

Abstract
Astrocytes play a key role in the progression of amyotrophic lateral sclerosis (ALS) by actively inducing the degeneration of motor neurons. Motor neurons isolated from receptor for advanced glycation end products (RAGE)‐knockout mice are resistant to the neurotoxic signal derived from ALS‐astrocytes. Here, we confirmed that in a co‐culture model, the neuronal death induced by astrocytes over‐expressing the ALS‐linked mutant hSOD1G93A is prevented by the addition of the RAGE inhibitors FPS‐ZM1 or RAP. These inhibitors also prevented the motor neuron death induced by spinal cord extracts from symptomatic hSOD1G93A mice. To evaluate the relevance of this neurotoxic mechanism in ALS pathology, we assessed the therapeutic potential of FPS‐ZM1 in hSOD1G93A mice. FPS‐ZM1 treatment significantly improved hind‐limb grip strength in hSOD1G93A mice during the progression of the disease, reduced the expression of atrophy markers in the gastrocnemius muscle, improved the survival of large motor neurons, and reduced gliosis in the ventral horn of the spinal cord. However, we did not observe a statistically significant effect of the drug in symptoms onset nor in the survival of hSOD1G93A mice. Maintenance of hind‐limb grip strength was also observed in hSOD1G93A mice with RAGE haploinsufficiency [hSOD1G93A;RAGE(+/‐)], further supporting the beneficial effect of RAGE inhibition on muscle function. However, no benefits were observed after complete RAGE ablation. Moreover, genetic RAGE ablation significantly shortened the median survival of hSOD1G93A mice. These results indicate that the advance of new therapies targeting RAGE in ALS demands a better understanding of its physiological role in a cell type/tissue‐specific context.
Keywords: advanced glycation end products receptor, amyotrophic lateral sclerosis, astrocytes, Atrogin‐1, motor neurons, MuRF1
Abbreviations
- ALS
amyotrophic lateral sclerosis
- BDNF
brain‐derived neurotrophic factor
- CNS
central nervous system
- Fbxo32
F‐box protein 32 or Atrogin‐1
- GDNF
glial‐derived neurotrophic factor
- HMGB1
high mobility group box 1 protein
- NGF
nerve growth factor
- RAGE
receptor for advanced glycation end products
- SOD1
superoxide dismutase 1
- TLR
toll‐like receptor
- Trim63
tripartite motif containing 63 or MuRF1
Significance statement.
This study demonstrates that RAGE pharmacological inhibition or RAGE haploinsufficiency improves muscle function during the progression of the ALS‐like pathology in hSOD1G93A mice. However, genetic RAGE ablation shortens the median survival of hSOD1G93A mice. These data highlight the complex role of RAGE signaling during neurodegeneration and identify protective effects of RAGE signaling in this ALS mouse model.
1. INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is characterized by the progressive degeneration of both upper and lower motor neurons, leading to paralysis and muscle atrophy. Most ALS cases are sporadic while ~10% are familial cases. 1 , 2 Superoxide dismutase 1 (SOD1) was the first ALS gene identified and over 170 genetic variants have been linked to ALS. 3 Mice over‐expressing ALS‐linked mutant human SOD1 variants, including hSOD1G93A, develop a progressive motor neuron disease that recapitulates key pathological hallmarks and clinical symptoms observed in ALS patients, 4 , 5 and are considered the standard mouse model for ALS pre‐clinical studies. 6 , 7
Motor neuron degeneration in ALS occurs through a non‐cell‐autonomous process. 8 Astrocytes, key regulators of central nervous system (CNS) homeostasis, play a major role in disease progression. 8 , 9 Accordingly, astrocytes isolated from rodents over‐expressing different mutant hSOD1 variants induce the death of co‐cultured motor neurons. 10 , 11 The relevance of this observation for the human pathology was confirmed using astrocytes differentiated from spinal cord autopsy‐derived neuronal progenitor cells, or fibroblast‐derived induced pluripotent stem cells (iPSCs), from sporadic and familial ALS patients. 12 , 13 We showed that astrocyte‐mediated motor neuron death involves receptor for advanced glycation end products (RAGE) signaling activation. The neuronal death induced by hSOD1G93A astrocytes was prevented by RAGE blocking antibodies, while motor neurons isolated from RAGE‐knockout mice were not sensitive to the neurotoxic signal derived from hSOD1G93A astrocytes. 14 Moreover, spinal cord extracts from symptomatic hSOD1G93A mice, but not from non‐transgenic littermates, induced the death of cultured hSOD1G93A motor neurons by a mechanism involving RAGE signaling. 14 Relevance of this neurotoxic mechanism in ALS is supported by the presence of increased levels of RAGE and many of its ligands, including oxidatively modified nerve growth factor (NGF), high mobility group box 1 protein (HMGB1), S100B and glycated proteins, in the spinal cord of ALS patients and hSOD1G93A mice. 14 , 15 , 16 , 17 , 18 , 19 Furthermore, ALS patients display decreased levels of soluble RAGE (sRAGE) in serum. 20 sRAGE variants comprise C‐terminally truncated isoforms that preserve the ligand‐binding domain and exert negative effects on RAGE activation, acting as decoy receptors by competing for ligand binding. 21 , 22 , 23 Collectively, these results suggest the involvement of RAGE signaling in ALS pathology and support the use of therapeutic approaches focused on RAGE inhibition.
Therapeutic strategies targeting astrocyte‐mediated neurotoxicity have proved to be effective in ALS mouse models, increasing motor neuron survival and improving motor performance. 24 , 25 , 26 , 27 Different approaches have been developed to target RAGE in vivo, including treatment with sRAGE, small molecule antagonists and neutralizing anti‐RAGE antibodies. 28 Treatment with sRAGE (intraperitoneal injections) modestly extended the survival of hSOD1G93A male mice. 29 However, sRAGE does not access the CNS after intraperitoneal administration, 30 and gaining access to the CNS is essential to prevent RAGE‐dependent astrocyte‐mediated neurotoxicity. Hence, the use of a pharmacological inhibitor with the ability to cross the blood‐brain barrier could result in improved therapeutic effects.
FPS‐ZM1 is a high‐affinity RAGE‐specific inhibitor that has been shown to cross the blood‐brain barrier to access the CNS. 31 FPS‐ZM1 has no toxicity in mice, even at high doses. Moreover, after intraperitoneal administration, FPS‐ZM1 gained access to the CNS and significantly reduced Aβ pathology, decreased neuroinflammation and improved cognitive impairment in an Alzheimer's disease mouse model. 31 Importantly, FPS‐ZM1 inhibits the binding of RAGE to different ligands and its effects are not restricted to Aβ‐RAGE interaction. 31
Here we sought to determine the effect of FPS‐ZM1 in the ALS‐like pathology developed by hSOD1G93A mice. Moreover, to further evaluate the effect of RAGE inhibition, we generated hSOD1G93A mice with genetic RAGE haploinsufficiency or complete RAGE ablation. Our results show that RAGE signaling could exert both neurotoxic and neuroprotective functions in hSOD1G93A mice. Hence, the advance of new therapies targeting RAGE signaling in ALS would require a better understanding of its physiological role in a cell type/tissue‐specific context.
2. MATERIALS AND METHODS
2.1. Reagents
All chemicals and reagents were acquired from Sigma‐Aldrich unless otherwise specified. Cell culture media, serum and supplements were obtained from Life Technologies unless otherwise indicated. The RAGE antagonist peptide RAP was obtained from Calbiochem‐EMD Millipore. FPS‐ZM1 was obtained from Selleck Chemicals LLC or Calbiochem‐EMD Millipore. Primers were obtained from Integrated DNA Technologies.
2.2. Animals
Transgenic mice over‐expressing hSOD1G93A were obtained from The Jackson Laboratory, strain B6.Cg‐Tg(SOD1*G93A)1Gur/J (stock # 004435) and were maintained as hemizygous animals in a C57BL/6J background. Littermates were randomly assigned to a vehicle (10% 2‐Hydroxypropyl‐β‐cyclodextrin in saline) or FPS‐ZM1 treatment group. Starting at 60 days of age mice received daily intraperitoneal (i.p.) injections of vehicle or FPS‐ZM1 at a dose of 1 mg/kg in a total volume of 0.1 mL. Treatment continued until endpoint was reached (see below).
RAGE‐knockout mice were previously described 32 and were also maintained in a C57BL/6J background. To generate the animals for this study, hemizygous hSOD1G93A males were mated with RAGE(−/−) or RAGE(+/‐) females to obtain breeders with the following genotype hSOD1G93A(+/‐);RAGE(+/‐) and hSOD1G93A(−/−);RAGE(+/‐). Then, hSOD1G93A(+/‐);RAGE(+/‐) males were mated with hSOD1G93A(−/−);RAGE(+/‐) females to obtain the genotypes analyzed in the study.
For lifespan studies, endpoint was determined by the lack of “righting reflex,” that is, the inability of the animal to right itself within 20 seconds after being placed on either side. Mice that were unable to right themselves within 20 seconds were euthanized and recorded as dead. Mice were weighed daily and disease onset was retrospectively determined as the time when mice reached peak body weight. Hind‐limb grip strength was measured using a grip strength meter (San Diego Instruments). Tests were performed by allowing the animal to grasp the grid with both hind limbs and then pulling the animal straight away from the grid until it released the platform. Grip strength was measured once a week, and in each session the average peak force of five attempts was recorded. Relative quantitative PCR was used to estimate hSOD1 gene copy number. One of the male breeders that fathered the litters used in this study was used as a reference for copy number estimation. Animals with a relative gene copy number lower than the reference breeder were not used in the study.
All animal procedures were carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the NIH. The Animal Care and Use Committee of MUSC (Animal Welfare Assurance number A3428‐01) and UW‐Madison (Animal Welfare Assurance number A3368‐01) approved the animal protocols pertinent to the experiments reported in this publication.
2.3. Histology and immunostaining
Mice were transcardially perfused with 0.1 M PBS (pH 7.4), followed by 4% paraformaldehyde in PBS. Spinal cords were removed and paraffin‐embedded using standard techniques. Antigen retrieval and immunostaining were performed as previously described. 33 Mice lumbar spinal cord sections were stained with anti‐GFAP (Novus, NBP2‐29415) and anti‐IBA1 (Fujifilm Wako, 013‐27691) antibodies. Secondary antibodies were Alexa Fluor 488‐conjugated goat anti‐mouse and Alexa Fluor 594‐conjugated goat anti‐rabbit (Invitrogen). Nuclei were counterstained with DAPI (4',6‐Diamidino‐2‐phenylindole dihydrochloride; Invitrogen). Each slide had sections from both treatment groups and were stained and imaged concurrently. Images were captured in a Zeiss LSM 880 NLO microscope (Carl Zeiss) with identical settings for both experimental groups. Image quantification was performed as previously described 27 using Imaris image analysis software 9.1.2 (Oxford Instruments).
To determine the number of large motor neurons in the ventral horn of the spinal cord, 10 μm serial sections across the lumbar spinal cord were stained with cresyl violet. Two independent observers blinded to the treatment group counted every fifth section and a total of 15 sections per animal were analyzed.
2.4. Cell culture
Primary astrocyte cultures were prepared from the spinal cord of 1‐day‐old mice as we previously described. 34 Astrocytes were maintained in Dulbecco's modified Eagle's medium supplemented with 10% FBS, HEPES (3.6 g/L), penicillin (100 IU/mL), and streptomycin (100 μg/mL). Cultures were >99% pure as determined by glial fibrillary acidic protein (GFAP) immunoreactivity and contained <1% of IBA1‐positive microglial cells.
Motor neuron cultures were prepared from mouse embryonic spinal cords (E12.5) as previously described. 35 To prepare hSOD1G93A motor neurons, genotyping was performed by real‐time PCR during dissection, and all embryos of the same genotype were pooled together for the rest of the preparation. Motor neurons were plated at a density of 500 cells/cm2 on four‐well multidishes (Nunclon) pre‐coated with polyornithine‐laminin. Cultures were maintained in Neurobasal medium supplemented with 2% horse serum, 25 μmol/L L‐glutamate, 25 μmol/L 2‐mercaptoethanol, 0.5 mmol/L L‐glutamine, 2% B‐27 supplement, and GDNF (1 ng/mL; Sigma‐Aldrich). Motor neuron death induced by trophic factor deprivation (NONE, without GDNF) was determined in all experiments as a control and was never >50%. Motor neuron survival was assessed by direct counting of all neurons displaying intact neurites longer than four cell bodies in diameter, as previously described. 36
For co‐culture experiments, motor neurons were plated on top of mouse astrocyte monolayers at a density of 300 cells/cm2. Co‐cultures were maintained in Leibovitz's L15 medium supplemented with 0.63 mg/mL bicarbonate, 5 μg/mL insulin, 0.1 mg/mL conalbumin, 0.1 mmol/L putrescine, 30 nmol/L sodium selenite, 20 nmol/L progesterone, 20 mmol/L glucose, 100 IU/mL penicillin, 100 μg/mmol/L streptomycin, and 2% horse serum, as previously described. 37 Motor neurons were identified by immunostaining with anti‐Tubulin βIII antibody (Clone 2G10; Sigma‐Aldrich) and survival was determined by direct counting over an area of 0.90 cm2 in 24‐well plates, as previously described. 37
2.5. Spinal cord extracts
Lumbar spinal cord extracts were prepared as previously described 14 from early symptomatic hSOD1G93A mice (120‐130 days old) or aged‐matched non‐transgenic littermates. Protein concentration was determined by the bicinchoninic acid method (BCA protein assay; Thermo Scientific‐Pierce). Extracts were collected and kept at −80°C until used. Aliquots were added to motor neuron cultures to reach a final protein concentration of 0.5 μg/mL.
2.6. Real‐time PCR analysis
RNA extraction, RNA retrotranscription, and real‐time PCR were performed as previously described. 38 Specific primers were as follows: Trim63 (5’‐GGACTACTTTACTCTGGACTTAGAAC‐3’ and 5’‐CAGCCTCCTCTTCTGTAAACTC‐3’), Fbxo32 (5’‐GCGCCATGGATACTGTACTT‐3’ and 5’‐ATCAGCTCCAACAGCCTTAC‐3’), and Rplp0 (5’‐CCTCCTTCTTCCAGGCTTTG‐3’ and 5’‐CCACCTTGTCTCCAGTCTTTATC‐3’).
2.7. Western blot analysis
Western blots were performed as previously described. 38 Membranes were incubated overnight with one of the following antibodies: rabbit monoclonal against FBX32 (1:1000; clone JE41‐27; Novus Biologicals Cat# NBP2‐76836), goat polyclonal against TRIM63 (1 μg/mL; Novus Biologicals Cat# AF5366), or rabbit monoclonal against pan‐ACTIN (1:1000; clone D18C11; Cell Signaling Cat# 8456). Membranes were developed using the ECL Prime chemiluminescent detection system (GE Healthcare‐Amersham Biosciences) and image acquisition was performed in a C‐DiGit chemiluminescence Western blot scanner (Li‐Cor Biosciences). Quantifications were performed using the Image Studio Software (Li‐Cor Biosciences).
2.8. Statistical analysis
Survival, onset, and weight‐loss data were analyzed with Kaplan‐Meier curves and log rank test. Groups of four animals were used for biochemical analysis and all data are reported as mean ± SD. Comparisons between two groups were performed with an unpaired t test. Hind‐limb grip strength data during the progression of the disease were analyzed by multiple t tests assuming populations with the same standard deviation. Cell culture experiments were repeated in at least three independent primary culture preparations, and values from each independent experiment were combined for data reporting. Multiple group comparisons were performed with one‐way ANOVA with Tukey's post‐test. Differences were declared statistically significant if P ≤ .05. All statistical computations were performed using GraphPad Prism 6.0 (GraphPad software).
3. RESULTS
We previously showed that in an astrocyte‐motor neuron co‐culture model, activation of RAGE signaling mediates motor neuron death induced by hSOD1G93A over‐expressing astrocytes. 14 Here we demonstrate that two RAGE pharmacological inhibitors effectively prevent the neuronal death induced by hSOD1G93A astrocytes (Figure 1A). Both, the small molecule RAGE antagonist FPS‐ZM1 31 and RAP, a RAGE antagonist peptide that inhibits the interaction of the receptor with multiple ligands, 39 prevented motor neuron death when added to the co‐cultures. In addition, we previously showed that lumbar spinal cord extracts from early symptomatic ALS mice induce the death of cultured hSOD1G93A‐expressing motor neurons by a mechanism involving RAGE signaling. 14 The RAGE antagonists FPS‐ZM1 and RAP also prevented the toxicity of ALS spinal cord extracts (Figure 1B), confirming the involvement of RAGE signaling in the neurotoxicity observed. The in vitro neuroprotective effect of FPS‐ZM1 and its ability to cross the blood‐brain barrier 31 prompted us to evaluate its therapeutic potential in vivo in hSOD1G93A mice.
FIGURE 1.
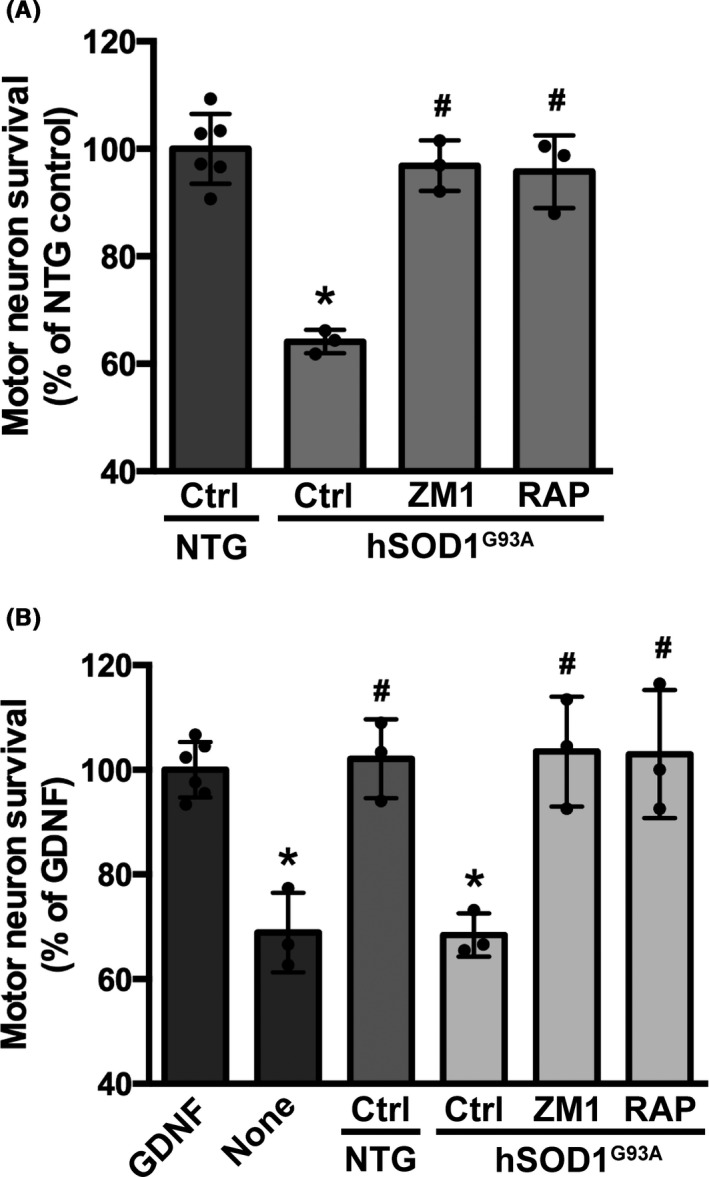
RAGE pharmacological inhibition prevented motor neuron death induced by hSOD1G93A astrocytes and hSOD1G93A lumbar spinal cord extracts. A, Non‐transgenic embryonic motor neurons were plated on top of astrocytes isolated from non‐transgenic (NTG) or hSOD1G93A mice. Vehicle (Control, Ctrl) or RAGE inhibitors, FPS‐ZM1 (100 nmol/L; ZM1) and RAP (20 μmol/L), were added 2 h after motor neuron plating. Neuronal survival was determined 72 h later. Data are expressed as percentage of motor neuron survival on top of NTG control astrocytes (mean ± SD). *Significantly different from NTG control (P ≤ .05); #Significantly different from hSOD1G93A control (P ≤ .05). B, Primary motor neuron cultures isolated from the spinal cord of E12.5 hSOD1G93A embryos were maintained with GDNF (1 ng/mL). Two hours after plating, cultures were treated with vehicle (control GDNF) or spinal cord extracts (0.5 μg protein/mL) from early symptomatic hSOD1G93A mice or aged‐matched non‐transgenic littermates (NTG). Vehicle (Ctrl) or RAGE pharmacological inhibitors, FPS‐ZM1 (100 nmol/L; ZM1) and RAP (10 μmol/L), were added 30 min before treatment with spinal cord extracts and motor neuron survival was determined 48 h later. Motor neuron death induced by trophic factor deprivation (NONE) was used as a control. Data are expressed as percentage of control GDNF (mean ± SD). *Significantly different from control GDNF (P ≤ .05); #Significantly different from hSOD1G93A control (P ≤ .05). For all panels, experiments were performed in three independent cultures performed in duplicate and the average of each experiment is shown. Each experimental replica is shown for control groups
We treated hSOD1G93A mice of both sexes with FPS‐ZM1 at a dose of 1 mg/kg/d, which was previously described to be effective in an Alzheimer's disease mouse model. 31 According to published guidelines for the preclinical in vivo evaluation of active drugs in hSOD1G93A mice, 40 we started treatment at the age of 60 days. At this age, hSOD1G93A mice already display histological evidence of neurodegeneration, although no overt decline in general locomotor function is evident. 4 , 5 Treatment continued until endpoint was reached. We originally performed a preliminary study in a small cohort of female mice (n = 7 in vehicle control and n = 13 in FPS‐ZM1 group) to rule out potential adverse effects of sequential daily injections in this model. Since no overt toxicity of the treatment was observed, we decided to perform an additional trial including a larger number of animals of both sexes. When combining the data from both trials, compared to vehicle treated mice, FPS‐ZM1 treatment did not significantly affect the onset of the disease in hSOD1G93A mice, as defined by the age at peak body weight (Figure 2). However, FPS‐ZM1‐treated mice showed a significant improvement in hind‐limb grip strength, compared to vehicle‐treated mice. This effect was observed in both sexes for several weeks during the progression of the disease (Figure 3A‐C). The beneficial effect of FPS‐ZM1 treatment in hSOD1G93A mice was accompanied by a significant decrease in the mRNA and protein levels of two important E3 ubiquitin ligases linked to muscle atrophy, Fbxo32 (FBX32, Atrogin‐1) and Trim63 (MuRF1), in the gastrocnemius muscle of 17‐week‐old FPS‐ZM1‐treated mice (Figure 3D‐H). The increase in hind‐limb grip strength observed in the FPS‐ZM1‐treated group was also associated with a ~24% increase in the number of large motor neurons in the ventral horn of the spinal cord (Figure 4A). In addition, when compared to vehicle‐treated mice, early symptomatic FPS‐ZM1‐treated mice displayed a significant decrease in astrogliosis and microgliosis in the ventral horn of the spinal cord, as evidenced by decreased GFAP and IBA1 immunostaining, respectively (Figure 4B‐D).
FIGURE 2.
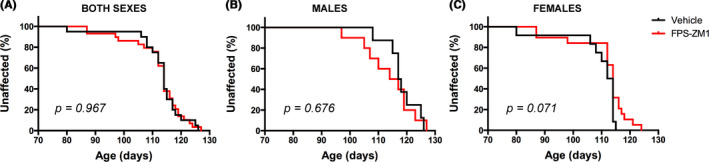
FPS‐ZM1 had no significant effect on the disease onset in hSOD1G93A mice. hSOD1G93A mice were treated with vehicle (black) or FPS‐ZM1 (1 mg/kg/d; red). Disease onset was retrospectively determined as the time when mice reached peak body weight. A, When both sexes were analyzed together, median onset was 114 d in both treatment groups (vehicle group n = 20 [12 females and 8 males], and FPS‐ZM1 group n = 29 [19 females and 10 males]). B, In males, median onset was 117.5 d in vehicle group (n = 8) and 115.5 d in FPS‐ZM1 group (n = 10). C, In females, median onset was 113 d in vehicle group (n = 12) and 114 d in FPS‐ZM1 group (n = 19). Onset curves were not significantly different
FIGURE 3.
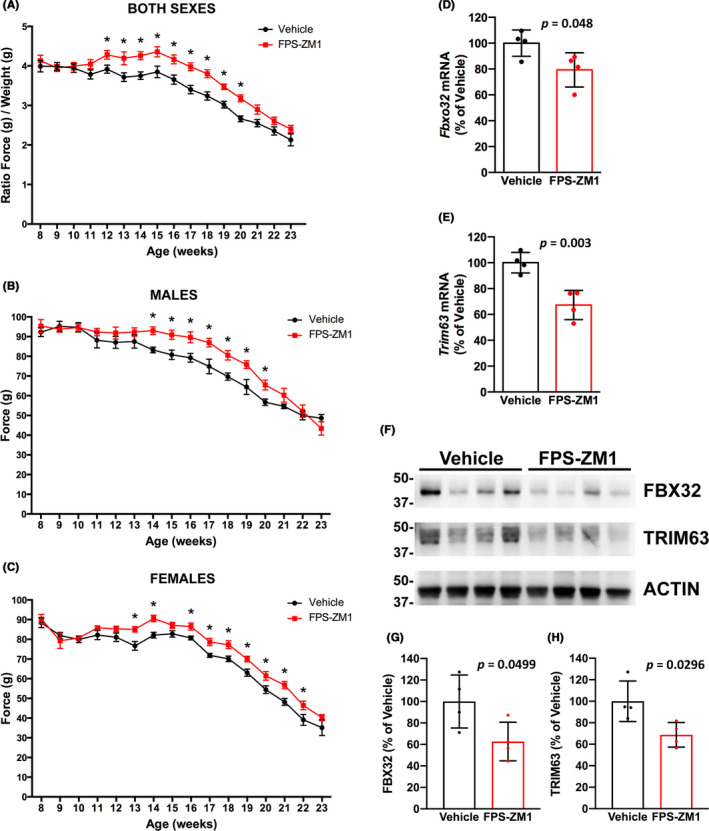
FPS‐ZM1 treatment preserved hind‐limb grip strength and decreased the expression of skeletal muscle atrophy markers in hSOD1G93A mice. A, Analysis of hind‐limb grip strength combining both sexes together (vehicle group n = 20 [12 females and 8 males], and FPS‐ZM1 group n = 29 [19 females and 10 males]). In order to combine both sexes together, data are presented as the ratio of hind‐limb grip strength to body weight (mean ± SEM). B, Analysis of hind‐limb grip strength in male mice (n = 8 in vehicle, n = 10 in FPS‐ZM1 group). C, Analysis of hind‐limb grip strength in female mice (n = 12 in vehicle, n = 19 in FPS‐ZM1 group). In (B and C), data are presented as mean ± SEM. *Significantly different from vehicle‐treated mice (P ≤ .05). For (A‐C), statistical significance was determined by multiple t tests assuming populations with the same standard deviation. D and E, FPS‐ZM1 treatment significantly decreased the expression of skeletal muscle atrophy markers (P ≤ .05). Total RNA was extracted from the gastrocnemius muscle of 17‐week‐old vehicle‐ or FPS‐ZM1‐treated mice and Fbxo32 (D) and Trim63 (E) mRNA levels were determined by real‐time PCR and corrected by Rplp0 mRNA levels (n = 4 mice per group). Data are expressed as percentage of vehicle‐treated mice (mean ± SD). F, Western blot analysis of FBX32 and TRIM63 protein levels in the gastrocnemius muscle of 17‐week‐old vehicle‐ or FPS‐ZM1‐treated mice. G and H, Quantification of FBX32 (G) and TRIM63 (H) protein expression after correction by ACTIN levels (n = 4 mice per group). Data are expressed as percentage of vehicle‐treated mice (mean ± SD)
FIGURE 4.
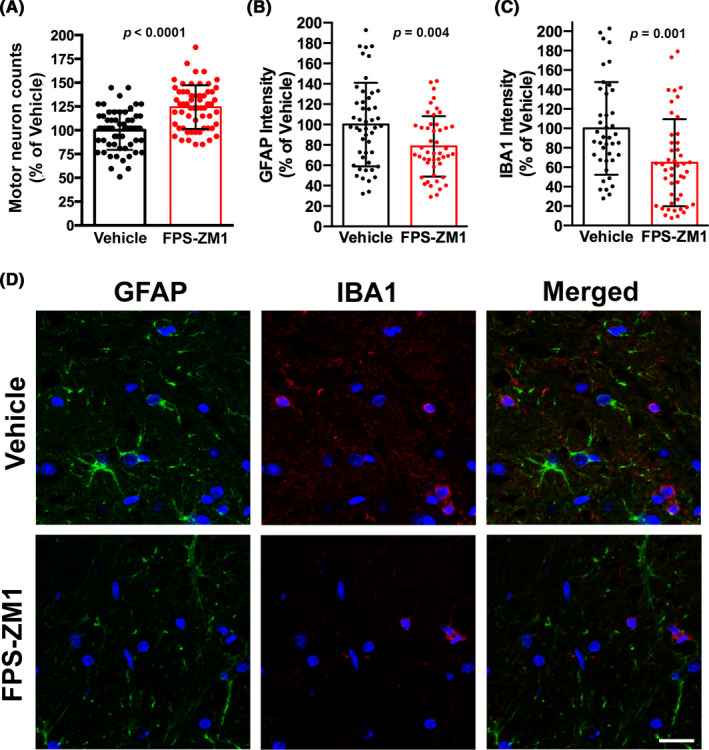
FPS‐ZM1 treatment delayed motor neuron loss and decreased glial activation in the spinal cord of hSOD1G93A mice. A, Number of large motor neurons in the ventral horn of the lumbar spinal cord of 17‐week‐old vehicle‐ or FPS‐ZM1‐treated mice. Data are expressed as percentage of vehicle‐treated mice (mean ± SD). Each data point corresponds to the value obtained from individual sections (15 sections per animal, n = 4 mice per treatment group). When compared with vehicle‐treated mice, the number of large motor neurons in FPS‐ZM1‐treated mice was increased by 24.3% (P ≤ .0001). B and C, Quantification of relative GFAP (B) and IBA1 (C) fluorescence intensity in images from the ventral horn of the lumbar spinal cord of vehicle‐ or FPS‐ZM1‐treated hSOD1G93A mice. Data are expressed as percentage of vehicle‐treated mice (mean ± SD). Each data point corresponds to the value obtained in an individual image (10‐12 images per animal, n = 4 mice per treatment group). D, Representative microphotographs showing GFAP (green) and IBA1 (red) immunofluorescence in the ventral horn of the lumbar spinal cord of 17‐week‐old vehicle‐ or FPS‐ZM1‐treated mice. Nuclei were counterstained with DAPI. Scale bar, 20 μm
Disease progression in the hSOD1G93A mouse model is accompanied by progressive weight loss. Although we did not observe a statistical difference in the onset of the disease determined by peak body weight, we did observe differences in the rate of the progressive weight loss experienced by vehicle‐ and FPS‐ZM1‐treated mice, which was statistically significant in female mice (Figure 5). As summarized in Figure 5D, female mice in FPS‐ZM1 group showed a statistically significant delay of 7.5 days in the age at which they displayed 5% and 10% body weight loss (as percentage of peak body weight). However, FPS‐ZM1 treatment appeared to accelerate disease progression at later stages, since the period of time elapsed after the animals lose 10% of the body weight until the endpoint is reached was significantly decreased in FPS‐ZM1‐treated female mice (median time of 24 days in vehicle‐treated group and 19 days in FPS‐ZM1‐treated group; Figure 5F). The observed accelerated weight loss rate at later stages of the disease likely contributes to the lack of significant effect of FPS‐ZM1 treatment in the survival of female hSOD1G93A mice (Figure 6B). On the other hand, male mice in the FPS‐ZM1 group showed an extension of 2 weeks in the time elapsed from 10% body weight loss until the endpoint was reached (median time of 17.5 days in vehicle‐treated group vs 31.5 days in FPS‐ZM1‐treated group; Figure 5E). This delay in the progression of the disease at later stages was reflected in an overall 14‐day extension in the survival of FPS‐ZM1‐treated male mice, although no statistical significance was reached (Figure 6A).
FIGURE 5.
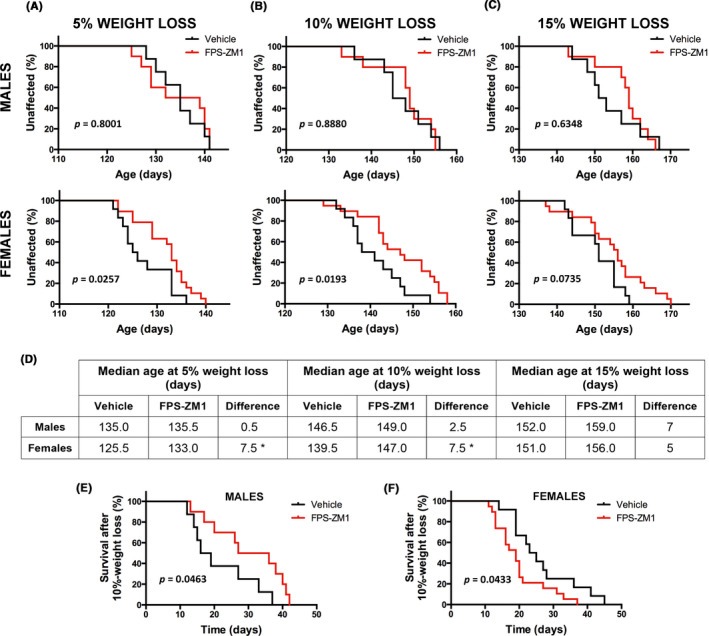
Sex‐specific effect of FPS‐ZM1‐treatment in the progressive weight loss observed in hSOD1G93A mice. Body weight was recorded daily and the age at which mice exhibited 5%, 10%, or 15% peak weight loss was determined (age at 95%, 90%, or 85% peak body weight, respectively). A‐C, Kaplan‐Meier curves showing the probability of losing 5% (A), 10% (B), or 15% (C) of the peak body weight. Both sexes were analyzed independently (top panel: males; lower panel: females; vehicle group n = 12 females and n = 8 males, and FPS‐ZM1 group n = 19 females and n = 10 males). A significant 7.5‐d delay in the median age at which female mice display 5% and 10% peak weight loss was observed in FPS‐ZM1 group. No significant effect of the treatment was observed in male mice. D, Table summarizing the median age at which mice display different percentages of weight loss. *Significant difference between vehicle and FPS‐ZM1 groups (P ≤ .05). E and F, Kaplan‐Meier curves representing the number of days elapsed from age at 10% peak weight loss until endpoint is reached. The median time elapsed until endpoint significantly increases in males (17.5 d in vehicle vs 31.5 d in FPS‐ZM1 group; χ 2 = 3.97) and decreases in females (24 d in vehicle vs 19 d in FPS‐ZM1 group; χ 2 = 4.09)
FIGURE 6.
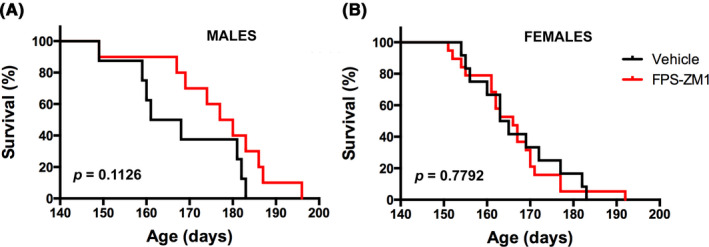
FPS‐ZM1 treatment has no statistically significant effect on the survival of hSOD1G93A mice. A, Median survival in male mice treated with vehicle (164.5 d, n = 8) or FPS‐ZM1 (178.5 d, n = 10). Curves are not significantly different. B, Median survival in female mice treated with vehicle (164 d, n = 12) or FPS‐ZM1 (166 d, n = 19). Curves are not significantly different
To further evaluate the effect of RAGE signaling inhibition in this ALS mouse model, we crossed hSOD1G93A mice with RAGE‐knockout mice in order to generate hSOD1G93A mice with RAGE haploinsufficiency or complete RAGE ablation. Further stressing the beneficial effect of RAGE inhibition on muscle function in hSOD1G93A mice, we observed the preservation of hind‐limb grip strength in hSOD1G93A mice after genetic RAGE haploinsufficiency (Figure 7A, hSOD1G93A;RAGE(+/‐)). However, this beneficial effect on grip strength was not observed in hSOD1G93A mice with complete RAGE ablation (Figure 7A, hSOD1G93A;RAGE(−/−)). It is worth noting that RAGE haploinsufficiency or complete ablation did not affect the grip strength of mice not expressing the mutant hSOD1 (not shown). We did not observe a significant effect of RAGE deletion in the onset of the disease, as determined by the age at peak body weight (Figure 7B). Unexpectedly, a significant decrease of 14 and 18 days in the median survival was observed in hSOD1G93A;RAGE(+/‐) and hSOD1G93A;RAGE(−/−) mice, respectively (Figure 7C). The detrimental effect of RAGE deletion in the survival of hSOD1G93A mice, together with the lack of beneficial effect in grip strength after complete RAGE ablation in hSOD1G93A;RAGE(−/−), suggests that RAGE signaling might not only exert a neurotoxic effect but also a stage‐specific neuroprotective effect during the development of the disease in this ALS mouse model.
FIGURE 7.
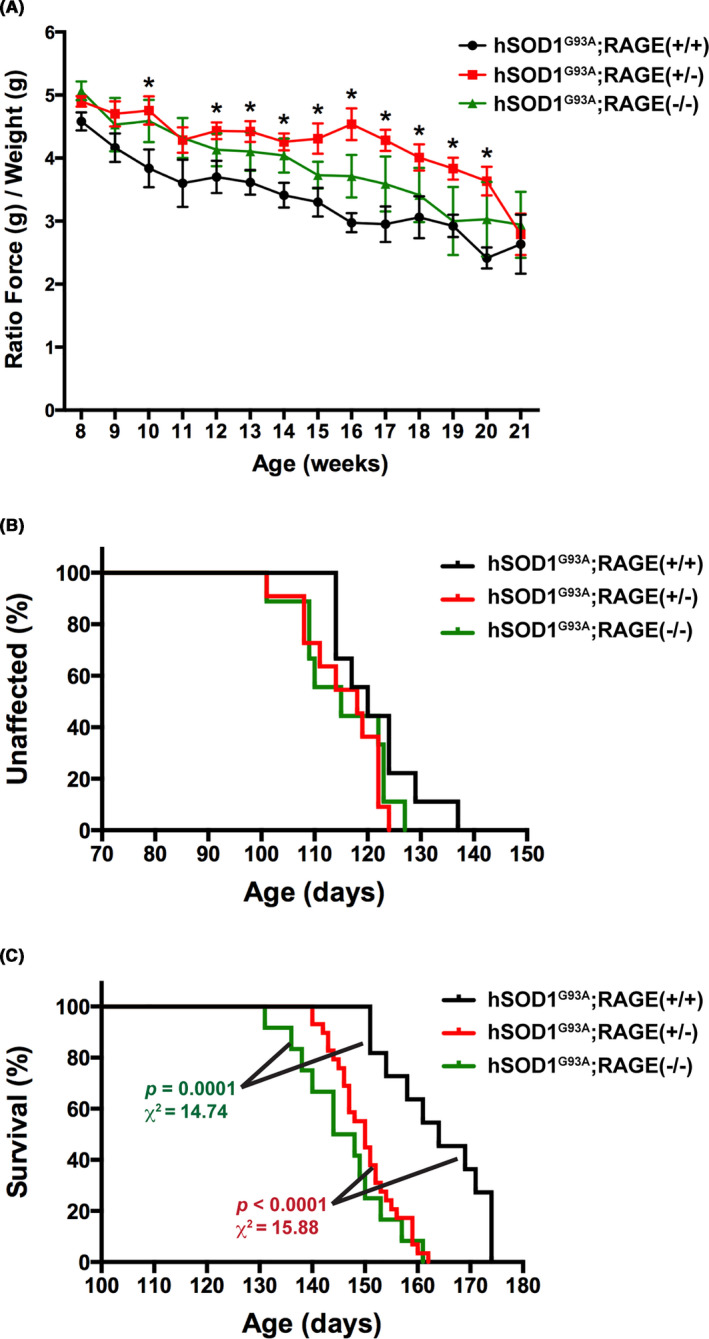
RAGE haploinsufficiency preserves hind‐limb grip strength but decreases the lifespan of hSOD1G93A mice. A, Analysis of hind‐limb grip strength combining both sexes together (hSOD1G93A;RAGE(+/+) group, black, n = 9 [6 females and 3 males], hSOD1G93A;RAGE(+/‐) group, red, n = 11 [7 females and 4 males], and hSOD1G93A;RAGE(−/−) group, green, n = 9 [4 females and 5 males]). Data are presented as the ratio of hind‐limb grip strength to body weight (mean ± SEM). *Significantly different from hSOD1G93A;RAGE(+/+) mice (P ≤ .05). Statistical significance was determined by multiple t tests assuming populations with the same standard deviation. B, No significant effect in the onset of the disease was observed after partial or complete genetic RAGE ablation. The onset of the disease was retrospectively determined as the time when mice reached peak body weight. The median onset was 120 d in hSOD1G93A;RAGE(+/+) mice (black), 118 d in hSOD1G93A;RAGE(+/‐) mice (red), and 115 d in hSOD1G93A;RAGE(−/−) mice (green). The number and sex of the animals are the same as in (A). C, A significant decrease in the median survival of hSOD1G93A mice was observed after genetic RAGE ablation. The median survival was 164 d in hSOD1G93A;RAGE(+/+) mice (black, n = 11 [7 females and 4 males]), 150 d in hSOD1G93A;RAGE(+/‐) mice (red, n = 29 [13 females and 16 males]), and 146 d in hSOD1G93A;RAGE(−/−) mice (green, n = 12 [6 females and 6 males]). No statistical difference was observed when comparing hSOD1G93A;RAGE(+/‐) versus hSOD1G93A;RAGE(−/−) mice
4. DISCUSSION
Inhibition of RAGE signaling has been shown to be neuroprotective in models of Alzheimer's disease, 41 , 42 , 43 Parkinson's disease, 44 and ALS. 29 Indeed, Phase 1 and 2 clinical trials of a RAGE inhibitor (PF‐04494700, TTP488, Azeliragon) for the treatment of Alzheimer's disease obtained promising beneficial effects, although they appeared to be restricted to patients with impaired glucose tolerance. 45 , 46 , 47 , 48 , 49 These clinical trials confirmed it is safe to target RAGE for therapeutic purposes. Our previous work identified RAGE as a potential mediator of astrocyte‐mediated neurotoxicity in ALS. 14 Here, we use RAGE pharmacological inhibitors to provide further evidence of the role of RAGE activation in the motor neuron death induced by ALS‐astrocytes and spinal cord extracts from symptomatic ALS mice. A previous study in hSOD1G93A mice showed that treatment with soluble RAGE, which does not gain access to the CNS, modestly extends the survival of hSOD1G93A male mice. 29 Since our data in cell culture models indicated the involvement of RAGE signaling in astrocyte‐mediated motor neuron death, we designed two experimental approaches to inhibit RAGE signaling at central level in hSOD1G93A mice. We used a small molecule inhibitor able to access the CNS, FPS‐ZM1, 31 and a genetic approach that generated hSOD1G93A mice in a background with complete or partial RAGE deletion. Our results showed that both approaches result in a significant improvement in hind‐limb grip strength. This agrees with previous reports showing that increased RAGE signaling is linked to the muscle atrophy occurring during aging and in pathological conditions such as diabetes, cancer, and myopathies. 50 , 51 , 52 , 53 Accordingly, the maintenance of hind‐limb grip strength in hSOD1G93A mice after FPS‐ZM1 treatment was associated with reduced expression of the atrophy markers TRIM63 and FBX32 in the gastrocnemius muscle. The ability of FPS‐ZM1 to decrease the expression of FBX32 in the skeletal muscle of hSOD1G93A mice is particularly relevant because increased expression of this muscle‐specific ubiquitin E3‐ligase was observed in atrophied muscles of ALS patients. 54 The maintenance of hind‐limb grip strength was also associated with improved survival of large motor neurons and reduced gliosis in the ventral horn of the spinal cord. The latter observation could be due to either a direct effect of FPS‐ZM1 in astrocyte and microglia biology or a secondary outcome due to delayed motor neuron degeneration. Nevertheless, these results confirm the beneficial functional outcome of inhibiting RAGE signaling in male and female hSOD1G93A mice.
However, we also identified a significant protective effect of RAGE expression in hSOD1G93A mice. FPS‐ZM1 treatment slowed down early disease progression in female mice, as reflected by a delay in the age at which they displayed 5% and 10% peak body weight loss. However, the treatment appeared to accelerate disease progression at late stages, leading to a lack of significant effect in overall survival. On the other hand, male mice showed a statistically significant extension in the duration of the disease in late stages, although the 2‐week extension in the median survival observed in male mice did not reach statistical significance. These results revealed an important role of sex in the final outcome of RAGE inhibition, and suggest that limiting the treatment window to an early disease stage could have a better clinical outcome in female hSOD1G93A mice. The protection conferred by RAGE expression in hSOD1G93A mice is also evidenced by the shorter lifespan of hSOD1G93A mice after partial or complete RAGE ablation. Although RAGE haploinsufficiency in hSOD1G93A;RAGE(+/‐) shortened the lifespan, mice preserved hind‐limb grip strength throughout most of the disease progression. However, animals with complete and ubiquitous RAGE deletion (hSOD1G93A;RAGE(−/−)) showed no significant improvement in grip strength, only a detrimental effect in the survival was observed. These results could be explained by the fact that RAGE signaling may not only promote neurodegeneration but may also promote homeostatic and reparative processes. 55 , 56
Accordingly, it has been shown that RAGE signaling can increase the trophic support provided by astrocytes. 57 It is worth noting that the neurotoxicity of ALS‐astrocytes in vivo could be explained not only by the secretion of toxic mediators but also by deficient supportive functions, 58 , 59 including secretion of trophic factors, 60 regulation of glutamatergic signaling, 61 , 62 and metabolic support. 63 Activation of RAGE and TLR4 signaling in astrocytes by HMGB1 released by damaged cells increases the expression of brain‐derived neurotrophic factor (BDNF) and GDNF, 57 two trophic factors with the ability to promote motor neuron survival. Although inhibition of RAGE signaling in motor neurons prevents the neuronal death induced by ALS‐astrocytes, inhibition of RAGE signaling in astrocytes could also negatively impact this neuroprotective mechanism mediated by astrocytes in response to HMGB1 released by damaged cells. Therefore, inhibition of RAGE signaling in a cell type‐specific manner could improve the overall neuroprotective effect in vivo.
RAGE is a pattern recognition receptor able to bind multiple ligands, most of which are oligomeric in nature. 64 Moreover, constitutive oligomerization of RAGE in the plasma membrane is required for ligand recognition and cell signaling. 65 Thus, the oligomerization state of RAGE and its ligands, which is dictated in part by structural properties and level of expression, together with the expression level of intracellular adaptor proteins involved in signal transduction, define the outcome of RAGE signaling in a context‐ and celltype‐specific manner. 56 , 64 Thus, the level of expression of RAGE and its ligands can significantly impact their oligomerization state and biological effect. Accordingly, RAGE activation in neurons can promote cell survival or cell death, depending on the ligand and its concentration, and on the cellular context. 55 , 56 Moreover, the detrimental effect of RAGE deletion in the survival of hSOD1G93A could also be explained by the physiological homeostatic functions of RAGE expression in peripheral tissues. For example, RAGE is constitutively expressed at high levels in the lung, where it plays an important protective role preventing the development of pulmonary fibrosis. 66 , 67 Furthermore, mutant hSOD1G93A retains catalytic activity, 68 and it was shown that the over‐expression of wild‐type SOD1 accelerates the development of asbestos‐induced pulmonary fibrosis. 69 Therefore, RAGE deletion and hSOD1G93A over‐expression could negatively impact pulmonary function and contribute to the detrimental effect of the genetic RAGE deletion in the survival of hSOD1G93A.
Contributing to the complexity of the RAGE signaling network, many RAGE ligands also signal through other receptors such as TLR4 and TLR2, whose expression is also altered in ALS patients. 16 Thus, RAGE antagonism or deletion can facilitate signaling through these other receptors. Moreover, soluble RAGE isoforms, generated by alternative splicing or proteolytic cleavage at the level of the plasma membrane, exert a decoy function competing for ligand binding and preventing signaling through RAGE and other receptors that share the same ligands. 21 , 22 , 23 Thus, pharmacological antagonism of RAGE or decreased expression below a certain threshold also eliminates this modulatory component of the intricate network of pattern recognition receptors, potentially explaining the limited neuroprotective effects of FPS‐ZM1 treatment and the detrimental effects observed after genetic RAGE ablation. Our results reveal that RAGE signaling exerts both neurotoxic and neuroprotective functions in hSOD1G93A ALS mice. The development of inhibitors to specifically counteract RAGE neurotoxic signaling, while preserving the potential beneficial effects of soluble RAGE isoforms, could improve the efficacy of RAGE‐targeting therapies in ALS.
CONFLICT OF INTEREST
The authors declare no competing interests.
AUTHORS' CONTRIBUTIONS
L. Liu, K. M. Killoy, M. R. Vargas, and M. Pehar conducted experiments. Y. Yamamoto contributed key reagents. L. Liu, K. M. Killoy, M. R. Vargas, and M. Pehar performed data analysis. L. Liu, M. R Vargas, and M. Pehar wrote or contributed to the writing of the manuscript.
ACKNOWLEDGEMENTS
This work was supported by the National Institutes of Health National Institute of Neurological Disorders and Stroke (Grant R01NS100835); and the ALS Association (Investigator Initiated Starter Grant 18‐IIA‐404). This work used resources and facilities of the William S. Middleton Memorial Veterans Hospital (Madison, WI, USA). This work used the Cell and Molecular Imaging Core supported by the Hollings Cancer Center, Medical University of South Carolina (P30 CA138313), the SC COBRE in Oxidants, Redox Balance, and Stress Signaling (P20 GM103542), and the Shared Instrumentation Grant S10 OD018113.
Liu L, Killoy KM, Vargas MR, Yamamoto Y, Pehar M. Effects of RAGE inhibition on the progression of the disease in hSOD1G93A ALS mice. Pharmacol Res Perspect. 2020;8:e00636 10.1002/prp2.636
This work was presented in part at the 2019 Society for Neuroscience Annual Meeting (October 2019).
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Brown RH, Al‐Chalabi A. Amyotrophic lateral sclerosis. N Engl J Med. 2017;377(2):162‐172. [DOI] [PubMed] [Google Scholar]
- 2. Hardiman O, Al‐Chalabi A, Chio A, et al. Amyotrophic lateral sclerosis. Nat Rev Dis Primers. 2017;3:17071. [DOI] [PubMed] [Google Scholar]
- 3. Taylor JP, Brown RH Jr, Cleveland DW. Decoding ALS: from genes to mechanism. Nature. 2016;539(7628):197‐206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vinsant S, Mansfield C, Jimenez‐Moreno R, et al. Characterization of early pathogenesis in the SOD1(G93A) mouse model of ALS: part I, background and methods. Brain Behav. 2013;3(4):335‐350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vinsant S, Mansfield C, Jimenez‐Moreno R, et al. Characterization of early pathogenesis in the SOD1(G93A) mouse model of ALS: part II, results and discussion. Brain Behav. 2013;3(4):431‐457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Scott S, Kranz JE, Cole J, et al. Design, power, and interpretation of studies in the standard murine model of ALS. Amyotroph Lateral Scler. 2008;9(1):4‐15. [DOI] [PubMed] [Google Scholar]
- 7. Ludolph AC, Bendotti C, Blaugrund E, et al. Guidelines for preclinical animal research in ALS/MND: a consensus meeting. Amyotroph Lateral Scler. 2010;11(1–2):38‐45. [DOI] [PubMed] [Google Scholar]
- 8. Ilieva H, Polymenidou M, Cleveland DW. Non‐cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J Cell Biol. 2009;187(6):761‐772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yamanaka K, Chun SJ, Boillee S, et al. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci. 2008;11(3):251‐253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vargas MR, Pehar M, Cassina P, Beckman JS, Barbeito L. Increased glutathione biosynthesis by Nrf2 activation in astrocytes prevents p75NTR‐dependent motor neuron apoptosis. J Neurochem. 2006;97(3):687‐696. [DOI] [PubMed] [Google Scholar]
- 11. Nagai M, Re DB, Nagata T, et al. Astrocytes expressing ALS‐linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci. 2007;10(5):615‐622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Meyer K, Ferraiuolo L, Miranda CJ, et al. Direct conversion of patient fibroblasts demonstrates non‐cell autonomous toxicity of astrocytes to motor neurons in familial and sporadic ALS. Proc Natl Acad Sci USA. 2014;111(2):829‐832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Haidet‐Phillips AM, Hester ME, Miranda CJ, et al. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat Biotechnol. 2011;29(9):824‐828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim MJ, Vargas MR, Harlan BA, et al. Nitration and glycation turn mature NGF into a toxic factor for motor neurons: a role for p75(NTR) and RAGE signaling in ALS. Antioxid Redox Signal. 2018;28(18):1587‐1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shibata N, Hirano A, Hedley‐Whyte TE, et al. Selective formation of certain advanced glycation end products in spinal cord astrocytes of humans and mice with superoxide dismutase‐1 mutation. Acta Neuropathol. 2002;104(2):171‐178. [DOI] [PubMed] [Google Scholar]
- 16. Casula M, Iyer AM, Spliet W, et al. Toll‐like receptor signaling in amyotrophic lateral sclerosis spinal cord tissue. Neuroscience. 2011;179:233‐243. [DOI] [PubMed] [Google Scholar]
- 17. Juranek JK, Daffu GK, Wojtkiewicz J, Lacomis D, Kofler J, Schmidt AM. Receptor for advanced glycation end products and its inflammatory ligands are upregulated in amyotrophic lateral sclerosis. Front Cell Neurosci. 2015;9:485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kikuchi S, Shinpo K, Ogata A, et al. Detection of N epsilon‐(carboxymethyl)lysine (CML) and non‐CML advanced glycation end‐products in the anterior horn of amyotrophic lateral sclerosis spinal cord. Amyotroph Lateral Scler Other Motor Neuron Disord. 2002;3(2):63‐68. [DOI] [PubMed] [Google Scholar]
- 19. Lee JY, Lee JD, Phipps S, Noakes PG, Woodruff TM. Absence of toll‐like receptor 4 (TLR4) extends survival in the hSOD1 G93A mouse model of amyotrophic lateral sclerosis. J Neuroinflammation. 2015;12:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ilzecka J. Serum‐soluble receptor for advanced glycation end product levels in patients with amyotrophic lateral sclerosis. Acta Neurol Scand. 2009;120(2):119‐122. [DOI] [PubMed] [Google Scholar]
- 21. Hudson BI, Carter AM, Harja E, et al. Identification, classification, and expression of RAGE gene splice variants. FASEB J. 2008;22(5):1572‐1580. [DOI] [PubMed] [Google Scholar]
- 22. Xie J, Mendez JD, Mendez‐Valenzuela V, Aguilar‐Hernandez MM. Cellular signalling of the receptor for advanced glycation end products (RAGE). Cell Signal. 2013;25(11):2185‐2197. [DOI] [PubMed] [Google Scholar]
- 23. Kierdorf K, Fritz G. RAGE regulation and signaling in inflammation and beyond. J Leukoc Biol. 2013;94(1):55‐68. [DOI] [PubMed] [Google Scholar]
- 24. Song S, Miranda CJ, Braun L, et al. Major histocompatibility complex class I molecules protect motor neurons from astrocyte‐induced toxicity in amyotrophic lateral sclerosis. Nat Med. 2016;22(4):397‐403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Miquel E, Cassina A, Martínez‐Palma L, et al. Neuroprotective effects of the mitochondria‐targeted antioxidant MitoQ in a model of inherited amyotrophic lateral sclerosis. Free Radic Biol Med. 2014;70:204‐213. [DOI] [PubMed] [Google Scholar]
- 26. Vargas MR, Johnson DA, Sirkis DW, Messing A, Johnson JA. Nrf2 activation in astrocytes protects against neurodegeneration in mouse models of familial amyotrophic lateral sclerosis. J Neurosci. 2008;28(50):13574‐13581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Harlan BA, Killoy KM, Pehar M, Liu L, Auwerx J, Vargas MR. Evaluation of the NAD(+) biosynthetic pathway in ALS patients and effect of modulating NAD(+) levels in hSOD1‐linked ALS mouse models. Exp Neurol. 2020;327:113219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shekhtman A, Ramasamy R, Schmidt AM. Glycation & the RAGE axis: targeting signal transduction through DIAPH1. Expert Rev Proteomics. 2017;14(2):147‐156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Juranek JK, Daffu GK, Geddis MS, et al. Soluble RAGE treatment delays progression of amyotrophic lateral sclerosis in SOD1 mice. Front Cell Neurosci. 2016;10:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Deane R, Du Yan S, Submamaryan RK, et al. RAGE mediates amyloid‐beta peptide transport across the blood‐brain barrier and accumulation in brain. Nat Med. 2003;9(7):907‐913. [DOI] [PubMed] [Google Scholar]
- 31. Deane R, Singh I, Sagare AP, et al. A multimodal RAGE‐specific inhibitor reduces amyloid beta‐mediated brain disorder in a mouse model of Alzheimer disease. J Clin Investig. 2012;122(4):1377‐1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Myint K‐M, Yamamoto Y, Doi T, et al. RAGE control of diabetic nephropathy in a mouse model: effects of RAGE gene disruption and administration of low‐molecular weight heparin. Diabetes. 2006;55(9):2510‐2522. [DOI] [PubMed] [Google Scholar]
- 33. Pehar M, O'Riordan KJ, Burns‐Cusato M, et al. Altered longevity‐assurance activity of p53:p44 in the mouse causes memory loss, neurodegeneration and premature death. Aging Cell. 2010;9(2):174‐190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pehar M, Cassina P, Vargas MR, et al. Astrocytic production of nerve growth factor in motor neuron apoptosis: implications for amyotrophic lateral sclerosis. J Neurochem. 2004;89(2):464‐473. [DOI] [PubMed] [Google Scholar]
- 35. Pehar M, Beeson G, Beeson CC, Johnson JA, Vargas MR. Mitochondria‐targeted catalase reverts the neurotoxicity of hSOD1G(9)(3)A astrocytes without extending the survival of ALS‐linked mutant hSOD1 mice. PLoS ONE. 2014;9(7):e103438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pehar M, Vargas MR, Robinson KM, et al. Peroxynitrite transforms nerve growth factor into an apoptotic factor for motor neurons. Free Radic Biol Med. 2006;41(11):1632‐1644. [DOI] [PubMed] [Google Scholar]
- 37. Cassina P, Peluffo H, Pehar M, et al. Peroxynitrite triggers a phenotypic transformation in spinal cord astrocytes that induces motor neuron apoptosis. J Neurosci Res. 2002;67(1):21‐29. [DOI] [PubMed] [Google Scholar]
- 38. Pehar M, Ball LE, Sharma DR, et al. Changes in protein expression and lysine acetylation induced by decreased glutathione levels in astrocytes. Mol Cell Proteomics. 2016;15(2):493‐505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Arumugam T, Ramachandran V, Gomez SB, Schmidt AM, Logsdon CD. S100P‐derived RAGE antagonistic peptide reduces tumor growth and metastasis. Clin Cancer Res. 2012;18(16):4356‐4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ludolph AC, Bendotti C, Blaugrund E, et al. Guidelines for the preclinical in vivo evaluation of pharmacological active drugs for ALS/MND: report on the 142nd ENMC international workshop. Amyotroph Lateral Scler. 2007;8(4):217‐223. [DOI] [PubMed] [Google Scholar]
- 41. Arancio O, Zhang HP, Chen X, et al. RAGE potentiates Abeta‐induced perturbation of neuronal function in transgenic mice. EMBO J. 2004;23(20):4096‐4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen X, Walker DG, Schmidt AM, Arancio O, Lue LF, Yan SD. RAGE: a potential target for Abeta‐mediated cellular perturbation in Alzheimer's disease. Curr Mol Med. 2007;7(8):735‐742. [DOI] [PubMed] [Google Scholar]
- 43. Fang F, Lue LF, Yan S, et al. RAGE‐dependent signaling in microglia contributes to neuroinflammation, Abeta accumulation, and impaired learning/memory in a mouse model of Alzheimer's disease. FASEB J. 2010;24(4):1043‐1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Teismann P, Sathe K, Bierhaus A, et al. Receptor for advanced glycation endproducts (RAGE) deficiency protects against MPTP toxicity. Neurobiol Aging. 2012;33(10):2478‐2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sabbagh MN, Agro A, Bell J, Aisen PS, Schweizer E, Galasko D. PF‐04494700, an oral inhibitor of receptor for advanced glycation end products (RAGE). Alzheimer disease. Alzheimer Dis Assoc Disord. 2011;25(3):206‐212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Galasko D, Bell J, Mancuso JY, et al. Clinical trial of an inhibitor of RAGE‐Abeta interactions in Alzheimer disease. Neurology. 2014;82(17):1536‐1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Walker D, Lue LF, Paul G, Patel A, Sabbagh MN. Receptor for advanced glycation endproduct modulators: a new therapeutic target in Alzheimer's disease. Expert Opin Investig Drugs. 2015;24(3):393‐399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Burstein AH, Sabbagh M, Andrews R, Valcarce C, Dunn I, Altstiel L. Development of Azeliragon, an oral small molecule antagonist of the receptor for advanced glycation endproducts, for the potential slowing of loss of cognition in mild Alzheimer's disease. J Prev Alzheimers Dis. 2018;5(2):149‐154. [DOI] [PubMed] [Google Scholar]
- 49. Valcarce C, Dunn I, Burstein AH. Linking diabetes and Alzheimer's disease through rage: a retrospective analysis of Azeliragon Phase 2 and Phase 3 studies Alzheimer's Association International Conference 2019. 2019;15(Issue 7S_Part_24):P1263. Abstract O1264–1211‐1204. [Google Scholar]
- 50. Chiu C‐Y, Yang R‐S, Sheu M‐L, et al. Advanced glycation end‐products induce skeletal muscle atrophy and dysfunction in diabetic mice via a RAGE‐mediated, AMPK‐down‐regulated, Akt pathway. J Pathol. 2016;238(3):470‐482. [DOI] [PubMed] [Google Scholar]
- 51. Sagheddu R, Chiappalupi S, Salvadori L, Riuzzi F, Donato R, Sorci G. Targeting RAGE as a potential therapeutic approach to Duchenne muscular dystrophy. Hum Mol Genet. 2018;27(21):3734‐3746. [DOI] [PubMed] [Google Scholar]
- 52. Riuzzi F, Sorci G, Sagheddu R, Chiappalupi S, Salvadori L, Donato R. RAGE in the pathophysiology of skeletal muscle. J Cachexia Sarcopenia Muscle. 2018;9(7):1213‐1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chiappalupi S, Sorci G, Vukasinovic A, et al. Targeting RAGE prevents muscle wasting and prolongs survival in cancer cachexia. J Cachexia Sarcopenia Muscle. 2020. 10.1002/jcsm.12561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Léger B, Vergani L, Sorarù G, et al. Human skeletal muscle atrophy in amyotrophic lateral sclerosis reveals a reduction in Akt and an increase in atrogin‐1. FASEB J. 2006;20(3):583‐585. [DOI] [PubMed] [Google Scholar]
- 55. Rong LL, Gooch C, Szabolcs M, et al. RAGE: a journey from the complications of diabetes to disorders of the nervous system ‐ striking a fine balance between injury and repair. Restor Neurol Neurosci. 2005;23(5–6):355‐365. [PubMed] [Google Scholar]
- 56. Sorci G, Riuzzi F, Giambanco I, Donato R. RAGE in tissue homeostasis, repair and regeneration. Biochim Biophys Acta. 2013;1833(1):101‐109. [DOI] [PubMed] [Google Scholar]
- 57. Brambilla L, Martorana F, Guidotti G, Rossi D. Dysregulation of astrocytic HMGB1 signaling in amyotrophic lateral sclerosis. Front Neurosci. 2018;12:622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pehar M, Harlan BA, Killoy KM, Vargas MR. Role and therapeutic potential of astrocytes in amyotrophic lateral sclerosis. Curr Pharm Des. 2017;23(33):5010‐5021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Valori CF, Guidotti G, Brambilla L, Rossi D. Astrocytes in motor neuron diseases. Adv Exp Med Biol. 2019;1175:227‐272. [DOI] [PubMed] [Google Scholar]
- 60. Ekestern E. Neurotrophic factors and amyotrophic lateral sclerosis. Neurodegener Dis. 2004;1(2–3):88‐100. [DOI] [PubMed] [Google Scholar]
- 61. Van Damme P, Bogaert E, Dewil M, et al. Astrocytes regulate GluR2 expression in motor neurons and their vulnerability to excitotoxicity. Proc Natl Acad Sci USA. 2007;104(37):14825‐14830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Rothstein JD. Excitotoxicity and neurodegeneration in amyotrophic lateral sclerosis. Clin Neurosci. 1995;3(6):348‐359. [PubMed] [Google Scholar]
- 63. Ferraiuolo L, Higginbottom A, Heath PR, et al. Dysregulation of astrocyte‐motoneuron cross‐talk in mutant superoxide dismutase 1‐related amyotrophic lateral sclerosis. Brain. 2011;134(Pt 9):2627‐2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Fritz G. RAGE: a single receptor fits multiple ligands. Trends Biochem Sci. 2011;36(12):625‐632. [DOI] [PubMed] [Google Scholar]
- 65. Xie J, Reverdatto S, Frolov A, Hoffmann R, Burz DS, Shekhtman A. Structural basis for pattern recognition by the receptor for advanced glycation end products (RAGE). J Biol Chem. 2008;283(40):27255‐27269. [DOI] [PubMed] [Google Scholar]
- 66. Song JS, Kang CM, Park CK, et al. Inhibitory effect of receptor for advanced glycation end products (RAGE) on the TGF‐beta‐induced alveolar epithelial to mesenchymal transition. Exp Mol Med. 2011;43(9):517‐524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kumar V, Fleming T, Terjung S, et al. Homeostatic nuclear RAGE‐ATM interaction is essential for efficient DNA repair. Nucleic Acids Res. 2017;45(18):10595‐10613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yim MB, Kang JH, Yim HS, Kwak HS, Chock PB, Stadtman ER. A gain‐of‐function of an amyotrophic lateral sclerosis‐associated Cu, Zn‐superoxide dismutase mutant: An enhancement of free radical formation due to a decrease in Km for hydrogen peroxide. Proc Natl Acad Sci USA. 1996;93(12):5709‐5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. He C, Ryan AJ, Murthy S, Carter AB. Accelerated development of pulmonary fibrosis via Cu, Zn‐superoxide dismutase‐induced alternative activation of macrophages. J Biol Chem. 2013;288(28):20745‐20757. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.