

Abstract
There has been a renewed interest in therapeutic small interfering RNAs (siRNAs) over the past few years. This is particularly the result of successful and efficient delivery of N-acetylgalactosamine (GalNAc)-conjugated siRNAs to the liver. In general, the lead selection process for siRNA drugs is faster and more straightforward than traditional small molecules. Nevertheless, many siRNAs of different sequences and chemical modification patterns must still be evaluated before arriving at a final candidate. One of the major difficulties in streamlining this workflow is the well-known phenomenon that the in vitro data obtained from oligonucleotides transfected into cells are not directly predictive of their in vivo activity. Consequently, all oligonucleotides with some degree of in vitro activity are typically screened in vivo before final lead selection. Here, we demonstrate that the stability of liver-targeting GalNAc-conjugated siRNAs in a mouse liver homogenate shows an acceptable correlation to their in vivo target knockdown efficacy. Therefore, we suggest the incorporation of an in vitro liver homogenate stability assay during the lead optimization process for siRNAs. The addition of this assay to a flow scheme may decrease the need for animal studies, and it could bring cost savings and increase efficiency in siRNA drug development.
Keywords: In vitro in vivo correlation, IVIVC, siRNA, liver homogenate stability, mass spectrometry, metabolite identification, in vitro metabolism, GalNAc-conjugated siRNA, oligonucleotide metabolite identification, LC-MS
Graphical Abstract
Basiri and colleagues have demonstrated that the stability of liver-targeting GalNAc-conjugated siRNAs in mouse liver homogenate shows an acceptable correlation to their in vivo target knockdown efficacy. Therefore, they suggest the incorporation of an in vitro liver homogenate stability assay during the lead optimization process for siRNAs.
Introduction
Gene silencing by RNA interference (RNAi) was first discovered over 20 years ago in nematode worm, Caenorhabditis elegans,1 and it soon generated high hopes for a revolutionary approach to drug design. However, researchers have faced major challenges for utilizing RNAi mechanism in a therapeutic context over the past two decades.2 Nevertheless, the US Food and Drug Administration (FDA) approval of two small interfering RNA (siRNA) drugs in recent years is an exciting indication that research on siRNA therapeutics is finally beginning to bear fruit. Patisiran was approved in 2018 to treat peripheral nerve disease (polyneuropathy) caused by hereditary transthyretin-mediated amyloidosis (hATTR),3 and givosiran was granted FDA approval in 2019 for the treatment of adult patients with acute hepatic porphyria.4
The excitement around siRNA therapeutics stems from the fact that siRNAs utilize a common cellular machinery with high potency and low toxicity to exert their sequence-specific effects.5 In theory, the generic nature of siRNA mechanism of action allows for targeting virtually any nucleic acid sequence, including disease targets that are usually deemed “undruggable”.6 Most approved drugs on the market generally fall within two major structural classes: small molecules and protein therapeutics. The targeting range of small molecules is limited to proteins with hydrophobic pockets, where the small molecules bind and modify the endogenous interactions of the target protein. Although protein-based drugs can recognize targets that are more structurally diverse, they cannot readily cross cell membranes and therefore they are largely restricted to extracellular targets.7 The remaining cellular proteins that are beyond the targeting capabilities of these established drug discovery technologies have been branded as “undruggable.” The so-called “druggable” proteins have been estimated to make up only about 2%–5% of the protein-coding human genome.8,9 It is expected that, upon successful implementation in drug discovery programs, RNAi will significantly increase the number of available drug targets. Another favorable attribute of siRNAs over traditional drug modalities is the speed at which new drug candidates can be developed. It takes a team of medicinal chemists a couple of years to develop a lead small molecule drug.10 In contrast, siRNAs can be designed and validated within months, drastically decreasing the turn-around cycle time for new drug development.
Despite the early promise of RNAi, challenges with delivery, safety, and efficacy of the newly developed siRNAs have hindered progress for the past two decades. However, the tides are turning, as exemplified by the above-mentioned approvals of patisiran and givosiran. As of early 2019, Hu et al.11 have listed more than 50 siRNAs at different phases of clinical trials for conditions as diverse as cardiometabolic and endocrinological diseases to cancer and infectious disease. At the time of this publication, more than 60 siRNA drugs could be found on the ClinicalTrials.gov website (https://clinicaltrials.gov/). Therefore, we should expect to see many more siRNA drugs in the coming years.
Delivery of the siRNA therapeutics is still the most challenging aspect of the RNAi drug development, despite significant progress in recent years. Although some of the current ongoing clinical trials utilize naked, albeit chemically modified, siRNAs that are delivered locally,6 the field has quickly honed in on encapsulation in nanoparticles or conjugation to bioactive ligands as more efficient methods of siRNA drug delivery.12 Lipid and polymer-based nanoparticles are a popular delivery method for siRNAs.13 Patisiran is an example of a siRNA encapsulated in a lipid nanoparticle.14 As demonstrated by givosiran, conjugation of siRNAs to N-acetylgalactosamine (GalNAc) is another successful approach that allows for robust and prolonged hepatic delivery of siRNAs following subcutaneous (SC) injection.15 Triantennary GalNAc-targeting moieties bind to the asialoglycoprotein receptor (ASGPR) that is highly and almost exclusively expressed on hepatocytes, resulting in potent and targeted siRNA delivery to the liver.16 Nevertheless, to date, liver remains the prime target organ for systemically delivered siRNAs.17
An under-studied area in the development process for siRNAs is the poor translation of siRNA efficacy from in vitro to in vivo. In order to discover the most efficacious siRNA sequence for a target mRNA, considering the limitations of currently available design rules and selection algorithms,18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28 the current best practice is to compare the in vivo potency of siRNAs tiling across the entire mRNA target.29,30 In addition to siRNA sequence, a variety of different chemical modification patterns of the backbone and nucleobases must be tested for each siRNA molecule to find the modification pattern that results in optimized delivery and pharmacodynamic response.31, 32, 33, 34, 35, 36 Interestingly, the number and position of chemical modifications necessary for any given sequence is variable and is usually empirically determined. Moreover, chemical modifications can influence potency, especially if they are placed on the antisense strand,37 and it has already been demonstrated that algorithms derived for non-modified siRNAs have very little predictive power for chemically-modified siRNAs.38 Given all these considerations, screening a large number of siRNAs with varying sequences and modification patterns is necessary for identifying potent lead siRNAs. The first round in this multi-step screening process begins with a single-point in vitro assessment of the siRNA activity in cultured cells,35,39,40 followed by in vitro dose response experiments in the same cell-based system.29,37 However, it is well established in the oligonucleotide therapeutics field that the data obtained from in vitro oligonucleotide transfection into cells is not predictive of their in vivo activity.30,41 Therefore, all siRNAs that show some gene silencing activity and a reasonable dose response curve in vitro must be dosed in vivo to determine their activity.29,37 This leads to many constructs (often 100s) being administered to rodent models, making the screening process long, costly, and inefficient in terms of animal use. Here, we present a simple in vitro screening assay based on the stability of siRNA constructs in a liver homogenate system that can satisfactorily predict pharmacodynamics (PD) efficacy in vivo. Based on our studies with a large panel of siRNAs, we concluded that poor siRNA candidates in terms of in vivo efficacy can be successfully identified in this assay. Eliminating such siRNAs from the future rounds of animal studies significantly reduces the effort and cost associated with siRNA screens for lead molecule identification, and, more importantly, it reduces the number of animals required for these experiments.
Results
Establishment of a Surrogate In Vitro System for siRNA Metabolite Identification
Our initial set of experiments involved the analysis of terminal liver samples from rat and cynomolgus monkey (cyno) toxicology studies to establish the pattern of siRNA metabolism in vivo. These animals had been dosed SC with internal GalNAc-conjugated siRNA molecules once weekly for 2 weeks and sacrificed at the end of the second week. A schematic representation of siRNAs used in this study is shown in Figure 1A. We observed metabolite formation for both sense and antisense strands in liver samples from the toxicology studies described above. For the sense strand, observed metabolites were mostly the result of cleavages in the triantennary GalNAc moiety. These included sense strand products resulting from the loss of GalNAcs, complete loss of the triantennary targeting moiety, or a cleavage in the middle of the targeting moiety (Figure 1B). Antisense (AS) strand metabolism was primarily the result of nucleolytic activity occurring on the 3′ side of the AS chain (Figure 1C). These observations were largely in agreement with previously published studies concerning metabolite identification of siRNAs.42, 43, 44, 45, 46, 47, 48, 49 An interesting observation with a few of our siRNA constructs was the asymmetric pattern in which antisense metabolites were formed from the 3′ end. For example, in the case of the siRNA shown in Figure 1D, while considerable amounts of 3′n-1, 3′n-4, and 3′n-5 shortmers of the antisense strand were observed, very little 3′n-2 and 3′n-3 were present. Therefore, because of its distinctive metabolite formation pattern, this molecule was chosen for distinguishing between the performance of in vitro models of siRNA metabolism described below.
Figure 1.

Several siRNA Metabolites Were Detected in Rat and NHP Liver Samples from a Toxicology Study
(A) Schematic structure of siRNAs used for the current study. The test constructs consisted of a 21-mer double-stranded RNA with three GalNAc molecules conjugated to the 5′ end of the sense strand through a polyethylene glycol (PEG) and a trivalent branching linker placed in tandem. (B) The antisense strand was primarily metabolized through the successive removal of nucleotides from its 3′ end. (C) Metabolism of the sense strand largely occurred within its triantennary GalNAc targeting moiety. (D) Total ion chromatogram of a representative siRNA molecule showing intact sense and antisense strands, as well as their metabolites. Note the lack of significant peaks for AS 3′n-2 and 3′n-3 for this particular siRNA construct.
Next, we investigated several in vitro incubation media with the aim of finding a suitable surrogate system that most closely resembles the metabolism of siRNAs in vivo. The previously mentioned siRNA was incubated for increasing durations of time in various biological matrices known for their drug-metabolizing capabilities. These in vitro biological systems included phosphodiesterase I from snake venom, RNase A/T1 enzyme mix cocktail, plasma and serum from different species (rodent, non-human primate [NHP], and human), hepatocytes from different species, rat tritosomes, human lysosomes, as well as liver homogenates from various species.
Out of all these different systems, the liver homogenate produced the best overall results. The siRNA molecules remained intact and did not generate any metabolites after 72 h of incubation in plasma, serum, or RNase cocktail enzyme mix. Snake venom phosphodiesterase only acted on the antisense strand and, even then, it failed to reproduce the previously described pattern with lower ratios of 3′n-2 and 3′n-3 compared to other AS metabolites. In contrast, only sense strand was metabolized by rat tritosome and human lysosome, with the antisense remaining intact in those systems. With hepatocytes, we observed siRNA uptake in hepatocytes derived from some species but not others. In addition, we did not observe significant siRNA metabolism in hepatocytes. These observations are summarized in Figure S1.
As shown in Figure 2, there is a strong relationship between metabolites generated in liver homogenate and those observed in vivo, especially for the antisense strand, where the previously mentioned asymmetric pattern of metabolite formation was replicated in liver homogenate. This particular pattern of AS metabolite formation from the selected siRNA molecule, where AS 3′n-2 and AS 3′n-3 had been skipped to a very large extent, drastically facilitated choosing among different in vitro systems. The lack of accumulation for AS 3′n-2 and 3′n-3 can be explained in two different ways. One possibility is that these metabolites are so unstable against exonucleases that they are rapidly converted to 3′n-4 and therefore they cannot accumulate. Another possibility is for AS 3′n-4 to be the product of an endonuclease and accumulate independent of 3′n-2 and 3′n-3. Based on time course data presented in Table 1 and our observations with snake venom phosphodiesterase, we favor the second explanation. Table 1 shows that there is no lag in AS 3′n-4 accumulation and it can be detected starting from early time points during incubation. By comparison, metabolites related to sequential loss of GalNAcs from sense strand show a clear trend, where −2 GalNAc accumulation follows −1 GalNAc by a lag and −3 GalNAc follows −2 GalNAc by a similar time lag. Also, AS 3′n-5 seems to follow AS 3′n-4 by a lag consistent with the idea that 3′n-4 is a putative endonuclease product of the AS strand while 3′n-5 is formed from 3′n-4 by an exonuclease. In addition, AS 3′n-2 and 3′n-3 did not show any significant instability compared to other metabolites in the presence of snake venom phosphodiesterase, where they accumulated to levels similar to other shortmers. Nevertheless, this is just circumstantial evidence, and confident distinction between those two possibilities can only be made after extensive nuclease reaction phenotyping.
Figure 2.

There Is a Strong Relationship between siRNA Metabolites Observed in Liver Homogenate and In Vivo
(A) The graphs on the top compare the metabolites observed in rat liver homogenate to those observed at three increasing in vivo dosing to rats, for both sense and antisense strands. (B) The graphs at the bottom show the correlation between metabolites identified from two different in vivo dose levels in cynos and the metabolites formed after siRNA incubation in cyno liver homogenate. The alignment between in vitro and in vivo observations is strong for the antisense strand. For sense strand, the in vivo samples generally demonstrate further metabolism than in vitro samples. Nevertheless, in both systems, metabolism is largely confined to the triantennary GalNAc moiety.
Table 1.
siRNA Metabolite Profile Change in Rat Liver Homogenate during a 48-h Incubation
| Time Point (h) | Sense Strand Metabolites (%) |
Antisense Strand Metabolites (%) |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Intact | −1 GalNAc | −2 GalNAc | −3 GalNAc | -PEG | -Branch | 3′n-1 -Branch | Intact | 3′ n-1 | 3′ n-2 | 3′ n-3 | 3′ n-4 | 3′ n-5 | 3′ n-6 | 5′ n-1 | |
| 1 | 98.0 | 1.6 | 0.3 | ND | ND | 0.1 | ND | 95.9 | 1.2 | 0.2 | 0.4 | 0.2 | ND | ND | 2.1 |
| 6 | 78.2 | 16.1 | 1.9 | ND | 0.1 | 0.1 | 3.6 | 76.8 | 11.7 | 0.4 | 0.6 | 4.9 | 1.9 | 1.6 | 2.1 |
| 24 | 31.8 | 32.8 | 20.4 | 10.5 | 0.1 | ND | 4.4 | 53.6 | 16.8 | 0.6 | 3.5 | 10.1 | 13.1 | 0.9 | 1.4 |
| 48 | 16.1 | 22.2 | 25.3 | 32.4 | 0.1 | ND | 3.9 | 49.5 | 17.4 | 0.6 | 2.3 | 12.8 | 14.6 | 1.6 | 1.2 |
For the sense strand, in vitro metabolites in liver homogenate were limited to GalNAc removal. Metabolites with the loss of 1, 2, or 3 GalNAcs from the triantenary ASGPR ligand were observed, with the metabolic profile shifting toward loss of all three GalNAcs over time (Table 1). This was different from in vivo samples, where further cleavages inside the linker were observed. Interestingly, sense strand linker cleavages were also observed in lysosome and tritosome incubations (data not shown), suggesting that they occur after siRNA internalization by cells. In contrast, nucleolytic cleavages of the antisense strand appear to happen within the extracellular hepatic matrix and hence they can be recapitulated by liver homogenate.
Identifying the Relationship between siRNA Stability in Liver Homogenate and In Vivo Efficacy
Considering that one of the main factors impeding oligonucleotide therapeutic efficacy is their susceptibility to nucleases, we sought to investigate whether the in vitro stability of siRNAs in our liver homogenate system can predict their in vivo PD efficacy. To this end, liver homogenates from different species (mouse, rat, cyno, and human) were incubated in the presence of varying concentrations of siRNAs and with different incubation times ranging from 24–96 h in pursuit of an optimized experimental protocol. As shown in Figure 3, our initial experiment with a panel of 20 siRNAs indicated that both mouse and cyno liver homogenates demonstrate a comparably strong correlation to human liver homogenate in terms of siRNA metabolism. Our follow-up study with a larger number of siRNAs further supported the alignment between results obtained from human and mouse liver homogenates (Figure 3B). Based on these observations, we chose mouse liver homogenate as our preferred incubation medium. This was partly due to the ease and low cost of acquiring mouse livers. But, more importantly, it was because we observed that by homogenizing whole mouse livers—instead of a piece of human or cyno liver—we had better reproducibility between the assays that were performed on different days. Following these optimization experiments, our final protocol consisted of incubation of 1 μM siRNA in mouse liver homogenate for 72 h. The details of the optimized workflow for the in vitro liver stability assay are as follows (Figure 4).
Figure 3.

The Stability of siRNAs in Human Liver Homogenate Has a Strong Correlation to Their Stability in Mouse Liver Homogenate
(A) The correlation between siRNA stability in mouse liver homogenate and human liver homogenate is comparable to the correlation between cyno and human. This can also be concluded from the strong correlation between siRNA stability in cyno and mouse liver homogenates. Therefore, mouse and cyno liver homogenates appear to be interchangeable for performing the liver stability assay. (B) The findings of (A) were further supported by employing a larger number of siRNAs and confirming the strong correlation between mouse and human liver homogenates for siRNA stability measurement.
Figure 4.

Summarized Workflow of the Liver Homogenate Stability Assay for siRNAs
Mouse livers were homogenized at 200 mg/mL tissue concentration in an aqueous buffer containing Tris (100 mM) and magnesium (1 mM) without any detergents or other reagents that might cause mass spectrometry (MS) signal suppression. It is worth mentioning that this liver homogenate system has been used previously by other groups for metabolite identification of single-stranded antisense oligonucleotides.50, 51, 52 200 μL of liver homogenate was added to each well of a 96-well plate. For each siRNA to be tested in this assay, two 200 μL aliquots of liver homogenate were required. One individual siRNA was added to each aliquot of liver homogenate in each well at 1 μM concentration. In this fashion, half of the already aliquoted liver homogenates were spiked with different siRNA constructs (1 siRNA per well), while the other half remained un-spiked. Then, all liver homogenates (spiked and un-spiked) were incubated for 72 h with gentle shaking (400 rpm) at 37°C. At the end of 72 h, the other half of liver homogenates were spiked with 1 μM of siRNA in the same manner that the rest of the samples were spiked at the beginning of the incubation (1 siRNA per well). Following this protocol, two distinct groups of samples in regards to siRNA incubation were attained: one group with siRNAs incubated in liver homogenate for 72 h (we called these 72-hr siRNA samples) and another group with the same siRNAs that were added to liver homogenates just before sample extraction without any incubation (we called them 0-hr siRNA samples). The purpose of adding siRNAs to incubated blank liver homogenate, instead of fresh liver homogenate, was to ensure that the matrix for 0-hr and 72-hr siRNA samples were matched as closely as possible. This was followed by adding equal amounts of an internal standard (we used 2.5 μg/mL of a different siRNA) to all samples. The samples were then processed by solid phase extraction (SPE) and analyzed by liquid chromatography-mass spectrometry (LC-MS). The stability of each siRNA was reported as percent of intact antisense remaining following 72 h of incubation using this simple formula (for an example, see Figure S2):
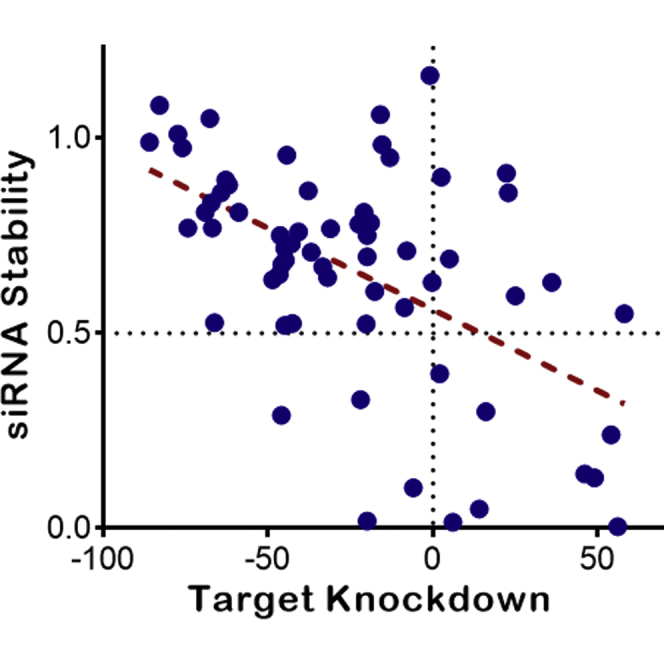
Subsequently, a large panel of GalNAc-conjugated siRNAs targeting the production of a hepatic protein was tested in the mouse liver homogenate stability assay as outlined above. Figure 5 shows a plot of antisense strand stability for these siRNAs versus their in vivo efficacy in knocking down the levels of serum biomarker for the target gene 4 weeks after SC administration. Please note that a few siRNAs generated stability values significantly greater than 100% in this assay, which was distinctively characterized as experimental failure. Therefore, 7 data points with siRNA stability values larger than 120% were excluded from further analysis and they are not shown in this plot. As demonstrated in this figure, a significant correlation (p < 0.0001) can be observed between the in vitro liver stability of the antisense strand of various siRNAs and their in vivo pharmacodynamics efficacy. Despite the fact that the absolute correlation is not extremely strong (as shown by the handful of very stable siRNAs in liver homogenate that didn’t demonstrate any knockdown activity), this assay shows huge promise as a means of screening out siRNAs with poor PD efficacy. Close examination of Figure 5 reveals that most siRNAs with stable antisense strands (AS stability greater than 50%) show at least some knockdown activity in vivo. Additionally, siRNAs with very stable antisense strands in this assay (AS stability close to 100%) are often capable of generating particularly strong target knockdown after in vivo administration to mice. More importantly, none of the siRNAs with less than 50% stability in liver homogenate have produced efficient target knockdown. Therefore, they can be confidently excluded from further in vivo evaluation. Based on these observations, the liver homogenate stability data have been incorporated as a decision-making factor in prioritizing early stage siRNA progress to in vivo animal studies in our flow scheme, and it has been a significant factor in reducing animal use for our siRNA programs.
Figure 5.

A Significant Correlation Is Observed between the In Vitro Stability of siRNAs in Mouse Liver Homogenate and Their In Vivo PD Efficacy
This plot demonstrates the antisense strand stability after 72 h of incubation in a mouse liver homogenate versus the target knockdown efficacy for various siRNAs. These two variables show a significant correlation to one another (p < 0.0001). Although the absolute correlation is modest (Pearson r: –0.54), the tremendous value of liver stability assay for decision making cannot be overlooked. Most siRNAs with in vitro stability values greater than 50% show target knockdown after in vivo administration to mice (data points in the upper left quadrant). In contrast, the majority of siRNA constructs with less than 50% in vitro stability of antisense strand do not show any knockdown when dosed to animals (data points at the lower right quadrant). It should be noted that a few siRNAs generated stability values significantly greater than 100% in this assay, which was distinctively characterized as experimental failure. Therefore, 7 data points with siRNA stability larger than 120% were excluded from further analysis and they are not shown in this plot.
Discussion
The current study reports the development and application of a stability assay for predicting the PD efficacy of liver-targeting siRNAs. Based on our investigation of a large panel of GalNAc-conjugated siRNAs, we established a strong relationship between the stability of siRNAs in mouse liver homogenate and in vivo target knockdown efficacy in transgenic mice. This suggests that siRNA stability in the extracellular matrix of liver tissue is a rate-limiting step for in vivo efficacy.
Given that intact or mostly intact siRNA is required for RNA-induced silencing complex (RISC) loading and RNAi specificity,53,54 it is not surprising that degradation-resistant siRNA constructs are more efficacious. However, considering the complex chain of events leading to an siRNA exerting its effect, it was initially surprising that liver homogenate stability alone appears to have an almost definitive predictive power. Before interacting with a target mRNA molecule and inducing its cleavage, GalNAc-conjguated siRNA must bind ASGPR, be taken up into the cell via endocytosis, escape from endosome, and be loaded into the RISC.55,56 To shed light on our potentially unlikely finding, we looked to the literature and to the specific panel of molecules we used to better understand why liver homogenate stability might be representative of in vivo efficacy.
It is well established that substitutions at the 2′ position on the ribose ring, as well as incorporation of terminal phosphorothioate backbone modifications, improve metabolic stability. During their optimization experiments for positioning of 2′-deoxy, 2′-fluoro and 2′-O-methyl substitutions across siRNA sequences, Foster et al.32 demonstrated that improvements to in vivo performance observed with the optimal designs are primarily due to enhanced metabolic stability rather than enhanced interactions with the RNAi machinery. Interestingly, these observations were in line with a mathematical model that Bartlett and Davis.57 developed for siRNA-mediated gene silencing over a decade ago. Based on this model, the experimental observations for siRNA efficacy were best explained by changing model parameters that affected the stability of siRNAs before they reach the cytosolic compartment, indicating that the stabilization advantages of chemically modified siRNAs primarily originate from the effects prior to cellular uptake and before the siRNAs can interact with the intracellular components.
If extracellular stability drives gene silencing, and we know that chemically modified siRNAs used in this study are highly stable in serum and plasma, it stands to reason that in vivo siRNA degradation must occur in a non-cytosolic, non-serum compartment in the body. Given that liver homogenate consists largely of liver interstitia populated with large cell fragments, it is plausible that the chemical or biochemical entities responsible for efficacy-dependent siRNA degradation reside in the liver interstitia. Furthermore, extracellular stability has functional implications for siRNA delivery and mechanism of action. If GalNAc is removed prior to ASGPR binding, receptor-mediated uptake cannot occur, and if exonucleases processively remove enough nucleobases, then the RISC no longer recognizes the siRNA molecule as a substrate.
In accordance with these considerations, our in vitro stability test in an extracellular matrix indeed enables prediction of GalNAc-conjugated siRNA in vivo efficacy, especially when testing different sequences and/or modification patterns. Our overall results indicate that liver homogenate is the best biological matrix for performing such stability assays on GalNAc-conjugated siRNAs that target liver, which are currently the primary class of siRNAs in research and development (R&D) stages of pharmaceutical development.
Although it is beyond the scope of this work, in the future, it will be important to perform reaction phenotyping to identify the molecular entities responsible for the biotransformation processes leading to the observed metabolites. In a recent review, Humphreys et al.56 compiled a list of known siRNA metabolites and the enzymes and chemical processes that are likely responsible for generating them. Major siRNA-metabolizing enzymes identified in the work include GalNAc cleavage by β-N-acetylglucosaminidase; catabolism by lysosomal hydrolase; internal esterification; cyclization; oxidation by alcohol dehydrogenase and aldehyde dehydrogenase; AGO2-mediated endonuclease hydrolysis; 5′→3′ exonuclease cleavage by XRN1 and XRN2; 3′→5′ exonuclease cleavage by RNR, DEDD, and PDX families and exosomes; and 5′ phosphorylation and dephosphorylation by kinases, such as CLP1 and phosphatases, respectively. Comprehensive cross-species phenotyping will inform species differences and improve translation.
In accordance with these considerations, we highly recommend the incorporation of liver homogenate stability assay in the siRNA lead selection workflow for GalNAc-conjugated siRNAs, as this simple and cheap assay can eliminate the need for a lot of costly rodent use for siRNA constructs with poor stability that are bound to have poor PD efficacy.
Materials and Methods
Chemicals and Reagents
Tris, EDTA, hydrochloric acid (HCl), triethylamine (TEA), hexafluoroisopropanol (HFIP), tris(2-carboxyethyl)phosphine (TCEP), magnesium chloride (MgCl2), methanol, acetonitrile, tetrahydrofuran (THF), and phosphodiesterase I from snake venom were purchased from Sigma-Aldrich (St. Louis, MO, USA). 10% phosphoric acid was purchased from Ricca Chemical Company (Arlington, TX, USA). RNAsecure RNase inactivation reagent and RNase cocktail enzyme mix were from Life Technologies (Thermo Fisher Scientific, Waltham, MA, USA). Clarity OTX lysis-loading buffer, as well as 96-well Clarity OTX SPE plates (100 mg/well), were purchased from Phenomenex (Torrance, CA, USA). C57BL/6 mouse, cyno and human liver, serum, and plasma samples were obtained from BioIVT (Westbury, NY, USA). Plateable hepatocytes were acquired from Lonza (Basel, Switzerland), and liver lysosomes and tritosomes were purchased from XenoTech (Osaka, Japan). 96-well nuclease-free plates for sample handling and autosampler induction and silicone sealing mats were purchased from Axygen (Corning, Corning, NY, USA). Nitrogen and Milli-Q water were generated in house. The following solid-phase extraction (SPE) buffers were also prepared in house.
Equilibration buffer: 50 mM NaH2PO4, 2 mM NaN3, 10 mM K2EDTA in water (pH = 5.5)
Wash buffer 1: 50 mM NaH2PO4 in 50:50 (v:v) water:acetonitrile (pH = 5.5)
Wash buffer 2: 50 mM NH4HCO3 in 20:80 (v:v) water:acetonitrile (pH = 5.5)
Elution buffer: 100 mM NH4HCO3, 10 mM TCEP in 50:40:10 (v:v:v) water:acetonitrile:tetrahydofuran (pH = 9)
The pH of all buffers and solutions were adjusted by adding MS-friendly volatile acids and bases, i.e., ammonium hydroxide or glacial acetic acid.
Tissue Homogenization and Incubation
Frozen mouse, cyno, or human livers were homogenized at 200 mg/mL tissue concentration in a homogenization buffer containing 100 mM Tris and 1 mM MgCl2 in water (pH = 6.0, adjusted with HCl). The homogenization was performed on a Bertin Technologies (Montigny-le-Bretonneux, France) Precellys24 bead mill tissue homogenizer. 200 μL of the homogenate was added to each well of a 96-well plate, and the plate was incubated at 37°C with gentle shaking (400 rpm) for 72 h using a Thermomixer-R from Eppendorf (Hauppauge, NY, USA). 4 μL of a 50 μM siRNA solution was added to each well for a final siRNA concentration of 1 μM either at the beginning (72-hr samples) or end (0-hr samples) of the 72h incubation. Also, an internal standard (IS) at a final concentration of 2.5 μg/mL was added to all samples (1 μL of 0.5 mg/mL IS solution to 200 μL of liver homogenate in each well) before starting the SPE sample preparation. For a schematic representation of the incubation protocol, see Figure 4.
Sample Preparation
Sample preparation was performed using the SPE method. 200 μL of 10% phosphoric acid was added to 200 μL of liver homogenate in each well, and the plates were vortexed for 5 min. After that, 600 μL of the Clarity lysis-loading buffer was added to each well, and the plates were vortexed for another 5 min followed by centrifugation at 3,500 rpm and 10°C for 8 min on an Eppendorf 3810R refrigerated centrifuge (Hamburg, Germany). During this time, Clarity OTX SPE plates (Phenomenex, Torrance, CA, USA) were equilibrated by sequentially adding 1 mL of methanol and 1 mL of the “equilibration buffer” to each well and draining the liquids through the SPE plate using a Waters (Milford, MA, USA) Positive Pressure-96 SPE processor. 800 μL of the supernatant from lysed and centrifuged liver homogenate samples was transferred to each well of the conditioned SPE plate and moved through the SPE resin by applying pressure. This was followed by washing the plate once with the “equilibration buffer” and four times with “wash buffer 1” (1 mL each time). The plates were then washed twice with 1 mL of “wash buffer 2” and dried by applying high pressure before eluting the absorbed siRNAs. Elution plates were prepared by adding 50 μL of 50% solution of RNAsecure reagent in 50 mM EDTA to each well. The samples were then eluted from the SPE plates to the elution plates by adding 500 μL of the “elution buffer” to each well two times (a total of 1 mL elution buffer). This was followed by drying the elution plate using a Caliper Life Sciences (Waltham, MA, USA) Turbovap 96 microplate nitrogen evaporator. The samples were dried at 60°C and 10 PSI nitrogen pressure for about 2 h until there was only ≈200 μL of liquid left in each well. The samples were then transferred to autosampler plates for LC-MS analysis.
LC-MS Analysis
The LC-MS analysis was performed using an Agilent Technologies (Santa Clara, CA, USA) 1290 Infinity series ultra-performance liquid chromatography (UPLC) system consisting of a binary pump, an autosampler, and a temperature-controlled column compartment (TCC) coupled to a Thermo Scientific (Waltham, MA, USA) Q Exactive HF-X Orbitrap mass spectrometer. Samples were separated on a Waters (Milford, MA) 2.1 × 50 mm Acquity UPLC Oligonucleotide BEH C18 Column (130 Å, 1.7 μm) at 80°C. Mobile phase A consisted of 15 mM TEA and 50 mM HFIP in water, and mobile phase B was 15 mM TEA and 50 mM HFIP in methanol. No pH adjustment was performed on mobile phase solutions. 20 μL of each sample was injected at a 0.4 mL/min flow rate under the following gradient condition (min – %B): 0 – 2, 1.5 – 2, 11.5 – 35, 12 – 95, 14 – 95, 14.5 – 2. The total runtime was 16 min, and the LC output was diverted to waste from 0–2 min and 12–16 min.
The mass spectrometer was operated with the following parameters: spray voltage (3.2 kV), capillary temperature (320°C), sheath gas flow rate (50 arbitrary units), sweep gas flow rate (1 arbitrary unit), auxillary gas flow rate (10 arbitrary units), auxillary gas temperature (425°C), and full scan negative-ion mode with 60,000 resolution. Instrument control and data acquisition was performed using the Xcalibur 4.0 from Thermo Scientific (Waltham, MA, USA), and data analysis was assisted with the Skyline software from the McCoss laboratory.58 The OMA & OPA software package was utilized for m/z prediction for the expected sense and antisense metabolites.59
Synthesis, Cleavage, Purification, and Annealing of the siRNA Duplex
Synthesis of chemically modified siRNA sequences was performed on the Bioautomation Mermade 12. Reagent solutions, dimethoxytrityl,2-cyanoethyl-phosphoramidite solutions, and solvents were attached to the instrument. The columns containing solid support (BioAutomation, Universal Support, 500Å Controlled Pore Glass [CPG]) were affixed to the synthesis block, loaded onto the instrument, and washed twice with acetonitrile (DNA Synthesis Grade, anhydrous, Sigma Aldrich). The phosphoramidite and reagent solution lines were purged. The synthesis was performed using Poseidon software and accomplished by repetition of the deprotection/coupling/capping/oxidation/capping synthesis cycle. After the final reaction cycle, the resin was first treated with diethylamine solution to remove the 2-cyanoethyl protecting groups from the phosphate backbone. The support was washed with acetonitrile, detritylation reagent (3% dichloroacetic acid in dichloromethane [DCM], EMD) and again washed with acetonitrile.
The monomethoxytrityl (MMT)-protected sense strands were first treated with 2% trifluoroacetic acid (TFA) in DCM to remove the MMT protecting group, followed by coupling with triantennary GalNAc carboxylic acid (4 eq) in the presence of N-[(7-aza-1H-benzotriazol-1-yl)(dimethylamino)methylene]-N-methylmethanaminium tetrafluoroborate N-oxide (TATU, 4 eq) and DIEA (8 eq). The solid support was washed with DMF and DCM after 18 h.
Concentrated ammonium hydroxide (1–1.5 mL, J.T. Baker) was added to the solid support in each column. The filtrates were collected by gravity into a 24-deep well plate. The cleavage process was repeated three times, and the eluent was combined. The plate was sealed, placed into a deprotection chuck, and incubated at 60°C for 6 h. The plate was cooled to room temperature before opening.
The cleavage solution was purified by anion exchange chromatography on an Agilent high-performance liquid chromatography (HPLC) system by elution using a linear gradient of sodium chloride in 20 mM sodium phosphate, 15% acetonitrile, pH 8.5. The pooled solutions were concentrated in Genevac to give the single strands.
Single strands were dissolved to an approximate concentration of 2 mM based on the calculated dry weight, and the concentration was quantified by UV absorbance at 260 nm. Single strands were diluted to 1 mM, and the sense and antisense strands were transferred in equal volumes to a Matrix tube to form the corresponding duplex. Duplexes were annealed at 90°C for 10 min and then allowed to cool to room temperature slowly. Single strands were titrated as necessary with heating and cooling to achieve >95% duplex purity.
Toxicology Studies in Rats and NHPs
14-day toxicology studies were performed at the Charles River Laboratories (Spencerville, OH, USA). The test and control articles were administered to male Sprague Dawley rats (5 per group) by once weekly SC injection into the scapular and mid-dorsal areas on days 1 and 8. The administered dose levels to rats were 0 (control article), 10, 50, and 200 mg/kg. Female cynos (3 per group) were dosed at 10 and 300 mg/kg of the test article at the same schedule by SC injection. All study animals underwent scheduled euthanasia on day 15 and collected tissue samples were snap frozen in liquid nitrogen and stored at −70°C for subsequent analysis.
In Vivo Mouse Pharmacology Experiments and Target Protein Measurement
All animal studies were approved and performed under guidance of the Amgen institutional animal care and use committee (IACUC). Transgenic mice expressing the human target of the siRNAs were bred at Taconic Laboratories (Germantown, NY, USA). Five mice per treatment group were bled via submandibular route at pre-dose baseline and week 4 following administration of a single SC dose of either siRNA or vehicle phosphate-buffered saline (pH 7.4). Serum was analyzed at time points described above, and the serum protein biomarker of target knockdown was measured by using commercially available ELISA kits from Mercodia (Winston Salem, NC, USA).
Author Contributions
B.B., B.M.R., F.X., and B.W. conceptualized this study. B.W. designed and synthesized the siRNA compounds. B.B. and F.X. performed the LC-MS experiments. B.M.R., J.M.L., S.C.H., and M.B.T. designed the toxicology studies, while P.Y. and M.F. generated the in vivo pharmacology data. B.B. and S.C.H. initially wrote the manuscript that was subsequently reviewed by all authors.
Conflicts of interest
All authors are employed by Amgen, which provided funding for this investigation.
Acknowledgments
Special thanks are given to Dan Rock, Omar Barnaby, Ed Hsieh, David Doherty, Jianxia Shi, and Angela Purcell.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2020.07.012.
Supplemental Information
References
- 1.Fire A., Xu S., Montgomery M.K., Kostas S.A., Driver S.E., Mello C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 2.Krieg A.M. Is RNAi dead? Mol Ther. 2011;19:1001–1002. doi: 10.1038/mt.2011.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adams D., Gonzalez-Duarte A., O’Riordan W.D., Yang C.-C., Ueda M., Kristen A.V., Tournev I., Schmidt H.H., Coelho T., Berk J.L. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018;379:11–21. doi: 10.1056/NEJMoa1716153. [DOI] [PubMed] [Google Scholar]
- 4.Scott L.J. Givosiran: First Approval. Drugs. 2020;80:335–339. doi: 10.1007/s40265-020-01269-0. [DOI] [PubMed] [Google Scholar]
- 5.Pratt A.J., MacRae I.J. The RNA-induced silencing complex: a versatile gene-silencing machine. J. Biol. Chem. 2009;284:17897–17901. doi: 10.1074/jbc.R900012200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaczmarek J.C., Kowalski P.S., Anderson D.G. Advances in the delivery of RNA therapeutics: from concept to clinical reality. Genome Med. 2017;9:60. doi: 10.1186/s13073-017-0450-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Verdine G.L., Walensky L.D. The challenge of drugging undruggable targets in cancer: lessons learned from targeting BCL-2 family members. Clin. Cancer Res. 2007;13:7264–7270. doi: 10.1158/1078-0432.CCR-07-2184. [DOI] [PubMed] [Google Scholar]
- 8.Hopkins A.L., Groom C.R. The druggable genome. Nat. Rev. Drug Discov. 2002;1:727–730. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- 9.Russ A.P., Lampel S. The druggable genome: an update. Drug Discov. Today. 2005;10:1607–1610. doi: 10.1016/S1359-6446(05)03666-4. [DOI] [PubMed] [Google Scholar]
- 10.Hughes J.P., Rees S., Kalindjian S.B., Philpott K.L. Principles of early drug discovery. Br. J. Pharmacol. 2011;162:1239–1249. doi: 10.1111/j.1476-5381.2010.01127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hu B., Weng Y., Xia X.-H., Liang X.J., Huang Y. Clinical advances of siRNA therapeutics. J. Gene Med. 2019;21:e3097. doi: 10.1002/jgm.3097. [DOI] [PubMed] [Google Scholar]
- 12.Martínez T., Jiménez A.I., Pañeda C. Short-interference RNAs: becoming medicines. EXCLI J. 2015;14:714–746. doi: 10.17179/excli2015-297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nikam R.R., Gore K.R. Journey of siRNA: Clinical Developments and Targeted Delivery. Nucleic Acid Ther. 2018;28:209–224. doi: 10.1089/nat.2017.0715. [DOI] [PubMed] [Google Scholar]
- 14.Suhr O.B., Coelho T., Buades J., Pouget J., Conceicao I., Berk J., Schmidt H., Waddington-Cruz M., Campistol J.M., Bettencourt B.R. Efficacy and safety of patisiran for familial amyloidotic polyneuropathy: a phase II multi-dose study. Orphanet J. Rare Dis. 2015;10:109. doi: 10.1186/s13023-015-0326-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nair J.K., Willoughby J.L.S., Chan A., Charisse K., Alam M.R., Wang Q., Hoekstra M., Kandasamy P., Kel’in A.V., Milstein S. Multivalent N-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J. Am. Chem. Soc. 2014;136:16958–16961. doi: 10.1021/ja505986a. [DOI] [PubMed] [Google Scholar]
- 16.Springer A.D., Dowdy S.F. GalNAc-siRNA Conjugates: Leading the Way for Delivery of RNAi Therapeutics. Nucleic Acid Ther. 2018;28:109–118. doi: 10.1089/nat.2018.0736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rondinone C.M. Therapeutic potential of RNAi in metabolic diseases. Biotechniques. 2006;40(Suppl):31–36. doi: 10.2144/000112163. [DOI] [PubMed] [Google Scholar]
- 18.Malhotra M., Nambiar S., Rengaswamy V., Prakash S. Small interfering ribonucleic acid design strategies for effective targeting and gene silencing. Expert Opin. Drug Discov. 2011;6:269–289. doi: 10.1517/17460441.2011.555394. [DOI] [PubMed] [Google Scholar]
- 19.ElHefnawi M., Hassan N., Kamar M., Siam R., Remoli A.L., El-Azab I., AlAidy O., Marsili G., Sgarbanti M. The design of optimal therapeutic small interfering RNA molecules targeting diverse strains of influenza A virus. Bioinformatics. 2011;27:3364–3370. doi: 10.1093/bioinformatics/btr555. [DOI] [PubMed] [Google Scholar]
- 20.Grinev V.V. Design and quality control of short interfering RNA. Mol. Biol. 2012;46:739–754. [PubMed] [Google Scholar]
- 21.Hagedorn P.H., Yakimov V., Ottosen S., Kammler S., Nielsen N.F., Høg A.M., Hedtjärn M., Meldgaard M., Møller M.R., Orum H. Hepatotoxic potential of therapeutic oligonucleotides can be predicted from their sequence and modification pattern. Nucleic Acid Ther. 2013;23:302–310. doi: 10.1089/nat.2013.0436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jayasena S.D. Designer siRNAs to overcome the challenges from the RNAi pathway. J. RNAi Gene Silencing. 2005;2:109–117. [PMC free article] [PubMed] [Google Scholar]
- 23.Mysara M., Garibaldi J.M., Elhefnawi M. MysiRNA-designer: a workflow for efficient siRNA design. PLoS ONE. 2011;6:e25642. doi: 10.1371/journal.pone.0025642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Naito Y., Ui-Tei K. siRNA Design Software for a Target Gene-Specific RNA Interference. Front. Genet. 2012;3:102. doi: 10.3389/fgene.2012.00102. 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pan W.-J., Chen C.-W., Chu Y.-W. siPRED: predicting siRNA efficacy using various characteristic methods. PLoS ONE. 2011;6:e27602. doi: 10.1371/journal.pone.0027602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pascut D., Bedogni G., Tiribelli C. Silencing efficacy prediction: a retrospective study on target mRNA features. Biosci. Rep. 2015;35:e00185. doi: 10.1042/BSR20140147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pei Y., Tuschl T. On the art of identifying effective and specific siRNAs. Nat. Methods. 2006;3:670–676. doi: 10.1038/nmeth911. [DOI] [PubMed] [Google Scholar]
- 28.Walton S.P., Wu M., Gredell J.A., Chan C. Designing highly active siRNAs for therapeutic applications. FEBS J. 2010;277:4806–4813. doi: 10.1111/j.1742-4658.2010.07903.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Fougerolles A., Vornlocher H.-P., Maraganore J., Lieberman J. Interfering with disease: a progress report on siRNA-based therapeutics. Nat. Rev. Drug Discov. 2007;6:443–453. doi: 10.1038/nrd2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Watts J.K., Corey D.R. Silencing disease genes in the laboratory and the clinic. J. Pathol. 2012;226:365–379. doi: 10.1002/path.2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Andersson S., Antonsson M., Elebring M., Jansson-Löfmark R., Weidolf L. Drug metabolism and pharmacokinetic strategies for oligonucleotide- and mRNA-based drug development. Drug Discov. Today. 2018;23:1733–1745. doi: 10.1016/j.drudis.2018.05.030. [DOI] [PubMed] [Google Scholar]
- 32.Foster D.J., Brown C.R., Shaikh S., Trapp C., Schlegel M.K., Qian K., Sehgal A., Rajeev K.G., Jadhav V., Manoharan M. Advanced siRNA Designs Further Improve In Vivo Performance of GalNAc-siRNA Conjugates. Mol. Ther. 2018;26:708–717. doi: 10.1016/j.ymthe.2017.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hassler M.R., Turanov A.A., Alterman J.F., Haraszti R.A., Coles A.H., Osborn M.F., Echeverria D., Nikan M., Salomon W.E., Roux L. Comparison of partially and fully chemically-modified siRNA in conjugate-mediated delivery in vivo. Nucleic Acids Res. 2018;46:2185–2196. doi: 10.1093/nar/gky037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jackson A.L., Burchard J., Leake D., Reynolds A., Schelter J., Guo J., Johnson J.M., Lim L., Karpilow J., Nichols K. Position-specific chemical modification of siRNAs reduces “off-target” transcript silencing. RNA. 2006;12:1197–1205. doi: 10.1261/rna.30706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kenski D.M., Butora G., Willingham A.T., Cooper A.J., Fu W., Qi N., Soriano F., Davies I.W., Flanagan W.M. siRNA-optimized Modifications for Enhanced In Vivo Activity. Mol. Ther. Nucleic Acids. 2012;1:e5. doi: 10.1038/mtna.2011.4. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nair J.K., Attarwala H., Sehgal A., Wang Q., Aluri K., Zhang X., Gao M., Liu J., Indrakanti R., Schofield S. Impact of enhanced metabolic stability on pharmacokinetics and pharmacodynamics of GalNAc-siRNA conjugates. Nucleic Acids Res. 2017;45:10969–10977. doi: 10.1093/nar/gkx818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vaishnaw A.K., Gollob J., Gamba-Vitalo C., Hutabarat R., Sah D., Meyers R., de Fougerolles T., Maraganore J. A status report on RNAi therapeutics. Silence. 2010;1:14. doi: 10.1186/1758-907X-1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shmushkovich T., Monopoli K.R., Homsy D., Leyfer D., Betancur-Boissel M., Khvorova A., Wolfson A.D. Functional features defining the efficacy of cholesterol-conjugated, self-deliverable, chemically modified siRNAs. Nucleic Acids Res. 2018;46:10905–10916. doi: 10.1093/nar/gky745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jensen K.K., Tadin-Strapps M., Wang S.P., Hubert J., Kan Y., Ma Y., McLaren D.G., Previs S.F., Herath K.B., Mahsut A. Dose-dependent effects of siRNA-mediated inhibition of SCAP on PCSK9, LDLR, and plasma lipids in mouse and rhesus monkey. J. Lipid Res. 2016;57:2150–2162. doi: 10.1194/jlr.M071498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tadin-Strapps M., Peterson L.B., Cumiskey A.-M., Rosa R.L., Mendoza V.H., Castro-Perez J., Puig O., Zhang L., Strapps W.R., Yendluri S. siRNA-induced liver ApoB knockdown lowers serum LDL-cholesterol in a mouse model with human-like serum lipids. J. Lipid Res. 2011;52:1084–1097. doi: 10.1194/jlr.M012872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Iwamoto N., Butler D.C.D., Svrzikapa N., Mohapatra S., Zlatev I., Sah D.W.Y., Meena, Standley S.M., Lu G., Apponi L.H. Control of phosphorothioate stereochemistry substantially increases the efficacy of antisense oligonucleotides. Nat. Biotechnol. 2017;35:845–851. doi: 10.1038/nbt.3948. [DOI] [PubMed] [Google Scholar]
- 42.Husser C., Brink A., Zell M., Müller M.B., Koller E., Schadt S. Identification of GalNAc-Conjugated Antisense Oligonucleotide Metabolites Using an Untargeted and Generic Approach Based on High Resolution Mass Spectrometry. Anal. Chem. 2017;89:6821–6826. doi: 10.1021/acs.analchem.7b01244. [DOI] [PubMed] [Google Scholar]
- 43.Yu R.Z., Gunawan R., Post N., Zanardi T., Hall S., Burkey J., Kim T.W., Graham M.J., Prakash T.P., Seth P.P. Disposition and Pharmacokinetics of a GalNAc3-Conjugated Antisense Oligonucleotide Targeting Human Lipoprotein (a) in Monkeys. Nucleic Acid Ther. 2016;26:372–380. doi: 10.1089/nat.2016.0623. [DOI] [PubMed] [Google Scholar]
- 44.Beverly M., Hartsough K., Machemer L. Liquid chromatography/electrospray mass spectrometric analysis of metabolites from an inhibitory RNA duplex. Rapid Commun. Mass Spectrom. 2005;19:1675–1682. doi: 10.1002/rcm.1972. [DOI] [PubMed] [Google Scholar]
- 45.Beverly M., Hartsough K., Machemer L., Pavco P., Lockridge J. Liquid chromatography electrospray ionization mass spectrometry analysis of the ocular metabolites from a short interfering RNA duplex. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2006;835:62–70. doi: 10.1016/j.jchromb.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 46.McGinnis A.C., Cummings B.S., Bartlett M.G. Ion exchange liquid chromatography method for the direct determination of small ribonucleic acids. Anal. Chim. Acta. 2013;799:57–67. doi: 10.1016/j.aca.2013.08.040. [DOI] [PubMed] [Google Scholar]
- 47.Zou Y., Tiller P., Chen I.-W., Beverly M., Hochman J. Metabolite identification of small interfering RNA duplex by high-resolution accurate mass spectrometry. Rapid Commun. Mass Spectrom. 2008;22:1871–1881. doi: 10.1002/rcm.3561. [DOI] [PubMed] [Google Scholar]
- 48.Studzińska S., Rola R., Buszewski B. Development of a method based on ultra high performance liquid chromatography coupled with quadrupole time-of-flight mass spectrometry for studying the in vitro metabolism of phosphorothioate oligonucleotides. Anal. Bioanal. Chem. 2016;408:1585–1595. doi: 10.1007/s00216-015-9266-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li J., Liu J., Enders J., Arciprete M., Tran C., Aluri K., Guan L.H., O’Shea J., Bisbe A., Charissé K. Discovery of a novel deaminated metabolite of a single-stranded oligonucleotide in vivo by mass spectrometry. Bioanalysis. 2019;11:1955–1965. doi: 10.4155/bio-2019-0118. [DOI] [PubMed] [Google Scholar]
- 50.Crooke R.M., Graham M.J., Martin M.J., Lemonidis K.M., Wyrzykiewiecz T., Cummins L.L. Metabolism of antisense oligonucleotides in rat liver homogenates. J. Pharmacol. Exp. Ther. 2000;292:140–149. [PubMed] [Google Scholar]
- 51.Baek M.-S., Yu R.Z., Gaus H., Grundy J.S., Geary R.S. In vitro metabolic stabilities and metabolism of 2′-O-(methoxyethyl) partially modified phosphorothioate antisense oligonucleotides in preincubated rat or human whole liver homogenates. Oligonucleotides. 2010;20:309–316. doi: 10.1089/oli.2010.0252. [DOI] [PubMed] [Google Scholar]
- 52.Kim J., Basiri B., Hassan C., Punt C., van der Hage E., den Besten C., Bartlett M.G. Metabolite Profiling of the Antisense Oligonucleotide Eluforsen Using Liquid Chromatography-Mass Spectrometry. Mol. Ther. Nucleic Acids. 2019;17:714–725. doi: 10.1016/j.omtn.2019.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nakanishi K. Anatomy of RISC: how do small RNAs and chaperones activate Argonaute proteins? Wiley Interdiscip. Rev. RNA. 2016;7:637–660. doi: 10.1002/wrna.1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gu S., Jin L., Zhang F., Huang Y., Grimm D., Rossi J.J., Kay M.A. Thermodynamic stability of small hairpin RNAs highly influences the loading process of different mammalian Argonautes. Proc. Natl. Acad. Sci. USA. 2011;108:9208–9213. doi: 10.1073/pnas.1018023108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dominska M., Dykxhoorn D.M. Breaking down the barriers: siRNA delivery and endosome escape. J. Cell Sci. 2010;123:1183–1189. doi: 10.1242/jcs.066399. [DOI] [PubMed] [Google Scholar]
- 56.Humphreys S.C., Thayer M.B., Campbell J., Chen W.L.K., Adams D., Lade J.M., Rock B.M. Emerging siRNA design principles and consequences for biotransformation and disposition in drug development. J. Med. Chem. 2020;63:6407–6422. doi: 10.1021/acs.jmedchem.9b01839. [DOI] [PubMed] [Google Scholar]
- 57.Bartlett D.W., Davis M.E. Effect of siRNA nuclease stability on the in vitro and in vivo kinetics of siRNA-mediated gene silencing. Biotechnol. Bioeng. 2007;97:909–921. doi: 10.1002/bit.21285. [DOI] [PubMed] [Google Scholar]
- 58.Schilling B., Rardin M.J., MacLean B.X., Zawadzka A.M., Frewen B.E., Cusack M.P., Sorensen D.J., Bereman M.S., Jing E., Wu C.C. Platform-independent and label-free quantitation of proteomic data using MS1 extracted ion chromatograms in skyline. Application to Protein Acetylation and Phosphorylation. Mol. Cell Proteomics. 2012;11:202–214. doi: 10.1074/mcp.M112.017707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nyakas A., Blum L.C., Stucki S.R., Reymond J.-L., Schürch S. OMA and OPA--software-supported mass spectra analysis of native and modified nucleic acids. J. Am. Soc. Mass Spectrom. 2013;24:249–256. doi: 10.1007/s13361-012-0529-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.