

Abstract
Cells have a remarkable ability to synthesize large amounts of protein in a very short period of time. Under these conditions, many hydrophobic surfaces on proteins may be transiently exposed, and the likelihood of deleterious interactions is quite high. To counter this threat to cell viability, molecular chaperones have evolved to help nascent polypeptides fold correctly and multimeric protein complexes assemble productively, while minimizing the danger of protein aggregation. Heat shock protein 90 (Hsp90) is an evolutionarily conserved molecular chaperone that is involved in the stability and activation of at least 300 proteins, also known as clients, under normal cellular conditions. The Hsp90 clients participate in the full breadth of cellular processes, including cell growth and cell cycle control, signal transduction, DNA repair, transcription, and many others. Hsp90 chaperone function is coupled to its ability to bind and hydrolyze ATP, which is tightly regulated both by co-chaperone proteins and post-translational modifications (PTMs). Many reported PTMs of Hsp90 alter chaperone function and consequently affect myriad cellular processes. Here, we review the contributions of PTMs, such as phosphorylation, acetylation, SUMOylation, methylation, O-GlcNAcylation, ubiquitination, and others, toward regulation of Hsp90 function. We also discuss how the Hsp90 modification state affects cellular sensitivity to Hsp90-targeted therapeutics that specifically bind and inhibit its chaperone activity. The ultimate challenge is to decipher the comprehensive and combinatorial array of PTMs that modulate Hsp90 chaperone function, a phenomenon termed the “chaperone code.”
Keywords: Molecular chaperone, heat shock protein 90 (Hsp90), post-translational modification (PTM), cancer, co-chaperone, phosphorylation, acetylation, methylation, O-GlcNAcylation, Hsp90 inhibitor sensitivity, chaperone code, chaperone
Molecular chaperones are necessary for the stability, folding, and activation of a wide array of “client proteins” (1). One such molecular chaperone, the 90-kDa heat shock protein 90 (Hsp90), has over 300 clients, including protein kinases, transcription factors, oncoproteins, and tumor suppressors. Tight regulation of Hsp90 chaperone function and the downstream activities of its client proteins is essential for the maintenance of proteostasis, execution of the full spectrum of normal cellular processes, and preservation of tissue and organismal health (2, 3). Hsp90 possesses an ATPase activity that is coupled to its chaperone function along with a series of Hsp90 conformational changes collectively known as the chaperone cycle (1). A group of proteins, called co-chaperones, and post-translational modifications (PTMs) of Hsp90 together regulate Hsp90 chaperone activity and fine-tune it to the needs of the client proteins and the cell (4–7). Cancer cells often use Hsp90 function to promote tumor growth and metastasis, proffering Hsp90 as an attractive therapeutic target (8–11). Specific Hsp90-targeted therapeutics can simultaneously affect a myriad of cellular processes through chaperone function inhibition. Despite strong preclinical evidence of Hsp90 inhibitor efficacy, several drugs have had only limited success in phase 3 clinical trials, and none has achieved FDA approval. Co-chaperone dynamics and Hsp90 PTM status contribute to the complex variables that determine Hsp90 inhibitor sensitivity (12–14). Effort toward elucidating the impact of post-translational modifications of Hsp90 on its chaperone function has found that these modification states alter Hsp90 ATPase activity, co-chaperone and client binding, client maturation, Hsp90 subcellular localization and degradation, and Hsp90 inhibitor sensitivity (15–17). Dozens of modification sites, including phosphorylation, acetylation, SUMOylation, methylation, and others, have been functionally and mechanistically studied, as will be reviewed here. Taken together, the literature suggests that we need to develop a holistic understanding of the combinatorial array of these PTMs that target Hsp90 and modulate its chaperone activity. This phenomenon is also known as the “chaperone code.”
Structure and chaperone function of Hsp90
The two cytosolic isoforms of Hsp90, Hsp90α and Hsp90β, share 85% sequence identity (18–20). Hsp90β is constitutively expressed under normal physiological conditions. Hsp90α, however, is stress-inducible, and increased levels of Hsp90α have been associated with poor prognosis in cancer (20). The chaperone function of Hsp90 encompasses an ordered series of conformational changes coupled to its ATPase activity, collectively known as the “chaperone cycle” (Fig. 1) (6, 21–24). The functional unit of Hsp90 is a dimer, and each protomer consists of three structural domains (25–27). The amino-terminal domain, or “N-domain,” contains the nucleotide-binding pocket, which also serves as the binding site for most Hsp90 inhibitors (28–30). The middle domain is connected to the N-domain by a flexible and highly charged linker region (31, 32). The middle domain harbors the catalytic loop that is necessary for ATP hydrolysis and also serves as the binding site for the majority of client proteins and many co-chaperones (1, 33, 34). The carboxyl-terminal domain, or “C-domain,” is the site of constitutive dimerization of the Hsp90 protomers (35, 36) and contains the extreme C-terminal MEEVD sequence that serves as an interaction site for tetratricopeptide repeat (TPR)-domain–containing co-chaperones (1). The Hsp90 chaperone cycle is considered to begin with an “open” dimer, which is only dimerized at the C-domain. ATP binding to the nucleotide pocket of the N-domain contributes to the large-scale conformational rearrangements that lead to transient dimerization of the Hsp90 N-domains. This transiently N-terminally dimerized state is referred to as the “closed” conformation. Upon ATP hydrolysis, Hsp90 returns to its “open” V-shaped conformation and is ready to begin another cycle (Fig. 1) (6, 24, 36–39). There is also a great deal of interdomain connectivity and communication across the Hsp90 protein (40–42). Progression through the chaperone cycle is required for client chaperoning and is tailored to individual client needs. In general, clients require chaperoning to either achieve a final active conformation, assemble into multiprotein complexes, or promote and stabilize a ligand-competent state that is awaiting activation (1).
Figure 1.

The Hsp90 chaperone cycle. Hsp90 begins its chaperone cycle in an open conformation that is dimerized only at the C-domain. ATP binding and an ordered series of conformational changes allow it to adopt a closed conformation, which is N-terminally dimerized. Upon ATP hydrolysis, Hsp90 returns back to the open conformation and is ready to begin another chaperone cycle. This allows for the activation of client proteins. This cycle is tightly regulated by co-chaperone proteins as well as PTMs, and Hsp90 inhibitors can also modulate the chaperone cycle.
Many factors contribute to Hsp90 chaperone cycle regulation and subsequent client chaperoning, including interacting co-chaperone proteins and post-translational modifications, and Hsp90 inhibitors can also affect the cycle. Co-chaperones, unlike client proteins, are not themselves dependent on Hsp90 chaperone function for their stability or activity. Generally, co-chaperones alter the progression of Hsp90 through the chaperone cycle by stabilizing different Hsp90 states and conformational intermediates. The TPR-containing co-chaperone Hsp70-Hsp90–organizing protein (HOP) binds to the open conformation of Hsp90, slows down its ATPase activity, and also helps transfer client proteins to Hsp90 (43–45). Client scaffolding or loading is another common attribute of Hsp90 co-chaperones. The co-chaperone cell division cycle 37 (Cdc37) specifically recruits kinase clients to Hsp90. The Cdc37-mediated chaperoning of kinases requires coordinated phosphorylation and subsequent dephosphorylation of Cdc37 by the phosphatase co-chaperone protein phosphatase 5 (PP5) (46–48). The co-chaperone activator of Hsp90 ATPase (Aha1), on the other hand, displaces HOP and helps to mediate Hsp90 N-domain dimerization and enhances the ATPase activity of Hsp90 (49–52). The co-chaperone prostaglandin E synthase 3 (p23) preferentially binds to and stabilizes Hsp90 in the closed conformation, slowing ATP hydrolysis and aiding client activation (26, 53). Co-chaperone dynamics, as well as several other processes, including Hsp90 ATPase activity, client activation, and Hsp90 inhibitor binding, are further altered by Hsp90 PTMs, as will be highlighted and discussed in the subsequent sections.
Post-translational modifications and Hsp90 function
Phosphorylation
Hsp90 is subject to various PTMs, as will be considered in the sections to follow. Phosphorylation of serine, threonine, and tyrosine residues is the most well-studied of these Hsp90 modifications. The earliest functional work demonstrated that treatment with the nonselective serine/threonine phosphatase inhibitor okadaic acid resulted in hyperphosphorylated Hsp90 and compromised chaperoning of the classic kinase client v-Src (54). As will be discussed, much of the functional work focuses on how different PTMs impact chaperone function via examination of ATPase activity of Hsp90, co-chaperone binding, and client stability. Additional works further detail how Hsp90 modification affects downstream cellular processes such as cell cycle control, DNA repair, and steroid hormone signaling as well as many others via effects on the stability and activity of Hsp90 client proteins (Fig. 2). Of note, a large number of potential Hsp90 modification sites in addition to those discussed below have been identified by high-throughput screening studies (RRID:SCR_001837). These studies will not be discussed here, as these sites have not been validated or linked to functional consequences; however, continued evaluation of these sites is necessary to further our understanding and application of the chaperone code.
Figure 2.

Modification by varied enzymes regulates Hsp90 function in biological processes. Enzymes known to modify Hsp90 regulate its PTM state, and this in turn influences various cellular functions as indicated. Arrows are color-coded to match the cellular functions. Yellow arrows indicate cell cycle and proliferation, purple indicates cytoskeleton remodeling and migration, light blue indicates transcription, red indicates angiogenesis and tumor formation, and navy blue indicates DNA repair, apoptosis, and metabolism. Enzymes that are also Hsp90 clients are shaded in green.
Serine/threonine phosphorylation
Chaperone cycle
As discussed above, Hsp90 chaperone function toward client maturation and downstream processes depends on the dynamics of the chaperone cycle, including Hsp90 ATPase activity and co-chaperone binding. The Hsp90 chaperone cycle is in part regulated by phosphorylation mediated by casein kinase 2 (CK2), a ubiquitously expressed and constitutively active protein kinase (55, 56). Phosphorylation of yeast Hsp90-T22 (human Hsp90α-T36) or Hsp90β-S365 by CK2 disrupts binding of Hsp90 with the kinase-specific co-chaperone Cdc37 (Table 1 and Fig. 3) (64, 93). Additionally, yHsp90-T22 phosphorylation compromises Hsp90 interaction with the activating co-chaperone Aha1 and subsequently results in decreased Hsp90 ATPase activity (64). Further, ATP binding is impacted by CK2 phosphorylation of the Hsp90β charged linker, which has been suggested to precede Hsp90:Cdc37 complex dissociation when ATP levels are low (84). Interestingly, CK2 phosphorylation of Cdc37-S13 promotes its association with kinases and recruitment of Hsp90 to kinase:Cdc37:Hsp90 ternary complexes for the chaperoning of kinase clients (46–48, 152). Furthermore, sequential and ordered phosphorylation of the co-chaperone folliculin-interacting protein 1 (FNIP1) on several adjacent serine residues promotes its binding to Hsp90 and facilitates kinase and nonkinase client chaperoning (153). CK2-mediated phosphorylation of both Cdc37 and FNIP1 is specifically reversed by the phosphatase co-chaperone PP5, highlighting the complex interplay of PTMs in the chaperone machinery (47, 153).
Table 1.
Hsp90 post-translational modifications, identified modifying enzymes, and functional consequences
Identified modification sites in Hsp90 are shown. Conservation between α and β isoforms is highlighted. Further indication of other species in which residues were found as well as other modifications that particular sites are subject to is as follows: boldface type, identified in that isoform; *, identified in Hsp82; †, identified in zebrafish Hsp90a1; §, identified in Candida albicans; a, acetylation; n, nitration; p, phosphorylation; s, SUMOylation; sc, succinylation; u, ubiquitination; g, glycation; o, O-GlcNAcylation; m, methylation; c, citrullination; sn, S-nitrosylation; t, thiocarbamlyation; ↑, binding increased (increased ATPase or increased inhibitor binding or sensitivity); ↓, binding decreased (decreased ATPase or decreased inhibitor binding or sensitivity); nc, no change; NA, not applicable/not conserved; no entry provided in the table if not determined or examined.
| PTM | Residue |
Enzyme |
Small-molecule binding |
References | ||
|---|---|---|---|---|---|---|
| Hsp90α | Hsp90β | Compound | ATP | Inhibitors | ||
| Serine/threonine phosphorylation | T5 | NA | ATM, DNA-PK | 57–62 | ||
| T7 | NA | ATM, DNA-PK | 57–63 | |||
| T36 (*,†) | T31 | CK2 | ↓ | ↑ | 64–66 | |
| S63 | S58 | CK2 | 67–69 | |||
| T65 | T60 | CK2 | 68, 70 | |||
| S68 | S63 | CK2 | 68 | |||
| S72 | S67 | CK2 | 68 | |||
| T88 | T83 | PKA | 69, 71, 72 | |||
| T90 | T85 | PKA | ↓ | 72–76 | ||
| S113 | S108 (*) | HopBF1 | ↓ | 77 | ||
| T115 | T110 | Mps1, Cdc14, PKCγ | ↓ | ↑ | 78, 79 | |
| S164 | S159 | Cdc7-Dbf4 | 80, 81 | |||
| S211 | S206 | PKA/PKG | 82, 83 | |||
| S231 | S226 | CK2, PP5 | 55, 84–89 | |||
| S263 | S255 | CK2, B-Raf, PP5 | 55, 84–92 | |||
| N373 | S365 | CK2 | 83, 93 | |||
| S399 (*) | S391 | ↓ | ↑ | 67, 94–96 | ||
| T425 | S417 | PKCγ | ↓ | 79 | ||
| S460 (o) | S452 (o) | PKA | 82 | |||
| S505 (*) | S497 | ↓ | ↑ | 69, 94 | ||
| S595 | S587 | Mitogen-activated protein kinase 12 (p38γ) | 97–99 | |||
| T603 | T595 | PKCγ | nc | 79, 99 | ||
| S623 (*) | S615 | ↓ | 94, 100, 101 | |||
| T624 | T616 | 102, 103 | ||||
| M625 (*) | M617 | ↓ | 94 | |||
| T725 | A717 | CK2, CK1, GSK3β | 104, 105 | |||
| S726 | S718 | CK2, CK1, GSK3β | 104, 105 | |||
| Tyrosine phosphorylation | Y38 (*,n) | Y33 (n) | Swe1 | ↓ | ↓ | 99, 106 |
| Y197 | Y192 | v-Src, Yes | 107–112 | |||
| Y309 | Y301 | c-Src | 110, 113, 114 | |||
| Y313 | Y305 | ↑ | 42, 69, 107, 115 | |||
| Y627 (*) | Y619 | 69, 107, 116, 117 | ||||
| Acetylation | K41 (§,u) | K36 (u) | ↑ | 118–120 | ||
| K69 (u) | K64 (u) | HAT p300 | ↓ | ↑ | 101, 121–124 | |
| K74 (u,g) | K69 (u) | 97, 99 | ||||
| K100 (u,g) | K95 | HAT p300 | ↓ | ↑ | 121 | |
| K292 (u) | K284 (u) | HAT p300 | ↑ | 121, 124 | ||
| K294 (†,§,u) | K286 (u) | HDAC6 | ↑ | 65, 118, 119, 125–129 | ||
| K327 (u) | K319 (u) | HAT p300 | ↓ | ↑ | 99, 121 | |
| K407 (u) | K399 (u) | ↑ | 124, 130 | |||
| K419 (u) | K411 | ↑ | 130, 131 | |||
| K478 (u,g) | S470 | HAT p300 | ↓ | ↑ | 121 | |
| K546 (u,g,sc) | K538 (u,sc) | HAT p300 | ↓ | ↑ | 121, 131 | |
| K558 (u) | K550 | HAT p300 | ↓ | ↑ | 121 | |
| Monomethylation | K209 (†,u) | K204 | SMYD2 | 65, 132 | ||
| K539 | K531 (a,u) | SMYD2 | 133 | |||
| K582 | K574 (a,u) | SMYD2 | 133 | |||
| K615 (*,†,a,u) | K607 | SMYD2 | 65, 132, 134–136 | |||
| Thiocarbamylation | C529 | C521 | 6-HITC-ME | 137 | ||
| C597 (sn) | C589 (sn) | STCA | 138 | |||
| S-Nitrosylation | C597 (t) | C589 (t) | Nitric oxide | 139, 140 | ||
| SUMOylation | K191 (a,u) | K186 | SUMO-1 | ↑ | 141 | |
| K559 (u) | K551 | SUMO peptidase sentrin/SUMO-specific protease 2 (SENP2) | 142 | |||
| Ubiquitination | K112 (g) | K107 | CHIP | 143–145 | ||
| K209 (m) | K204 (a) | CHIP | 143, 144, 146 | |||
| K224 (a) | K219 (a) | CHIP | 143, 144, 146 | |||
| K283 (a,g) | K275 (a) | CHIP | 143, 144, 146 | |||
| K292 (a) | K284 (a) | CHIP | 143, 144, 146 | |||
| R355 | K347 (a,m,sc) | CHIP | 144 | |||
| K407 (a) | K399 (a) | CHIP | 143, 144, 146 | |||
| K485 (a) | K477 (a) | CHIP | 143, 144 | |||
| K489 (a,m,sc) | K481 (a,sc) | CHIP | 143, 144, 146 | |||
| K546 (a,g,sc) | K538 (a,sc) | CHIP | 143–145 | |||
| K558 (a) | K550 | CHIP | 143, 144 | |||
| K615 (a,m) | K607 (a) | CHIP | 143, 144, 147 | |||
| K631 (a,sc) | K623 (a,sc) | CHIP | 143, 144, 148 | |||
| Nitration | Y38 (p) | Y33 (p) | 149, 150 | |||
| Y61 (p) | Y56 (p) | 149 | ||||
| O-GlcNAcylation | S442 (p) | S434 (p) | 151 | |||
| S460 (p) | S452 (p) | 151 | ||||
Figure 3.

Schematic representation of Hsp90α PTM sites. Residues that have been functionally studied and found to contribute to the chaperone code are shown in red with colored circles indicating effects on cellular function (blue), co-chaperone binding (orange), ATP binding (yellow), and Hsp90 inhibitor binding/sensitivity (green). Residues in black have been reported to be modified but have not yet been validated to affect chaperone function.
Phosphorylation of Hsp90α-T90 by protein kinase A (PKA) also alters the complement of co-chaperones bound to Hsp90 and decreases Hsp90 affinity for ATP (73). Phosphomimetic mutation of Hsp90α-T90 amplified binding to the co-chaperones Aha1, p23, PP5, and C terminus of Hsp70-interacting protein (CHIP) but diminished binding to Hsp70, Cdc37, and HOP. In contrast, phosphorylation of residues in the Hsp90α C-domain by the kinases CK2, CK1, and glycogen synthase kinase 3β (GSK3β) increased interaction between Hsp90α and HOP while decreasing interaction with CHIP (104).
Hsp90 phosphorylation also has a consequent impact on client binding and activation. Hsp90α-T90 phosphorylation abrogated binding to the client kinases Src, Akt, and PKCγ likely as a result of diminished interaction between Hsp90 and Cdc37 (73). Interestingly, phosphorylation of yHsp90-T101 (hHsp90-T115) by the dual-specificity kinase Mps1 promoted kinase client activation, whereas the nonphosphorylatable alanine mutation favored nonkinase clients, possibly due to altered co-chaperone interactions (78). Furthermore, phosphorylation of two residues in the charged linker, Hsp90α-S231 and -S263, was reduced in cells expressing a truncated form of the co-chaperone p23 (85). Phosphorylation at these residues was important for telomerase activity through stability of the hTERT telomerase catalytic subunit, which is an Hsp90 client (85).
Phosphatases also play a critical role in modulating Hsp90 phosphorylation status and therefore the chaperone cycle. The co-chaperone Ppt1 (yeast ortholog of PP5) was shown to directly dephosphorylate Hsp90 and consequently affected its chaperone activity (154). Another study found 10 residues of yHsp90 that are dephosphorylated by Ppt1 (94). The phosphorylation status of these residues differentially influenced ATPase activity as well as client activity. Interestingly, Hsp90-S485E phosphomimetic mutant had the most robustly decreased ATPase activity despite being distant from the ATP-binding pocket, suggesting that phosphorylation of this residue may cause distant structural changes (94).
Taken together, phosphorylation is a dynamic regulatory mechanism on the Hsp90 chaperone cycle. Of note, when these sites are modified there is generally a resultant decrease in Hsp90 ATPase activity. Phosphorylation can additionally allosterically affect co-chaperone and client dynamics at binding sites far from the modified site, suggesting a complex interplay of communication across the chaperone protein. Furthermore, these allosteric effects on co-chaperone dynamics often have further consequences on Hsp90 ATPase activity, and the details of these regulatory mechanisms are not yet fully understood.
Cell cycle control
Hsp90 interacts with numerous cell cycle regulators, including the kinases CDK2, -4, and -6; Mps1 and Swe1 (yeast ortholog of human Wee1); and cyclin B (78, 106, 155–157). Hsp90α-T90 phosphorylation was found to be more abundant in actively proliferating cells (73). Phosphorylation specifically by PKA on Hsp90-T90 also promoted prostate cancer cell proliferation (74). Additionally, Hsp90α-T725 and -S726 and Hsp90β-S718 phosphomimetic mutations decreased the doubling time of HEK293 cells (104). Furthermore, Hsp90 threonine phosphorylation levels fluctuate throughout the cell cycle (78). Yeast Hsp90-T101 (hHsp90α-T115) was phosphorylated throughout mitosis, but Hsp90-T101 phosphorylation was absent in G1 phase. The cell cycle mediators Mps1 and Cdc14 phosphorylate and dephosphorylate Hsp90-T101, respectively. Together, Mps1 and Cdc14 regulate mitotic arrest and exit from mitosis, at least in part, via Hsp90 phosphorylation (78).
DNA repair and apoptosis
DNA-dependent protein kinase (DNA-PK) is both a client and a regulator of Hsp90α that phosphorylates Hsp90α on Thr-5 and -7 (57, 58, 63). Phosphorylation of these residues occurred early in apoptosis and was critical for histone γH2AX formation at dsDNA breaks, DNA fragmentation, and apoptotic body formation (57). Furthermore, Thr-7 phosphorylation in the cytosol was a prerequisite for Hsp90α accumulation at DNA double-strand breaks and subsequent formation of DNA repair foci. Hsp90α-T7 phosphorylation level was also found to correlate with the apoptotic marker pH2AX in tumors, suggesting that Hsp90α-T7 phosphorylation could serve as a potential marker for DNA damage (63). It is unclear precisely how the role of Hsp90-T7 phosphorylation is integrated across the spectrum of DNA damage recognition and repair to apoptosis or whether these roles are context-dependent. It is noteworthy that nuclear yHsp90 was recently discovered to be important for chromatin remodeling through regulation of the Arp2/3-dependent actin filaments (158), which were found to be clustered at double-strand DNA breaks (159, 160). Interestingly, levels of pThr-5/7 increase with age and correlate with a decline in AMPK activity (58). There is evidence of a role for Hsp90β in apoptosis as well. Hypophosphorylation of Hsp90β-S226 and -S255 was found to prevent cytochrome c–induced apoptosome activation (86).
Transcription factor regulation
Numerous transcription factors have been found to interact with Hsp90, including vertebrate steroid hormone receptors and PAS domain family members. It has been shown that Hsp90 phosphorylation increased glucocorticoid receptor (GR) activity (64, 93). Mutation of yeast Hsp90-T22 to phosphomimetic glutamate increased GR activity over 4-fold compared with WT Hsp90, whereas mutation to the phospho-null Hsp90-T22A significantly decreased GR activity (64). Interestingly, phosphomimetic mutation of Hsp90β-S365E, a residue that is not conserved in Hsp90α, also significantly increased GR activity when expressed as the sole Hsp90 in yeast (93). Additionally, Hsp90-T90 phosphorylation by PKA is important for androgen receptor (AR)-mediated transcription (74). Phosphorylation of Hsp90-T90 released AR from Hsp90, leading to AR nuclear localization and subsequent transcription (74). Work by Ogiso et al. (87) demonstrated phosphorylation of Hsp90 residues in the charged linker (Hsp90α-S230 and Hsp90β-S226 and -S255) that impaired Hsp90 binding to the bHLH/PAS family transcription factor arylhydrocarbon receptor. Nonphosphorylatable mutation of these residues restored Hsp90 interaction with arylhydrocarbon receptor, subsequently increasing its transcriptional activity toward xenobiotic-responsive elements (87). These works highlight the importance of Hsp90 PTMs for regulating chaperoning of transcription factors and demonstrate the need for further investigation into this area.
Cancer development and progression
Consistent with cancer cells' reliance on Hsp90 chaperone machinery, Hsp90 is differentially modified in cancer versus normal cells, and Hsp90 phosphorylation has been linked to numerous cancer-driving processes. In addition to some examples seen above, early work in the field posited a role for Hsp90 phosphorylation in cellular transformation. Phosphatase inhibitor treatment led to hyperphosphorylation of Hsp90 and decreased complex formation between Hsp90 and the oncogenic tyrosine kinase client, v-Src, and suggested that cycling of Hsp90 PTMs could direct v-Src trafficking and cellular transformation (54). Hsp90 phosphorylation has been implicated to contribute to many of the hallmarks of cancer, including processes such as cancer cell migration (79), proliferation (74, 104), invasion (75), tumor regression and immunogenicity (90), and therapeutic resistance (86). Furthermore Hsp90α-S164 is hyperphosphorylated in oral cancer (80), and total phosphorylated Hsp90 was found to be increased in breast tumor samples compared with normal tissue (104). In contrast, Hsp90β-S226 and -S255 are hypophosphorylated in leukemic cells, further demonstrating the regulatory importance of Hsp90β PTMs (86). The mechanisms that contribute to these differential degrees of Hsp90 phosphorylation in specific instances are largely unknown. It is likely that this reflects a combination of alterations in kinase or phosphatase expression as well as changes to kinase regulation. Of note, many of the kinases that modify Hsp90 are themselves clients. Mps1, one of the client kinases, was increased in kidney cancer tissue relative to adjacent normal tissue, and this correlated with Hsp90 inhibitor sensitivity, suggesting that Hsp90 phosphorylation plays a role in kidney cancer as well (78). The effect of Hsp90 PTMs on drug sensitivity will be further discussed below.
Tyrosine phosphorylation
Chaperone cycle and kinase maturation
Numerous cellular pathways are also regulated by Hsp90 tyrosine phosphorylation. Significant work has been done detailing a series of tyrosine phosphorylation events that affect the Hsp90 chaperone cycle and chaperoning of kinase clients. Nonreceptor tyrosine kinase Yes plays a prominent role in Hsp90 function through phosphorylation of both Hsp90α and co-chaperone Cdc37. Yes first phosphorylates Cdc37-Y298, inducing a conformational change that primes the client:Cdc37:Yes complex to bind to Hsp90α (107). Next, Yes phosphorylates Hsp90-Y197, disrupting the Hsp90:Cdc37 complex (107, 108). This process is essential to kinase client maturation (108). Perhaps this is due to the subsequent phosphorylation of Hsp90-Y313 on only one protomer of an Hsp90 dimer (i.e. asymmetric phosphorylation), which recruits Aha1, thereby stimulating Hsp90 ATP hydrolysis and client maturation (42, 107). Interestingly, whereas phosphomimetic mutation Hsp90-Y313E enhances Aha1 association with Hsp90, it abrogates interaction with the competing co-chaperone tuberous sclerosis complex protein 1 (Tsc1) (161). Phosphorylation of Hsp90-Y627 by Yes then triggers dissociation of Aha1 and the mature client, “resetting” Hsp90 for the next cycle (107). Additionally, Hsp90-Y627 phosphorylation has been proposed as a mechanism to functionally replace the evolutionarily lost yeast Aha1-like co-chaperone, Hch1 (88). This series of Hsp90 tyrosine phosphorylation events profoundly affects the dynamics of the chaperone cycle, including the binding and release of co-chaperone proteins. Determination of precisely how this translates to client activation, however, requires further work. One example that has been elucidated is in B-cell antigen receptor (BCR) signaling. Signals transduced through BCR signaling play a role in a subset of B-cell lymphomas. Spleen tyrosine kinase (SYK), a key effector of BCR signaling, is an Hsp90 client and depends on Hsp90α-Y197 phosphorylation for its function (109). The Hsp90 client Swe1 also mediates Hsp90 tyrosine phosphorylation that affects Hsp90:co-chaperone dynamics. Phosphorylation of yHsp90-Y24 (human Hsp90α-Y38) was important for Hsp90 interaction with the co-chaperones Aha1 and p23; knockout of Swe1 or mutation of Tyr-24 to the nonphosphorylatable phenylalanine abolished Hsp90 interaction with Aha1 and decreased interaction with p23 (106).
Cell cycle control
Phosphorylation of the conserved yHsp90-Y24 (human Hsp90α-Y38) has diverse implications on not only Hsp90 chaperone function but also the cell cycle. Swe1 (yeast ortholog of human Wee1) is a key component of the G2-M cell cycle checkpoint (162). Swe1 phosphorylated yHsp90-Y24 in a cell cycle–dependent manner, and Hsp90 tyrosine phosphorylation was observed only in S phase–arrested cells (106). It was also shown that yHsp90-Y24 phosphorylation serves as a prerequisite to Hsp90 ubiquitination and proteasomal degradation (106). Interestingly, yHsp90-Y24 phosphorylation is also important for chaperoning of Swe1, which is an Hsp90 client. Swe1-mediated phosphorylation enhanced the association of the kinase with Hsp90 and its stability and function (163). Expression of the nonphosphorylatable yHsp90-Y24F mutant caused yeast to undergo premature nuclear division and made them insensitive to G2/M checkpoint arrest (163).
Angiogenesis and inflammation
Additionally, Hsp90 tyrosine phosphorylation regulates cell proliferation, angiogenesis, and inflammation through endothelial nitric oxide synthase (eNOS). eNOS produces nitric oxide (NO), which mediates angiogenic and vasodilatory effects, and eNOS signaling is affected by Hsp90 phosphorylation (113). Duval et al. (113) found that, upon vascular endothelial growth factor receptor 2 (VEGFR2) stimulation, VEGFR2-bound Hsp90β is phosphorylated by the kinase c-Src at Tyr-300. This phosphorylation event is critical to subsequent Hsp90 interaction with eNOS and VEGF-stimulated eNOS signaling and angiogenesis. Furthermore, c-Src–mediated phosphorylation of Hsp90α-Y309 (Hsp90β-Y301) in endothelial cells could also be induced by the inflammation-inducing bacterial wall component lipopolysaccharide, and this was needed to mediate the pro-inflammatory actions of Hsp90 in this context (164). Overall, Hsp90 phosphorylation has a spectrum of effects on chaperone function and cellular processes, but there are roles for the regulatory functions of other PTMs as well.
Acetylation–deacetylation
Hsp90 chaperone function is also affected by acetylation. Acetylation is a post-translational modification that results from the addition of acetyl groups generally to lysine residues. As this modification was first identified and described on histones in the nucleus, the classes of enzymes responsible were named histone acetyltransferases (HATs) and histone deacetylases (HDACs). These enzymes, however, can modify numerous other cellular proteins, including Hsp90. Treatment with HDAC inhibitors (HDACi) has been demonstrated to lead to hyperacetylation of Hsp90 in numerous reports (165, 166). In general, Hsp90 acetylation alters its ability to bind ATP and Hsp90 inhibitors and its interaction with both client and co-chaperone proteins (125, 166). This has particular consequences for the stability and function of kinases and steroid hormone receptors as well as cell migration. Hsp90 inhibitor sensitivity and fungal virulence are also impacted and will be discussed later. Additionally, there are studies examining the role of Hsp90 acetylation in mediating glucocerebrosidase activity in Gaucher disease and tau phosphorylation in Alzheimer's disease (126, 167).
Oncogenic kinase chaperoning
Kinases are one of the main classes of Hsp90 clients and are needed to support growth and survival in many cancers. Early work demonstrated that inhibition of HDAC6 led to the hyperacetylation of Hsp90 and reduced the association of Hsp90 with its kinase clients, including Bcr-Abl, Akt, and c-Raf (165). Destabilization and degradation of kinases as a result of HDACi and decreased kinase association with Hsp90 has also been demonstrated for VEGFR1, VEGFR2, erythroblastic oncogene B (ErbB1), ErbB2, Raf-1, and extracellular signal–regulated protein kinase (ERK1/2) (168, 169). Hsp90-K294 acetylation specifically has been shown to affect both kinase and nonkinase client binding (125). Acetylated lysine mimic mutations K294A and K294Q disrupted interaction of Hsp90 with its clients ErbB2 and v-Src as well as the kinase-specific co-chaperone Cdc37, whereas nonacetylatable mutation K294R either enhanced binding or had no effect (125). Another interesting study demonstrated that Hsp90 acetylation can also be increased by inhibition of the transcription factor heat shock factor 1 (HSF1) as a result of disruption of a cytosolic complex containing HSF1, p97, Hsp90, and HDAC6 (170). This further resulted in reduced association of Hsp90 and Cdc37 and consequent depletion of kinase clients, including Bruton's tyrosine kinase, c-Raf, and CDK4 (170). Hsp90 function and kinase stability were also reduced following HSF1-targeted treatment in a leukemia mouse model, suggesting possible therapeutic benefit of targeting this mechanism (170).
Steroid hormone receptor stability and activity
Steroid hormone receptors are another well-described class of Hsp90 clients that are affected by Hsp90 acetylation. Some of the initial work demonstrating regulation of Hsp90 acetylation by HDAC6 showed compromised Hsp90-dependent GR maturation in HDAC6-deficient cells (166). GR ligand binding, nuclear translocation, and transcriptional activity were impaired with HDAC6 inhibition or loss (166, 171). This stemmed from a defect of hyperacetylated Hsp90 in functional heterocomplex formation with GR and not from an intrinsic change to GR (172). As a result of HDAC6 inhibition, hyperacetylated Hsp90 also dissociated from the co-chaperone p23 (95). Hsp90 acetylation–dependent GR signaling has also been implicated in stress resilience. Selective depletion of HDAC6 in serotonergic neurons decreased GR signaling and led to reduced angiogenic effects of corticosterone and also blocked the expression of social avoidance behavior in mice exposed to chronic social defeat (173). Similarly, lower levels of Hsp90 acetylation and consequent enhanced GR translocation have been seen in the dorsal raphe nucleus after chronic social defeat stress (127). Treatment with an HDAC6i, ACY-738, increased relative association of Hsp90 with the co-chaperone FKBP52 and inhibited hormone-induced GR translocation in cells and in mice promoted resilience to chronic social defeat stress. The effects of HDACi on stress resilience could also be replicated with a Lys-294 acetylation mimetic, suggesting these effects are mediated through modulation of acetylation on this specific lysine residue (127). In contrast to the effects of Hsp90 acetylation on GR function, specifically Hsp90-K294 acetylation, another group demonstrated that acetylation of the equivalent lysine, Lys-295, in mouse Hsp90α alters the cellular localization of the mineralocorticoid receptor (MR). Interestingly, it did not alter MR transcriptional activity and therefore suggests that Hsp90 acetylation could assist in balancing MR and GR activity when both are expressed (128).
Modulation of Hsp90 acetylation also has effects on the estrogen and androgen receptors. Hsp90 hyperacetylation as a result of HDACi decreased binding of Hsp90 to the estrogen receptor (ERα) leading to its ubiquitination and proteasomal degradation (174). This led to a subsequent decrease in ER-mediated transcription. HDACi in breast cancer cells also decreased the stability of other prosurvival Hsp90 clients, including Akt and c-Raf (174). Similarly, AR dissociated from Hsp90 and was degraded in the proteasome upon treatment with sulforaphane, a derivative of glucoraphanin that has been shown to mediate HDAC6 inhibition (175). Sulforaphane increased Hsp90 acetylation, leading to AR instability and reduced AR target gene expression in an HDAC6-specific manner. HDAC6 knockdown (KD) similarly impaired AR localization and inhibited prostate-specific antigen expression in the absence or presence of the AR ligand dihydrotestosterone (176). The effect on AR localization in this study was also mediated by acetylation on Lys-294 of Hsp90 (176), and AR has been seen to dissociate from K294A or K294Q acetyl mimics (125). Overall, Hsp90 acetylation, particularly at Lys-294, plays an important role in the chaperoning and function of steroid hormone receptors. Modulation of these dynamics may present a therapeutic target in stress resilience as well as hormone-dependent cancers, such as breast and prostate cancer.
Cytoskeletal dynamics and cell migration and invasion
Several studies have examined the role of Hsp90 acetylation in affecting cytoskeletal remodeling dynamics as well as cell migration. He et al. (65) examined the role of various Hsp90α PTMs on myosin thick filament organization in zebrafish skeletal muscle. Hsp90 plays a key role in myosin folding and thick filament assembly, but this role was repressed by acetylation mimic K287Q (Lys-294 in hHsp90α) mutation, whereas the nonacetylatable K287R was able to rescue thick filament defects in Hsp90α KD embryos (65).
Hsp90 acetylation also plays a role in actin cytoskeleton dynamics. An unexpected finding of HDAC6 localizing to actin-enriched membrane ruffles led to the identification of Hsp90 recruitment to membrane ruffles and macropinosomes as well (177). The acetylation-resistant K294R mutant enhanced dorsal ruffle formation in HDAC6 knockout mouse embryonic fibroblasts, whereas acetyl mimic K294Q did not. Treatment with the Hsp90 inhibitor geldanamycin also inhibited ruffle formation as well as cell migration (177). Hsp90 further plays a role in mediating HDAC1-regulated cell migration through stability of TAp73, a member of the p53 family (178). HDAC1 KD resulted in hyperacetylation of Hsp90 and subsequent proteasomal degradation of TAp73, which was required for the enhanced cell migration seen with HDAC1 KD (178). Hsp90 is also a target of SIRT2, a tumor suppressor and class III histone deacetylase. SIRT2 was found to deacetylate Hsp90, and this was a necessary prerequisite to ubiquitination and degradation of Hsp90 (179). The regulation of Hsp90 by SIRT2 deacetylase activity was required for SIRT2 inhibition of actin polymerization and cell migration (179). Additionally, Hsp90 acetylation plays a role in tumor cell invasiveness. Three acetylation mimics of Hsp90α, K69Q, K100Q, and K558Q, were found to promote the export of Hsp90 and increased the amount found extracellularly (121). These acetyl mimics further facilitated binding to the matrix metalloproteinase MMP2 and enhanced invasiveness of breast cancer cells in a Matrigel invasion assay. Treatment with pan-HDACi promoted cell invasiveness; however, treatment with an anti-acetylated-K69-Hsp90 antibody inhibited invasiveness to a greater degree than a polyclonal anti-Hsp90 antibody, suggesting that specifically targeting acetylated Hsp90 in the extracellular space could decrease tumor invasion and metastasis (121). Further potential therapeutic benefits utilizing Hsp90 acetylation will be discussed below.
Other reported PTMs
As seen above, the literature examining the effects of phosphorylation and acetylation on Hsp90 function is quite robust. Many other types of PTMs, however, have also been reported to impact Hsp90 function. Here we summarize those studies, and of note, these sections are not further subdivided by functional consequence because the reports are less numerous than those examined above.
Methylation
Protein methylation has most notably been studied in nuclear histone proteins, where the so-called “histone code” regulates gene expression (180). Despite this focus, protein methylation is also a common cytosolic modification. Catalyzed by methyltransferases, this enzymatic process frequently targets lysine and arginine side chains. SET and MYND domain–containing 2 (SMYD2) is one such lysine methyltransferase, reported to methylate Hsp90α at Lys-209 and conserved Lys-615 (Hsp90β-K607, yHsp90-K594) (132, 134). Methylation of yHsp90-K594 causes allosteric conformational changes that impact Hsp90 ATP hydrolysis, Aha1 and p23 binding, and client activity (134). Specifically, monomethylated yHsp90-K594 was generated via amber suppression and demonstrated impaired N-terminal dimerization kinetics compared with WT Hsp90 (134). Additionally, SMYD2 is also reported to interact with p23 and HOP co-chaperones, suggesting varied roles for methylation in the regulation of Hsp90 function (132). Hsp90-K615 methylation in skeletal and cardiac muscle has been shown to promote the stability of the sarcomeric protein titin and myofibril organization, providing a mechanistic explanation for the role of Hsp90 in skeletal and cardiac muscle function (135, 181). SMYD2 also methylated Hsp90β at the conserved residues Lys-531 and -574. These methylation events promoted Hsp90 chaperone function and served to increase the growth of bladder cancer cell lines (133). In summary, lysine methylation serves to enhance Hsp90 function through a wide array of modification sites and can act as a switch point to allosterically regulate distant regions of Hsp90.
Thiocarbamylation and S-nitrosylation
Thiocarbamylation of Hsp90 results from binding of the small molecules sulfoxylthiocarbamate alkyne (STCA) and 6-methylsulfinylhexyl isothiocyanate-β-mercaptoethanol (6-HITC-ME) to Hsp90 after the exogenous addition of these compounds. Treatment with these small molecules has been reported to inhibit Hsp90 function by modification of the middle domain residues Cys-597 in Hsp90α (Cys-589 in Hsp90β; STCA) (138) as well as Cys-521 in Hsp90β (6-HITC-ME) (137). Both small molecules were found to inhibit Hsp90 activity and induce degradation of client proteins. This was accompanied by the induction and nuclear translocation of the prosurvival transcription factor HSF-1, potentially blunting the cytotoxic effects of these compounds (137, 138).
Similarly to thiocarbamylation, S-nitrosylation results from chemical modification of cysteine residues. NO induces reversible S-nitrosylation by forming stable thionitrite groups on cysteine residues (139). NO is produced by the Hsp90 client eNOS in cells and is an important signal transduction molecule (182). Hsp90α-C597 was subject to nitrosylation as a result of eNOS activity, which decreased Hsp90 chaperone activity, providing a reciprocal mechanism for the regulation of eNOS in cells. It has been posited that this contributes to the anti-cancer effects of nitric oxide (139, 140).
SUMOylation and citrullination
SUMOylation is the addition of a small ubiquitin-like modifier (SUMO) protein to lysine residues. Asymmetric SUMOylation of Hsp90α-K191 by SUMO-1 recruited Aha1 to the chaperone complex and sensitized Hsp90 to its inhibitors (141). SUMOylated Hsp90 was enriched in cells that had undergone malignant transformation, providing one potential explanation for the preferential accumulation of Hsp90 inhibitors in cancer cells (141). In vitro cross-linking of SUMO to an introduced cysteine at the target site in yeast Hsp90 (yHsp90-K178) demonstrated an ATPase activity similar to that of the unmodified Hsp90 (183). Chemically SUMOylated Hsp90 displayed enhanced interaction with Aha1. Additionally, Sba1 (yeast p23 ortholog) was less able to compete Aha1 off and slow down Hsp90 ATPase activity (183). Interestingly, SUMOylated Hsp90α-K559 was shown to serve as an immunogen and biomarker in monoclonal gammopathies of undetermined significance, multiple myelomas, and Waldenstrom's macroglobulinemias (142). This SUMOylation state is the result of a heritable defect in sentrin/SUMO-specific protease 2 (SENP2) that renders it unable to deSUMOylate Hsp90 (142). This specific modification event may contribute to the relative success of Hsp90 inhibition in multiple myelomas (184–187).
Citrullination of Hsp90 has also been implicated in immunogenicity. Examination of sera from rheumatoid arthritis–associated interstitial lung disease patients identified citrullinated Hsp90α/β as autoantigens (188). Citrullination results from the deamination of arginine into citrulline. Numerous residues in Hsp90β have been reported as citrullinated, including the “minimum citrullination set” of Arg-392, -604, -612, and -679 (189). In the context of Hsp90, citrullination induced local unfolding, exposing cryptic epitopes that contributed to its immunogenicity (189). This almost certainly results in decreased chaperone function, although no specific impact on Hsp90 has been reported.
Ubiquitination
One of the many quality control mechanisms in cells involves the ubiquitination and proteasomal degradation of proteins. Indeed, post-translational cross-talk between phosphorylation and ubiquitination has been previously shown to induce Hsp90 turnover (66, 106).
HECT domain E3 ubiquitin protein ligase (Hectd1) participates in the ubiquitination and degradation of Hsp90 (190). Functionally, Hectd1 mutation increased Hsp90α secretion and Hsp90-dependent migration of cranial mesenchyme cells (190). Indeed, ubiquitination of Hsp90 was recently identified at numerous lysine residues using a novel detection technique (143). Interestingly, the Hsp90 co-chaperone CHIP is also an E3 ubiquitin ligase. CHIP was shown to ubiquitinate Hsp90β on 13 lysine residues, leading to Hsp90β degradation (Table 1) (144). It is noteworthy that phosphorylation of Hsp90α-T725 and -S726 and Hsp90β-S718 diminished interaction with CHIP, further demonstrating the cross-talk between Hsp90 phosphorylation and ubiquitination (104). These data point to Hsp90 ubiquitination as a critical cellular regulatory control module.
Oxidation
Tubocapsenolide A (TA) is a bioactive compound isolated from Tubocapsicum anomalum that demonstrates toxicity against a variety of cancer cell lines. TA induces degradation of Hsp90 client proteins, suggesting an Hsp90-inhibitory function. Mechanistically, TA treatment perturbed redox homeostasis, promoting reactive oxygen species generation and oxidation-induced aggregation of the chaperone proteins Hsp90 and Hsp70 (191). Similarly, 4-hydroxy-2-nonenal was shown to inhibit Hsp90 activity by forming thiol-adducts with Hsp90-C572 (192), further confirming redox homeostasis as a crucial factor of proteome maintenance.
Nitration
Hsp90 activity can also be inhibited by peroxynitrite, through the process of nitration (149). Nitration of two conserved N-domain tyrosine residues (Hsp90α-Y38/Y61; Hsp90β-Y33/Y56) induced apoptosis in neuronal models and was also found in spinal cord sections of ALS patients (149). These residues are in close proximity to the N-domain ATP-binding pocket of Hsp90, providing a potential mechanism for the inhibition of Hsp90 activity. Interestingly, Tyr-33–nitrated Hsp90β has also been shown to preferentially associate with mitochondria, exerting a detrimental effect on mitochondrial metabolism (150). Taken together, the down-regulation of mitochondrial function provides an explanation for the association between nitrated Hsp90 and neurodegenerative disease.
Glycosylation
O-GlcNAcylation of Hsp90 has been reported at several sites in both Hsp90α and β using a novel biotin-cystamine approach (151). Although no functional assays were performed, O-GlcNAc antagonizes phosphorylation of serine residues (88), suggesting specific redundant regulatory modules for the identified modification sites. O-GlcNAcylation of Hsp90 was later found to preferentially occur under hyperglycemic conditions. This modification decreased the ability of Hsp90 to support developmental competence in mouse oocytes in an in vitro maturation assay. Interestingly, this phenotype was reversed by 17-AAG treatment, suggesting a detrimental function for O-GlcNAcylated Hsp90, although the precise mechanism remains unknown (193). Notably, O-GlcNAc of the Hsp90 co-chaperone FNIP1 was recently reported, demonstrating multiple layers of Hsp90 regulation by one type of modification (153).
Other forms of glycosylation are distinct from O-GlcNAcylation in that they are typically irreversible. Nonenzymatic glycosylation, or glycation, involves the modification of lysine and arginine side chains with reactive sugar moieties such as methylglyoxal (Fig. 3). Several Hsp90 residues spanning all domains are reported to be glycated, inducing degradation of the Hsp90 client LATS1 and functionally inhibiting Hsp90 activity, while supporting the tumorigenesis of breast cancer cell lines (194).
Hsp90 post-translational modification and drug sensitivity
Sensitivity to Hsp90 inhibitors
As mentioned above, Hsp90 is an attractive therapeutic target, particularly in cancer, due to the reliance of key processes such as cell proliferation and survival as well as cell migration on Hsp90 chaperone function. Inhibiting Hsp90 therefore provides a unique way to simultaneously target divergent signaling pathways, including those mutated or up-regulated in cancers, such as oncogenic kinase pathways (195). Multiple studies have demonstrated increased sensitivity of cancer cells to Hsp90 inhibition relative to normal cells (196). It has also been shown that Hsp90 inhibitors preferentially accumulate in tumor tissue compared with normal tissue; however, the mechanism for this selectivity is not yet fully understood (197–199). Post-translational modification of Hsp90 is one modulatory mechanism that has been explored for Hsp90 inhibitor sensitivity and selectivity (Fig. 4, A and B).
Figure 4.
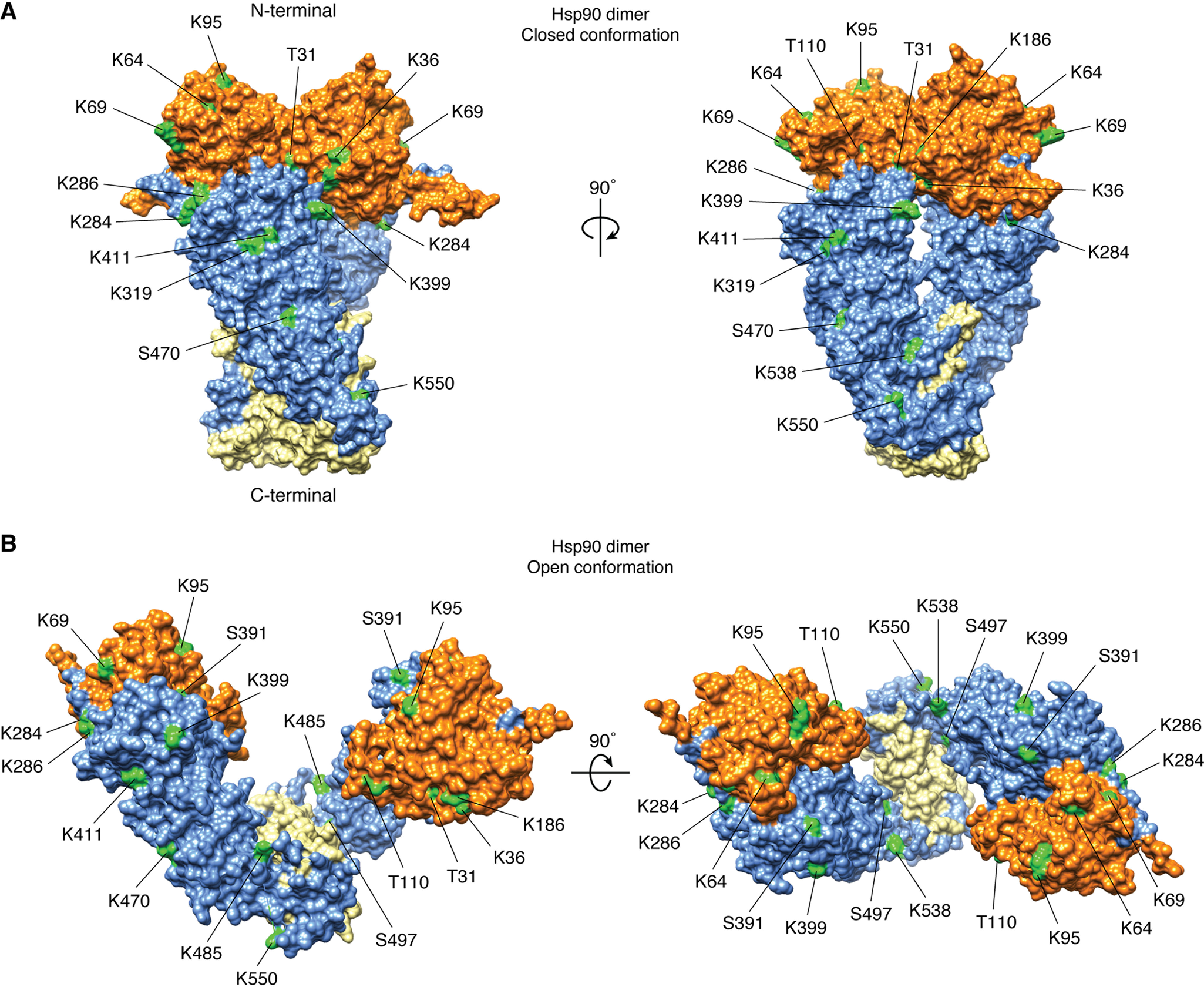
PTM sites known to alter Hsp90 inhibitor sensitivity. Shown is the Hsp90 dimer structure in the closed (A; Protein Data Bank entry 5FWP) and open (B; Protein Data Bank entry 2IOQ) conformation with PTM sites known to alter sensitivity to Hsp90 inhibitors highlighted (green), demonstrating that modifications throughout Hsp90 can profoundly influence the efficacy of N-domain inhibition. Of note, some of these residues are subject to various PTMs, only one of which has been shown to impact drug binding as detailed in Table 1.
As mentioned earlier, SUMOylation of Hsp90-K191 increased binding to and enhanced cell sensitivity to Hsp90 inhibitors (141). Additionally, cells expressing transforming oncogenic kinases exhibited an increase in Hsp90 SUMOylation and concomitant increase in Hsp90 inhibitor sensitivity.
Hsp90 phosphorylation status is often altered in cancer and has also been shown to affect sensitivity to Hsp90 inhibitors, as seen by growth inhibition or arrest or induction of apoptosis (66, 78, 80, 86, 104, 106). Phosphorylation of yHsp90-T22 increased sensitivity to various Hsp90 inhibitors, particularly at higher concentrations (66). Effects of phosphorylation on Hsp90 inhibitor binding can also vary depending on the inhibitor in question and the Hsp90 isoform. Beebe et al. (110) showed that PU-H71 bound Hsp90 phosphorylated on various tyrosine residues better than geldanamycin (GA). Furthermore, whereas phosphomimetic mutations on conserved Hsp90α-Y197E and Hsp90β-Y192E both disrupted binding to GA, only Hsp90β-Y301E and not Hsp90α-Y309E decreased binding to GA (110). Our group also demonstrated that Mps1-mediated phosphorylation of Hsp90α-T115 conferred sensitivity to Hsp90 inhibitors SNX-2112 and ganetespib. Additionally, Mps1 levels were elevated in renal cell carcinoma tissue, and ganetespib selectively accumulated in renal cell carcinoma tumor tissue with elevated levels of Mps1 as compared with adjacent normal renal tissue (78). Phosphorylation of Hsp90α-Y38 (yHsp90-Y24), on the other hand, decreased sensitivity to Hsp90 inhibitors, and nonphosphorylatable yHsp90-Y24F bound more inhibitor (106). Knockdown or inhibition of Wee1, the kinase responsible for Tyr-38 phosphorylation, sensitized cancer cells to Hsp90 inhibition. Dual pharmacologic treatment caused induction of the intrinsic apoptotic pathway and led to down-regulation of an inhibitor of apoptosis, Survivin, as well as Wee1 (200). Taken together, residue-specific Hsp90 phosphorylation may serve as a biomarker for the efficacy of Hsp90 inhibitors, and combination therapies may be of benefit in some cases. It is noteworthy that Tyr-38 phosphorylation is the only PTM reported to decrease Hsp90 inhibitor sensitivity (Table 1).
The potential role for combination treatments is further highlighted when examining how Hsp90 acetylation impacts Hsp90 binding to its inhibitors and inhibitor sensitivity. Overall, acetylation of Hsp90 appears to largely decrease the ATP-binding and chaperone function of Hsp90 but increases Hsp90 binding to its inhibitors (121, 201). Acetylation mimic mutations at several specific residues (Lys-69, -100, -292, -327, -478, -546, and -558) have also been shown to enhance binding of Hsp90 to biotinylated geldanamycin (121). Furthermore, co-treatment of leukemic cells with Hsp90 inhibitor 17-AAG and an HDAC6i led to greater cell death than treatment with either agent alone (201). Additionally, leukemic cells resistant to pan-HDAC inhibitors have been shown to have increased expression of HDAC1, -2, and -4, with loss of HDAC6 expression but hyperacetylation of Hsp90 (122). These cells retain sensitivity to Hsp90 inhibition (122). Recent work by our group has also demonstrated that Hsp90 acetylation affects bladder cancer cell sensitivity to Hsp90 inhibitors (130). Loss of the tumor suppressor and co-chaperone Tsc1 was associated with decreased binding to and uptake of Hsp90 inhibitors and with a decrease of Hsp90 acetylation but not phosphorylation (130, 161). Hsp90 was found to be acetylated on Lys-407 and -419, and HDACi treatment rescued Hsp90 acetylation in Tsc1 knockout cells. Ultimately, Tsc1-null bladder cancer cells were more sensitive to HDACi, and treatment with HDACi resensitized the cells and synergized with Hsp90 inhibitor treatment (130). Overall, there is a growing body of evidence that acetylation of Hsp90 diminishes its chaperone function and increases sensitivity to Hsp90 inhibitors. As a result, there may be a therapeutic advantage to combination treatment with HDAC and Hsp90 inhibitors.
As can be seen in the above examples, reported PTMs in general tend to increase Hsp90 inhibitor sensitivity (Table 1). This must, however, be taken into context. Whereas some modifications, such as Hsp90-T115 phosphorylation, are elevated in cancer and confer sensitivity, elevation of others like Hsp90-Y38 confers resistance. Similarly, whereas Hsp90 acetylation may serve as a marker for sensitivity, its absence provides the opportunity for co-treatment with an HDACi to restore sensitivity in an otherwise resistant cell. Collectively, these results paint a complex landscape that requires further study for mechanistic insight.
Resistance to other therapeutics
In addition to affecting sensitivity to Hsp90 inhibitors, Hsp90 modifications have also been demonstrated to affect sensitivity to other drugs and therapies. In one example, rifampin-induced CK2-mediated phosphorylation of Hsp90β-S226/255 in the charged linker led to the induction of P-glycoprotein, a drug-exporting transporter involved in multidrug resistance (89). In contrast, hypophosphorylation of the same residues conferred resistance to imatinib (Gleevec) in leukemic cells (86). This may be due to inhibition of cytochrome c–induced apoptosome formation through Hsp90-mediated Apaf-1 sequestration, but the exact mechanism of imatinib resistance is unknown (86). Reduced phosphorylation at the conserved Hsp90α-S231 and -S263 also correlated with a down-regulation of telomerase activity and subsequent sensitization to cisplatin (85). These examples highlight the complexity of identifying the impact of Hsp90 PTMs, as the same sites can lead to very different consequences, depending on the context. There are also instances where Hsp90 phosphorylation is exploitable. ATM-mediated phosphorylation of Hsp90α-T5 and -T7 sensitized cells to ionizing radiation (59). Furthermore, point mutation to phosphomimetic Hsp90-S370E and -S640E increased susceptibility of cells to UV irradiation (94).
Finally, Hsp90 acetylation has also been identified in fungal species and was found to play a role in fungal virulence and drug resistance (202). In fungi, particularly in pathogenic species, fungal Hsp90 mediates resistance to anti-fungals, such as azoles, by enabling resistance mutations and stabilizing resistance-mediating protein products (203, 204). Lysine deacetylase (KDAC) inhibition blocked the emergence of Hsp90-dependent resistance to azoles in Candida albicans and Saccharomyces cerevisiae (118, 205). KDAC inhibition compromised the stability and interaction with clients, including calcineurin, which has been shown to be important for drug resistance (205). Knockdown and inhibition of KDACs also increased sensitivity to Hsp90 inhibitors or phenocopied Hsp90 inhibition in modulating resistance to azoles (118, 205). Hsp90 was found to be acetylated on Lys-27 and -270 in baker's yeast (Lys-30 and -271 in C. albicans), and there was functional redundancy between the KDACs Hda1, Rpd3, Hos2, and Rpd31 (118, 205). Acetylation status at Lys-27 has also been shown to be important for azole and echinocandin resistance in Aspergillus fumigatus (119). Acetyl mimic mutations at Lys-27 increased susceptibility to antifungals, and nonacetylatable K27R attenuated these effects (119). Ultimately, targeting Hsp90 acetylation may represent a target for antifungal resistance and understanding the role of Hsp90 acetylation may assist in the development of new antifungal therapeutics. In summary, there is strong evidence that a complete understanding of the effects of Hsp90 PTMs may allow us to better utilize a variety of therapeutics in addition to Hsp90 inhibitors.
Concluding remarks and future perspectives
Post-translational modification often impacts intracellular localization of Hsp90 and its ability to bind clients, nucleotide, co-chaperones, and also inhibitors. Collectively, these PTMs are critical for regulating Hsp90 chaperone activity and, ultimately, cellular functions mediated by affected client proteins. It has been demonstrated that these cellular effects are far-reaching and impact processes including but not limited to cell cycle control, DNA repair, cell signaling, and cell migration. Despite the vast number of reports available with regard to Hsp90 PTMs, there are still several gaps in our knowledge.
First, the vast majority of PTMs have only been explored in the context of cytosolic Hsp90. Translating these findings to organelle- and compartment-specific Hsp90 regulation will promote a more complete understanding of chaperone-mediated signaling pathways. Further, the growing appreciation for extracellular Hsp90 necessitates further research into the mechanisms regulating its secretion activity and how PTMs affect Hsp90 function extracellularly. Additionally, it is well-established that whereas Hsp90 functions as a dimer, there is a great deal of asymmetry between the protomers, and this is essential for its function. This is highlighted by one Aha1 molecule interacting asymmetrically with a dimer and being influenced by asymmetric modifications, such as Hsp90-K191 SUMOylation and Hsp90-Y313 phosphorylation (42, 141, 161). We therefore need to consider whether both protomers are modified concurrently and how modification conservation between the α and β isoforms affects chaperone function. Furthermore, outside of some reports of cross-talk between Hsp90 phosphorylation and ubiquitination, there is a void in understanding the relationships between different modifications and how they influence one another and work in concert or opposition. Last, methods for detecting Hsp90 PTMs have recently been reviewed by our group (206), but more specific methods, such as the development of phosphospecific antibodies, are warranted and likely necessary to fully unravel the chaperone code.
Ultimately, however, we need to use several orthogonal approaches to fully decipher the chaperone code (Fig. 5). Knowledge obtained from the tools used in this field to date leaves us with information on how a limited number of modifications affect consequences in specific settings. We are also lacking robust mechanistic detail of how PTMs lead to the observed changes in client chaperoning. Looking forward, we must gain an appreciation for the many different populations of Hsp90 with respect to their modification states and how they function together or independently to ultimately influence the protein landscape within cells. Understanding the cross-talk between different modifications is paramount to unraveling the regulation of chaperone function. Implementation of methods such as artificial intelligence, bioinformatics, and molecular modeling allows data processing on a much larger scale. Synthesis of a detailed, mechanistic understanding of Hsp90 modifications with population-wide analysis will allow us to fully decipher the chaperone code and translate it to therapeutic benefit.
Figure 5.

The Hsp90 chaperone code. Collectively, the landscape of Hsp90 PTMs, or the “chaperone code,” affects Hsp90 chaperone function as defined by Hsp90 ATPase activity, co-chaperone binding, drug sensitivity, and client function.
Acknowledgments
We are grateful to our colleagues and collaborators Drs. Johannes Buchner, Len Neckers, Timothy A. Haystead, Dimitra Bourboulia, and Gennady Bratslavsky for scientific contributions and discussions.
Funding and additional information—This work was supported in part by NIGMS, National Institutes of Health, Grant R01GM124256 (to M. M.). This work was also supported by funds from SUNY Upstate Medical University, the Upstate Foundation, Upstate Cancer Center, and the Carol M. Baldwin Breast Cancer Fund (to M. M.) The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- PTM
- post-translational modification
- TPR
- tetratricopeptide repeat
- HOP
- Hsp70-Hsp90–organizing protein
- PKA
- protein kinase A
- CHIP
- C terminus of Hsp70-interacting protein
- DNA-PK
- DNA-dependent protein kinase
- GR
- glucocorticoid receptor
- AR
- androgen receptor
- BCR
- B-cell antigen receptor
- eNOS
- endothelial nitric oxide synthase
- HAT
- histone acetyltransferase
- HDAC
- histone deacetylase
- HDACi
- HDAC inhibitor
- MR
- mineralocorticoid receptor
- ER
- estrogen receptor
- STCA
- sulfoxylthiocarbamate alkyne
- 6-HITC-ME
- 6-methylsulfinylhexyl isothiocyanate-β-mercaptoethanol
- SUMO
- small ubiquitin-like modifier
- TA
- tubocapsenolide A
- 17-AAG
- 17-allylamino-17-demethoxygeldanamycin
- GA
- geldanamycin
- KDAC
- lysine deacetylase
- KD
- knockdown.
References
- 1. Schopf F. H., Biebl M. M., and Buchner J. (2017) The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 18, 345–360 10.1038/nrm.2017.20 [DOI] [PubMed] [Google Scholar]
- 2. O'Brien D., and van Oosten-Hawle P. (2016) Regulation of cell-non-autonomous proteostasis in metazoans. Essays Biochem. 60, 133–142 10.1042/EBC20160006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. O'Brien D., Jones L. M., Good S., Miles J., Vijayabaskar M. S., Aston R., Smith C. E., Westhead D. R., and van Oosten-Hawle P. (2018) A PQM-1-mediated response triggers transcellular chaperone signaling and regulates organismal proteostasis. Cell Rep. 23, 3905–3919 10.1016/j.celrep.2018.05.093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cox M. B., and Johnson J. L. (2018) Evidence for Hsp90 co-chaperones in regulating Hsp90 function and promoting client protein folding. Methods Mol. Biol. 1709, 397–422 10.1007/978-1-4939-7477-1_28 [DOI] [PubMed] [Google Scholar]
- 5. Sahasrabudhe P., Rohrberg J., Biebl M. M., Rutz D. A., and Buchner J. (2017) The plasticity of the Hsp90 co-chaperone system. Mol. Cell 67, 947–961.e5 10.1016/j.molcel.2017.08.004 [DOI] [PubMed] [Google Scholar]
- 6. Zierer B. K., Rübbelke M., Tippel F., Madl T., Schopf F. H., Rutz D. A., Richter K., Sattler M., and Buchner J. (2016) Importance of cycle timing for the function of the molecular chaperone Hsp90. Nat. Struct. Mol. Biol. 23, 1020–1028 10.1038/nsmb.3305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Röhl A., Rohrberg J., and Buchner J. (2013) The chaperone Hsp90: changing partners for demanding clients. Trends Biochem. Sci. 38, 253–262 10.1016/j.tibs.2013.02.003 [DOI] [PubMed] [Google Scholar]
- 8. Neckers L., and Workman P. (2012) Hsp90 molecular chaperone inhibitors: are we there yet? Clin. Cancer Res. 18, 64–76 10.1158/1078-0432.CCR-11-1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Barrott J. J., and Haystead T. A. (2013) Hsp90, an unlikely ally in the war on cancer. FEBS J. 280, 1381–1396 10.1111/febs.12147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rodina A., Wang T., Yan P., Gomes E. D., Dunphy M. P., Pillarsetty N., Koren J., Gerecitano J. F., Taldone T., Zong H., Caldas-Lopes E., Alpaugh M., Corben A., Riolo M., Beattie B., et al. (2016) The epichaperome is an integrated chaperome network that facilitates tumour survival. Nature 538, 397–401 10.1038/nature19807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang H., Lu M., Yao M., and Zhu W. (2016) Effects of treatment with an Hsp90 inhibitor in tumors based on 15 phase II clinical trials. Mol. Clin. Oncol. 5, 326–334 10.3892/mco.2016.963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Walton-Diaz A., Khan S., Bourboulia D., Trepel J. B., Neckers L., and Mollapour M. (2013) Contributions of co-chaperones and post-translational modifications towards Hsp90 drug sensitivity. Future Med. Chem. 5, 1059–1071 10.4155/fmc.13.88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Woodford M. R., Dunn D., Miller J. B., Jamal S., Neckers L., and Mollapour M. (2016) Impact of posttranslational modifications on the anticancer activity of Hsp90 inhibitors. Adv. Cancer Res. 129, 31–50 10.1016/bs.acr.2015.09.002 [DOI] [PubMed] [Google Scholar]
- 14. Soroka J., and Buchner J. (2012) Mechanistic aspects of the Hsp90 phosphoregulation. Cell Cycle 11, 1870–1871 10.4161/cc.20418 [DOI] [PubMed] [Google Scholar]
- 15. Cloutier P., and Coulombe B. (2013) Regulation of molecular chaperones through post-translational modifications: decrypting the chaperone code. Biochim. Biophys. Acta 1829, 443–454 10.1016/j.bbagrm.2013.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cloutier P., Lavallée-Adam M., Faubert D., Blanchette M., and Coulombe B. (2013) A newly uncovered group of distantly related lysine methyltransferases preferentially interact with molecular chaperones to regulate their activity. PLoS Genet. 9, e1003210 10.1371/journal.pgen.1003210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nitika and Truman A. W. (2017) Cracking the chaperone code: cellular roles for Hsp70 phosphorylation. Trends Biochem. Sci. 42, 932–935 10.1016/j.tibs.2017.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Csermely P., Schnaider T., Sőti C., Prohaszka Z., and Nardai G. (1998) The 90-kDa molecular chaperone family: structure, function, and clinical applications: a comprehensive review. Pharmacol. Ther. 79, 129–168 10.1016/S0163-7258(98)00013-8 [DOI] [PubMed] [Google Scholar]
- 19. Johnson J. L. (2012) Evolution and function of diverse Hsp90 homologs and cochaperone proteins. Biochim. Biophys. Acta 1823, 607–613 10.1016/j.bbamcr.2011.09.020 [DOI] [PubMed] [Google Scholar]
- 20. Zuehlke A. D., Beebe K., Neckers L., and Prince T. (2015) Regulation and function of the human HSP90AA1 gene. Gene 570, 8–16 10.1016/j.gene.2015.06.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Panaretou B., Prodromou C., Roe S. M., O'Brien R., Ladbury J. E., Piper P. W., and Pearl L. H. (1998) ATP binding and hydrolysis are essential to the function of the Hsp90 molecular chaperone in vivo. EMBO J. 17, 4829–4836 10.1093/emboj/17.16.4829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Obermann W. M., Sondermann H., Russo A. A., Pavletich N. P., and Hartl F. U. (1998) In vivo function of Hsp90 is dependent on ATP binding and ATP hydrolysis. J. Cell Biol. 143, 901–910 10.1083/jcb.143.4.901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Graf C., Stankiewicz M., Kramer G., and Mayer M. P. (2009) Spatially and kinetically resolved changes in the conformational dynamics of the Hsp90 chaperone machine. EMBO J. 28, 602–613 10.1038/emboj.2008.306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mickler M., Hessling M., Ratzke C., Buchner J., and Hugel T. (2009) The large conformational changes of Hsp90 are only weakly coupled to ATP hydrolysis. Nat. Struct. Mol. Biol. 16, 281–286 10.1038/nsmb.1557 [DOI] [PubMed] [Google Scholar]
- 25. Wayne N., and Bolon D. N. (2007) Dimerization of Hsp90 is required for in vivo function: design and analysis of monomers and dimers. J. Biol. Chem. 282, 35386–35395 10.1074/jbc.M703844200 [DOI] [PubMed] [Google Scholar]
- 26. Ali M. M., Roe S. M., Vaughan C. K., Meyer P., Panaretou B., Piper P. W., Prodromou C., and Pearl L. H. (2006) Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature 440, 1013–1017 10.1038/nature04716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Verba K. A., Wang R. Y., Arakawa A., Liu Y., Shirouzu M., Yokoyama S., and Agard D. A. (2016) Atomic structure of Hsp90-Cdc37-Cdk4 reveals that Hsp90 traps and stabilizes an unfolded kinase. Science 352, 1542–1547 10.1126/science.aaf5023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Prodromou C., Roe S. M., O'Brien R., Ladbury J. E., Piper P. W., and Pearl L. H. (1997) Identification and structural characterization of the ATP/ADP-binding site in the Hsp90 molecular chaperone. Cell 90, 65–75 10.1016/S0092-8674(00)80314-1 [DOI] [PubMed] [Google Scholar]
- 29. Prodromou C., Roe S. M., Piper P. W., and Pearl L. H. (1997) A molecular clamp in the crystal structure of the N-terminal domain of the yeast Hsp90 chaperone. Nat. Struct. Biol. 4, 477–482 10.1038/nsb0697-477 [DOI] [PubMed] [Google Scholar]
- 30. Tsutsumi S., Mollapour M., Prodromou C., Lee C. T., Panaretou B., Yoshida S., Mayer M. P., and Neckers L. M. (2012) Charged linker sequence modulates eukaryotic heat shock protein 90 (Hsp90) chaperone activity. Proc. Natl. Acad. Sci. U. S. A. 109, 2937–2942 10.1073/pnas.1114414109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hainzl O., Lapina M. C., Buchner J., and Richter K. (2009) The charged linker region is an important regulator of Hsp90 function. J. Biol. Chem. 284, 22559–22567 10.1074/jbc.M109.031658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jahn M., Rehn A., Pelz B., Hellenkamp B., Richter K., Rief M., Buchner J., and Hugel T. (2014) The charged linker of the molecular chaperone Hsp90 modulates domain contacts and biological function. Proc. Natl. Acad. Sci. U. S. A. 111, 17881–17886 10.1073/pnas.1414073111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Meyer P., Prodromou C., Hu B., Vaughan C., Roe S. M., Panaretou B., Piper P. W., and Pearl L. H. (2003) Structural and functional analysis of the middle segment of hsp90: implications for ATP hydrolysis and client protein and cochaperone interactions. Mol. Cell 11, 647–658 10.1016/S1097-2765(03)00065-0 [DOI] [PubMed] [Google Scholar]
- 34. Biebl M. M., and Buchner J. (2019) Structure, Function, and Regulation of the Hsp90 Machinery. Cold Spring Harb. Perspect. Biol. 11, a034017 10.1101/cshperspect.a034017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Harris S. F., Shiau A. K., and Agard D. A. (2004) The crystal structure of the carboxy-terminal dimerization domain of htpG, the Escherichia coli Hsp90, reveals a potential substrate binding site. Structure 12, 1087–1097 10.1016/j.str.2004.03.020 [DOI] [PubMed] [Google Scholar]
- 36. Prodromou C., and Pearl L. H. (2003) Structure and functional relationships of Hsp90. Curr. Cancer Drug Targets 3, 301–323 10.2174/1568009033481877 [DOI] [PubMed] [Google Scholar]
- 37. Hessling M., Richter K., and Buchner J. (2009) Dissection of the ATP-induced conformational cycle of the molecular chaperone Hsp90. Nat. Struct. Mol. Biol. 16, 287–293 10.1038/nsmb.1565 [DOI] [PubMed] [Google Scholar]
- 38. Neckers L., Mollapour M., and Tsutsumi S. (2009) The complex dance of the molecular chaperone Hsp90. Trends Biochem. Sci. 34, 223–226 10.1016/j.tibs.2009.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shiau A. K., Harris S. F., Southworth D. R., and Agard D. A. (2006) Structural analysis of E. coli hsp90 reveals dramatic nucleotide-dependent conformational rearrangements. Cell 127, 329–340 10.1016/j.cell.2006.09.027 [DOI] [PubMed] [Google Scholar]
- 40. Stetz G., Tse A., and Verkhivker G. M. (2018) Dissecting structure-encoded determinants of allosteric cross-talk between post-translational modification sites in the Hsp90 chaperones. Sci. Rep. 8, 6899 10.1038/s41598-018-25329-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cunningham C. N., Krukenberg K. A., and Agard D. A. (2008) Intra- and intermonomer interactions are required to synergistically facilitate ATP hydrolysis in Hsp90. J. Biol. Chem. 283, 21170–21178 10.1074/jbc.M800046200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Xu W., Beebe K., Chavez J. D., Boysen M., Lu Y., Zuehlke A. D., Keramisanou D., Trepel J. B., Prodromou C., Mayer M. P., Bruce J. E., Gelis I., and Neckers L. (2019) Hsp90 middle domain phosphorylation initiates a complex conformational program to recruit the ATPase-stimulating cochaperone Aha1. Nat. Commun. 10, 2574 10.1038/s41467-019-10463-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wegele H., Wandinger S. K., Schmid A. B., Reinstein J., and Buchner J. (2006) Substrate transfer from the chaperone Hsp70 to Hsp90. J. Mol. Biol. 356, 802–811 10.1016/j.jmb.2005.12.008 [DOI] [PubMed] [Google Scholar]
- 44. Li J., Richter K., and Buchner J. (2011) Mixed Hsp90-cochaperone complexes are important for the progression of the reaction cycle. Nat. Struct. Mol. Biol. 18, 61–66 10.1038/nsmb.1965 [DOI] [PubMed] [Google Scholar]
- 45. Prodromou C., Siligardi G., O'Brien R., Woolfson D. N., Regan L., Panaretou B., Ladbury J. E., Piper P. W., and Pearl L. H. (1999) Regulation of Hsp90 ATPase activity by tetratricopeptide repeat (TPR)-domain co-chaperones. EMBO J. 18, 754–762 10.1093/emboj/18.3.754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Miyata Y., and Nishida E. (2005) CK2 binds, phosphorylates, and regulates its pivotal substrate Cdc37, an Hsp90-cochaperone. Mol. Cell. Biochem. 274, 171–179 10.1007/s11010-005-2949-8 [DOI] [PubMed] [Google Scholar]
- 47. Vaughan C. K., Mollapour M., Smith J. R., Truman A., Hu B., Good V. M., Panaretou B., Neckers L., Clarke P. A., Workman P., Piper P. W., Prodromou C., and Pearl L. H. (2008) Hsp90-dependent activation of protein kinases is regulated by chaperone-targeted dephosphorylation of Cdc37. Mol. Cell 31, 886–895 10.1016/j.molcel.2008.07.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Keramisanou D., Aboalroub A., Zhang Z., Liu W., Marshall D., Diviney A., Larsen R. W., Landgraf R., and Gelis I. (2016) Molecular mechanism of protein kinase recognition and sorting by the Hsp90 kinome-specific cochaperone Cdc37. Mol. Cell 62, 260–271 10.1016/j.molcel.2016.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Panaretou B., Siligardi G., Meyer P., Maloney A., Sullivan J. K., Singh S., Millson S. H., Clarke P. A., Naaby-Hansen S., Stein R., Cramer R., Mollapour M., Workman P., Piper P. W., Pearl L. H., et al. (2002) Activation of the ATPase activity of hsp90 by the stress-regulated cochaperone aha1. Mol. Cell 10, 1307–1318 10.1016/S1097-2765(02)00785-2 [DOI] [PubMed] [Google Scholar]
- 50. Meyer P., Prodromou C., Liao C., Hu B., Mark Roe S., Vaughan C. K., Vlasic I., Panaretou B., Piper P. W., and Pearl L. H. (2004) Structural basis for recruitment of the ATPase activator Aha1 to the Hsp90 chaperone machinery. EMBO J. 23, 511–519 10.1038/sj.emboj.7600060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Retzlaff M., Hagn F., Mitschke L., Hessling M., Gugel F., Kessler H., Richter K., and Buchner J. (2010) Asymmetric activation of the hsp90 dimer by its cochaperone aha1. Mol. Cell 37, 344–354 10.1016/j.molcel.2010.01.006 [DOI] [PubMed] [Google Scholar]
- 52. Mercier R., Wolmarans A., Schubert J., Neuweiler H., Johnson J. L., and LaPointe P. (2019) The conserved NxNNWHW motif in Aha-type co-chaperones modulates the kinetics of Hsp90 ATPase stimulation. Nat. Commun. 10, 1273 10.1038/s41467-019-09299-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Richter K., Walter S., and Buchner J. (2004) The co-chaperone Sba1 connects the ATPase reaction of Hsp90 to the progression of the chaperone cycle. J. Mol. Biol. 342, 1403–1413 10.1016/j.jmb.2004.07.064 [DOI] [PubMed] [Google Scholar]
- 54. Mimnaugh E. G., Worland P. J., Whitesell L., and Neckers L. M. (1995) Possible role for serine/threonine phosphorylation in the regulation of the heteroprotein complex between the hsp90 stress protein and the pp60v-src tyrosine kinase. J. Biol. Chem. 270, 28654–28659 10.1074/jbc.270.48.28654 [DOI] [PubMed] [Google Scholar]
- 55. Lees-Miller S. P., and Anderson C. W. (1989) Two human 90-kDa heat shock proteins are phosphorylated in vivo at conserved serines that are phosphorylated in vitro by casein kinase II. J. Biol. Chem. 264, 2431–2437 [PubMed] [Google Scholar]
- 56. Franchin C., Borgo C., Zaramella S., Cesaro L., Arrigoni G., Salvi M., and Pinna L. A. (2017) Exploring the CK2 paradox: restless, dangerous, dispensable. Pharmaceuticals (Basel) 10, 11 10.3390/ph10010011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Solier S., Kohn K. W., Scroggins B., Xu W., Trepel J., Neckers L., and Pommier Y. (2012) Heat shock protein 90α (HSP90α), a substrate and chaperone of DNA-PK necessary for the apoptotic response. Proc. Natl. Acad. Sci. U. S. A. 109, 12866–12872 10.1073/pnas.1203617109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Park S. J., Gavrilova O., Brown A. L., Soto J. E., Bremner S., Kim J., Xu X., Yang S., Um J. H., Koch L. G., Britton S. L., Lieber R. L., Philp A., Baar K., Kohama S. G., et al. (2017) DNA-PK promotes the mitochondrial, metabolic, and physical decline that occurs during aging. Cell Metab. 25, 1135–1146.e7 10.1016/j.cmet.2017.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Elaimy A. L., Ahsan A., Marsh K., Pratt W. B., Ray D., Lawrence T. S., and Nyati M. K. (2016) ATM is the primary kinase responsible for phosphorylation of Hsp90α after ionizing radiation. Oncotarget 7, 82450–82457 10.18632/oncotarget.12557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Pennisi R., Antoccia A., Leone S., Ascenzi P., and di Masi A. (2017) Hsp90α regulates ATM and NBN functions in sensing and repair of DNA double-strand breaks. FEBS J. 284, 2378–2395 10.1111/febs.14145 [DOI] [PubMed] [Google Scholar]
- 61. Hasan M., Hama S., and Kogure K. (2019) Low electric treatment activates Rho GTPase via heat shock protein 90 and protein kinase C for intracellular delivery of siRNA. Sci. Rep. 9, 4114 10.1038/s41598-019-40904-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lees-Miller S. P., and Anderson C. W. (1989) The human double-stranded DNA-activated protein kinase phosphorylates the 90-kDa heat-shock protein, hsp90 α at two NH2-terminal threonine residues. J. Biol. Chem. 264, 17275–17280 [PubMed] [Google Scholar]
- 63. Quanz M., Herbette A., Sayarath M., de Koning L., Dubois T., Sun J. S., and Dutreix M. (2012) Heat shock protein 90α (Hsp90α) is phosphorylated in response to DNA damage and accumulates in repair foci. J. Biol. Chem. 287, 8803–8815 10.1074/jbc.M111.320887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Mollapour M., Tsutsumi S., Truman A. W., Xu W., Vaughan C. K., Beebe K., Konstantinova A., Vourganti S., Panaretou B., Piper P. W., Trepel J. B., Prodromou C., Pearl L. H., and Neckers L. (2011) Threonine 22 phosphorylation attenuates Hsp90 interaction with cochaperones and affects its chaperone activity. Mol. Cell 41, 672–681 10.1016/j.molcel.2011.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. He Q., Liu K., Tian Z., and Du S. J. (2015) The effects of Hsp90α1 mutations on myosin thick filament organization. PLoS ONE 10, e0142573 10.1371/journal.pone.0142573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Mollapour M., Tsutsumi S., Kim Y. S., Trepel J., and Neckers L. (2011) Casein kinase 2 phosphorylation of Hsp90 threonine 22 modulates chaperone function and drug sensitivity. Oncotarget 2, 407–417 10.18632/oncotarget.272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Huttlin E. L., Jedrychowski M. P., Elias J. E., Goswami T., Rad R., Beausoleil S. A., Villén J., Haas W., Sowa M. E., and Gygi S. P. (2010) A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143, 1174–1189 10.1016/j.cell.2010.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Rose D. W., Wettenhall R. E., Kudlicki W., Kramer G., and Hardesty B. (1987) The 90-kilodalton peptide of the heme-regulated eIF-2 α kinase has sequence similarity with the 90-kilodalton heat shock protein. Biochemistry 26, 6583–6587 10.1021/bi00395a003 [DOI] [PubMed] [Google Scholar]
- 69. Kettenbach A. N., Schweppe D. K., Faherty B. K., Pechenick D., Pletnev A. A., and Gerber S. A. (2011) Quantitative phosphoproteomics identifies substrates and functional modules of Aurora and Polo-like kinase activities in mitotic cells. Sci. Signal. 4, rs5 10.1126/scisignal.2001497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Sharma K., D'Souza R. C. J., Tyanova S., Schaab C., Wiśniewski J. R., Cox J., and Mann M. (2014) Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr-based signaling. Cell Rep. 8, 1583–1594 10.1016/j.celrep.2014.07.036 [DOI] [PubMed] [Google Scholar]
- 71. Lei H., Venkatakrishnan A., Yu S., and Kazlauskas A. (2007) Protein kinase A-dependent translocation of Hsp90 α impairs endothelial nitric-oxide synthase activity in high glucose and diabetes. J. Biol. Chem. 282, 9364–9371 10.1074/jbc.M608985200 [DOI] [PubMed] [Google Scholar]
- 72. Molina H., Horn D. M., Tang N., Mathivanan S., and Pandey A. (2007) Global proteomic profiling of phosphopeptides using electron transfer dissociation tandem mass spectrometry. Proc. Natl. Acad. Sci. U. S. A. 104, 2199–2204 10.1073/pnas.0611217104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wang X., Lu X. A., Song X., Zhuo W., Jia L., Jiang Y., and Luo Y. (2012) Thr90 phosphorylation of Hsp90α by protein kinase A regulates its chaperone machinery. Biochem. J. 441, 387–397 10.1042/BJ20110855 [DOI] [PubMed] [Google Scholar]
- 74. Dagar M., Singh J. P., Dagar G., Tyagi R. K., and Bagchi G. (2019) Phosphorylation of HSP90 by protein kinase A is essential for the nuclear translocation of androgen receptor. J. Biol. Chem. 294, 8699–8710 10.1074/jbc.RA119.007420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wang X., Song X., Zhuo W., Fu Y., Shi H., Liang Y., Tong M., Chang G., and Luo Y. (2009) The regulatory mechanism of Hsp90α secretion and its function in tumor malignancy. Proc. Natl. Acad. Sci. U. S. A. 106, 21288–21293 10.1073/pnas.0908151106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Schweppe D. K., Rigas J. R., and Gerber S. A. (2013) Quantitative phosphoproteomic profiling of human non-small cell lung cancer tumors. J. Proteomics 91, 286–296 10.1016/j.jprot.2013.07.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Lopez V. A., Park B. C., Nowak D., Sreelatha A., Zembek P., Fernandez J., Servage K. A., Gradowski M., Hennig J., Tomchick D. R., Pawłowski K., Krzymowska M., and Tagliabracci V. S. (2019) A bacterial effector mimics a host HSP90 client to undermine immunity. Cell 179, 205–218.e21 10.1016/j.cell.2019.08.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Woodford M. R., Truman A. W., Dunn D. M., Jensen S. M., Cotran R., Bullard R., Abouelleil M., Beebe K., Wolfgeher D., Wierzbicki S., Post D. E., Caza T., Tsutsumi S., Panaretou B., Kron S. J., et al. (2016) Mps1 mediated phosphorylation of Hsp90 confers renal cell carcinoma sensitivity and selectivity to Hsp90 inhibitors. Cell Rep. 14, 872–884 10.1016/j.celrep.2015.12.084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Lu X. A., Wang X., Zhuo W., Jia L., Jiang Y., Fu Y., and Luo Y. (2014) The regulatory mechanism of a client kinase controlling its own release from Hsp90 chaperone machinery through phosphorylation. Biochem. J. 457, 171–183 10.1042/BJ20130963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Cheng A. N., Fan C. C., Lo Y. K., Kuo C. L., Wang H. C., Lien I. H., Lin S. Y., Chen C. H., Jiang S. S., Chang I. S., Juan H. F., Lyu P. C., and Lee A. Y. (2017) Cdc7-Dbf4-mediated phosphorylation of HSP90-S164 stabilizes HSP90-HCLK2-MRN complex to enhance ATR/ATM signaling that overcomes replication stress in cancer. Sci. Rep. 7, 17024 10.1038/s41598-017-17126-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Tsai C. F., Wang Y. T., Yen H. Y., Tsou C. C., Ku W. C., Lin P. Y., Chen H. Y., Nesvizhskii A. I., Ishihama Y., and Chen Y. J. (2015) Large-scale determination of absolute phosphorylation stoichiometries in human cells by motif-targeting quantitative proteomics. Nat. Commun. 6, 6622 10.1038/ncomms7622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Huang S. Y., Tsai M. L., Chen G. Y., Wu C. J., and Chen S. H. (2007) A systematic MS-based approach for identifying in vitro substrates of PKA and PKG in rat uteri. J. Proteome Res. 6, 2674–2684 10.1021/pr070134c [DOI] [PubMed] [Google Scholar]
- 83. Klammer M., Kaminski M., Zedler A., Oppermann F., Blencke S., Marx S., Müller S., Tebbe A., Godl K., and Schaab C. (2012) Phosphosignature predicts dasatinib response in non-small cell lung cancer. Mol. Cell. Proteomics 11, 651–668 10.1074/mcp.M111.016410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Olesen S. H., Ingles D. J., Zhu J. Y., Martin M. P., Betzi S., Georg G. I., Tash J. S., and Schönbrunn E. (2015) Stability of the human Hsp90-p50Cdc37 chaperone complex against nucleotides and Hsp90 inhibitors, and the influence of phosphorylation by casein kinase 2. Molecules 20, 1643–1660 10.3390/molecules20011643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Woo S. H., An S., Lee H. C., Jin H. O., Seo S. K., Yoo D. H., Lee K. H., Rhee C. H., Choi E. J., Hong S. I., and Park I. C. (2009) A truncated form of p23 down-regulates telomerase activity via disruption of Hsp90 function. J. Biol. Chem. 284, 30871–30880 10.1074/jbc.M109.052720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Kurokawa M., Zhao C., Reya T., and Kornbluth S. (2008) Inhibition of apoptosome formation by suppression of Hsp90β phosphorylation in tyrosine kinase-induced leukemias. Mol. Cell Biol. 28, 5494–5506 10.1128/MCB.00265-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Ogiso H., Kagi N., Matsumoto E., Nishimoto M., Arai R., Shirouzu M., Mimura J., Fujii-Kuriyama Y., and Yokoyama S. (2004) Phosphorylation analysis of 90 kDa heat shock protein within the cytosolic arylhydrocarbon receptor complex. Biochemistry 43, 15510–15519 10.1021/bi048736m [DOI] [PubMed] [Google Scholar]
- 88. Wang Z., Gucek M., and Hart G. W. (2008) Cross-talk between GlcNAcylation and phosphorylation: site-specific phosphorylation dynamics in response to globally elevated O-GlcNAc. Proc. Natl. Acad. Sci. U. S. A. 105, 13793–13798 10.1073/pnas.0806216105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Kim S. W., Hasanuzzaman M., Cho M., Heo Y. R., Ryu M. J., Ha N. Y., Park H. J., Park H. Y., and Shin J. G. (2015) Casein kinase 2 (CK2)-mediated phosphorylation of Hsp90β as a novel mechanism of rifampin-induced MDR1 expression. J. Biol. Chem. 290, 17029–17040 10.1074/jbc.M114.624106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Negroni L., Samson M., Guigonis J. M., Rossi B., Pierrefite-Carle V., and Baudoin C. (2007) Treatment of colon cancer cells using the cytosine deaminase/5-fluorocytosine suicide system induces apoptosis, modulation of the proteome, and Hsp90β phosphorylation. Mol. Cancer Ther. 6, 2747–2756 10.1158/1535-7163.MCT-07-0040 [DOI] [PubMed] [Google Scholar]
- 91. Beranova-Giorgianni S., Zhao Y., Desiderio D. M., and Giorgianni F. (2006) Phosphoproteomic analysis of the human pituitary. Pituitary 9, 109–120 10.1007/s11102-006-8916-x [DOI] [PubMed] [Google Scholar]
- 92. Old W. M., Shabb J. B., Houel S., Wang H., Couts K. L., Yen C. Y., Litman E. S., Croy C. H., Meyer-Arendt K., Miranda J. G., Brown R. A., Witze E. S., Schweppe R. E., Resing K. A., and Ahn N. G. (2009) Functional proteomics identifies targets of phosphorylation by B-Raf signaling in melanoma. Mol. Cell 34, 115–131 10.1016/j.molcel.2009.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Nguyen M. T. N., Knieß R. A., Daturpalli S., Le Breton L., Ke X., Chen X., and Mayer M. P. (2017) Isoform-specific phosphorylation in human Hsp90β affects interaction with clients and the cochaperone Cdc37. J. Mol. Biol. 429, 732–752 10.1016/j.jmb.2017.01.011 [DOI] [PubMed] [Google Scholar]
- 94. Soroka J., Wandinger S. K., Mäusbacher N., Schreiber T., Richter K., Daub H., and Buchner J. (2012) Conformational switching of the molecular chaperone Hsp90 via regulated phosphorylation. Mol. Cell 45, 517–528 10.1016/j.molcel.2011.12.031 [DOI] [PubMed] [Google Scholar]
- 95. Mayya V., Lundgren D. H., Hwang S. I., Rezaul K., Wu L., Eng J. K., Rodionov V., and Han D. K. (2009) Quantitative phosphoproteomic analysis of T cell receptor signaling reveals system-wide modulation of protein-protein interactions. Sci. Signal. 2, ra46 10.1126/scisignal.2000007 [DOI] [PubMed] [Google Scholar]
- 96. Wiśniewski J. R., Nagaraj N., Zougman A., Gnad F., and Mann M. (2010) Brain phosphoproteome obtained by a FASP-based method reveals plasma membrane protein topology. J. Proteome Res. 9, 3280–3289 10.1021/pr1002214 [DOI] [PubMed] [Google Scholar]
- 97. Mertins P., Qiao J. W., Patel J., Udeshi N. D., Clauser K. R., Mani D. R., Burgess M. W., Gillette M. A., Jaffe J. D., and Carr S. A. (2013) Integrated proteomic analysis of post-translational modifications by serial enrichment. Nat. Methods 10, 634–637 10.1038/nmeth.2518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Qi X., Xie C., Hou S., Li G., Yin N., Dong L., Lepp A., Chesnik M. A., Mirza S. P., Szabo A., Tsai S., Basir Z., Wu S., and Chen G. (2014) Identification of a ternary protein-complex as a therapeutic target for K-Ras-dependent colon cancer. Oncotarget 5, 4269–4282 10.18632/oncotarget.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Hornbeck P. V., Zhang B., Murray B., Kornhauser J. M., Latham V., and Skrzypek E. (2015) PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 43, D512–D520 10.1093/nar/gku1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Zhou H., Di Palma S., Preisinger C., Peng M., Polat A. N., Heck A. J., and Mohammed S. (2013) Toward a comprehensive characterization of a human cancer cell phosphoproteome. J. Proteome Res. 12, 260–271 10.1021/pr300630k [DOI] [PubMed] [Google Scholar]
- 101. Beli P., Lukashchuk N., Wagner S. A., Weinert B. T., Olsen J. V., Baskcomb L., Mann M., Jackson S. P., and Choudhary C. (2012) Proteomic investigations reveal a role for RNA processing factor THRAP3 in the DNA damage response. Mol. Cell 46, 212–225 10.1016/j.molcel.2012.01.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Deb T. B., Zuo A. H., Wang Y., Barndt R. J., Cheema A. K., Sengupta S., Coticchia C. M., and Johnson M. D. (2011) Pnck induces ligand-independent EGFR degradation by probable perturbation of the Hsp90 chaperone complex. Am. J. Physiol. Cell Physiol. 300, C1139–1154 10.1152/ajpcell.00167.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Shiromizu T., Adachi J., Watanabe S., Murakami T., Kuga T., Muraoka S., and Tomonaga T. (2013) Identification of missing proteins in the neXtProt database and unregistered phosphopeptides in the PhosphoSitePlus database as part of the Chromosome-centric Human Proteome Project. J. Proteome Res. 12, 2414–2421 10.1021/pr300825v [DOI] [PubMed] [Google Scholar]
- 104. Muller P., Ruckova E., Halada P., Coates P. J., Hrstka R., Lane D. P., and Vojtesek B. (2013) C-terminal phosphorylation of Hsp70 and Hsp90 regulates alternate binding to co-chaperones CHIP and HOP to determine cellular protein folding/degradation balances. Oncogene 32, 3101–3110 10.1038/onc.2012.314 [DOI] [PubMed] [Google Scholar]
- 105. Dephoure N., Zhou C., Villén J., Beausoleil S. A., Bakalarski C. E., Elledge S. J., and Gygi S. P. (2008) A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. U. S. A. 105, 10762–10767 10.1073/pnas.0805139105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Mollapour M., Tsutsumi S., Donnelly A. C., Beebe K., Tokita M. J., Lee M. J., Lee S., Morra G., Bourboulia D., Scroggins B. T., Colombo G., Blagg B. S., Panaretou B., Stetler-Stevenson W. G., Trepel J. B., et al. (2010) Swe1Wee1-dependent tyrosine phosphorylation of Hsp90 regulates distinct facets of chaperone function. Mol. Cell 37, 333–343 10.1016/j.molcel.2010.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Xu W., Mollapour M., Prodromou C., Wang S., Scroggins B. T., Palchick Z., Beebe K., Siderius M., Lee M. J., Couvillon A., Trepel J. B., Miyata Y., Matts R., and Neckers L. (2012) Dynamic tyrosine phosphorylation modulates cycling of the HSP90-P50(CDC37)-AHA1 chaperone machine. Mol. Cell 47, 434–443 10.1016/j.molcel.2012.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Bachman A. B., Keramisanou D., Xu W., Beebe K., Moses M. A., Vasantha Kumar M. V., Gray G., Noor R. E., van der Vaart A., Neckers L., and Gelis I. (2018) Phosphorylation induced cochaperone unfolding promotes kinase recruitment and client class-specific Hsp90 phosphorylation. Nat. Commun. 9, 265 10.1038/s41467-017-02711-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Walter R., Pan K. T., Doebele C., Comoglio F., Tomska K., Bohnenberger H., Young R. M., Jacobs L., Keller U., Bönig H., Engelke M., Rosenwald A., Urlaub H., Staudt L. M., Serve H., et al. (2017) HSP90 promotes Burkitt lymphoma cell survival by maintaining tonic B-cell receptor signaling. Blood 129, 598–608 10.1182/blood-2016-06-721423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Beebe K., Mollapour M., Scroggins B., Prodromou C., Xu W., Tokita M., Taldone T., Pullen L., Zierer B. K., Lee M. J., Trepel J., Buchner J., Bolon D., Chiosis G., and Neckers L. (2013) Posttranslational modification and conformational state of heat shock protein 90 differentially affect binding of chemically diverse small molecule inhibitors. Oncotarget 4, 1065–1074 10.18632/oncotarget.1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Gu T. L., Goss V. L., Reeves C., Popova L., Nardone J., Macneill J., Walters D. K., Wang Y., Rush J., Comb M. J., Druker B. J., and Polakiewicz R. D. (2006) Phosphotyrosine profiling identifies the KG-1 cell line as a model for the study of FGFR1 fusions in acute myeloid leukemia. Blood 108, 4202–4204 10.1182/blood-2006-06-026666 [DOI] [PubMed] [Google Scholar]
- 112. Rikova K., Guo A., Zeng Q., Possemato A., Yu J., Haack H., Nardone J., Lee K., Reeves C., Li Y., Hu Y., Tan Z., Stokes M., Sullivan L., Mitchell J., et al. (2007) Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 131, 1190–1203 10.1016/j.cell.2007.11.025 [DOI] [PubMed] [Google Scholar]
- 113. Duval M., Le Boeuf F., Huot J., and Gratton J. P. (2007) Src-mediated phosphorylation of Hsp90 in response to vascular endothelial growth factor (VEGF) is required for VEGF receptor-2 signaling to endothelial NO synthase. Mol. Biol. Cell 18, 4659–4668 10.1091/mbc.e07-05-0467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Desjardins F., Delisle C., and Gratton J. P. (2012) Modulation of the cochaperone AHA1 regulates heat-shock protein 90 and endothelial NO synthase activation by vascular endothelial growth factor. Arterioscler. Thromb. Vasc. Biol. 32, 2484–2492 10.1161/ATVBAHA.112.256008 [DOI] [PubMed] [Google Scholar]
- 115. Ballif B. A., Carey G. R., Sunyaev S. R., and Gygi S. P. (2008) Large-scale identification and evolution indexing of tyrosine phosphorylation sites from murine brain. J. Proteome Res. 7, 311–318 10.1021/pr0701254 [DOI] [PubMed] [Google Scholar]
- 116. Zuehlke A. D., Reidy M., Lin C., LaPointe P., Alsomairy S., Lee D. J., Rivera-Marquez G. M., Beebe K., Prince T., Lee S., Trepel J. B., Xu W., Johnson J., Masison D., and Neckers L. (2017) An Hsp90 co-chaperone protein in yeast is functionally replaced by site-specific posttranslational modification in humans. Nat. Commun. 8, 15328 10.1038/ncomms15328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Li J., Rix U., Fang B., Bai Y., Edwards A., Colinge J., Bennett K. L., Gao J., Song L., Eschrich S., Superti-Furga G., Koomen J., and Haura E. B. (2010) A chemical and phosphoproteomic characterization of dasatinib action in lung cancer. Nat. Chem. Biol. 6, 291–299 10.1038/nchembio.332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Li X., Robbins N., O'Meara T. R., and Cowen L. E. (2017) Extensive functional redundancy in the regulation of Candida albicans drug resistance and morphogenesis by lysine deacetylases Hos2, Hda1, Rpd3 and Rpd31. Mol. Microbiol. 103, 635–656 10.1111/mmi.13578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Lamoth F., Juvvadi P. R., Soderblom E. J., Moseley M. A., Asfaw Y. G., and Steinbach W. J. (2014) Identification of a key lysine residue in heat shock protein 90 required for azole and echinocandin resistance in Aspergillus fumigatus. Antimicrob. Agents Chemother. 58, 1889–1896 10.1128/AAC.02286-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Wu D., Gu Q., Zhao N., Xia F., and Li Z. (2015) Structure-based rational design of peptide hydroxamic acid inhibitors to target tumor necrosis factor-α converting enzyme as potential therapeutics for hepatitis. J. Drug Target 23, 936–942 10.3109/1061186X.2015.1043916 [DOI] [PubMed] [Google Scholar]
- 121. Yang Y., Rao R., Shen J., Tang Y., Fiskus W., Nechtman J., Atadja P., and Bhalla K. (2008) Role of acetylation and extracellular location of heat shock protein 90α in tumor cell invasion. Cancer Res. 68, 4833–4842 10.1158/0008-5472.CAN-08-0644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Fiskus W., Rao R., Fernandez P., Herger B., Yang Y., Chen J., Kolhe R., Mandawat A., Wang Y., Joshi R., Eaton K., Lee P., Atadja P., Peiper S., and Bhalla K. (2008) Molecular and biologic characterization and drug sensitivity of pan-histone deacetylase inhibitor-resistant acute myeloid leukemia cells. Blood 112, 2896–2905 10.1182/blood-2007-10-116319 [DOI] [PubMed] [Google Scholar]
- 123. Wang Y., Fiskus W., Chong D. G., Buckley K. M., Natarajan K., Rao R., Joshi A., Balusu R., Koul S., Chen J., Savoie A., Ustun C., Jillella A. P., Atadja P., Levine R. L., et al. (2009) Cotreatment with panobinostat and JAK2 inhibitor TG101209 attenuates JAK2V617F levels and signaling and exerts synergistic cytotoxic effects against human myeloproliferative neoplastic cells. Blood 114, 5024–5033 10.1182/blood-2009-05-222133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Choudhary C., Kumar C., Gnad F., Nielsen M. L., Rehman M., Walther T. C., Olsen J. V., and Mann M. (2009) Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840 10.1126/science.1175371 [DOI] [PubMed] [Google Scholar]
- 125. Scroggins B. T., Robzyk K., Wang D., Marcu M. G., Tsutsumi S., Beebe K., Cotter R. J., Felts S., Toft D., Karnitz L., Rosen N., and Neckers L. (2007) An acetylation site in the middle domain of Hsp90 regulates chaperone function. Mol Cell 25, 151–159 10.1016/j.molcel.2006.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Yang C., Rahimpour S., Lu J., Pacak K., Ikejiri B., Brady R. O., and Zhuang Z. (2013) Histone deacetylase inhibitors increase glucocerebrosidase activity in Gaucher disease by modulation of molecular chaperones. Proc. Natl. Acad. Sci. U. S. A. 110, 966–971 10.1073/pnas.1221046110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Jochems J., Teegarden S. L., Chen Y., Boulden J., Challis C., Ben-Dor G. A., Kim S. F., and Berton O. (2015) Enhancement of stress resilience through histone deacetylase 6-mediated regulation of glucocorticoid receptor chaperone dynamics. Biol. Psychiatry 77, 345–355 10.1016/j.biopsych.2014.07.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Jiménez-Canino R., Lorenzo-Diaz F., Jaisser F., Farman N., Giraldez T., and Alvarez de la Rosa D. (2016) Histone deacetylase 6-controlled Hsp90 acetylation significantly alters mineralocorticoid receptor subcellular dynamics but not its transcriptional activity. Endocrinology 157, 2515–2532 10.1210/en.2015-2055 [DOI] [PubMed] [Google Scholar]
- 129. Deskin B., Lasky J., Zhuang Y., and Shan B. (2016) Requirement of HDAC6 for activation of Notch1 by TGF-β1. Sci. Rep. 6, 31086 10.1038/srep31086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Woodford M. R., Hughes M., Sager R. A., Backe S. J., Baker-Williams A. J., Bratslavsky M. S., Jacob J. M., Shapiro O., Wong M., Bratslavsky G., Bourboulia D., and Mollapour M. (2019) Mutation of the co-chaperone Tsc1 in bladder cancer diminishes Hsp90 acetylation and reduces drug sensitivity and selectivity. Oncotarget 10, 5824–5834 10.18632/oncotarget.27217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Weinert B. T., Schölz C., Wagner S. A., Iesmantavicius V., Su D., Daniel J. A., and Choudhary C. (2013) Lysine succinylation is a frequently occurring modification in prokaryotes and eukaryotes and extensively overlaps with acetylation. Cell Rep. 4, 842–851 10.1016/j.celrep.2013.07.024 [DOI] [PubMed] [Google Scholar]
- 132. Abu-Farha M., Lanouette S., Elisma F., Tremblay V., Butson J., Figeys D., and Couture J. F. (2011) Proteomic analyses of the SMYD family interactomes identify HSP90 as a novel target for SMYD2. J. Mol. Cell Biol. 3, 301–308 10.1093/jmcb/mjr025 [DOI] [PubMed] [Google Scholar]
- 133. Hamamoto R., Toyokawa G., Nakakido M., Ueda K., and Nakamura Y. (2014) SMYD2-dependent HSP90 methylation promotes cancer cell proliferation by regulating the chaperone complex formation. Cancer Lett. 351, 126–133 10.1016/j.canlet.2014.05.014 [DOI] [PubMed] [Google Scholar]
- 134. Rehn A., Lawatscheck J., Jokisch M. L., Mader S. L., Luo Q., Tippel F., Blank B., Richter K., Lang K., Kaila V. R. I., and Buchner J. (2020) A methylated lysine is a switch point for conformational communication in the chaperone Hsp90. Nat. Commun. 11, 1219 10.1038/s41467-020-15048-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Donlin L. T., Andresen C., Just S., Rudensky E., Pappas C. T., Kruger M., Jacobs E. Y., Unger A., Zieseniss A., Dobenecker M. W., Voelkel T., Chait B. T., Gregorio C. C., Rottbauer W., Tarakhovsky A., et al. (2012) Smyd2 controls cytoplasmic lysine methylation of Hsp90 and myofilament organization. Genes Dev. 26, 114–119 10.1101/gad.177758.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Olsen J. B., Cao X. J., Han B., Chen L. H., Horvath A., Richardson T. I., Campbell R. M., Garcia B. A., and Nguyen H. (2016) Quantitative profiling of the activity of protein lysine methyltransferase SMYD2 using SILAC-based proteomics. Mol. Cell. Proteomics 15, 892–905 10.1074/mcp.M115.053280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Shibata T., Kimura Y., Mukai A., Mori H., Ito S., Asaka Y., Oe S., Tanaka H., Takahashi T., and Uchida K. (2011) Transthiocarbamoylation of proteins by thiolated isothiocyanates. J. Biol. Chem. 286, 42150–42161 10.1074/jbc.M111.308049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Zhang Y., Dayalan Naidu S., Samarasinghe K., Van Hecke G. C., Pheely A., Boronina T. N., Cole R. N., Benjamin I. J., Cole P. A., Ahn Y. H., and Dinkova-Kostova A. T. (2014) Sulphoxythiocarbamates modify cysteine residues in HSP90 causing degradation of client proteins and inhibition of cancer cell proliferation. Br. J. Cancer 110, 71–82 10.1038/bjc.2013.710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Martínez-Ruiz A., Villanueva L., González de Orduña C., López-Ferrer D., Higueras M. A., Tarín C., Rodríguez-Crespo I., Vázquez J., and Lamas S. (2005) S-Nitrosylation of Hsp90 promotes the inhibition of its ATPase and endothelial nitric oxide synthase regulatory activities. Proc. Natl. Acad. Sci. U. S. A. 102, 8525–8530 10.1073/pnas.0407294102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Retzlaff M., Stahl M., Eberl H. C., Lagleder S., Beck J., Kessler H., and Buchner J. (2009) Hsp90 is regulated by a switch point in the C-terminal domain. EMBO Rep. 10, 1147–1153 10.1038/embor.2009.153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Mollapour M., Bourboulia D., Beebe K., Woodford M. R., Polier S., Hoang A., Chelluri R., Li Y., Guo A., Lee M. J., Fotooh-Abadi E., Khan S., Prince T., Miyajima N., Yoshida S., et al. (2014) Asymmetric Hsp90 N domain SUMOylation recruits Aha1 and ATP-competitive inhibitors. Mol. Cell 53, 317–329 10.1016/j.molcel.2013.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Preuss K. D., Pfreundschuh M., Weigert M., Fadle N., Regitz E., and Kubuschok B. (2015) Sumoylated HSP90 is a dominantly inherited plasma cell dyscrasias risk factor. J. Clin. Invest. 125, 316–323 10.1172/JCI76802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Akimov V., Barrio-Hernandez I., Hansen S. V. F., Hallenborg P., Pedersen A. K., Bekker-Jensen D. B., Puglia M., Christensen S. D. K., Vanselow J. T., Nielsen M. M., Kratchmarova I., Kelstrup C. D., Olsen J. V., and Blagoev B. (2018) UbiSite approach for comprehensive mapping of lysine and N-terminal ubiquitination sites. Nat. Struct. Mol. Biol. 25, 631–640 10.1038/s41594-018-0084-y [DOI] [PubMed] [Google Scholar]
- 144. Kundrat L., and Regan L. (2010) Identification of residues on Hsp70 and Hsp90 ubiquitinated by the cochaperone CHIP. J. Mol. Biol. 395, 587–594 10.1016/j.jmb.2009.11.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Povlsen L. K., Beli P., Wagner S. A., Poulsen S. L., Sylvestersen K. B., Poulsen J. W., Nielsen M. L., Bekker-Jensen S., Mailand N., and Choudhary C. (2012) Systems-wide analysis of ubiquitylation dynamics reveals a key role for PAF15 ubiquitylation in DNA-damage bypass. Nat. Cell Biol. 14, 1089–1098 10.1038/ncb2579 [DOI] [PubMed] [Google Scholar]
- 146. Wagner S. A., Beli P., Weinert B. T., Nielsen M. L., Cox J., Mann M., and Choudhary C. (2011) A proteome-wide, quantitative survey of in vivo ubiquitylation sites reveals widespread regulatory roles. Mol. Cell. Proteomics 10, M111.013284 10.1074/mcp.M111.013284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Danielsen J. M., Sylvestersen K. B., Bekker-Jensen S., Szklarczyk D., Poulsen J. W., Horn H., Jensen L. J., Mailand N., and Nielsen M. L. (2011) Mass spectrometric analysis of lysine ubiquitylation reveals promiscuity at site level. Mol. Cell. Proteomics 10, M110.003590 10.1074/mcp.M110.003590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Udeshi N. D., Svinkina T., Mertins P., Kuhn E., Mani D. R., Qiao J. W., and Carr S. A. (2013) Refined preparation and use of anti-diglycine remnant (K-ε-GG) antibody enables routine quantification of 10,000s of ubiquitination sites in single proteomics experiments. Mol. Cell. Proteomics 12, 825–831 10.1074/mcp.O112.027094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Franco M. C., Ye Y., Refakis C. A., Feldman J. L., Stokes A. L., Basso M., Melero Fernández de Mera R. M., Sparrow N. A., Calingasan N. Y., Kiaei M., Rhoads T. W., Ma T. C., Grumet M., Barnes S., Beal M. F., et al. (2013) Nitration of Hsp90 induces cell death. Proc. Natl. Acad. Sci. U. S. A. 110, E1102–E1111 10.1073/pnas.1215177110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Franco M. C., Ricart K. C., Gonzalez A. S., Dennys C. N., Nelson P. A., Janes M. S., Mehl R. A., Landar A., and Estévez A. G. (2015) Nitration of Hsp90 on tyrosine 33 regulates mitochondrial metabolism. J. Biol. Chem. 290, 19055–19066 10.1074/jbc.M115.663278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Overath T., Kuckelkorn U., Henklein P., Strehl B., Bonar D., Kloss A., Siele D., Kloetzel P. M., and Janek K. (2012) Mapping of O-GlcNAc sites of 20 S proteasome subunits and Hsp90 by a novel biotin-cystamine tag. Mol. Cell. Proteomics 11, 467–477 10.1074/mcp.M111.015966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Stetz G., Astl L., and Verkhivker G. M. (2020) Exploring mechanisms of communication switching in the Hsp90-Cdc37 regulatory complexes with client kinases through allosteric coupling of phosphorylation sites: perturbation-based modeling and hierarchical community analysis of residue interaction networks. J. Chem. Theory Comput. 10.1021/acs.jctc.0c00280 [DOI] [PubMed] [Google Scholar]
- 153. Sager R. A., Woodford M. R., Backe S. J., Makedon A. M., Baker-Williams A. J., DiGregorio B. T., Loiselle D. R., Haystead T. A., Zachara N. E., Prodromou C., Bourboulia D., Schmidt L. S., Linehan W. M., Bratslavsky G., and Mollapour M. (2019) Post-translational regulation of FNIP1 creates a rheostat for the molecular chaperone Hsp90. Cell Rep. 26, 1344–1356.e5 10.1016/j.celrep.2019.01.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Wandinger S. K., Suhre M. H., Wegele H., and Buchner J. (2006) The phosphatase Ppt1 is a dedicated regulator of the molecular chaperone Hsp90. EMBO J. 25, 367–376 10.1038/sj.emboj.7600930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Hallett S. T., Pastok M. W., Morgan R. M. L., Wittner A., Blundell K., Felletar I., Wedge S. R., Prodromou C., Noble M. E. M., Pearl L. H., and Endicott J. A. (2017) Differential regulation of G1 CDK complexes by the Hsp90-Cdc37 chaperone system. Cell Rep. 21, 1386–1398 10.1016/j.celrep.2017.10.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Prince T., Sun L., and Matts R. L. (2005) Cdk2: a genuine protein kinase client of Hsp90 and Cdc37. Biochemistry 44, 15287–15295 10.1021/bi051423m [DOI] [PubMed] [Google Scholar]
- 157. Basto R., Gergely F., Draviam V. M., Ohkura H., Liley K., and Raff J. W. (2007) Hsp90 is required to localise cyclin B and Msps/ch-TOG to the mitotic spindle in Drosophila and humans. J. Cell Sci. 120, 1278–1287 10.1242/jcs.000604 [DOI] [PubMed] [Google Scholar]
- 158. Wang A., Kolhe J. A., Gioacchini N., Baade I., Brieher W. M., Peterson C. L., and Freeman B. C. (2020) Mechanism of long-range chromosome motion triggered by gene activation. Dev. Cell 52, 309–320.e5 10.1016/j.devcel.2019.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Caridi C. P., D'Agostino C., Ryu T., Zapotoczny G., Delabaere L., Li X., Khodaverdian V. Y., Amaral N., Lin E., Rau A. R., and Chiolo I. (2018) Nuclear F-actin and myosins drive relocalization of heterochromatic breaks. Nature 559, 54–60 10.1038/s41586-018-0242-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Schrank B. R., Aparicio T., Li Y., Chang W., Chait B. T., Gundersen G. G., Gottesman M. E., and Gautier J. (2018) Nuclear ARP2/3 drives DNA break clustering for homology-directed repair. Nature 559, 61–66 10.1038/s41586-018-0237-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Woodford M. R., Sager R. A., Marris E., Dunn D. M., Blanden A. R., Murphy R. L., Rensing N., Shapiro O., Panaretou B., Prodromou C., Loh S. N., Gutmann D. H., Bourboulia D., Bratslavsky G., Wong M., et al. (2017) Tumor suppressor Tsc1 is a new Hsp90 co-chaperone that facilitates folding of kinase and non-kinase clients. EMBO J. 36, 3650–3665 10.15252/embj.201796700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Matheson C. J., Backos D. S., and Reigan P. (2016) Targeting WEE1 kinase in cancer. Trends Pharmacol. Sci. 37, 872–881 10.1016/j.tips.2016.06.006 [DOI] [PubMed] [Google Scholar]
- 163. Mollapour M., Tsutsumi S., and Neckers L. (2010) Hsp90 phosphorylation, Wee1 and the cell cycle. Cell Cycle 9, 2310–2316 10.4161/cc.9.12.12054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Barabutis N., Handa V., Dimitropoulou C., Rafikov R., Snead C., Kumar S., Joshi A., Thangjam G., Fulton D., Black S. M., Patel V., and Catravas J. D. (2013) LPS induces pp60c-src-mediated tyrosine phosphorylation of Hsp90 in lung vascular endothelial cells and mouse lung. Am. J. Physiol. Lung Cell Mol. Physiol. 304, L883–L893 10.1152/ajplung.00419.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Bali P., Pranpat M., Bradner J., Balasis M., Fiskus W., Guo F., Rocha K., Kumaraswamy S., Boyapalle S., Atadja P., Seto E., and Bhalla K. (2005) Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90. J. Biol. Chem. 280, 26729–26734 10.1074/jbc.C500186200 [DOI] [PubMed] [Google Scholar]
- 166. Kovacs J. J., Murphy P. J., Gaillard S., Zhao X., Wu J. T., Nicchitta C. V., Yoshida M., Toft D. O., Pratt W. B., and Yao T. P. (2005) HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol. Cell 18, 601–607 10.1016/j.molcel.2005.04.021 [DOI] [PubMed] [Google Scholar]
- 167. Fan S. J., Huang F. I., Liou J. P., and Yang C. R. (2018) The novel histone de acetylase 6 inhibitor, MPT0G211, ameliorates tau phosphorylation and cognitive deficits in an Alzheimer's disease model. Cell Death Dis. 9, 655 10.1038/s41419-018-0688-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Park J. H., Kim S. H., Choi M. C., Lee J., Oh D. Y., Im S. A., Bang Y. J., and Kim T. Y. (2008) Class II histone deacetylases play pivotal roles in heat shock protein 90-mediated proteasomal degradation of vascular endothelial growth factor receptors. Biochem. Biophys. Res. Commun. 368, 318–322 10.1016/j.bbrc.2008.01.056 [DOI] [PubMed] [Google Scholar]
- 169. Yu X., Guo Z. S., Marcu M. G., Neckers L., Nguyen D. M., Chen G. A., and Schrump D. S. (2002) Modulation of p53, ErbB1, ErbB2, and Raf-1 expression in lung cancer cells by depsipeptide FR901228. J. Natl. Cancer Inst. 94, 504–513 10.1093/jnci/94.7.504 [DOI] [PubMed] [Google Scholar]
- 170. Ganguly S., Home T., Yacoub A., Kambhampati S., Shi H., Dandawate P., Padhye S., Saluja A. K., McGuirk J., and Rao R. (2015) Targeting HSF1 disrupts HSP90 chaperone function in chronic lymphocytic leukemia. Oncotarget 6, 31767–31779 10.18632/oncotarget.5167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Zhang Y., Kwon S., Yamaguchi T., Cubizolles F., Rousseaux S., Kneissel M., Cao C., Li N., Cheng H. L., Chua K., Lombard D., Mizeracki A., Matthias G., Alt F. W., Khochbin S., et al. (2008) Mice lacking histone deacetylase 6 have hyperacetylated tubulin but are viable and develop normally. Mol. Cell Biol. 28, 1688–1701 10.1128/MCB.01154-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Murphy P. J., Morishima Y., Kovacs J. J., Yao T. P., and Pratt W. B. (2005) Regulation of the dynamics of hsp90 action on the glucocorticoid receptor by acetylation/deacetylation of the chaperone. J. Biol. Chem. 280, 33792–33799 10.1074/jbc.M506997200 [DOI] [PubMed] [Google Scholar]
- 173. Espallergues J., Teegarden S. L., Veerakumar A., Boulden J., Challis C., Jochems J., Chan M., Petersen T., Deneris E., Matthias P., Hahn C. G., Lucki I., Beck S. G., and Berton O. (2012) HDAC6 regulates glucocorticoid receptor signaling in serotonin pathways with critical impact on stress resilience. J. Neurosci. 32, 4400–4416 10.1523/JNEUROSCI.5634-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Fiskus W., Ren Y., Mohapatra A., Bali P., Mandawat A., Rao R., Herger B., Yang Y., Atadja P., Wu J., and Bhalla K. (2007) Hydroxamic acid analogue histone deacetylase inhibitors attenuate estrogen receptor-α levels and transcriptional activity: a result of hyperacetylation and inhibition of chaperone function of heat shock protein 90. Clin. Cancer Res. 13, 4882–4890 10.1158/1078-0432.CCR-06-3093 [DOI] [PubMed] [Google Scholar]
- 175. Gibbs A., Schwartzman J., Deng V., and Alumkal J. (2009) Sulforaphane destabilizes the androgen receptor in prostate cancer cells by inactivating histone deacetylase 6. Proc. Natl. Acad. Sci. U. S. A. 106, 16663–16668 10.1073/pnas.0908908106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Ai J., Wang Y., Dar J. A., Liu J., Liu L., Nelson J. B., and Wang Z. (2009) HDAC6 regulates androgen receptor hypersensitivity and nuclear localization via modulating Hsp90 acetylation in castration-resistant prostate cancer. Mol. Endocrinol. 23, 1963–1972 10.1210/me.2009-0188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Gao Y. S., Hubbert C. C., Lu J., Lee Y. S., Lee J. Y., and Yao T. P. (2007) Histone deacetylase 6 regulates growth factor-induced actin remodeling and endocytosis. Mol. Cell Biol. 27, 8637–8647 10.1128/MCB.00393-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Zhang J., Xu E., and Chen X. (2013) TAp73 protein stability is controlled by histone deacetylase 1 via regulation of Hsp90 chaperone function. J. Biol. Chem. 288, 7727–7737 10.1074/jbc.M112.429522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Min J. S., Kim J. C., Kim J. A., Kang I., and Ahn J. K. (2018) SIRT2 reduces actin polymerization and cell migration through deacetylation and degradation of HSP90. Biochim. Biophys. Acta Mol. Cell Res. 1865, 1230–1238 10.1016/j.bbamcr.2018.06.005 [DOI] [PubMed] [Google Scholar]
- 180. Jenuwein T., and Allis C. D. (2001) Translating the histone code. Science 293, 1074–1080 10.1126/science.1063127 [DOI] [PubMed] [Google Scholar]
- 181. Voelkel T., Andresen C., Unger A., Just S., Rottbauer W., and Linke W. A. (2013) Lysine methyltransferase Smyd2 regulates Hsp90-mediated protection of the sarcomeric titin springs and cardiac function. Biochim. Biophys. Acta 1833, 812–822 10.1016/j.bbamcr.2012.09.012 [DOI] [PubMed] [Google Scholar]
- 182. Averna M., Stifanese R., De Tullio R., Passalacqua M., Salamino F., Pontremoli S., and Melloni E. (2008) Functional role of HSP90 complexes with endothelial nitric-oxide synthase (eNOS) and calpain on nitric oxide generation in endothelial cells. J. Biol. Chem. 283, 29069–29076 10.1074/jbc.M803638200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Wolmarans A., Kwantes A., and LaPointe P. (2019) A novel method for site-specific chemical SUMOylation: SUMOylation of Hsp90 modulates co-chaperone binding in vitro. Biol. Chem. 400, 487–500 10.1515/hsz-2018-0251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Cavenagh J., Oakervee H., Baetiong-Caguioa P., Davies F., Gharibo M., Rabin N., Kurman M., Novak B., Shiraishi N., Nakashima D., Akinaga S., and Yong K. (2017) A phase I/II study of KW-2478, an Hsp90 inhibitor, in combination with bortezomib in patients with relapsed/refractory multiple myeloma. Br. J. Cancer 117, 1295–1302 10.1038/bjc.2017.302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Richardson P. G., Chanan-Khan A. A., Alsina M., Albitar M., Berman D., Messina M., Mitsiades C. S., and Anderson K. C. (2010) Tanespimycin monotherapy in relapsed multiple myeloma: results of a phase 1 dose-escalation study. Br. J. Haematol. 150, 438–445 10.1111/j.1365-2141.2010.08265.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Richardson P. G., Mitsiades C. S., Laubach J. P., Lonial S., Chanan-Khan A. A., and Anderson K. C. (2011) Inhibition of heat shock protein 90 (HSP90) as a therapeutic strategy for the treatment of myeloma and other cancers. Br. J. Haematol. 152, 367–379 10.1111/j.1365-2141.2010.08360.x [DOI] [PubMed] [Google Scholar]
- 187. Siegel D., Shieh B., Yan C., Kepa J. K., and Ross D. (2011) Role for NAD(P)H:quinone oxidoreductase 1 and manganese-dependent superoxide dismutase in 17-(allylamino)-17-demethoxygeldanamycin-induced heat shock protein 90 inhibition in pancreatic cancer cells. J. Pharmacol. Exp. Ther. 336, 874–880 10.1124/jpet.110.176438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Harlow L., Rosas I. O., Gochuico B. R., Mikuls T. R., Dellaripa P. F., Oddis C. V., and Ascherman D. P. (2013) Identification of citrullinated hsp90 isoforms as novel autoantigens in rheumatoid arthritis-associated interstitial lung disease. Arthritis Rheum. 65, 869–879 10.1002/art.37881 [DOI] [PubMed] [Google Scholar]
- 189. Travers T. S., Harlow L., Rosas I. O., Gochuico B. R., Mikuls T. R., Bhattacharya S. K., Camacho C. J., and Ascherman D. P. (2016) Extensive citrullination promotes immunogenicity of HSP90 through protein unfolding and exposure of cryptic epitopes. J. Immunol. 197, 1926–1936 10.4049/jimmunol.1600162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Sarkar A. A., and Zohn I. E. (2012) Hectd1 regulates intracellular localization and secretion of Hsp90 to control cellular behavior of the cranial mesenchyme. J. Cell Biol. 196, 789–800 10.1083/jcb.201105101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Chen W. Y., Chang F. R., Huang Z. Y., Chen J. H., Wu Y. C., and Wu C. C. (2008) Tubocapsenolide A, a novel withanolide, inhibits proliferation and induces apoptosis in MDA-MB-231 cells by thiol oxidation of heat shock proteins. J. Biol. Chem. 283, 17184–17193 10.1074/jbc.M709447200 [DOI] [PubMed] [Google Scholar]
- 192. Carbone D. L., Doorn J. A., Kiebler Z., Ickes B. R., and Petersen D. R. (2005) Modification of heat shock protein 90 by 4-hydroxynonenal in a rat model of chronic alcoholic liver disease. J. Pharmacol. Exp. Ther. 315, 8–15 10.1124/jpet.105.088088 [DOI] [PubMed] [Google Scholar]
- 193. Frank L. A., Sutton-McDowall M. L., Brown H. M., Russell D. L., Gilchrist R. B., and Thompson J. G. (2014) Hyperglycaemic conditions perturb mouse oocyte in vitro developmental competence via β-O-linked glycosylation of heat shock protein 90. Hum. Reprod. 29, 1292–1303 10.1093/humrep/deu066 [DOI] [PubMed] [Google Scholar]
- 194. Nokin M. J., Durieux F., Peixoto P., Chiavarina B., Peulen O., Blomme A., Turtoi A., Costanza B., Smargiasso N., Baiwir D., Scheijen J. L., Schalkwijk C. G., Leenders J., De Tullio P., Bianchi E., et al. (2016) Methylglyoxal, a glycolysis side-product, induces Hsp90 glycation and YAP-mediated tumor growth and metastasis. Elife 5, e19375 10.7554/eLife.19375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Butler L. M., Ferraldeschi R., Armstrong H. K., Centenera M. M., and Workman P. (2015) Maximizing the therapeutic potential of HSP90 inhibitors. Mol. Cancer Res. 13, 1445–1451 10.1158/1541-7786.MCR-15-0234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Echeverria P. C., Bhattacharya K., Joshi A., Wang T., and Picard D. (2019) The sensitivity to Hsp90 inhibitors of both normal and oncogenically transformed cells is determined by the equilibrium between cellular quiescence and activity. PLoS ONE 14, e0208287 10.1371/journal.pone.0208287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Neckers L., Blagg B., Haystead T., Trepel J. B., Whitesell L., and Picard D. (2018) Methods to validate Hsp90 inhibitor specificity, to identify off-target effects, and to rethink approaches for further clinical development. Cell Stress Chaperones 23, 467–482 10.1007/s12192-018-0877-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Chiosis G., and Neckers L. (2006) Tumor selectivity of Hsp90 inhibitors: the explanation remains elusive. ACS Chem. Biol. 1, 279–284 10.1021/cb600224w [DOI] [PubMed] [Google Scholar]
- 199. Kamal A., Thao L., Sensintaffar J., Zhang L., Boehm M. F., Fritz L. C., and Burrows F. J. (2003) A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature 425, 407–410 10.1038/nature01913 [DOI] [PubMed] [Google Scholar]
- 200. Iwai A., Bourboulia D., Mollapour M., Jensen-Taubman S., Lee S., Donnelly A. C., Yoshida S., Miyajima N., Tsutsumi S., Smith A. K., Sun D., Wu X., Blagg B. S., Trepel J. B., Stetler-Stevenson W. G., et al. (2012) Combined inhibition of Wee1 and Hsp90 activates intrinsic apoptosis in cancer cells. Cell Cycle 11, 3649–3655 10.4161/cc.21926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Rao R., Fiskus W., Yang Y., Lee P., Joshi R., Fernandez P., Mandawat A., Atadja P., Bradner J. E., and Bhalla K. (2008) HDAC6 inhibition enhances 17-AAG–mediated abrogation of hsp90 chaperone function in human leukemia cells. Blood 112, 1886–1893 10.1182/blood-2008-03-143644 [DOI] [PubMed] [Google Scholar]
- 202. O'Meara T. R., Robbins N., and Cowen L. E. (2017) The Hsp90 chaperone network modulates Candida virulence traits. Trends Microbiol. 25, 809–819 10.1016/j.tim.2017.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Cowen L. E., Carpenter A. E., Matangkasombut O., Fink G. R., and Lindquist S. (2006) Genetic architecture of Hsp90-dependent drug resistance. Eukaryot. Cell 5, 2184–2188 10.1128/EC.00274-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Cowen L. E., and Lindquist S. (2005) Hsp90 potentiates the rapid evolution of new traits: drug resistance in diverse fungi. Science 309, 2185–2189 10.1126/science.1118370 [DOI] [PubMed] [Google Scholar]
- 205. Robbins N., Leach M. D., and Cowen L. E. (2012) Lysine deacetylases Hda1 and Rpd3 regulate Hsp90 function thereby governing fungal drug resistance. Cell Rep. 2, 878–888 10.1016/j.celrep.2012.08.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Sager R. A., Woodford M. R., Neckers L., and Mollapour M. (2018) Detecting posttranslational modifications of Hsp90. Methods Mol. Biol. 1709, 209–219 10.1007/978-1-4939-7477-1_16 [DOI] [PMC free article] [PubMed] [Google Scholar]