

Abstract
Chemokines mediate leukocyte migration and homeostasis and are key targets in inflammatory diseases including atherosclerosis, cytokine storm, and chronic autoimmune disease. Chemokine redundancy and ensuing network robustness has frustrated therapeutic development. Salivary evasins from ticks bind multiple chemokines to overcome redundancy and are effective in several preclinical disease models. Their clinical development has not progressed because of concerns regarding potential immunogenicity, parenteral delivery, and cost. Peptides mimicking protein activity can overcome the perceived limitations of therapeutic proteins. Here we show that peptides possessing multiple chemokine-binding and anti-inflammatory activities can be developed from the chemokine-binding site of an evasin. We used hydrogen–deuterium exchange MS to map the binding interface of the evasin P672 that physically interacts with C–C motif chemokine ligand (CCL) 8 and synthesized a 16-mer peptide (BK1.1) based on this interface region in evasin P672. Fluorescent polarization and native MS approaches showed that BK1.1 binds CCL8, CCL7, and CCL18 and disrupts CCL8 homodimerization. We show that a BK1.1 derivative, BK1.3, has substantially improved ability to disrupt P672 binding to CCL8, CCL2, and CCL3 in an AlphaScreen assay. Using isothermal titration calorimetry, we show that BK1.3 directly binds CCL8. BK1.3 also has substantially improved ability to inhibit CCL8, CCL7, CCL2, and CCL3 chemotactic function in vitro. We show that local as well as systemic administration of BK1.3 potently blocks inflammation in vivo. Identification and characterization of the chemokine-binding interface of evasins could thus inspire the development of novel anti-inflammatory peptides that therapeutically target the chemokine network in inflammatory diseases.
Keywords: chemokine, C–C motif chemokine ligand (CCL), chemotaxis, mass spectrometry, inflammation, protein–protein interaction, evasin, tick, immune response, innate immunity, hydrogen exchange mass spectrometry, protein structure, host–pathogen interaction, peptide interaction, peptides
The disease burden created by inflammation ranges from acute multiorgan failure in influenza or coronavirus COVID-19–induced cytokine storm (1–3), to chronic autoimmune diseases such as rheumatoid arthritis, and to inflammatory diseases such as atherosclerosis (4). Chemoattractant cytokines or chemokines are key players in cytokine storm hyperinflammation syndromes (1–3, 5), in diverse autoimmune diseases (6), and in atherosclerosis (7). Chemokines are classified as CCL, CXCL, CX3CL, or XCL based on the spacing of their N-terminal cysteine residues (8). The binding of chemokines to G protein–coupled receptors expressed on leukocytes causes their directed migration to sites of inflammation and also maintains leukocyte homeostasis (8). Although chemokines are promising therapeutic targets, clinical trials of agents that target single chemokine ligands or receptors have not been successful (9, 10). The reason for this is thought to lie, at least in part, in the apparent redundancy within the chemokine network, which creates robustness (11). Natural selection in diverse pathogens including viruses (12), helminths (13), and ticks (14) has resulted in the convergent evolution of structurally unrelated proteins that bind multiple chemokines. This phenomenon suggests that the ability to target multiple chemokines is an effective strategy to disable the chemokine network and host defense mechanisms such as inflammation. The application of such chemokine-binding proteins in diverse preclinical models of inflammation has been well-documented (15, 16). Of particular interest in the development of therapeutics that target chemokines are the evasin proteins from ticks. Three of these proteins were initially identified in seminal studies from the Proudfoot laboratory (reviewed in Ref. 16), and we and others have since identified and characterized over 40 evasins to date (17–21). Evasins fall into two classes: class A, exclusively binding CCL chemokines; and class B, exclusively binding CXCL chemokines (reviewed in Ref. 14). When administered parenterally, evasins have potent anti-inflammatory efficacy in preclinical disease models including myocardial ischemia and reperfusion injury, intestinal ischemia, colitis, acute pancreatitis, lung inflammation, arthritis, psoriasis, and graft-versus-host disease (reviewed in Ref. 16). Unfortunately, the clinical translation of evasins has not progressed, in part because of the perceived limitations of using foreign proteins as biological therapeutics such as immunogenicity, requirement for parenteral delivery, and relatively high manufacturing costs (22, 23). Peptidomimetic and peptide therapeutics developed from foreign proteins and mimicking their activity can overcome some of these limitations (24), and creating such agents from evasins is the driver of this work.
We have recently shown that a class A evasin EVA-P672 (here referred to as P672), identified from the tick Rhipicephalus pulchellus, binds several CC-class chemokines and contains a CCL8-binding region in its N terminus (20). Here we report the experimental mapping of the P672–CCL8 interface using hydrogen–deuterium exchange MS and biophysical analyses and identify a linear sequence in the N terminus of P672 that binds CCL8. Using this information, we designed a series of short synthetic peptides that demonstrate promiscuous chemokine binding and neutralization activity in vitro. One of these peptides, BK1.3, was shown to be able to block inflammatory recruitment of neutrophils, eosinophils, monocytes, and T-cells in an in vivo air-pouch model, induced by the pathogen-associated molecular pattern (PAMP), zymosan. Taken together, these experiments provide proof of concept that small biologically active peptides that target multiple chemokines and have anti-inflammatory activity can be engineered through the analysis of evasin–chemokine interactions.
Results
Hydrogen–deuterium exchange MS reveals the P672–CCL8 complex interface
We performed peptide-resolution hydrogen–deuterium exchange (HDX) MS to characterize the interaction between P672 and CCL8. HDX-MS measures the rate of exchange of protein backbone hydrogen atoms with deuterium atoms in the solvent (25). Changes in deuterium uptake between free and complexed proteins can inform on protein–protein interfaces and conformational dynamics (26). Regions that are protected from deuterium uptake upon complex formation are shielded from the solvent typically because of involvement in interprotein hydrogen-bonding networks that stabilize the complex (27). We measured the deuterium uptake of free P672, of free CCL8, and of each protein upon complex formation. After confirming satisfactory sequence mapping and coverage of each protein (100% for P672, and 96.9% for CCL8; Fig. S1), we compared the deuterium uptake of the free species with that of the P672–CCL8 complex species (5-s, 30-s, 5-min, and 60-min incubation time points; Fig. S2). The results were mapped on to a homology model of the P672–CCL8 complex (Fig. 1, A and B). Although large parts of P672 and CCL8 showed no significant changes in H/D exchange rates, we observed a significant decrease in H/D exchange in Arg18–Ser27 of CCL8 (% relative deuterium uptake (%D) ranging from −6 to −18), which lies in the N-terminal extended loop/β1 region (28), and in the N-terminal unstructured (predicted) region of P672 (Glu22–Phe32, %D up to −58%), indicating protection of these regions from solvent exposure when in complex (Fig. 1, B–D, and Fig. S2). All residues in CCL8 and most in P672 (Phe25–Cys30, Phe32) from these regions were protected at all time points (Fig. S2). Spectra of two representative P672 peptides showing a reduction in deuterium incorporation for this protected region upon complex formation are shown in Fig. 1E. An increase in relative deuterium uptake was observed for the C-terminal region of P672 (Gly87–Cys94, %D ranging from 15 to 18%), indicating higher exposure to solvent water after complex formation (Fig. 1C and Fig. S2). All HDX-MS uptake data and plots are shown in Table S1. These results indicate that the P672 (Glu22–Phe32) and CCL8 (Arg18–Ser27) regions are likely involved in P672–CCL8 complex formation. The protected regions of P672 and CCL8 overlap the binding interface predicted by the homology model of P672–CCL8 (20), suggesting that these residues are involved in protein–protein interactions. Changes in the deuterium uptake in these regions show little time-dependent change (5 s to 60 min; Fig. S2), in agreement with the tight-binding kinetics of P672–CCL8 interaction (Kd = 8.5 nm, residency time = 27 min) (20).
Figure 1.
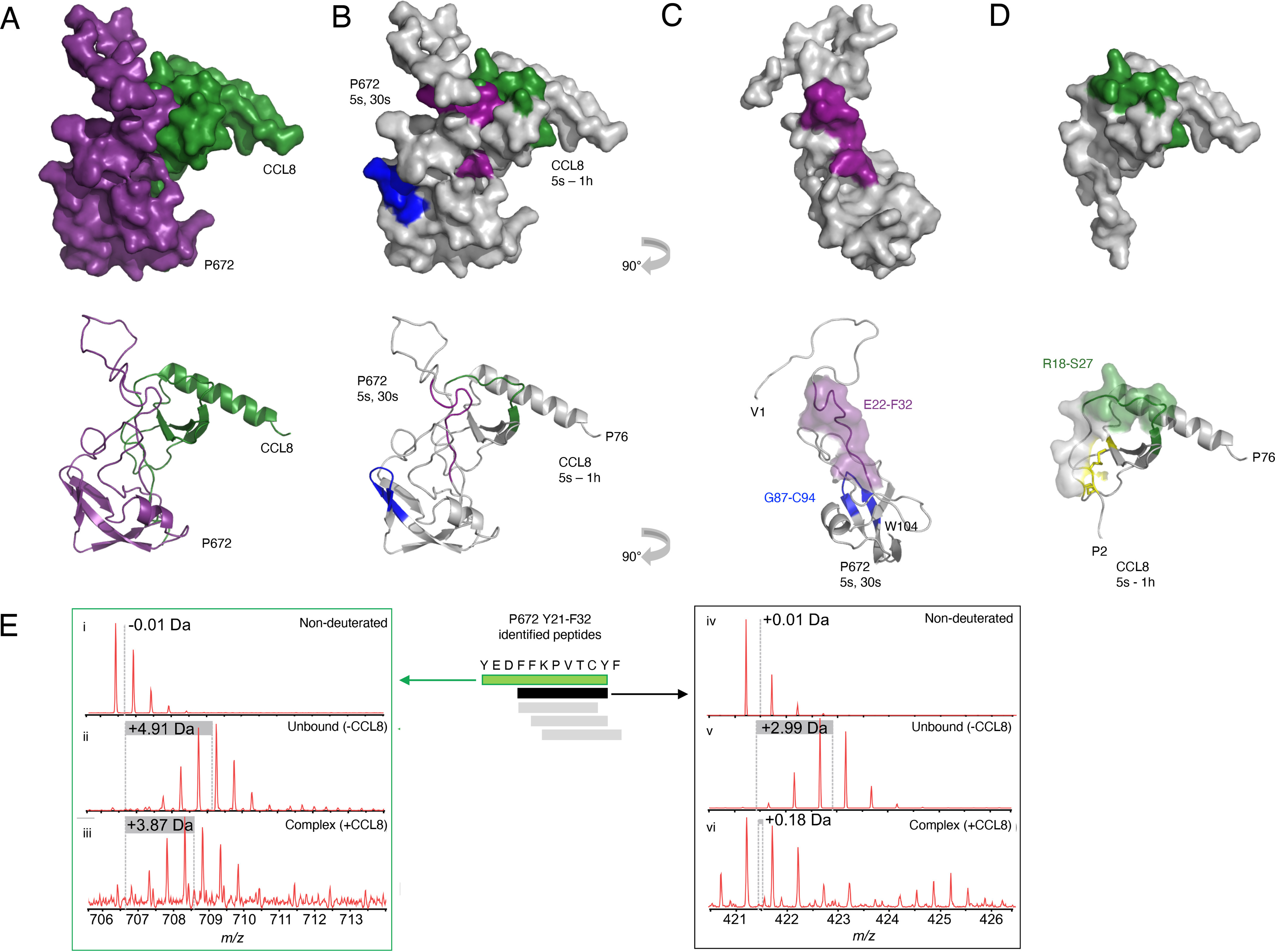
Characterization of CCL8/P672 interface by HDX-MS. A, surface representation (top panel) and ribbon diagram (bottom panel) of a homology model of P672 (purple) and CCL8 (green) complex (see “Experimental procedures” for details of model generation). P672 and CCL8 in 1:1 ratio were preincubated for 1 h, diluted in D2O-containing buffer, and quenched at different time intervals (5 s, 30 s, 5 min, and 60 min). B, surface representation (top panel) and ribbon diagram (bottom panel) of P672 and CCL8 complex at the time points indicated. Residues with statistically significant increased HDX rates (exposed residues; see Table S1) are shown in blue. Regions with statistically significant decreased HDX rates (protected residues; see Table S1) are shown in purple for P672 and in green for CCL8. All analyses were performed in triplicate. C, surface representations (top panel) and ribbon diagrams (bottom panel) of P672 (−90° rotated view along the y axis of B). Residues protected at the 5- and 30-s time points (Glu22–Phe32) are indicated in purple. Exposed residues (Gly87–Cys94) are indicated in blue. The surface of the protected residues (Glu22–Phe32) is also shown in the bottom panel. D, surface representation (top panel) and ribbon diagram (bottom panel) of CCL8, with residues protected at all time points (Arg18–Ser27) indicated in green. Disulfide bonds are indicated in yellow. The surface of the N-loop (residues Cys12–Arg24) is also shown in the bottom panel to show the overlap with protected residues. E, spectra of two representative peptides from the Tyr21–Phe32 region in P672 (green and black bars) that are protected from deuterium uptake upon complex formation. H/D exchange mass spectra were measured at t = 5 s. These peptides display reduced relative deuterium uptake upon complex formation. Other peptides from this region are indicated as gray bars. Mass spectra are shown for control nondeuterated peptides (panels i and iv), unbound P672 deuterated peptides (panels ii and v), and P672 deuterated peptides when in complex with CCL8 (panels iii and vi).
Residues Glu22–Phe32 in P672 contain a transferable CCL8-binding activity
To explore the function of P672 (Glu22–Phe32), we swapped this region with the corresponding segment of EVA1, which is a related CC-chemokine–binding evasin that does not bind CCL8 (20) (Fig. 2A). We analyzed the CCL8-binding activity of the resulting hybrid protein EVA1(P67222-32) (Fig. 2B) using biolayer interferometry and found that it bound CCL8, whereas consistent with previously reported results (20), the parental evasin EVA1 did not. Dose-titration experiments indicated that EVA1(P67222–32) bound CCL8 with modest affinity (Kd = 490 nm). Taken together with the HDX-MS analysis, these experiments confirmed that P672(Glu22–Phe32) is involved in forming protein–protein interactions with CCL8 and that this function can be transferred to another evasin.
Figure 2.

Design and biophysical analysis of an EVA1/P672 hybrid protein. A, alignment of EVA1, P672, and EVA1/P672 (EVA1 containing P672E22–E32) hybrid protein using MUSCLE algorithm. Amino acids are color-coded according to physicochemical properties: yellow, aromatic (Phe, Trp, and Try); red, acidic (Asp and Glu); blue, basic (Arg, His, and Lys); orange, nonpolar aliphatic (Ala, Gly, Ile, Leu, Met, Pro, and Val); and green, polar neutral (Cys, Asn, Gln, and Thr). Amino acids that were protected from deuterium uptake in P672 are indicated with a red box. The N-terminal acidic region is enclosed in a black box. B, biolayer interferometry sensorgram obtained when either P672, EVA1/P672, or EVA1 is loaded onto the biolayer interferometry sensor and exposed to 600 nm CCL8. Plots display wavelength shift (y axis, nm) versus time (x axis, s). C, biolayer interferometry sensorgram for EVA1/P672 hybrid binding to CCL8. Dotted lines indicate collected data, and solid lines indicate modeled data. Plots display wavelength shift (y axis, nm) versus time (x axis, s).
Development of BK1.1, a CCL8-binding peptide
Guided by the HDX-MS and swapping experiments, we tested a number of tiled peptide fragments spanning the Glu17–Phe32 region in P672 for CCL8 binding (Fig. 3A). Tyr21 and the four acidic residues N-terminal to Tyr21 were also included in this array, because both P672 and EVA1 share this region. To prevent disulfide bond formation, all peptides were synthesized with Cys30 replaced by Ala. These peptides were synthesized with N-terminal FITC to assess their chemokine binding affinities using a fluorescence polarization assay (BK1–6; Fig. 3B). The longest test peptide P672(Glu17–Phe32) was termed BK1.1FITC, and a corresponding scrambled sequence was generated as a negative control (SCRFITC). Only the full contiguous peptide (BK1.1FITC) displayed an increase in anisotropy upon incubation with CCL8 (at a concentration of 1 μm) compared with control, indicating a binding interaction. Interestingly, no changes in anisotropy were observed for Tyr21–Phe32 (BK6FITC) under the conditions tested. We next used fluorescent polarization and dose titration of CCL8 to estimate its affinity for BK1.1FITC (Fig. 3C) and found that this was relatively high (Kd =156 ± 7 nm, mean ± S.E.). To further explore the mechanism of BK1.1 binding, we performed alanine-scanning mutagenesis in which each residue of BK1.1FITC was replaced with Ala. We tested each mutant for binding to CCL8 using the fluorescent anisotropy assay to measure binding affinity. This revealed a number of key residues that contribute to CCL8 binding (Fig. 3D and Table S2). Significant differences were observed when aromatic residues Tyr and Phe (Tyr21, Tyr31, Phe25, and Phe32) were mutated. The Asp18 mutation also showed reduced affinity, supporting the peptide tiling data and indicating the importance of interactions outside of the Tyr21–Phe32 region. Notably, mutation of Pro27 completely abolished binding to CCL8, indicating that it has a key function.
Figure 3.
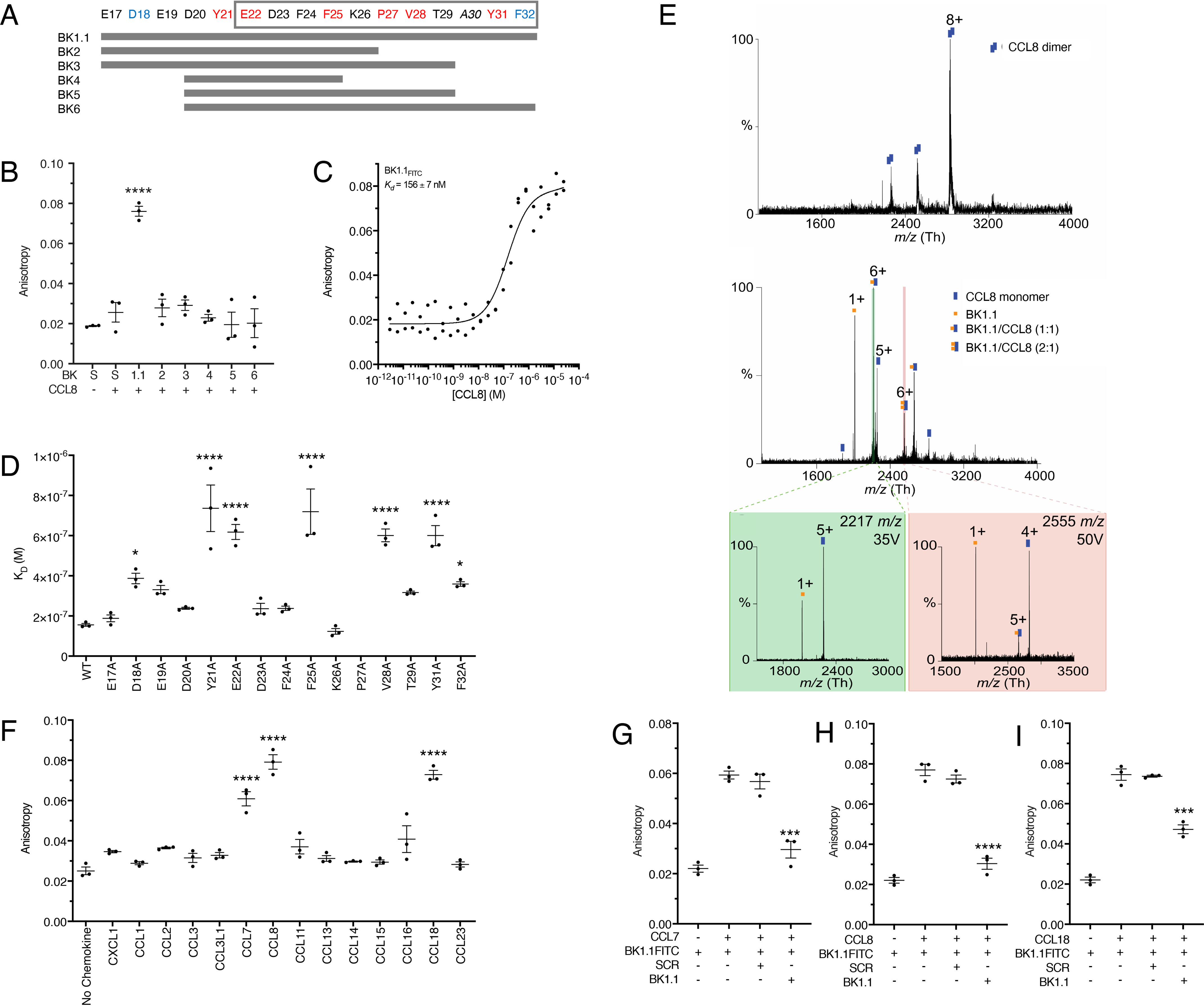
Development and biophysical analysis of P672-derived peptides. A, design of a P672 peptide tiling array to identify CCL8-binding peptides. Positions of each residue within P672 are indicated, and the gray box indicates the CCL8-binding region identified by HDX-MS. P672 residues are colored according to CCL8-binding affinity from the Ala-scanning mutagenesis (see text and below). Red indicates either complete or highly significant loss of activity (p < 0.0001), and blue indicates moderately significant loss of activity (p < 0.05). Peptides synthesized (BK1.1–BK6) are indicated as gray bars. B, fluorescent peptides BK1.1–BK6 (50 nm) were incubated with CCL8 (1 μm), and the resulting anisotropy was determined. A scrambled peptide (S, SCRFITC) was used as a negative control. The anisotropy of each peptide after being incubated with CCL8 was compared with scrambled peptide using one-way ANOVA with Sidak's correction for multiple comparisons. ****, p ≤ 0.0001. C, fluorescent polarization assay to determine binding of BK1.1FITC to CCL8. The y axis shows anisotropy, and the x axis shows the dose of CCL8. Individual data points are indicated for one data set. The curve was fitted as described under “Experimental procedures” to calculate Kd. The mean Kd and S.E. values of three independent experiments are shown. D, fluorescent polarization assay to assess effect of alanine-scanning mutagenesis of BK1.1FITC on CCL8 binding. Kd values for each BK1.1FITC Ala mutant are shown as means ± S.E. of three biological replicates, which are individually indicated as points. The data for each mutant were compared with WT BK1.1, using a one-way ANOVA with Sidak's correction for multiple comparisons. ****, p ≤ 0.0001; *, p ≤ 0.05. The mutant P27A showed no detectable binding. E, MS to assess effect of BK1.1 on CCL8. Top panel, native MS of CCL8 homodimer. Middle panel, in-solution dissociation of CCL8 dimer and further binding of CCL8 to one and two BK-1. Confirmation of CCL8/BK-1 complex by HCD gas-phase dissociation of isolated precursor ions is shown in the bottom panel: left panel, 2217 m/z corresponding to CCL8/BK1.1 (1:1); and right panel 2555 m/z corresponding to CCL8/BK1.1 (1:2). Buffers contained up to 0.5% DMSO. All analyses were performed in triplicate. F, fluorescent polarization assay to assess the binding of BK1.1FITC against a CC-chemokine panel. The data are presented as means ± S.E. of three biological replicates, which are individually indicated as points. Each biological replicate was performed as technical duplicate. CXCL1 was used as a negative control. CC-chemokine binding compared with the negative control using a one-way ANOVA with Sidak's correction for multiple comparisons. ****, p ≤ 0.0001; *, p < 0.05. G–I, fluorescence polarization competition assay for BK1.1FITC and CC-chemokine interactions. BK1.1FITC (50 nm) was incubated with the indicated chemokine (1 μm) with or without unlabeled BK1.1 or SCR (BK1.1 scrambled) peptides (50 μm) for 30 min, and the resulting anisotropy was measured. The data are presented as means ± S.E. of three biological replicates, which are individually indicated as points. Each biological replicate was performed as technical duplicate. Statistical significance of differences (SCR versus BK1.1) were calculated using a one-way ANOVA. ****, p ≤ 0.0001; ***, p ≤ 0.001.
BK1.1 disrupts CCL8 homodimerization
To further investigate the binding of BK1.1 to CCL8, we employed native MS. Under native conditions, CCL8 exists as a homodimer (Fig. 3E). However, after incubation with BK1.1, both 1:1 and 2:1 species of the BK1.1–CCL8 complex were observed, together with CCL8 monomer and BK1.1. The presence of CCL8 monomer and BK1.1 species is likely due to partial dissociation of the complex. The stoichiometry observed was supported by dissociation of these complexes using higher-energy collisional dissociation (HCD). BK1.1 can thus form a stable 1:1 complex with CCL8 and disrupt CCL8 homodimerization in line with our P672–CCL8 native MS analysis (20). Interestingly, the presence of low levels of 2:1 BK1.1–CCL8 complex indicates a possible second site of BK1.1 binding.
BK1.1 promiscuously binds three CC-class chemokines
We next screened BK1.1FITC for binding against the 13 CC-chemokines known to bind to P672 (20) (Fig. 3F). CCL7, CCL8, and CCL18 caused significant increases in anisotropy of the emitted light compared with the negative control CXCL1, a chemokine that does not bind P672 (20), suggesting a binding interaction between BK1.1FITC and these chemokines. Fluorescent polarization displacement assays with unlabeled BK1.1 confirmed its binding to CCL7, CCL8, and CCL18 (Fig. 3, G–I).
Engineering of peptides with improved potency and promiscuous CC–chemokine binding
We next explored the role of the four acidic residues N-terminal to Tyr21 and the impact of Cys-Ala mutation introduced in BK1.1. We designed two shorter peptides from P672 residues Tyr21–Phe32, with either Ala (peptide Y21F32, C30A) or Cys (peptide Y21F32) at position 30 (Fig. 4A) and compared them with BK1.1 in their ability to disrupt the interaction between P672 and CCL8 using an AlphaScreen assay. We found that all three peptides significantly disrupted the interaction, with the effects of Y21F32 and BK1.1 far exceeding that of peptide Y21F32, C30A (Fig. 4B). We found that only Y21F32 and BK1.1 disrupted the P672–CCL2 interaction and that only Y21F32 disrupted the P672–CCL3 interaction (Fig. 4, C and D). These results implied that the four acidic residues N-terminal to Tyr21 and the Cys residue were important for chemokine binding. To improve chemokine-binding affinity, we designed a series of peptides (BK1.2–BK1.5) based on BK1.1 (Fig. 4A), in which we maintained the four acidic residues N-terminal to Tyr21 and also Cys at position 30. Because cyclization is known to improve conformational stability, a cyclic version BK1.2 was designed, with Cys30 cyclized to a N-terminal Tyr residue that was introduced (29, 30). As a control, we also created a noncyclized version of this peptide, BK1.3. We assayed the binding of these peptides to CCL8 by examining their ability to disrupt the P672–CCL8 interaction using an AlphaScreen assay (Fig. 4, E and F). We found that both BK1.2 (IC50 = 729 nm) and BK1.3 (IC50 = 238 nm) had significantly improved ability to disrupt the P672–CCL8 interaction compared with BK1.1 (IC50 = 59.1 μm). To explore the mechanism of enhanced activity, we created further peptides BK1.4 (cyclized) and BK1.5 (linear) that lacked the N-terminal Tyr. BK1.4 is cyclized to Cys30 through the N-terminal Glu17 residue. We found that these modifications resulted in a significant reduction of binding activity compared with BK1.3 (Fig. 4, E and F). These results suggested that cyclization itself is not critical but instead that the N-terminal Tyr is important. Examination of the peptides by MS revealed that BK1.3 readily oxidized to form a disulfide-bonded dimer, whereas BK1.5 was monomeric (Fig. S3). To examine the binding profile of BK1 derivatives for other CC-chemokines, we tested their ability to inhibit P672 interactions with CCL2 and CCL3 using AlphaScreen (Fig. 4, G–J). Although BK1.1 did not inhibit, in line with the lack of binding observed against CCL2 and CCL3 in fluorescent polarization assays (Fig. 3F), all other BK derivatives showed good inhibition against CCL2 and weaker inhibition against CCL3. The IC50 of BK1.3 against CCL2 was 5.7 μm, and that against CCL3 was 43 μm. Isothermal titration calorimetry (ITC) (31) confirmed direct binding of BK1.3 to CCL8 (Kd = 217 nm, stoichiometry = 0.78; Fig. 4, K and L).
Figure 4.
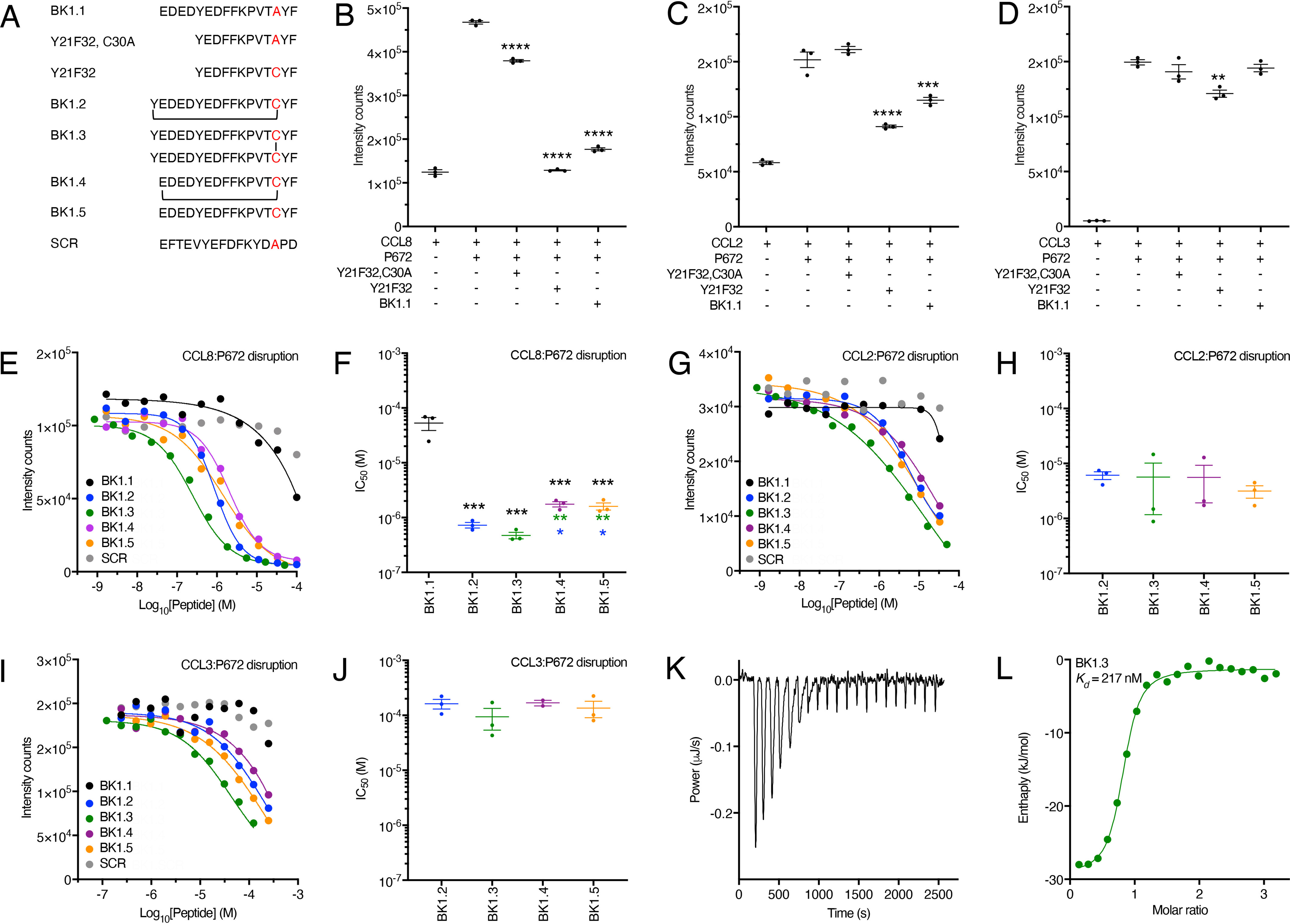
Development and biophysical analysis of the BK1.1 peptide series. A, sequences of peptides studied with disulfide bond (BK1.3) or thioether cyclization (BK1.2 and BK1.4) indicated by lines. SCR is a scrambled peptide based on the sequence of BK1.1. B–D, effect of indicated peptides at a concentration of 100 μm on a His-tagged P672-biotinylated CCL8, CCL2, or CCL3 interaction, respectively, using an AlphaScreen assay. In each panel, the y axis shows intensity counts, and the x axis shows the peptide. The data are presented as means ± S.E. of three independent experiments, shown as individual data points. Statistically significant differences (compared with chemokine + P672) using a one-way ANOVA with Sidak's multiple comparisons test are indicated by asterisks: ****, p ≤ 0.0001; ***, p ≤ 0.001; **, p ≤ 0.01. E, G, and I, representative dose-response AlphaScreen assay curves showing disruption of His-tagged P672 interactions with biotinylated human CCL8, CCL2, and CCL3, respectively, by each member of the BK1.1-derived series. The y axis shows intensity counts, and the x axis the peptide concentration (Log10 molar). The data are shown as means of two technical replicates. The curves were fitted with four parameters to estimate IC50. F, H, and J, summary IC50 values for inhibition of His-tagged P672 binding to human CCL8, CCL2, and CCL3, respectively, by each member of the BK1.1-derived series, where these could be calculated. The y axis shows IC50 (m). The data are presented as the means ± S.E. of three independent experiments, each shown as individual data points. Each independent experiment was conducted as two technical replicates. Summary IC50 values and Hill slopes are provided in Table S3. Statistically significant differences (compared with BK1.1), using a one-way ANOVA with Sidak's multiple comparisons test, are indicated by black asterisks. Statistically significant differences (pairwise comparisons of BK1.2, BK1.3, BK1.4, and BK1.5) using one-way ANOVA with Tukey's multiple comparisons test are indicated with blue asterisks (comparisons with BK1.2) or green asterisks (comparisons to BK1.3). ***, p ≤ 0.001; **, p ≤ 0.01; *, p ≤ 0.05. K, isothermal calorimetry measurements of BK1.3 binding to CCL8. The y axis shows the thermal power applied during sequential injections of BK1.3 to maintain constant temperature, and the x axis shows the time. L, binding isotherm of BK1.3 binding to CCL8. Each point represents a single injection. Binding enthalpy (kJ/mol) is shown on the y axis, and the molar ratio of BK1.3 to CCL8 is shown on the x axis. An independent single-site model (green line) was fitted to the data. The calculated thermodynamic binding parameters (± 95% confidence interval) are Kd = 217 ± 83 nm, stoichiometry n = 0.776 ± 0.024, enthalpy ΔH= −28.12 ± 1.587 kJ/mol, and entropy ΔS = 33.26 J/mol/K. Values for the blank model were 0.309 ± 0.109 ΔJ.
Engineered peptides promiscuously neutralize chemokine function
We next explored the effect of BK1.1, 1.2, and 1.3 on CCL8-, CCL7-, CCL3-, and CCL2-induced cell migration. These chemokines were chosen because P672 has previously been shown to neutralize them in analogous experiments (20). We used the acute monocytic leukemia cell line THP-1 (32) in these studies because they express CCR1, CCR2, and CCR5 (33, 34). All three receptors are activated by CCL8, CCL7, and CCL2, whereas CCL3 activates CCR1 and CCR5 (35). We performed these experiments with a single concentration of peptide (10 μm; Fig. 5, A–D). P672 (300 nm) was included as a positive control, and a scrambled version of BK1.1 (SCR) was included as a negative control. We observed that BK1.1 reduced CCL8-induced migration to background levels and had a modest but significant effect on CCL7-induced migration, consistent with its ability to bind these chemokines in the fluorescent polarization assay (Fig. 5, A and B). There was no significant effect on CCL3-induced migration (Fig. 5C). However, we found that it had a modest but significant effect in inhibiting CCL2-induced cell migration (Fig. 5D). Like BK1.1, BK1.2 and 1.3 also reduced CCL8-induced cell migration to baseline levels and had a stronger effect on CCL7- and CCL2-induced migration (Fig. 5, A, B, and D). However, unlike BK1.1 they also significantly reduced CCL3-induced cell migration (Fig. 5C). We next performed dose-titration experiments to establish the relative potencies (IC50) of the engineered peptides against CCL8 (Fig. 5, E and F). We found that BK1.1, BK1.2, and BK1.3 had IC50 values for CCL8 inhibition of 458, 19, and 6.7 nm, respectively, correlating well with the increased binding affinity. In comparison, the positive control, P672 had an IC50 of 2.6 nm. Taken together, these results indicated that the engineered peptides promiscuously neutralize different CC-class chemokines, with BK1.3 possessing the most potent activity.
Figure 5.
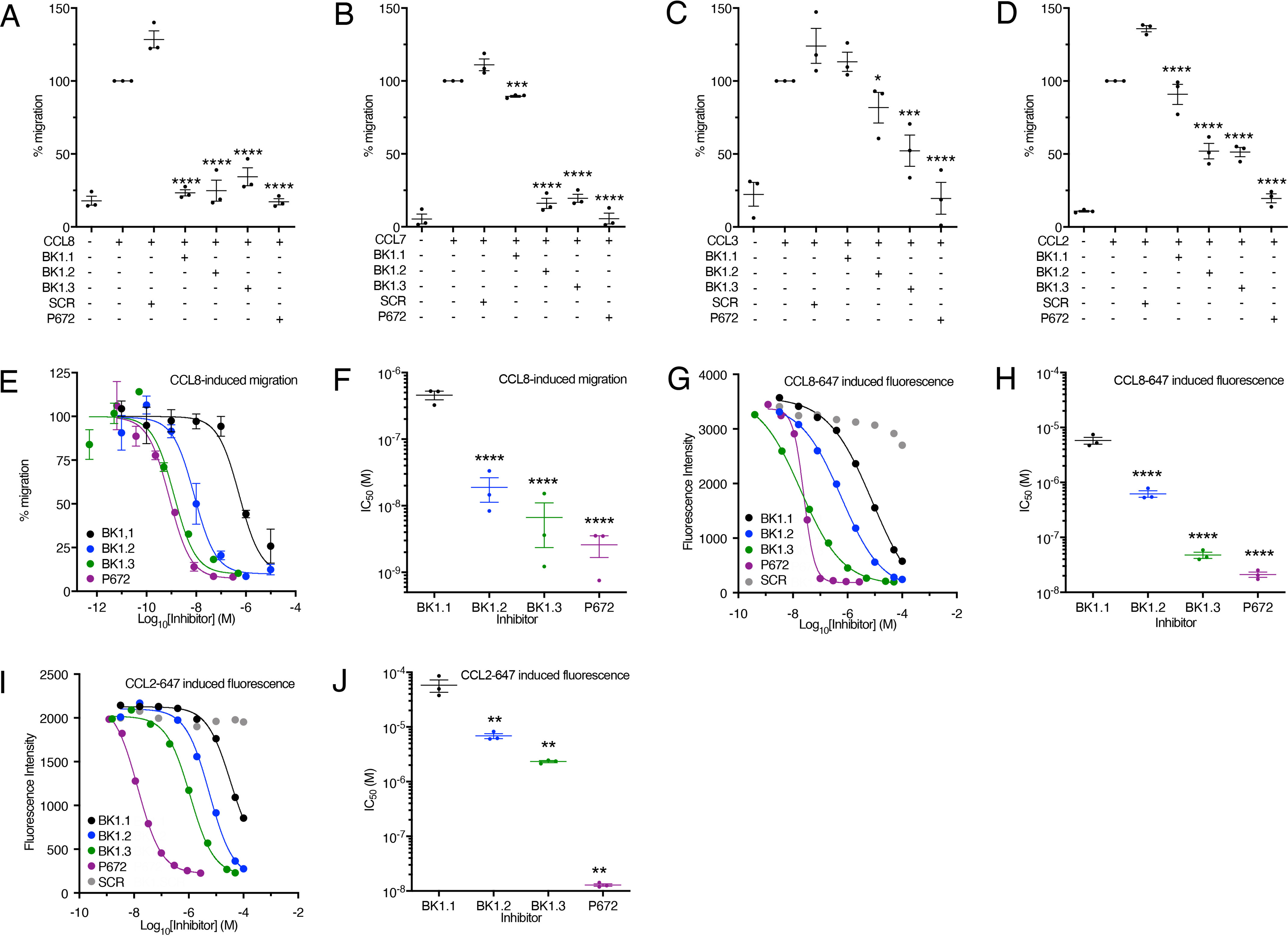
Cell-based assessment of P672-derived peptide activity. A–D, inhibition of human chemokine induced THP-1 cell migration by BK1.1, BK1.2, BK1.3, and SCR (BK1.1 scrambled, negative control) peptides, each at 10 μm, and by P672 protein (positive control, 300 nm). The y axis in each panel shows the percentage of migration of THP-1 cells normalized to chemokine alone, which was set at 100%. All experiments were performed at EC80 doses of CCL8 (5.8 nm), CCL7 (7.2 nm), CCL3 (3.5 nm), and CCL2 (1.2 nm), respectively. The data are shown as means ± S.E. of three independent biological replicates, shown as individual data points. Each biological experiment was performed as three technical replicates. Statistically significant differences (compared with SCR), using a one-way ANOVA with Sidak's correction for multiple comparisons, are indicated by asterisks: ***, p ≤ 0.0001; ***, p ≤ 0.001; **, p ≤ 0.01; *, p ≤ 0.05. E, representative dose-response curves showing inhibition of human CCL8 induced THP-1 cell migration by BK1.1 (black), BK1.2 (blue), BK1.3 (green), and SCR (scrambled, negative control, gray) peptides and by P672 protein (positive control, magenta). The y axis shows the percentage of migration of THP-1 cells normalized to CCL8 alone, which was set at 100%. The data are shown as means ± S.E. of three technical replicates. The x axis shows inhibitor concentration (Log10 molar). The curves were fitted with four parameters to estimate IC50. F, summary IC50 values for inhibition of human CCL8 induced THP-1 cell migration by BK1.1, BK1.2, BK1.3, and P672 protein. The y axis shows IC50 (m). The data are shown as means ± S.E. of three biological replicates. Summary IC50 values and Hill slopes are provided in Table S3. Statistically significant differences (compared with BK1.1) using a one-way ANOVA with Sidak's correction for multiple comparisons, are indicated by asterisks: ****, p ≤ 0.0001; ***, p ≤ 0.001; **, p ≤ 0.01; *, p ≤ 0.05. G and I, representative dose-response curves showing inhibition of human CCL8-647 (G) and human CCL2-647 (I) induced THP-1 cell fluorescence by BK1.1 (black), BK1.2 (blue), BK1.3 (green), and SCR (scrambled, negative control, gray) peptides and by P672 protein (positive control, magenta). The y axis shows fluorescence (arbitrary units). The data are shown as means of two technical replicates. The x axis shows inhibitor concentration (Log10 molar). The curves were fitted with four parameters to estimate IC50. H and J, summary IC50 values for inhibition of human CCL8-647–induced (H) or CCL2-647–induced (I) THP-1 cell fluorescence by BK1.1, BK1.2, BK1.3, and P672 protein. The y axis shows IC50 (m). The data are shown as means ± S.E. of three biological replicates, shown as individual data points. Each biological experiment was conducted as two technical replicates. Statistically significant differences (compared with BK1.1) using a one-way ANOVA with Sidak's correction for multiple comparisons, are indicated by asterisks: ****, p ≤ 0.0001; ***, p ≤ 0.001; **, p ≤ 0.01; *, p ≤ 0.05.
Engineered peptides prevent cellular chemokine binding
To explore the effect of BK1.1 and derivatives on chemokine ligand–cell interactions, we developed a fluorescent-chemokine cell-binding assay. Fluorescent-chemokine (conjugated to Alexa Fluor 647) binding to THP-1 cells results in an increase in the cellular fluorescence intensity, which is quantitatively measured using flow cytometry. In dose-response assays, we found that increasing doses of peptide suppressed CCL8–647– and CCL2–647–induced cellular fluorescence (Fig. 5, G–J). IC50 values for BK1.1, BK1.2, and BK1.3 against CCL8–647 were found to be 5.8 μm, 630 nm, and 47 nm respectively, whereas P672 had an IC50 of 21 nm (Fig. 5, G and H). In similar assays, IC50 values for BK1.1, BK1.2, and BK1.3 against CCL2-647 were found to be 45, 6.3, and 2.2 μm, respectively, whereas P672 had an IC50 of 21 nm (Fig. 5, I and J). Taken together, these results indicate that the engineered peptides not only bind chemokines promiscuously but neutralize their chemotactic function by preventing them from binding to cells.
Engineered peptide BK1.3 has in vivo anti-inflammatory activity
The above results suggested that the chemokine-neutralizing properties of the engineered peptides may translate into anti-inflammatory activity in vivo. To study this, we tested the lead peptide BK1.3 in a mouse short-term inflammation model. In this model, zymosan, a yeast cell wall–derived PAMP, activates cytokine and chemokine production and leukocyte infiltration when injected into an artificially created subcutaneous air pouch (36, 37). Characterization of this model showed that Ccl9 is expressed at a high basal level but is not induced by zymosan. Ccl2, 5, 11, 12, 20, 22, and 24 and Cxcl1, 2, 4, 5, 11, 13, and 16 are expressed (>3-fold) at 4 h following zymosan, and Ccl2, 5, and 12 and Cxcl2, 4, 13, and 16 are expressed (>3-fold) at 24 h (Fig. S4). We injected BK1.3 and control SCR peptides and the positive control P672 directly into the air-pouch at 0 and 9 h following zymosan injection. We characterized the air-pouch exudate using flow cytometry at 24 h after zymosan injection to assess the severity and nature of inflammation. Both BK1.3 and P672 showed a strong and significant reduction in the number of neutrophils, eosinophils, monocytes, and T-cells recruited to the air pouch (Fig. 6, A–F). We next determined whether systemic administration of BK1.3 peptide would have anti-inflammatory activity. We injected BK1.3 and control SCR peptides and the positive control P672 intraperitoneally at 0 and 9 h following zymosan injection, and characterized the air-pouch exudate at 24 h after zymosan injection as before. Again, both BK1.3 and P672 showed a substantial and significant reduction in the number of neutrophils, eosinophils, monocytes, and T-cells recruited to the air pouch (Fig. 6, G–L and Fig. S5). These results show that the engineered peptide BK1.3 has in vivo local and systemic anti-inflammatory activity.
Figure 6.
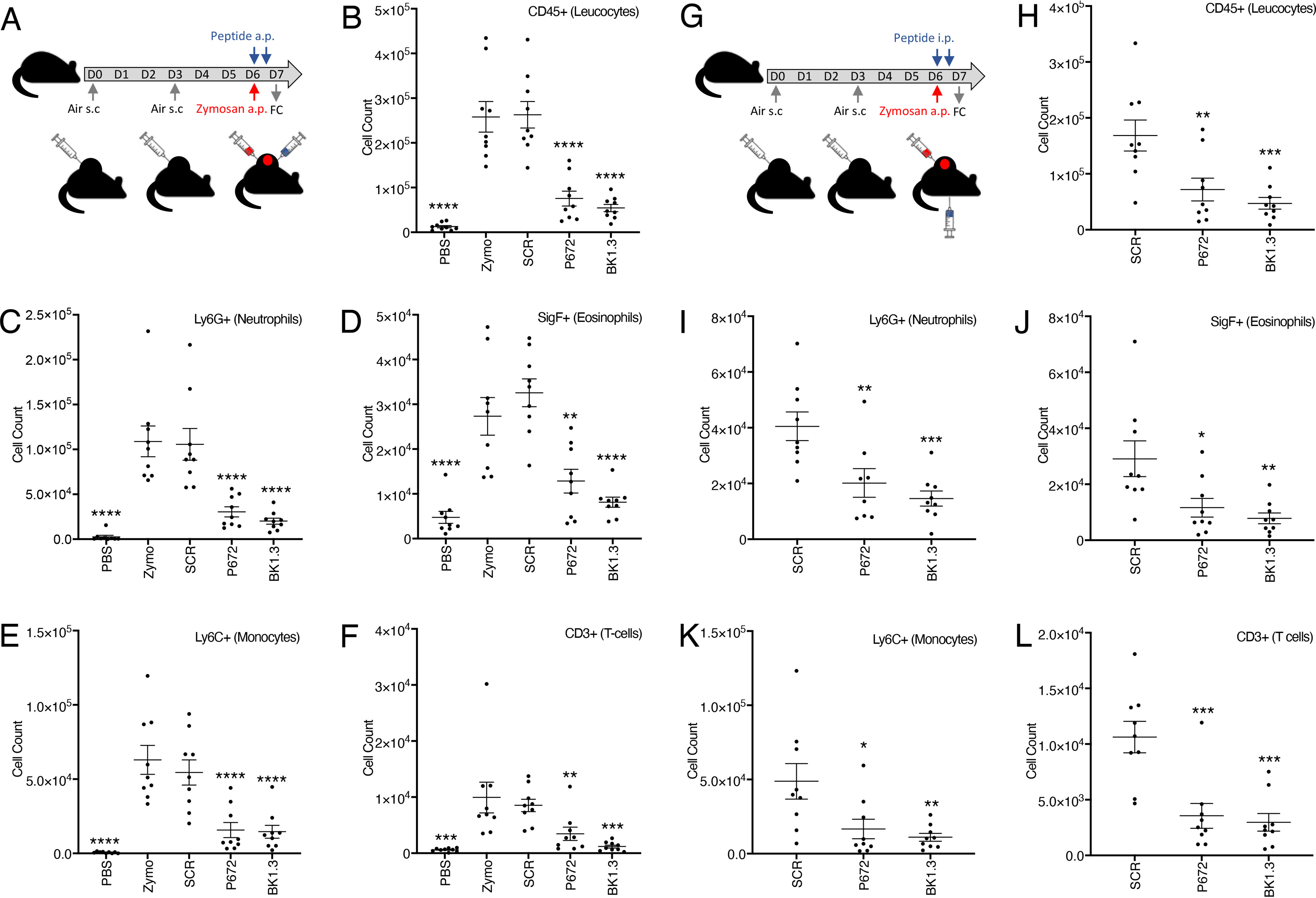
Assessment of anti-inflammatory activity of locally or systemically administered peptide in a mouse dorsal air-pouch model. A, experimental design to assess efficacy of locally administered peptide. A dorsal air pouch (a.p.) was created by subcutaneous (s.c.) injection of air on days 0 and 3. Zymosan (red) or PBS (control) was injected into the air pouch on day 6. Peptide or protein (blue) was injected into the air pouch on day 6 at the time of zymosan injection and repeated 9 h later. Air-pouch exudate was collected and analyzed on day 7 by flow cytometry (FC). Nine mice were studied in each of five study arms: PBS alone (PBS), zymosan (zymo), zymosan + scrambled peptide (SCR), zymosan + P672 (P672), and zymosan + BK1.3 (BK1.3). B–F, summary data for flow cytometry analysis for locally administered peptide. The y axis shows cell counts of total leukocytes (B), neutrophils (C), eosinophils (D), monocytes (E), and T-cells (F). The data are presented for each arm as means ± S.E. and with individual data points. Statistically significant differences (compared with zymosan) using a one-way ANOVA with Dunnett's correction for multiple comparisons, are indicated by asterisks: ****, p ≤ 0.0001; ***, p ≤ 0.001; **, p ≤ 0.01; *, p ≤ 0.05. G, experimental design to assess the efficacy of intraperitoneally administered peptide. This is identical to that used for locally administered peptide (above) except that peptide or protein was administered intraperitoneally. Nine mice were studied in each of three study arms: zymosan + SCR (SCR), zymosan + P672 (P672), and zymosan + BK1.3 (BK1.3). H and I, summary data for flow cytometry analysis for intraperitoneally administered peptide. The y axis shows cell counts of total leukocytes (H), neutrophils (I), eosinophils (J), monocytes (K), and T-cells (L). The data are presented for each arm as means ± S.E. and with individual data points. Statistically significant differences (compared with SCR) using a one-way ANOVA with Dunnett's correction for multiple comparisons, are indicated by asterisks: ****, p ≤ 0.0001; ***, p ≤ 0.001; **, p ≤ 0.01; *, p ≤ 0.05.
Discussion
Efforts to develop peptide or peptidomimetic agents that bind and inhibit multiple chemokines have to date been based on the sequences of chemokine-binding regions of receptors (38–40) and the unbiased identification by phage display of peptides that bind anti-receptor antibodies (41). These approaches have resulted in the identification of peptides that, where reported, bind one or more chemokines with relatively low (micromolar to millimolar) affinity. Individual peptides designed from the CCL5 heterodimer interface have been developed that efficiently disrupt heterodimerization of CCL5 with CCL17, CXCL4, and CXCL12, but are, however, specific for the heterodimer pair (42), and lack ability to target chemokines promiscuously. We have developed an alternative approach that starts with the identification of promiscuous chemokine-binding proteins that have evolved in parasitic organisms to evade the host chemokine network. We followed these initial discoveries by mapping the chemokine-binding segment of one of these proteins and then designing small peptides based on the mapped segment that not only promiscuously bind chemokines with relatively high (nanomolar) affinity but also have anti-inflammatory activity in vivo.
In this study we used HDX-MS and identified a 11-residue region (Glu22–Phe32) of P672 that was protected from deuterium uptake upon complexing with CCL8. Swapping this region into EVA1, an evasin that does not bind CCL8, transferred CCL8-binding activity to the hybrid protein. These results indicate that this 11-residue region binds CCL8. Whereas the structural modeling informed the potential protein–protein interaction interface, the HDX-MS provided experimental validation and confidence that accelerated the discovery process. The HDX-MS result also indicated that CCL8 residues Arg18–Ser27, which overlap the N-terminal loop (C12-R24) (28), interact with P672. A key function of the N-terminal loop of CC chemokines is receptor binding, and it is targeted by several pathogenic chemokine-binding proteins (43). For example, the viral chemokine-binding protein VV-35kDa targets Lys19 and Arg24 of CCL2 (44), and the viral chemokine-binding protein vCCI targets Arg18 and Arg24 of CCL2 (45). This common mechanism suggests the convergent evolution of these proteins to target the residues found in this region. The binding of P672 to this region would competitively prevent CCL8 binding to its receptor, explaining how CCL8 function is neutralized. The N-terminal loop of CCL8 and other CC chemokines is also part of the homodimerization interface (28, 43), and binding to this loop explains the prevention of CCL8 dimerization by P672 reported previously (20).
To develop chemokine-binding peptides based on the 11-residue segment identified by HDX-MS, we initially screened a tiled array covering this segment. We found that addition of four acidic N-terminal residues and Tyr21 was necessary to be able to detect binding under these conditions, suggesting that these acidic residues may be needed for increased affinity or that the shorter peptides were sterically hindered from binding by the FITC moiety. Alanine-scanning mutagenesis of the 16-residue peptide BK1.1 indicated that binding to CCL8 was mediated by Tyr and Phe residues and also by the acidic residues at the N terminus. Notably, Tyr and Phe are both found in protein interaction “hot spots” (46, 47), and complementarity in surface charge mediated by acidic residues can modulate protein interactions (48, 49). A notable finding was that the Pro residue is critical for binding. Pro residues are found in turns (50) and can undergo cis-trans–isomerization (51), making it likely that the Pro residue is of structural importance for BK1.1. We observed that BK1.1 prevents CCL8 homodimerization, suggesting that it likely employs a similar mechanism as P672 in binding CCL8, i.e. to the N-loop region. The fluorescent polarization studies reported indicate that BK1.1 also binds the chemokines CCL7 and CCL18, but not several others. The molecular mechanism of promiscuous chemokine binding by this short peptide is unclear at present and will require structural approaches for a more complete understanding.
Given the role of Pro in protein conformation, we decided to employ cyclization as a strategy for restricting conformational flexibility. A surprising finding was that the addition of an N-terminal Tyr residue led to enhanced potency. This finding may be explained by the observation that Tyr residues are found in protein interaction hot spots (46), and indeed Tyr and sulfo-Tyr are used by chemokine receptors to target the chemokine N-loop region (52). The role of the reintroduced Cys30 is supported by BK1.5, which differs by a single residue compared with BK1.1, and has marked improvement in affinity. The substantial enhancement of activity of BK1.3 thus likely arises from addition of Tyr and reintroduction of Cys30. In addition, it is likely that the unpredicted formation of a Cys-linked dimer in BK1.3 enhances the functional affinity or avidity of the molecule. The isothermal calorimetry experiment assessing binding of BK1.3 to CCL8 indicated an unusual stoichiometry (n = 0.78). BK1.3 is a dimer with two presumed CCL8-binding sites, and after the first binding event, we assume that the second site might be partially sterically hindered and not effectively bind to a second molecule of CCL8. This could explain how the estimated BK1.3:CCL8 stoichiometry (n = 0.78) lies between 1:1 (n = 1) and 2:1 (n = 0.5).
Peptide cyclization did not appear to enhance affinity, as evidenced by the lack of improvement of BK1.4 compared with BK1.5 or BK1.2 compared with BK1.3. This may be, in part, due to nonoptimized cyclization points and/or forced constraint. The serendipitous discoveries reported here—the addition of N-terminal Tyr and the dimerization consequent to the presence of an unpaired Cys residue—may be exploited in the rational design of other chemokine N-loop–binding peptides.
The improvement in binding to CCL8 observed in the BK1.1–BK1.3 peptide series, as well as their ability to inhibit P672–CC-chemokine interactions (CCL8, CCL2, and CCL3), correlated with increased chemokine neutralization potency and promiscuity. In cell-based chemotaxis assays, we found that the improvement in binding affinity for CCL8 translated into increased potency for inhibiting CCL8-induced cell migration, as evidenced by the reduced IC50. In addition to neutralizing CCL8 and CCL7, which was predicted by the BK1.1 fluorescent polarization binding study, the peptides BK1.2 and BK1.3 were also able to neutralize CCL2- and CCL3-induced chemotaxis. The inhibition of chemokine binding to cells indicates that the mechanism of neutralization is the prevention of chemokine binding to the cells, likely by preventing chemokine–receptor interactions.
A critical step in the clinical translation of novel anti-inflammatory therapeutics is the demonstration of efficacy in vivo, using a model where many components of the immune-inflammatory network are activated. We used a short-term inflammation model using the well-characterized PAMP, zymosan, which activates TLR2 signaling (53), and results in the production of cytokines, chemokines, and complement (36, 37). Our data indicate that zymosan-induced inflammation is significantly inhibited by both local and systemic administration of BK1.3. It is likely that the in vivo mechanism of action of BK1.3 includes the inhibition of CC-class chemokines, which not only are chemoattractants for leukocyte recruitment but also heterodimerize and synergize with certain CXC-class chemokines (42).
In conclusion, we have elucidated the molecular mechanism for the interaction between the tick salivary protein P672 and a target chemokine, CCL8, and have used the information to design promiscuous CC-chemokine–binding peptides that bind with high affinity, neutralize chemokine action by preventing receptor binding, and have anti-inflammatory activity in vivo. Our work indicates that peptides with promiscuous chemokine-binding and anti-inflammatory activity can be developed by studying evasin–chemokine interactions. Such peptides could provide a route to the development of new anti-inflammatory therapeutics that have relevance not only to chronic diseases such as atherosclerosis and rheumatoid arthritis but also to acute illnesses such as influenza or COVID-19–induced cytokine storm.
Experimental procedures
Reagents
All chemokines, unless otherwise stated, were purchased from Peprotech. Fluorescent chemokines were purchased from Almac. THP-1 cells (ECACC 88081201) were maintained in RPMI 1640 medium supplemented with 10% fetal calf serum and 4 mm l-glutamine. The cultures were maintained between 3 × 105 and 1 × 106 cells/ml in a 37 °C incubator with 5% CO2. HEK 293F cells (Thermo Fisher) were maintained between 3 × 105 and 1 × 106 cells/ml in a 37 °C incubator with 8% CO2 and 130 rpm agitation in FreeStyleTM 293 expression medium.
Plasmids
Evasins were cloned in the expression vector pHLSec (54). P672 (N-terminal His8–StrepII tag) expression vector and EVA1 (C-terminal StrepII–His8 tag) have been described previously (20). The expression vector EVA1(P67221-32) was constructed using PCR and infusion cloning as described (17), and has a N-terminal His8–StrepII tag. Plasmid sequences were confirmed by Sanger sequencing (Source Bioscience). The CCL8 expression plasmid in vector pNIC-BIO3 has been described previously (20).
Protein expression
Evasin proteins were expressed as described previously using a mammalian expression system (20). Recombinant CCL8 was expressed as described previously as a small ubiquitin-like modifier fusion protein from Escherichia coli RosettaGamiTM 2 (DE3) cells (Novagen) (20).
Hydrogen–deuterium exchange analysis
Working solutions of CCL8 and P672 were prepared at a concentration of 35 μm in 50 mm ammonium bicarbonate buffer, pH 6.5. For estimation of HDX in the heterodimer state, solutions of CCL8 and P672 were mixed in a (1:1) ratio to reach a final concentration of 17.5 μm and incubated at 4 °C for 1 h (20). For estimation of HDX in the unbound state, working solution were diluted to 17.5 μm with 50 mm ammonium bicarbonate, pH 6.5. Aliquots of 4.3 μl of heterodimer or unbound proteins were mixed with 48.2 μl of D2O containing 50 mm ammonium bicarbonate buffer adjusted to pH 6.5 with DCl (final content of D2O of 91.8%) and incubated for 5 s, 30 s, 5 min, and 60 min at room temperature. HDX was quenched by adding 22.5 μl of 10% formic acid to reach a final volume of 75 μl and a pH level of 2.5, corresponding to a final concentration of 1 μm. The samples were then rapidly flash frozen in liquid nitrogen and stored at −80 °C for up to 5 days before analysis.
An Acquity M class ultra-high-performance liquid chromatographer with a nanoAcquity HDX manager coupled to a Synapt G2-Si TOF mass spectrometer (Waters) was used and controlled using the MassLynx 4.1 software. The samples were loaded at 200 μl/min into an Enzymate pepsin column (2.1 mm × 30 mm, 5 μm particle size) in which the proteins were quickly digested at 20 °C. The peptides were then captured for 2 min into a BEH C18 trap column (300 μm × 30 mm, 1.7-μm particle size) at 0 °C and then separated in a BEH C18 analytical column (2.1 mm × 50 mm, 1.7-μm particle size) at 40 μl/min and 0 °C under a 12-min linear gradient from 4 to 85% of acetonitrile with 0.1% formic acid. The MSE approach was used for peptide mapping of nondeuterated proteins with trap collision energies of 15–35 V. Deuterated samples were analyzed in scan mode only. Source parameters included: cone voltage, 30 V; capillary voltage, 2.8 KV; source temperature, 80 °C; desolvation temperature, 150 °C; gas cone flow rate, 80 liter/h; and desolvation gas, 250 liter/h.
The ProteinLynx Global Server 3.0.2 software was used for peptide mapping. The spectra were searched against a custom database containing the protein sequence of interest, requiring a nonspecific digestion enzyme and allowing for variable modifications (i.e. N terminus pyroglutamic acid from glutamine and deamidation or N-acetylhexosamine of asparagine present in a NX(S/T) motif). Peptide identification required at least three fragment ion matches, the peptide presence in four of five replicates, a retention time relative standard deviation of ≤5%, a precursor ion mass tolerance of 10 ppm, and peptide maximum length of 30 residues. Relative deuterium uptake percentages at the peptide level were estimated using Dynamix 3.0 as the difference between the uptake (Da) observed for the complex species and the free species divided by the maximum possible uptake of the peptide. Manual check of peptide retention time, charge state, and possible peak overlap were also performed. Statistical analysis included a Student's t test and HDX rate differences of ≥5%, with a p value of ≤0.05 considered significant. Residues with statistically significant increased or decreased HDX rates (see Table S1; % relative uptake) were mapped on to a homology model (see below) of the P672–CCL8 complex. Note that in the case of overlapping peptides, Dynamix 3.0 displays the percentage of relative uptake for any given residue as the percentage of relative uptake of the shortest peptide. Additionally, in the particular case of overlapping peptides of equal length, the percentage of relative uptake refers to that of the peptide in which the residue is closest to the peptide C terminus.
Homology modeling
The previously reported P672–CCL8 homology model (20), which was generated using the EVA1:CCL3 complex 3FPU (56) as template, was modified by replacing the CCL8 (homology modeled) structure with the CCL8 X-ray crystal structure (Protein Data Bank entry 1ESR) (28), using the align function in PyMOL 2.3.4.
Biolayer interferometry
This was carried out as described previously using an OctetRed® system (17). Briefly, affinity determination was evaluated with chemokine concentrations typically ranging from 300 to 0.4 nm, using a noninteracting reference protein to allow for nonspecific binding to the sensor. We used ForteBio 9 data analysis software to process the data and calculate association (kon), dissociation (koff), and affinity (Kd) constants. The data with poor curve fits (R2 < 0.9) were excluded. All biolayer interferometry experiments were performed at least three times.
Fluorescent peptides
All fluorescent peptides and scrambled (SCR) peptide were purchased from GL Biochem (Shanghai, China) and were synthesized using standard Fmoc solid-phase synthesis to give peptides with a C-terminal amide. The scrambled peptide sequence EFTEVYEFDFKYDAPD is based on BK1.1. They were all deemed to be >90% pure by HPLC analysis and verified by LC–MS. The peptides were dissolved in DMSO, and the concentration was determined using NMR with TSP as an internal standard (57). All peptides were analyzed using a Bruker Microflex LRF MALDI-TOF mass spectrometer.
Peptide synthesis in-house
Amino acids were purchased from CEM. Peptides were purified by HPLC using a Waters SFO system with a Kinetex® 5-mm EVO C18 100 Å (150 × 21.2 mm) column. All peptides were synthesized with a C-terminal amide on a 0.05-mmol scale using standard Fmoc protection chemistry on a CEM Liberty Blue automated peptide synthesizer. Detailed methods are provided in the supporting information.
Fluorescence polarization assays
Fluorescence polarization (FP) assays were performed using a Clariostar (BMG Tech) plate reader with the supplied FITC excitation and emission filters using 96-well half area plates (Corning). The buffer used (FP assay buffer) was 50 mm HEPES, 150 mm NaCl, 0.1% BSA, 0.002% Tween 20, 0.2% DMSO, pH 7.4, and the final volume in each well was 30 μl. Polarization was converted to anisotropy using the following equation: A = (2 × P)/(3 – P), where P is polarization, and A is anisotropy. For each peptide tested, the gain was set to 35 mP and adjusted to a well containing fluorescent peptide only. The polarization of the emitted light in the FITC emission channel was then determined. Experiments were performed as two technical and three biological replicates. Screening of P672 peptide fragments was achieved through incubation of each peptide (50 nm) with 1 μm CCL8 (Peprotech) for 30 min in FP assay buffer, and the resulting anisotropy of the emitted light was determined as above. The chemokine cross-binding screen was performed by incubating 1 μm chemokine (Peprotech) with 50 nm BK1.1FITC for half an hour in FP assay buffer, and the resulting anisotropy of the emitted light was determined as above. To monitor the binding of BK1.1FITC Ala mutants to CCL8, 50 nm labeled peptide was incubated with varying concentrations of recombinant CCL8 (final concentration, 0–25 μm) in 30 μl of FP assay buffer for 30 min, and the resulting anisotropy of the emitted light was determined as above. The anisotropy was plotted as a function of CCL8 concentration and fitted to the equation: Y = Bmax × X/(Kd + X) + NS × X + background, where Y is the measured anisotropy, X is the concentration of CCL8 added, Bmax is the maximum binding, Kd is the equilibrium dissociation constant, NS is the slope of the nonlinear regression, and “background” is the anisotropy when no CCL8 is present, in GraphPad Prism. Displacement assays were carried out with CCL7, CCL8, and CCL18 (Peprotech, 1 μm). The chemokines were incubated with BK1.1 (50 μm) or SCR (50 μm) and BK1.1FITC (50 nm) for 30 min in FP buffer, and the resulting anisotropy of the emitted light was determined as above. For all FP assays, the experiments were carried out as two technical and three biological replicates.
Native MS analyses
The samples were analyzed using a modified Q-Exactive mass spectrometer (Thermo Fisher Scientific) for high-mass range measurements (58). CCL8 was buffer exchanged into 200 mm ammonium acetate solution (pH 6.5). BK1.1 obtained in DMSO was then added to the CCL8 homodimer solution in a 1:1 (CCL8 monomer:BK1.1) ratio. In all cases no more than 0.5% DMSO was present in the final mixture, and a control sample of CCL8 homodimer containing 0.5% DMSO was also analyzed. Instrumental parameters were set to a capillary voltage of 1.2 KV, a source temperature of 50 °C, and 60 V of source-induced dissociation. Gas-phase dissociation was carried out by applying 35 and 55 V of HCD to the most intense charge state after isolation (25 m/z window). Spectra were acquired using a mass resolution of 60,000 for both precursor and dissociated product ions. All measurements were done in triplicate.
AlphaScreen assay
The AlphaScreen® Histidine detection kit was purchased from PerkinElmer (6760619M lot 2457886), and the assay was set up in white-bottomed Proxiplate™ 384 Plus microplates (PerkinElmer) following the manufacturer's instructions. The assay buffer used was 50 mm HEPES, 150 mm NaCl, 0.1% BSA, 0.01% Tween 20, 1% DMSO, pH 7.5, and the final volume in each well was 20 μl. Briefly, biotinylated chemokine (recombinant; final concentrations, 1.25 nm (CCL8, produced in-house), 5 nm (CCL2, Almac), and 2.5 nm (CCL3, Almac) was preincubated at room temperature for 15 min with different concentrations of each peptide. His-tagged P672 (final concentrations, 2.5 nm (CCL8), 5 nm (CCL2), and 1.25 nm (CCL3)) was then added to each well, and the plate was incubated at room temperature for 30 min. Finally, acceptor and donor beads were added as a 1:1 suspension in buffer to each well, and the plate was further incubated at room temperature for 1 h. The data were obtained by reading the plate using a Pherastar FSX plate reader (excitation, 680 nm; emission, 570 nm) and was analyzed using GraphPad Prism.
Isothermal calorimetry
The experiments were carried out on a TA Instruments Affinity ITC with low volume cells at 25 °C and stirring at 250 rpm using a buffer system of 50 mm HEPES, 150 mm NaCl, pH 7.5. Twenty sequential 2-μl injections of BK1.3 (125 μm) into CCL8 (10 μm) were used to generate a binding isotherm for the interaction, which was fitted to the independent (single-site) model using NanoAnalyze (version 3.11.0).
Fluorescent chemokine/receptor blocking assay
CCL8-647 (final concentration, 2.5 nm) or CCL2-657 (final concentration, 1.2 nm) was incubated for 30 min with varying doses of peptide (final concentration, 0–100 μm) in 50 μl of assay buffer (RPMI 1640 + l-glutamine (4 mm) + 10% heat-treated fetal bovine serum + 0.2% DMSO) at 37 °C. This mixture was then added to 50,000 THP-1 cells in a 96-well v-bottomed plated to give a final volume of 100 μl, and everything was incubated together for 30 min more at 37 °C. Following this time, the plate was centrifuged, the supernatant was flicked off, and the cells were resuspended in 150 μl of ice-cold PBSA (PBS + 0.1% BSA). This was repeated twice more, and the cells were finally resuspended in 150 μl of ice-cold PBSA. The median fluorescence intensity of 10,000 cells on the RL-1 channel was determined using an ATTUNE flow cytometer and plotted as function of peptide concentration and the data fitted to an inhibitor-response curve with four parameters using GraphPad Prism. The experiments were performed as two technical and three biological replicates.
THP-1 cell migration assays
THP-1 monocyte cell migration assays were carried out as described (17). IC50 was calculated by fitting an inhibitor response curve with four parameters in GraphPad Prism. The experiments were performed as three technical and three biological replicates.
Subcutaneous dorsal air pouch model
C57BL/6J male mice (25-30 g, 8–10 weeks old) were obtained from Charles River. Air pouches were established at the dorsal side of the mice as described (59). Detailed methods are provided in the supporting information. All animal procedures were approved by the UK Home Office and carried out in accordance with the UK Animals (Scientific Procedures) Act 1986, under project license PPL P973A60F5.
Statistical analysis
All statistical analyses were performed using GraphPad Prism 8. The statistical significance was evaluated by one-way analysis of variance (ANOVA). p values (probability of a type I error) were adjusted for multiple comparisons with threshold (α) for a type I error of <0.05. Unless otherwise indicated, all data are represented as the means ± S.E. of three independent experiments.
Data availability
The MS proteomics data have been deposited to the ProteomeXchange Consortium via PRIDE (55) with the data set identifier PXD019199. All other data are contained within the article or in the supporting information. Plasmids and sequences are available on request from S. B. (sbhattac@well.ox.ac.uk).
Supplementary Material
Acknowledgments
We thank Tom McAllister for advice on fluorescence polarization and ITC assays and Christopher Lynch for advice on endotoxin-free protein production.
This article contains supporting information.
Author contributions—B. D., J. R. O. E., L. G.-A., G. K. Y., A. K., and S. B. conceptualization; B. D., J. R. O. E., L. G.-A., G. K. Y., K. K., C. V. R., and A. K. data curation; B. D., J. R. O. E., L. G.-A., G. K. Y., G. D., C. V. R., A. K., and S. B. formal analysis; B. D., J. R. O. E., L. G.-A., G. K. Y., K. K., G. D., C. V. R., A. K., and S. B. supervision; B. D., L. G.-A., A. K., and S. B. funding acquisition; B. D., J. R. O. E., L. G.-A., G. K. Y., K. K., G. D., C. V. R., A. K., and S. B. investigation; B. D., J. R. O. E., L. G.-A., G. K. Y., K. K., G. D., C. V. R., A. K., and S. B. methodology; B. D. and S. B. writing-original draft; B. D., L. G.-A., K. K., C. V. R., A. K., and S. B. project administration; B. D., J. R. O. E., L. G.-A., G. K. Y., K. K., G. D., C. V. R., A. K., and S. B. writing-review and editing.
Funding and additional information—This work was supported by British Heart Foundation Chair Award CH/09/003/26631 (to S. B.), British Heart Foundation Program Grant RG/18/1/33351 (to S. B. and A. K.) and the Oxford British Heart Foundation Centre of Research Excellence Grant RE/13/1/30181 (to J. R. O. E., A. K., and S. B.). C. V. R. is supported by Medical Research Council Grant MR/N020413/1. B. D. is supported by Grant EP/L015838/1 from the Engineering and Physical Sciences Research Council Centre for Doctoral Training in Synthesis for Biology and Medicine. J. R. O. E. is supported by a studentship from the Oxford British Heart Foundation Centre of Research Excellence, A. K. is supported by a Royal Society Dorothy Hodgkin Fellowship.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- ITC
- isothermal titration calorimetry
- CCL
- C–C motif chemokine ligand
- PAMP
- pathogen-associated molecular pattern
- HDX
- hydrogen–deuterium exchange
- HCD
- higher-energy collisional dissociation
- Fmoc
- N-(9-fluorenyl)methoxycarbonyl
- FP
- fluorescence polarization
- ANOVA
- analysis of variance
- H/D
- hydrogen–deuterium.
References
- 1. Tisoncik J. R., Korth M. J., Simmons C. P., Farrar J., Martin T. R., and Katze M. G. (2012) Into the eye of the cytokine storm. Microbiol. Mol. Biol. Rev. 76, 16–32 10.1128/MMBR.05015-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mehta P., McAuley D. F., Brown M., Sanchez E., Tattersall R. S., Manson J. J., and HLH Across Speciality Collaboration, UK (2020) COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet 395, 1033–1034 10.1016/S0140-6736(20)30628-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Huang C., Wang Y., Li X., Ren L., Zhao J., Hu Y., Zhang L., Fan G., Xu J., Gu X., Cheng Z., Yu T., Xia J., Wei Y., Wu W., et al. (2020) Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395, 497–506 10.1016/S0140-6736(20)30183-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tabas I., and Glass C. K. (2013) Anti-inflammatory therapy in chronic disease: challenges and opportunities. Science 339, 166–172 10.1126/science.1230720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brandes M., Klauschen F., Kuchen S., and Germain R. N. (2013) A systems analysis identifies a feedforward inflammatory circuit leading to lethal influenza infection. Cell 154, 197–212 10.1016/j.cell.2013.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Garin A., and Proudfoot A. E. (2011) Chemokines as targets for therapy. Exp. Cell Res. 317, 602–612 10.1016/j.yexcr.2010.12.021 [DOI] [PubMed] [Google Scholar]
- 7. Zernecke A., and Weber C. (2014) Chemokines in atherosclerosis: proceedings resumed. Arterioscler. Thrombo. Vasc. Biol. 34, 742–750 10.1161/ATVBAHA.113.301655 [DOI] [PubMed] [Google Scholar]
- 8. Zlotnik A., and Yoshie O. (2012) The chemokine superfamily revisited. Immunity 36, 705–716 10.1016/j.immuni.2012.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Horuk R. (2009) Chemokine receptor antagonists: overcoming developmental hurdles. Nat. Rev. Drug Discov. 8, 23–33 10.1038/nrd2734 [DOI] [PubMed] [Google Scholar]
- 10. Szekanecz Z., and Koch A. E. (2016) Successes and failures of chemokine-pathway targeting in rheumatoid arthritis. Nat Rev Rheumatol 12, 5–13 10.1038/nrrheum.2015.157 [DOI] [PubMed] [Google Scholar]
- 11. Mantovani A. (2018) Redundancy and robustness versus division of labour and specialization in innate immunity. Semin. Immunol. 36, 28–30 10.1016/j.smim.2017.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Heidarieh H., Hernaez B., and Alcamí A. (2015) Immune modulation by virus-encoded secreted chemokine binding proteins. Virus Res. 209, 67–75 10.1016/j.virusres.2015.02.028 [DOI] [PubMed] [Google Scholar]
- 13. Maizels R. M., Smits H. H., and McSorley H. J. (2018) Modulation of host immunity by helminths: the expanding repertoire of parasite effector molecules. Immunity 49, 801–818 10.1016/j.immuni.2018.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bhusal R. P., Eaton J. R. O., Chowdhury S. T., Power C. A., Proudfoot A. E. I., Stone M. J., and Bhattacharya S. (2020) Evasins: tick salivary proteins that inhibit mammalian chemokines. Trends Biochem. Sci. 45, 108–122 10.1016/j.tibs.2019.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sharif S., Nakatani Y., Wise L., Corbett M., Real N. C., Stuart G. S., Lateef Z., Krause K., Mercer A. A., and Fleming S. B. (2016) A broad-spectrum chemokine-binding protein of bovine papular stomatitis virus inhibits neutrophil and monocyte infiltration in inflammatory and wound models of mouse skin. PLoS One 11, e0168007 10.1371/journal.pone.0168007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bonvin P., Power C. A., and Proudfoot A. E. (2016) Evasins: therapeutic potential of a new family of chemokine-binding proteins from ticks. Front. Immunol. 7, 208 10.3389/fimmu.2016.00208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Singh K., Davies G., Alenazi Y., Eaton J. R. O., Kawamura A., and Bhattacharya S. (2017) Yeast surface display identifies a family of evasins from ticks with novel polyvalent CC chemokine-binding activities. Sci. Rep. 7, 4267 10.1038/s41598-017-04378-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hayward J., Sanchez J., Perry A., Huang C., Rodriguez Valle M., Canals M., Payne R. J., and Stone M. J. (2017) Ticks from diverse genera encode chemokine-inhibitory evasin proteins. J. Biol. Chem. 292, 15670–15680 10.1074/jbc.M117.807255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Alenazi Y., Singh K., Davies G., Eaton J. R. O., Elders P., Kawamura A., and Bhattacharya S. (2018) Genetically engineered two-warhead evasins provide a method to achieve precision targeting of disease-relevant chemokine subsets. Sci. Rep. 8, 6333 10.1038/s41598-018-24568-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Eaton J. R. O., Alenazi Y., Singh K., Davies G., Geis-Asteggiante L., Kessler B., Robinson C. V., Kawamura A., and Bhattacharya S. (2018) The N-terminal domain of a tick evasin is critical for chemokine binding and neutralization and confers specific binding activity to other evasins. J. Biol. Chem. 293, 6134–6146 10.1074/jbc.RA117.000487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lee A. W., Deruaz M., Lynch C., Davies G., Singh K., Alenazi Y., Eaton J. R. O., Kawamura A., Shaw J., Proudfoot A. E. I., Dias J. M., and Bhattacharya S. (2019) A knottin scaffold directs the CXC-chemokine–binding specificity of tick evasins. J. Biol. Chem. 294, 11199–11212 10.1074/jbc.RA119.008817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Boehncke W. H., and Brembilla N. C. (2018) Immunogenicity of biologic therapies: causes and consequences. Exp. Rev. Clin. Immunol. 14, 513–523 10.1080/1744666X.2018.1468753 [DOI] [PubMed] [Google Scholar]
- 23. Antosova Z., Mackova M., Kral V., and Macek T. (2009) Therapeutic application of peptides and proteins: parenteral forever?. Trends Biotechnol. 27, 628–635 10.1016/j.tibtech.2009.07.009 [DOI] [PubMed] [Google Scholar]
- 24. Lau J. L., and Dunn M. K. (2018) Therapeutic peptides: historical perspectives, current development trends, and future directions. Bioorg. Med. Chem. 26, 2700–2707 10.1016/j.bmc.2017.06.052 [DOI] [PubMed] [Google Scholar]
- 25. Konermann L., Pan J., and Liu Y.-H. (2011) Hydrogen exchange mass spectrometry for studying protein structure and dynamics. Chem. Soc. Rev. 40, 1224–1234 10.1039/c0cs00113a [DOI] [PubMed] [Google Scholar]
- 26. Brown K. A., and Wilson D. J. (2017) Bottom-up hydrogen deuterium exchange mass spectrometry: data analysis and interpretation. Analyst 142, 2874–2886 10.1039/c7an00662d [DOI] [PubMed] [Google Scholar]
- 27. Marcsisin S. R., and Engen J. R. (2010) Hydrogen exchange mass spectrometry: what is it and what can it tell us? Anal. Bioanal. Chem. 397, 967–972 10.1007/s00216-010-3556-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Blaszczyk J., Coillie E. V., Proost P., Damme J. V., Opdenakker G., Bujacz G. D., Wang J. M., and Ji X. (2000) Complete crystal structure of monocyte chemotactic protein-2, a CC chemokine that interacts with multiple receptors. Biochemistry 39, 14075–14081 10.1021/bi0009340 [DOI] [PubMed] [Google Scholar]
- 29. McAllister T. E., Yeh T. L., Abboud M. I., Leung I. K. H., Hookway E. S., King O. N. F., Bhushan B., Williams S. T., Hopkinson R. J., Münzel M., Loik N. D., Chowdhury R., Oppermann U., Claridge T. D. W., Goto Y., et al. (2018) Non-competitive cyclic peptides for targeting enzyme–substrate complexes. Chem. Sci. 9, 4569–4578 10.1039/c8sc00286j [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kawamura A., Münzel M., Kojima T., Yapp C., Bhushan B., Goto Y., Tumber A., Katoh T., King O. N., Passioura T., Walport L. J., Hatch S. B., Madden S., Müller S., Brennan P. E., et al. (2017) Highly selective inhibition of histone demethylases by de novo macrocyclic peptides. Nat. Commun. 8, 14773 10.1038/ncomms14773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Velazquez-Campoy A., Leavitt S. A., and Freire E. (2015) Characterization of protein–protein interactions by isothermal titration calorimetry. Methods Mol. Biol. 1278, 183–204 10.1007/978-1-4939-2425-7_11 [DOI] [PubMed] [Google Scholar]
- 32. Tsuchiya S., Yamabe M., Yamaguchi Y., Kobayashi Y., Konno T., and Tada K. (1980) Establishment and characterization of a human acute monocytic leukemia cell line (THP-1). Int. J. Cancer 26, 171–176 10.1002/ijc.2910260208 [DOI] [PubMed] [Google Scholar]
- 33. Parker L. C., Whyte M. K., Vogel S. N., Dower S. K., and Sabroe I. (2004) Toll-like receptor (TLR)2 and TLR4 agonists regulate CCR expression in human monocytic cells. J. Immunol. 172, 4977–4986 10.4049/jimmunol.172.8.4977 [DOI] [PubMed] [Google Scholar]
- 34. Achour L., Scott M. G., Shirvani H., Thuret A., Bismuth G., Labbé-Jullié C., and Marullo S. (2009) CD4-CCR5 interaction in intracellular compartments contributes to receptor expression at the cell surface. Blood 113, 1938–1947 10.1182/blood-2008-02-141275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Harding S. D., Sharman J. L., Faccenda E., Southan C., Pawson A. J., Ireland S., Gray A. J. G., Bruce L., Alexander S. P. H., Anderton S., Bryant C., Davenport A. P., Doerig C., Fabbro D., Levi-Schaffer F., et al. NC-IUPHAR, (2018) The IUPHAR/BPS Guide to Pharmacology in 2018: updates and expansion to encompass the new guide to immunopharmacology. Nucleic Acids Res. 46, D1091–D1106 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Coates N. J., and McColl S. R. (2001) Production of chemokines in vivo in response to microbial stimulation. J. Immunol. 166, 5176–5182 10.4049/jimmunol.166.8.5176 [DOI] [PubMed] [Google Scholar]
- 37. El-Achkar G. A., Mrad M. F., Mouawad C. A., Badran B., Jaffa A. A., Motterlini R., Hamade E., and Habib A. (2019) Heme oxygenase-1–dependent anti-inflammatory effects of atorvastatin in zymosan-injected subcutaneous air pouch in mice. PLoS One 14, e0216405 10.1371/journal.pone.0216405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Skelton N. J., Quan C., Reilly D., and Lowman H. (1999) Structure of a CXC chemokine-receptor fragment in complex with interleukin-8. Structure 7, 157–168 10.1016/S0969-2126(99)80022-7 [DOI] [PubMed] [Google Scholar]
- 39. Ezerzer C., Dolgin M., Skovorodnikova J., and Harris N. (2009) Chemokine receptor–derived peptides as multi-target drug leads for the treatment of inflammatory diseases. Peptides 30, 1296–1305 10.1016/j.peptides.2009.03.016 [DOI] [PubMed] [Google Scholar]
- 40. Girrbach M., Meliciani I., Waterkotte B., Berthold S., Oster A., Brurein F., Strunk T., Wadhwani P., Berensmeier S., Wenzel W., and Schmitz K. (2014) A fluorescence polarization assay for the experimental validation of an in silico model of the chemokine CXCL8 binding to receptor-derived peptides. Phys. Chem. Chem. Phys. 16, 8036–8043 10.1039/c3cp53850h [DOI] [PubMed] [Google Scholar]
- 41. Houimel M., and Mazzucchelli L. (2013) Chemokine CCR3 ligands-binding peptides derived from a random phage-epitope library. Immunol. Lett. 149, 19–29 10.1016/j.imlet.2012.11.006 [DOI] [PubMed] [Google Scholar]
- 42. von Hundelshausen P., Agten S. M., Eckardt V., Blanchet X., Schmitt M. M., Ippel H., Neideck C., Bidzhekov K., Leberzammer J., Wichapong K., Faussner A., Drechsler M., Grommes J., van Geffen J. P., Li H., et al. (2017) Chemokine interactome mapping enables tailored intervention in acute and chronic inflammation. Sci. Transl. Med. 9, eaah6650 10.1126/scitranslmed.aah6650 [DOI] [PubMed] [Google Scholar]
- 43. Kufareva I. (2016) Chemokines and their receptors: insights from molecular modeling and crystallography. Curr. Opin. Pharmacol. 30, 27–37 10.1016/j.coph.2016.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Seet B. T., Singh R., Paavola C., Lau E. K., Handel T. M., and McFadden G. (2001) Molecular determinants for CC-chemokine recognition by a poxvirus CC-chemokine inhibitor. Proc. Natl. Acad. Sci. U.S.A. 98, 9008–9013 10.1073/pnas.171069398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Beck C. G., Studer C., Zuber J. F., Demange B. J., Manning U., and Urfer R. (2001) The viral CC chemokine-binding protein vCCI inhibits monocyte chemoattractant protein-1 activity by masking its CCR2B-binding site. J. Biol. Chem. 276, 43270–43276 10.1074/jbc.M106305200 [DOI] [PubMed] [Google Scholar]
- 46. Moreira I. S., Fernandes P. A., and Ramos M. J. (2007) Hot spots–a review of the protein–protein interface determinant amino-acid residues. Proteins 68, 803–812 10.1002/prot.21396 [DOI] [PubMed] [Google Scholar]
- 47. Ma B., and Nussinov R. (2007) Trp/Met/Phe hot spots in protein–protein interactions: potential targets in drug design. Curr. Top. Med. Chem. 7, 999–1005 10.2174/156802607780906717 [DOI] [PubMed] [Google Scholar]
- 48. Chothia C., and Janin J. (1975) Principles of protein–protein recognition. Nature 256, 705–708 10.1038/256705a0 [DOI] [PubMed] [Google Scholar]
- 49. Lua R. C., Marciano D. C., Katsonis P., Adikesavan A. K., Wilkins A. D., and Lichtarge O. (2014) Prediction and redesign of protein–protein interactions. Prog. Biophys. Mol. Biol. 116, 194–202 10.1016/j.pbiomolbio.2014.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. MacArthur M. W., and Thornton J. M. (1991) Influence of proline residues on protein conformation. J. Mol. Biol. 218, 397–412 10.1016/0022-2836(91)90721-H [DOI] [PubMed] [Google Scholar]
- 51. Schmidpeter P. A., Koch J. R., and Schmid F. X. (2015) Control of protein function by prolyl isomerization. Biochim. Biophys. Acta 1850, 1973–1982 10.1016/j.bbagen.2014.12.019 [DOI] [PubMed] [Google Scholar]
- 52. Ludeman J. P., and Stone M. J. (2014) The structural role of receptor tyrosine sulfation in chemokine recognition. Br. J. Pharmacol. 171, 1167–1179 10.1111/bph.12455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Underhill D. M., Ozinsky A., Hajjar A. M., Stevens A., Wilson C. B., Bassetti M., and Aderem A. (1999) The Toll-like receptor 2 is recruited to macrophage phagosomes and discriminates between pathogens. Nature 401, 811–815 10.1038/44605 [DOI] [PubMed] [Google Scholar]
- 54. Zhao Y., Bishop B., Clay J. E., Lu W., Jones M., Daenke S., Siebold C., Stuart D. I., Jones E. Y., and Aricescu A. R. (2011) Automation of large scale transient protein expression in mammalian cells. J. Struct. Biol. 175, 209–215 10.1016/j.jsb.2011.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Vizcaíno J. A., Csordas A., del-Toro N., Dianes J. A., Griss J., Lavidas I., Mayer G., Perez-Riverol Y., Reisinger F., Ternent T., Xu Q. W., Wang R., and Hermjakob H. (2016) 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 44, D447–D456 10.1093/nar/gkv1145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Dias J. M., Losberger C., Déruaz M., Power C. A., Proudfoot A. E. I., and Shaw J. P. (2009) Structural basis of chemokine sequestration by a tick chemokine binding protein: the crystal structure of the complex between evasin-1 and CCL3. PLoS One 4, e8514 10.1371/journal.pone.0008514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Larive C. K., Jayawickrama D., and Orfi L. (1997) Quantitative analysis of peptides with NMR spectroscopy. Appl. Spectrosc. 51, 1531–1536 10.1366/0003702971939055 [DOI] [Google Scholar]
- 58. Gault J., Donlan J. A., Liko I., Hopper J. T., Gupta K., Housden N. G., Struwe W. B., Marty M. T., Mize T., Bechara C., Zhu Y., Wu B., Kleanthous C., Belov M., Damoc E., et al. (2016) High-resolution mass spectrometry of small molecules bound to membrane proteins. Nat. Methods 13, 333–336 10.1038/nmeth.3771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Duarte D. B., Vasko M. R., and Fehrenbacher J. C. (2016) Models of inflammation: carrageenan air pouch. Curr. Protoc. Pharmacol. 72, 5.6.1–5.6.9 10.1002/0471141755.ph0506s72 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The MS proteomics data have been deposited to the ProteomeXchange Consortium via PRIDE (55) with the data set identifier PXD019199. All other data are contained within the article or in the supporting information. Plasmids and sequences are available on request from S. B. (sbhattac@well.ox.ac.uk).