by which mouse endothelial cells were identified among dissociated tissue homogenate and analyzed for FA uptake by flow cytometry. Below the schematic, a legend indicates the chemical inhibitors used and their corresponding colors in graphs G-I (DMSO, white; niclosamide, red; ibipinabant, cyan).
(G-I). Effects of 1-hour treatment of 10 μM niclosamide and 100 μM ibipinabant on the FA uptake of endothelial cells from male mouse hearts (G), livers (H), and skeletal muscle from the hindlimb (I). Median fluorescent BODIPY-C12 signal was measured as a readout of FA uptake. Left of each subfigure is the corresponding representative histogram. Cell number for each condition was normalized to mode. Right, bar graphs depicted such that all niclosamide or ibipinabant treated samples were measured relative to each corresponding DMSO treated sample in the same mouse (all DMSO samples were thus set to one). 4 mice were used to test each organ.
BODIPY-C12 and BSA were used at 2 and 1 μM, respectively, in all FA uptake assays unless otherwise specified. For B-E, data are means and error bars are ± SEM. Statistics were determined using ordinary one-way ANOVA with Dunnett’s test for multiple comparisons. For G-I, data are measures of median BODIPY-C12 signal and error bars are ± SEM. Statistics were determined using paired one-way ANOVA with Dunnett’s test for multiple comparisons, with DMSO controls set to one for each mouse used. *p<0.05, **p<0.01, **p<0.001, ****p<0.0001 (compared to DMSO treated control).
One thing that all of these perturbagens did have in common was their suppression of mitochondrial production of ATP. To test if mitochondrial ATP production was the key driver of FA uptake, we inhibited the adenine nucleotide translocator (ANT) complex, responsible for transporting mitochondrial ATP from the matrix to the cytoplasm. The structurally dissimilar ANT inhibitors bongkrekic acid and ibipinabant both reduced FA uptake in a dose-dependent fashion (Figures 2C–D, S2A). Moreover, ibipinabant also reduced trans-endothelial transport of FA (Figure 2E), much like niclosamide (Figure 1D). Both niclosamide and ibipinabant had some effects on nonendothelial cell types as well, though this varied depending on the specific cell line (Figure S2E). Additionally, it is important to note that while ibipinabant also targets the CB1 receptor CNR1 (while bongkrekic acid does not), this gene is not expressed in our ECs. Taken together, these data indicate that FA uptake in endothelial cells requires intact mitochondrial electron transport, oxidative phosphorylation, and production of ATP.
Perturbation of ex vivo mouse mitochondria reduces FA uptake
Cultured primary endothelial cells are notoriously different from those endothelia that still reside in the mouse, having undergone significant transcriptional changes; thus, they may respond to stimuli differently than while still in vivo. Therefore, we carried out studies in which we harvested organs directly from mice and, within 3–4 hours of sacrifice, analyzed the ECs in each sample for differences in FA uptake in response to niclosamide and ibipinabant. The organs were enzymatically dissociated, and the resultant conglomerate of cells were incubated with antibodies against the EC-specific markers CD31 and CDH5, as well as the aforementioned inhibitors. This was quickly followed by flow cytometry analysis, double-gating for the endothelial markers (Figure 2F). Both niclosamide and ibipinabant reduced FA uptake in ECs from cardiac, hepatic, and skeletal muscle tissue by up to 50% (Figures 2G–I and S2F–H), recapitulating in vitro results. Additionally, niclosamide reduced lipid droplet accumulation in ECs of en face aortas treated with oleic acid (Figures S2I–J). The requirement for mitochondrial ATP production to sustain FA uptake is thus also apparent in ECs freshly prepared ex vivo.
Specifically mitochondrial ATP production regulates FA uptake
The requirement of mitochondrial ATP production for endothelial FA uptake was unexpected because most of the ATP in endothelial cells derives from glycolysis (Culic et al., 1997; De Bock et al., 2013). Indeed, 2-deoxyglucose, a competitive inhibitor of glycolysis, strongly decreased the cellular ATP/ADP ratio, whereas niclosamide and FCCP had no effect, consistent with minimal contribution of mitochondrial ATP production to overall cellular ATP (Figure 3A). However, 2-deoxyglucose had no effect on FA uptake, unlike niclosamide and FCCP (Figure 3B). AMPK activation, a frequent response to low cellular ATP/ADP ratio, also had no effect on FA uptake (Figure S3A). Glycolytic ATP production thus does not contribute to FA uptake, which appears to depend specifically on ATP emanating from mitochondria. Consistent with this conclusion, ibipinabant reduced mitochondrial ATP production by approximately 34%, as measured by the difference between the basal and oligomycin-depressed oxygen consumption rates (OCR) (Figures 3C–D), with little effect on glycolysis as determined by extracellular acidification rate (ECAR) (Figures S3B), underscoring the importance of ATP derived specifically from mitochondria for FA uptake.
Figure 3. Specifically mitochondrial, not glycolytic, ATP production is necessary and sufficient to promote endothelial FA uptake.
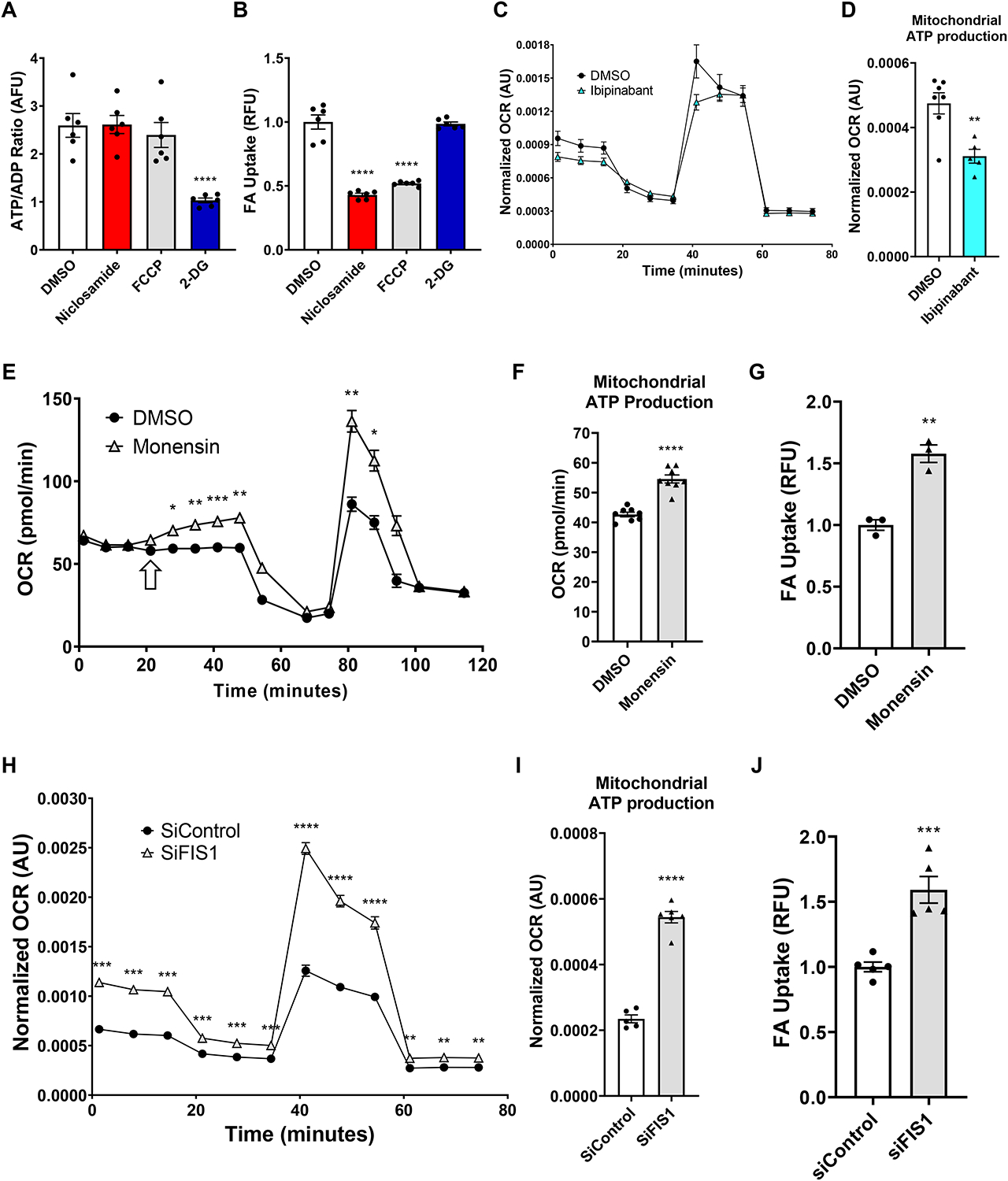
(A-B) 5.5 mM 2-deoxyglucose (2-DG) reduces cellular ATP/ADP ratio (arbitrary fluorescence units, AFU) (A) but does not affect FA uptake (B); in contrast, the uncouplers, FCCP and niclosamide, do not affect cellular ATP/ADP ratio while strongly suppressing FA uptake.
(C-D) Pre-treatment of cells with 50 μM ibipinabant reduced basal oxygen consumption rate (OCR, normalized using CyQUANT fluorescence, arbitrary units, AU) (C) as measured during the Seahorse Mito Stress Test, as well as mitochondrial ATP production (D) as determined by subtraction of the oligomycin-depressed OCR from basal OCR.
(E-G) Seahorse Mito Stress test modified to include a primary injection of 5 μM monensin, which steadily increased OCR (pmol O2 consumed per minute) (E) and mitochondrial ATP production (F). This correlates with monensin’s effect on FA uptake (G).
(H-J) 72-hour knockdown of FIS1 with 25 nM siRNA (siFIS1) increased overall cellular OCR (H) and mitochondrial ATP production (I) as compared to scrambled siRNA control (siControl). The same relation is seen when measuring FA uptake (J).
BODIPY-C12 and BSA were used at 2 and 1 μM, respectively, in all FA uptake assays unless otherwise specified. Data are means and error bars are ± SEM. Statistics were determined as such:
For A-B and I-J, one-way ANOVA with Dunnett’s test for multiple comparisons was used. For C, E, and H, 2-way ANOVA with Dunnett’s test for multiple comparisons was used. For D and F-G, unpaired, two-tailed Student’s t-test was used. *p<0.05, **p<0.01, **p<0.001, ****p<0.0001 (compared to DMSO treated control or siControl).
We next tested if, conversely, boosting ATP production specifically from mitochondria is sufficient to increase FA uptake. Monensin is a polyether ionophore that can transport monovalent cations such as Na+ across lipid membranes (Lichtshtein et al., 1979). Treating cells with monensin leads to an increase in intracellular sodium, likely boosting activity from the Na+/K+ ATPase pump to export Na+ and maintain sodium homeostasis. This higher demand for ATP then led to stimulated mitochondrial ATP production (Figures 3E–F). The rise in mitochondrial ATP production is accompanied by a significant increase in FA uptake (Figure 3G). Of additional importance is the fact that there was no accompanying change in ECAR (Figure S3C), nor of cellular ATP/ADP ratio (Figure S3D). Therefore, the ability of monensin to increase endothelial FA uptake likely stemmed from its effect on mitochondrial, not glycolytic, ATP production.
To further test this notion genetically, we turned to mitochondrial fusion/fission dynamics. It has been observed that cells with more fused mitochondria in general have greater production of ATP and respiratory capacity (Westermann, 2012; Yao et al., 2019). SiRNA-mediated knockdown (KD) of FIS1, a critical fission protein, led to increased OCR in ECFCs under all conditions and increased mitochondrial ATP production as compared to those given scrambled siRNA control (Figure 3H–I). As with monensin, KD of FIS1 also led to increased FA uptake (Figure 3J). KD of the fusion proteins MFN1 and MFN2, which causes fragmented mitochondria, had only a minor effect on ATP production and exhibited no change in FA uptake (Figure S3E–G). Thus, under both pharmaceutical and genetic approaches, increasing mitochondrial ATP production leads to increased FA uptake, independently of glycolytic or overall cellular ATP.
Mitochondrial ATP enables FATP4-mediated endothelial FA uptake
The reliance on ATP specifically from mitochondria suggested both that endothelial FA uptake is an ATP-requiring process, and that it must occur in microdomains that are in close proximity to mitochondria, exclusive of glycolysis-derived ATP pools. The family of so-called fatty acid transporter proteins (FATPs) have been implicated in mediating FA uptake in various cells. We and others have shown that ECs express FATP3 and FATP4, and that these two transporters are required for efficient uptake of FAs (Hagberg et al., 2010; Jang et al., 2016a). FATP4, like all FATPs, contains intrinsic ATP-dependent acyl-CoA synthetase (ACS) activity (Jia et al., 2007). SiRNA-mediated KD of FATP3 and FATP4 led to a 33–45% decrease in FA uptake in ECFCs (Figures 4A, S4A–B). Knockdown of ACSL1, another protein exhibiting ACS activity, also decreased FA uptake, while knockdown of ACSL3, ACSL4 and CD36 had no effect (Figure S4A–B). Conversely, overexpression of FATP4 is sufficient to substantially increase both basal and stimulated endothelial FA uptake, as well as intracellular neutral BODIPY staining, in both human and mouse ECs, and in the presence or absence of endogenous FATP3/4 (Figures 4B–C, S4C–D), and without impacting cellular respiration in the presence or absence of exogenous fatty acids (Figures S4E). Mutation of serine 247 to alanine in this domain renders FATP4 unable to convert FAs to acyl-CoAs (Milger et al., 2006; Stuhlsatz-Krouper et al., 1998). Overexpression of this S247A mutant failed entirely to increase endothelial FA uptake or neutral BODIPY staining (Figures 4B–C, S4C–D). Thus, FA uptake in ECs requires ACS activity, at least in part provided by FATP4, in an ATP-dependent fashion.
Figure 4. FATP4 mediates endothelial FA uptake in a manner dependent on ATP and proximity to mitochondria.
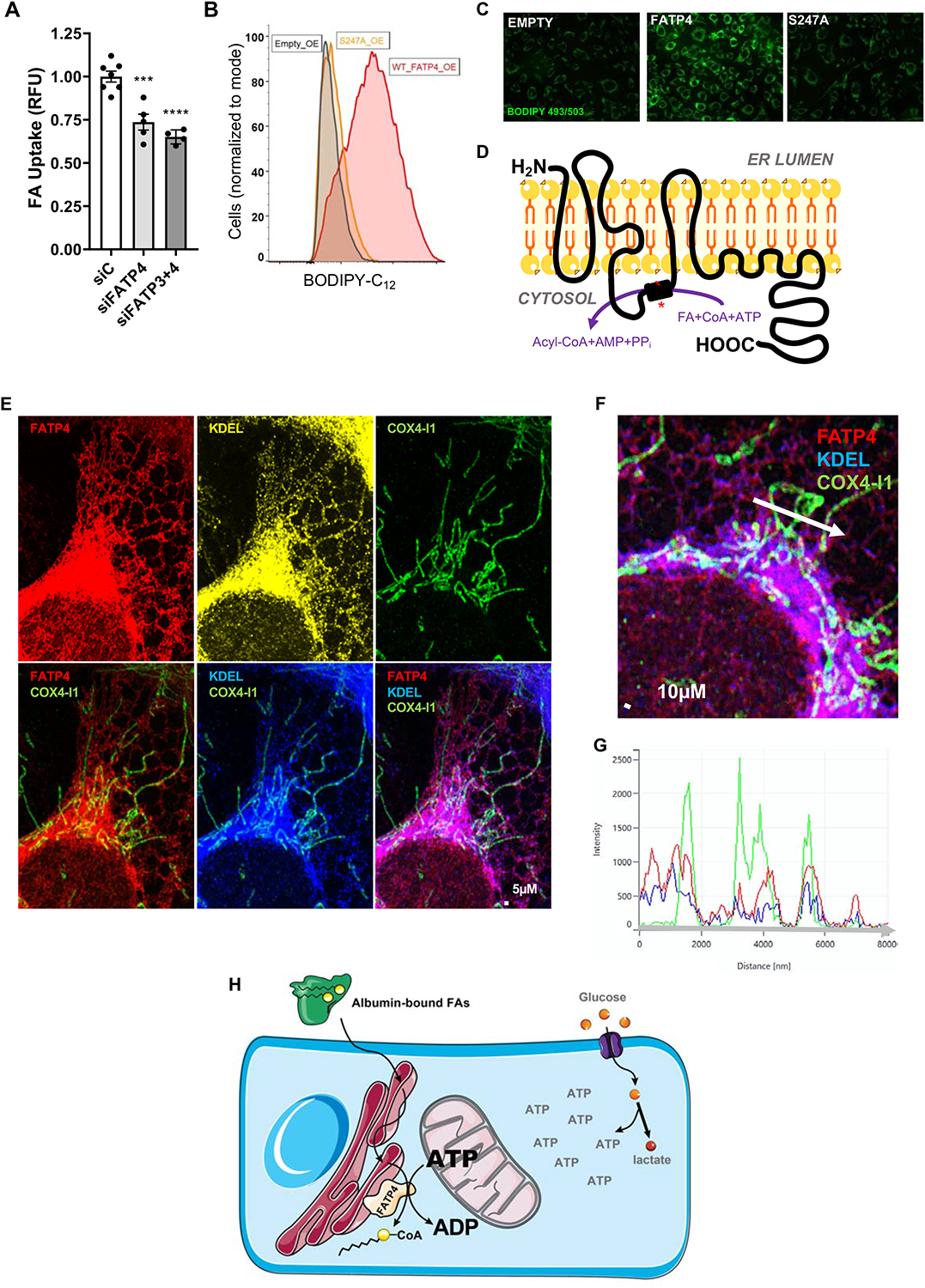
(A) 72-hour knockdown of FATP3 and FATP4 with 25 nM siRNA reduced FA uptake 25–33% as compared to scrambled control siRNA (siC).
(B) Basal FA uptake of cells over-expressing empty vector (black), wildtype FATP4 (red) and mutant S247A FATP4 (orange).
(C) Overexpressing cells were incubated with 500 μM oleic acid overnight and then stained with BODIPY 493/503, which detects accumulation of neutral lipids in lipid droplets.
(D) Proposed model for the topology of human FATP4 in the endoplasmic reticulum (ER) membrane, based on the hydropathy analysis, software prediction, and similarity to murine FATP1 (Lewis et al., 2001). The acyl-CoA synthetase domain (black box) mediates the conversion of FAs to Acyl-CoAs at the cost of one ATP. Mutating serine 247 to alanine in this domain (the red line and asterisk) renders FATP4 unable to promote FA uptake, as shown above.
(E) FATP4 localizes to ER and co-localizes with mitochondria. Anti-FATP4 antibody was used to stain FATP4 (red, left), anti-KDEL antibody was used to stain ER (yellow and blue, middle) and anti-COX4-I1 was used to stain mitochondria (green).
(F) Overlap of mitochondrial staining (COX4-I1, green) with both FATP4 (red) and ER (KDEL, blue) staining.
(G) Line analysis quantitation of green, red, and blue signal along the white arrow in the image depicted in (F).
(H) Model depicting the use of local mitochondrially-derived ATP (separate from the glycolytic ATP pool) for FATP4-mediated vectorial acylation, driving endothelial FA uptake.
BODIPY-C12 and BSA were used at 2 and 1 μM, respectively, in all FA uptake assays unless otherwise specified. Data are means and error bars are ± SEM. All statistics were determined using ordinary one-way ANOVA with Dunnett’s test for multiple comparisons. **p<0.001, ****p<0.0001 (compared to siC).
Based on a series of protease protection and immunofluorescence experiments, as well as hydropathy analysis, Lewis et al. proposed a topological model for murine FATP1, in which a single N-terminal transmembrane domain consisting of multiple helical loops is followed by an intracellular ACS domain, a peripherally-associated membrane-bound section, and ending with a cytoplasmic tail (Lewis et al., 2001). Comparison of mouse FATP1 and human FATP4 by BLAST indicate that they are 62% identical and share 77% similarity in primary sequence (Altschul et al., 1997). Additionally, these proteins exhibit quite similar hydropathy plots (Figure S4F). Based on these facts and additional computational analyses garnered through online protein prediction software (see Methods), we propose a similar, hypothetical model for the topology of FATP4 (Figure 4D). Lewis et al. proposed that the N-terminus of FATP1 lies in the extracellular space. Other studies have also indicated that FATP1 resides in the plasma membrane under certain circumstances (Stahl et al., 2002). In contrast, using Airyscan confocal microscopy, we found that FATP4 does not localize to the plasma membrane (Figure S4G–I). Instead, FATP4 colocalizes most strongly with endothelial ER (Figure 4E), as other groups have noted in other cells (Li et al., 2013; Milger et al., 2006). Thus, FATP4 mediates FA uptake in ECs not at the plasma membrane, but from the ER, and the N-terminal domain of FATP4 most likely faces the lumen of the ER.
The observations that: FATP4 requires ATP to promote FA uptake, that FA uptake in ECs requires mitochondrial-derived ATP, and that FATP4 resides in the ER, all suggest that FATP4-containing ER networks may lie in close proximity to mitochondria. Indeed, we found striking evidence that endothelial mitochondrial networks in ECs are unerringly found in apposition to FATP4-containing ER (Figures 4E–G). Using COX4-I1 as a marker of mitochondria, we find that mitochondria in ECs form extended continuous networks that are primarily perinuclear and extending into the cytoplasm but largely avoiding the periphery. COX4-I1 staining invariably coincided with KDEL staining, a marker of ER, which in turn is nearly identical to FATP4 staining; thus, we observed that every instance of endothelial mitochondria overlapped with the presence of FATP4 (Figure 4F–G). Together, these data indicate that endothelial FATP4 resides in close proximity to mitochondria and relies on locally produced mitochondrial ATP to fuel its ACS activity, thereby promoting FA uptake.
DISCUSSION:
Here, we performed a small-molecule screen to glean insight into the mechanisms by which ECs take up and transport FAs. Our data has led us to the conclusion that specifically mitochondrially derived ATP is required for the process of FA uptake, via provision of ATP to FATPs within microdomains of ER juxtaposed to mitochondria (Figure 4H). Importantly, ATP derived from glycolysis, the dominant source of ATP in ECs, has no impact on FA uptake.
Mitochondria have not traditionally been thought to play major roles in endothelial biology. Despite abundant access to oxygen, ECs generate >75% of their ATP via glycolysis (Culic et al., 1997; De Bock et al., 2013). Recent work, however, has demonstrated the importance of mitochondria in EC proliferation, and has suggested that the primary role of endothelial mitochondria is to serve as biosynthetic organelles for cell proliferation (Diebold et al., 2019). While proliferation of ECs is absolutely required for angiogenesis in contexts such as development or wound healing, the vast majority of ECs in adult organisms are quiescent and perform critical homeostatic tasks such as nutrient transport into underlying parenchyma. Our data demonstrate the key role that mitochondria play in such quiescent ECs.
The strong requirement for mitochondrial ATP, and of ACS activity of the FATPs and other similar proteins, is consistent with the idea of vectorial transport, a model for cellular FA uptake first hypothesized over 50 years ago (Mitchell and Moyle, 1958; Overath et al., 1969; Black and DiRusso, 2007; Arias-Barrau et al., 2009). In this model, FAs are “trapped” within the cytoplasm by enzyme-driven, ATP-dependent covalent attachment to Coenzyme A (CoA), a large hydrophilic group. The resulting “activated” acyl-CoA, now cytoplasmic, can now participate in various metabolic pathways e.g., β-oxidation or storage in lipid droplets (LDs). Moreover, this continuing shift in the equilibrium of intracellular acyl-CoA to FA promotes further FA uptake. This process is analogous to how phosphorylation of glucose to G6P not only traps the glucose in the cell but also promotes further glucose uptake (Wasserman et al., 2011; Adeva-Andany et al., 2016). The model is also consistent with the localization of FATP4 in the endothelial ER, rather than in the plasma membrane, because proximity to mitochondrial ATP is critical. In this model, access to extracellular FAs is conveyed by interconnected cellular lipid routes, i.e., the bilayer membrane network of the plasma membrane and ER. Importantly, despite their name, FATPs are unlikely to physically transport FAs, as their predicted topology does not contain channels. Indeed, in contrast to hydrophilic molecules that require active transport, the mobility of fats within the lipid bilayer is unlikely to need facilitation through aqueous channels. Instead, FATPs rely on their ACS activity to promote vectorial transport of FAs from membranes into the cytoplasm.
The fate of acyl-CoAs, once generated by FATP and other similar enzymes, is of interest and will require further study. One possibility is that they are shuttled into the EC triglyceride pool. Sessa and colleagues have shown that a large bolus of FAs, such as with an olive oil gavage, promotes transient formation of LDs in the aortic endothelium and likely elsewhere (Kuo et al., 2017). This temporary storage of fats in the body’s large EC compartment may serve to protect the underlying parenchyma from sudden surges of potentially damaging free FAs (Ibrahim and Arany, 2017). Formation of LDs requires esterification of FAs into triglycerides, a process that necessitates activation of the fatty acids by linkage to CoA. It will be of future interest to determine if FA transport across the endothelium obligatorily necessitates transient passage through the EC triglyceride pool.
Mitochondria have been found adjacent to the ER in multiple cell types, forming so-called mitochondrial associated membranes (MAMs) (Raturi and Simmen, 2013; Rutter and Pinton, 2014; van Vliet and Agostinis, 2018). The exact protein components and their functions within these MAMs remains debated and are often conflicting (Brito and Scorrano, 2008; Filadi et al., 2015; Lee and Min, 2018). Our data suggest that active FATP4 may reside in such MAMs of ECs, and that a key function of these MAMs may be to create microdomains within the cytoplasm that allow for selective and local transfer of ATP without intermixing with the rest of the cytoplasm. Analogous metabolic compartmentalization can be found in metabolons: multi-enzyme complexes that channel reaction products from one enzyme to another in spatially confined multi-step metabolic processes (Srere, 1987). ECs, for example, likely contain glycolytic metabolons within lamellipodia, capable of rapid generation of ATP to sustain migration (De Bock et al., 2013; Jang and Arany, 2013). Similarly, MAMs may represent mitochondrial metabolons, capable of local generation of ATP to sustain ACS activity of FATPs and related enzymes, which in turn drive FA uptake.
In summary, we find that endothelial FA uptake and transport, a process critical to highly oxidative organs such as the skeletal muscle and heart, surprisingly depends on ATP specifically generated from mitochondria, which locally drives ATP-dependent acyl-CoA formation to promote vectorial FA transport.
Limitations of Study
Our study is limited to in vitro and ex vivo experiments in freshly isolated mouse organs, and the full physiological relevance of our findings will require further studies in intact organisms, e.g., live rodent models. Another limitation of our work is the poorly understood molecular nature of the mitochondria/ER interaction; thus, we could not successfully design experiments to interfere with that interaction. Finally, we focused on FATP4 in the ER as a paradigm for control of endothelial FA uptake, but the complete picture may be more complex. It likely includes the involvement of other acyl-CoA synthetases, possibly in subcellular locations outside of the mitochondria/ER interface.
STAR METHODS:
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Zoltan Arany (zarany@pennmedicine.upenn.edu).
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact without restriction.
Data and Code Availability
This study did not generate any unique datasets or code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Isolation and culture of primary human endothelial cells
Endothelial colony forming cells (ECFCs) were obtained from human umbilical cord blood as previously described (Lin and Melero-Martin 2012). Briefly, cord blood was drawn from the umbilical vein. Mononuclear cells were separated by using Ficoll-Pacque solution and plated in a dish coated with 1% gelatin. After the cells reached 80% confluency, the CD31-positive cell fraction was purified by using Dynabeadconjugated anti-CD31 capture. These were cultured on 100-mm dishes coated with 0.1% gelatin (dissolved in PBS filtered through a 0.22 μM filter). Media used was EBM-2 with EGM-2 SingleQuots Supplements, 10% FBS and 1% Penn/Strep antibiotic solution (heretofore termed 10% EGM-2). ECFCs were used between passage 4 and 16.
Culture of HEK293T cells, bEnd.3 cells, 10T1/2 and C2C12 cells
All four cell lines were obtained from ATCC and cultured with DMEM GlutaMax, 10% FBS, and 1% Penn/Strep. For the bEnd.3 cells, dishes were coated with 0.1% gelatin prior to plating. To differentiate the C2C12 cells into myotubes, they were cultured in high glucose DMEM without pyruvate, 2% horse serum, and 1% Penn/Strep.
Culture of adipocyte cell line
Isolation, maintenance and differentiation of cultured adipocytes were carried out as previously described (Wada et al., 2016).
Creation of LUCEs
pLenti CMV Puro LUC (w168–1) (gift from Eric Campeau & Paul Kaufman, addgene # 17477) was used as the transfer vector encoding luciferase. Virus production and spinfection onto ECFC was carried out as detailed above in Retroviral over-expression albeit with one difference: psPAX2 (gift from Didier Trono, addgene # 12260) was used as the packaging plasmid instead of gag/pol. Selection here was carried out with 10 μg/mL puromycin. qPCR expression confirmed successful transduction. To determine functionality of the luciferase vis-à-vis fatty acid uptake, LUCEs were incubated with divalent PBS or 3-HIB for one hour and then given differing concentrations of the Luc-SS-FFA reagent (complexed to differing concentrations of BSA). Resulting basal and stimulated intracellular luminescence was measured using the SpectraMax M5 microplate reader.
Animal use
All mouse experiments were performed according to procedures approved by the University of Pennsylvania Institute for Animal Care and Use Committees (Philadelphia, PA). Mice (all of C57BL/6J genetic background) were housed under standard light/conditions. They were given food and water ad libitum. Both male and female mice were used; all were approximately 12–15 weeks old at time of experiment.
METHOD DETAILS
RT-qPCR
Qiagen’s TurboCapture mRNA kit was used to isolate mRNA from cells and synthesize cDNA via reverse transcription. qPCR was then performed on the cDNA using the CFX384 Bio-Rad Touch Real-Time PCR Detection System and iQ SYBR Green Supermix. Primers used for mRNA analysis can be found in Supplementary Table S3.
siRNA transfection
siRNA transfections were carried out using Invitrogen’s Lipofectamine RNAiMAX reagent. Cells at 70–90% confluency were kept in serum-free Opti-MEM media for the 6-hour duration of the transfection, after which they were refreshed with 10% EGM-2. Confirmation of siRNA-mediated genetic knockdown was determined using RT-qPCR.
Retroviral cloning
LbNOX was obtained as an addgene plasmid (gift from Vamsi Mootha, addgene # 75285) while Human FATP4 cDNA was synthesized as an IDT gBlock. PCR was done to add 5’ and 3’ extensions that were homologous to a modified pMSCV retroviral parent vector originally obtained from addgene (gift from David Mu, addgene # 75085). The PCR products was run on a 2% agarose (in TAE buffer) gel containing 0.01% ethidium bromide. After the correct size band was excised out under UV illumination, the DNA was obtained using the Qiagen gel extraction kit.
Meanwhile, restriction digest was performed on the pMSCV parent vector using EcoR1 and Xho1 restriction enzymes (37°C, 30 minutes). This removed the filler insert and the linearized vector DNA was obtained through gel extraction as well. Ligation of the linearized pMSCV vector and the desired insert (LbNOX or FATP4) was carried out using the Takara In-Fusion HD cloning system (50°C, 15 minutes). Transformation of the in-fusion product was carried out in Top10 chemically competent E.coli cells plated onto LB plates containing 50 ug/mL ampicillin. Colonies were selected, inoculated overnight, and purified via the Machery-Nagel NucleoSpin plasmid miniprep kit. The resulting purified plasmid DNA was then confirmed to be correct by submission for gene sequencing to GeneWiz.
Retroviral over-expression
HEK293T cells were given the retroviral envelope plasmid pMD2.G (gift from Didier Trono, addgene # 12259), packaging plasmid gag/pol (gift from Tannishtha Reya, addgene # 14887) and the pMSCV transfer vector containing FATP4 or LbNOX cDNA, as well as Fugene HD Transfection Reagent. Media was refreshed the next morning. Media was collected 48- and 72-hours post-transfection; this solution contained the virus of interest and was then “spinfected” onto ECFCs. This method of transduction was carried out as follows: viral solution supplemented with 8 μg/mL polybrene was administered to 75% confluent ECFCs, after which the plate was spun in a centrifuge at 800g for 40 minutes. Immediately afterward, viral media was aspirated and 10% EGM-2 was given to cells. After one day of growth, cells were split and given fresh media containing 10 μg/mL blastomycin. Un-transduced ECFCs were also given the same media as a negative control. After 2–3 days, the latter cells were 100% dead, and survivors of the transduced cells were expanded and analyzed for expression of gene of interest via qPCR.
Fatty acid uptake assay of adherent cells
ECFCs were plated onto Corning 96-well black-walled, clear-bottom plates (Millipore Sigma, CLS3603) at 40,000 cells/well and cultured for 48 hours or until confluent. For the assay, cells were treated with varying chemical perturbagens in divalent PBS or serum-free EGM-2 for varying times depending on the experiment, after which they were given 2 μM BODIPY-C12 (with 1 μM BSA) for four minutes. After two washes with 0.1% BSA (in PBS), cells were given 0.08% Trypan Blue to quench extracellular fluorescence. Intracellular fluorescence was then swiftly measured using a Spectramax microplate reader or Synergy H1 Multi-Mode microplate reader (bottom-read, excitation: 488 nm, emission: 520 nm). Signal from wells containing cells given no BODIPY-C12 was used as background noise and subtracted from experimental values.
Niclosamide structure/activity relationship studies
Compounds 1 through 4 were found and purchased through MolPort, an online software that searches for chemical compounds similar in structure to the desired parent compound (i.e., niclosamide).
Fatty acid transport assay
bEnd.3 cells were seeded at 50,000 cells/transwell onto 0.4 μM inserts (pre-coated with 0.1% gelatin). Two days later, another 50,000 cells/well were seeded into the same inserts and allowed to grow for another two days. In order to ascertain that a tight monolayer of cells had formed, 70 kDa Texas-Red Dextran was added to the lower chamber of these transwells, as well as to “empty” transwells (whose inserts had been coated with gelatin but were not seeded with cells). This Dextran, being about the size of BSA, should not pass through a tight cellular monolayer as well as it passes through empty inserts. This is exactly what we observed after periodically sampling the media in the upper chamber over an hour and then reading that on a Spectramax microplate reader (bottom-read, excitation: 488 nm, emission: 520 nm).
Meanwhile, C2C12 myoblasts were differentiated into myotubes over five days. For the assay, DMSO, niclosamide or ibipinabant was added to the top chamber for one hour. Additionally, inserts were placed on top of the newly formed myotubes. The top chamber’s drug solutions were then replaced with media containing 20 μM BODIPY-C12 (with 10 μM BSA) while the bottom chamber was given only 10 μM BSA. The bottom chambers were sampled periodically over an hour; each sample was kept in a black-walled 96-well plate until conclusion of the assay. Finally, BODIPY-C12 signal from these samples was measured using the Synergy H1 Multi-Mode microplate reader (bottom-read, excitation: 488 nm, emission: 520 nm).
High throughput chemical screening
LUCEs were plated onto 384-well plates at 6,000 cells per well and allowed to grow completely confluent over two days in cell culture incubators housed at the University of Pennsylvania High Throughput Screening Core. For the assay, media from each plate was aspirated and replaced by 25 μL/well divalent PBS. Immediately afterward, the SelleckChem drug library was robotically administered such that each inhibitor was present at a 1 μM concentration in each well. Columns 1 and 23 of each 384-well plate was given equivalent volume of DMSO and columns 2 and 24 were given 300 μM sulfosuccinimidyl oleate, a drug shown previously to inhibit endothelial fatty acid uptake (Jang et al., 2016b). After 30 minutes, 25 μL of 50 mM 3-HIB was added to each well, resulting in a final concentration of 25 mM 3-HIB per well. After another 30 minutes, plates were loaded into the FLIPR Tetra High-Throughput Cellular Screening system. The machine then used a 384-pin pipettor head to inject 12.5 μL of a solution containing 20 μM Luc-SS-FFA and 15 μM BSA into each well of a plate simultaneously, resulting in a 4:3 μM final concentration. Reading of luminescence began within a few seconds and continued for 310 seconds. The entire chemical library was screened twice more in exactly the same manner and the resulting data was collected and analyzed by the Core. The normalized percent inhibition (NPI) and Z-score for each inhibitor was obtained and averaged across all three runs. This resulting data was plotted in Figures 1B and S1D. The raw primary screening data can be found in Supplementary Table S1. The top and bottom 20 compounds (highest and lowest average NPI, respectively), were then tested in a focused secondary screen. The methods here were very similar to those in the primary screen with two key differences. Firstly, the effects of the drugs on both basal and stimulated fatty acid uptake were tested (whereas in the primary screen, only stimulated uptake was measured as 3-HIB had been given to all samples). Secondly, dosage was tested with an 8-point serial dilution: 20, 6.67, 2.22, 0.74, 0.25, 0.08, 0.03, and 0.01 μM. The secondary screen was done in duplicate. The raw data can be found in (Supplementary Table S2).
Flow cytometric analysis of cultured cells
ECFCs given siRNA or chemical inhibitors were incubated with 2 μM BODIPY-C12 (with 1 μM BSA) while still adherent. If also analyzing mitochondrial membrane potential, 75 nM TMRE was given for 15 minutes prior to incubation with BODIPY-C12. 0.25% trypsin was used to lift cells off dishes and 0.1% BSA was used to wash them before centrifugation. Cells were eventually resuspended in flow buffer (comprising 0.5% BSA, 2 mM EDTA, 0.1 μg/mL DAPI) and kept on ice until flow cytometry, which was carried out on a BD LSRII machine. Live cells were identified by gating for DAPI and choosing those events with low DAPI signal. Measure of BODIPY-C12 signal was used to determine fatty acid uptake. Data was analyzed on flowJo.
Flow cytometric analysis of ex vivo endothelial fatty acid uptake
12-week old male and female C57BL/6J mice were purchased from Jackson Laboratories and used over the span of three weeks for ex vivo work. For each run, either the heart, liver, or skeletal muscle (from the leg) from three mice were dissected and digested in 2 mg/mL collagenase and dispase in serum-free DMEM (that latter enzyme was used only for heart and skeletal muscle). Heart and liver tissue dissociated well within 20–25 minutes while skeletal muscle digestion took at least 45 minutes. All tissue homogenates were then triturated through a metal cannula and 60 mL syringe. They were then filtered through a 100- and 40-micron filter, in that order. After washing with PBS and centrifugation at 300g for five minutes, cell pellets were resuspended in 0.5% BSA in PBS and incubated with anti-CD31 (BioLegend, 102427) and anti-CD144 (ThermoFisher, 50-1441-82) antibodies for 20 minutes on ice, with gentle vortexing carried out at regular intervals. After another wash, each sample was split into three and incubated with 0.2% DMSO for 45 minutes, 10 μM niclosamide for 20 minutes, and 100 μM ibipinabant for 45 minutes at room temperature in divalent PBS. After drug incubation, all samples were washed and given 2 μM BODIPY-C12 (with 1 μM BSA) for four minutes. These were then washed with 0.1% BSA. After the final centrifugation, cells were resuspended in flow buffer and kept on ice until flow cytometry analysis on a BD LSRII machine. Live cells were identified by gating for DAPI. The endothelial cell population was selected by double-gating with the aforementioned antibodies, and their BODIPY-C12 signal was measured to determine fatty acid uptake. Data was analyzed on FlowJo.
Oxygen Consumption and Extracellular Acidification analysis
ECFCs were plated at 40,000 cells per well onto a gelatin coated 96-well Seahorse cell culture plate and grown for 48 hours in 10% EGM-2. For the experiments using ECFCs in which Fis1 or MFN1+MFN2 were knocked down (or scrambled siRNA control was used), siRNA transfection was carried out on a different 100-mm dish or 6-well plate three days prior to the Seahorse assay. 1 day before the assay, these cells were split into a 96-well Seahorse cell culture plate as detailed above. As a negative control, four corners of the plate were kept devoid of cells and given only Seahorse media, which is comprised of basal XF media, 5.5 mM glucose, 1 mM sodium pyruvate, and 4 mM glutamine (additionally, the pH was adjusted to 7.4). For Supplemental Figure S4E, the only nutrient provided in the Seahorse media was 167 μM palmitate (conjugated to BSA, as provided by the manufacturer). At least 12 hours prior to running a plate, the Seahorse sensor cartridge was incubated with Seahorse Calibrant solution as according to manufacturer’s protocol in a 37°C, CO2 free incubator.
On the day of an assay, cells were washed and incubated with Seahorse media. For the monensin treatment and siRNA knockdown experiments (Figures 3E–F, 3H–I, S3C, S3E–F, S3H), this media formulation contained no additional perturbagen. For the ANT inhibition experiment (Figures 3C–D, S3B), the media contained 0.5% DMSO or 50 μM ibipinabant. The sensor cartridge was fitted onto the cell culture plate, which was then placed into a 37°C, CO2 free incubator for one hour. During the assay, which was run on the Seahorse XF96 Analyzer, the following inhibitors were injected sequentially, as is standard for the Mito Stress Test: oligomycin (1 μM), FCCP (0.5 μM), and rotenone/antimycin (0.5 μM). For the monensin treatment experiment, 0.1% DMSO or 5 μM monensin was injected as well, prior to oligomycin injections as listed above.
CyQUANT Analysis
Data obtained from the Seahorse Oxygen Consumption and Extracellular Acidification assays done with ANT inhibition or siRNA knockdown (Figures 3C–D, 3H–I, S3B, S3E–F and S3H) were normalized using ThermoFisher Scientific’s fluorescent CyQUANT Kit according to manufacturer’s instruction.
ATP/ADP Ratio Analysis
Cellular ATP/ADP ratio was measured using the BioVision ADP/ATP Ratio Bioluminescence Kit according to manufacturer’s instruction.
NAD+/NADH Ratio Analysis
Cellular NAD+/NADH ratio was measured on ECFCs expressing LbNOX (or empty vector) using the Promega NAD/NADH-Glo Assay Kit according to manufacturer’s instruction.
Site-Directed Mutagenesis
The pMSCV-based retroviral plasmid containing FATP4 was used to generate the S247A point mutant via NEB’s Q5 Site-Directed Mutagenesis kit according to manufacturer’s instruction.
Neutral lipid staining
For Figure 4C, Confluent cells plated onto glass coverslips were loaded with 500 μM oleic acid (Sigma, O3008) overnight. For Figure S2I, isolated mouse aorta dissected en face were incubated with 500 μM oleic acid ± 5 μM niclosamide overnight. The next day, samples were fixed with 3.7% paraformaldehyde, washed, permeabilized with 0.3% Triton X-100. After blocking with 2% BSA, samples were incubated with anti-mouse CD31 antibody overnight (Millipore Sigma, MAB1398Z), with secondary antibody for two hours at room temperature, and then treated with BODIPY 493/503 for 10 minutes. Finally, they were washed with PBS and mounted onto glass slides for imaging. Cells were imaged with a Nikon DS-Qi1Mc camera while aorta were imaged using the Zeiss LSM 880 with Fast Airyscan module.
Image Acquisition and Analysis
ECFCs overexpressing FATP4 were fixed in 3.7% paraformaldehyde for 10 min. After fixation, the cells were permeabilized in 0.3% Triton X-100 for 5 min and blocked with 3% BSA for 30 min. For Figures 4E–F, the cells were then incubated with primary antibodies overnight at 4°C: anti-COX4-I1 (R&D, AF5814), anti-KDEL (Abcam, ab12223), and anti-FATP4 (Abcam, ab200353). For Figure S4H, cells were incubated with anti-FATP4 and anti-CDH5 (Thermofisher Scientific, 12-1449-82). After washing with PBS, the cells were incubated with secondary antibodies for two hours at room temperature. After washing with PBS, cells were mounted with VectaShield mounting medium (VectorLabs). Imaging was done using the Zeiss LSM 880 with Fast Airyscan module (laser excitation 405 nm for KDEL, 488nm for COX4-I1 (and CDH5), and 560nm for FATP4). Image processing and the intensity values of the line profile analysis were done by using Zen 3.0 blue software.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistics
Most data in this paper are presented as mean ± SEM. For flow cytometry experiments, BODIPY-C12 signal was measured as median values. P-value < 0.05 was considered statistically significant (*p<0.05, **p<0.01, **p<0.001, ****p<0.0001). For comparison between two treatments, unpaired, two-tailed Student’s t-test was used. For multiple comparisons, ordinary 1-way ANOVA with Dunnett’s correction was used. For the ex vivo experiments, paired 1-way ANOVA with Dunnett’s correction was used. For the Seahorse experiments involving monensin or ibipinabant, multiple t-tests with Dunnett’s correction was used. For the Seahorse experiments involving genetic knockdown of FIS1 and MFN1+MFN2, ordinary 2-way ANOVA with Dunnett’s correction was used.
Supplementary Material
Supplementary Table S1. Results of the primary chemical screening experiment done in triplicate. Related to Figures 1 and S1.
Supplementary Table S2. Results of the secondary screening efforts of top hits from the initial chemical screening results. Related to Figures 1 and S1.
Supplementary Table S3. Sequences of primers used for RT-qPCR. Related to STAR Methods.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-SLC27A4/FATP4 | Abcam | Cat#Ab200353 |
| Mouse monoclonal anti-KDEL | Abcam | Cat#Ab12223; RRID:AB_298945 |
| Goat polyclonal anti-COX4-I1 | R&D | Cat#AF5814; RRID:AB_2085286 |
| Brilliant Violet 605 anti-mouse monoclonal CD31 | BioLegend | Cat#102427; RRID:AB_2563982 |
| CD144 (VE-cadherin) monoclonal, eFluor 660 | ThermoFisher Scientific | Cat#50-1441-82; RRID:AB_11219483 |
| Purified rat anti-mouse CD31 | BD Pharmigen | Cat#553370; RRID:AB_394816 |
| Dynabeads Sheep polyclonal anti-rat IgG | ThermoFisher Scientific | Cat#11035 |
| Armenian Hamster anti-mouse CD31 | MilliporeSigma | Cat#MAB1398Z; RRID:AB_94207 |
| Mouse anti-human CDH5 (CD144, VE-Cadherin) | ThermoFisher Scientific | Cat#12-1449-82; RRID:AB_763438 |
| Anti-rabbit IgG, Alexa Fluor 555 | Cell Signaling Technology | Cat#4413S; RRID: AB_10694110 |
| Anti-mouse IgG, Pacific Blue | ThermoFisher Scientific | Cat#P31582; RRID: AB_10374586 |
| Anti-goat IgG, Alexa Fluor 488 | ThermoFisher Scientific | Cat#A-11055; RRID:AB_2534102 |
| Anti-mouse IgG, Alexa Fluor 488 | ThermoFisher Scientific | Cat#4408S; RRID: AB_10694704 |
| Anti-hamster IgG, Alexa Fluor 647 | ThermoFisher Scientific | Cat#A-21451; RRID: AB_2535868 |
| Bacterial and Virus Strains | ||
| One Shot TOP10 Chemical Competent E. coli | ThermoFisher Scientific | Cat#C404003 |
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Lipofectamine RNAiMAX Transfection Reagent | ThermoFisher Scientific | Cat#13778150 |
| Restriction enzyme: EcoRI-HF | New England BioLabs | Cat#R3101S |
| Restriction enzyme: XhoI | New England BioLabs | Cat#R0146S |
| Fugene HD Transfection Reagent | Promega | Cat#E2311 |
| Polybrene Infection/Transfection Reagent | MilliporeSigma | Cat#TR-1003-G |
| Fatty acid free bovine serum albumin solution | MilliporeSigma | Cat#A9205 |
| BODIPY FL C12 | ThermoFisher Scientific | Cat#D3822 |
| BODIPY 493/503 | ThermoFisher Scientific | Cat#D3922 |
| Dextran Texas Red, 70,000 MW, Neutral | Fisherscientific | Cat#D1830; CAS: 9004-54-0 |
| FFA-SS-Luciferase probe | Intrace Medical | N/A |
| (±)-Sodium β-hydroxyisobutyrate |
MilliporeSigma | Cat#36105; CAS: 1219589-99-7 |
| Sulfo-N-succinimidyl oleate (SSO) | Cayman Chemical Company |
Cat#11211; CAS: 135661-44-8 (free acid) |
| Tetramethylrhodamine ethyl ester perchlorate (TMRE) | MilliporeSigma | Cat#87917; CAS: 115532-52-0 |
| 4′,6-diamidino-2-phenylindole (DAPI) | MilliporeSigma | Cat#D9542; CAS: 28718-90-3 |
| Collagenase, Type I | Fisherscientific | Cat#AAJ1382003; CAS: 9001-12-1 |
| Dispase II | MilliporeSigma | Cat#D4693; CAS: 42613-33-2 |
| Niclosamide (Niclocide) | Selleckchem | Cat#S3030; CAS: 50-65-7 |
| N-(2-chloro-4-nitrophenyl)-2-hydroxybenzamide (Compound 1) | Specs | AG-690/10526046; MolPort-000-639-505 |
| 5-Chloro-N-(2,6-dichloro-4-nitrophenyl)-2-methoxybenzamide (Compound 2) | Specs | AF-399/08800034; MolPort-001-667-150 |
| 5-Chloro-N-(2,6-dichloro-4-nitrophenyl)-2-hydroxybenzamide (Compound 3) | Vitas-M Laboratory | STK055196; MolPort-000-839-501 |
| 4-chloro-N-(2-chloro-4-nitrophenyl)-1-hydroxynaphthalene-2-carboxamide (Compound 4) | Vitas-M Laboratory | STK365880; MolPort-002-319-058 |
| Ibipinabant (SLV-319) | Cayman Chemical Company | Cat#10009226; CAS: 362519-49-1 |
| Bongkrekic Acid (ammonium salt) | Cayman Chemical Company | Cat#19079; CAS: 11076-19-0 |
| Monensin (sodium salt) | Cayman chemical Company | Cat#16488; CAS: 22373-78-0 |
| Carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP) | MilliporeSigma | Cat#C2920; CAS: 370-86-5 |
| 2,4-Dinitrophenol (DNP) | MilliporeSigma | Cat#D198501; CAS: 51-28-5 |
| Oligomycin A | MilliporeSigma | Cat#75351; CAS: 579-13-5 |
| Rotenone | MilliporeSigma | Cat#R8875; CAS: 83-79-4 |
| β-etomoxir | Cayman Chemical Company | Cat#11969; CAS: 828934-41-4 |
| 2-Deoxy-D-glcuose | MilliporeSigma | Cat#25972; CAS: 154-17-6 |
| 5-Aminoimidazole-4-carboxamide-1-β-d-ribofuranoside (AICAR) | Cayman Chemical Company | Cat#10010241; CAS: 2627-69-2 |
| Seahorse XF Calibrant Solution 500 mL | Agilent | Cat#100840-000 |
| Seahorse XF base medium 500 mL | Agilent | Cat#103334-100 |
| Seahorse XF Palmitate-BSA FAO Substrate | Agilent | Cat#102720-100 |
| Oleic Acid-Albumin from bovine serum | MilliporeSigma | Cat#O3008; MDL: MFCD00284022 |
| VECTASHIELD HardSet Antifade Mounting Medium | Vector Laboratories | Cat#H-1400 |
| Selleckchem Bioactive Compound Library (96-well) | Selleckchem | L1700 |
| Critical Commercial Assays | ||
| Turbocapture 384 mRNA kit | Qiagen | Cat#72271 |
| QIAEX II Gel Extraction kit | Qiagen | Cat#20021 |
| In-Fusion HD Cloning Plus | Takara | Cat#638909 |
| NucleoSpin Plasmid Miniprep Kit | Machery-Nagel | Cat#740588.250 |
| Seahorse XF Cell Mito Stress Test Kit | Agilent | Cat#103015-100 |
| ADP/ATP Ratio Bioluminescence Assay Kit, ApoSENSOR | BioVision | Cat#K255 |
| NAD/NADH-Glo Assay | Promega | Cat#G9071 |
| Q5 Site-Directed Mutagenesis Kit | New England BioLabs | Cat#E0552S |
| CyQUANT Cell Proliferation Assay | ThermoFisher Scientific | Cat#C7026 |
| Deposited Data | ||
| Experimental Models: Cell Lines | ||
| Endothelial colony forming cells (ECFCs) | Obtained from pooled umbilical cord blood | N/A |
| Human: HEK293T cells | ATCC | CRL-3216; RRID: CVCL_0063 |
| Mouse: bEnd.3 cells | ATCC | CRL-2299; RRID: CVCL_0170 |
| Mouse: C2C12 cells | ATCC | CRL-1772; RRID: CVCL_0188 |
| Mouse: 10T1/2 cells | ATCC | CCL-226; RRID: CVCL_0190 |
| Mouse: Preadipocytes | Isolated from white adipose tissue and immortalized with SV40 TLA | Wada et al., 2016 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J | The Jackson Laboratory | JAX: 000664; RRID: IMSR_JAX:000664 |
| Oligonucleotides | ||
| siRNA universal negative control: proprietary sequence | MilliporeSigma | Cat#SIC001 |
| siRNA targeting human FATP3 | MilliporeSigma | SASI_Hs01_00100092 |
| SiRNA targeting human FATP4 | MilliporeSigma | SASI_Hs01_00047531 |
| SiRNA targeting human FIS1 | MilliporeSigma | SASI_Hs01_00171952 |
| SiRNA targeting human MFN1 | MilliporeSigma | SASI_Hs01_00057999 |
| SiRNA targeting human MFN2 | MilliporeSigma | SASI_Hs02_00330014 |
| siRNA targeting mouse FATP3 | MilliporeSigma | SASI_Mm01_00093273 |
| SiRNA targeting mouse FATP4 | MilliporeSigma | SASI_Mm01_00151569 |
| SiRNA targeting human ACSL1 | MilliporeSigma | SASI_Hs01_00202187 |
| SiRNA targeting human ACSL3 | MilliporeSigma | SASI_Hs01_00034737 |
| SiRNA targeting human ACSL4 | MilliporeSigma | SASI_Hs01_00114667 |
| SiRNA targeting human CD36 | MilliporeSigma | SASI_Hs01_00075562 |
| RT-qPCR primers | This paper | See Table S3 |
| Recombinant DNA | ||
| pMD2.G | Addgene | Cat#12259 |
| gag/pol | Addgene | Cat#14887 |
| psPAX2 | Addgene | Cat#12260 |
| pMSCV-Blasticidin | Addgene | Cat#75085 |
| pUC57-LbNOX | Addgene | Cat#75285 |
| pLenti CMV Puro LUC (w186-1) | Addgene | Cat#17477 |
| Synthesized human FATP4 CCDS with 5’ and 3’ homology to pMSCV (gBlock Gene Fragments) | IDT | N/A |
| Software and Algorithms | ||
| MolPort Structure Search | MolPort | https://www.molport.com/shop/index |
| FlowJo | FlowJo, LLC | https://www.flowjo.com/; RRID:SCR_008520 |
| Graphpad Prism | Graphpad Prism, Inc. | https://www.graphpad.com/scientific-software/prism/; RRID:SCR_002798 |
| Zen 3.0 Blue for line profile analysis | Zeiss | https://www.zeiss.com/microscopy/us/products/microscope-software/zen.html |
| NCBI Protein-Protein BLAST | Altschul et al., 1997 | https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE=Proteins& |
| Adobe Illustrator | Adobe | https://www.adobe.com/products/illustrator.html; RRID:SCR_010279 |
| ExPASy ProtScale for Hydropathy Plot | Gasteiger et al., 2005 | https://web.expasy.org/protscale/ |
| PredictProtein server | Rost et al., 2004 | https://www.predictprotein.org/home |
| PSIPRED 4.0 (Predict Secondary Structure)/MEMSAT-SVM (Membrane Helix Prediction) | Jones, 1999; Nugent and Jones, 2009; Buchan and Jones, 2019 | http://bioinf.cs.ucl.ac.uk/psipred/; RRID:SCR_010246 |
| Other | ||
| Corning 96-well black polystyrene microplate | MilliporeSigma | CAT#CLS3603 |
| Corning 6.5 mm, 0.4 μm pore transwell inserts | MilliporeSigma | CAT#CLS3413 |
Highlights.
A chemical screen identifies novel inhibitors of endothelial fatty acid uptake
Endothelial fatty acid uptake and transport requires specifically mitochondrial ATP production
Endothelial mitochondria are proximally positioned to the ER
ER-borne FATP4 uses mitochondrial ATP to mediate vectorial acylation of fatty acids
Context and Significance.
Numerous tissues, including skeletal muscle, depend on fatty acids as fuels. But excess influx of fatty acids into skeletal muscle can lead to insulin resistance and type 2 diabetes. Control of fat influx into muscle is therefore important. To reach skeletal muscle, fatty acids must first be taken up and transported through the endothelial barrier of the capillary walls, a process that is poorly understood. Here, researchers from the University of Pennsylvania find that perturbing mitochondrial ATP generation (but not glycolysis) in endothelial cells decreases fatty acid uptake and transport. They also demonstrate that endothelial mitochondria line up almost precisely with the endoplasmic reticulum, and that ATP locally derived from mitochondria is used by an ER-resident protein, FATP4, which requires ATP to promote fatty acid uptake. These findings reveal a new mechanism mediating endothelial fatty acid biology and thus open new doors for therapeutic prevention of lipid-induced insulin resistance.
ACKNOWLEDGEMENTS:
We acknowledge Drs. David Schultz and Sara Cherry and the High-throughput Screening Core at the University of Pennsylvania for their assistance in carrying out the chemical screen. This work was supported by funding from the NIDDK (DK111091 to A.I and DK114103 to ZA) and the NHLBI (HL126797 to ZA).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS:
The authors declare no competing interests.
REFERENCES:
- Adeva-Andany MM, Pérez-Felpete N, Fernández-Fernández C, Donapetry-García C, and Pazos-García C (2016). Liver glucose metabolism in humans. Biosci. Rep 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aird WC (2010). Proximate and evolutionary causation of endothelial heterogeneity. Semin. Thromb. Hemost 36, 276–285. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, and Lipman DJ (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson CM, and Stahl A (2013). SLC27 fatty acid transport proteins. Mol. Aspects Med 34, 516–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arany Z, Foo S-Y, Ma Y, Ruas JL, Bommi-Reddy A, Girnun G, Cooper M, Laznik D, Chinsomboon J, Rangwala SM, et al. (2008). HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1α. Nature 451, 1008–1012. [DOI] [PubMed] [Google Scholar]
- Arias-Barrau E, Dirusso CC, and Black PN (2009). Methods to monitor Fatty Acid transport proceeding through vectorial acylation. Methods Mol. Biol. Clifton NJ 580, 233–249. [DOI] [PubMed] [Google Scholar]
- Black PN, and DiRusso CC (2007). Vectorial acylation: linking fatty acid transport and activation to metabolic trafficking. Novartis Found. Symp 286, 127–138; discussion 138–141, 162–163, 196–203. [DOI] [PubMed] [Google Scholar]
- Black PN, Sandoval A, Arias-Barrau E, and DiRusso CC (2009). Targeting the fatty acid transport proteins (FATP) to understand the mechanisms linking fatty acid transport to metabolism. Immunol. Endocr. Metab. Agents Med. Chem 9, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brito O.M. de, and Scorrano L (2008). Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456, 605–610. [DOI] [PubMed] [Google Scholar]
- Culic O, Gruwel ML, and Schrader J (1997). Energy turnover of vascular endothelial cells. Am. J. Physiol.-Cell Physiol 273, C205–C213. [DOI] [PubMed] [Google Scholar]
- De Bock K, Georgiadou M, Schoors S, Kuchnio A, Wong BW, Cantelmo AR, Quaegebeur A, Ghesquière B, Cauwenberghs S, Eelen G, et al. (2013). Role of PFKFB3-Driven Glycolysis in Vessel Sprouting. Cell 154, 651–663. [DOI] [PubMed] [Google Scholar]
- Diebold LP, Gil HJ, Gao P, Martinez CA, Weinberg SE, and Chandel NS (2019). Mitochondrial complex III is necessary for endothelial cell proliferation during angiogenesis. Nat. Metab 1, 158–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filadi R, Greotti E, Turacchio G, Luini A, Pozzan T, and Pizzo P (2015). Mitofusin 2 ablation increases endoplasmic reticulum–mitochondria coupling. Proc. Natl. Acad. Sci 112, E2174–E2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frayn KN, and Langin D (2003). Triacylglycerol metabolism in adipose tissue In Advances in Molecular and Cell Biology, (Elsevier; ), pp. 337–356. [Google Scholar]
- Glatz JFC, Luiken JJFP, and Bonen A (2010). Membrane Fatty Acid Transporters as Regulators of Lipid Metabolism: Implications for Metabolic Disease. Physiol. Rev 90, 367–417. [DOI] [PubMed] [Google Scholar]
- Goldberg IJ (1996). Lipoprotein lipase and lipolysis: central roles in lipoprotein metabolism and atherogenesis. J. Lipid Res 37, 693–707. [PubMed] [Google Scholar]
- Hagberg CE, Falkevall A, Wang X, Larsson E, Huusko J, Nilsson I, van Meeteren LA, Samen E, Lu L, Vanwildemeersch M, et al. (2010). Vascular endothelial growth factor B controls endothelial fatty acid uptake. Nature 464, 917–921. [DOI] [PubMed] [Google Scholar]
- Henkin AH, Cohen AS, Dubikovskaya EA, Park HM, Nikitin GF, Auzias MG, Kazantzis M, Bertozzi CR, and Stahl A (2012). Real-Time Noninvasive Imaging of Fatty Acid Uptake in Vivo. ACS Chem. Biol 7, 1884–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim A, and Arany Z (2017). Does Endothelium Buffer Fat? Circ. Res 120, 1219–1221. [DOI] [PubMed] [Google Scholar]
- Iso T, Maeda K, Hanaoka H, Suga T, Goto K, Syamsunarno MRAA, Hishiki T, Nagahata Y, Matsui H, Arai M, et al. (2013). Capillary Endothelial Fatty Acid Binding Proteins 4 and 5 Play a Critical Role in Fatty Acid Uptake in Heart and Skeletal Muscle. Arterioscler. Thromb. Vasc. Biol 33, 2549–2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang C, and Arany Z (2013). Metabolism: Sweet enticements to move. Nature 500, 409–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang C, Oh SF, Wada S, Rowe GC, Liu L, Chan MC, Rhee J, Hoshino A, Kim B, Ibrahim A, et al. (2016a). A branched-chain amino acid metabolite drives vascular fatty acid transport and causes insulin resistance. Nat. Med 22, 421–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang C, Oh SF, Wada S, Rowe GC, Liu L, Chan MC, Rhee J, Hoshino A, Kim B, Ibrahim A, et al. (2016b). A branched-chain amino acid metabolite drives vascular fatty acid transport and causes insulin resistance. Nat. Med 22, 421–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Z, Pei Z, Maiguel D, Toomer CJ, and Watkins PA (2007). The fatty acid transport protein (FATP) family: very long chain acyl-CoA synthetases or solute carriers? J. Mol. Neurosci. MN 33, 25–31. [DOI] [PubMed] [Google Scholar]
- Kampf JP, Cupp D, and Kleinfeld AM (2006). Different Mechanisms of Free Fatty Acid Flip-Flop and Dissociation Revealed by Temperature and Molecular Species Dependence of Transport across Lipid Vesicles. J. Biol. Chem 281, 21566–21574. [DOI] [PubMed] [Google Scholar]
- Kim B, Li J, Jang C, and Arany Z (2017). Glutamine fuels proliferation but not migration of endothelial cells. EMBO J. 36, 2321–2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komarova Y, and Malik AB (2010). Regulation of Endothelial Permeability via Paracellular and Transcellular Transport Pathways. Annu. Rev. Physiol 72, 463–493. [DOI] [PubMed] [Google Scholar]
- Kuo A, Lee MY, and Sessa WC (2017). Lipid Droplet Biogenesis and Function in the Endothelium. Circ. Res 120, 1289–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, and Min K-T (2018). The Interface Between ER and Mitochondria: Molecular Compositions and Functions. Mol. Cells 41, 1000–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis SE, Listenberger LL, Ory DS, and Schaffer JE (2001). Membrane topology of the murine fatty acid transport protein 1. J. Biol. Chem 276, 37042–37050. [DOI] [PubMed] [Google Scholar]
- Li S, Lee J, Zhou Y, Gordon WC, Hill JM, Bazan NG, Miner JH, and Jin M (2013). Fatty Acid Transport Protein 4 (FATP4) Prevents Light-Induced Degeneration of Cone and Rod Photoreceptors by Inhibiting RPE65 Isomerase. J. Neurosci 33, 3178–3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtshtein D, Dunlop K, Kaback HR, and Blume AJ (1979). Mechanism of monensin-induced hyperpolarization of neuroblastoma-glioma hybrid NG108–15. Proc. Natl. Acad. Sci. U. S. A 76, 2580–2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin R-Z, and Melero-Martin JM (2012). Fibroblast growth factor-2 facilitates rapid anastomosis formation between bioengineered human vascular networks and living vasculature. Methods 56, 440–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehrotra D, Wu J, Papangeli I, and Chun HJ (2014). Endothelium as a gatekeeper of fatty acid transport. Trends Endocrinol. Metab 25, 99–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milger K, Herrmann T, Becker C, Gotthardt D, Zickwolf J, Ehehalt R, Watkins PA, Stremmel W, and Fullekrug J (2006). Cellular uptake of fatty acids driven by the ER-localized acyl-CoA synthetase FATP4. J. Cell Sci 119, 4678–4688. [DOI] [PubMed] [Google Scholar]
- Minshall RD, and Malik AB (2006). Transport across the endothelium: regulation of endothelial permeability. Handb. Exp. Pharmacol 107–144. [DOI] [PubMed] [Google Scholar]
- Mitchell P, and Moyle J (1958). Group-Translocation: A Consequence of Enzyme-Catalysed Group-Transfer. Nature 182, 372–373. [DOI] [PubMed] [Google Scholar]
- Niot I, and Besnard P (2003). Intestinal uptake and transport of fatty acids In Advances in Molecular and Cell Biology, (Elsevier; ), pp. 9–28. [Google Scholar]
- Overath P, Pauli G, and Schairer HU (1969). Fatty Acid Degradation in Escherichia coli. Eur. J. Biochem 7, 559–574. [PubMed] [Google Scholar]
- Pepino MY, Kuda O, Samovski D, and Abumrad NA (2014). Structure-Function of CD36 and Importance of Fatty Acid Signal Transduction in Fat Metabolism. Annu. Rev. Nutr 34, 281–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohl J, Ring A, Hermann T, and Stremmel W (2004). Role of FATP in parenchymal cell fatty acid uptake. Biochim. Biophys. Acta 1686, 1–6. [DOI] [PubMed] [Google Scholar]
- Raturi A, and Simmen T (2013). Where the endoplasmic reticulum and the mitochondrion tie the knot: The mitochondria-associated membrane (MAM). Biochim. Biophys. Acta BBA - Mol. Cell Res 1833, 213–224. [DOI] [PubMed] [Google Scholar]
- Ren X, Duan L, He Q, Zhang Z, Zhou Y, Wu D, Pan J, Pei D, and Ding K (2010). Identification of Niclosamide as a New Small-Molecule Inhibitor of the STAT3 Signaling Pathway. ACS Med. Chem. Lett 1, 454–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose CP, and Goresky CA (1977). Constraints on the uptake of labeled palmitate by the heart. The barriers at the capillary and sarcolemmal surfaces and the control of intracellular sequestration. Circ. Res 41, 534–545. [DOI] [PubMed] [Google Scholar]
- Rutter GA, and Pinton P (2014). Mitochondria-Associated Endoplasmic Reticulum Membranes in Insulin Signaling. Diabetes 63, 3163–3165. [DOI] [PubMed] [Google Scholar]
- Son N-H, Basu D, Samovski D, Pietka TA, Peche VS, Willecke F, Fang X, Yu S-Q, Scerbo D, Chang HR, et al. (2018). Endothelial cell CD36 optimizes tissue fatty acid uptake. J. Clin. Invest 128, 4329–4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srere PA (1987). Complexes of Sequential Metabolic Enzymes. Annu. Rev. Biochem 56, 89–124. [DOI] [PubMed] [Google Scholar]
- Stahl A, Evans JG, Pattel S, Hirsch D, and Lodish HF (2002). Insulin causes fatty acid transport protein translocation and enhanced fatty acid uptake in adipocytes. Dev. Cell 2, 477–488. [DOI] [PubMed] [Google Scholar]
- Stuhlsatz-Krouper SM, Bennett NE, and Schaffer JE (1998). Substitution of alanine for serine 250 in the murine fatty acid transport protein inhibits long chain fatty acid transport. J. Biol. Chem 273, 28642–28650. [DOI] [PubMed] [Google Scholar]
- Sukriti S, Tauseef M, Yazbeck P, and Mehta D (2014). Mechanisms regulating endothelial permeability. Pulm. Circ 4, 535–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao H, Zhang Y, Zeng X, Shulman GI, and Jin S (2014). Niclosamide ethanolamine-induced mild mitochondrial uncoupling improves diabetic symptoms in mice. Nat. Med 20, 1263–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Vusse GJ, Glatz JFC, Van Nieuwenhoven FA, Reneman RS, and Bassingthwaighte JB (1998). Transport of Long-Chain Fatty Acids across the Muscular Endothelium In Skeletal Muscle Metabolism in Exercise and Diabetes, Richter EA, Kiens B, Galbo H, and Saltin B, eds. (Boston, MA: Springer US; ), pp. 181–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Vliet AR, and Agostinis P (2018). Mitochondria-Associated Membranes and ER Stress. Curr. Top. Microbiol. Immunol 414, 73–102. [DOI] [PubMed] [Google Scholar]
- van der Vusse GJ (2009). Albumin as Fatty Acid Transporter. Drug Metab. Pharmacokinet 24, 300–307. [DOI] [PubMed] [Google Scholar]
- Wada S, Neinast M, Jang C, Ibrahim YH, Lee G, Babu A, Li J, Hoshino A, Rowe GC, Rhee J, et al. (2016). The tumor suppressor FLCN mediates an alternate mTOR pathway to regulate browning of adipose tissue. Genes Dev. 30, 2551–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasserman DH, Kang L, Ayala JE, Fueger PT, and Lee-Young RS (2011). The physiological regulation of glucose flux into muscle in vivo. J. Exp. Biol 214, 254–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermann B (2012). Bioenergetic role of mitochondrial fusion and fission. Biochim. Biophys. Acta BBA - Bioenerg 1817, 1833–1838. [DOI] [PubMed] [Google Scholar]
- Yao C-H, Wang R, Wang Y, Kung C-P, Weber JD, and Patti GJ (2019). Mitochondrial fusion supports increased oxidative phosphorylation during cell proliferation. ELife 8, e41351. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table S1. Results of the primary chemical screening experiment done in triplicate. Related to Figures 1 and S1.
Supplementary Table S2. Results of the secondary screening efforts of top hits from the initial chemical screening results. Related to Figures 1 and S1.
Supplementary Table S3. Sequences of primers used for RT-qPCR. Related to STAR Methods.
Data Availability Statement
This study did not generate any unique datasets or code.