

Abstract
CopG is an uncharacterized protein ubiquitous in Gram-negative bacteria whose gene frequently occurs in clusters of copper resistance genes and can be recognized by the presence of a conserved CxCC motif. To investigate its contribution to copper resistance, here we undertook a structural and biochemical characterization of the CopG protein from Pseudomonas aeruginosa. Results from biochemical analyses of CopG purified under aerobic conditions indicate that it is a green copper-binding protein that displays absorbance maxima near 411, 581, and 721 nm and is monomeric in solution. Determination of the three-dimensional structure by X-ray crystallography revealed that CopG consists of a thioredoxin domain with a C-terminal extension that contributes to metal binding. We noted that adjacent to the CxCC motif is a cluster of four copper ions bridged by cysteine sulfur atoms. Structures of CopG in two oxidation states support the assignment of this protein as an oxidoreductase. On the basis of these structural and spectroscopic findings and also genetic evidence, we propose that CopG has a role in interconverting Cu(I) and Cu(II) to minimize toxic effects and facilitate export by the Cus RND transporter efflux system.
Keywords: copper, metalloprotein, oxidation-reduction, structure-function, thioredoxin, metal homeostasis, metal resistance, Pseudomonas aeruginosa, oxidation-reduction (redox), P. aeruginosa, metal ion homeostasis
Copper ions are an essential cofactor for many enzymes and, thus, must be retained in the cell at sufficient levels to support metabolism. This cellular requirement is balanced by the potential for copper to show toxic effects when copper levels are too high. Bacteria can be exposed to various levels of copper through their environments; therefore, their ability to adapt to changing conditions is essential for their survival. When copper homeostasis systems are overwhelmed, growth inhibition or cell death occurs. This outcome forms the basis for the use of copper ions in a variety of clinical and environmental settings to control microbial growth (1, 2) and is part of the macrophage response to intracellular pathogens (3). Copper ions have shown promise in combating infections, including those from multidrug resistance organisms; however, bacterial resistance systems counter human efforts to use metal ions as broad-spectrum biocides. Therefore, a thorough understanding of how bacteria sense and respond to metal ions is needed to maintain this avenue to fight infection in this era of rapidly increasing multidrug resistance.
The exact mechanisms of copper toxicity are still under debate (reviewed in reference 4) but appear to involve two main mechanisms. Metalloenzymes can be inactivated by metal ion displacement with Cu(I), particularly those containing Fe-S clusters (5). Free copper ions can generate damaging hydroxyl radicals and reactive oxygen species through Fenton-like chemistry (6, 7). Cu(I) is also thought to be capable of passing through the cell membrane, whereas Cu(II) cannot. Therefore, decreasing the intracellular pool of free copper ions protects cells from toxicity.
Bacteria maintain metal ion homeostasis through a coordinated network of systems to detect, import, detoxify, traffic, sequester, and export metal ions. In response to elevated metal ion concentrations, transcription factors or two-component systems detect metal ions and then upregulate appropriate genes. Primary or secondary transporters are commonly used to export metal ions, and these systems may also include metallochaperones that maintain the metal ion in a sequestered state before removal from the cell. Under aerobic conditions, enzymatic detoxification of copper ions by oxidation to Cu(II) via a multicopper oxidase also provides protection. Well-characterized systems that provide these responses to elevated levels of copper ions include the transcription factor and two-component systems, CueR and CusRS, respectively, of Escherichia coli; P-type ATPases, such as E. coli CopA; tripartite efflux systems (RND transporters), such as CusCBA; CopZ chaperones (8); and the multicopper oxidases, typified by E. coli CueO and PcoA (9, 10).
Metal ion homeostasis systems are commonly organized into operons and/or clustered within metal resistance islands. These regions also include additional genes encoding proteins of unknown function. Among these uncharacterized ORFs is a small gene, copG, also annotated as encoding the Pfam domain DUF411. The copG gene encodes a protein of ∼14 kDa with limited homology to previously characterized proteins. To investigate the role for the CopG protein in metal homeostasis, we carried out structural and biochemical characterization of the CopG protein from Pseudomonas aeruginosa in conjunction with genetic studies of its role in copper resistance. We show that CopG possesses a tetranuclear copper cluster in a modified thioredoxin domain. The structural details suggest that the protein contributes to copper resistance by harnessing the redox properties of this fold to reduce copper, adding to our knowledge of the adaptive strategies bacteria utilize in response to elevated metal ions.
Results
CopG homologs are associated with metal export components and contain a conserved CxCC motif
A homology search using the Pseudomonas aeruginosa CopG sequence reveals over 8000 distinct proteins containing putative CopG domains from diverse Gram-negative bacterial species. The presence of localization sequences indicates that these are periplasmic proteins. Comparison of the genomic context of genes encoding CopG homologs shows that CopG is frequently associated with known metalloproteins. Genes encoding CopG are also found in metal resistance islands and other mobile genetic elements associated with metal resistance (11, 12) and, in particular, are frequently found adjacent to copper resistance genes (Fig. 1). These include CueO-like multicopper oxidases, CopA-like P-type copper translocases, and CusCBA-like RND efflux systems. (Note that in P. aeruginosa, however, the copG gene is not in the immediate vicinity of any of these.)
Figure 1.
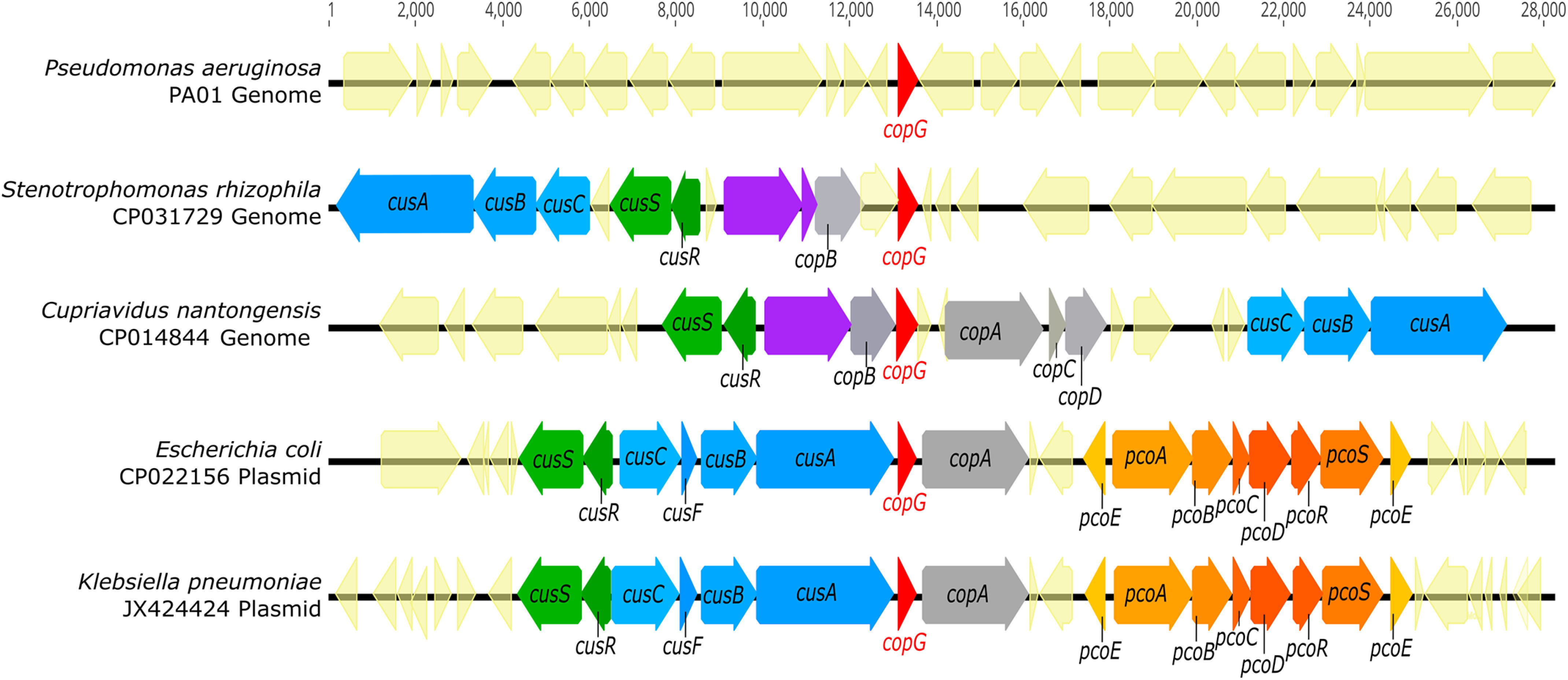
copG is located within metal resistance gene clusters in diverse organisms. Genomic DNA sequences (CP031729 and CP014844) were retrieved from NCBI, and plasmid DNA sequences (CP022156 and JX424424) were retrieved from the PATRIC annotated bacterial plasmid database. Genomes containing CopG homologs were identified via tblastn search using the CopG amino acid sequence from Pseudomonas aeruginosa strain PA01 as a query. All identified sequences displayed at least 40% amino acid sequence identity and at least 90% query coverage to PA01 CopG. Purple gene annotations indicate copper oxidases.
Sequence comparison of CopG homologs revealed a subset of highly conserved residues that includes potential metal ligands. Fig. 2 shows the CopG sequences from the organisms shown in Fig. 1 extracted from an alignment of 1000 homologs of P. aeruginosa CopG. Among the absolutely conserved residues are four cysteines, three of which are found in an unusual CxCC motif (residues 13–16). Additional conserved residues include His 55, His 67, Cys 54, Met 90, and Met 97.
Figure 2.

Conserved residues in CopG sequences. Comparison of the sequences of CopG homologs from Fig. 1. Alignment of the top-scoring 1,000 homologs to PAO1 CopG was used to generate the consensus sequence at the 99% level. Metal-binding residues within these highly conserved sites in the sequence are marked with arrows.
Deletion of copG confers sensitivity to copper under anaerobic conditions
The role of copG in conferring copper resistance was tested by comparison of the growth of P. aeruginosa PAO1 and PAO1/ΔcopG strains in the presence of differing amounts of copper. Plate assays show that the ΔcopG strain grows less well in the presence of increasing copper concentrations under anaerobic conditions but has growth similar to that of PAO1 under aerobic conditions (Fig. 3).
Figure 3.

Copper resistance assay. Differences in copper resistance between the PAO1 and ΔcopG Pseudomonas aeruginosa strains were assayed by serial dilution on copper-containing agar plates. A, single plate with 4 replicates of each dilution series. Under anaerobic conditions, the copG knockout shows increased sensitivity to copper. B, comparison of growth on plates containing 0–4 mm copper sulfate for PAO1 and ΔcopG strains under aerobic and anaerobic conditions. The average of the highest dilution allowing growth from 8 replicates (2 plates) is plotted, and error bars indicate 1 standard deviation.
CopG is a green copper-binding protein
To investigate the mechanism by which CopG contributes to copper resistance, we cloned, expressed, and purified the protein and subjected it to biochemical characterization. Purified CopG is dark green in color to the naked eye, displays absorbance maxima near 411 nm, 581 nm, and 721 nm (Fig. 4), and is monomeric in solution (Fig. S1). When purified from cells grown in the absence of supplemental copper, CopG has a CD spectrum indicating that the protein is predominantly unfolded, but when cells are grown in the presence of 1 mm copper sulfate, a spectrum of the resulting protein shows features consistent with a folded configuration (Fig. S2). Analysis of the secondary structure content predicts a mixed alpha/beta structure for the copper-bound state. As determined by atomic absorption spectroscopy, CopG contains 3.4 ± 1.0 molar equivalents of copper when purified from cells grown in media containing 1 mm copper sulfate. Metal could be removed from CopG by dialysis in the presence of the copper chelator phenanthroline, yielding samples with <0.1 molar equivalents. The CD spectrum of CopG depleted of metals shows that the majority of secondary structure features are lost (Fig. S3). Attempts to restore metal to the protein treated with chelators caused the protein to precipitate.
Figure 4.

Absorption spectrum of CopG. Spectrum of CopG (purified from medium containing supplemental copper) under aerobic conditions. The absorbance data (black) are fitted with a sum of gaussian functions (cyan). The spectrum can be approximated by three main peaks (dashed lines) in the visible range centered at 411 nm (violet), 581 nm (yellow), and 721 nm (red) in addition to the near-UV absorbance from aromatic side chains. For fitting purposes, these maxima are each treated as single peaks (but see the text). The inset shows the visual appearance of the purified CopG protein solution.
Structure of CopG
To gain insight into CopG function and copper binding mode, we carried out structural characterization of CopG by X-ray crystallography. In the crystallization trials, two crystal forms of CopG with space groups P61 (form I) and P212121 (form II) were obtained. Form I crystals appear green in color, whereas form II crystals appear clear. The structure of CopG was determined by SAD using data from selenomethionine-substituted protein in crystal form I, and the resulting model was used to solve form II via molecular replacement. The crystal forms contain two and six independent copies of CopG in the asymmetric unit, respectively.
The structure of CopG consists of a single domain with a central 7-stranded mixed β-sheet with segments A–D connecting individual strands 1–2 (segment A), 2–3 (B), 4–5 (C), and 5–6 (D) (Fig. 5A). The connections between strands 3–4 and 6–7 are short beta-turns. The N-terminal 77 residues comprising the first 4 strands along with the three helix-containing segments form a thioredoxin-like fold, and the remaining 49 residues comprise a C-terminal extension that consists of the three remaining strands and segment D. Segment A contains a helical region (helix A) from residues 14–26, at the N terminus of which is located the conserved CxCC motif (residues 13–16). Segments B and C each contain helical regions (residues 38–45, helix B; residues 70–79, helix C) and connections to the β-strands. Segment B also contains a short region consistent with a single helical turn.
Figure 5.

Structure of CopG. A, secondary structure diagram of CopG, with the dashed line outlining the thioredoxin domain. The C-terminal extension consists of strands 5, 6 and 7 and the extended loop of segment D (cyan and orange). B, ribbon diagram of CopG using the same color scheme. Copper ions are shown in orange. Electron density is from an anomalous difference Fourier map contoured at 4σ. The map was calculated from data collected at 1.2894 Å using phases obtained from the refined model with metal ions omitted.
CopG contains a tetranuclear copper cluster
Several potential copper-binding residues are highly conserved between CopG sequences, including the CxCC motif of residues 13–16, Cys 54, His 55, His 67, Met 90, and Met 97. In the CopG structure, these conserved residues are located at the junction of helix A, the end of strands 3 and 4, and in segment D. The CxCC motif of residues 13–16 is seen to be a variant of the CxxC motif common in the active site of thioredoxin domains and is located at the N terminus of helix A. The strongest features in the electron density map are four peaks corresponding to a tetranuclear metal cluster, with the conserved CxCC motif of residues 13–16, Cys 54, His 55, His 67, Met 90, and Met 97 serving as direct ligands. An anomalous difference map showed these peaks arise from copper ions (Fig. 5B) (13).
In the metal cluster, three copper ions (Cu1, Cu2, and Cu3) are arranged in a triangle ligated by three equatorial cysteine sulfur atoms (Fig. 6). Below the plane of the triangle are three nitrogen ligands from two histidine side chain residues and a backbone amide nitrogen. Thus, each of these three copper ions has 2 adjacent sulfur ligands and a nitrogen ligand. A fourth cysteine sulfur is located above the plane formed by the triangle, serving as a ligand only to the copper ion (Cu3) that has the backbone amide ligand. The primary ligands to copper ions 1–3 are from the thioredoxin domain, whereas the two Met ligands to Cu4, located on the protein's surface, are from segment D in the C-terminal extension. Cu1 and Cu2 have trigonal planar geometry, whereas Cu3 is square planar. Distances and ligands are summarized in Table 1.
Figure 6.
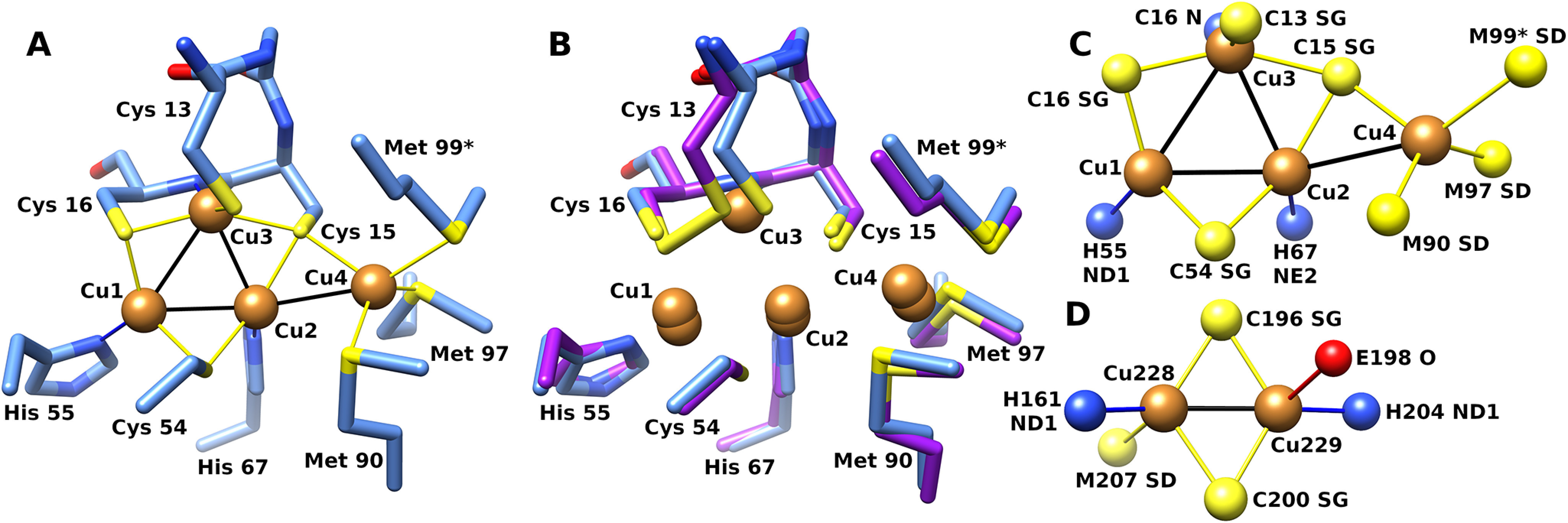
Structure of the CopG copper cluster. A, CopG copper cluster with side chain ligands from crystal form I (blue). B, superposition of corresponding atoms from form II (purple). Met 99 from a symmetry-related molecule is indicated with an asterisk. C, top view of copper cluster with ligand atoms. D, CuA site from cytochrome c oxidase (PDB code 1OCC [25]). Atom color: sulfur, yellow; copper, orange; nitrogen, blue; oxygen, red.
Table 1.
Interatomic distances in the copper clustera
| Cu ion | Cu1 | Cu2 | Cu3 | Cu4 |
|---|---|---|---|---|
| Form I | ||||
| Cu1 | 2.85, 2.81 | 3.05, 3.05 | ||
| Cu2 | 2.79, 2.76 | 3.10, 3.13 | ||
| H55 Nδ1 | 2.19, 2.14 | |||
| H67 Nϵ1 | 2.21, 2.12 | |||
| C16 N | 2.12, 2.10 | |||
| C54 Sγ | 2.26, 2.20 | 2.29, 2.23 | ||
| C16 Sγ | 2.17, 2.19 | 2.24, 2.36 | ||
| C15 Sγ | 2.26, 2.37 | 2.34, 2.41 | ||
| C13 Sγ | 2.26, 2.30 | |||
| M90 Sδ | 2.42, 2.43 | |||
| M97 Sδ | 2.41, 2.46 | |||
| M99 Sδ | 2.62, 2.57 | |||
| Form II | ||||
| Cu1 | 2.81 ± 0.04 | 2.84 (C), 3.05 (D) | ||
| Cu2 | 2.68 (C), 2.68 (D) | 3.17 ± 0.13 | ||
| H55 Nδ1 | 2.07 ± 0.03 | |||
| H67 Nϵ1 | 2.08 ± 0.06 | |||
| C16 N | 2.67 (C), 2.24 (D) | |||
| C54 Sγ | 2.21 ± 0.07 | 2.18 ± 0.10 | ||
| C16 Sγ | 2.23 ± 0.12 | 2.25 (C), 2.17 (D) | ||
| C15 Sγ | 2.14 ± 0.09 | 2.23 (C), 2.14 (D) | 2.42 ± 0.04 | |
| C13 Sγ | 2.86 (C), 2.29 (D) | |||
| M90 Sδ | 2.39 ± 0.10 | |||
| M97 Sδ | 2.41 ± 0.10 | |||
| M99 Sδ | 2.67 ± 0.08 | |||
| OMT99 OD1 | 2.79 (C), 3.41 (D) | |||
| OMT99 OD2 | 2.52 (C), 2.53 (D) |
a Distances between the four copper ions (listed at left) and adjacent atoms (listed at top of cell), in Angstroms. For form I, individual distances for chains A and B are given. For form II, distances are listed as the average and standard deviation seen in the 6 chains. Distances for Met 99 Sδ are to the residue from an adjacent chain. Ligand distances for Cu3 are listed where present.
The Cu4 ion is in a distinct environment and lies on the surface of the protein at the base of a shallow depression. The surrounding surface is hydrophobic in the immediate vicinity of the copper ion. The Cu4 ion is tetrahedral, with two sulfur ligands coming from methionine side chains within segment D, a third ligand supplied by a cysteine (Cys 15), and, in all eight molecules in both crystal forms, a fourth ligand to Cu4 supplied by a nonconserved methionine sulfur atom (Met 99) from a symmetry-related molecule.
In addition to residues serving as direct ligands to metals, conserved sites include three glycines and two proline residues within the irregular loop region of segment D. These likely provide important structural constraints to position the Met 90 and 97 side chains for interaction with Cu4. The backbone carbonyl of Pro 95 interacts with Nϵ of His 67, which positions the His 67 Nδ for interactions with Cu2. Trp 19, which is also highly conserved, appears to form a hydrogen–π interaction with His 55, where the indole π ring interacts with the protonated Nϵ of His 55, orienting the deprotonated Nδ for interaction with Cu1. Another set of conserved residues form a buried salt bridge (Glu 65, Lys 42) that also involves Thr 56. On the opposite side of the molecule from the exposed copper is an internal pocket (Fig. 7A), the walls of which are hydrophobic and formed mainly by Leu 23 and the absolutely conserved Trp 19.
Figure 7.

Structural consequences of disulfide formation. A, superposition of the Cα traces of the 2 form I models (blue) representing the reduced state and 6 form II models (purple) representing the oxidized state. Disulfide formation between sites 13 and 16 is associated with displacement of helix B and a bending of the loop between strand 1 and helix A relative to the reduced state. The loop between strands 6 and 7 also shows structural variability, but there does not appear to be a discrete difference in this region between the two crystal forms. B and C, detail of residues 13 to 16 showing reduced thiolates on Cys 13 and 16 (form I, chain A) (B) and disulfide (form II, chain A) (C). These are superimposed on the corresponding residues (C32-I38, in black) from the structure of oxidized E. coli thioredoxin (PDB code 2TRX [69]).
The two crystal forms represent different oxidation states of the protein
Comparison of the structures of CopG from the two crystal forms shows some conformational rearrangements as well as differences near the metal cluster (Fig. 7). These structural changes are associated with the formation of a disulfide cross-link between the side chains of Cys 13 and 16 in crystal form II (Fig. 7B). Cys 13 is located in the loop between strand 1 and the N terminus of helix A, and formation of the disulfide with Cys 16 alters the loop conformation. This change propagates to the adjacent helix B, which undergoes a 1.5-Å axial shift relative to its position in the form I structure. In addition, disulfide formation sequesters two of the Cu3 ligands and pulls the Cys 13 side chain closer to Cys 16 such that it lies about 1.5 Å from the Cu3 site. These changes displace the copper ion at the Cu3 site in 4 of the 6 chains in form II (Fig. S8). In the remaining two chains, some weak density at the Cu3 site is present, suggesting these locations in the crystal have a mixed population comprised of a superposition of a reduced state in which Cu3 is present and an oxidized state where Cu3 is not present and a disulfide bond has formed between the side chains of Cys 13 and 16.
Comparing all 8 chains, the regions of the molecule with the largest structural variability are segment B, the loop between strand 1 and helix A, and the turn between strands 6 and 7. These regions lie on one face of the structure, on the opposite side from the pocket adjacent to W19. As the chains from form I lie within the set of conformations spanned by the chains from form II, the variability seen at these sides appears to be independent of disulfide formation.
Both crystal forms contain a similar arrangement in which the Met 99 side chain from one chain is reciprocally shared as the ligand to Cu4, such that the asymmetric unit in crystal forms I and II are comprised of one and three such pairs, respectively. Superposition of one chain from each pair indicates that the relative orientation of each pair of chains differs significantly from the others (Fig. S7). Analysis by PDBePISA (14) of the interface between the two chains results in a complex formation significance score of 0.013, indicating it is a result of crystal packing. This is consistent with the size-exclusion chromatography data, which indicated the protein is monomeric.
In chains A and B from form I and chains A, B, E, and F from form II, the Met99 sulfur atom serves as a ligand to Cu4. In chains C and D from form II, which are the two showing partial occupancy at the Cu3 site, additional density adjacent to the Met99 side chain is present (Fig. S10). In these chains, the side chain is modeled as S-dioxymethionine and is positioned such that the two oxygens on these side chains interact with Cu4 and Cu3. Both crystal forms also contain a number of bound zinc ions, which are present in the crystallization buffer. These could be distinguished from copper through the use of anomalous difference maps (13) (Figs. S4 and S5). The amino acids serving as ligands at these sites are not conserved (Fig. S6), suggesting that zinc binding is an artifact of the crystallization conditions.
Discussion
CopG is widely present in diverse bacteria and contributes to metal resistance
CopG-like proteins are currently classified as uncharacterized conserved proteins and are variously annotated as members of COG3019 (15), Pfam 04214 (16), and conserved domain DUF411 with accession code cl23841 (17). Computational modeling of a COG3019 family member from Campylobacter jejuni predicted the presence of a thioredoxin fold in this family (18). (Note that the name CopG is also used to refer to a transcriptional repressor protein [19] and a putative P-type ATPase [20]. These are unrelated to the protein described here.)
Several prior lines of evidence have previously implicated CopG as being involved in copper metabolism in Pseudomonas aeruginosa. Transcript levels for the copG gene were found to be elevated after copper exposure (21), although no significant change was observed in a related subsequent study (22). In a recent study of protein levels associated with copper exposure in P. aeruginosa PA01, the CopG protein showed the second highest fold increase in abundance (16.2-fold) of the observed proteins following copper treatment (23), suggesting an involvement in copper resistance. In Vibrio cholerae, copG also was shown to contribute to copper resistance under anaerobic conditions (24).
Here, we have provided direct experimental data showing that CopG from Pseudomonas aeruginosa is a copper-binding protein and, in this organism, contributes to copper resistance under anaerobic conditions. The broader mechanism by which CopG does so remains to be delineated, but the structure offers some insights into the likely biochemical function of this protein.
The CopG tetranuclear copper cluster and its relation to other copper sites
CopG contains a copper site with a unique arrangement, providing the first observation of a new type of copper center (Fig. 6A). In crystal form I, copper ions 1–3 form a triangle with three bridging cysteine sulfur atoms arranged equatorially, with each copper sharing 2 sulfur ligands. Ligands below the plane of the triangle in CopG are all nitrogens, two from histidines and a third, on Cu3, from a backbone amide. Cu3 also has another unshared ligand from Cys 13 above the plane of the triangle. This arrangement suggests a 3-metal generalization of the diamond-core 2-metal CuA site found in cytochrome c oxidase (25), nitrous oxide reductase (26), and methane monooxygenase (27), where two cysteines bridge the pair of copper ions (28–31) (Fig. 6, C and D). Additional side chain ligands in CuA sites are two histidines, methionine, and a backbone carbonyl oxygen. The oxidized CuA site has the unusual property of forming a mixed-valence metal–metal bond with a single electron delocalized over both atoms to yield two Cu(+1.5) ions, and these centers appear purple (28, 31).
Complete specification of the oxidation state of CopG requires not only assigning the charge on the 4 copper ions but also the presence or absence of the disulfide bond between Cys 13 and 16. Formally, a fully oxidized CopG with a disulfide and 4 Cu(II) ions could accept up to 6 electrons, and intermediate numbers of electrons could be distributed in a number of ways. Thus, it will be of interest to define the range of redox states accessed by the protein. Crystallographic data cannot assign the charge states on the copper ions in the two crystal forms, but the spectroscopic properties can provide some insight. Cu(I) is spectroscopically silent in the visible range, so these absorbances in the visible spectrum likely arise from ligand-to-metal charge transfer interactions to oxidized Cu sites. Individual ligand interactions observed crystallographically include 8 to thiolate sulfurs, 2 to imidazole nitrogens, 1 to a backbone amide nitrogen, and 3 to thioether sulfurs, including the external ligand. The spectrum in Fig. 3 indicates the presence of at least 3 absorption regions at 411 nm (24,331 cm−1), 581 (17,211 cm−1), and 721 nm (13,870 cm−1), which correspond to violet, yellow, and red light, respectively. Other spectroscopically characterized green copper proteins contain perturbed type I blue copper sites (32–34) where the relative intensity of transitions near 450 nm and 600 nm govern the apparent color. These are attributed to cysteine sulfur σ- or π-to-Cu charge transfer transitions (30). The spectroscopic properties of CopG resemble these perturbed type I copper sites, but the local structure of Cu1–3 more closely resembles the CuA sites (Fig. 6, C and D). The binding sites for Cu1–3 are comprised of both sulfur (a soft ligand) and nitrogen (a harder ligand) and, thus, would be consistent with the environments observed for either Cu(I) or Cu(II) (29, 35). Further studies will be required to rigorously assign the electronic structure and range of oxidation states of the copper cluster. The Cu4 site has 4 sulfur ligands, with only one from cysteine. Cu(I) sites most frequently utilize the soft ligands cysteine and methionine (29); therefore, the Cu4 site would be expected to bind Cu(I) preferentially.
CopG as a Cu(I)/Cu(II) oxidoreductase
The thioredoxin fold is utilized in many biological roles, where its active-site CxxC motif enables it to perform redox chemistry by forming a disulfide between the two cysteines and reducing an oxidized substrate (36, 37). For example, thioredoxin itself utilizes this mechanism to maintain cytoplasmic proteins in a reduced state (38). In E. coli, the inner membrane protein DsbD transfers reducing equivalents from cytoplasmic thioredoxin to periplasmic DsbC, which reduces incorrectly formed disulfides in misfolded periplasmic proteins. DsbD and DsbC both utilize the CxxC motif within their thioredoxin domains for this purpose (39, 40), but other thioredoxin-like proteins can serve as oxidases, extracting electrons from the substrate to reduce the active-site disulfide. An example is DsbA from E. coli, which catalyzes disulfide formation in the bacterial periplasm (39). Therefore, the biological roles of thioredoxins include reductases and oxidases, with the redox potential of the active-site disulfide being tuned to facilitate these different functions (41, 42).
CopG, a periplasmic protein, includes a clearly identifiable thioredoxin domain. Further, in crystal form I the side chains of Cys 13 and 16 are in the thiolate form, whereas in form II a disulfide has formed between them, representing reduced and oxidized states of CopG, respectively (Fig. 7). Taken together, these findings imply that CopG is a redox enzyme that utilizes the catalytic cycle common to other thioredoxin proteins. Whereas an active-site disulfide is a defining feature of this structural class, the presence of the copper cluster is unique to CopG. The physical proximity of the disulfide and the copper cluster leads to the hypothesis that the biological function of CopG involves action as a copper oxidoreductase. The formation of the disulfide in the oxidized state displaces a bound copper ion, also consistent with the interpretation that copper is a substrate that is bound and released during the catalytic cycle. The copper content of CopG in solution was measured to be 3.4 ± 1 equivalents per CopG. A noninteger value between 3 and 4 may be because of a loss of some copper during the purification process but also may indicate a mixed population of oxidized and reduced molecules with 3 and 4 bound coppers, respectively.
To ascertain whether this activity is possible, it is instructive to consider the range of redox potentials of thioredoxin-like proteins relative to that of copper ions. The standard reduction potential for the reduction of Cu(II) to Cu(I) is 159 mV, so if the reduction potential of CopG is below −159 mV, it should be capable of reducing Cu(II). The potentials measured for thioredoxin proteins range from E°′ = −270 mV for E. coli thioredoxin to E°′ = −124 mV for DsbA (43). These values bracket that for copper reduction, indicating that sufficient reducing power can be achieved by the thioredoxin fold. However, if CopG is an enzyme that catalyzes reduction of Cu(II), it must also be capable of catalyzing the reverse reaction, oxidation of Cu(I). Whereas a lower redox potential would favor reduction over oxidation, the concentrations of Cu(I) and Cu(II) will dictate whether there is a driving force for the reaction to proceed. The relative concentrations of these ions are subject to dramatic reversals (see below), which could provide conditions for CopG to operate alternately as a reductase and as an oxidase.
If CopG does function in this manner, it would require reducing equivalents to reach (or be removed from) the Cys 13–16 disulfide to carry out successive rounds of catalysis. The availability of these could then serve a regulatory function. Structurally, reducing equivalents could be transferred via the pocket lined by Trp 19 and Leu 23. A plausible electron transfer path from this pocket to the copper cluster would utilize the absolutely conserved residues Trp 19 and His 55 (Fig. 8). To reduce a disulfide, 2 electrons are needed. A possible reason for the tetranuclear cluster would be to accommodate electrons delivered singly but donated in pairs through oxidation of a disulfide. Two coppers would then be substrates for the enzyme, and the other two could serve as cofactors to store reducing equivalents. Based on this reasoning, we propose that the reaction catalyzed by CopG is 2 Cu(II) +2e− 2 Cu(I).
Figure 8.

Proposed electron transfer path. A, section through CopG solvent-accessible surface bisecting pocket at left and showing Cu4 (red) on opposite face of protein. B, protein interior showing potential relay system (black) for reducing equivalents between pocket, disulfide, and copper cluster. Note the visible surface shows the interior face.
Structural features of the metal-binding sites also appear consistent with a role as an oxidoreductase. When functioning as a Cu(II) reductase, crystal form I would represent an enzyme-substrate complex with Cu(II) at the Cu3 site adjacent to the reduced Cys 13 and 16 and at the Cu4 site (Fig. 9A). The oxidation of 2 thiolate side chains to disulfide liberates 2 electrons. The most plausible immediate acceptor of the first electron would be the copper ion bound at the adjacent site. Thus, Cu3 would be reduced to Cu(I), and upon dissociation because of displacement by the disulfide, one electron would be carried off. Dissociation of Cu3 would also require the uptake of a proton to reform the backbone amide on Cys 16. The second electron could be used to reduce Cu4 to Cu(I). The more polarizable Cu(I) at site 4 would be stabilized relative to Cu(II) because of interactions with sulfurs from Met 90 and 97, which act as soft ligands (35). There is not an obvious path for direct dissociation of Cu3, but upon disulfide formation and concomitant reduction of two of the coppers, the ion at site 4 could dissociate and be replaced by the one displaced from the Cu3 site. Form II would then represent a resulting enzyme-product complex where Cu3 has been displaced and Cu4 has been reduced to Cu(I) (Fig. 9B).
Figure 9.

Models for CopG action. A and B, proposed oxidation states of metal cluster in crystal forms I and II. In crystal form I, the disulfide is reduced and Cu3 and Cu4 are oxidized. In form II, Cu3 is displaced by the disulfide after being reduced, and the Cu4 site remains occupied by Cu(I). C, proposed cellular roles for CopG. Under aerobic conditions, CopG acts as a reductase to facilitate Cu(I) export by the CusCBA efflux pump. Under anaerobic conditions, CopG acts as an oxidase to detoxify Cu(I).
Proposed biological role
Genes encoding CopG are frequently found in association with genes encoding CueO (a multicopper oxidase), CopA [a Cu(I)-transporting P-type ATPase], and CusCBA [a Cu(I)-transporting RND efflux pump], suggesting both a functional role in copper handling and, more specifically, that CopG works in conjunction with these proteins. (However, in P. aeruginosa, copG is not in close proximity to any known metal resistance genes.) In Gram-negative bacteria, the RND pumps include a variety of divalent metal cation transporters as well as the Cus system, which transports monovalent ions Cu(I) and Ag(I) from the periplasm across the outer membrane. None of the divalent transporters have been shown to export Cu(II). A cus operon has not been unambiguously assigned in P. aeruginosa, but an RND system upregulated in the presence of copper was recently described (22).
The presence of an exogenous methionine ligand on each Cu4 site in both crystal structures suggests that CopG hands off this copper to some other protein that utilizes methionine for copper ligation. As all the metal sites characterized in the Cus system fit this description, a direct handoff from CopG to components of the Cus efflux system seems appropriate for further study. (A caveat is that CopG is present in some organisms that lack the cus locus [24], but there are other copper resistance systems that could also serve as acceptors [44].) In E. coli, the Cus system consists of the outer membrane protein CusC, the metallochaperone CusF, the adapter protein CusB, and the pump CusA (45). CusF utilizes 2 methionines, a histidine, and a tryptophan to bind Cu(I) and has been shown to activate the efflux channel by donating bound Cu(I) to the adapter protein CusB (46). CusB binds copper with three methionines (47). One copper binding site on CusA consists of three methionines (48), and the mechanism of copper export likely involves relay of Cu(I) along additional sites featuring methionine pairs (49). The Cus export pathway utilizes metal handoff from CusF to CusB and CusA, and these processes suggest how CopG might do the same (46, 50). Metal transfer from the CusF/Cu(I) complex to CusB involves a tripartite complex where ligands from both CusF and CusB are simultaneously present on the transferred metal (51), suggesting metal transfer is achieved by successive ligand displacement while maintaining Cu(I) in a protein-bound state. CusF directly donates Cu(I) to an entry site on CusA, most likely containing 2 methionine sulfur ligands (50). Pseudomonas aeruginosa, in common with many other bacteria, lacks a CusF gene. It may be that P. aeruginosa CopG carries out a role similar to that of CusF by passing off bound copper to CusB and/or CusA.
Fig. 9C depicts a working model for CopG action based on the hypothesis that it acts as a reversible oxidoreductase. The model is in accordance with the results of the copper resistance assays shown in Fig. 2. These assays show that the deletion of copG has no effect on copper survival under aerobic conditions but is deleterious under anaerobic conditions. Before considering the proposed role of CopG, it is useful to consider the oxidation states of copper ions under the two conditions and the current understanding of how these ions are handled. The cytoplasm is a reducing environment, so cytoplasmic copper is found in the Cu(I) state. From the cytoplasm, Cu(I) is transported to the periplasm by the P-type ATPase CopA (52). The two primary mechanisms for coping with rising periplasmic copper levels are detoxification and export. Under aerobic conditions, copper resistance is achieved by utilizing both strategies (Fig. 9C, left). Cu(I) can be exported directly by the Cus system (45, 53) and is oxidized to Cu(II) by the multicopper oxidase CueO, using oxygen as an electron acceptor (9, 10). The periplasm is an oxidizing environment, so the pool of periplasmic copper, once oxidized, would accumulate in the Cu(II) oxidation state. In contrast, under anaerobic conditions, detoxification by the oxygen-requiring CueO oxidase cannot occur. Copper will accumulate in the periplasm in the form of Cu(I), leaving export by the Cus system as the means to survive copper stress. (Fig. 9C, right).
Under aerobic conditions, periplasmic copper accumulates in the form of Cu(II). No dedicated periplasmic copper reductase has been identified to date, leaving unanswered how the periplasmic Cu(II) pool produced by the CopA/CueO system ultimately could be exported from the cell by the Cu(I)-specific CusCBA export pump. The model proposes that, driven by high Cu(II) concentrations, CopG could act as a Cu(II) reductase to carry out this function. This would occur when Cu(I) levels have been reduced by the action of the Cus system to levels that enable mass action to drive the reductase reaction. During aerobic copper stress, both the detoxification and export pathways can function, allowing the cell to reach its maximum tolerance independently of the presence of copG. This is consistent with the results of the copper resistance assay, which showed no difference in copper tolerance between ΔcopG and its parent strain in the presence of oxygen.
Under anaerobic conditions, Cu(I) will be the primary form of periplasmic copper and there will be negligible amounts of Cu(II). The model proposes that under these conditions, mass action would cause CopG to act as a Cu(I) oxidase. Subject to the availability of oxidizing equivalents, CopG in this role would complement the action of CueO by providing an alternative path to oxidation of Cu(I) that can operate anaerobically. When Cu(I) builds up to levels that exceed the transport capacity of the Cus system, CopG protects the cell by carrying out a detoxification function. This proposed function is supported by the results of the copper resistance assay: with the loss of this additional protection, the ΔcopG strain becomes more sensitive to copper than the parent strain.
Experimental procedures
Bioinformatic analysis
Homologous sequences were identified using a BLAST (54) search, excluding P. aeruginosa sequences. The top-scoring 1000 sequences were aligned using Clustal Omega (55). Site-specific residue conservation was determined as the percentage of residues identical to the P. aeruginosa CopG sequence in this alignment.
Copper resistance assay
Strains PAO1 and ΔcopG were obtained from the University of Washington transposon mutant collection as MPAO1 and PW8743, respectively (56, 57). Copper resistance was assessed by comparison of the growth of these strains on LB-agar plates with 0, 1, 2, 3, 3.5, 4, and 5 mm CuSO4. Cultures were grown to log phase in LB media, diluted to an optical density of 0.1, and plated in serial dilution from 1 (i.e., 100)- to 107-fold. Identical plates were prepared and grown in a standard incubator to assess aerobic growth or enclosed in an airtight container with GazPak EZ sachets to assess anaerobic growth. Plates were incubated for 18 h at 37 °C to assess growth under aerobic conditions or for 24 h at 37 °C under anaerobic conditions. Individual assays used 2 plates at each copper concentration, each of which had 4 replicates of the PAO1 and ΔcopG strains. Assays were scored by noting the maximum dilution showing visible growth for each dilution series.
Cloning and expression of CopG
The full-length CopG protein sequence was analyzed using SignalP (58, 59) to search for the presence of a localization sequence and to delineate the domain boundary. A synthetic gene for P. aeruginosa CopG lacking a leader sequence (consisting of residues 23–148) was obtained from ATUM Bio. This gene was amplified using primers 5′-AGATTGGTGGCGAGCCGCTGGCAATTGACG-3′ and 5′-GAGGAGAGTTTAGACGTACTCGGCCAGGACGCT-3′, subcloned into the expression vector petHSUL (a kind gift from P. Loll) using ligation-independent cloning (60), and verified by sequencing. The resulting expression vector was transformed into BL21/DE3 for protein production. Protein was expressed in overnight cultures of ZYP5052 (61) at 37 °C supplemented with 1 mm CuSO4. All subsequent steps were done on ice or at 4 °C. Cells were lysed by sonication in 50 mm Tris, pH 8.0, 300 mm NaCl (buffer A), and insoluble material was removed by centrifugation. Clarified lysate was applied to a Ni2+-NTA column, and the column was washed with 10 column volumes of buffer A with 30 mm imidazole. The fusion protein was eluted with buffer A with 400 mm imidazole, digested overnight with sumo protease with dialyzing against buffer A with 5 mm beta-mercaptoethanol (BME). BME and residual imidazole were removed by dialysis against buffer A, and the digested mixture was applied to a second Ni2+-NTA column. The eluted protein was precipitated with 4 M ammonium sulfate, resuspended in 10 mm Tris, pH 8.0, 100 mm NaCl (buffer B) supplemented with 5 mm BME, applied to a Sephacryl S-100 column, and then eluted in buffer B. Fractions were analyzed with SDS-PAGE, and those that were >95% pure were pooled and used for experiments. Selenomethionine-labeled CopG was produced in M9 minimal medium supplemented with 0.1 mm CuSO4 and purified as described above. Sumo protease was produced as described previously (60).
Determination of metal content
To determine the metal content, purified protein samples were first dialyzed against 10 mm MES, pH 6.0, 1 mm tris(2-carboxyethyl)phosphine (TCEP). To obtain metal-free protein, protein was dialyzed twice against 10 mm MES, pH 6.0, 1 mm TCEP, 5 mm phenanthroline and then twice against 10 mm MES, pH 6.0, 1 mm TCEP. Samples were then mineralized in 40% nitric acid at 80 °C for 1 h, diluted to 4% nitric acid, and analyzed for copper content using an iCE 3000 (Thermo) graphite furnace atomic absorption spectrometer calibrated with standard solutions.
Crystallization
Crystals were grown using hanging drops from a 1:2 mixture of well solution with protein solution at 18.9 mg/ml in buffer B with 5 mm BME. Form I and II crystals grew under similar conditions. Data were collected from crystals with well solution containing 100 mm Na cacodylate, pH 6.5, 17% ethanol, and 0.2 m zinc acetate (form I) and 100 mm Na cacodylate, pH 6.8, 19% ethanol, and 0.2 m zinc acetate (form II). Crystals grew for 1–3 weeks at room temperature. Diffraction-quality crystals were equilibrated in a cryoprotectant solution containing a 2:1 mixture of well solution and glycerol, flash-frozen, and stored in liquid nitrogen.
Structure determination
Data collection was carried out at 100K on beamlines 24-ID-E and 24-ID-C at the Advanced Photon Source and processed with XDS (62). The structure was solved using single anomalous diffraction using data collected at the Se edge from P61 crystals. Initial phasing and model construction were carried out with SHELX (63) and Phenix (64). The model obtained was completed manually using iterative manual rebuilding with Coot (65) and refinement with Phenix (64). The residue numbering used in the model uses residue 1 to denote the N-terminal residue of the mature form of the protein without the periplasmic leader sequence. The resulting structure was used as a search model to determine the structure from the P212121 crystal form, using Phaser and programs from the CCP4 suite (66, 67), and refined using Phenix. Anomalous difference Fourier maps from data collected at the Se, Cu, and Zn edges, phased with a model lacking metal ions, were used to discriminate between bound Cu and Zn atoms (see the supporting information). Data collection and refinement statistics are reported in Tables 2 and 3. Figures were prepared with Chimera (68).
Table 2.
Crystallographic statistics
| Parameter | Value(s) for: |
|||
|---|---|---|---|---|
| Form I | Form II | Cu anomalous | Zn anomalous | |
| Space group | ||||
| Cell dimensions(Å) | a = b = 86.13, c = 58.56 | a = 63.70, b = 87.46, c = 143.27 | a = b = 86.25, c = 58.52 | a = b = 86.26, c = 58.03 |
| Beamline | APS 24-ID-E | APS 24-ID-E | APS 24-ID-C | APS 24-ID-C |
| Wavelength (Å) | 0.9792 | 0.9792 | 1.2894 | 1.2827 |
| Resolution (Å) | 74.6–2.0 | 55.4–2.5 | 74.7–2.4 | 74.7–2.5 |
| No. of observations (unique) | 32888 | 28435 | 18931 | 16721 |
| No. of observations (total) | 243010 | 188433 | 199394 | 81885 |
| Completeness (%) | 99.9 (99.7) | 99.8 (99.2) | 99.5 (98.3) | 99.9 (98.8) |
| Redundancy | 7.4 (7.4) | 6.6 (6.1) | 10.5 (10.5) | 4.9 (4.5) |
| I/σ | 10.7 (2.2) | 8.9 (1.9) | 12.6 (1.8) | 10.1 (1.9) |
| CC1/2a | 99.6 (59.4) | 99.6 (56.6) | 99.9 (76.4) | 99.5 (63.3) |
| Rmeas (%) | 12.3 (86.2) | 13.6 (103.1) | 12.7 (98.9) | 15.1 (117.6) |
| Anomalous completeness | 99.8 | 99.4 | 99.7 | |
| Anomalous multiplicity | 7.1 | 10.4 | 4.8 | |
| Δanom corr. half-setsb | 0.389 | 0.626 | 0.477 | |
| Se sites | 10 | |||
| Figure of merit | 0.47 | |||
a CC1/2, Correlation percentage between intensities from randomly chosen half-sets.
b Δanom corr. half-sets, Correlation between anomalous differences from randomly chosen half-sets.
Table 3.
Refinement statistics
| Data set namea | Value(s) for form: |
|
|---|---|---|
| I | II | |
| Rwork (%) | 17.8 | 19.2 |
| Rfree (%) | 22.3 | 24.7 |
| Molecules in AU | 2 | 6 |
| No. of protein atoms | 1837 | 5482 |
| No. of solvent atoms | 91 | 124 |
| Bond length RMSD (Å) | 0.010 | 0.009 |
| Bond angle RMSD (°) | 1.099 | 1.286 |
| Ramachandran favored/outliers (%) | 96.3/0 | 98.1/0 |
| Avg B-value (Å2) | 35.0 | 53.0 |
| Avg B-value, coppers (Å2) | 28.6 | 49.8 |
| Avg B-value, waters (Å2) | 44.4 | 53.3 |
a RMSD, root mean square deviations.
Data availability
Atomic coordinates of CopG and crystallographic data were deposited in the Protein Data Bank (PDB) with accession codes 6WIS (form I) and 6WJE (form II). All other data are available within the article.
Supplementary Material
Acknowledgments
We thank M. Collazo from the UCLA crystallization facility for assistance with crystallization trials and advice on crystallization strategies and D. Cascio and M. Sawaya for assistance with crystallographic data collection and for many helpful discussions. We also thank K. Gorecki and our anonymous reviewers for insightful comments on the manuscript.
This article contains supporting information.
Author contributions—A. C. H. and M. M. M. conceptualization; A. C. H. and M. M. M. formal analysis; A. C. H. and M. M. M. supervision; A. C. H., N. A. R., and A. T. L. investigation; A. C. H., N. A. R., and A. T. L. visualization; A. C. H. methodology; A. C. H. writing-original draft; A. C. H. and M. M. M. project administration; A. C. H. and M. M. M. writing-review and editing; M. M. M. funding acquisition.
Funding and additional information—We thank the National Institutes of Health (GM079192) for support to M. M. M. Diffraction data were collected at the Northeastern Collaborative Access Team beamlines 24-ID-E and -C, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P41 GM103403). The Pilatus 6M detector on 24-ID-C beam line is funded by an NIH-ORIP HEI grant (S10 RR029205). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under contract no. DE-AC02-06CH11357. Pseudomonas strains were obtained from the University of Washington Transposon Mutant Collection, supported by NIH P30 DK089507. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- BME
- beta-mercaptoethanol
- TCEP
- tris(2-carboxyethyl)phosphine.
References
- 1. Casey A. L., Adams D., Karpanen T. J., Lambert P. A., Cookson B. D., Nightingale P., Miruszenko L., Shillam R., Christian P., and Elliott T. S. J. (2010) Role of copper in reducing hospital environment contamination. J. Hosp. Infect. 74, 72–77 10.1016/j.jhin.2009.08.018 [DOI] [PubMed] [Google Scholar]
- 2. Lazary A., Weinberg I., Vatine J. J., Jefidoff A., Bardenstein R., Borkow G., and Ohana N. (2014) Reduction of healthcare-associated infections in a long-term care brain injury ward by replacing regular linens with biocidal copper oxide impregnated linens. Int. J. Infect. Dis. 24, 23–29 10.1016/j.ijid.2014.01.022 [DOI] [PubMed] [Google Scholar]
- 3. White C., Lee J., Kambe T., Fritsche K., and Petris M. J. (2009) A role for the ATP7A copper-transporting ATPase in macrophage bactericidal activity. J. Biol. Chem. 284, 33949–33956 10.1074/jbc.M109.070201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vincent M., Duval R. E., Hartemann P., and Engels-Deutsch M. (2018) Contact killing and antimicrobial properties of copper. J. Appl. Microbiol. 124, 1032–1046 10.1111/jam.13681 [DOI] [PubMed] [Google Scholar]
- 5. Macomber L., and Imlay J. A. (2009) The iron-sulfur clusters of dehydratases are primary intracellular targets of copper toxicity. Proc. Natl. Acad. Sci. U S A 106, 8344–8349 10.1073/pnas.0812808106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Espírito Santo C., Lam E. W., Elowsky C. G., Quaranta D., Domaille D. W., Chang C. J., and Grass G. (2011) Bacterial killing by dry metallic copper surfaces. Appl. Environ. Microbiol. 77, 794–802 10.1128/AEM.01599-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Halliwell B., and Gutteridge J. M. C. (1984) Oxygen-toxicity, oxygen radicals, transition-metals and disease. Biochem. J. 219, 1–14 10.1042/bj2190001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Novoa-Aponte L., Ramirez D., and Arguello J. M. (2019) The interplay of the metallosensor CueR with two distinct CopZ chaperones defines copper homeostasis in Pseudomonas aeruginosa. J. Biol. Chem. 294, 4934–4945 10.1074/jbc.RA118.006316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Grass G., and Rensing C. (2001) CueO is a multi-copper oxidase that confers copper tolerance in Escherichia coli. Biochem. Biophys. Res. Commun. 286, 902–908 10.1006/bbrc.2001.5474 [DOI] [PubMed] [Google Scholar]
- 10. Lee S. M., Grass G., Rensing C., Barrett S. R., Yates C. J. D., Stoyanov J. V., and Brown N. L. (2002) The Pco proteins are involved in periplasmic copper handling in Escherichia coli. Biochem. Biophys. Res. Commun. 295, 616–620 10.1016/S0006-291X(02)00726-X [DOI] [PubMed] [Google Scholar]
- 11. Hao X., Lüthje F. L., Qin Y., McDevitt S. F., Lutay N., Hobman J. L., Asiani K., Soncini F. C., German N., Zhang S., Zhu Y.-G., and Rensing C. (2015) Survival in amoeba-a major selection pressure on the presence of bacterial copper and zinc resistance determinants? Identification of a copper pathogenicity island. Appl. Microbiol. Biotechnol. 99, 5817–5824 10.1007/s00253-015-6749-0 [DOI] [PubMed] [Google Scholar]
- 12. Staehlin B. M., Gibbons J. G., Rokas A., O'Halloran T. V., and Slot J. C. (2016) Evolution of a heavy metal homeostasis/resistance island reflects increasing copper stress in Enterobacteria. Genome Biol. Evol. 8, 811–826 10.1093/gbe/evw031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sommerhalter M., Lieberman R. L., and Rosenzweig A. C. (2005) X-ray crystallography and biological metal centers: is seeing believing? Inorg. Chem. 44, 770–778 10.1021/ic0485256 [DOI] [PubMed] [Google Scholar]
- 14. Krissinel E., and Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 10.1016/j.jmb.2007.05.022 [DOI] [PubMed] [Google Scholar]
- 15. Tatusov R. L., Koonin E. V., and Lipman D. J. (1997) A genomic perspective on protein families. Science 278, 631–637 10.1126/science.278.5338.631 [DOI] [PubMed] [Google Scholar]
- 16. El-Gebali S., Mistry J., Bateman A., Eddy S. R., Luciani A., Potter S. C., Qureshi M., Richardson L. J., Salazar G. A., Smart A., Sonnhammer E. L. L., Hirsh L., Paladin L., Piovesan D., Tosatto S. C. E., et al. (2019) The Pfam protein families database in 2019. Nucleic Acids Res. 47, D427–D432 10.1093/nar/gky995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Marchler-Bauer A., Bo Y., Han L., He J., Lanczycki C. J., Lu S., Chitsaz F., Derbyshire M. K., Geer R. C., Gonzales N. R., Gwadz M., Hurwitz D. I., Lu F., Marchler G. H., Song J. S., et al. (2017) CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 45, D200–D203 10.1093/nar/gkw1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kinch L. N., Baker D., and Grishin N. V. (2003) Deciphering a novel thioredoxin-like fold family. Proteins Struct. Funct. Bioinformatics 52, 323–331 10.1002/prot.10425 [DOI] [PubMed] [Google Scholar]
- 19. Gomis-Ruth F. X., Sola M., Acebo P., Parraga A., Guasch A., Eritja R., Gonzalez A., Espinosa M., del Solar G., and Coll M. (1998) The structure of plasmid-encoded transcriptional repressor CopG unliganded and bound to its operator. EMBO J. 17, 7404–7415 10.1093/emboj/17.24.7404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gutierrez-Barranquero J. A., de Vicente A., Carrion V. J., Sundin G. W., and Cazorla F. M. (2013) Recruitment and rearrangement of three different genetic determinants into a conjugative plasmid increase copper resistance in Pseudomonas syringae. Appl. Environ. Microbiol. 79, 1028–1033 10.1128/AEM.02644-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Teitzel G. M., Geddie A., De Long S. K., Kirisits M. J., Whiteley M., and Parsek M. R. (2006) Survival and growth in the presence of elevated copper: transcriptional profiling of copper-stressed Pseudomonas aeruginosa. J. Bacteriol. 188, 7242–7256 10.1128/JB.00837-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Quintana J., Novoa-Aponte L., and Arguello J. M. (2017) Copper homeostasis networks in the bacterium Pseudomonas aeruginosa. J. Biol. Chem. 292, 15691–15704 10.1074/jbc.M117.804492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wright B. W., Kamath K. S., Krisp C., and Molloy M. P. (2019) Proteome profiling of Pseudomonas aeruginosa PAO1 identifies novel responders to copper stress. BMC Microbiol. 19, 69 10.1186/s12866-019-1441-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Marrero K., Sanchez A., Gonzalez L. J., Ledon T., Rodriguez-Ulloa A., Castellanos-Serra L., Perez C., and Fando R. (2012) Periplasmic proteins encoded by VCA0261-0260 and VC2216 genes together with copA and cueR products are required for copper tolerance but not for virulence in Vibrio cholerae. Microbiology 158, 2005–2016 10.1099/mic.0.059345-0 [DOI] [PubMed] [Google Scholar]
- 25. Iwata S., Ostermeier C., Ludwig B., and Michel H. (1995) Structure at 2.8-angstrom resolution of cytochrome-c-oxidase from paracoccus-denitrificans. Nature 376, 660–669 10.1038/376660a0 [DOI] [PubMed] [Google Scholar]
- 26. Brown K., Tegoni M., Prudencio M., Pereira A. S., Besson S., Moura J. J., Moura I., and Cambillau C. (2000) A novel type of catalytic copper cluster in nitrous oxide reductase. Nat. Struct. Biol. 7, 191–195 10.1038/73288 [DOI] [PubMed] [Google Scholar]
- 27. Fisher O. S., Kenney G. E., Ross M. O., Ro S. Y., Lemma B. E., Batelu S., Thomas P. M., Sosnowski V. C., DeHart C. J., Kelleher N. L., Stemmler T. L., Hoffman B. M., and Rosenzweig A. C. (2018) Characterization of a long overlooked copper protein from methane- and ammonia-oxidizing bacteria. Nat. Commun. 9, 4276 10.1038/s41467-018-06681-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Savelieff M. G., and Lu Y. (2010) Cu-A centers and their biosynthetic models in azurin. J. Biol. Inorg. Chem. 15, 461–483 10.1007/s00775-010-0625-2 [DOI] [PubMed] [Google Scholar]
- 29. Rubino J. T., and Franz K. J. (2012) Coordination chemistry of copper proteins: how nature handles a toxic cargo for essential function. J. Inorg. Biochem. 107, 129–143 10.1016/j.jinorgbio.2011.11.024 [DOI] [PubMed] [Google Scholar]
- 30. Solomon E. I. (2006) Spectroscopic methods in bioinorganic chemistry: blue to green to red copper sites. Inorg. Chem. 45, 8012–8025 10.1021/ic060450d [DOI] [PubMed] [Google Scholar]
- 31. Liu J., Chakraborty S., Hosseinzadeh P., Yu Y., Tian S., Petrik I., Bhagi A., and Lu Y. (2014) Metalloproteins containing cytochrome, iron-sulfur, or copper redox centers. Chem. Rev. 114, 4366–4469 10.1021/cr400479b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Roger M., Biaso F., Castelle C. J., Bauzan M., Chaspoul F., Lojou E., Sciara G., Caffarri S., Giudici-Orticoni M. T., and Ilbert M. (2014) Spectroscopic characterization of a green copper site in a single-domain cupredoxin. PLoS ONE 9, e98941 10.1371/journal.pone.0098941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. King J. D., Harrington L., Lada B. M., He G., Cooley J. W., and Blankenship R. E. (2014) Site-directed mutagenesis of the highly perturbed copper site of auracyanin D. Arch. Biochem. Biophys. 564, 237–243 10.1016/j.abb.2014.10.003 [DOI] [PubMed] [Google Scholar]
- 34. LaCroix L. B., Shadle S. E., Wang Y. N., Averill B. A., Hedman B., Hodgson K. O., and Solomon E. I. (1996) Electronic structure of the perturbed blue copper site in nitrite reductase: Spectroscopic properties, bonding, and implications for the entatic/rack state. J. Am. Chem. Soc. 118, 7755–7768 10.1021/ja961217p [DOI] [Google Scholar]
- 35. Pearson R. G. (1963) Hard and soft acids and bases. J. Am. Chem. Soc. 85, 3533–3539 10.1021/ja00905a001 [DOI] [Google Scholar]
- 36. Qi Y., and Grishin N. V. (2005) Structural classification of thioredoxin-like fold proteins. Proteins Struct. Funct. Bioinformatics 58, 376–388 10.1002/prot.20329 [DOI] [PubMed] [Google Scholar]
- 37. Atkinson H. J., and Babbitt P. C. (2009) An atlas of the thioredoxin fold class reveals the complexity of function-enabling adaptations. PLoS Comput. Biol. 5, e1000541 10.1371/journal.pcbi.1000541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Arner E. S. J., and Holmgren A. (2000) Physiological functions of thioredoxin and thioredoxin reductase. Eur. J. Biochem. 267, 6102–6109 10.1046/j.1432-1327.2000.01701.x [DOI] [PubMed] [Google Scholar]
- 39. Denoncin K., and Collet J. F. (2013) Disulfide bond formation in the bacterial periplasm: major achievements and challenges ahead. Antioxidants Redox Signal. 19, 63–71 10.1089/ars.2012.4864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fabianek R. A., Hennecke H., and Thony-Meyer L. (2000) Periplasmic protein thiol: disulfide oxidoreductases of Escherichia coli. FEMS Microbiol. Rev. 24, 303–316 10.1111/j.1574-6976.2000.tb00544.x [DOI] [PubMed] [Google Scholar]
- 41. Jacobi A., Huber-Wunderlich M., Hennecke J., and Glockshuber R. (1997) Elimination of all charged residues in the vicinity of the active-site helix of the disulfide oxidoreductase DsbA–influence of electrostatic interactions on stability and redox properties. J. Biol. Chem. 272, 21692–21699 10.1074/jbc.272.35.21692 [DOI] [PubMed] [Google Scholar]
- 42. Bewley K. D., Dey M., Bjork R. E., Mitra S., Chobot S. E., Drennan C. L., and Elliott S. J. (2015) Rheostat re-wired: alternative hypotheses for the control of thioredoxin reduction potentials. PLoS ONE 10, e0122466 10.1371/journal.pone.0122466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Aslund F., Berndt K. D., and Holmgren A. (1997) Redox potentials of glutaredoxins and other thiol-disulfide oxidoreductases of the thioredoxin superfamily determined by direct protein-protein redox equilibria. J. Biol. Chem. 272, 30780–30786 10.1074/jbc.272.49.30780 [DOI] [PubMed] [Google Scholar]
- 44. Pontel L. B., and Soncini F. C. (2009) Alternative periplasmic copper-resistance mechanisms in Gram negative bacteria. Mol. Microbiol. 73, 212–225 10.1111/j.1365-2958.2009.06763.x [DOI] [PubMed] [Google Scholar]
- 45. Mealman T. D., Blackburn N. J., and McEvoy M. M. (2012) Metal export by CusCFBA, the periplasmic Cu(I)/Ag(I) transport system of Escherichia coli. Curr. Top. Membr. 69, 163–196 10.1016/B978-0-12-394390-3.00007-0 [DOI] [PubMed] [Google Scholar]
- 46. Bagai I., Rensing C., Blackburn N. J., and McEvoy M. M. (2008) Direct metal transfer between periplasmic proteins identifies a bacterial copper chaperone. Biochemistry 47, 11408–11414 10.1021/bi801638m [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bagai I., Liu W., Rensing C., Blackburn N. J., and McEvoy M. M. (2007) Substrate-linked conformational change in the periplasmic component of a Cu(I)/Ag(I) efflux system. J. Biol. Chem. 282, 35695–35702 10.1074/jbc.M703937200 [DOI] [PubMed] [Google Scholar]
- 48. Long F., Su C. C., Zimmermann M. T., Boyken S. E., Rajashankar K. R., Jernigan R. L., and Yu E. W. (2010) Crystal structures of the CusA efflux pump suggest methionine-mediated metal transport. Nature 467, 484–U140 10.1038/nature09395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Su C. C., Long F., and Yu E. W. (2011) The Cus efflux system removes toxic ions via a methionine shuttle. Protein Sci. 20, 6–18 10.1002/pro.532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chacon K. N., Perkins J., Mathe Z., Alwan K., Ho E. N., Ucisik M. N., Merz K. M., and Blackburn N. J. (2018) Trapping intermediates in metal transfer reactions of the CusCBAF export pump of Escherichia coli. Commun. Biol. 1, 192 10.1038/s42003-018-0181-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chacon K. N., Mealman T. D., McEvoy M. M., and Blackburn N. J. (2014) Tracking metal ions through a Cu/Ag efflux pump assigns the functional roles of the periplasmic proteins. Proc. Natl. Acad. Sci. U S A 111, 15373–15378 10.1073/pnas.1411475111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Padilla-Benavides T., Thompson A. M. G., McEvoy M. M., and Arguello J. M. (2014) Mechanism of ATPase-mediated Cu+ export and delivery to periplasmic chaperones the interaction of Escherichia coli CopA and CusF. J. Biol. Chem. 289, 20492–20501 10.1074/jbc.M114.577668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kim E. H., Rensing C., and McEvoy M. M. (2010) Chaperone-mediated copper handling in the periplasm. Nat. Product Rep. 27, 711–719 10.1039/b906681k [DOI] [PubMed] [Google Scholar]
- 54. Boratyn G. M., Camacho C., Cooper P. S., Coulouris G., Fong A., Ma N., Madden T. L., Matten W. T., McGinnis S. D., Merezhuk Y., Raytselis Y., Sayers E. W., Tao T., Ye J., and Zaretskaya I. (2013) BLAST: a more efficient report with usability improvements. Nucleic Acids Res. 41, W29–W33 10.1093/nar/gkt282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Madeira F., Park Y. M., Lee J., Buso N., Gur T., Madhusoodanan N., Basutkar P., Tivey A. R. N., Potter S. C., Finn R. D., and Lopez R. (2019) The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 47, W636–W641 10.1093/nar/gkz268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Jacobs M. A., Alwood A., Thaipisuttikul I., Spencer D., Haugen E., Ernst S., Will O., Kaul R., Raymond C., Levy R., Liu C. R., Guenthner D., Bovee D., Olson M. V., and Manoil C. (2003) Comprehensive transposon mutant library of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U S A 100, 14339–14344 10.1073/pnas.2036282100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Held K., Ramage E., Jacobs M., Gallagher L., and Manoil C. (2012) Sequence-verified two-allele transposon mutant library for Pseudomonas aeruginosa PAO1. J. Bacteriol. 194, 6387–6389 10.1128/JB.01479-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Nielsen H., Engelbrecht J., Brunak S., and von Heijne G. (1997) Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng. 10, 1–6 10.1093/protein/10.1.1 [DOI] [PubMed] [Google Scholar]
- 59. Armenteros J. J. A., Tsirigos K. D., Sonderby C. K., Petersen T. N., Winther O., Brunak S., von Heijne G., and Nielsen H. (2019) SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 37, 420–423 10.1038/s41587-019-0036-z [DOI] [PubMed] [Google Scholar]
- 60. Weeks S. D., Drinker M., and Loll P. J. (2007) Ligation independent cloning vectors for expression of SUMO fusions. Protein Expr. Purif. 53, 40–50 10.1016/j.pep.2006.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Studier F. W. (2005) Protein production by auto-induction in high-density shaking cultures. Protein Expr. Purif. 41, 207–234 10.1016/j.pep.2005.01.016 [DOI] [PubMed] [Google Scholar]
- 62. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 10.1107/S0907444909047337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sheldrick G. M. (2008) A short history of SHELX. Acta Crystallogr. A Found. Adv. 64, 112–122 10.1107/S0108767307043930 [DOI] [PubMed] [Google Scholar]
- 64. Adams P. D., Afonine P. V., Bunkoczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Struct. Biol. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G. W., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Struct. Biol. 67, 235–242 10.1107/S0907444910045749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- 69. Katti S. K., Lemaster D. M., and Eklund H. (1990) Crystal-structure of thioredoxin from Escherichia coli at 1.68A resolution. J. Mol. Biol. 212, 167–184 10.1016/0022-2836(90)90313-B [DOI] [PubMed] [Google Scholar]
- 70. Johnson W. C. (1992) Analysis of circular-dichroism spectra. Methods Enzymol. 210, 426–447 10.1016/0076-6879(92)10022-6 [DOI] [PubMed] [Google Scholar]
- 71. Lees J. G., Miles A. J., Wien F., and Wallace B. A. (2006) A reference database for circular dichroism spectroscopy covering fold and secondary structure space. Bioinformatics 22, 1955–1962 10.1093/bioinformatics/btl327 [DOI] [PubMed] [Google Scholar]
- 72. Liebschner D., Afonine P. V., Moriarty N. W., Poon B. K., Sobolev O. V., Terwilliger T. C., and Adams P. D. (2017) Polder maps: improving OMIT maps by excluding bulk solvent. Acta Crystallogr. D Struct. Biol. 73, 148–157 10.1107/S2059798316018210 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Atomic coordinates of CopG and crystallographic data were deposited in the Protein Data Bank (PDB) with accession codes 6WIS (form I) and 6WJE (form II). All other data are available within the article.