

Abstract
l-Lysine oxidase/monooxygenase (l-LOX/MOG) from Pseudomonas sp. AIU 813 catalyzes the mixed bioconversion of l-amino acids, particularly l-lysine, yielding an amide and carbon dioxide by an oxidative decarboxylation (i.e. apparent monooxygenation), as well as oxidative deamination (hydrolysis of oxidized product), resulting in α-keto acid, hydrogen peroxide (H2O2), and ammonia. Here, using high-resolution MS and monitoring transient reaction kinetics with stopped-flow spectrophotometry, we identified the products from the reactions of l-lysine and l-ornithine, indicating that besides decarboxylating imino acids (i.e. 5-aminopentanamide from l-lysine), l-LOX/MOG also decarboxylates keto acids (5-aminopentanoic acid from l-lysine and 4-aminobutanoic acid from l-ornithine). The reaction of reduced enzyme and oxygen generated an imino acid and H2O2, with no detectable C4a-hydroperoxyflavin. Single-turnover reactions in which l-LOX/MOG was first reduced by l-lysine to form imino acid before mixing with various compounds revealed that under anaerobic conditions, only hydrolysis products are present. Similar results were obtained upon H2O2 addition after enzyme denaturation. H2O2 addition to active l-LOX/MOG resulted in formation of more 5-aminopentanoic acid, but not 5-aminopentamide, suggesting that H2O2 generated from l-LOX/MOG in situ can result in decarboxylation of the imino acid, yielding an amide product, and extra H2O2 resulted in decarboxylation only of keto acids. Molecular dynamics simulations and detection of charge transfer species suggested that interactions between the substrate and its binding site on l-LOX/MOG are important for imino acid decarboxylation. Structural analysis indicated that the flavoenzyme oxidases catalyzing decarboxylation of an imino acid all share a common plug loop configuration that may facilitate this decarboxylation.
Keywords: l-amino acid monooxygenase, l-amino acid oxidase, 5-aminopentanamide, flavin-dependent monooxygenase, flavin-dependent oxidase, decarboxylation, imino acid, keto acid, dual activity enzyme, flavin enzyme mechanism, oxidase, flavin adenine dinucleotide (FAD), flavoprotein, monooxygenase
Enzymes are indispensable for living cells, as they are required to catalyze the reactions that serve physiological needs. Most enzymes catalyze a single specific reaction with the ability to use a certain set of compounds as substrates that are compatible with the binding and catalytic machinery. However, enzymes that employ a single active site to catalyze two types of reactions also exist. Their origin and the principles underlying their ability to accommodate dual activities are currently not well-understood. Nevertheless, the existence of these enzymes in nature is becoming more recognized. Understanding the general principles dictating their activities is useful for enzyme engineering or protein evolution to devise biocatalysts relevant for future industrial and biotechnology applications (1–4).
The characteristics of enzymes with dual activities in a single active site are diverse and can be found in a wide range of enzyme classes. Tetrachlorohydroquinone dehalogenase from Sphingomonas chlorophenolica catalyzes dehalogenation and isomerization of maleylactone and its analogues within a single active site (5). Acetohydroxyacid synthase (AHAS) from Thermotoga maritime catalyzes dual reactions with a major activity (∼90%) of converting pyruvate to acetoacetate and a minor (10%) pyruvate decarboxylase (PDC) activity that can convert pyruvate to acetaldehyde. Understanding how the PDC activity in AHAS is controlled is useful for the biofuel industry (6). Dual activities in a single active site have also been recognized in many flavoenzyme oxidases. However, insights into the mechanisms of these flavoenzymes and the factors dictating their activities are still lacking.
Flavoenzyme oxidases generally catalyze oxidation of substrates with reduction of O2 to generate H2O2. Their reactions are useful for a wide range of applications, such as the production of valuable chemicals (7). Amino acid oxidases comprise a large class of flavin-dependent oxidases with two structural families, including monoamine oxidase (MAO) and d-amino acid oxidase (d-AAO) families (8). Despite catalyzing the same conversion of amino acids to keto acids and H2O2, d-amino acid oxidases and l-amino acid oxidases (l-AAO) are classified into different structural families, with d-AAO in the d-AAO structural family and l-AAO in the MAO family (7). Enzymes in both d-AAO and l-AAO have been developed and applied as biosensors for detection of food contamination in nutritional industries (9, 10), as indicators for metabolic and neurological disorders (11), and as biocatalysts in pharmaceutical industries (12, 13). For example, l-lysine α-oxidase from Trichoderma viride (14–16), l-glutamate oxidase (17), l-aspartate oxidase (18), and l-phenylalanine oxidase (19, 20) has been used for l-amino acid detection. Many l-AAOs also show antiproliferative (21) and antimicrobial (22) activities, suggesting their roles as protein drugs.
Many l-AAOs, such as l-phenylalanine oxidase (PAO) from Pseudomonas sp. P-501 (23), tryptophan 2-monooxygenase (TMO) from Pseudomonas savastanoi (24, 25), and l-lysine monooxygenase (LMO) from Pseudomonas sp. (26), are known to contain two activities, oxidases and monooxygenases, in the same active sites. The monooxygenase is an apparent activity resulting from decarboxylation of an imino acid to form an amide product (Scheme 1). For some enzymes, such as LMO and TMO, which have structural characteristics classifying them as l-AAOs, their native major activity is monooxygenation of l-amino acid substrates (decarboxylation of imino acids) (Scheme 1) (26–29). It should be noted that the monooxygenation activities of these l-AAO enzymes are different from true flavin-dependent monooxygenases, such as various phenolic monooxygenases (30–32) or dechlorinating monooxygenase (33, 34), in which one atom of a molecular O2 is truly incorporated into the substrates via formation of a reactive C4a-(hydro)peroxyflavin (32, 35, 36). Although the reactions of flavin-dependent oxidases also involve an initial step of oxygen activation, most oxidases cannot facilitate the formation of a kinetically stable C4a-(hydro)peroxyflavin. The only exception is found in the reaction of pyranose 2-oxidase, which belongs to the class of glucose-methanol-choline oxidases in which C4a-hydroperoxyflavin first forms before H2O2 elimination takes place (37). For l-AAOs mentioned above, which can catalyze decarboxylation in addition to oxidation, their ability to form the C4a-hydroperoxyflavin intermediate is not clear.
Scheme 1.

General scheme of oxidase and monooxygenase activities catalyzed by l-amino acid oxidases. Oxidase activity results in deamination of an imino acid to form a keto acid and ammonium (blue), whereas monooxygenase activity results in decarboxylation of an imino acid to form an amide product (red). Thus, the monooxygenation catalyzed by l-amino acid oxidases is apparent monooxygenation resulting from decarboxylation—not true monooxygenase activity resulting from oxygen insertion.
l-Amino acid oxidase/monooxygenase (l-AAO/MOG) from Pseudomonas sp. AIU 813 is an FAD-dependent oxidase that displays dual activities of oxidase and monooxygenase. l-Lysine is the most preferred substrate and is considered a native substrate for this enzyme. l-AAO/MOG showed significant oxidase activity (19%) and it is a different enzyme from the previously explored LMO, which displayed only monooxygenation activity (27). To avoid confusion, in this report, we will refer to l-AAO/MOG from Pseudomonas sp. AIU 813 as l-lysine oxidase/monooxygenase (l-LOX/MOG). l-LOX/MOG is an ideal system for investigating the mechanism controlling the dual activities of a flavoenzyme oxidase. l-LOX/MOG catalyzes the minority reaction of oxidative deamination of l-lysine to produce ammonia, H2O2, and 2-keto-6-aminohexanoic acid (KH) and the majority reaction (81%) of oxidative decarboxylation of l-lysine to form CO2, H2O, and 5-aminopentanamide (5-APNM) (Fig. 1, gray panel). The oxidase activity of l-LOX/MOG could be increased by treating the enzyme with p-chloromercuribenzoate or by site-directed mutagenesis of C254I (38). However, rather than direct detection of product, the previous reports measured only the formation of ammonium in the presence and absence of amidase as a readout for the first liberation of ammonia or for the cleavage of the amide product (Fig. 1) (38, 39). Recently, new X-ray structures of l-LOX/MOG in complex with l-lysine, l-ornithine, and l-arginine have been reported (40). Nevertheless, how l-LOX/MOG controls the two oxidase and monooxygenase activities, structurally and mechanistically, is unclear.
Figure 1.
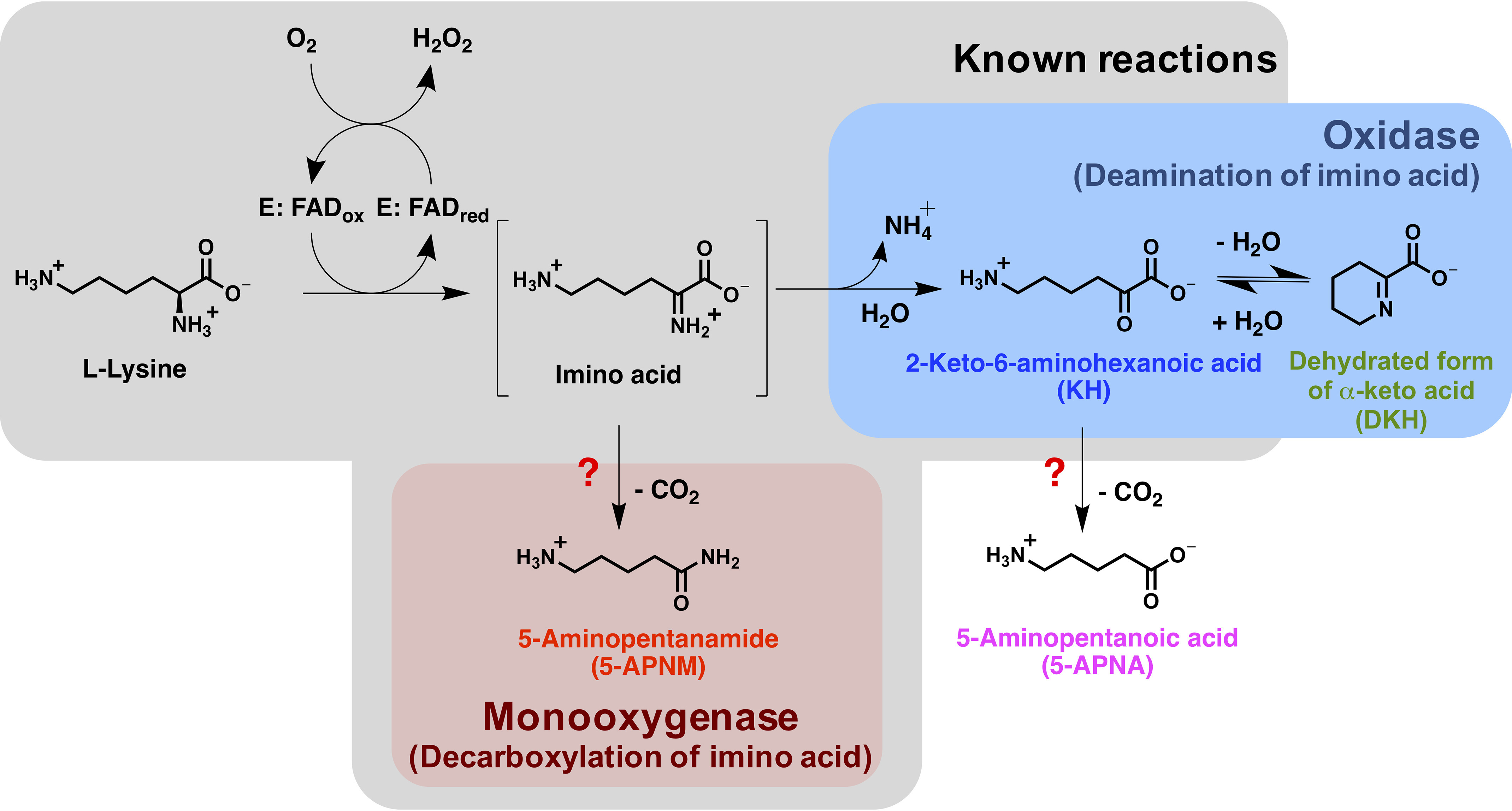
The oxidase and monooxygenase reactions of l-LOX/MOG with l-lysine. Known products from previous studies are shown in the gray box. This report newly identified 5-APNA and DKH as products from the reaction of l-LOX/MOG with l-lysine.
In this study, we investigated the mechanistic features controlling the dual activities of l-LOX/MOG from Pseudomonas sp. AIU 813 by analyzing the products from the reactions of l-lysine and l-ornithine using high-resolution MS and monitoring the transient kinetics of the reactions using stopped-flow spectrophotometry. The single-turnover reactions performed under anaerobic conditions were added H2O2 under various setups to identify key catalytic features enabling decarboxylation of 5-APNM and 5-aminopentanoic acid (5-APNA). Binding interactions of l-lysine and l-ornithine with the enzyme were explored by quantum mechanics/molecular mechanics (QM/MM) molecular dynamics (MD) simulations and by detection of charge transfer of the reduced FAD and the oxidized l-lysine complex by stopped-flow spectrophotometry. The common structural feature that facilitates decarboxylation of imino acids and allows the oxidases to catalyze apparent monooxygenation was also explored.
Results
Screening for l-LOX/MOG substrate specificity using flavin reduction
To identify amino acids that can reduce the enzyme-bound oxidized FAD, a solution of l-LOX/MOG–bound oxidized FAD was added to various types of l-amino acids under anaerobic conditions. The results (Fig. S1) showed that only three l-amino acids (l-lysine, l-ornithine, and l-arginine) could reduce the enzyme-bound oxidized FAD to generate reduced FAD, which could be detected by the decrease in absorbance at 462 nm. Among the three amino acids, only the reactions of l-lysine showed complete reduction after 20 min. In contrast, l-ornithine and l-arginine showed significantly slower flavin reduction rates.
Catalytic reactions of l-LOX/MOG
Product analysis of the overall turnovers of l-LOX/MOG with l-lysine and l-ornithine
Previous studies of l-LOX/MOG reported KH and 5-APNM as products from l-LOX/MOG with l-lysine and 2-keto-5-aminovaleric acid (KV) and 4-aminobutanamide (4-ABNM) as products from the reaction with l-ornithine. These data were based on HPLC identification of products and a combination of enzyme assays for both activities that measured free before and after deamination by amide hydrolase (38). In this work, we wanted to establish and clarify whether the enzyme can generate other products in addition to these compounds and determine the ratios of compounds produced from the dual activities of l-LOX/MOG. Therefore, products from the multiple-turnover reactions of l-LOX/MOG with l-lysine and l-ornithine were identified by HPLC coupled with high-resolution MS quadrupole-time-of-flight (HPLC/QTOF-MS). Quantitative measurements were carried out by HPLC coupled with triple-quadrupole or single-quadrupole MS. Control reactions without enzyme added were also carried out to verify that the stability of the substrates was constant.
Multiple-turnover reactions of l-LOX/MOG and l-lysine were carried out for 24 h, and the resulting products were analyzed. Results (Table 1) showed that all l-lysine was completely consumed, and three compounds were detected with the major product being 5-APNM (85 ± 1%), consistent with previous reports (38). However, we did not detect the expected product, KH, but detected the dehydrated form of KH (DKH) instead. Unexpectedly, we also detected a very small amount of 5-APNA (0.32 ± 0.02%). This newly detected 5-APNA may be derived from decarboxylation of KH generated in the deamination path (Figs. 1 and 2 and Table 1). Mass spectra of all these compounds are shown in Table S1 and Fig. S2. Based on these data, it can be firmly established that l-LOX/MOG catalyzes the decarboxylation of imino lysine as a major reaction (85%) and deamination of imino lysine as a minor reaction (14.7%). HPLC chromatograms of the control and turnover reactions and all standard compounds are shown in Figs. S3–S5.
Table 1.
Percentage of utilized substrates and product yields from multiple-turnover reactions
| Substrate | Compounds | Percentage |
|---|---|---|
| l-Lysine | l-Lysine | 100.0 ± 0.1 (utilized) |
| 5-APNM | 85 ± 1 | |
| 5-APNA | 0.32 ± 0.02 | |
| KH | Detected | |
| DKH | Detected | |
| l-Ornithine | l-Ornithine | 100 ± 2 (utilized) |
| 4-ABNM | 0.021 ± 0.002 | |
| 4-ABNA | 32.8 ± 0.2 | |
| KV | Detected | |
| DKV | Detected |
Figure 2.

Multiple-turnover reactions of l-LOX/MOG with l-lysine. Percentages of l-lysine utilization and product formation were plotted versus reaction times for up to 24 h. At 24 h, filled black circles represent utilization of l-lysine (up to 100%). Filled red diamonds represent formation of 5-APNM (up to 85%). Filled green triangles represent peak area of DΚΗ formation. Empty blue circles represent the peak area of KH, and filled pink squares represent peak area of 5-APNA acid formation (up to 0.32%). The error bars are SD.
Multiple-turnover reactions of l-LOX/MOG and l-ornithine were dramatically slower than that of l-lysine (data not shown). For 24 h, all l-ornithine was consumed, and unlike the reaction of l-lysine, only 0.021 ± 0.002% of 4-ABNM from the decarboxylation of imino ornithine was detected, whereas a majority of the compounds produced was KV, as expected. Different from the previously reported data (38) but consistent with the reaction of l-lysine discussed above, a new product, 4-aminobutanoic acid (4-ABNA), resulting from decarboxylation of KV, was also detected (in significant amount, 32.8 ± 0.2%) (Fig. 3 and Table 1). Interestingly, the ratio of products from the l-ornithine reaction was different from that of l-lysine. The reaction of l-ornithine prefers to proceed via deamination to form KV and 4-ABNA more than to proceed via direct decarboxylation of imino ornithine to form 4-ABNM. Nevertheless, the decarboxylation of KV also exists. HPLC chromatograms and mass spectra of all compounds are shown in Figs. S6–S9 and Table S2.
Figure 3.
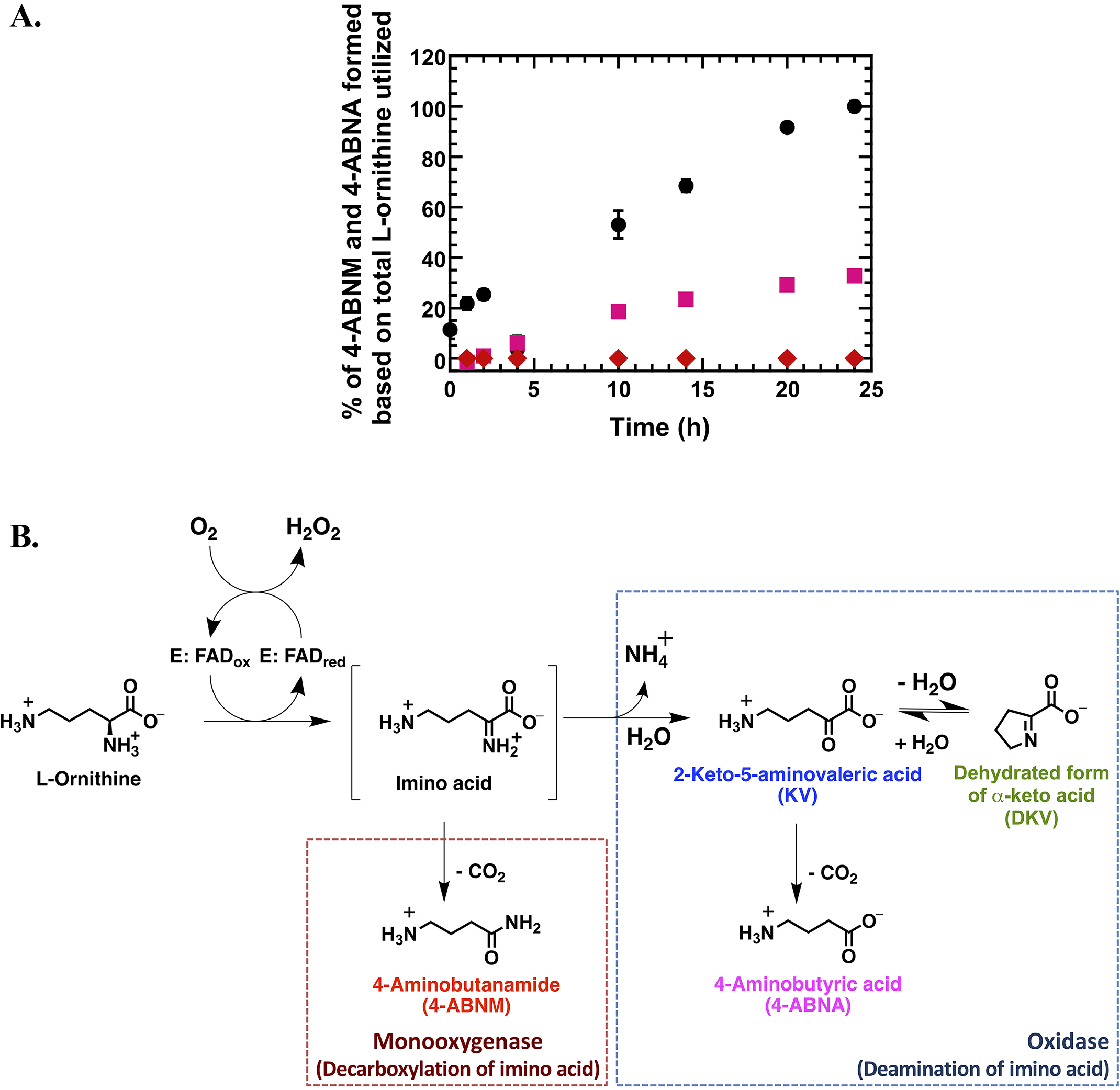
Multiple-turnover reactions of l-LOX/MOG with l-ornithine. A, percentages of l-ornithine utilization and product formation were plotted versus reaction times for up to 24 h. Filled black circles represent utilization of l-ornithine (up to 100%). Filled pink squares represent formation of 4-ABNA (up to 32.8%) newly identified in this work. Filled red diamonds represent formation of 4-ABNM (up to 0.021%). B, scheme of deamination and decarboxylation of l-LOX/MOG with l-ornithine. Error bars are SD. The data are also shown in Table 1.
Kinetics of flavin reduction in the reactions of l-AAO/MOG–bound oxidized FAD by l-lysine and l-ornithine
The kinetics of the l-LOX/MOG–bound oxidized FAD reduction by l-lysine and l-ornithine were investigated by monitoring absorbance changes at 462 nm (see “Experimental procedures”). The resultant kinetic traces showed that the flavin reduction by l-lysine started much quicker (0.02 s) than the reduction by l-ornithine (2 s). The enzyme-bound oxidized FAD could be completely reduced by l-lysine after 0.6 h, whereas 1.6 h was required for the reaction with l-ornithine. The kinetic traces were triphasic in both reactions of l-lysine and l-ornithine. Observed rate constants (kobs) for the reactions of l-lysine and l-ornithine are shown in Table 2. For l-lysine, kobs of the first phase is 0.540 ± 0.002 s−1, which is 22-fold greater than that of l-ornithine (0.025 ± 0.002 s−1). Therefore, the flavin reduction of l-LOX/MOG with l-lysine was dramatically faster than that of l-ornithine (Fig. 4). This indicates that l-LOX/MOG clearly prefers to use l-lysine rather than l-ornithine.
Table 2.
kobs of the reduction of l-LOX/MOG–bound FAD by 16 mm l-lysine and l-ornithine monitored by stopped-flow spectrophotometry
|
kobs |
|||
|---|---|---|---|
| 1st phase | 2nd phase | 3rd phase | |
| s−1 | |||
| l-Lysine | 0.540 ± 0.002 | 53.2 ± 0.4 × 10−3 | 7.78 ± 0.05 × 10−3 |
| l-Ornithine | 0.025 ± 0.002 | 3.18 ± 0.03 × 10−3 | 6.5 ± 0.5 × 10−4 |
Figure 4.

Flavin reduction of l-LOX/MOG. The reduction of l-LOX/MOG–bound FAD by 16 mm l-lysine (solid line) and l-ornithine (dashed line) was monitored by stopped-flow spectrophotometry.
Investigation of C4a-(hydro)peroxyflavin formation in the l-LOX/MOG reaction
As C4a-(hydro)peroxyflavin is a reactive intermediate commonly found in all flavin-dependent monooxygenases (32, 41), we investigated whether this flavin intermediate is involved in the decarboxylation reaction of l-LOX/MOG using stopped-flow spectrophotometry. Absorbance changes at 385 nm and at various other wavelengths in the regions where C4a-(hydro)peroxyflavin absorbs were monitored. Typically, when C4a-(hydro)peroxyflavin is formed, kinetic traces at 385 nm show an absorbance initial increase followed by a decrease when the intermediate eliminates H2O2 to form oxidized FAD. These kinetic traces would be clearly different from the data measured in the 450–460 nm region in which only flavin oxidation is monitored (33, 42–44). A solution of reduced l-LOX/MOG was mixed with l-lysine in the presence of oxygen (details under “Experimental procedures”). Results showed that absorbance changes at 385 nm and other wavelengths in that area increase with the same kinetic patterns as those at 462 nm (Fig. 5). Consequently, the oxidation of reduced l-LOX/MOG by oxygen does not stabilize C4a-(hydro)peroxyflavin as an intermediate, and the reaction of the enzyme with oxygen generates H2O2 directly.
Figure 5.

Kinetic traces of the flavin oxidation. The oxidation of l-LOX/MOG–bound reduced FAD by O2 was monitored by absorbance at 462 and 385 nm (solid and light gray lines, respectively).
Identification of key catalytic features enabling formation of 5-APNM and 5-APNA (decarboxylation)
The necessity of H2O2 bound at the active site for formation of 5-APNM
Three sets of single-turnover experiments (Scheme 2, E1–E3) were carried out to investigate whether the decarboxylation of the imino acid (Fig. 1) to result in 5-APNM requires H2O2 bound in the l-LOX/MOG active site. The enzyme-bound reduced FAD and the imino lysine were generated by mixing the oxidized enzyme and l-lysine under anaerobic conditions. For E1, oxygen (260 μm) was added into the solution after the flavin reduction was complete (Scheme 2, E1). Based on the concentration of l-LOX/MOG as the limiting reagent, the results showed 102 ± 4% consumption of l-lysine and the production of 5-APNM (around 63 ± 5%) and higher amount of products from the oxidase path (KH and DKH) than those in E2 and E3 (Table 3). These results agree well with the product analysis done for the above multiple-turnover reactions of l-lysine (Table 1). For E2, the reaction was kept anaerobic with no addition of oxygen after the flavin reduction or before product analysis (no H2O2 would be generated in this case). The results showed only the products KH and DKH from the oxidase path, and no 5-APNA and 5-APNM was detected (Scheme 2 (E2) and Table 3). These data clearly confirm that formation of the oxidase products, KH and DKH, does not require H2O2, and only the hydrolysis by H2O enables generation of KH and DKH from the imino acid. For E3, the enzyme was denatured after the flavin reduction, and free H2O2 was added into the solution (Scheme 2, E3). Only oxidase products (KH and DKH) were produced at amounts comparable with those from E2, again confirming that the oxidase path only requires H2O for imino lysine deamination and that free H2O2 could not decarboxylate the imino acid. Interestingly, whereas no 5-APNM from decarboxylation of imino acid was detected, E3 showed production of a small amount of 5-APNA (0.54 ± 0.05%), which is the result of decarboxylation of KH (Table 3). These findings suggest that free H2O2 does not lead to formation of 5-APNM but rather promotes formation of the decarboxylation product of α-keto acids to form 5-APNA instead. The data also imply that the decarboxylation of the imino acid to form 5-APNM needs H2O2 bound in the active site of l-LOX/MOG, whereas reaction of free H2O2 with KH can lead to the decarboxylation to form 5-APNA. (HPLC chromatograms of products from all reactions are shown in Figs. S10–S12).
Scheme 2.

Workflow of the reactions from each condition to determine the effect of H2O2 added after the flavin reduction and formation of imino acid. After the flavin reductions, the active enzyme reactions were added into aerobic buffer (E1), anaerobic buffer (E2), formic acid to denature the enzyme and H2O2 in aerobic buffer (E3), H2O2 in anaerobic buffer (E4), and H2O2 in aerobic buffer (E5). The effect of H2O2 preincubated with the enzyme before flavin reduction was carried out under all anaerobic conditions (E6).
Table 3.
Product analysis after the flavin reduction with the active enzyme reactions in the presence of O2 (E1), the absence of O2 (E2), the presence of H2O2 without O2 (E4), and the presence of H2O2 with O2 (E5)
The reaction of free H2O2 was explored with the reaction in which the enzyme was denatured and in the presence of O2 (E3). The effect of H2O2 preincubated with the enzyme before the flavin reduction was explored in the absence O2 (E6). Expt., experiment.
| Expt. | Conditions | l-Lysine utilized | 5-APNM | 5-APNA | Peak area |
|||
|---|---|---|---|---|---|---|---|---|
| KH | DKH | |||||||
| % | % | % | × 105 | × 106 | ||||
| E1 | Active | Buffer | O2 | 102 ± 4 | 63 ± 5 | 1.46 ± 0.02 | 2.82 ± 0.03 | |
| E2 | Active | Buffer | No O2 | 77 ± 9 | 1.41 ± 0.03 | 2.8 ± 0.1 | ||
| E3 | Denatured | H2O2 | O2 | 78 ± 8 | 0.54 ± 0.05% | 1.36 ± 0.05 | 2.8 ± 0.5 | |
| E4 | Active | H2O2 | No O2 | 74 ± 9 | 1.06 ± 0.01% | 1.37 ± 0.05 | 2.77 ± 0.09 | |
| E5 | Active | H2O2 | O2 | 95 ± 9 | 61 ± 2 | 1.00 ± 0.03% | 1.34 ± 0.03 | 2.67 ± 0.03 |
| E6 | Preincubation with H2O2 | 100 ± 13 | 0.34 ± 0.04 | 7.5 ± 1.3% | 1.48 ± 0.31 | 2.8 ± 0.3 | ||
The role of H2O2 in decarboxylation activity
We investigated whether extra H2O2 added may enhance the decarboxylation activity of l-LOX/MOG to form more 5-APNM. In E4 (Scheme 2, E4), free H2O2 in the absence of O2 was added into a solution of reduced l-LOX/MOG and analyzed for 5-APNM formation. The analysis did not detect significant formation of 5-APNM, and most product was found as KH and DKH (∼74 ± 9%) (Table 3). Interestingly, 5-APNA (1.06 ± 0.01%) was detected under this condition (Table 3), indicating that adding extra H2O2 after l-lysine oxidation could enhance KH decarboxylation to form 5-APNA. For E5, the reaction was the same as E4 but additionally exposed to oxygen after the imino acid was generated (Scheme 2, E5). It was carried out to provide the system with both types of H2O2—the one produced in situ from the enzymatic reaction and free added H2O2. The results (Table 3) indicate that both 5-APNM (61 ± 2%) and 5-APNA (1.00 ± 0.03%) can be generated in this case, demonstrating that H2O2 generated in situ in the enzyme active site and extra H2O2 added function differently. The addition of H2O2 to the enzyme could not produce 5-APNM but led to formation of 5-APNA instead (Table 3). To generate 5-APNM, H2O2 must come from the reaction of the l-LOX/MOG–bound reduced FAD with O2. HPLC chromatograms of the E4 and E5 reactions are shown in Figs. S13 and S14.
The reaction of extra H2O2 pre-bound at the l-LOX/MOG active site
Based on the previous results, which indicated that H2O2 produced in situ (not extra H2O2 added) is required for the generation of 5-APNM, we further investigated in E6 (Scheme 2, E6) whether preincubation of H2O2 in the active site could lead to formation of 5-APNM. The results show that the preincubation of H2O2 with the enzyme before carrying out the experiments did not increase the amount of 5-APNM formed (0.34 ± 0.04%), but instead significantly increased the amount of 5-APNA, the decarboxylation product of KH, by about 7-fold (Scheme 2 (E5 and E6) and Table 3). These results agree with the data in E3, E5, and E6 which indicate that extra H2O2 added only reacts with KH to enhance the generation of 5-APNA, not 5-APNM. The catalytic reaction generating 5-APNM, the native major product, mainly requires H2O2 generated from the reaction of the enzyme-bound reduced FAD with oxygen. This is possibly due to the proper orientation of H2O2 to be suitable to readily attack the imino acid of l-lysine generated during the flavin reduction (see Fig. 11A and see below). HPLC chromatograms of E6 reactions are shown in Fig. S15.
Figure 11.
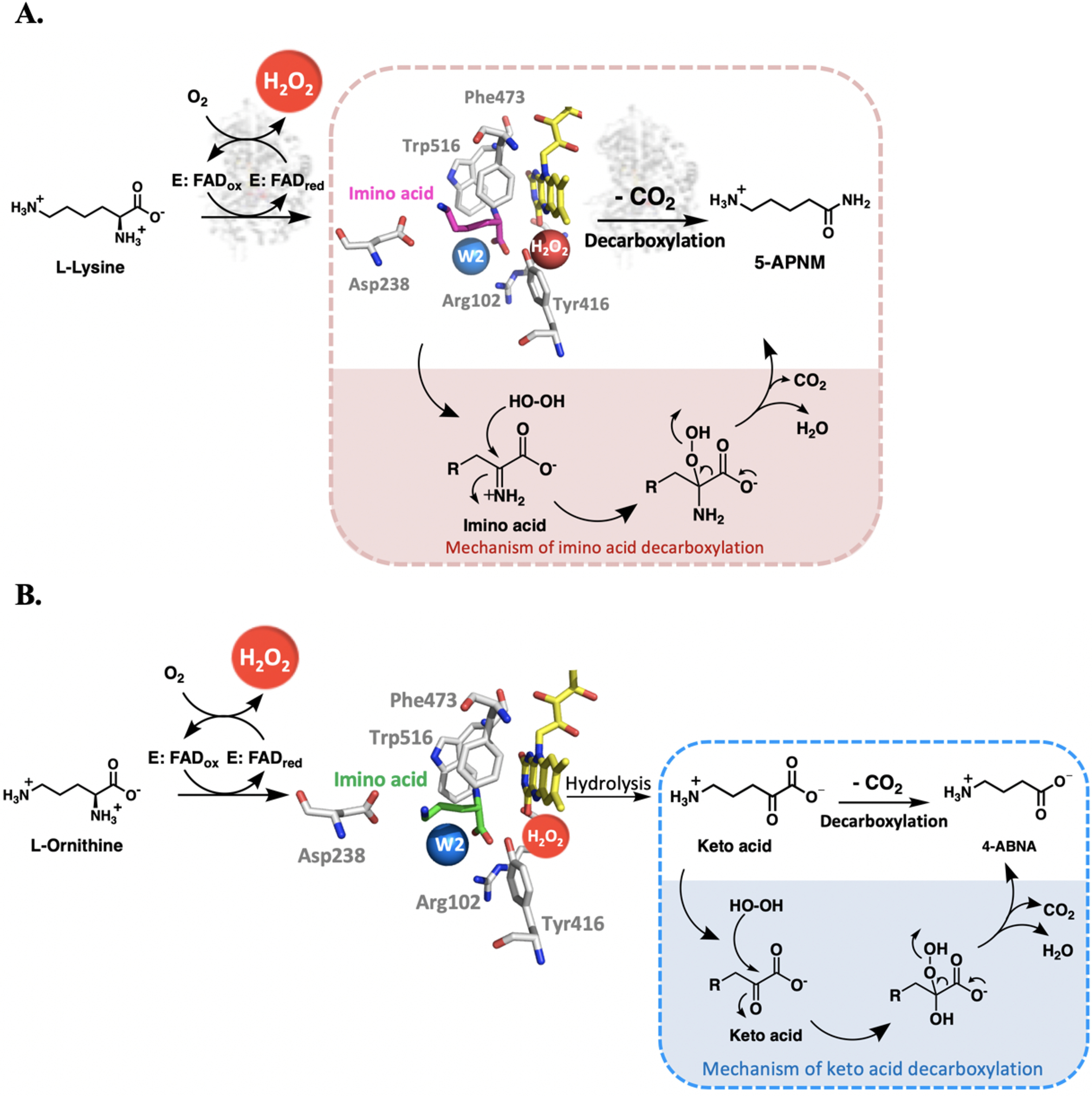
Reaction mechanisms of l-LOX/MOG with l-lysine and l-ornithine. A, mechanism of imino acid decarboxylation to yield 5-APNM. B, mechanism of keto acid decarboxylation to yield 4-ABNA.
In addition, we also carried out the multiple-turnover reactions of l-LOX/MOG in the presence of catalase to remove extra H2O2 outside the active site. The results indicate that adding catalase does not affect the amount of 5-APNM formed (Fig. S16). Altogether, our data reported here illustrate that formation of 5-APNM from the decarboxylation of the imino lysine via the monooxygenase activity results from the proper binding of H2O2 and timing of its generation in situ in the active site of the enzyme (see Fig. 11A).
Investigation into the proper interactions required to catalyze the decarboxylation of imino acids from l-lysine and l-ornithine
Results from the multiple-turnover reactions (Table 1) indicate that the ratios of oxidase and monooxygenase products are different between the reactions using l-lysine and l-ornithine as substrates. l-Ornithine resulted in 0.021% of decarboxylation of imino ornithine, whereas l-lysine resulted in 85% of decarboxylation of imino lysine. This is interesting because l-lysine and l-ornithine have similar structures, but l-ornithine is one carbon shorter than l-lysine. These data imply that the proper binding distance and orientation of H2O2 may be important for the reaction of H2O2 and these imino acids. We therefore probed the distance of imino acid binding to active-site residues of l-LOX/MOG using two approaches—MD simulations and measurement of charge transfer formation.
Distances between Cα of imino lysine and imino ornithine to N5 of isoalloxazine ring investigated by MD simulations
MD simulations monitored during 8.0 ns in equilibrium were carried out to estimate the distance between the N5 position of FAD and Cα of imino lysine or imino ornithine bound in the active site of l-LOX/MOG (see “Experimental procedures”). The geometry of the complex was also probed. The enzyme structure used for this calculation was based on the structure of l-LOX/MOG with l-lysine and l-ornithine bound (40). Results indicate that the distance between the Ν5 of FAD and the Cα of imino lysine (7.1 Å) (Fig. 6) is shorter than the distance between the corresponding atoms in the complex with imino ornithine (10.2 Å). It correlates with the distance of the N5 of FAD to Cα from crystal structures of l-lysine and l-ornithine (Fig. 7). In addition, the characteristics of the enzyme l-lysine complex also shows less flexibility in movement compared with that of l-ornithine (data not shown). Altogether, the data indicate that the binding of imino lysine results in closer interaction with the enzyme-bound flavin than that of imino ornithine. This is likely a key factor explaining why the reaction of l-lysine can form more of the decarboxylation product of the imino acid than the reaction of l-ornithine.
Figure 6.

Distances between N5 of FAD and Cα of the imino acid of l-lysine (filled blue diamonds) and l-ornithine (filled orange squares). The data were analyzed using MD simulations of l-LOX/MOG for 8 ns.
Figure 7.

Crystal structures of the active sites of l-LOX/MOG with l-lysine (A) and l-ornithine (B) bound. Distances of Ν5−Cα to l-lysine and l-ornithine are 3.7 and 3.9 Å, respectively.
In addition, results from MD simulations can also explain the larger rate constant of flavin reduction in l-lysine than in l-ornithine (Fig. 4). With regard to the N5-Hα-Cα angle, the l-lysine complex has a slightly wider angle (137 ± 9 degrees) than the l-ornithine complex (128 ± 10 degrees) (Fig. S17), implying that l-lysine has a greater possibility of transferring the Hα to the N5 of the isoalloxazine ring of FAD. Results from the MD simulations agree well with the results from monitoring of charge transfer complex (see below).
Probing the distance of l-amino acid binding by charge transfer complex
To probe the difference in the ligand binding environment in l-LOX/MOG with l-lysine and l-ornithine, we explored formation of a charge transfer complex that can be used to indicate a close distance between the ligand and flavin in the active site of flavoenzymes (45, 46). A solution of an l-LOX/MOG-bound oxidized FAD was mixed with solutions of l-lysine, l-ornithine, and l-arginine under anaerobic conditions, and the spectra of the reactions were monitored. The spectra indicate that only the reaction of l-lysine showed the characteristics of a charge transfer complex (around 550–700 nm). It likely indicates close interactions between reduced FAD and the imino acid (Fig. 8A). However, no charge transfer complex could be detected for the reactions of l-ornithine and l-arginine (Fig. 8B and Fig. S18). The data suggest that the imino acid of l-lysine likely binds closer to the N5 position of the reduced FAD, consistent with the MD simulation results described in the previous section. These results strongly suggest that the decarboxylation of imino acids in the decarboxylation path requires proper binding positioning of H2O2. Otherwise, the reaction will proceed via the oxidase path as in the reaction of l-ornithine.
Figure 8.

Spectra of flavin reduction during the reaction of l-LOX/MOG with l-lysine (A) and l-ornithine (B). Shown are spectra of the enzyme-bound oxidized FAD (solid line), the reduced flavin (dashed line), and the species detected midway through the reduction, which indicates formation of a charge transfer species in the case of the l-lysine reaction (dotted line).
Structural analysis
The previous structural studies of l-LOX/MOG identified a flexible loop designated as a “plug loop”. The plug loop is encoded by eight residues near the substrate-binding site, from Val-228 to Trp-235, consisting of VGFGTGGW. Previous work suggests that the plug loop may play a role in substrate binding and product release (40). However, the location of the plug loop in the complex of l-LOX/MOG with l-lysine is different from that found in the ligand-free l-LOX/MOG (Fig. S19A) (40). We hypothesize that the position of this plug loop may play a role in governing whether the reaction will proceed via the decarboxylation or deamination paths for the imino acid. Therefore, we carried out the alignment of structures of l-LOX/MOG bound with l-lysine, l-ornithine, and l-arginine (Fig. 9A). The data indicate that the position of the plug loop of ligand-free l-LOX/MOG or the bound forms with l-ornithine and l-arginine are similar and are positioned in the “opened” conformation. However, the position of the loop in these complexes is clearly different from the conformation found in the l-LOX/MOG with l-lysine bound. The distinctive plug loop conformation shows more of a “closed” conformation in the l-lysine–bound structure, which is the only decarboxylation of imino acid that takes place. It suggests that the “closed” plug loop conformation as in the l-LOX/MOG and l-lysine complex may be a factor promoting the decarboxylation of the imino acid (monooxygenase activity) (Fig. 9A and Table 4).
Figure 9.

The crystal structures of l-LOX/MOG showing the plug loop with eight residues from Val-228 to Trp-235 (VGFGTGGW). A, l-LOX/MOG with l-lysine bound (gray), l-ornithine bound (orange), and l-arginine bound (cyan). The plug loop of the l-lysine–bound structure is shown in magenta. B, structure of l-LOX/MOG with l-lysine bound (gray) and plug loop (magenta) is aligned with PAO from Pseudomonas sp. P-501 with l-phenylalanine bound (purple) and TMO from P. savastanoi with IAM bound (orange). These two enzymes can also catalyze decarboxylation of their imino acids. C, the structure of l-LOX/MOG with l-lysine bound (gray) is aligned with the structure of l-AAO from C. rhodostoma with l-phenylalanine bound (yellow). l-AAO cannot catalyze the decarboxylation of imino acids, and the alignment does not show any plug loop in the l-AAO structure.
Table 4.
Conformation of the plug loop based on the crystal structures of l-LOX/MOG with l-lysine, l-ornithine, and l-arginine bound; PAO with l-phenylalanine bound, TMO with IAM bound, l-AAO with l-phenylalanine bound, and MAO B with N-[(E)-methyl](phenyl)-N-[(E)-2-propenylidene]methanaminium (NYP) (MAO B inhibitor) bound
Cα-RMSDs from alignments are shown in comparison with the structure of l-LOX/MOG and l-lysine (5YB6).
| PDB | Enzyme | Ligand | Conformation of the plug loop | Product from imino acid |
RMSD | Reference | |
|---|---|---|---|---|---|---|---|
| Decarboxylation | Deamination | ||||||
| Å | |||||||
| 3WE0 | l-LOX/MOG | Opened | − | − | 0.531 | 38 | |
| 5YB6 | l-LOX/MOG | l-Lys | Closed | ++ | + | 40 | |
| 5YB7 | l-LOX/MOG | l-Orn | Opened | + | ++ | 0.206 | 40 |
| 5YB8 | l-LOX/MOG | l-Arg | Opened | NDa | NDa | 0.417 | 40 |
| 3AYJ | PAO | l-Phe | Closed | ++ | + | 4.384 | 23 |
| 4IV9 | TMO | IAM | Closed | +++ | − | 1.177 | 24, 47 |
| 2IID | l-AAO | l-Phe | No plug loop | − | +++ | 2.525 | 48 |
| 1GOS | MAO B | NYP | No plug loop | − | +++ | 8.857 | 49 |
aND, not determined.
To explore how widespread and common the features of this plug loop conformation are, we explored structures of other l-AAOs that are known to carry out monooxygenation of l-amino acids, such as PAO with l-phenylalanine bound and TMO with indole-3-acetamide (IAM) bound. The data indicate that all of these enzymes have their plug loops in the “closed” conformation as in the structure of l-LOX/MOG with l-lysine bound (Fig. 9B). Interestingly, l-AAO, which cannot catalyze decarboxylation, does not contain any loop with a similar conformation as the l-LOX/MOG plug loop (Fig. 9C). A similar situation was also found in MAO B (49) (Fig. S19B). These data imply that the presence of the plug loop in the “closed” conformation may be a key factor governing the ability of amino acid oxidases to catalyze decarboxylation of imino acids, such as in the reaction of l-LOX/MOG.
Discussion
Our studies reported here elucidate the mechanistic features important for allowing the single active site of l-LOX/MOG to catalyze the two reactions of apparent monooxygenation and the (classical) oxidation of flavoprotein oxidase. We have shown that the monooxygenation, which is essentially decarboxylation of imino acid to form an amide product, requires H2O2 generated in situ. Extra addition of H2O2 results in decarboxylation of α-keto acid instead. Only the reaction of l-LOX/MOG with the native substrate, l-lysine, results in a majority of decarboxylation to yield 5-APNM. In contrast, the reaction with the smaller substrate analog, l-ornithine, results in more processing through the oxidation path to form keto acid followed by decarboxylation of the keto acid to form 4-ABNA. Analysis by MD simulations and charge transfer complex formation studies indicated that this may be due to the larger distance between the FAD cofactor and l-ornithine. Structural analysis of l-LOX/MOG and other flavoprotein oxidases indicate the presence of an interesting plug loop in a closed conformation in l-LOX/MOG, which may be important for decarboxylation by H2O2.
The properties of l-LOX/MOG that allow it to catalyze different reactions with different substrates (i.e. more decarboxylation with l-lysine and more deamination with l-ornithine) (Figs. 3 and 4 and Table 1) are similar to those of many l-amino acid oxidases. The reaction of TMO with l-tryptophan yields only products of the decarboxylation path, but mutation of Arg-98 resulted in a variant that can catalyze both deamination and decarboxylation of imino acids (25, 50). Similar to TMO, ORF7, which is a gene in a polyene polyketide biosynthesis gene cluster from Amycolatopsis arientalis ATCC 43491, shows only an oxidase activity with arginine, but with Nδ-methylarginine it mainly catalyzes decarboxylation of the imino acid to form an amide product (51). For PAO, the enzyme mainly catalyzes decarboxylation with l-phenylalanine and mainly deamination (oxidase activity) with l-methionine. Based on a crystal structure of the enzyme, a shorter distance between the Cα of l-methionine and H2O in the active site may be a factor promoting the oxidase activity (52). For LMO, the reaction with l-lysine showed only decarboxylation of imino acid to form 5-APNM, whereas the reaction with l-ornithine generated only α-keto acid (26, 28). The reaction of LMO is clearly different from the reaction of l-LOX/MOG reported here.
The reaction of reduced l-LOX/MOG with oxygen does not stabilize C4a-(hydro)peroxyflavin or another flavin-oxygen adduct intermediate (Fig. 5). In addition to C4a-(hydro)peroxyflavin, it has been found that a flavin-N5-(hydro)peroxide intermediate can also act as an oxygen-transferring species for monooxygenases such as in the reactions of RutA (amide monooxygenase) and EncM (1,3-diketone oxidation) (53, 54). Both C4a-(hydro)peroxyflavin and flavin-N5-(hydro)peroxide have spectral characteristics different from oxidized and reduced flavin species. As the flavin oxidation of l-LOX/MOG with l-lysine did not show any intermediate that resembled the flavin-oxygen adducts mentioned, it is unlikely that these flavin intermediates are involved with the decarboxylation of imino acid in the l-LOX/MOG reaction.
We propose a mechanism for the decarboxylation of imino acid in which the H2O2 generated from the flavin oxidation near N5 of the isoalloxazine ring can attack the Cα of imino lysine to form an intermediate and release CO2 and H2O to yield 5-APNM (Figs. 6 and 10A). This result contradicts the previously proposed mechanism of PAO in which C4a-hydroperoxyflavin was proposed to be involved in the process of oxidative decarboxylation (23). The mechanism of l-LOX/MOG proposed here is also different from the mechanism of oxidative denitrification in nitronate monooxygenase, in which an anionic flavosemiquinone and a radical superoxide anion are proposed to be involved in the oxidative denitrification (36, 55). The H2O2 in the l-LOX/MOG reaction must also be in the bound state because free H2O2 could not react with imino acid to generate decarboxylated product (Scheme 2, E3 and E4). The addition of catalase does not interfere with product formation (Fig. S16). These findings are similar to a previous report of l-lactate monooxygenase (LaMO), in which mixing free H2O2 with keto acid does not generate acetate as a decarboxylated product (56), and addition of catalase does not interfere with the reaction (57).
Figure 10.

The active sites of l-LOX/MOG with imino acids of l-lysine (A) and l-ornithine (B) bound as analyzed by MD simulations for 8 ns.
Interestingly, extra addition of H2O2 either by supplementing l-LOX/MOG (E3–E5) with H2O2 directly into the reaction mixture, or by preincubation of the H2O2 with the enzyme (E6) prior to the addition of the other substrates enhances the decarboxylation of α-keto acid to yield 5-APNA (Table 1). These results are similar to the decarboxylation of pyruvate by LaMO. The reaction of reduced FAD with oxygen results in formation of H2O2 that reacts with the enzyme-bound pyruvate (generated from the flavin reduction) (58). The mechanism of decarboxylation of keto acid by l-LOX/MOG can be explained as H2O2 attacking the Cα of the keto acid after hydrolysis of the imino acid to yield acid, CO2, and H2O (Fig. 11B).
For the partition reactions between the two paths of l-LOX/MOG to form decarboxylated product and hydrolyzed imino acid, it can be seen that plentiful H2O can react to form the keto acid product. The active site of l-LOX/MOG is fine-tuned to facilitate the decarboxylation of imino acid by H2O2 to form 5-APNM. In E2 and E4, where oxygen is absent and, thus, no in situ H2O2 could be generated, the 5-APNM almost could not form. Most of the products were keto acids from the hydrolysis of imino acid. These data indicate that there are plenty of H2O molecules bound at the active site, but under regular turnover conditions, such as in experiment E1, this H2O cannot compete with H2O2 generated by the enzyme in the decarboxylation path. However, the preincubation of H2O2 in the absence of oxygen (E6) could not yield 5-APNM, as under regular turnover conditions. The data indicate that H2O2 must be bound at the proper position to react with an imino acid. In addition, when free H2O2 was added to a free imino acid liberated after the enzyme was denatured (E3), no 5-APNM was formed, and only products from the hydrolysis path could be detected. These data all suggest that the fine-tuned positioning of H2O2 at the active site of l-LOX/MOG is important for controlling the decarboxylation reaction.
Proper distance between the bound substrate and FAD must be a key factor controlling the decarboxylation activity of l-LOX/MOG. A close distance of interaction between reduced flavin and imino acid indicates that only the reaction of l-lysine—not l-ornithine (Fig. 8) nor l-arginine (Fig. S18)—could form the charge transfer species. This indicated that only the ligand that can bind closely to the FAD binding site is poised for the decarboxylation reaction. A short distance between H2O2 and the imino acid may allow H2O2 to react quickly, resulting in the achievement of decarboxylation before the hydrolysis reaction can take place (Figs. 6, 10, and 11A). The interaction of ligand bound at the active site is mediated by the negatively charged side chain of Asp-238, which can interact with a positive charge of the amino or imino acid (Figs. 7 and 10). The previous mutation of Asp-238 also confirmed that the residue is important for substrate binding (40). The interactions between the substrate and active site in PAO are also similar to those of l-LOX/MOG with regard to water bound in the active site. In PAO, one water molecule, W1, forms a hydrogen bond with Lys-478, whereas another one, W2, is located near Trp-660. Similar water molecules were found in the active site of l-LOX/MOG. Both l-LOX/MOG and PAO structures have a tunnel structure, and W1 is located at the end of the tunnel and close to the surface of the enzyme. It has been suggested that this tunnel may be a tunnel for O2 diffusion into the active site (Fig. S20) (23, 40).
Structural analysis has shown that the plug loop conformation may be an important feature promoting some flavoprotein oxidases to catalyze monooxygenation of l-amino acids. The “closed” conformation of the plug loop found in l-LOX/MOG may slow down H2O2 diffusion and force it to react with the imino acid. This hypothesis is supported by the mutation of C254I of l-LOX/MOG located far from the active site but close to the plug loop area, which resulted in only oxidase activity (38). These results are also consistent with results of TMO (23, 25, 59), PAO (23), and LaMO (60) mutations, which may affect the conformation of the plug loop. Interestingly, the effects of sulfhydryl reagents, such as p-chloromercuribenzoate, which react with the thiol groups of cysteine residues to induce a conformational change in l-LOX/MOG, demonstrated that this led to modulation of the dual activities of l-LOX/MOG by increasing oxidase activity (38, 61). For LaMO, Loop 4 was proposed to serve as a lid for the active site when it is in a specific conformation, acting to prevent the release of imino acid after flavin reduction (Fig. S21) (62). In the case of l-LOX/MOX, the plug loop near a substrate-binding site may play an important role as a gate to prevent or enhance the release of product, depending on the reaction steps. For a totally different enzyme system, a similar phenomenon can be found in AHAS which catalyzes not only the conversion of pyruvate to acetoacetate, but also the minor conversion of pyruvate to acetaldehyde. The ratio of the two activities depends upon the substrate concentration, because a high concentration of pyruvate causes a conformational change in the enzyme that promotes only the PDC activity (6). Therefore, a particular conformation of the active site loop, such as a plug loop in l-LOX/MOG, may be a key factor in controlling dual activities of the enzyme by keeping the H2O2 locally confined and thereby maintaining a high local concentration.
As the reaction of l-LOX/MOG can be useful for biotechnology, the knowledge gained from this work is important for future work on enzyme engineering of this system. For example, engineering to gain only the oxidase activity of l-LOX/MOG will allow the enzyme to be applied in biosensors for detection in food industries (63, 64). Engineering to gain an enzyme with only decarboxylation activity can be used in biocatalysis applications such as to synthesize 4-ABNA, which is an inhibitor of neurotransmitters (65, 66).
In conclusion, our findings here have expanded the mechanistic understanding of catalysis by flavoenzyme oxidases, particularly those that can catalyze apparent monooxygenation of decarboxylation by H2O2. We have elucidated how two activities can be catalyzed in the same single active site of l-LOX/MOG. This understanding will be useful for future enzyme engineering and directed evolution to reconstruct or repurpose enzymes of interest to be compatible with these useful applications.
Experimental procedures
Chemical reagents
Commercially available substances were purchased from Merck, Sigma–Aldrich, and Tokyo Chemical Industry Co., Ltd. Standard compounds of 5-APNM and 5-ABNM were purchased from Enamine, Ltd. Solvents (HPLC grade) were purchased from Honeywell Burdick & Jackson™.
Plasmids and bacterial strain
The l-LOX/MOG gene from Pseudomonas sp. AIU 813 in pET15b-LAAO plasmid containing the l-lox/mog gene was obtained as a gift from Toyama Prefectural University (39).
Spectroscopic studies
A HP8453 diode-array spectrophotometer (Agilent Technologies), 2501PC spectrophotometer (Shimadzu), or Cary 300Bio spectrophotometer (Agilent Technologies) was used to record UV-visible absorption spectra. All spectrophotometers were equipped with thermostated cell compartments.
Determination of the molar absorption coefficient of l-LOX/MOG–bound FAD
Previous studies have used the same molar absorption coefficient of l-LOX/MOG–bound FAD as for free FAD (ε462 of 11.3 mm−1 cm−1) (39). To determine the molar absorption coefficient of the l-LOX/MOG–bound FAD, the UV-visible spectrum of a solution of l-LOX/MOG complex was recorded. The enzyme absorbs a visible light maximum at 462 nm. The solution was then denatured by adding 2% (w/v) SDS to obtain the spectrum of free FAD. A molar absorption coefficient of l-LOX/MOG–bound FAD was determined based on the liberated free FAD (λmax at 450 nm and ε450 of 11.3 mm−1 cm−1) (67). The molar absorption coefficient of l-LOX/MOG–bound FAD was thus determined as ε462 of 11.15 ± 0.09 mm−1 cm−1 (data not shown).
Substrate specificity studies of flavin reduction
To investigate the substrate specificity for the flavin reduction of l-LOX/MOG, spectra of the enzymes upon the addition of substrates under anaerobic conditions were used to identify which amino acid can reduce the flavin. The spectrum of l-LOX/MOG–bound FAD (E:FADox) solution (20 μm) in 50 mm sodium phosphate buffer, pH 7.0, was recorded. Various l-amino acids (18.18 mm), including l-lysine, l-arginine, l-ornithine, l-asparagine, l-aspartic, l-glutamine, l-glutamic acid, and l-histidine, representing positively charged, negatively charged, and polar uncharged side chains were then added into the enzyme solution. The reaction without substrates was carried out as a control. The addition of l-lysine into a solution of free FAD was carried out to determine a dilution effect in the spectra measured. The UV-visible spectra of the mixed solutions were then recorded to identify which amino acids could reduce l-LOX/MOG–bound FAD.
Product analysis
Products from multiple-turnover reactions obtained from mixing 1 and 10 μm l-LOX/MOG–bound FAD with 2 mm l-lysine or 20 mm l-ornithine, respectively, in 50 mm sodium phosphate buffer, pH 7.0, were analyzed with HPLC/MS.
To determine the effect of catalase on 5-APNM formation, multiple-turnover reactions containing l-LOX/MOG–bound FAD (1 μm) with l-lysine (300 μm) and catalase (2%, w/v) in 50 mm sodium phosphate buffer, pH 7.0, were carried out. Positive and negative controls were reactions without catalase and without l-LOX/MOG, respectively.
To determine the effect of O2 and H2O2 on product formation, single-turnover reactions under anaerobic conditions were carried out by mixing a solution that contained l-LOX/MOG–bound FAD (100 μm) as a limiting reagent with l-lysine (400 μm) inside an anaerobic glove box (Belle Technology Ltd.) for 6 h to fully reduce the enzyme. The reaction was divided into two portions. To the first portion was added 0.4 m formic acid at a ratio of 1:1 to denature l-LOX/MOG and quench the reaction (E3). To the second portion, 50 mm sodium phosphate buffer, pH 7.0, was added, and the solution was kept under anaerobic conditions the entire time so that the l-LOX/MOG-bound FAD was maintained in the reduced FAD form. For E1 and E2, 50 mm sodium phosphate buffers at pH 7.0 with (E1) or without O2 (E2) were added. For E4 and E5, 400 μm H2O2 with (E5) or without O2 (E4) were added and incubated for 1 h. In the final step, the previously denatured l-LOX/MOG–bound reduced FAD (E3) was added to 50 mm sodium phosphate buffer, pH 7.0, whereas solutions of l-LOX/MOG from other all other experiments were quenched with 0.4 m formic acid at a ratio of 1:1 to denature l-LOX/MOG. All reactions were filtered using an ultrafiltration device (size 0.5 ml and 10,000 kDa, Amicon® Ultra from Merck Millipore Ltd.) to separate small molecules from enzyme samples.
To determine the effect of extra H2O2 on the decarboxylation reaction, 400 μm H2O2 was preincubated with 100 μm l-LOX/MOG–bound FAD for 1 h before adding 400 μm l-lysine (final concentration) for 6 h. The control without l-LOX/MOG–bound FAD was also carried out in comparison. All reactions were quenched with 0.4 m formic acid at a ratio of 1:1 to denature l-LOX/MOG and then filtered using an ultrafiltration device to remove proteins from solutions.
Products from multiple-turnover reactions were analyzed using HPLC (Agilent Technologies 1100, 1260 or 1290 Infinity series) with an electrospray ionization mass spectroscopic detector (MSD) (Agilent Technologies 6120 Quadrupole LC–MS, InfinityLab LC/MSD) and an Ultimate 3000 HPLC (Thermo Fisher Scientific) equipped with electrospray ionization and combined with QTOF (Bruker). A ZORBAX Eclipse Plus C18 reverse phase column containing 5-μm particle size in 4.6 × 250-mm column size (Agilent Technologies) was used as a stationary phase. Mobile phases were 0.5% formic acid (Sigma–Aldrich) in an H2O/acetonitrile (Burdick & Jackson Scientific Co., Ltd.) gradient. For HPLC/MS, the flow rate and injection volume were 0.5 ml/min and 1 µl, respectively. A positive mode of MSD was used for scanning a range of mass to charge ratios (m/z) from 50 to 500, and specific m/z values were selected to monitor substrates and expected products. HPLC/MSD profiles of substrates and products were compared with those of standard compounds. For determination of products from single-turnover reactions (E1–E6), a UPLC (Agilent Technologies 1290 Infinity series) with an Agilent Technologies 6470 Triple Quad (LC–MS QQQ-MS) was used. MS conditions with each compound were set to MRM mode to monitor l-lysine, and all products are shown in Table S3.
Transient kinetics of the l-LOX/MOG reactions
A characteristic spectrum of the charge transfer complex in the flavin reduction was determined by mixing l-amino acids (50 mm l-lysine or l-ornithine) with l-LOX/MOG–bound oxidized FAD (36 μm for l-lysine and 20 μm for l-ornithine) in anaerobic buffer, pH 7.0, and the reactions were monitored by a SF-61SX stopped-flow spectrophotometer using diode array and photomultiplier tube detectors from TgK Scientific at 25 °C. To remove oxygen from the stopped-flow unit and solutions, anaerobic buffer (50 mm sodium phosphate buffer, pH 7.0) and an oxygen-scrubbing solution (d-glucose (400 μm), glucose oxidase (1 mg/ml), catalase (4.8 mg/ml) in 50 mm sodium phosphate buffer, pH 7.0) were flushed through the flow systems, and the oxygen-scrubbing solution was allowed to stand in the flow system overnight. To monitor flavin reduction and flavin oxidation, 20 μm oxidized and reduced l-LOX/MOG solutions were prepared in an anaerobic glove box, respectively. To prepare reduced l-LOX/MOG, 0.25 mm l-lysine or an equivalent reducing amount of dithionite was added into a solution of oxidized l-LOX/MOG in 50 mm sodium phosphate buffer, pH 7.0. Oxidized and reduced enzyme solutions were placed into tonometers under anaerobic conditions, and the solution was loaded into the stopped-flow machine. To monitor the flavin reduction, l-LOX/MOG–bound oxidized FAD was mixed with 32 mm of anaerobic l-lysine and l-ornithine solutions in 50 mm sodium phosphate buffer, pH 7.0. Kinetic traces were observed, and the absorbance was monitored at 462 nm at 25 °C. To monitor flavin oxidation, the reduced l-LOX/MOG solution was mixed with buffers containing oxygen concentrations of 0.13 mm. Kinetic traces were observed, and the absorbance at 462 and 385 nm was monitored at 4 °C. Traces were plotted as a function of l-lysine and oxygen concentrations using KaleidaGraph software. Observed rate constants were analyzed by the ProgramA software (C. J. Chiu, R. Chang, J. Diverno, and D. P. Ballou, University of Michigan, Ann Arbor, MI, USA).
Computational chemistry
To investigate formation of a charge transfer complex in the active site of l-LOX/MOG with l-lysine, the location of imino acids of l-lysine and l-ornithine and their interactions with FAD were characterized by computational chemistry. The structures of l-LOX/MOG with l-lysine bound were obtained from the Protein Data bank (PDB) with code 5YB6. Chain A was selected to be a center of the model system in this study. The system was truncated to a 25-Å sphere with the center on carbon atom Cα of the l-lysine molecule (Scheme 2). The positions of the hydrogen atoms were located in the enzyme using the CHARMM procedure HBUILD (68). Hydrogen atoms of amino acid residues were added by considering results from Propka (69). The atom types in the topology files were assigned based on the CHARMM27 parameter set (70).
For investigation of the charge transfer complex, the system was divided into two parts, which are QM and MM parts for QM/MM simulations. The QM part consisted of the l-lysine and FAD molecules. The system was minimized using 1,000 steps of adopted basis Newton–Raphson minimization with the AM1/CHARMM27 method. Next, AM1/CHARMM27 molecular dynamics using the leapfrog Langevin dynamics with a time step of 0.001 ps was performed at 300 K. The rest of the protein and water molecules were treated as the MM part. The system was equilibrated with QM/MM MD for 120 ps. The structures of this equilibration were saved every 20 ps. To investigate the dynamics of different substrates, l-lysine was replaced by l-ornithine and l-arginine. The distance between the flavin ring and reacting atoms was determined.
For the location of imino acids with FAD in the active site, the structure of l-LOX/MOG was solvated in a cubic box of TIP3P with water extending at least 15 Å in each direction from the solute. The dimensions of the solvated system are 85 × 100 × 100 Å. MD (71) simulations were carried out using the NAMD program (72) with simulation protocols adapted from NAMD tutorials (73, 74) and our previous work (75). The simulations were started by minimizing hydrogen atom positions for 3,000 steps followed by water minimization for 6,000 steps. The system water was heated to 300 K for 5 ps and then equilibrated for 15 ps. The whole system was minimized for 10,000 steps and heated to 300 K for 20 ps. After that, the whole system was equilibrated for 180 ps followed by production stage for 8 ns. To investigate the substrate effect on binding in the active site of the enzyme, imino lysine was replaced by imino ornithine, and then the same procedure as applied on the imino lysine substrate was also applied on imino ornithine.
Structural analysis
Crystal structures of all enzymes were aligned together using PyMOL to identify a common structural factor that may allow l-LOX/MOG to carry out decarboxylation by H2O2. Structures of l-LOX/MOG from Pseudomonas sp. AIU 813 with free-ligand, l-lysine, l-ornithine, and l-arginine bound were obtained from PDB with codes 3WE0, 5YB6, 5YB7, and 5YB8, respectively. Structures of lactate monooxygenase from Mycobacterium smegmatis and lactate oxidase from Aerococcus viridans obtained from PDB entries 6DVI and 2E77, respectively, were aligned. Crystal structures of l-phenylalanine oxidase from Pseudomonas sp. P-501 with l-phenylalanine bound (PDB code 3AYJ), tryptophan 2-monooxygenase from P. savastanoi with IAM bound (PDB code 4IV9), l-AAO from Calloselasma rhodostoma with l-phenylalanine bound (PDB code 2IID), and MAO B from Homo sapiens with N-[(E)-methyl](phenyl)-N-[(E)-2-propenylidene] methanaminium bound (PDB code 1GOS) were aligned with l-LOX/MOG to identify common and divergent structural features.
Data availability
All data are included in the article and supporting information.
Supplementary Material
Acknowledgments
The gene for l-LOX/MOG in pET15b-LAAO was received from ERATO Asano Active Enzyme Molecule Project, Toyama Prefectural University. We acknowledge help from members of the Chaiyen laboratory, Department of Biochemistry and Center for Excellence in Protein and Enzyme Technology at Faculty of Science, Mahidol University, and School of Biomolecular Science and Engineering and Frontier Research Center at Vidyasirimedhi Institute of Science and Technology (VISTEC). We thank Panu Pimviriyakul for helpful suggestions.
This article contains supporting information.
Author contributions—D. T. data curation; D. T. and P. Chaiyen formal analysis; D. T. and P. Chaiyen validation; D. T., N. L., and P. Chaiyen investigation; D. T. visualization; D. T. and P. Chaiyen methodology; D. T. and P. Chaiyen writing-original draft; D. T. and P. Chaiyen project administration; D. T. writing-review and editing; P. Chenprakhon and P. Chaiyen supervision; D. M., Y. A., and P. Chaiyen resources; Y. A. and P. Chaiyen conceptualization; P. Chaiyen funding acquisition.
Funding and additional information—This work was supported by the Development and Promotion of Science and Technology Talents Project (DPST) (to D. T.) and Thailand Research Fund Grant RTA5980001 and the Thailand Science Research and Innovation and Program Management Unit-B Global Partnership program (to P. Chaiyen). This research work was also supported in part by Chiang Mai University (to N. L.) and ERATO Asano Active Enzyme Molecule Project of Japan Science and Technology Agency Grant JPMJER1102 (to Y. N. and D. M.).
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- AHAS
- acetohydroxyacid synthase
- 4-ABNA
- 4-aminobutanoic acid
- 4-ABNM
- 4-aminobutanamide
- 5-APNA
- 5-aminopentanoic acid
- 5-APNM
- 5-aminopentanamide
- d-AAO
- d-amino acid oxidase
- DKH
- dehydrated form of 2-keto-6-aminohexanoic acid
- DKV
- dehydrated form of 2-keto-5-aminovaleric acid
- E:FADox
- l-LOX/MOG–bound oxidized FAD
- E:FADred
- l-LOX/MOG–bound reduced FAD
- IAM
- indole-3-acetamide
- KH
- 2-keto-6-aminohexanoic acid
- KV
- 2-keto-5-aminovaleric acid
- l-AAO
- l-amino acid oxidase
- MOG
- monooxygenase
- l-LOX
- l-lysine oxidase
- LaMO
- lactate monooxygenase
- LMO
- l-lysine monooxygenase
- MAO
- monoamine oxidase
- MD
- molecular dynamics
- MSD
- mass spectroscopic detector
- PAO
- l-phenylalanine oxidase
- PDB
- Protein Data Bank
- PDC
- pyruvate decarboxylase
- QM
- quantum mechanics
- MM
- molecular mechanics
- QQQ
- triple quadrupole
- QTOF
- quadrupole-time-of-flight
- RMSD
- root mean square deviation
- TMO
- tryptophan 2-monooxygenase.
References
- 1. Copley S. D. (2003) Enzymes with extra talents: moonlighting functions and catalytic promiscuity. Curr. Opin. Chem. Biol. 7, 265–272 10.1016/S1367-5931(03)00032-2 [DOI] [PubMed] [Google Scholar]
- 2. Khersonsky O., Roodveldt C., and Tawfik D. S. (2006) Enzyme promiscuity: evolutionary and mechanistic aspects. Curr. Opin. Chem. Biol. 10, 498–508 10.1016/j.cbpa.2006.08.011 [DOI] [PubMed] [Google Scholar]
- 3. Copley S. D. (2015) An evolutionary biochemist's perspective on promiscuity. Trends Biochem. Sci. 40, 72–78 10.1016/j.tibs.2014.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Copley S. D. (2017) Shining a light on enzyme promiscuity. Curr. Opin. Struct. Biol. 47, 167–175 10.1016/j.sbi.2017.11.001 [DOI] [PubMed] [Google Scholar]
- 5. Anandarajah K., Kiefer P. M. Jr., Donohoe B. S., and Copley S. D. (2000) Recruitment of a double bond isomerase to serve as a reductive dehalogenase during biodegradation of pentachlorophenol. Biochemistry 39, 5303–5311 10.1021/bi9923813 [DOI] [PubMed] [Google Scholar]
- 6. Eram M. S., and Ma K. (2016) Pyruvate decarboxylase activity of the acetohydroxyacid synthase of Thermotoga maritima. Biochem. Biophys. Rep. 7, 394–399 10.1016/j.bbrep.2016.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dijkman W. P., de Gonzalo G., Mattevi A., and Fraaije M. W. (2013) Flavoprotein oxidases: classification and applications. Appl. Microbiol. Biotechnol. 97, 5177–5188 10.1007/s00253-013-4925-7 [DOI] [PubMed] [Google Scholar]
- 8. Fitzpatrick P. F. (2010) Oxidation of amines by flavoproteins. Arch. Biochem. Biophys. 493, 13–25 10.1016/j.abb.2009.07.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Civitelli R., Villareal D. T., Agnusdei D., Nardi P., Avioli L. V., and Gennari C. (1992) Dietary l-lysine and calcium metabolism in humans. Nutrition 8, 400–405 [PubMed] [Google Scholar]
- 10. Mora M. F., Giacomelli C. E., and Garcia C. D. (2009) Interaction of d-amino acid oxidase with carbon nanotubes: implications in the design of biosensors. Anal. Chem. 81, 1016–1022 10.1021/ac802068n [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Turpin F., Dallerac G., and Mothet J. P. (2012) Electrophysiological analysis of the modulation of NMDA-receptors function by d-serine and glycine in the central nervous system. Methods Mol. Biol. 794, 299–312 10.1007/978-1-61779-331-8_20 [DOI] [PubMed] [Google Scholar]
- 12. Pollegioni L., and Molla G. (2011) New biotech applications from evolved d-amino acid oxidases. Trends. Biotechnol. 29, 276–283 10.1016/j.tibtech.2011.01.010 [DOI] [PubMed] [Google Scholar]
- 13. Füller J. J., Röpke R., Krausze J., Rennhack K. E., Daniel N. P., Blankenfeldt W., Schulz S., Jahn D., and Moser J. (2016) Biosynthesis of violacein, structure and function of l-tryptophan oxidase VioA from Chromobacterium violaceum. J. Biol. Chem. 291, 20068–20084 10.1074/jbc.M116.741561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kelly S. C., O'Connell P. J., O'Sullivan C. K., and Guilbault G. G. (2000) Development of an interferent free amperometric biosensor for determination of l-lysine in food. Anal. Chim. Acta 412, 111–119 10.1016/S0003-2670(00)00767-4 [DOI] [Google Scholar]
- 15. Olschewski H., Erlenkötter A., Zaborosch C., and Chemnitius G. C. (2000) Screen-printed enzyme sensors for l-lysine determination. Enzyme Microb. Technol. 26, 537–543 10.1016/S0141-0229(99)00192-1 [DOI] [PubMed] [Google Scholar]
- 16. Chauhan N., Narang J., Sunny, and Pundir C. S. (2013) Immobilization of lysine oxidase on a gold-platinum nanoparticles modified Au electrode for detection of lysine. Enzyme Microb. Technol. 52, 265–271 10.1016/j.enzmictec.2013.01.006 [DOI] [PubMed] [Google Scholar]
- 17. Böhmer A., Müller A., Passarge M., Liebs P., Honeck H., and Müller H. G. (1989) A novel l-glutamate oxidase from Streptomyces endus. Purification and properties. Eur. J. Biochem 182, 327–332 10.1111/j.1432-1033.1989.tb14834.x [DOI] [PubMed] [Google Scholar]
- 18. Leese C., Fotheringham I., Escalettes F., Speight R., and Grogan G. (2013) Cloning, expression, characterisation and mutational analysis of l-aspartate oxidase from Pseudomonas putida. J. Mol. Catal. B Enzym. 85–86, 17–22 10.1016/j.molcatb.2012.07.008 [DOI] [Google Scholar]
- 19. Ida K., Kurabayashi M., Suguro M., Hiruma Y., Hikima T., Yamomoto M., and Suzuki H. (2008) Structural basis of proteolytic activation of l-phenylalanine oxidase from Pseudomonas sp. P-501. J. Biol. Chem. 283, 16584–16590 10.1074/jbc.M800366200 [DOI] [PubMed] [Google Scholar]
- 20. Koyama H. (1982) Purification and characterization of a novel l-phenylalanine oxidase (deaminating and decarboxylating) from Pseudomonas sp. P-501. J. Biochem. 92, 1235–1240 10.1093/oxfordjournals.jbchem.a134041 [DOI] [PubMed] [Google Scholar]
- 21. Li Lee M., Chung I., Yee Fung S., Kanthimathi M. S., and Hong Tan N. (2014) Antiproliferative activity of king cobra (Ophiophagus hannah) venom l-amino acid oxidase. Basic Clin. Pharmacol. Toxicol. 114, 336–343 10.1111/bcpt.12155 [DOI] [PubMed] [Google Scholar]
- 22. Yang H., Johnson P. M., Ko K. C., Kamio M., Germann M. W., Derby C. D., and Tai P. C. (2005) Cloning, characterization and expression of escapin, a broadly antimicrobial FAD-containing l-amino acid oxidase from ink of the sea hare Aplysia californica. J. Exp. Biol. 208, 3609–3622 10.1242/jeb.01795 [DOI] [PubMed] [Google Scholar]
- 23. Ida K., Suguro M., and Suzuki H. (2011) High resolution X-ray crystal structures of l-phenylalanine oxidase (deaminating and decarboxylating) from Pseudomonas sp. P-501. Structures of the enzyme-ligand complex and catalytic mechanism. J. Biochem. 150, 659–669 10.1093/jb/mvr103 [DOI] [PubMed] [Google Scholar]
- 24. Emanuele J. J., and Fitzpatrick P. F. (1995) Mechanistic studies of the flavoprotein tryptophan 2-monooxygenase. 2. pH and kinetic isotope effects. Biochemistry 34, 3716–3723 10.1021/bi00011a029 [DOI] [PubMed] [Google Scholar]
- 25. Sobrado P., and Fitzpatrick P. F. (2003) Analysis of the role of the active site residue Arg98 in the flavoprotein tryptophan 2-monooxygenase, a member of the l-amino oxidase family. Biochemistry 42, 13826–13832 10.1021/bi035299n [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Flashner M. I., and Massey V. (1974) Purification and properties of l-lysine monooxygenase from Pseudomonas fluorescens. J. Biol. Chem. 249, 2579–2586 [PubMed] [Google Scholar]
- 27. Takeda H., Yamamoto S., Kojima Y., and Hayaishi O. (1969) Studies on monooxygenases. I. General properties of crystalline l-lysine monooxygenase. J. Biol. Chem. 244, 2935–2941 [PubMed] [Google Scholar]
- 28. Nakazawa T., Hori K., and Hayaishi O. (1972) Studies on monooxygenases. V. Manifestation of amino acid oxidase activity by l-lysine monooxygenase. J. Biol. Chem. 247, 3439–3444 [PubMed] [Google Scholar]
- 29. Yamamoto S., Yamauchi T., and Hayaishi O. (1972) Alkylamine-dependent amino-acid oxidation by lysine monooxygenase–fragmented substrate of oxygenase. Proc. Natl. Acad. Sci. U. S. A. 69, 3723–3726 10.1073/pnas.69.12.3723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Joosten V., and van Berkel W. J. (2007) Flavoenzymes. Curr. Opin. Chem. Biol. 11, 195–202 10.1016/j.cbpa.2007.01.010 [DOI] [PubMed] [Google Scholar]
- 31. Thotsaporn K., Chenprakhon P., Sucharitakul J., Mattevi A., and Chaiyen P. (2011) Stabilization of C4a-hydroperoxyflavin in a two-component flavin-dependent monooxygenase is achieved through interactions at flavin N5 and C4a atoms. J. Biol. Chem. 286, 28170–28180 10.1074/jbc.M111.241836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chaiyen P., Fraaije M. W., and Mattevi A. (2012) The enigmatic reaction of flavins with oxygen. Trends Biochem. Sci. 37, 373–380 10.1016/j.tibs.2012.06.005 [DOI] [PubMed] [Google Scholar]
- 33. Pimviriyakul P., Thotsaporn K., Sucharitakul J., and Chaiyen P. (2017) Kinetic mechanism of the dechlorinating flavin-dependent monooxygenase HadA. J. Biol. Chem. 292, 4818–4832 10.1074/jbc.M116.774448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pimviriyakul P., and Chaiyen P. (2018) A complete bioconversion cascade for dehalogenation and denitration by bacterial flavin-dependent enzymes. J. Biol. Chem. 293, 18525–18539 10.1074/jbc.RA118.005538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Huijbers M. M., Montersino S., Westphal A. H., Tischler D., and van Berkel W. J. (2014) Flavin dependent monooxygenases. Arch. Biochem. Biophys. 544, 2–17 10.1016/j.abb.2013.12.005 [DOI] [PubMed] [Google Scholar]
- 36. Romero E., Gómez Castellanos J. R., Gadda G., Fraaije M. W., and Mattevi A. (2018) Same substrate, many reactions: oxygen activation in flavoenzymes. Chem. Rev. 118, 1742–1769 10.1021/acs.chemrev.7b00650 [DOI] [PubMed] [Google Scholar]
- 37. Chuaboon L., Wongnate T., Punthong P., Kiattisewee C., Lawan N., Hsu C. Y., Lin C. H., Bornscheuer U. T., and Chaiyen P. (2019) One-pot bioconversion of l-arabinose to l-ribulose in an enzymatic cascade. Angew. Chem. 58, 2428–2432 10.1002/anie.201814219 [DOI] [PubMed] [Google Scholar]
- 38. Matsui D., Im D. H., Sugawara A., Fukuta Y., Fushinobu S., Isobe K., and Asano Y. (2014) Mutational and crystallographic analysis of l-amino acid oxidase/monooxygenase from Pseudomonas sp. AIU 813: Interconversion between oxidase and monooxygenase activities. FEBS Open Bio 4, 220–228 10.1016/j.fob.2014.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Isobe K., Sugawara A., Domon H., Fukuta Y., and Asano Y. (2012) Purification and characterization of an l-amino acid oxidase from Pseudomonas sp. AIU 813. J. Biosci. Bioeng. 114, 257–261 10.1016/j.jbiosc.2012.04.020 [DOI] [PubMed] [Google Scholar]
- 40. Im D., Matsui D., Arakawa T., Isobe K., Asano Y., and Fushinobu S. (2018) Ligand complex structures of l-amino acid oxidase/monooxygenase from Pseudomonas sp. AIU 813 and its conformational change. FEBS Open Bio 8, 314–324 10.1002/2211-5463.12387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. van Berkel W. J., Kamerbeek N. M., and Fraaije M. W. (2006) Flavoprotein monooxygenases, a diverse class of oxidative biocatalysts. J. Biotechnol. 124, 670–689 10.1016/j.jbiotec.2006.03.044 [DOI] [PubMed] [Google Scholar]
- 42. Suadee C., Nijvipakul S., Svasti J., Entsch B., Ballou D. P., and Chaiyen P. (2007) Luciferase from Vibrio campbellii is more thermostable and binds reduced FMN better than its homologues. J. Biochem. 142, 539–552 10.1093/jb/mvm155 [DOI] [PubMed] [Google Scholar]
- 43. Sucharitakul J., Prongjit M., Haltrich D., and Chaiyen P. (2008) Detection of a C4a-hydroperoxyflavin intermediate in the reaction of a flavoprotein oxidase. Biochemistry 47, 8485–8490 10.1021/bi801039d [DOI] [PubMed] [Google Scholar]
- 44. Ruangchan N., Tongsook C., Sucharitakul J., and Chaiyen P. (2011) pH-dependent studies reveal an efficient hydroxylation mechanism of the oxygenase component of p-hydroxyphenylacetate 3-hydroxylase. J. Biol. Chem. 286, 223–233 10.1074/jbc.M110.163881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pollegioni L., Fukui K., and Massey V. (1994) Studies on the kinetic mechanism of pig kidney d-amino acid oxidase by site-directed mutagenesis of tyrosine 224 and tyrosine 228. J. Biol. Chem. 269, 31666–31673 [PubMed] [Google Scholar]
- 46. Pollegioni L., Blodig W., and Ghisla S. (1997) On the mechanism of d-amino acid oxidase. Structure/linear free energy correlations and deuterium kinetic isotope effects using substituted phenylglycines. J. Biol. Chem. 272, 4924–4934 10.1074/jbc.272.8.4924 [DOI] [PubMed] [Google Scholar]
- 47. Gaweska H. M., Taylor A. B., Hart P. J., and Fitzpatrick P. F. (2013) Structure of the flavoprotein tryptophan 2-monooxygenase, a key enzyme in the formation of galls in plants. Biochemistry 52, 2620–2626 10.1021/bi4001563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Moustafa I. M., Foster S., Lyubimov A. Y., and Vrielink A. (2006) Crystal structure of LAAO from Calloselasma rhodostoma with an l-phenylalanine substrate: insights into structure and mechanism. J. Mol. Biol. 364, 991–1002 10.1016/j.jmb.2006.09.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Binda C., Newton-Vinson P., Hubálek F., Edmondson D. E., and Mattevi A. (2002) Structure of human monoamine oxidase B, a drug target for the treatment of neurological disorders. Nat. Struct. Biol. 9, 22–26 10.1038/nsb732 [DOI] [PubMed] [Google Scholar]
- 50. Emanuele J. J., Heasley C. J., and Fitzpatrick P. F. (1995) Purification and characterization of the flavoprotein tryptophan 2-monooxygenase expressed at high levels in Escherichia coli. Arch. Biochem. Biophys. 316, 241–248 10.1006/abbi.1995.1034 [DOI] [PubMed] [Google Scholar]
- 51. Kingston N. L., Liu Y., and Calderone C. T. (2017) ORF7 from Amycolatopsis orientalis catalyzes decarboxylation of N(d)-methylarginine and amine oxidation of arginine: biosynthetic implications. Biochim. Biophys. Acta Proteins Proteom. 1865, 99–106 10.1016/j.bbapap.2016.09.01-8 [DOI] [PubMed] [Google Scholar]
- 52. Blank L. M., Ebert B. E., Buehler K., and Bühler B. (2010) Redox biocatalysis and metabolism: molecular mechanisms and metabolic network analysis. Antioxid. Redox Signal. 13, 349–394 10.1089/ars.2009.2931 [DOI] [PubMed] [Google Scholar]
- 53. Teufel R., Miyanaga A., Michaudel Q., Stull F., Louie G., Noel J. P., Baran P. S., Palfey B., and Moore B. S. (2013) Flavin-mediated dual oxidation controls an enzymatic Favorskii-type rearrangement. Nature 503, 552–556 10.1038/nature12643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Matthews A., Saleem-Batcha R., Sanders J. N., Stull F., Houk K. N., and Teufel R. (2020) Aminoperoxide adducts expand the catalytic repertoire of flavin monooxygenases. Nat. Chem. Biol. 16, 556–563 10.1038/s41589-020-0476-2 [DOI] [PubMed] [Google Scholar]
- 55. Su D., Kabir M. P., Orozco-Gonzalez Y., Gozem S., and Gadda G. (2019) Fluorescence properties of flavin semiquinone radicals in nitronate monooxygenase. Chembiochem 20, 1646–1652 10.1002/cbic.201900016 [DOI] [PubMed] [Google Scholar]
- 56. Lockridge O., Massey V., and Sullivan P. A. (1972) Mechanism of action of the flavoenzyme lactate oxidase. J. Biol. Chem. 247, 8097–8106 [PubMed] [Google Scholar]
- 57. Sutton W. B. (1957) Mechanism of action and crystallization of lactic oxidative decarboxylase from Mycobacterium phlei. J. Biol. Chem. 226, 395–405 [PubMed] [Google Scholar]
- 58. Ghisla S., and Massey V. (1980) Studies on the catalytic mechanism of lactate oxidase. Formation of enantiomeric flavin-N(5)-glycollyl adducts via carbanion intermediates. J. Biol. Chem. 255, 5688–5696 [PubMed] [Google Scholar]
- 59. Sobrado P., and Fitzpatrick P. F. (2003) Identification of Tyr413 as an active site residue in the flavoprotein tryptophan 2-monooxygenase and analysis of its contribution to catalysis. Biochemistry 42, 13833–13838 10.1021/bi035300i [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sanders S. A., Williams C. H. Jr., and Massey V. (1999) The roles of two amino acid residues in the active site of l-lactate monooxygenase. Mutation of arginine 187 to methionine and histidine 240 to glutamine. J. Biol. Chem. 274, 22289–22295 10.1074/jbc.274.32.22289 [DOI] [PubMed] [Google Scholar]
- 61. Yamauchi T., Yamamoto S., and Hayaishi O. (1975) A possible involvement of sulfhydryl groups in the conversion of lysine monooxygenase to an oxidase. J. Biol. Chem. 250, 7127–7133 [PubMed] [Google Scholar]
- 62. Kean K. M., and Karplus P. A. (2019) Structure and role for active site lid of lactate monooxygenase from Mycobacterium smegmatis. Protein Sci. 28, 135–149 10.1002/pro.3506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Asano Y., and Matsui D. (2015) l-amino acid oxidase, method for measuring l-lysine, kit and enzyme sensor. Patent US20140335553
- 64. Asano Y., and Matsui D. (August 8, 2013) Novel l-amino acid oxidase, method for measuring l-lysine, kit, and enzyme sensor. International Patent WO/2013/115180
- 65. Phillips A. T. (1986) Biosynthetic and catabolic features of amino acid metabolism in Pseudomonas. in The Biology of Pseudomonas (Sokatch J., ed) pp. 385–437, Academic Press, Inc., New York [Google Scholar]
- 66. Adkins J., Pugh S., McKenna R., and Nielsen D. R. (2012) Engineering microbial chemical factories to produce renewable “biomonomers”. Front. Microbiol. 3, 313 10.3389/fmicb.2012.00313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Chapman S. K., and Reid G. A. (1999) Flavoprotein Protocols, pp. 1–7, Humana Press, Totowa, NJ [Google Scholar]
- 68. Brünger A. T., and Karplus M. (1988) Polar hydrogen positions in proteins: empirical energy placement and neutron diffraction comparison. Proteins 4, 148–156 10.1002/prot.340040208 [DOI] [PubMed] [Google Scholar]
- 69. Dolinsky T. J., Nielsen J. E., McCammon J. A., and Baker N. A. (2004) PDB2PQR: an automated pipeline for the setup of Poisson–Boltzmann electrostatics calculations. Nucleic Acids Res. 32, W665–W667 10.1093/nar/gkh381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. MacKerell A. D., Bashford D., Bellott M., Dunbrack R. L., Evanseck J. D., Field M. J., Fischer S., Gao J., Guo H., Ha S., Joseph-McCarthy D., Kuchnir L., Kuczera K., Lau F. T. K., Mattos C., et al. (1998) All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616 10.1021/jp973084f [DOI] [PubMed] [Google Scholar]
- 71. Karplus M., and Petsko G. A. (1990) Molecular dynamics simulations in biology. Nature 347, 631–639 10.1038/347631a0 [DOI] [PubMed] [Google Scholar]
- 72. Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kalé L., and Schulten K. (2005) Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802 10.1002/jcc.20289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Isgro T., Phillips J., Sotomayor M., Villa E., Yu H., Tanner D., and Liu Y. (2012) NAMD Tutorial, University of Illinois at Urbana-Champaign, Champaign, IL [Google Scholar]
- 74. Isgro T., Phillips J., Sotomayor M., Villa E., Yu H., Tanner D., Liu Y., Wu Z., and Hardy D. (2017) NAMD Tutorial, University of Illinois at Urbana-Champaign, Champaign, IL [Google Scholar]
- 75. Pongpamorn P., Watthaisong P., Pimviriyakul P., Jaruwat A., Lawan N., Chitnumsub P., and Chaiyen P. (2019) Identification of a hotspot residue for improving the thermostability of a flavin-dependent monooxygenase. Chembiochem 20, 3020–3031 10.1002/cbic.201900413 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are included in the article and supporting information.