

Abstract
Sirtuin 6 (SIRT6) is a nuclear NAD+-dependent deacetylase of histone H3 that regulates genome stability and gene expression. However, nonhistone substrates and additional catalytic activities of SIRT6, including long-chain deacylation and mono-ADP-ribosylation of other proteins, have also been reported, but many of these noncanonical roles remain enigmatic. Genetic studies have revealed critical homeostatic cellular functions of SIRT6, underscoring the need to better understand which catalytic functions and molecular pathways are driving SIRT6-associated phenotypes. At the physiological level, SIRT6 activity promotes increased longevity by regulating metabolism and DNA repair. Recent work has identified natural products and synthetic small molecules capable of activating the inefficient in vitro deacetylase activity of SIRT6. Here, we discuss the cellular functions of SIRT6 with a focus on attributing its catalytic activity to its proposed biological functions. We cover the molecular architecture and catalytic mechanisms that distinguish SIRT6 from other NAD+-dependent deacylases. We propose that combining specific SIRT6 amino acid substitutions identified in enzymology studies and activity-selective compounds could help delineate SIRT6 functions in specific biological contexts and resolve the apparently conflicting roles of SIRT6 in processes such as tumor development. We further highlight the recent development of small-molecule modulators that provide additional biological insight into SIRT6 functions and offer therapeutic approaches to manage metabolic and age-associated diseases.
Keywords: sirtuin, histone deacetylase (HDAC), cell metabolism, small molecule, cancer, chromatin, gene expression, aging, metabolic disorder, longevity, sirtuin, activator, SIRT6
Sirtuin 6 (SIRT6) is a histone deacetylase with critical homeostatic roles in many cellular pathways. SIRT6-deficient mice display spontaneous genomic instability, dysregulated glycolysis, increased tumorigenesis, and early onset aging-like degeneration, ultimately dying at around 4 weeks (1, 2). Loss-of-function homozygous mutation of SIRT6 led to brain and muscle developmental defects and ultimately to late fetal loss, marking the first instance of a mutated chromatin factor causing perinatal lethality in humans (3). Similar phenotypes were observed in a SIRT6-knockout study using CRISPR-Cas9–generated cynomolgus monkeys, indicating a conserved and obligatory role for SIRT6 function in primate development (4). Loss-of-function mutations were identified in naturally arising human cancers and later shown to be sufficient to drive tumor formation in mice (5). These genetic insights illustrate critical cellular functions of SIRT6 and establish the need to understand which catalytic functions and molecular pathways are driving SIRT6-associated phenotypes.
SIRT6 is a member of a highly conserved family of enzymes that couple NADH (NAD+) cleavage to the removal of an acyl group from the lysine of protein substrates, producing deacylated protein, nicotinamide, and O-acyl-ADP-ribose (Fig. 1). Prior to the discovery of enzymatic activity, the founding member of this family, Sir2 (silent information regulator 2), was identified in Saccharomyces cerevisiae from a screen for genome silencing factors (6–8) and was later shown to promote longevity by suppressing the formation of extrachromosomal rDNA circles (9–11). Compelling evidence indicates that Sir2 represses transcription at silent mating type loci and subtelomeric sequences via its histone deacetylase activity (8, 12–14).
Figure 1.

SIRT6 is an NAD+-dependent protein deacylase. SIRT6 couples the cleavage of NAD+ to the removal of an acyl group from substrate protein. Catalysis produces deacylated protein, O-acyl-ADP-ribose, and nicotinamide. Acyl modifications (-R) of lysine can be but are not limited to acetyl, propionyl, hexanoyl, octanoyl, decanoyl, dodecanoyl, myristoyl, or palmitoyl moieties.
Sirtuin homologs exist in all phyla of life (15–17) and comprise class III histone deacetylases (HDACs), which uniquely utilize NAD+ as a cofactor of catalysis rather than a catalytic zinc as observed in class I, II, and IV HDACs (18). The catalytic requirement for NAD+ has led the field to regard sirtuins as critical factors linking energy metabolism to cellular homeostasis (19). Although initially defined as histone deacetylases, sirtuins have many nonhistone substrates and can catalyze the removal of acetyl, long-chain acyl, and acid acyl moieties from lysine substrates. The seven mammalian sirtuins (SIRT1–7) show varying cellular localization and regulate diverse functions. All sirtuins can catalyze the removing of an acyl group of the ε-amino of lysine, although the specificities for distinct acyl groups and catalytic efficiencies vary greatly.
Mechanistic studies of SIRT6 activity lagged behind those of other sirtuins owing in part to the difficulty in detecting in vitro deacetylase activity. As with the early reports on Sir2 and other sirtuins, the possibility that SIRT6 acts as a protein ADP-ribosyltransferase was reported (20–22). The development of more sensitive assays revealed that SIRT6 is a deacetylase with catalytic efficiencies nearly ∼100–1,000 times lower than the most active sirtuins (23). Strong support for enzymatic deacylation activity was provided when it was revealed that SIRT6 is a more efficient long-chain deacylase and can be catalytically activated toward deacetylation by small molecules, many of which are long-chain fatty acids (24, 25). SIRT6 is implicated in many diverse biological functions, although the specific biochemical activity essential for each phenotype remains rather murky. Identifying which activity mediates which biological phenotype is therefore critical to understand the molecular functions of SIRT6 and to accurately develop small-molecule effectors specific to appropriate activities of this emerging therapeutic target. Here we highlight cellular functions ascribed to SIRT6, where evidence allows the inherent biochemical activities of SIRT6 to be matched with the observed phenotype. The first section of this review covers the physiological and cellular phenotypes noted when SIRT6 is removed, mutated, or overexpressed. We comment on the strength of the evidence linking biological functions with biochemical activity and provide substrate identities when possible. The prospect of targeting SIRT6 necessitates a detailed understanding of its structure and biochemical activities. Accordingly, we devote much of this review to describing the unique structural and catalytic features of SIRT6, with particular attention paid to small-molecule modulators that have the potential to resolve SIRT6 functions and provide a pathway for SIRT6-targeted therapeutics.
Cellular and physiological functions
Overview of SIRT6 molecular functions
SIRT6 is localized to the nucleus, where it mediates DNA repair, regulates the expression of metabolic genes, and maintains genomic stability (Fig. 2). Through these functions, SIRT6 has been described as a tumor suppressor and positive regulator of longevity. Evidence indicates that SIRT6 regulates these processes primarily via deacetylation of histone and nonhistone proteins, although long-chain deacylation, ADP-ribosylation, and recruitment of proteins to chromatin are other biochemical functions ascribed to some SIRT6-dependent phenotypes. This review will focus on processes in which direct SIRT6 substrates or protein-binding partners have been identified.
Figure 2.
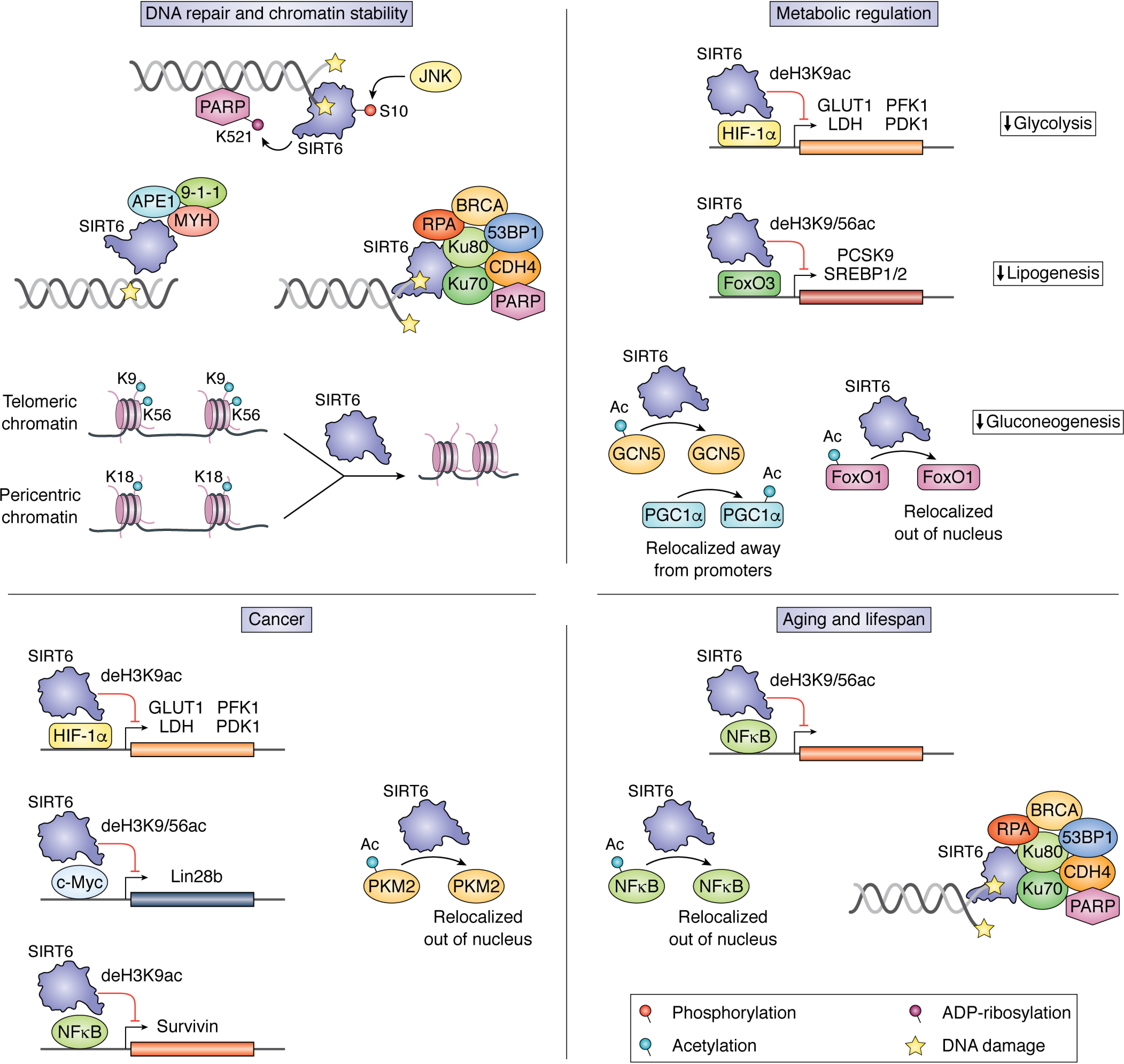
Cellular and molecular functions of SIRT6. SIRT6 is a critical regulator of cellular homeostasis with roles in mediating DNA repair, regulating expression of genes, and maintaining genomic stability. This figure outlines mechanistically understood functions of SIRT6 that contribute to DNA repair and chromatin stability, metabolic regulation, cancer, and aging and lifespan. These functions can be broadly separated into three classes: (i) recruitment of factors to chromatin, (ii) corepression of transcription factors through histone deacetylation, and (iii) deacetylation of nonhistone proteins, resulting in a change of localization or activity. In addition to these functions, SIRT6 also acts as a pioneer factor through direct binding of DNA damage and performs global deacetylation of pericentric and telomeric DNA to prevent aberrant transcription and telomeric damage.
DNA repair and chromatin maintenance
Some of the earliest phenotypic studies on SIRT6 knockout cells demonstrated increased susceptibility to DNA-damaging agents and higher levels of chromosomal aberrations, suggesting a critical role for SIRT6 in maintaining genomic stability (1). Later reports demonstrated that SIRT6 is recruited to double-strand breaks (DSBs) within seconds and further recruits DNA repair factors to initiate DNA damage response (DDR) (26, 27). This fast response is attributed to the ability of SIRT6 to directly bind to open-ended DSBs, where Onn et al. (28) postulate that each monomer of a SIRT6 dimer binds one loose ssDNA end of a DSB. Onn et al. suggested a potential DNA-binding tunnel along the core of SIRT6 that mediates ssDNA binding independent of the N and C termini. Another study demonstrated that c-Jun N-terminal kinase phosphorylates SIRT6 on Ser10 in response to oxidative stress, leading to binding of SIRT6 to DSBs and subsequent recruitment and mono-ADP-ribosylation of poly(ADP-ribose) polymerase 1 (PARP1) (29). The mechanism by which the DNA repair factor, PARP1, is recruited to DSBs following SIRT6 phosphorylation and whether this is mediated by direct SIRT6 interactions are unknown. Serine 10 is located on the N terminus of SIRT6, which was previously shown to be dispensable for DNA binding, suggesting multiple pathways of passive and active recruitment to DSBs (30).
Following recruitment of SIRT6 to sites of DNA damage, multiple studies have indicated contexts in which SIRT6 either deacetylates nucleosomes or actively recruits repair factors to mediate DDR. SIRT6 knockout mouse embryonic fibroblasts and knockdown human fibroblasts show impaired nonhomologous end joining (NHEJ) and homologous recombination efficiencies, respectively, whereas SIRT6 overexpression improves both processes (21). Mao et al. (21) demonstrated that this effect was facilitated by SIRT6-mediated recruitment of repair factors 53BP1 and NBS1 to sites of DNA damage, and loss-of-function mutants suggested that both deacetylase and ADP-ribosyltransferase activities of SIRT6 were required to facilitate NHEJ. The requirement of SIRT6 ADP-ribosyltransferase activity was attributed to the modification of PARP1 on Lys521, resulting in its activation; however, the target of the requisite deacetylase activity in this study was not identified. Separately, SIRT6 has been shown to globally deacetylate H3K9 in response to DNA damage and co-localize with DNA-dependent protein kinase, Ku70, and Ku80 at DSBs (27), although it remains unknown which factor pioneers DSB binding. Hou et al. (31) showed that SIRT6 can recruit chromodomain helicase DNA-binding protein 4 (CDH4) to sites of DNA damage to facilitate chromatin relaxation and efficient homologous recombination. This effect was lost with the SIRT6 catalytic mutant H133Y, implicating a role for SIRT6 catalysis in this DDR pathway. Toiber et al. (26) reported that SIRT6 deacetylates H3K56 in response to DNA damage and recruits the SNF2H chromatin remodeler, making chromatin more accessible and allowing for efficient recruitment of DNA repair factors RPA, 53BP1, and BRCA1 (26). In this study, sites of DNA damage were identified by the presence of γH2AX, a phosphorylated form of the histone subtype H2AX present at sites of DSBs (32, 33). A complex containing SIRT6 and SNF2H was later reported to stabilize γH2AX at DSB sites (34). In contrast to the well-studied roles of SIRT6 in DSB repair, limited data also suggest that SIRT6 mediates base excision repair (BER). Overexpression of SIRT6 exhibited improved BER efficiency in mouse fibroblasts, whereas deletion increased sensitivity to BER-associated DNA-damaging agents (1, 35). Separate studies demonstrated that SIRT6 interacts with and stimulates BER factors, including APE1, MYH, and Rad9-Rad1-Hus1, following oxidative damage (36). A recent study compared SIRT6 proteins from rodents with varying lifespan and found a correlation between SIRT6-mediated DSB repair efficiency and the lifespan of the host organism (37). The difference between repair efficiencies of orthologous SIRT6 enzymes was largely isolated to five amino acids that were predicted to be surface-exposed based on homology modeling with human SIRT6. Substitution of these five amino acids from a short-lived mouse SIRT6 sequence to those of long-lived beaver SIRT6 sequence was sufficient to increase NHEJ and decrease global acetylation in culture and improve the survivability of human fibroblasts exposed to γ-irradiation (37).
Several studies implicated roles for SIRT6 in maintaining genome stability through the repression of telomeric and pericentric regions of chromatin. SIRT6 was shown to deacetylate telomeric chromatin in mammalian cells at H3K9 and H3K56 (38, 39). Michishita et al. (38) reported that SIRT6 deacetylation of H3K9 was required for stable association of WRN protein, and depletion of SIRT6 resulted in telomeric defects that resemble those observed in Werner syndrome, an aging disorder characterized by mutated WRN. Furthermore, SIRT6-deficient HeLa cells displayed telomeric sequence loss, genomic instability, and chromosomal end-to-end fusions (40). Following oxidative damage, Gao et al. (41) observed that SIRT6 was required for directional telomere movement, a process that facilitates efficient DNA damage repair at telomeric chromatin. Additionally, SIRT6 overexpression in mice was reported to up-regulate telomerase reverse transcriptase and telomere repeat binding factor, although this study did not explore the mechanism of up-regulation or potential downstream effects on telomere length (42). Proper mitosis requires condensed and silenced peri-centric heterochromatin at centromeres to protect against genomic instability and cellular senescence during cell division. Tasselli et al. (43) showed that SIRT6 repressed transcription at these regions through the deacetylation of H3K18 and also noted that knockdown of SIRT6 resulted in hyperacetylation and aberrant transcription of these pericentric chromatin domains. Collectively, SIRT6's ability to deacetylate proteins, ADP-ribosylate PARP1, or mediate protein-protein interactions with repair proteins has been implicated in controlling DNA stability and repair. Clearly, better genetic and biochemical tools are needed to provide a deeper molecular understanding of which biochemical functions of SIRT6 control various aspects of these processes and dictate histone substrate specificity. The development of better tools will require a robust understanding of catalytic mechanism, regulated activity, and structure-function relationships. Therefore, our current knowledge in these areas is presented later in the review. Despite these outstanding questions, the extensive research summarized here depicts a clear role for SIRT6 in facilitating DNA repair and stabilizing heterochromatic regions.
Metabolic regulation
A number of studies have demonstrated the ability of SIRT6 to maintain cellular homeostasis through the regulation of glucose and lipid metabolism. Early studies characterizing SIRT6 knockout mice showed a lethal phenotype at 2–3 weeks of age, likely resulting from hypoglycemia (1, 44). This hypoglycemic phenotype was later attributed to misregulation of glycolytic genes in the absence of SIRT6. Specifically, SIRT6 was found to co-repress the transcription factor hypoxia-inducible factor 1α (HIF-1α), in effect down-regulating glycolytic genes, including glucose transporter-1 (GLUT1), lactate dehydrogenase (LDH), phosphofructokinase-1 (PFK1), and pyruvate dehydrogenase kinase-1 (PDK1) (45). In the same study, SIRT6 was reported to directly inhibit expression of these genes by deacetylating H3K9 at promoters of targeted genes. SIRT6 knockout resulted in the derepression of these glycolytic genes and increased glucose uptake, reminiscent of the “Warburg effect,” a metabolic hallmark often observed in cancer cells (46). Accordingly, SIRT6 loss-of-function mutation and down-regulation is observed in several naturally arising human cancers. Mouse model studies by Sebastián et al. (2) and Kugel et al. (5) demonstrated that loss-of-function mutation in SIRT6 was sufficient to form tumors and was independent of known oncogene activation. A detailed summary of the roles of SIRT6 in cancer are discussed in a following section. Separately, Gertman et al. (47) generated two SIRT6 mutants with enhanced in vitro rates of kcat against long-chain myristoyl substrate. Stable transfection of these mutants in mouse embryonic fibroblasts demonstrated an increased ability to inhibit the expression of GLUT1, PDK1, and PFK1 relative to WT SIRT6. Interestingly, a global increase in acetylation of H3K9 and H3K56 (47) was noted, suggesting that the demyristoylase activity of SIRT6 may regulate glycolysis and may be distinct from H3 deacetylase activity. However, these mutants also bound HIF-1α more tightly in vitro, making it difficult to definitively conclude whether more efficient recruitment to HIF-1α genes or higher demyristoylase activity was responsible for the down-regulation of these glycolytic genes (47).
SIRT6 also regulates glucose metabolism by inhibiting gluconeogenesis, a metabolic process that generates glucose from noncarbohydrate sources. Mostoslavsky et al. (1) found that SIRT6 knockout mice are hypoglycemic and display elevated expression of gluconeogenic genes. Although this observation alone could simply be dismissed as a proper liver response to hypoglycemia, work by Dominy et al. demonstrated active regulation of gluconeogenesis in the liver by SIRT6. Mechanistically, SIRT6 was described to deacetylate general control nonrepressed protein 5 (GCN5), which enhanced its acetyl-transferase activity toward peroxisome proliferator-activated receptor-γ coactivator 1α (PGC-1α) (48). PGC-1α is the primary regulator of hepatic gluconeogenesis and induces the expression of gluconeogenic genes (49). Following acetylation by GCN5, PGC-1α was found to relocate away from the promoters of gluconeogenic genes, resulting in decreased expression (48). Another study demonstrated that SIRT6 overexpression reduced gluconeogenic gene expression in WT, but not Forkhead box O1 (FoxO1) knockout mice (50). FoxO1 is a transcription factor required for activation of gluconeogenic genes in hepatic cells (49). Subsequent work by Zhang et al. (51) mechanistically described the FoxO1/SIRT6 regulatory axis by demonstrating that SIRT6 directly deacetylates FoxO1, leading to its nuclear export, thus resulting in decreased gluconeogenic gene expression.
SIRT6 has also been implicated in the regulation of lipid metabolism with studies reporting roles in both lipolysis and lipogenesis. Early work in mice demonstrated that liver-specific knockout of SIRT6 resulted in fatty liver formation owing to increased triglyceride synthesis and reduced β-oxidation, whereas mice overexpressing SIRT6 were protected from the accumulation of visceral fat, LDL-cholesterol, and triglycerides when fed high-fat diets (52, 53). Fat-specific SIRT6 knockout increased obesity-related phenotypes in mice (54, 55). Mechanistically, SIRT6 was found to represses the expression of proprotein convertase subtilisin/kexin type 9 (PCSK9) and sterol-regulatory element–binding protein 1 and 2 (SREBP1 and SREBP2). This was demonstrated to occur through SIRT6 recruitment to the promoter of Pcsk9, a critical regulator of LDL-cholesterol, via interactions with the transcription factor FoxO3 where SIRT6 deacetylates H3K9 and H3K56 to reduce expression of Pcsk9, resulting in lowered LDL-cholesterol levels (56). FoxO3 was also shown to recruit SIRT6 to promoters of SREBP genes, positive regulators of lipogenic genes, resulting in inhibition of expression by promoter-specific SIRT6 deacetylation of H3K9 and H3K56 (57–59). Interestingly, miRNAs expressed from the introns of SREBP, miR-33a and miR-33b, as well as the most common hepatic miRNA, miR-122, were shown by Elhanati et al. (58, 60) to down-regulate SIRT6 expression, indicating intricate regulation of SIRT6 toward lipid metabolism. Separately, SIRT6 was noted to promote β-oxidation of fatty acids during fasting through regulation of the PPARα coactivator, NCOA2. In this distinct mechanism, SIRT6 was found to decrease NCOA2 acetylation at Lys780, resulting in the activation of PPARα and increased expression of hepatic β-oxidation genes (61). It remains to be determined whether NCOA2 Lys780 is a direct substrate of SIRT6 deacetylation.
Aging and lifespan
The yeast homolog and founding member of the sirtuin family, Sir2, is implicated in lifespan extension through repression of tandem rRNA gene arrays (rDNA) (62, 63). Subsequent studies have solidified links between aging and the mammalian homolog, SIRT6. SIRT6 overexpression in mice was found to extend the lifespan of male mice by 15% in a mechanism thought to be driven through reduced insulin-like growth factor 1 (IGF1) signaling. Zhang et al. (64) demonstrated that caloric restriction prevented aging-related renal insufficiency and suggested that this effect was mediated via the up-regulation of SIRT6 in response to caloric restriction, which resulted in the repression of nuclear factor-κB (NF-κB). NF-κB is a nuclear transcription factor associated with inflammatory responses, aging, and apoptosis (65). SIRT6 was found to represses NF-κB activity both by deacetylating H3K9 at NF-κB promoters and by deacetylating Lys310 of the NF-κB p65 subunit (RelA), resulting in its nuclear export (64). Xu et al. (35) observed a decrease in SIRT6 levels with age in human fibroblasts. In separate studies, aging-related decreases in SIRT6 levels impaired the reprogramming potential of iPSCs from old mice and decrease DNA repair efficiency (66, 67). Another study demonstrated that increased copy number of Sirt6 was sufficient to restore lifespan in Caenorhabditis elegans deficient in Atm, a DNA repair factor (68). Studies on human populations have discovered various SNPs that correlate both positively and negatively with longevity (69–71). The mechanistic outcomes of these polymorphisms are yet unknown; however, they present a potentially powerful tool for further probing the link between SIRT6 and aging. Work by Tian et al. (37), discussed previously, compared long- and short-lived rodents and demonstrated a SIRT6-mediated increase in DNA-repair efficiency among long-lived species corresponding to increased in vitro deacetylase and self-mono-ADP-ribosyltransferase activities. Transgenic Drosophila expressing beaver (long-lived) SIRT6 showed increased lifespan relative to those expressing mouse (short-lived) SIRT6, directly linking SIRT6 activity to increased lifespan.
Cancer
With dysregulated DNA damage repair and metabolic homeostasis as hallmarks of cancer, SIRT6 has been implicated in oncogenesis, proliferation, and poor prognosis. Down-regulation of SIRT6 in human cancers has been reported in hepatocellular carcinomas, ovarian cancers, and colon cancers (72–75). Furthermore, the Cancer Cell Line Encyclopedia reports that the SIRT6 gene is deleted in 35% of all cancer cell lines in their database (76, 77). SIRT6 knockout in mouse embryonic fibroblasts led to tumor formation in mouse xenograft studies independent of known oncogene activation (2). This tumorigenesis was attributed to derepression of glycolytic genes resulting in a Warburg-like phenotype characterized by increased glucose uptake and anaerobic glycolysis. Similarly, SIRT6 point mutations identified in human cancers were shown to be loss-of-function, and xenograft studies by Kugel et al. (5) demonstrated that these loss-of-function mutations were sufficient to drive tumor formation in mice. Copy loss number of SIRT6 has been observed in 60% of pancreatic cancer cell lines, and studies on low SIRT6-expressing cell lines demonstrated accelerated tumorigenesis in a mechanism independent of enhanced glycolysis (78). Rather, SIRT6 was shown to deacetylate H3K9 and H3K56 at the promoter of Lin28b, an RNA-binding fetal oncogene that promotes transformation and tumor progression (78, 79). It was therefore concluded by Kugel et al. that loss of SIRT6 leads to c-Myc transcription factor–driven overexpression of Lin28b and subsequent tumor growth. Additionally, SIRT6 was shown to deacetylate H3K9 at the promoter of survivin, an oncoprotein up-regulated in most cancers that impairs tumorigenesis (72, 80). Positive outcomes for breast cancer patients correlated with SIRT6 levels and inversely correlated with SIRT6 phosphorylation at Ser338 (81). This was mechanistically supported through the demonstration that Ser338 phosphorylation of SIRT6 by the kinase AKT1 induced SIRT6 ubiquitination by MDM2, targeting SIRT6 for protease degradation. In colon cancer cells, the tumor suppressor USP10, a ubiquitin-specific peptidase, was found to be critical for deubiquitinating SIRT6 and stabilizing protein levels to negatively regulate the transcriptional activity of the c-Myc oncogene (82). In separate studies, SIRT6 SUMOylation was shown to promote interaction with c-Myc, resulting in the deacetylation of H3K56, but not H3K9, and transcriptional repression at the promoters of c-Myc genes (83). The site of SIRT6 SUMOylation was localized to the C terminus and isolated to either Lys296, Lys300, Lys316, or Lys332. Furthermore, Bhardwaj et al. (84) demonstrated that SIRT6 interacts with and deacetylates nuclear pyruvate kinase M2 (PKM2), an oncogenic transcriptional coactivator, on Lys433, which resulted in its nuclear export and a downstream reduction in cell proliferation, migration potential, and invasiveness.
Despite the vast majority of data supporting the roles of SIRT6 as a tumor suppressor, some evidence suggests that SIRT6 may promote tumor survival in a context-dependent manner. It has been observed that SIRT6 is up-regulated in cases of squamous cell carcinoma, chronic lymphocytic leukemia, multiple myeloma, and acute myeloid leukemia (85–88). Three of the aforementioned diseases are hematopoietic cancer lines, which curiously are the only tumor type exhibiting gain of SIRT6 gene copies according to the Cancer Cell Line Encyclopedia (2, 77). In the case of multiple myeloma, a disease characterized by a highly unstable genome, Cea et al. (85) noted increased SIRT6 expression as an adaptive response to combat genomic instability. In acute myeloid leukemia, SIRT6 overexpression was demonstrated to protect tumors from DNA damage through the deacetylation and activation of DNA-dependent protein kinase, a DDR element that facilitates DSB repair (85, 89). In the same study, SIRT6 depletion in multiple myeloma cells was found to potentiate genotoxic treatment and increase proliferation rate. Mechanistically, SIRT6 was shown to interact with the transcription factor ELK1 to deacetylate H3K9 and suppress the expression of ERK signaling–related genes and reduce proliferation (85). Bauer et al. (90) reported that SIRT6 promotes metastasis through the up-regulation of pro-inflammatory cytokines, including IL-8 and TNF-α. This effect was noted to occur through SIRT6 catalytic activity, resulting in increased intracellular ADP-ribose levels, an activator of the Ca2+ channel TRPM2 required for LPS-induced production of IL-8 and TNF-α (90, 91). In hepatocellular carcinoma (HCC), Lee et al. (92) identified SIRT6 as a tumor promoter that prevented DNA damage and cellular senescence. Separate studies of HCC by Ran et al. (93) identified a separate oncogenic role for SIRT6. Namely, SIRT6 was found to deacetylate H3K9 at the promoter of Bcl-2–associated X protein (Bax), an apoptotic activator, resulting in HCC protection from apoptosis. Contrasting research by Zhang et al. (94) suggested a tumor-suppressing role of SIRT6 toward HCC cells by blocking the ERK1/2 signaling pathway, although the mechanism of SIRT6/ERK cross-talk is unknown.
Clearly, SIRT6 is an important regulator of metabolism and genome maintenance, and the ability to suppress tumor formation is well-documented. Nevertheless, cell- and context-dependent examples of SIRT6-driven tumor survival have also been reported. These contrasting studies suggest a complex role for SIRT6 in tumor development and survival that is still being evaluated. One interpretation of current data are that within the context of a healthy cell, DNA repair and genomic stability prevents oncogenesis; however, following specific types of transformation, these same functions can protect the aberrant cells from genotoxic therapies or the accumulation of excess deleterious mutations. The context-dependent role of SIRT6 as either a tumor suppressor or tumor driver opens the door to informed disease-specific pharmacological activation or inhibition. Likewise, the development of small-molecule SIRT6 modulators will aid the study of SIRT6 activity in varying disease contexts. To provide context for a subsequent review of small-molecule modulators, we will first discuss the overall structure of SIRT6 and unique features that differentiate this sirtuin from other family members.
Molecular architecture of SIRT6
Domain overview
Sirtuins 1–7 are an evolutionarily conserved family of enzymes that share a core catalytic domain spanning ∼250 amino acids (Fig. 3). The catalytic core comprises an NAD+-binding Rossmann fold domain and a structural zinc-binding domain containing four conserved cysteine residues, which coordinate a zinc ion (95). Catalysis occurs in a hydrophobic cleft situated between these two domains that varies in size and composition between sirtuin family members to confer specificity toward acyl substrates (24, 96, 97). Additionally, sirtuins contain diverse N and C termini that can direct cellular localization and protein-protein interactions.
Figure 3.
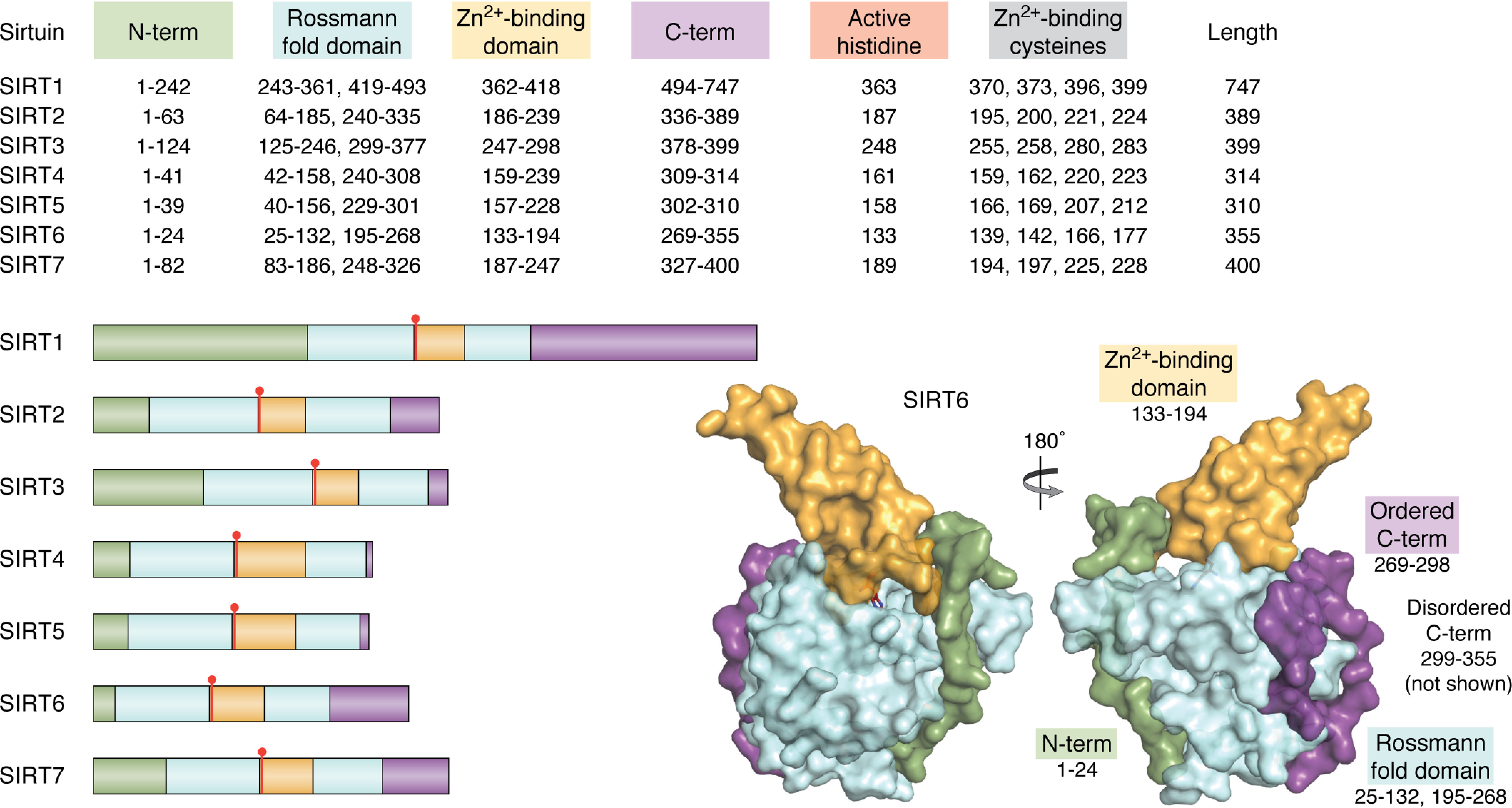
Overall domain architecture of SIRT1-7. Sirtuins share a conserved catalytic core consisting of a Rossmann fold domain (light blue), Zn2+-binding domain (orange), and catalytic histidine (red). Human sirtuins have unique N-terminal (green) and C-terminal (purple) domains that vary in length and sequence. The overall architecture of SIRT6 shows that the Rossmann fold domain, although separated by primary sequence, forms a single globular domain that the N terminus and part of the C terminus (residues 269-294) are structured around. The majority of the C terminus (residues 295-355) is disordered and has not been structurally solved. This structure was produced using entry 3ZG6 from the RCSB Protein Data Bank.
Rossmann fold domain and NAD+ binding
SIRT6 contains a Rossmann fold structural motif responsible for NAD+ binding that is common among nucleotide-binding proteins (Fig. 3) (98). This domain spans residues 27–132 and 195–268 and is structurally represented by a six-stranded parallel β-sheet in between two helices on one side and four helices on the other (23). The Rossmann fold domain forms a platform for the NAD+-binding site, whereas the flanking cofactor binding loop forms additional contacts with NAD+ to stabilize the bound form (99). Structural analysis of bound adenosine diphosphate ribose (ADPr) revealed 18 possible polar contacts between primarily the Rossmann fold domain and ADPr (Fig. 4C). ADPr is an NAD+ metabolite lacking the nicotinamide moiety that inhibits SIRT6 catalysis, suggesting that it similarly occupies the NAD+-binding pocket (23, 100). Substrate binding by SIRT1-3 has been reported to follow an ordered binding mechanism in which acetyl peptide precedes that of NAD+ (101, 102). SIRT6 is the only known sirtuin to bind NAD+ in the absence of acyl substrate (23, 103). The ability to bind NAD+ in the absence of acyl peptide substrate is structurally explained by the NAD+ binding loop of SIRT6 maintaining an ordered helix in the absence of peptide substrate (23), whereas this loop is disordered in the absence of substrate for SIRT2,3,5 (101, 104, 105). Interestingly, a naturally occurring point mutation (D63Y) in human cancer exists in the NAD+-binding loop and has been shown to abolish deacetylase activity (5). Additionally, a study by Mao et al. (21) reported that S56Y and R65A variants lacked cellular deacetylase activity. Subsequent studies further demonstrated that Arg65 is critical for in vitro activation of deacetylation (103). These studies suggest that the NAD+-binding domain of SIRT6 is a hotspot for loss-of-function mutation.
Figure 4.

SIRT6 structural features of ligand binding and overall architecture. SIRT6 sequentially binds NAD+ followed by acyl-substrate prior to initiating chemical catalysis. The noncatalytic NAD+ surrogate lacking nicotinamide, ADP-ribose, is often used in structural studies to prevent catalytic turnover. A, ADP-ribose (pink) binds to the interior of the Rossman fold domain with the nicotinamide-ribose proximal to the catalytic His-133 (red). H3K9 myristoyl peptide (yellow) binds to the solvent-exposed exterior of both the Rossmann fold and Zn2+-binding domain with the substrate lysine inserted into the catalytic site proximal to the opposing side of His-133. The N terminus has been excluded to more clearly demonstrate the relative positioning of ligands. B, polar contacts between the H3K9 peptide substrate (yellow) and residues SIRT6 (light blue). C, extensive polar contacts between residues of SIRT6 (tan) and ADP-ribose (pink). D, SIRT6 contains an extended hydrophobic tunnel, relative to the latent tunnels observed in other structurally solved sirtuins, that accommodates the myristoyl chain (yellow spheres) of an H3K9myr peptide (yellow sticks). This hydrophobic tunnel is formed between the Rossmann fold domain (light blue surface) and Zn2+-binding domain (orange surface), is composed of numerous nonpolar residues (magenta sticks), and is sealed by the N terminus (green surface). E, the Zn2+-binding domain of SIRT6 utilizes four conserved cysteines (magenta sticks) to coordinate a zinc-ion (magenta sphere). This domain contains a 10-amino acid extension between the third and fourth cysteines, resulting in an unordered loop (blue) unique to SIRT6. These structures were produced using entry 3ZG6 from the RCSB Protein Data Bank.
Zinc-binding domain
The Zn2+-binding domain of the sirtuins (class III HDACs) is thought to be strictly structural and does not participate in catalysis, in contrast to the active-site zinc ion in the zinc-dependent class I, II, and IV HDACs (99). The sirtuin zinc domain utilizes four conserved cysteine residues to coordinate Zn2+ (SIRT6: Cys141, Cys144, Cys167, and Cys178). Most sirtuins contain the conserved sequence motif Cys-X-X-Cys-X15-20-Cys-X-X-Cys; however, SIRT6 incorporates a 10-amino acid insert between the third and fourth cysteine, resulting in a unique extended flexible loop (Fig. 4E) (23). Interestingly, C terminus of Hsp70-interacting protein (CHIP) has been shown to noncanonically ubiquitinate SIRT6 on Lys170, a residue that resides in the extended flexible loop on the Zn2+-binding domain (106). This ubiquitination prevented proteasome-mediated degradation of SIRT6 and led to increased protein t1/2. Studies involving SIRT1 and the yeast homolog, Sir2, demonstrated that cysteine S-nitrosation or mutation of these zinc-coordinating cysteines to alanine results in loss of deacetylase function (107–109). Molecular dynamics simulations suggested that loss of activity was due to repositioning of the Zn2+-binding domain away from the catalytic domain following zinc release, resulting in allosteric changes that disrupt NAD+ and acyl substrate binding (110). Trichostatin A (TSA) is well-documented as a Class I and II HDAC inhibitor that functions by chelating the active-site zinc of these enzymes (111). Curiously, it was demonstrated that TSA specifically inhibits SIRT6 (Ki = 2 μm) over SIRT1–3 and SIRT5 (Ki > 50 μm) against H3K9ac peptide substrate (110). Subsequent kinetic analyses suggested that TSA competitively inhibited SIRT6 with respect to H3K9ac substrate (110). A structure of SIRT6 bound to TSA was solved in a separate study revealing binding to a SIRT6 specific region at the distal end of the hydrophobic cleft, explaining its isoform selectivity (112). TSA is often used in an effort to specifically inhibit class I and II HDACs in cellular experiments at concentrations ranging from 0.1 to 2 μm. Given this expanded role of TSA as a zinc chelation–independent inhibitor of SIRT6 with a Ki of 2 μm, methodological caution should be taken to rule out “nonspecific” inhibition of SIRT6.
Hydrophobic pocket
A conserved salt bridge between the Rossmann fold and zinc-binding domains of SIRT5 and many Sir2 homologs is thought to contribute to the stabilized positioning of these domains (105, 109). SIRT6 lacks this salt bridge (predicted by homology modeling to occur between Arg182 and Asp190), resulting in a structure that is splayed open relative to other sirtuins (23). This expanded cleft between domains contains an elongated hydrophobic pocket that accommodates long-chain acyl substrates. A structure of SIRT6 bound to H3K9myr revealed nonpolar contacts of the myristoyl chain with Ala13, Pro62, Phe64, Trp71, Pro80, Phe82, Phe86, Val115, Leu132, Met157, Leu186, and Ile219 (Fig. 4D) (24). Furthermore, structures containing bound myristoyl peptide are the only ones to date in which the SIRT6 N terminus became structured, revealing that it covers part of this hydrophobic pocket at the interface of the Rossmann fold and zinc-binding domains (24, 113). Together, this suggests that acyl substrate occupation of the hydrophobic pocket can lead to an ordered structure and provides one possible explanation for why long-chain deacylation is more efficient than short-chain deacetylation. This hydrophobic pocket has also been implicated as an activator-binding site through both kinetic and structural studies. Kinetically, the activators CL5D and myristic acid displayed competitive inhibition toward myristoyl substrate (25, 103). Crystal structures of SIRT6 in the presence of the activators UBCS039, MDL-801, and cyanidin demonstrated their occupation of the distal hydrophobic pocket away from the active site (114, 115). Structural alignment of these deacetylase activators with myristoylated peptide demonstrates varying levels of overlap, resulting in differing effects toward competitive inhibition of long-chain deacylation. These kinetics will be discussed further in subsequent sections of this review. Structural analysis of H3K9 peptide binding reveals primarily hydrogen bonds between the main chain of the peptide and the surface of SIRT6 with just Thr11 and Gln6 of the peptide substrate participating in side-chain interactions (Fig. 4B). Microarray analysis using ∼6,800 acetyllysine substrates demonstrated a SIRT6 preference for uncharged residues around acetyllysine except for negatively charged residues at –4 and +2 (116); however, none of the three identified in vivo histone substrates (H3K9/18/56ac) satisfy these reported in vitro preferences. This suggests that selectivity for histone substrate may be dependent on other factors such as protein-protein binding and optimized orientation between SIRT6 and target sites on nucleosomes. The nature of the acyl group modification on lysine might also facilitate SIRT6-substrate interactions. Consistent with this notion, SIRT6 performs more efficient deacylation in vitro against substrates containing acyl groups ranging from 8 to 14 carbons in length (25); however, identification and validation of long-chain acyl substrates in vivo remains limited (117, 118). Directed evolution of SIRT6 conducted using SIRT6 ancestral libraries identified multiple point mutations that resulted in enhanced demyristoylation of peptide substrate (47). The mutation M157H, located in the hydrophobic pocket, was identified in two separately evolved SIRT6 variants and resulted in improved kcat of demyristoylation. In silico modeling led the authors to propose improved catalysis through better positioning of NAD+ and peptide substrate; however, the precise contributions of this mutation are unknown. Despite the structural and biochemical evidence that SIRT6 preferentially hydrolyzes long-chain acyl groups, compelling cellular evidence suggests that SIRT6 exerts biological regulation primarily through the direct deacetylation of specific sites on histone and nonhistone proteins (38, 39, 43).
N- and C-terminal extensions
In addition to the conserved catalytic core consisting of a Rossmann fold and zinc-binding domain, individual sirtuins harbor unique N- and C-terminal extensions (Fig. 3). Structurally, the 24-amino acid N terminus of SIRT6 is surface-exposed and lacks secondary structural elements. The second half of the N terminus (Asp14–Phe24) is ordered in crystal structures and is adjacent to the Rossmann fold domain. The first half of the N terminus (Met1–Ala13) has only been observed to be ordered upon H3K9myr peptide binding, which makes contacts with both the Rossmann fold domain and zinc-binding domain to seal the hydrophobic pocket. Given that SIRT6 performs more efficient long-chain deacylation in vitro, it follows that the ordering of this domain may facilitate catalysis. The N terminus of SIRT6 has been shown to be critical for H3K9 and H3K56 deacetylase activity and for chromatin association in cells; however, this region is dispensable for nuclear localization (30). No SIRT6 structures to date have been solved in the presence of acetylated substrates, so it remains to be seen whether canonical acetyl substrates can induce ordering of the N terminus. The N-terminal domain of SIRT6 is highly conserved among mammalian homologs and can be phosphorylated at Ser10 to facilitate DNA repair (29, 119).
The C terminus of SIRT6 (Leu269–Ser355) is significantly larger than the N terminus and is reported to contain a nuclear localization signal (30). This C terminus is predicted to be an intrinsically disordered region (IDR) and is composed of 24.7% prolines with a pI of 10.1 (119). To date, only a short region (Leu269–Glu298) of this C terminus has been solved by crystallography, with the rest remaining unstructured (Fig. 3). Given the difficulty in crystallizing this inherently disordered domain, some researchers have opted to remove this region to facilitate crystal formation (23, 24). Interestingly, it has recently been suggested that heterochromatin can form through phase separation into liquid droplets, and many of the proteins that form these condensates contain IDRs (120, 121). Given the role of SIRT6 in maintaining heterochromatin and telomeres (38, 40, 122), it is an emerging and exciting question whether the C-terminal IDR participates in heterochromatic condensates. In vivo phosphorylation of the C terminus at Thr294, Ser303, Ser330, and Ser338 has been reported (119, 123, 124). The SIRT6 variant S338A disrupted interactions with nuclear pore complexes as determined by immunoprecipitation-MS, suggesting that phosphorylation of the C terminus may modulate protein-protein interactions (119). However, there are no additional data suggesting the cellular function for this potentially regulated interaction.
Catalytic activities
Catalytic mechanism
Sirtuins remove acyl moieties from the ε-amino group of lysines and, in the process, consume one molecule of NAD+ for every one molecule of acyl chain removed (125). Three products are generated: nicotinamide, deacylated peptide/protein, and acylated ADP-ribose. After both substrates are bound, the glycosidic bond between nicotinamide and ribose is cleaved, and the acyl oxygen attacks the C1 of ribose, resulting in an adduct between the two substrates (Fig. 5). Kinetic isotope effect experiments suggest that the glycoside bond is largely broken before the addition of the acyl oxygen (126), although the net rate constant for this first chemical step is greatly influenced by the nucleophilicity of the attacking oxygen (127). The lifetime of the resulting alkylimidate intermediate can be extended by replacing a catalytic histidine (His133 in SIRT6), which would normally facilitate (via general base action) the attack of the ribose 2′-OH on the imidate intermediate (Fig. 5), forming a bicyclic adduct (128, 129). During catalysis, the histidine to alanine mutant protein is partially stalled at the imidate intermediate, allowing water or other solvent nucleophiles (methanol/ethanol) to react, essentially turning the enzyme into an acetyl substrate–dependent NAD+ hydrolase, with no net deacetylation (128). These results have implications for the weak ability of sirtuins to mediate protein ADP-ribosylation under some circumstances (discussed further below). During the normal catalytic cycle for deacylation, formation of the bicyclic intermediate (Fig. 5, species iii) is followed by attack of a water molecule to form intermediate iv, followed by cleavage of the C–N bond to liberate the final two products, O-acyl-ADP-ribose and deacetylated lysine. Evidence for these catalytic steps and intermediates has come from biochemical, structural, and theoretical experiments (128, 130–133).
Figure 5.
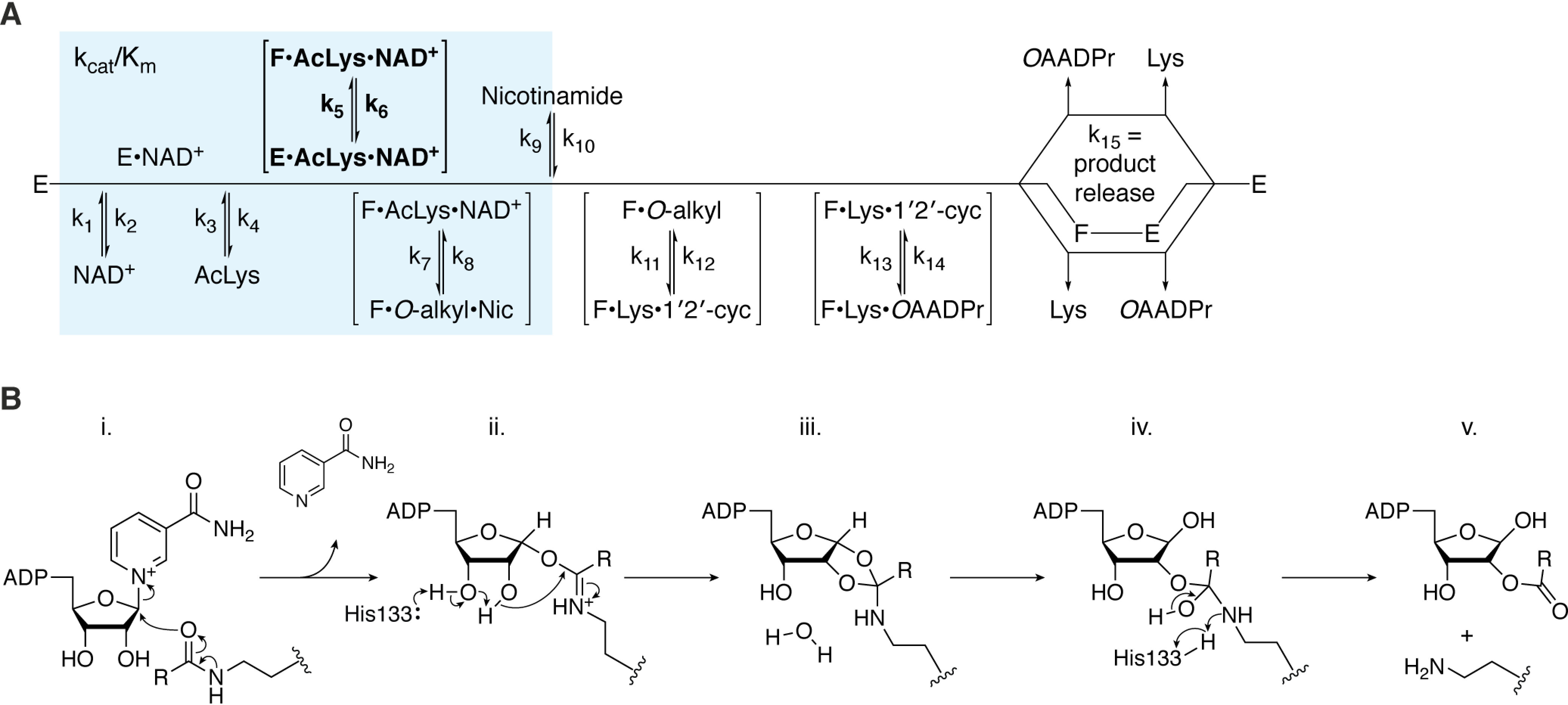
Mechanism of SIRT6 catalysis. SIRT6 performs NAD+-dependent deacylation of a substrate lysine (AcLys), resulting in the formation of nicotinamide (Nic), O-acyl-ADP-ribose (OAADPr), and deacylated lysine (Lys). A, the kinetic scheme of SIRT6 (E) begins with ordered sequential binding of NAD+ followed by acyl-lysine substrate. Next, the complex undergoes a conformational change (boldface, E→F) that has been shown to be activated by some small-molecule SIRT6 activators of deacetylation or directly by long-chain acyl substrates. Chemical catalysis is initiated by formation of the alkylimidate intermediate (O-alkyl) and release of nicotinamide. Subsequently, SIRT6 catalyzes the formation of the 1′,2′-cyclic intermediate (1′2′-cyc) and then its subsequent water-mediated collapse to form the final products. After release of deacylated lysine and O-acyl-ADP-ribose, SIRT6 is ready for its next round of catalysis. B, chemical catalysis; i, substrate acyl-oxygen performs nucleophilic addition on the 1′-carbon of the nicotinamide ribose, resulting in the C1′-O-alkylamidate intermediate and release of nicotinamide. ii, His133 acts as a general base to facilitate the intramolecular nucleophilic attack of the nicotinamide ribose 2′-hydroxyl on the O-alkylamidate carbon affording the 1′,2′-cyclic intermediate. iii, water-catalyzed hydrolysis of the 1′,2′-cyclic intermediate yields the tetrahedral intermediate. iv, positively charged His133 donates a proton to the imino group of the tetrahedral intermediate, resulting in cleavage of the C–N bond and yielding the final products. v, O-Acyl-ADPr and deacylated lysine products are released from SIRT6.
Early reports on the possible reactions catalyzed by sirtuins included both deacetylation and ADP-ribosylation (13–15, 125, 130, 134). To date, a preponderance of data supports the main catalytic activity as NAD+-dependent deacylation, with unique specificities between sirtuins for various acyl groups on the lysine ε-amine. Whereas most studies have ascribed deacylation as the biological function of sirtuins, a few others have suggested that the protein ADP-ribosylation activity of some sirtuins is responsible for certain cellular functions. Such reports suggest that SIRT4 can ADP-ribosylate GDH (135), that SIRT2 can target BSA (134), and that SIRT6 can ADP-ribosylate PARP1 (21). The role of sirtuin-dependent ADP-ribosylation remains enigmatic. From a biochemical perspective, protein ADP-ribosylation can be explained through the intrinsic catalytic properties of sirtuins (22, 125, 136). However, parsing deacylation from ADP-ribosylation in cellular assays remains challenging, largely because both activities occur through the same active site. In vitro, ADP-ribosylation activity pales compared with deacylation, and the ability of SIRT6 to perform multiple rounds of catalytic turnover for ADP-ribosylation has not been reported. Nevertheless, in cellular contexts, it is possible that there are molecular circumstances that greatly promote ADP-ribosylation over deacylation. With SIRT6, the unique kinetic mechanism might allow SIRT6 to facilitate such a proposed switch toward more favorable ADP-ribosylation.
Detailed kinetic mechanisms for a few sirtuins are reported. Generally, sirtuins bind NAD+ after acylated substrates are bound (101, 102, 137). Nicotinamide is the first product produced and released by the enzymes (Fig. 5). After completing the final chemical steps of catalysis, deacylated product and O-acyl-ADPr are released, with O-acyl-ADPr likely the last product to be released. Interestingly, SIRT6 displays a distinct kinetic mechanism that permits NAD+ binding in the absence of acylated substrate (Fig. 5) (23, 103). Also, after both substrates bind SIRT6, reports suggest a slow conformational step that enables formation of the first bisubstrate adduct, the alkylimidate intermediate (103). This conformational step was greatly enhanced when long-chain acylated substrates were used or when SIRT6 was activated by fatty acids (FAs) or other small-molecule activators (103). Among sirtuins, the rate-limiting steps in turnover appear to vary, depending on the intrinsic properties of each enzyme, choice of acylated substrate, and whether the enzyme is activated. Accordingly, these factors affect the Km for NAD+ as well as the acylated substrate (138). These observations have important implications for the differential NAD+ dependence for sirtuin-catalyzed deacylation. Current data suggest that low NAD+ levels will favor SIRT1, SIRT2, and SIRT6 long-chain deacylation over deacetylation, whereas higher NAD+ levels will preferentially enhance deacetylation (138). The idea of stimulating cellular sirtuin activity has long been a goal of many researchers and pharma industries. Two main strategies have been explored: (i) the use of vitamin B3 compounds to boost cellular NAD+ levels and (ii) the use of allosteric activators that enhance catalytic efficiency. SIRT1 is the most studied human sirtuin and the first sirtuin targeted for therapeutic development with natural products and synthetic small molecules as allosteric activators (139–141). In mouse studies, promising evidence supports the therapeutic potential of targeting SIRT1 activation (142–144). These studies provided proof of concept for therapeutically targeting sirtuin activation.
Activation
Sirtuins have been targets of drug discovery efforts since the discovery that their biological functions are implicated in age-related diseases (145). Investigations into the molecular function of SIRT6 had lagged that of SIRT1 in part because of SIRT6's poor in vitro deacetylase activity, which is nearly ∼1,000 times lower than that of other sirtuins (23). It was later demonstrated that SIRT6 is hundreds-fold more efficient at removing long-chain acyl modifications, such as myristoyl, compared with short-chain deacetylation, and structural studies revealed that the myristoyl group of this peptide is accommodated in a hydrophobic pocket extending past the catalytic site (24, 25). However, there was a discrepancy between cellular data suggesting that SIRT6 acted as a H3K9 deacetylase and the poor in vitro activity of SIRT6 against acetylated substrates versus long-chain acylated peptides.
A major advance in our understanding of in vitro SIRT6 deacetylation came with the discovery that this activity could be greatly enhanced in the presence of certain long-chain free fatty acids. Studies by Feldman et al. (25) demonstrated that free fatty acids ranging from 12 to 18 carbons were capable of improving the catalytic efficiency of SIRT6 deacetylation up to 35-fold. This evidence suggests that SIRT6 deacetylation can be regulated by small molecules and provides a possible mechanism for cellular SIRT6 to achieve efficient levels of deacetylase activity. Whereas we predict that SIRT6 can be activated by endogenous FAs, direct evidence of cellular SIRT6 activation by FAs has yet to be reported. A recent study provided compelling evidence that monounsaturated FAs activate SIRT1 specifically toward PGC-1a and FOXO3, but not histone H3, both in vitro and in vivo (146). With SIRT6, Michaelis–Menten kinetic analysis revealed that FAs drive activation primarily through an improved kcat/Km, suggesting that one or more steps prior to and including the release of nicotinamide are enhanced during activation (Fig. 5) (25). This same study showed that free myristic acid acts as a competitive inhibitor of long-chain demyristoylation. Together, these biochemical studies suggest that the hydrophobic pocket of SIRT6 is an activation site that can be either accommodated directly by long-chain acyl substrates or by FA activators of deacetylation. These findings and others have led to the development of structurally diverse small-molecule activators of SIRT6 (Table 1) that will be discussed in the following sections.
Table 1.
Identified activators of SIRT6 deacetylation
A select set of SIRT6 activators are presented in which detailed studies have been conducted or that demonstrate the diversity of small-molecule activators. Half-maximal effective concentration (EC50) values and kinetic parameters describing maximal observed activation were collected using the adjacent peptide substrate, with “+WW” referring to the addition of two tryptophans not native to the histone sequence used for the accurate quantification of substrate. Effects on SIRT6 demyristoylation of peptide substrate are also reported when available. NA, not available; the referenced study did not provide pertinent data.

Natural compounds as activators
Utilizing magnetic beads coated with catalytically active SIRT6, the flavonoids quercetin and vitexin were initially identified from fenugreek seed extract as in vitro inhibitors of SIRT6 deacetylation (147). Later studies of quercetin as well as an additional flavonoid, luteolin, demonstrated IC50 values of 24 and 1.9 μm, respectively (148). Curiously, dose-response assays of quercetin and luteolin showed 6- and 10-fold activation at higher concentrations with EC50 values of 990 and 270 μm, respectively. The concentration-dependent dual function of these flavonoids opens the possibility of multiple modes of small-molecule binding, although this has not been further explored. This same study demonstrated N-acylethanolamines, namely oleoylethanolamide and myristoyl-ethanolamide (EC50 of 3.1 and 7.5 μm), as more potent activators, albeit with lower maximum -fold activation of SIRT6, than their fatty acid cousins, oleic and myristic acid (EC50 of 90 and 246 μm) (25, 148).
Additional flavonoids were subsequently investigated, resulting in the identification of multiple anthocyanidins that stimulated SIRT6 activity, the most effective of which, cyanidin, activated SIRT6 55-fold against acetyl peptide with an EC50 of 460 μm as determined under proper steady-state dose-response conditions (115). Delphinidin, which contains three rather than two hydroxyl groups on the B-benzene ring of the flavonoid scaffold, yielded a similar EC50; however, it showed a weaker maximum activation of 6.3-fold. In silico docking of these compounds suggested surface contacts with the β6/α6 loop that capped the distal end of the hydrophobic pocket. The A- and B-benzene rings of cyanidin are predicted to rotate slightly compared with that of delphinidin, resulting in a closer contact and conformational change of Trp186 of SIRT6, providing a structural rationale for its increased effectiveness. A subsequent X-ray crystal structure of SIRT6 bound with cyanidin confirmed this binding orientation (Fig. 6) (149). Interestingly, treatment of Caco-2 cells with cyanidin showed a dose-dependent increase (3.5-fold) in SIRT6 protein levels (115). It is entirely possible that cyanidin bound to SIRT6 stabilizes the protein against degradation, providing both catalytic activation and decreased protein turnover. Anthocyanidins, including cyanidin, have been implicated in reducing the risk of aging-related diseases thought to be mediated by their protective effect against oxidative stress (150, 151). Given the protective role of SIRT6 against aging-related diseases, it is compelling to speculate that some anthocyanidines act through SIRT6. Additionally, it raises the point that small molecules eliciting increased SIRT6 protein levels should be considered as a potential therapeutic strategy in addition to those that directly activate SIRT6.
Figure 6.

SIRT6 activators bind to the terminus of the hydrophobic pocket. Crystallization of SIRT6 small-molecule activators reveals a conserved site of activation located at the terminal end of the hydrophobic pocket. A, a structure of SIRT6 bound to myristoyl peptide (yellow) and ADP-ribose (pink) overlaid with MDL-801 (green), UBCS039 (magenta), and cyanidin (blue) shows that these activators bind away from the active-site His-133 (red). B, UBCS039 and cyanidin cap the hydrophobic pocket and directly overlap with the myristoyl chain of peptide substrate. MDL-801 also caps this hydrophobic pocket; however, it is shifted back, allowing for the accommodation of myristoyl substrate. This structure was produced using entries 5Y2F, 5MF6, 6QCH, and 3ZG6 from the RCSB Protein Data Bank.
Screening of brown algae extracts led to the identification of fucoidan, a highly abundant sulfated polysaccharide, as an activator of SIRT6 deacetylation (152). Fucoidan is a complex heterogeneous polysaccharide found in various brown algae and seaweeds, making structural determination of this compound difficult. However, fucoidan isolated from Fucus vesiculosus exhibited a ∼355-fold activation of SIRT6 at 100 μg/ml under steady-state conditions. Dose-response assays of fucoidan did not reach saturation due to limitations of available fucoidan, suggesting that maximum activation may be higher than reported. Fucoidan was additionally shown to activate SIRT6 in vitro against histone substrate and demonstrated strong specificity for SIRT6 over SIRT1–3. This finding marks the first identification of a high molecular weight polysaccharide activator of SIRT6; however, a lack of detailed kinetics of activation, in particular the effects on kcat or kcat/Km, prevent the mechanism of activation from being compared with that of other small-molecule activators.
Synthetic activators
The first validated set of synthetic SIRT6 deacetylase activators, pyrrolo[1,2-α]quinoxaline derivatives, were characterized by You et al. (114). This work was motivated by a previous study in which virtual docking and subsequent in vitro studies suggested a weak stimulation of SIRT6 by the compound 4-(pyridin-3-yl)-5-((3-(trifluoromethyl)-phenyl)sulfonyl)-4,5-dihydropyrrolo[1,2-α]quinoxaline (153). This lead compound was ultimately derivatized to 4-(pyridin-3-yl)-4,5-dihydropyrrolo[1,2-α]quinoxaline (renamed UBCS039), which demonstrated a 3.5-fold increase in SIRT6 activity against an acetyl peptide with an EC50 of 38 μm as determined under steady-state conditions utilizing a coupled assay that monitors the formation of nicotinamide (114, 154). UBCS039 was further validated as an activator of SIRT6 against endogenously sourced calf thymus histones and HeLa mononucleosomes; however no kinetic data were presented to clarify whether activation occurred via an improved kcat or kcat/Km. The structure of UBCS039/SIRT6 was solved and showed the activator occupying the distal pocket of the hydrophobic channel, consistent with the previous notion of this pocket as an activation site (Fig. 6). When compared with the structure of SIRT6 bound to a myristoylated peptide (3ZG6), UBCS039 overlaps with the terminal seven carbons of the myristoyl chain (114, 138). In line with these structural data, the presence of myristoylated peptide weakened the binding of UBCS039 by an order of magnitude. However, no inhibition of demyristoylation was observed in the presence of UBCS039, likely due to the weak binding of UBCS039 being unable to compete with the tight-binding myristoyl peptide. No structural rearrangement of SIRT6 was observed in the presence of UBCS039 compared with apo-SIRT6, perhaps due to preformed SIRT6 crystals soaked with UBCS039 being unable to adopt potential allosteric changes. In a subsequent study, UBCS039 was shown to elicit cellular activation of SIRT6 that resulted in decreased levels of H3K9ac and H3K56ac and SIRT6-dependent autophagy-related cell death (155). This study was the first reported instance of a cellularly active small-molecule SIRT6 activator, thus providing proof-of-principle for the potential therapeutic targeting of SIRT6.
An additional SIRT6 activator, MDL-800, with cellular activity was reported by Huang et al. (113). Researchers began by computationally predicting an allosteric site on SIRT6 and virtually docking five million compounds. In vitro testing of the top predicted binders validated lead compounds as SIRT6 activators with demonstrated EC50 values in the 170-220 μm range as determined under steady-state conditions. Chemical optimization of the substituents of the two terminal phenyl rings resulted in the compound MDL-800, which displayed a 22-fold increase in SIRT6 activity with an EC50 value of 10.3 μm. Activation was further validated against additional peptide substrates, and HeLa extracted heterogeneously modified nucleosomes. Michaelis–Menten kinetic analysis against peptide substrate demonstrated an 18-fold increase in kcat/Km for H3K9ac, driven by a 7.4-fold increase in kcat and a 2.2-fold decrease in Km. Interestingly, these kinetics of activation vary from other synthetic activators in which detailed kinetic analyses have been performed. CL5D (discussed next) and myristic acid, for example, drive activation of SIRT6 primarily by reducing the Km for H3K9ac and show minimal effects on kcat (25, 103). These varied kinetics of activation are potentially explained by subsequent structural studies of MDL-801, a demethylated derivative of MDL-800, which revealed a unique activation pocket peripheral to that of UBCS039 (Fig. 6). A complex of SIRT6/MDL-801 could only be solved in the presence of myristoylated peptide and exhibited nonoverlapping binding pockets of the two ligands. MDL-801 was located near the surface-exposed distal region of the hydrophobic pocket and made contacts with the N terminus and π-stacking interactions with Phe82 and Phe86 of SIRT6. Mutation of Phe86 resulted in decreased potency of activation, demonstrating the importance of this π-stacking interaction. Despite the ability of myristoyl peptide and MDL-801 to co-occupy SIRT6, no enhancement or inhibition of demyristoylation was observed. Importantly, MDL-801 and UBCS039 exhibited synergistic activation of SIRT6 deacetylation, substantiating separate binding sites and suggesting multiple modes of allosteric activation. MDL-800 treatment of multiple human cancer cell lines showed a dose-dependent and SIRT6-dependent cell-cycle arrest and corresponding decrease in global H3K9ac and H3K56ac levels, confirming MDL-800 as a cellularly active SIRT6 activator. Together, studies of UBCS039 and MDL-800 demonstrate the viability of pharmacological activation of SIRT6 and suggest multiple modes of allosteric activation. The presence of distinct binding sites also opens the door to potential development of multivalent activators with improved affinity for SIRT6.
Recent work by Klein et al. (103) identified the novel SIRT6 activator, CL5D, and used this compound to probe the mechanism of activation, resulting in the identification of a conformational step preceding chemical catalysis that is enhanced by small-molecule activators of deacetylation or directly by long-chain myristoyl substrates. CL5D was derived through chemical optimization of a lead compound identified through targeted screening of lipid-like molecules. Kinetic analysis demonstrated an EC50 value of 15.5 μm with a maximum 50-fold activation in kcat/Km under steady-state conditions. Although no structural data were presented, CL5D exhibited competitive inhibition of demyristoylation, suggesting occupation of the hydrophobic pocket, as had been described previously for free myristic acid (25). Extensive kinetic analyses excluded the possibility of any previously identified catalytic steps being enhanced by CL5D. Kinetic modeling proposed that a slow conformational step following substrate binding but prior to the first chemical step—formation of the alkylimidate intermediate—could be activated by CL5D to account for the empirically determined 50-fold increase in kcat/Km. The SIRT6 variant, R65A, maintained base-level deacetylase activity but was unable to be activated by CL5D toward deacetylation and demonstrated inefficient demyristoylation. Although SIRT6 R65A could bind NAD+, this variant lacked the ability to mediate the conformational change observed for WT SIRT6 upon NAD+ binding. Together, this suggested that CL5D activated SIRT6 deacetylation via an Arg65-mediated conformational change prior to chemical catalysis and that this mechanism of enhanced catalysis is shared by long-chain demyristoylation.
Kinetic implications of SIRT6 activation
The therapeutic promise of pharmacological SIRT6 activation in combating aging-related diseases has motivated the identification of novel SIRT6 activators with diverse structures and varying levels of corresponding biochemical data. Studies that resolve the structural interaction of SIRT6 and activator have universally identified a region at the distal site of the hydrophobic pocket responsible for ligand interaction (113–115). Anthocyanidins, including cyanidin and delphinidin, as well as UBCS039 partially occupy the hydrophobic interior, in effect capping this pocket from the solvent-exposed exterior (Fig. 6). MDL-801 similarly binds the distal region of this pocket; however, it appears shifted back toward the solvent-exposed exterior when compared with previously mentioned compounds (Fig. 6B). Curiously, MDL-801 is the only activator to date that catalytically activates SIRT6 primarily by driving an increased kcat. Other activators, including myristic acid and CL5D, have been shown to primarily enhance kcat/Km. Therefore, it is of interest whether varying binding orientations within this distal hydrophobic pocket elicit varying allosteric mechanisms of SIRT6 activation.
The steady-state kcat/Km parameter describes the ability of the enzyme to capture substrate at low concentration and commit it to catalysis; therefore, activation via an improved kcat/Km implicates the improvement of substrate binding or a catalytic step including or prior to the first irreversible step, the release of nicotinamide. These steps include NAD+ and acetyl peptide binding, formation of an alkylimidate intermediate, and nicotinamide release and are universal to sirtuin-mediated catalysis (Fig. 5). As discussed previously, a recent study reported an additional conformational step in SIRT6 catalysis following substrate binding mediated by Arg65 that was enhanced by CL5D (103). The rate of conformational change is slow in the absence of activator and occurs prior to nicotinamide release and is therefore also reflected in the kcat/Km term of SIRT6. It remains to be determined whether other activators that improve kcat/Km similarly increase the rate of conformational change or whether this conformational step occurs in other sirtuins. MDL-801 uniquely activates SIRT6 primarily through an improved kcat, which reflects an enhancement of a rate-limiting step. The terms reflected in kcat can include any of the chemical steps or product release. The rate-limiting step of sirtuin catalysis varies depending on isoform as well as acyl-substrate and remains unknown in the case of SIRT6 deacetylation due to the inability to saturate SIRT6 with acetyl substrate, a requisite condition for single-turnover kinetic analysis (138). Studies involving other sirtuins have suggested that either the formation of the 1′,2′-cyclic intermediate, water-mediated collapse of the 1′,2′-cyclic intermediate, or product release can be rate-limiting (128, 133, 138, 156). Further biochemical studies of MDL-801 are required to reach a full mechanistic understanding of its unique activation.
Kinetic studies used to characterize in vitro activation of SIRT6 thus far have been limited to the use of histone H3 tail-mimetic peptides and endogenously sourced, heterogeneously modified nucleosomes. Multiple activators have been identified that improve kcat/Km for H3K9ac peptide substrates. In cells, SIRT6 has been reported to be almost exclusively bound to chromatin (1). Because kcat/Km describes the ability of SIRT6 to capture substrate for catalysis, it is important to understand how improving kcat/Km will affect an enzyme that is tightly bound to a nucleosome substrate. Perhaps SIRT6 is capable of undergoing activation during internucleosomal reaction, where SIRT6 bound to one nucleosome can accept acetylated tail substrates from adjacent nucleosomes. Activators that improve kcat could be imagined to affect both the activities of intra- and internucleosomal deacetylation of nucleosomes. How in vitro derived kinetic parameters influence SIRT6 toward physiologically complex polynucleosome substrates and whether SIRT6 performs intra- or internucleosomal deacetylation are therefore of importance in evaluating small-molecule activators. Because endogenously sourced nucleosomes display as little as 2% acetylated H3K9 (157), additional studies will be needed to accurately ascertain the role of activation in SIRT6-dependent chromatin deacetylation.
Inhibition
Context-dependent inhibition of SIRT6 is thought to have pharmacological value and has been successfully applied in vitro as a chemosensitizing and prodifferentiating strategy (158–160). Inhibitors of SIRT6 fall into four classes: product inhibitors, thioacyl-lysine warheads, isoform-specific small-molecule inhibitors of deacetylation, and competitive inhibitors of long-chain deacylation.
The end products of SIRT6 catalysis are deacylated protein, O-acyl-ADP-ribose, and nicotinamide. Nicotinamide is a noncompetitive inhibitor of sirtuins, binding to the enzyme and reacting with the substrate–ADP-ribose adduct to reform substrates (132, 161–164). Subsequent studies validated nicotinamide as an inhibitor of SIRT6 deacylation (138, 165). Studies in yeast demonstrated that nicotinamide depletion via overexpression of the nicotinamide-metabolizing enzyme, PNC1, results in activation of Sir2 and lifespan extension (166). This suggests that inhibition of sirtuins by nicotinamide may serve as an endogenous regulator of activity, although this effect has not been studied in mammalian cells. Both ADP-ribose and the nonhydrolyzable analog of O-acetyl ADP-ribose, N-acetyl ADP-ribose, have also been shown to bind to SIRT6 at low to mid-micromolar affinities (23). Kinetic studies demonstrated ADP-ribose as an inhibitor of SIRT6 deoctanoylation and demyristoylation with less potent IC50 values compared with nicotinamide (100). The full potential for cellular regulation by end-product inhibition has not been evaluated.
Thioacyl-lysine substrates of sirtuins demonstrate mechanism-based inhibition where, following the first chemical step of catalysis, the corresponding 1′-S-alkylamidate intermediate displays impaired catalysis, in effect stalling the enzyme after the first chemical step (129, 167). This mechanism-based inhibition has been exploited to create isoform-specific thioacyl peptides, which specifically inhibit SIRT6 at micromolar levels (168, 169). Subsequent work created cyclic peptide thioacyl-lysine inhibitors with nanomolar IC50 values with specificity for SIRT6 (170). Although cyclic peptides tend to show improved stability against protease/peptidase activity, cellular targeting of SIRT6 with these compounds has not been realized.
Isoform-specific inhibition of sirtuins is an attractive therapeutic strategy and has been utilized successfully in targeting SIRT1 in mouse models. Specifically, small-molecule inhibitors of SIRT1 have been shown to sensitize cells to DNA-damaging agents, and inhibition of SIRT1 and SIRT2 appeared to slow tumor growth (171–174). The first identified small-molecule inhibitors of SIRT6 were identified from an in silico screen; however, subsequent biochemical analysis suggested a lack of isoform specificity (153). Quercetin and vitexin were isolated from plant extracts and demonstrated SIRT6 inhibition; however, these compounds were later shown to activate SIRT6 at higher concentrations, and isoform specificity was never reported (147, 148). Research by Parenti et al. (175) first identified a set of synthetic small-molecule inhibitors with in vitro specificity for SIRT6 over SIRT1 and SIRT2. These compounds elicited a time-dependent increase of H3K9ac levels in cultured cells; however, SIRT6 dependence was not evaluated. One of the quinazolinedione inhibitors identified by Parenti et al. was subsequently derivatized successfully to increase specificity over SIRT1 and SIRT2 (176). Kinetic analysis of these quinazolinedione derivatives revealed competitive inhibition with respect to both NAD+ and acetyl peptide substrate. Separate studies later determined that SIRT6 follows an ordered binding mechanism in which NAD+ binding precedes acetyl peptide, suggesting that the inhibition noted by Sociali et al. (103) is likely competitive with respect to NAD+ binding. Cell-based assays further demonstrated that these quinazolinedione compounds mimic the effect of SIRT6 knockdown in potentiating the activity of PARP inhibitors and chemotherapeutics (21, 90, 177). A following study reported that one of these quinazolinediones, named “compound 1” for the purposes of the study, improved glucose tolerance in a type 2 diabetes mellitus mouse model, the first study implicating in vivo pharmacological SIRT6 inhibition (159). An additional salicylate-like compound, originally identified by Parenti et al., was further derivatized (renamed OSS_128167), showed specificity for SIRT6 over SIRT1 and SIRT2, and elicited immunosuppressive and chemosensitizing effects in cell-based assays (158). Although OSS_128167 was not tested in type 2 diabetes mellitus mouse models, pharmacokinetic studies in mice demonstrated a short t1/2 of ∼10 min. A separate study demonstrated that SIRT6 overexpression in cell culture resulted in a decrease of H3K9ac levels that could be reversed by OSS_128167 treatment, suggesting cellular inhibition of SIRT6 (178). Curiously, this same study reported that OSS_128167 treatment resulted in inhibition of hepatitis B viral transcription and replication in mice despite the compound, with a t1/2 of ∼10 min, only being administered every 4 days. Further studies are required to determine whether cellular t1/2 rates vary from previously reported serum t1/2 and whether this antiviral effect is due directly to intermittent SIRT6 inhibition.
As mentioned previously, various small-molecule activators of SIRT6 deacetylation simultaneously inhibit long-chain deacylation. Both myristic acid and CL5D demonstrate competitive inhibition against demyristoylation, likely resulting from these compounds occupying the hydrophobic pocket and sterically occluding the myristoyl but not the acetyl group of substrate (Table 1) (25, 103). Other deacetylase activators, including MDL-800 and UBC039, are unable to inhibit demyristoylation because the binding pocket is distinct from the hydrophobic pocket or such compounds have weak binding relative to myristoylated peptide, respectively (113, 114). To date, no inhibitor specific to long-chain deacylation of SIRT6 has been evaluated in cellular models.
Perspective
Research progress over the past few years has described critical roles for SIRT6 in DNA repair, metabolic regulation, tumor suppression, and longevity (179, 180). Given these biological roles, SIRT6 has emerged as a candidate for small molecule activation to extend lifespan through the treatment or prevention of age-associated disease, including nonalcoholic fatty liver disease, cancer, fibrosis, and cardiac hypertrophy (52, 181–183). Early work demonstrated the ability of SIRT6 to be activated in vitro by various small molecules, including free fatty acids and natural products (25, 115, 147, 152). In recent years, new synthetic small molecule activators have appeared to exert cellular effects via SIRT6 stimulation (113, 155). The compound UBCS039 was reported to stimulate SIRT6 deacetylation of H3K9 and H3K56 and ultimately led to autophagy-dependent cell death in human cancer cells (155). However, autophagy has been shown to serve as a prosurvival response in some cancer cell contexts (184), suggesting that therapeutic targeting of SIRT6 in cancer to induce autophagy would need to be carefully considered. Huang et al. reported that MDL-800 reduced H3K9 and H3K56 acetylation in a SIRT6-dependent fashion and resulted in cell-cycle arrest of hepatocellular carcinoma cells (113). Administration of MDL-800 modestly decreased xenograft tumor growth in immunocompromised mice. These preliminary studies demonstrate proof of principle for the cellular targeting of SIRT6 and mark the beginning of a new phase of research in which small-molecule probes aid in the biological evaluation of SIRT6 activities and therapeutic opportunities are assessed.
The therapeutic targeting of SIRT6 is still in its infancy, and although several compounds have exhibited SIRT6-dependent cellular effects, questions regarding mechanisms of action and appropriate disease contexts remain. Our biochemical understanding of SIRT6 has lagged that of phenotypic studies in which loss of function mutants or manipulation of SIRT6 levels were correlated with altered cellular or organismal physiology. As an understanding of in vitro activities of SIRT6 advances, our knowledge of the catalytic mechanisms and ability to modulate activity by small molecules have provided an important foundation for accelerated development of such molecules. Most cellular studies reporting the modulation of SIRT6 activity by small molecules utilize complementary in vitro data to support the activation or inhibition of SIRT6; however, future studies will need to confirm direct engagement of SIRT6 with these small-molecule modulators. Evidence of cellular engagement is critical to exclude the possibility of off-target effects that may otherwise be easily misattributed to SIRT6. Moreover, all catalytic activities ascribed to SIRT6 likely utilize a portion or all of the catalytic steps depicted in Fig. 5 but employ distinct substrate-binding regions just beyond the shared active site. Accordingly, loss-of-function mutations in SIRT6 could manifest phenotypes that represent the combination of all SIRT6 cellular functions. Similar thoughts would apply to increased expression of SIRT6. To target only one activity, small-molecule modulators will need to preferentially bind these unique structural regions. Highly specific and validated cellular SIRT6 activators/inhibitors have the potential to serve as powerful probes for elucidating the effects of modulating SIRT6 activity, including deacetylation, ADP-ribosylation, or long-chain deacylation. Combining select SIRT6 amino acid substitutions, gleaned from enzymology studies, and activity-selective compounds could provide the needed selectivity to parse out SIRT6 functions in context-specific settings, such as the potentially conflicting roles of SIRT6 in carcinogenesis and tumor development. SIRT6 is a complex enzyme with diverse functions implicated in human health and disease. With a detailed understanding of SIRT6 functions there exists an opportunity for precise selection of SIRT6 modulators to combat a host of common age-associated human diseases.
Acknowledgments
We thank every scientist who has contributed to our collective understanding of sirtuins.
Funding and additional information—This work was supported by National Institutes of Health Grant GM065386 (to J. M. D.). M. A. K. was supported by the Chemistry-Biology Interface Training Program (NIGMS, National Institutes of Health, Grant T32GM008505). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—J. M. D. is a consultant for Evrys Bio and cofounder of Galilei BioSciences.
- HDAC
- histone deacetylase
- DSB
- double-strand break
- DDR
- DNA damage response
- NHEJ
- nonhomologous end joining
- H3
- histone H3
- ac
- acetylation
- BER
- base excision repair
- LDL
- low-density lipoprotein
- IL
- interleukin
- TNF
- tumor necrosis factor
- HCC
- hepatocellular carcinoma
- ADPr
- adenosine diphosphate ribose
- TSA
- trichostatin A
- IDR
- intrinsically disordered region
- FA
- fatty acid
- ERK
- extracellular signal–regulated kinase
- SREBP
- sterol-regulatory element–binding protein
- miRNA
- microRNA.
References
- 1. Mostoslavsky R., Chua K. F., Lombard D. B., Pang W. W., Fischer M. R., Gellon L., Liu P., Mostoslavsky G., Franco S., Murphy M. M., Mills K. D., Patel P., Hsu J. T., Hong A. L., Ford E., et al. (2006) Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell 124, 315–329 10.1016/j.cell.2005.11.044 [DOI] [PubMed] [Google Scholar]
- 2. Sebastián C., Zwaans B. M. M., Silberman D. M., Gymrek M., Goren A., Zhong L., Ram O., Truelove J., Guimaraes A. R., Toiber D., Cosentino C., Greenson J. K., MacDonald A. I., McGlynn L., Maxwell F., et al. (2012) The histone deacetylase SIRT6 is a tumor suppressor that controls cancer metabolism. Cell 151, 1185–1199 10.1016/j.cell.2012.10.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ferrer C. M., Alders M., Postma A. V., Park S., Klein M. A., Cetinbas M., Pajkrt E., Glas A., van Koningsbruggen S., Christoffels V. M., Mannens M. M. A. M., Knegt L., Etchegaray J.-P., Sadreyev R. I., Denu J. M., et al. (2018) An inactivating mutation in the histone deacetylase SIRT6 causes human perinatal lethality. Genes Dev. 32, 373–388 10.1101/gad.307330.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhang W., Wan H., Feng G., Qu J., Wang J., Jing Y., Ren R., Liu Z., Zhang L., Chen Z., Wang S., Zhao Y., Wang Z., Yuan Y., Zhou Q., et al. (2018) SIRT6 deficiency results in developmental retardation in cynomolgus monkeys. Nature 560, 661–665 10.1038/s41586-018-0437-z [DOI] [PubMed] [Google Scholar]
- 5. Kugel S., Feldman J. L., Klein M. A., Silberman D. M., Sebastián C., Mermel C., Dobersch S., Clark A. R., Getz G., Denu J. M., and Mostoslavsky R. (2015) Identification of and molecular basis for SIRT6 loss-of-function point mutations in cancer. Cell Rep. 13, 479–488 10.1016/j.celrep.2015.09.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rine J., Strathern J. N., Hicks J. B., and Herskowitz I. (1979) A suppressor of mating-type locus mutations in Saccharomyces cerevisiae: evidence for and identification of cryptic mating-type loci. Genetics 93, 877–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Klar A. J., Strathern J. N., Broach J. R., and Hicks J. B. (1981) Regulation of transcription in expressed and unexpressed mating type cassettes of yeast. Nature 289, 239–244 10.1038/289239a0 [DOI] [PubMed] [Google Scholar]
- 8. Rine J., and Herskowitz I. (1987) Four genes responsible for a position effect on expression from HML and HMR in Saccharomyces cerevisiae. Genetics 116, 9–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gottlieb S., and Esposito R. E. (1989) A new role for a yeast transcriptional silencer gene, SIR2, in regulation of recombination in ribosomal DNA. Cell 56, 771–776 10.1016/0092-8674(89)90681-8 [DOI] [PubMed] [Google Scholar]
- 10. Smith J. S., and Boeke J. D. (1997) An unusual form of transcriptional silencing in yeast ribosomal DNA. Genes Dev. 11, 241–254 10.1101/gad.11.2.241 [DOI] [PubMed] [Google Scholar]
- 11. Sinclair D. A., and Guarente L. (1997) Extrachromosomal rDNA circles–a cause of aging in yeast. Cell 91, 1033–1042 10.1016/S0092-8674(00)80493-6 [DOI] [PubMed] [Google Scholar]
- 12. Aparicio O. M., Billington B. L., and Gottschling D. E. (1991) Modifiers of position effect are shared between telomeric and silent mating-type loci in S. cerevisiae. Cell 66, 1279–1287 10.1016/0092-8674(91)90049-5 [DOI] [PubMed] [Google Scholar]
- 13. Imai S., Armstrong C. M., Kaeberlein M., and Guarente L. (2000) Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403, 795–800 10.1038/35001622 [DOI] [PubMed] [Google Scholar]
- 14. Landry J., Sutton A., Tafrov S. T., Heller R. C., Stebbins J., Pillus L., and Sternglanz R. (2000) The silencing protein SIR2 and its homologs are NAD-dependent protein deacetylases. Proc. Natl. Acad. Sci. U.S.A. 97, 5807–5811 10.1073/pnas.110148297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Smith J. S., Brachmann C. B., Celic I., Kenna M. A., Muhammad S., Starai V. J., Avalos J. L., Escalante-Semerena J. C., Grubmeyer C., Wolberger C., and Boeke J. D. (2000) A phylogenetically conserved NAD+-dependent protein deacetylase activity in the Sir2 protein family. Proc. Natl. Acad. Sci. U.S.A. 97, 6658–6663 10.1073/pnas.97.12.6658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Frye R. A. (2000) Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem. Biophys. Res. Commun. 273, 793–798 10.1006/bbrc.2000.3000 [DOI] [PubMed] [Google Scholar]
- 17. Blander G., and Guarente L. (2004) The Sir2 family of protein deacetylases. Annu. Rev. Biochem. 73, 417–435 10.1146/annurev.biochem.73.011303.073651 [DOI] [PubMed] [Google Scholar]
- 18. Seto E., and Yoshida M. (2014) Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 6, a018713 10.1101/cshperspect.a018713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Imai S.-I., and Guarente L. (2016) It takes two to tango: NAD+ and sirtuins in aging/longevity control. NPJ Aging Mech. Dis. 2, 16017 10.1038/npjamd.2016.17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liszt G., Ford E., Kurtev M., and Guarente L. (2005) Mouse Sir2 homolog SIRT6 is a nuclear ADP-ribosyltransferase. J. Biol. Chem. 280, 21313–21320 10.1074/jbc.M413296200 [DOI] [PubMed] [Google Scholar]
- 21. Mao Z., Hine C., Tian X., Van Meter M., Au M., Vaidya A., Seluanov A., and Gorbunova V. (2011) SIRT6 promotes DNA repair under stress by activating PARP1. Science 332, 1443–1446 10.1126/science.1202723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Du J., Jiang H., and Lin H. (2009) Investigating the ADP-ribosyltransferase activity of sirtuins with NAD analogues and 32P-NAD. Biochemistry 48, 2878–2890 10.1021/bi802093g [DOI] [PubMed] [Google Scholar]
- 23. Pan P. W., Feldman J. L., Devries M. K., Dong A., Edwards A. M., and Denu J. M. (2011) Structure and biochemical functions of SIRT6. J. Biol. Chem. 286, 14575–14587 10.1074/jbc.M111.218990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jiang H., Khan S., Wang Y., Charron G., He B., Sebastian C., Du J., Kim R., Ge E., Mostoslavsky R., Hang H. C., Hao Q., and Lin H. (2013) SIRT6 regulates TNF-α secretion through hydrolysis of long-chain fatty acyl lysine. Nature 496, 110–113 10.1038/nature12038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Feldman J. L., Baeza J., and Denu J. M. (2013) Activation of the protein deacetylase SIRT6 by long-chain fatty acids and widespread deacylation by mammalian sirtuins. J. Biol. Chem. 288, 31350–31356 10.1074/jbc.C113.511261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Toiber D., Erdel F., Bouazoune K., Silberman D. M., Zhong L., Mulligan P., Sebastian C., Cosentino C., Martinez-Pastor B., Giacosa S., D'Urso A., Näär A. M., Kingston R., Rippe K., and Mostoslavsky R. (2013) SIRT6 recruits SNF2H to DNA break sites, preventing genomic instability through chromatin remodeling. Mol. Cell. 51, 454–468 10.1016/j.molcel.2013.06.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. McCord R. A., Michishita E., Hong T., Berber E., Boxer L. D., Kusumoto R., Guan S., Shi X., Gozani O., Burlingame A. L., Bohr V. A., and Chua K. F. (2009) SIRT6 stabilizes DNA-dependent protein kinase at chromatin for DNA double-strand break repair. Aging (Albany NY) 1, 109–121 10.18632/aging.100011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Onn L., Portillo M., Ilic S., Cleitman G., Stein D., Kaluski S., Shirat I., Slobodnik Z., Einav M., Erdel F., Akabayov B., and Toiber D. (2020) SIRT6 is a DNA double-strand break sensor. Elife 9, e51636 10.7554/elife.51636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Van Meter M., Simon M., Tombline G., May A., Morello T. D., Hubbard B. P., Bredbenner K., Park R., Sinclair D. A., Bohr V. A., Gorbunova V., and Seluanov A. (2016) JNK phosphorylates SIRT6 to stimulate DNA double-strand break repair in response to oxidative stress by recruiting PARP1 to DNA breaks. Cell Rep. 16, 2641–2650 10.1016/j.celrep.2016.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tennen R. I., Berber E., and Chua K. F. (2010) Functional dissection of SIRT6: identification of domains that regulate histone deacetylase activity and chromatin localization. Mech. Ageing Dev. 131, 185–192 10.1016/j.mad.2010.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hou T., Cao Z., Zhang J., Tang M., Tian Y., Li Y., Lu X., Chen Y., Wang H., Wei F.-Z., Wang L., Yang Y., Zhao Y., Wang Z., Wang H., et al. (2020) SIRT6 coordinates with CHD4 to promote chromatin relaxation and DNA repair. Nucleic Acids Res. 48, 2982–3000 10.1093/nar/gkaa006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rogakou E. P., Pilch D. R., Orr A. H., Ivanova V. S., and Bonner W. M. (1998) DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 273, 5858–5868 10.1074/jbc.273.10.5858 [DOI] [PubMed] [Google Scholar]
- 33. Podhorecka M., Skladanowski A., and Bozko P. (2010) H2AX phosphorylation: its role in DNA damage response and cancer therapy. J. Nucleic Acids 2010, 920161 10.4061/2010/920161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Atsumi Y., Minakawa Y., Ono M., Dobashi S., Shinohe K., Shinohara A., Takeda S., Takagi M., Takamatsu N., Nakagama H., Teraoka H., and Yoshioka K. (2015) ATM and SIRT6/SNF2H mediate transient H2AX stabilization when DSBs form by blocking HUWE1 to allow efficient γH2AX foci formation. Cell Rep. 13, 2728–2740 10.1016/j.celrep.2015.11.054 [DOI] [PubMed] [Google Scholar]
- 35. Xu Z., Zhang L., Zhang W., Meng D., Zhang H., Jiang Y., Xu X., Van Meter M., Seluanov A., Gorbunova V., and Mao Z. (2015) SIRT6 rescues the age related decline in base excision repair in a PARP1-dependent manner. Cell Cycle 14, 269–276 10.4161/15384101.2014.980641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hwang B.-J., Jin J., Gao Y., Shi G., Madabushi A., Yan A., Guan X., Zalzman M., Nakajima S., Lan L., and Lu A.-L. (2015) SIRT6 protein deacetylase interacts with MYH DNA glycosylase, APE1 endonuclease, and Rad9-Rad1-Hus1 checkpoint clamp. BMC Mol. Biol. 16, 12 10.1186/s12867-015-0041-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tian X., Firsanov D., Zhang Z., Cheng Y., Luo L., Tombline G., Tan R., Simon M., Henderson S., Steffan J., Goldfarb A., Tam J., Zheng K., Cornwell A., Johnson A., et al. (2019) SIRT6 is responsible for more efficient DNA double-strand break repair in long-lived species. Cell 177, 622–638.e22 10.1016/j.cell.2019.03.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Michishita E., McCord R. A., Berber E., Kioi M., Padilla-Nash H., Damian M., Cheung P., Kusumoto R., Kawahara T. L. A., Barrett J. C., Chang H. Y., Bohr V. A., Ried T., Gozani O., and Chua K. F. (2008) SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature 452, 492–496 10.1038/nature06736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Michishita E., McCord R. A., Boxer L. D., Barber M. F., Hong T., Gozani O., and Chua K. F. (2009) Cell cycle-dependent deacetylation of telomeric histone H3 lysine K56 by human SIRT6. Cell Cycle 8, 2664–2666 10.4161/cc.8.16.9367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tennen R. I., Bua D. J., Wright W. E., and Chua K. F. (2011) SIRT6 is required for maintenance of telomere position effect in human cells. Nat. Commun. 2, 433 10.1038/ncomms1443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gao Y., Tan J., Jin J., Ma H., Chen X., Leger B., Xu J., Spagnol S. T., Dahl K. N., Levine A. S., Liu Y., and Lan L. (2018) SIRT6 facilitates directional telomere movement upon oxidative damage. Sci. Rep. 8, 5407 10.1038/s41598-018-23602-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li Y., Meng X., Wang W., Liu F., Hao Z., Yang Y., Zhao J., Yin W., Xu L., Zhao R., and Hu J. (2017) Cardioprotective effects of SIRT6 in a mouse model of transverse aortic constriction-induced heart failure. Front. Physiol. 8, 394 10.3389/fphys.2017.00394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tasselli L., Xi Y., Zheng W., Tennen R. I., Odrowaz Z., Simeoni F., Li W., and Chua K. F. (2016) SIRT6 deacetylates H3K18ac at pericentric chromatin to prevent mitotic errors and cellular senescence. Nat. Struct. Mol. Biol. 23, 434–440 10.1038/nsmb.3202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Xiao C., Kim H.-S., Lahusen T., Wang R.-H., Xu X., Gavrilova O., Jou W., Gius D., and Deng C.-X. (2010) SIRT6 deficiency results in severe hypoglycemia by enhancing both basal and insulin-stimulated glucose uptake in mice. J. Biol. Chem. 285, 36776–36784 10.1074/jbc.M110.168039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhong L., D'Urso A., Toiber D., Sebastian C., Henry R. E., Vadysirisack D. D., Guimaraes A., Marinelli B., Wikstrom J. D., Nir T., Clish C. B., Vaitheesvaran B., Iliopoulos O., Kurland I., Dor Y., et al. (2010) The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1α. Cell 140, 280–293 10.1016/j.cell.2009.12.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Warburg O. (1956) On the origin of cancer cells. Science 123, 309–314 10.1126/science.123.3191.309 [DOI] [PubMed] [Google Scholar]
- 47. Gertman O., Omer D., Hendler A., Stein D., Onn L., Khukhin Y., Portillo M., Zarivach R., Cohen H. Y., Toiber D., and Aharoni A. (2018) Directed evolution of SIRT6 for improved deacylation and glucose homeostasis maintenance. Sci. Rep. 8, 3538 10.1038/s41598-018-21887-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dominy J. E., Lee Y., Jedrychowski M. P., Chim H., Jurczak M. J., Camporez J. P., Ruan H.-B., Feldman J., Pierce K., Mostoslavsky R., Denu J. M., Clish C. B., Yang X., Shulman G. I., Gygi S. P., et al. (2012) The deacetylase Sirt6 activates the acetyltransferase GCN5 and suppresses hepatic gluconeogenesis. Mol. Cell 48, 900–913 10.1016/j.molcel.2012.09.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Puigserver P., Rhee J., Donovan J., Walkey C. J., Yoon J. C., Oriente F., Kitamura Y., Altomonte J., Dong H., Accili D., and Spiegelman B. M. (2003) Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1α interaction. Nature 423, 550–555 10.1038/nature01667 [DOI] [PubMed] [Google Scholar]
- 50. Xiong X., Tao R., DePinho R. A., and Dong X. C. (2013) Deletion of hepatic FoxO1/3/4 genes in mice significantly impacts on glucose metabolism through downregulation of gluconeogenesis and upregulation of glycolysis. PLoS ONE 8, e74340 10.1371/journal.pone.0074340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhang P., Tu B., Wang H., Cao Z., Tang M., Zhang C., Gu B., Li Z., Wang L., Yang Y., Zhao Y., Wang H., Luo J., Deng C.-X., Gao B., et al. (2014) Tumor suppressor p53 cooperates with SIRT6 to regulate gluconeogenesis by promoting FoxO1 nuclear exclusion. Proc. Natl. Acad. Sci. U.S.A. 111, 10684–10689 10.1073/pnas.1411026111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kim H.-S., Xiao C., Wang R.-H., Lahusen T., Xu X., Vassilopoulos A., Vazquez-Ortiz G., Jeong W.-I., Park O., Ki S. H., Gao B., and Deng C.-X. (2010) Hepatic-specific disruption of SIRT6 in mice results in fatty liver formation due to enhanced glycolysis and triglyceride synthesis. Cell Metab. 12, 224–236 10.1016/j.cmet.2010.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kanfi Y., Peshti V., Gil R., Naiman S., Nahum L., Levin E., Kronfeld-Schor N., and Cohen H. Y. (2010) SIRT6 protects against pathological damage caused by diet-induced obesity. Aging Cell 9, 162–173 10.1111/j.1474-9726.2009.00544.x [DOI] [PubMed] [Google Scholar]
- 54. Kuang J., Zhang Y., Liu Q., Shen J., Pu S., Cheng S., Chen L., Li H., Wu T., Li R., Li Y., Zou M., Zhang Z., Jiang W., Xu G., et al. (2017) Fat-specific Sirt6 ablation sensitizes mice to high-fat diet-induced obesity and insulin resistance by inhibiting lipolysis. Diabetes 66, 1159–1171 10.2337/db16-1225 [DOI] [PubMed] [Google Scholar]
- 55. He J., Zhang G., Pang Q., Yu C., Xiong J., Zhu J., and Chen F. (2017) SIRT6 reduces macrophage foam cell formation by inducing autophagy and cholesterol efflux under ox-LDL condition. FEBS J. 284, 1324–1337 10.1111/febs.14055 [DOI] [PubMed] [Google Scholar]
- 56. Tao R., Xiong X., DePinho R. A., Deng C.-X., and Dong X. C. (2013) FoxO3 transcription factor and Sirt6 deacetylase regulate low density lipoprotein (LDL)-cholesterol homeostasis via control of the proprotein convertase subtilisin/kexin type 9 (Pcsk9) gene expression. J. Biol. Chem. 288, 29252–29259 10.1074/jbc.M113.481473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tao R., Xiong X., DePinho R. A., Deng C.-X., and Dong X. C. (2013) Hepatic SREBP-2 and cholesterol biosynthesis are regulated by FoxO3 and Sirt6. J. Lipid Res. 54, 2745–2753 10.1194/jlr.M039339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Elhanati S., Kanfi Y., Varvak A., Roichman A., Carmel-Gross I., Barth S., Gibor G., and Cohen H. Y. (2013) Multiple regulatory layers of SREBP1/2 by SIRT6. Cell Rep. 4, 905–912 10.1016/j.celrep.2013.08.006 [DOI] [PubMed] [Google Scholar]
- 59. Shimano H., and Sato R. (2017) SREBP-regulated lipid metabolism: convergent physiology - divergent pathophysiology. Nat. Rev. Endocrinol. 13, 710–730 10.1038/nrendo.2017.91 [DOI] [PubMed] [Google Scholar]
- 60. Elhanati S., Ben-Hamo R., Kanfi Y., Varvak A., Glazz R., Lerrer B., Efroni S., and Cohen H. Y. (2016) Reciprocal regulation between SIRT6 and miR-122 controls liver metabolism and predicts hepatocarcinoma prognosis. Cell Rep. 14, 234–242 10.1016/j.celrep.2015.12.023 [DOI] [PubMed] [Google Scholar]
- 61. Naiman S., Huynh F. K., Gil R., Glick Y., Shahar Y., Touitou N., Nahum L., Avivi M. Y., Roichman A., Kanfi Y., Gertler A. A., Doniger T., Ilkayeva O. R., Abramovich I., Yaron O., et al. (2019) SIRT6 promotes hepatic β-oxidation via activation of PPARα. Cell Rep. 29, 4127–4143.e8 10.1016/j.celrep.2019.11.067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kaeberlein M., McVey M., and Guarente L. (1999) The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 13, 2570–2580 10.1101/gad.13.19.2570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Fritze C. E., Verschueren K., Strich R., and Easton Esposito R. (1997) Direct evidence for SIR2 modulation of chromatin structure in yeast rDNA. EMBO J. 16, 6495–6509 10.1093/emboj/16.21.6495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zhang N., Li Z., Mu W., Li L., Liang Y., Lu M., Wang Z., Qiu Y., and Wang Z. (2016) Calorie restriction-induced SIRT6 activation delays aging by suppressing NF-κB signaling. Cell Cycle. 15, 1009–1018 10.1080/15384101.2016.1152427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Perkins N. D. (2015) The importance of the p50 NF-κB subunit. Cell Cycle. 14, 2877–2878 10.1080/15384101.2015.1010952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sharma A., Diecke S., Zhang W. Y., Lan F., He C., Mordwinkin N. M., Chua K. F., and Wu J. C. (2013) The role of SIRT6 protein in aging and reprogramming of human induced pluripotent stem cells. J. Biol. Chem. 288, 18439–18447 10.1074/jbc.M112.405928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Chen Q., Hao W., Xiao C., Wang R., Xu X., Lu H., Chen W., and Deng C.-X. (2017) SIRT6 is essential for adipocyte differentiation by regulating mitotic clonal expansion. Cell Rep. 18, 3155–3166 10.1016/j.celrep.2017.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Qian M., Liu Z., Peng L., Tang X., Meng F., Ao Y., Zhou M., Wang M., Cao X., Qin B., Wang Z., Zhou Z., Wang G., Gao Z., Xu J., et al. (2018) Boosting ATM activity alleviates aging and extends lifespan in a mouse model of progeria. Elife 7, e34836 10.7554/elife.34836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. TenNapel M. J., Lynch C. F., Burns T. L., Wallace R., Smith B. J., Button A., and Domann F. E. (2014) SIRT6 minor allele genotype is associated with >5-year decrease in lifespan in an aged cohort. PLoS ONE 9, e115616 10.1371/journal.pone.0115616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Li Y., Qin J., Wei X., Liang G., Shi L., Jiang M., Xia T., Liang X., He M., and Zhang Z. (2016) Association of SIRT6 gene polymorphisms with human longevity. Iran J. Public Health 45, 1420–1426 [PMC free article] [PubMed] [Google Scholar]
- 71. Hirvonen K., Laivuori H., Lahti J., Strandberg T., Eriksson J. G., and Hackman P. (2017) SIRT6 polymorphism rs117385980 is associated with longevity and healthy aging in Finnish men. BMC Med. Genet. 18, 41 10.1186/s12881-017-0401-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Min L., Ji Y., Bakiri L., Qiu Z., Cen J., Chen X., Chen L., Scheuch H., Zheng H., Qin L., Zatloukal K., Hui L., and Wagner E. F. (2012) Liver cancer initiation is controlled by AP-1 through SIRT6-dependent inhibition of survivin. Nat. Cell Biol. 14, 1203–1211 10.1038/ncb2590 [DOI] [PubMed] [Google Scholar]
- 73. Marquardt J. U., Fischer K., Baus K., Kashyap A., Ma S., Krupp M., Linke M., Teufel A., Zechner U., Strand D., Thorgeirsson S. S., Galle P. R., and Strand S. (2013) Sirtuin-6-dependent genetic and epigenetic alterations are associated with poor clinical outcome in hepatocellular carcinoma patients. Hepatology 58, 1054–1064 10.1002/hep.26413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zhang J., Yin X.-J., Xu C.-J., Ning Y.-X., Chen M., Zhang H., Chen S.-F., and Yao L.-Q. (2015) The histone deacetylase SIRT6 inhibits ovarian cancer cell proliferation via down-regulation of Notch 3 expression. Eur. Rev. Med. Pharmacol. Sci. 19, 818–824 [PubMed] [Google Scholar]
- 75. Zhang Y., Nie L., Xu K., Fu Y., Zhong J., Gu K., and Zhang L. (2019) SIRT6, a novel direct transcriptional target of FoxO3a, mediates colon cancer therapy. Theranostics. 9, 2380–2394 10.7150/thno.29724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Qi J., Cui C., Deng Q., Wang L., Chen R., Zhai D., Xie L., and Yu J. (2018) Downregulated SIRT6 and upregulated NMNAT2 are associated with the presence, depth and stage of colorectal cancer. Oncol. Lett. 16, 5829–5837 10.3892/ol.2018.9400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Barretina J., Caponigro G., Stransky N., Venkatesan K., Margolin A. A., Kim S., Wilson C. J., Lehár J., Kryukov G. V., Sonkin D., Reddy A., Liu M., Murray L., Berger M. F., Monahan J. E., et al. (2012) The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607 10.1038/nature11003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kugel S., Sebastián C., Fitamant J., Ross K. N., Saha S. K., Jain E., Gladden A., Arora K. S., Kato Y., Rivera M. N., Ramaswamy S., Sadreyev R. I., Goren A., Deshpande V., Bardeesy N., et al. (2016) SIRT6 suppresses pancreatic cancer through control of Lin28b. Cell 165, 1401–1415 10.1016/j.cell.2016.04.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Tu H.-C., Schwitalla S., Qian Z., LaPier G. S., Yermalovich A., Ku Y.-C., Chen S.-C., Viswanathan S. R., Zhu H., Nishihara R., Inamura K., Kim S. A., Morikawa T., Mima K., Sukawa Y., et al. (2015) LIN28 cooperates with WNT signaling to drive invasive intestinal and colorectal adenocarcinoma in mice and humans. Genes Dev. 29, 1074–1086 10.1101/gad.256693.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Wheatley S. P., and Altieri D. C. (2019) Survivin at a glance. J. Cell Sci. 4, 132 10.1242/jcs.223826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Thirumurthi U., Shen J., Xia W., LaBaff A. M., Wei Y., Li C.-W., Chang W.-C., Chen C.-H., Lin H.-K., Yu D., and Hung M.-C. (2014) MDM2-mediated degradation of SIRT6 phosphorylated by AKT1 promotes tumorigenesis and trastuzumab resistance in breast cancer. Sci. Signal. 7, ra71 10.1126/scisignal.2005076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Lin Z., Yang H., Tan C., Li J., Liu Z., Quan Q., Kong S., Ye J., Gao B., and Fang D. (2013) USP10 antagonizes c-Myc transcriptional activation through SIRT6 stabilization to suppress tumor formation. Cell Rep. 5, 1639–1649 10.1016/j.celrep.2013.11.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Cai J., Zuo Y., Wang T., Cao Y., Cai R., Chen F.-L., Cheng J., and Mu J. (2016) A crucial role of SUMOylation in modulating Sirt6 deacetylation of H3 at lysine 56 and its tumor suppressive activity. Oncogene 35, 4949–4956 10.1038/onc.2016.24 [DOI] [PubMed] [Google Scholar]
- 84. Bhardwaj A., and Das S. (2016) SIRT6 deacetylates PKM2 to suppress its nuclear localization and oncogenic functions. Proc. Natl. Acad. Sci. U.S.A. 113, E538–E547 10.1073/pnas.1520045113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Cea M., Cagnetta A., Adamia S., Acharya C., Tai Y.-T., Fulciniti M., Ohguchi H., Munshi A., Acharya P., Bhasin M. K., Zhong L., Carrasco R., Monacelli F., Ballestrero A., Richardson P., et al. (2016) Evidence for a role of the histone deacetylase SIRT6 in DNA damage response of multiple myeloma cells. Blood 127, 1138–1150 10.1182/blood-2015-06-649970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Lai C.-C., Lin P.-M., Lin S.-F., Hsu C.-H., Lin H.-C., Hu M.-L., Hsu C.-M., and Yang M.-Y. (2013) Altered expression of SIRT gene family in head and neck squamous cell carcinoma. Tumour Biol. 34, 1847–1854 10.1007/s13277-013-0726-y [DOI] [PubMed] [Google Scholar]
- 87. Wang J. C., Kafeel M. I., Avezbakiyev B., Chen C., Sun Y., Rathnasabapathy C., Kalavar M., He Z., Burton J., and Lichter S. (2011) Histone deacetylase in chronic lymphocytic leukemia. Oncology 81, 325–329 10.1159/000334577 [DOI] [PubMed] [Google Scholar]
- 88. Cagnetta A., Soncini D., Orecchioni S., Talarico G., Minetto P., Guolo F., Retali V., Colombo N., Carminati E., Clavio M., Miglino M., Bergamaschi M., Nahimana A., Duchosal M., Todoerti K., et al. (2018) Depletion of SIRT6 enzymatic activity increases acute myeloid leukemia cells' vulnerability to DNA-damaging agents. Haematologica 103, 80–90 10.3324/haematol.2017.176248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Jackson S. P. (1997) DNA-dependent protein kinase. Int. J. Biochem. Cell Biol. 29, 935–938 10.1016/S1357-2725(97)00006-X [DOI] [PubMed] [Google Scholar]
- 90. Bauer I., Grozio A., Lasigliè D., Basile G., Sturla L., Magnone M., Sociali G., Soncini D., Caffa I., Poggi A., Zoppoli G., Cea M., Feldmann G., Mostoslavsky R., Ballestrero A., et al. (2012) The NAD+-dependent histone deacetylase SIRT6 promotes cytokine production and migration in pancreatic cancer cells by regulating Ca2+ responses. J. Biol. Chem. 287, 40924–40937 10.1074/jbc.M112.405837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Wehrhahn J., Kraft R., Harteneck C., and Hauschildt S. (2010) Transient receptor potential melastatin 2 is required for lipopolysaccharide-induced cytokine production in human monocytes. J. Immunol. 184, 2386–2393 10.4049/jimmunol.0902474 [DOI] [PubMed] [Google Scholar]
- 92. Lee N., Ryu H. G., Kwon J.-H., Kim D.-K., Kim S. R., Wang H. J., Kim K.-T., and Choi K. Y. (2016) SIRT6 depletion suppresses tumor growth by promoting cellular senescence induced by DNA damage in HCC. PLoS ONE 11, e0165835 10.1371/journal.pone.0165835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Ran L.-K., Chen Y., Zhang Z.-Z., Tao N.-N., Ren J.-H., Zhou L., Tang H., Chen X., Chen K., Li W.-Y., Huang A.-L., and Chen J. (2016) SIRT6 overexpression potentiates apoptosis evasion in hepatocellular carcinoma via BCL2-associated X protein-dependent apoptotic pathway. Clin. Cancer Res. 22, 3372–3382 10.1158/1078-0432.CCR-15-1638 [DOI] [PubMed] [Google Scholar]
- 94. Zhang Z.-G., and Qin C.-Y. (2014) Sirt6 suppresses hepatocellular carcinoma cell growth via inhibiting the extracellular signal–regulated kinase signaling pathway. Mol Med Rep. 9, 882–888 10.3892/mmr.2013.1879 [DOI] [PubMed] [Google Scholar]
- 95. Greiss S., and Gartner A. (2009) Sirtuin/Sir2 phylogeny, evolutionary considerations and structural conservation. Mol. Cells 28, 407–415 10.1007/s10059-009-0169-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Zhu A. Y., Zhou Y., Khan S., Deitsch K. W., Hao Q., and Lin H. (2012) Plasmodium falciparum Sir2A preferentially hydrolyzes medium and long chain fatty acyl lysine. ACS Chem. Biol. 7, 155–159 10.1021/cb200230x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Du J., Zhou Y., Su X., Yu J. J., Khan S., Jiang H., Kim J., Woo J., Kim J. H., Choi B. H., He B., Chen W., Zhang S., Cerione R. A., Auwerx J., et al. (2011) Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 334, 806–809 10.1126/science.1207861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Rossmann M. G., Moras D., and Olsen K. W. (1974) Chemical and biological evolution of nucleotide-binding protein. Nature 250, 194–199 10.1038/250194a0 [DOI] [PubMed] [Google Scholar]
- 99. Sanders B. D., Jackson B., and Marmorstein R. (2010) Structural basis for sirtuin function: what we know and what we don't. Biochim. Biophys. Acta 1804, 1604–1616 10.1016/j.bbapap.2009.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Madsen A. S., Andersen C., Daoud M., Anderson K. A., Laursen J. S., Chakladar S., Huynh F. K., Colaço A. R., Backos D. S., Fristrup P., Hirschey M. D., and Olsen C. A. (2016) Investigating the sensitivity of NAD+-dependent sirtuin deacylation activities to NADH. J. Biol. Chem. 291, 7128–7141 10.1074/jbc.M115.668699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Jin L., Wei W., Jiang Y., Peng H., Cai J., Mao C., Dai H., Choy W., Bemis J. E., Jirousek M. R., Milne J. C., Westphal C. H., and Perni R. B. (2009) Crystal structures of human SIRT3 displaying substrate-induced conformational changes. J. Biol. Chem. 284, 24394–24405 10.1074/jbc.M109.014928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Borra M. T., Langer M. R., Slama J. T., and Denu J. M. (2004) Substrate specificity and kinetic mechanism of the Sir2 family of NAD+-dependent histone/protein deacetylases. Biochemistry 43, 9877–9887 10.1021/bi049592e [DOI] [PubMed] [Google Scholar]
- 103. Klein M. A., Liu C., Kuznetsov V. I., Feltenberger J. B., Tang W., and Denu J. M. (2020) Mechanism of activation for the sirtuin 6 protein deacylase. J. Biol. Chem. 295, 1385–1399 10.1074/jbc.RA119.011285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Finnin M. S., Donigian J. R., and Pavletich N. P. (2001) Structure of the histone deacetylase SIRT2. Nat. Struct. Biol. 8, 621–625 10.1038/89668 [DOI] [PubMed] [Google Scholar]
- 105. Schuetz A., Min J., Antoshenko T., Wang C.-L., Allali-Hassani A., Dong A., Loppnau P., Vedadi M., Bochkarev A., Sternglanz R., and Plotnikov A. N. (2007) Structural basis of inhibition of the human NAD+-dependent deacetylase SIRT5 by suramin. Structure 15, 377–389 10.1016/j.str.2007.02.002 [DOI] [PubMed] [Google Scholar]
- 106. Ronnebaum S. M., Wu Y., McDonough H., and Patterson C. (2013) The ubiquitin ligase CHIP prevents SirT6 degradation through noncanonical ubiquitination. Mol. Cell Biol. 33, 4461–4472 10.1128/MCB.00480-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Kalous K. S., Wynia-Smith S. L., Olp M. D., and Smith B. C. (2016) Mechanism of Sirt1 NAD+-dependent protein deacetylase inhibition by cysteine S-nitrosation. J. Biol. Chem. 291, 25398–25410 10.1074/jbc.M116.754655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Sherman J. M., Stone E. M., Freeman-Cook L. L., Brachmann C. B., Boeke J. D., and Pillus L. (1999) The conserved core of a human SIR2 homologue functions in yeast silencing. Mol. Biol. Cell 10, 3045–3059 10.1091/mbc.10.9.3045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Min J., Landry J., Sternglanz R., and Xu R. M. (2001) Crystal structure of a SIR2 homolog-NAD complex. Cell 105, 269–279 10.1016/S0092-8674(01)00317-8 [DOI] [PubMed] [Google Scholar]
- 110. Wood M., Rymarchyk S., Zheng S., and Cen Y. (2018) Trichostatin A inhibits deacetylation of histone H3 and p53 by SIRT6. Arch. Biochem. Biophys. 638, 8–17 10.1016/j.abb.2017.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Finnin M. S., Donigian J. R., Cohen A., Richon V. M., Rifkind R. A., Marks P. A., Breslow R., and Pavletich N. P. (1999) Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature 401, 188–193 10.1038/43710 [DOI] [PubMed] [Google Scholar]
- 112. You W., and Steegborn C. (2018) Structural basis of sirtuin 6 inhibition by the hydroxamate trichostatin A: implications for protein deacylase drug development. J. Med. Chem. 61, 10922–10928 10.1021/acs.jmedchem.8b01455 [DOI] [PubMed] [Google Scholar]
- 113. Huang Z., Zhao J., Deng W., Chen Y., Shang J., Song K., Zhang L., Wang C., Lu S., Yang X., He B., Min J., Hu H., Tan M., Xu J., et al. (2018) Identification of a cellularly active SIRT6 allosteric activator. Nat. Chem. Biol. 14, 1118–1126 10.1038/s41589-018-0150-0 [DOI] [PubMed] [Google Scholar]
- 114. You W., Rotili D., Li T.-M., Kambach C., Meleshin M., Schutkowski M., Chua K. F., Mai A., and Steegborn C. (2017) Structural basis of sirtuin 6 activation by synthetic small molecules. Angew. Chem. Int. Ed. Engl. 56, 1007–1011 10.1002/anie.201610082 [DOI] [PubMed] [Google Scholar]
- 115. Rahnasto-Rilla M., Tyni J., Huovinen M., Jarho E., Kulikowicz T., Ravichandran S., A Bohr V., Ferrucci L., Lahtela-Kakkonen M., and Moaddel R. (2018) Natural polyphenols as sirtuin 6 modulators. Sci. Rep. 8, 4163 10.1038/s41598-018-22388-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Rauh D., Fischer F., Gertz M., Lakshminarasimhan M., Bergbrede T., Aladini F., Kambach C., Becker C. F. W., Zerweck J., Schutkowski M., and Steegborn C. (2013) An acetylome peptide microarray reveals specificities and deacetylation substrates for all human sirtuin isoforms. Nat. Commun. 4, 1–10 10.1038/ncomms3327 [DOI] [PubMed] [Google Scholar]
- 117. Zhang X., Khan S., Jiang H., Antonyak M. A., Chen X., Spiegelman N. A., Shrimp J. H., Cerione R. A., and Lin H. (2016) Identifying the functional contribution of the defatty-acylase activity of SIRT6. Nat. Chem. Biol. 12, 614–620 10.1038/nchembio.2106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Zhang X., Spiegelman N. A., Nelson O. D., Jing H., and Lin H. (2017) SIRT6 regulates Ras-related protein R-Ras2 by lysine defatty-acylation. eLife 6, e25158 10.7554/eLife.25158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Miteva Y. V., and Cristea I. M. (2014) A proteomic perspective of Sirtuin 6 (SIRT6) phosphorylation and interactions and their dependence on its catalytic activity. Mol. Cell. Proteomics 13, 168–183 10.1074/mcp.M113.032847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Strom A. R., Emelyanov A. V., Mir M., Fyodorov D. V., Darzacq X., and Karpen G. H. (2017) Phase separation drives heterochromatin domain formation. Nature 547, 241–245 10.1038/nature22989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Guthmann M., Burton A., and Torres-Padilla M.-E. (2019) Expression and phase separation potential of heterochromatin proteins during early mouse development. EMBO Rep. 20, e47952 10.15252/embr.201947952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Tennen R. I., and Chua K. F. (2011) Chromatin regulation and genome maintenance by mammalian SIRT6. Trends Biochem. Sci. 36, 39–46 10.1016/j.tibs.2010.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Dephoure N., Zhou C., Villén J., Beausoleil S. A., Bakalarski C. E., Elledge S. J., and Gygi S. P. (2008) A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 105, 10762–10767 10.1073/pnas.0805139105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Olsen J. V., Vermeulen M., Santamaria A., Kumar C., Miller M. L., Jensen L. J., Gnad F., Cox J., Jensen T. S., Nigg E. A., Brunak S., and Mann M. (2010) Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci Signal. 3, ra3 10.1126/scisignal.2000475 [DOI] [PubMed] [Google Scholar]
- 125. Tanner K. G., Landry J., Sternglanz R., and Denu J. M. (2000) Silent information regulator 2 family of NAD- dependent histone/protein deacetylases generates a unique product, 1-O-acetyl-ADP-ribose. Proc. Natl. Acad. Sci. U.S.A. 97, 14178–14182 10.1073/pnas.250422697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Cen Y., and Sauve A. A. (2010) Transition state of ADP-ribosylation of acetyllysine catalyzed by Archaeoglobus fulgidus Sir2 determined by kinetic isotope effects and computational approaches. J. Am. Chem. Soc. 132, 12286–12298 10.1021/ja910342d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Smith B. C., and Denu J. M. (2007) Sir2 deacetylases exhibit nucleophilic participation of acetyl-lysine in NAD+ cleavage. J. Am. Chem. Soc. 129, 5802–5803 10.1021/ja070162w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Smith B. C., and Denu J. M. (2006) Sir2 protein deacetylases: evidence for chemical intermediates and functions of a conserved histidine. Biochemistry 45, 272–282 10.1021/bi052014t [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Smith B. C., and Denu J. M. (2007) Mechanism-based inhibition of Sir2 deacetylases by thioacetyl-lysine peptide. Biochemistry 46, 14478–14486 10.1021/bi7013294 [DOI] [PubMed] [Google Scholar]
- 130. Sauve A. A., Celic I., Avalos J., Deng H., Boeke J. D., and Schramm V. L. (2001) Chemistry of gene silencing: the mechanism of NAD+-dependent deacetylation reactions. Biochemistry 40, 15456–15463 10.1021/bi011858j [DOI] [PubMed] [Google Scholar]
- 131. Hawse W. F., Hoff K. G., Fatkins D. G., Daines A., Zubkova O. V., Schramm V. L., Zheng W., and Wolberger C. (2008) Structural insights into intermediate steps in the Sir2 deacetylation reaction. Structure 16, 1368–1377 10.1016/j.str.2008.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Jackson M. D., Schmidt M. T., Oppenheimer N. J., and Denu J. M. (2003) Mechanism of nicotinamide inhibition and transglycosidation by Sir2 histone/protein deacetylases. J. Biol. Chem. 278, 50985–50998 10.1074/jbc.M306552200 [DOI] [PubMed] [Google Scholar]
- 133. Shi Y., Zhou Y., Wang S., and Zhang Y. (2013) Sirtuin deacetylation mechanism and catalytic role of the dynamic cofactor binding loop. J. Phys. Chem. Lett. 4, 491–495 10.1021/jz302015s [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Frye R. A. (1999) Characterization of five human cDNAs with homology to the yeast SIR2 gene: Sir2-like proteins (sirtuins) metabolize NAD and may have protein ADP-ribosyltransferase activity. Biochem. Biophys. Res. Commun. 260, 273–279 10.1006/bbrc.1999.0897 [DOI] [PubMed] [Google Scholar]
- 135. Haigis M. C., Mostoslavsky R., Haigis K. M., Fahie K., Christodoulou D. C., Murphy A. J., Valenzuela D. M., Yancopoulos G. D., Karow M., Blander G., Wolberger C., Prolla T. A., Weindruch R., Alt F. W., and Guarente L. (2006) SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell 126, 941–954 10.1016/j.cell.2006.06.057 [DOI] [PubMed] [Google Scholar]
- 136. Kowieski T. M., Lee S., and Denu J. M. (2008) Acetylation-dependent ADP-ribosylation by Trypanosoma brucei Sir2. J. Biol. Chem. 283, 5317–5326 10.1074/jbc.M707613200 [DOI] [PubMed] [Google Scholar]
- 137. Milne J. C., Lambert P. D., Schenk S., Carney D. P., Smith J. J., Gagne D. J., Jin L., Boss O., Perni R. B., Vu C. B., Bemis J. E., Xie R., Disch J. S., Ng P. Y., Nunes J. J., et al. (2007) Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature 450, 712–716 10.1038/nature06261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Feldman J. L., Dittenhafer-Reed K. E., Kudo N., Thelen J. N., Ito A., Yoshida M., and Denu J. M. (2015) Kinetic and structural basis for acyl-group selectivity and NAD+ dependence in sirtuin-catalyzed deacylation. Biochemistry 54, 3037–3050 10.1021/acs.biochem.5b00150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Howitz K. T., Bitterman K. J., Cohen H. Y., Lamming D. W., Lavu S., Wood J. G., Zipkin R. E., Chung P., Kisielewski A., Zhang L.-L., Scherer B., and Sinclair D. A. (2003) Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 425, 191–196 10.1038/nature01960 [DOI] [PubMed] [Google Scholar]
- 140. Baur J. A., Pearson K. J., Price N. L., Jamieson H. A., Lerin C., Kalra A., Prabhu V. V., Allard J. S., Lopez-Lluch G., Lewis K., Pistell P. J., Poosala S., Becker K. G., Boss O., Gwinn D., et al. (2006) Resveratrol improves health and survival of mice on a high-calorie diet. Nature 444, 337–342 10.1038/nature05354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Hubbard B. P., Gomes A. P., Dai H., Li J., Case A. W., Considine T., Riera T. V., Lee J. E., E S. Y., Lamming D. W., Pentelute B. L., Schuman E. R., Stevens L. A., Ling A. J. Y., Armour S. M., et al. (2013) Evidence for a common mechanism of SIRT1 regulation by allosteric activators. Science 339, 1216–1219 10.1126/science.1231097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Mercken E. M., Mitchell S. J., Martin-Montalvo A., Minor R. K., Almeida M., Gomes A. P., Scheibye-Knudsen M., Palacios H. H., Licata J. J., Zhang Y., Becker K. G., Khraiwesh H., González-Reyes J. A., Villalba J. M., Baur J. A., et al. (2014) SRT2104 extends survival of male mice on a standard diet and preserves bone and muscle mass. Aging Cell 13, 787–796 10.1111/acel.12220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Mitchell S. J., Martin-Montalvo A., Mercken E. M., Palacios H. H., Ward T. M., Abulwerdi G., Minor R. K., Vlasuk G. P., Ellis J. L., Sinclair D. A., Dawson J., Allison D. B., Zhang Y., Becker K. G., Bernier M., et al. (2014) The SIRT1 activator SRT1720 extends lifespan and improves health of mice fed a standard diet. Cell Rep. 6, 836–843 10.1016/j.celrep.2014.01.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Dai H., Sinclair D. A., Ellis J. L., and Steegborn C. (2018) Sirtuin activators and inhibitors: promises, achievements, and challenges. Pharmacol. Ther. 188, 140–154 10.1016/j.pharmthera.2018.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Lavu S., Boss O., Elliott P. J., and Lambert P. D. (2008) Sirtuins—novel therapeutic targets to treat age-associated diseases. Nat. Rev. Drug Discov. 7, 841–853 10.1038/nrd2665 [DOI] [PubMed] [Google Scholar]
- 146. Najt C. P., Khan S. A., Heden T. D., Witthuhn B. A., Perez M., Heier J. L., Mead L. E., Franklin M. P., Karanja K. K., Graham M. J., Mashek M. T., Bernlohr D. A., Parker L., Chow L. S., and Mashek D. G. (2020) Lipid droplet-derived monounsaturated fatty acids traffic via PLIN5 to allosterically activate SIRT1. Mol. Cell. 77, 810–824.e8 10.1016/j.molcel.2019.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Yasuda M., Wilson D. R., Fugmann S. D., and Moaddel R. (2011) Synthesis and characterization of SIRT6 protein coated magnetic beads: identification of a novel inhibitor of SIRT6 deacetylase from medicinal plant extracts. Anal. Chem. 83, 7400–7407 10.1021/ac201403y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Rahnasto-Rilla M., Kokkola T., Jarho E., Lahtela-Kakkonen M., and Moaddel R. (2016) N-Acylethanolamines bind to SIRT6. Chembiochem 17, 77–81 10.1002/cbic.201500482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. You W., Zheng W., Weiss S., Chua K. F., and Steegborn C. (2019) Structural basis for the activation and inhibition of Sirtuin 6 by quercetin and its derivatives. Sci. Rep. 9, 19176 10.1038/s41598-019-55654-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Kumar S., and Pandey A. K. (2013) Chemistry and biological activities of flavonoids: an overview. ScientificWorldJournal 2013, 162750 10.1155/2013/162750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. He J., and Giusti M. M. (2010) Anthocyanins: natural colorants with health-promoting properties. Annu. Rev. Food Sci. Technol. 1, 163–187 10.1146/annurev.food.080708.100754 [DOI] [PubMed] [Google Scholar]
- 152. Rahnasto-Rilla M. K., McLoughlin P., Kulikowicz T., Doyle M., Bohr V. A., Lahtela-Kakkonen M., Ferrucci L., Hayes M., and Moaddel R. (2017) The identification of a SIRT6 activator from brown algae Fucus distichus. Mar. Drugs. 15, 190 10.3390/md15060190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Schlicker C., Boanca G., Lakshminarasimhan M., and Steegborn C. (2011) Structure-based development of novel sirtuin inhibitors. Aging (Albany NY) 3, 852–872 10.18632/aging.100388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Smith B. C., Hallows W. C., and Denu J. M. (2009) A continuous microplate assay for sirtuins and nicotinamide-producing enzymes. Anal. Biochem. 394, 101–109 10.1016/j.ab.2009.07.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Iachettini S., Trisciuoglio D., Rotili D., Lucidi A., Salvati E., Zizza P., Di Leo L., Del Bufalo D., Ciriolo M. R., Leonetti C., Steegborn C., Mai A., Rizzo A., and Biroccio A. (2018) Pharmacological activation of SIRT6 triggers lethal autophagy in human cancer cells. Cell Death Dis. 9, 996 10.1038/s41419-018-1065-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Guan X., Upadhyay A., Munshi S., and Chakrabarti R. (2018) Biophysical characterization of hit compounds for mechanism-based enzyme activation. PLoS ONE 13, e0194175 10.1371/journal.pone.0194175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Krautkramer K. A., Reiter L., Denu J. M., and Dowell J. A. (2015) Quantification of SAHA-dependent changes in histone modifications using data-independent acquisition mass spectrometry. J. Proteome Res. 14, 3252–3262 10.1021/acs.jproteome.5b00245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Damonte P., Sociali G., Parenti M. D., Soncini D., Bauer I., Boero S., Grozio A., Holtey M. V., Piacente F., Becherini P., Sanguineti R., Salis A., Damonte G., Cea M., Murone M., et al. (2017) SIRT6 inhibitors with salicylate-like structure show immunosuppressive and chemosensitizing effects. Bioorg. Med. Chem. 25, 5849–5858 10.1016/j.bmc.2017.09.023 [DOI] [PubMed] [Google Scholar]
- 159. Sociali G., Magnone M., Ravera S., Damonte P., Vigliarolo T., Von Holtey M., Vellone V. G., Millo E., Caffa I., Cea M., Parenti M. D., Del Rio A., Murone M., Mostoslavsky R., Grozio A., et al. (2017) Pharmacological Sirt6 inhibition improves glucose tolerance in a type 2 diabetes mouse model. FASEB J. 31, 3138–3149 10.1096/fj.201601294R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Khan R. I., Nirzhor S. S. R., and Akter R. (2018) A review of the recent advances made with SIRT6 and its implications on aging related processes, major human diseases, and possible therapeutic targets. Biomolecules 8, 44 10.3390/biom8030044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Bitterman K. J., Anderson R. M., Cohen H. Y., Latorre-Esteves M., and Sinclair D. A. (2002) Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J. Biol. Chem. 277, 45099–45107 10.1074/jbc.M205670200 [DOI] [PubMed] [Google Scholar]
- 162. Avalos J. L., Bever K. M., and Wolberger C. (2005) Mechanism of sirtuin inhibition by nicotinamide: altering the NAD+ cosubstrate specificity of a Sir2 enzyme. Mol. Cell. 17, 855–868 10.1016/j.molcel.2005.02.022 [DOI] [PubMed] [Google Scholar]
- 163. Zhao K., Harshaw R., Chai X., and Marmorstein R. (2004) Structural basis for nicotinamide cleavage and ADP-ribose transfer by NAD+-dependent Sir2 histone/protein deacetylases. Proc. Natl. Acad. Sci. U.S.A. 101, 8563–8568 10.1073/pnas.0401057101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Sauve A. A., and Schramm V. L. (2003) Sir2 regulation by nicotinamide results from switching between base exchange and deacetylation chemistry. Biochemistry 42, 9249–9256 10.1021/bi034959l [DOI] [PubMed] [Google Scholar]
- 165. Hu J., He B., Bhargava S., and Lin H. (2013) A fluorogenic assay for screening Sirt6 modulators. Org. Biomol. Chem. 11, 5213–5216 10.1039/c3ob41138a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Anderson R. M., Bitterman K. J., Wood J. G., Medvedik O., and Sinclair D. A. (2003) Nicotinamide and PNC1 govern lifespan extension by calorie restriction in Saccharomyces cerevisiae. Nature 423, 181–185 10.1038/nature01578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Fatkins D. G., Monnot A. D., and Zheng W. (2006) Nε-thioacetyl-lysine: a multi-facet functional probe for enzymatic protein lysine Nε-deacetylation. Bioorg. Med. Chem. Lett. 16, 3651–3656 10.1016/j.bmcl.2006.04.075 [DOI] [PubMed] [Google Scholar]
- 168. Kokkonen P., Rahnasto-Rilla M., Kiviranta P. H., Huhtiniemi T., Laitinen T., Poso A., Jarho E., and Lahtela-Kakkonen M. (2012) Peptides and pseudopeptides as SIRT6 deacetylation inhibitors. ACS Med Chem Lett. 3, 969–974 10.1021/ml300139n [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Zang W., Hao Y., Wang Z., and Zheng W. (2015) Novel thiourea-based sirtuin inhibitory warheads. Bioorg. Med. Chem. Lett. 25, 3319–3324 10.1016/j.bmcl.2015.05.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Liu J., and Zheng W. (2016) Cyclic peptide-based potent human SIRT6 inhibitors. Org. Biomol. Chem. 14, 5928–5935 10.1039/c5ob02339d [DOI] [PubMed] [Google Scholar]
- 171. Heltweg B., Gatbonton T., Schuler A. D., Posakony J., Li H., Goehle S., Kollipara R., Depinho R. A., Gu Y., Simon J. A., and Bedalov A. (2006) Antitumor activity of a small-molecule inhibitor of human silent information regulator 2 enzymes. Cancer Res. 66, 4368–4377 10.1158/0008-5472.CAN-05-3617 [DOI] [PubMed] [Google Scholar]
- 172. Lain S., Hollick J. J., Campbell J., Staples O. D., Higgins M., Aoubala M., McCarthy A., Appleyard V., Murray K. E., Baker L., Thompson A., Mathers J., Holland S. J., Stark M. J. R., Pass G., et al. (2008) Discovery, in vivo activity, and mechanism of action of a small-molecule p53 activator. Cancer Cell 13, 454–463 10.1016/j.ccr.2008.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Rotili D., Tarantino D., Carafa V., Lara E., Meade S., Botta G., Nebbioso A., Schemies J., Jung M., Kazantsev A. G., Esteller M., Fraga M. F., Altucci L., and Mai A. (2010) Identification of tri- and tetracyclic pyrimidinediones as sirtuin inhibitors. ChemMedChem 5, 674–677 10.1002/cmdc.201000030 [DOI] [PubMed] [Google Scholar]
- 174. Mai A., Massa S., Lavu S., Pezzi R., Simeoni S., Ragno R., Mariotti F. R., Chiani F., Camilloni G., and Sinclair D. A. (2005) Design, synthesis, and biological evaluation of sirtinol analogues as class III histone/protein deacetylase (Sirtuin) inhibitors. J. Med. Chem. 48, 7789–7795 10.1021/jm050100l [DOI] [PubMed] [Google Scholar]
- 175. Parenti M. D., Grozio A., Bauer I., Galeno L., Damonte P., Millo E., Sociali G., Franceschi C., Ballestrero A., Bruzzone S., Del Rio A., and Nencioni A. (2014) Discovery of novel and selective SIRT6 inhibitors. J. Med. Chem. 57, 4796–4804 10.1021/jm500487d [DOI] [PubMed] [Google Scholar]
- 176. Sociali G., Galeno L., Parenti M. D., Grozio A., Bauer I., Passalacqua M., Boero S., Donadini A., Millo E., Bellotti M., Sturla L., Damonte P., Puddu A., Ferroni C., Varchi G., et al. (2015) Quinazolinedione SIRT6 inhibitors sensitize cancer cells to chemotherapeutics. Eur. J. Med. Chem. 102, 530–539 10.1016/j.ejmech.2015.08.024 [DOI] [PubMed] [Google Scholar]
- 177. Liu Y., Xie Q. R., Wang B., Shao J., Zhang T., Liu T., Huang G., and Xia W. (2013) Inhibition of SIRT6 in prostate cancer reduces cell viability and increases sensitivity to chemotherapeutics. Protein Cell. 4, 702–710 10.1007/s13238-013-3054-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Jiang H., Cheng S.-T., Ren J.-H., Ren F., Yu H.-B., Wang Q., Huang A.-L., and Chen J. (2019) SIRT6 inhibitor, OSS_128167 restricts hepatitis B virus transcription and replication through targeting transcription factor peroxisome proliferator-activated receptors α. Front. Pharmacol. 10, 1270 10.3389/fphar.2019.01270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Chang A. R., Ferrer C. M., and Mostoslavsky R. (2020) SIRT6, a mammalian deacylase with multitasking abilities. Physiol. Rev. 100, 145–169 10.1152/physrev.00030.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Tasselli L., Zheng W., and Chua K. F. (2017) SIRT6: novel mechanisms and links to aging and disease. Trends Endocrinol. Metab. 28, 168–185 10.1016/j.tem.2016.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Hall J. A., Dominy J. E., Lee Y., and Puigserver P. (2013) The sirtuin family's role in aging and age-associated pathologies. J. Clin. Invest. 123, 973–979 10.1172/JCI64094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Maity S., Muhamed J., Sarikhani M., Kumar S., Ahamed F., Spurthi K. M., Ravi V., Jain A., Khan D., Arathi B. P., Desingu P. A., and Sundaresan N. R. (2019) Sirtuin 6 deficiency transcriptionally up-regulates TGF-β signaling and induces fibrosis in mice. J. Biol. Chem. 295, 415–434 10.1074/jbc.ra118.007212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Sundaresan N. R., Vasudevan P., Zhong L., Kim G., Samant S., Parekh V., Pillai V. B., Ravindra P. V., Gupta M., Jeevanandam V., Cunningham J. M., Deng C.-X., Lombard D. B., Mostoslavsky R., and Gupta M. P. (2012) The sirtuin SIRT6 blocks IGF-Akt signaling and development of cardiac hypertrophy by targeting c-Jun. Nat. Med. 18, 1643–1650 10.1038/nm.2961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Fulda S., and Kögel D. (2015) Cell death by autophagy: emerging molecular mechanisms and implications for cancer therapy. Oncogene 34, 5105–5113 10.1038/onc.2014.458 [DOI] [PubMed] [Google Scholar]