

Abstract
Most characterized protein methylation events encompass arginine and lysine N-methylation, and only a few cases of protein methionine thiomethylation have been reported. Newly discovered oncohistone mutations include lysine-to-methionine substitutions at positions 27 and 36 of histone H3.3. In these instances, the methionine substitution localizes to the active-site pocket of the corresponding histone lysine methyltransferase, thereby inhibiting the respective transmethylation activity. SET domain–containing 3 (SETD3) is a protein (i.e. actin) histidine methyltransferase. Here, we generated an actin variant in which the histidine target of SETD3 was substituted with methionine. As for previously characterized histone SET domain proteins, the methionine substitution substantially (76-fold) increased binding affinity for SETD3 and inhibited SETD3 activity on histidine. Unexpectedly, SETD3 was active on the substituted methionine, generating S-methylmethionine in the context of actin peptide. The ternary structure of SETD3 in complex with the methionine-containing actin peptide at 1.9 Å resolution revealed that the hydrophobic thioether side chain is packed by the aromatic rings of Tyr312 and Trp273, as well as the hydrocarbon side chain of Ile310. Our results suggest that placing methionine properly in the active site—within close proximity to and in line with the incoming methyl group of SAM—would allow some SET domain proteins to selectively methylate methionine in proteins.
Keywords: protein methylation, S-adenosylmethionine (SAM), methionine, protein structure, enzyme catalysis, actin, methionine methylation, methyltransferases, S-methylmethionine, SETD3
Post-translational methylation on proteins, particularly histones, plays a fundamental role in regulating gene expression and chromatin organization (1–3). For example, histone H3 methylation at lysine residues 4, 9, 27, 36, and 79 signals for gene activation or repression of transcription. Dysregulation of these epigenetic signals has been correlated with and mechanistically linked to many human disorders including cancer (4–6). Recurrent mutations in histone H3.3, first discovered in pediatric high-grade glioma and since found in multiple other cancer types, have been found to lead to alterations in the epigenetic landscape that are associated with the pathology of these diseases (7, 8) (reviewed in Ref. 9 and the references therein). The most heavily characterized oncohistone mutations are the lysine-to-methionine mutants at K27M and K36M, which perturb the methylation site for specific lysine methyltransferases (MTases) and dominantly inhibit methylation of corresponding lysine residues in other histone H3 proteins. Structural characterization of histone MTases in complex with the methionine-substituted histone H3 peptides, including G9a-H3K9M (10, 11), PRC2-H3K27M (12), and SETD2-H3K36M (13), revealed that the methionine residue localizes to the active-site pocket that normally accommodates the target lysine residue, thereby inhibiting the respective transmethylation activity. In vitro, it was the study of LSD1 (a histone H3K4me2/1 demethylase) that first used a H3 peptide bearing methionine in place of methyl-Lys4 (PDB code 2V1D), which showed that the K4M peptide is a strong inhibitor of LSD1 because the methionine substitution led to a 30-fold increase in binding affinity (14). Interestingly, EZHIP (enhancer of zeste homologs inhibitory protein) inhibits PRC2 methylation activity by presenting a methionine-containing, naturally occurring, H3K27M-like sequence (15–18), suggesting a broader role for these sequences in normal regulatory processes.
Biochemically, the methyl donor S-adenosyl-l-methionine (SAM) is the ATP-activated form of methionine (19), which contains a reactive positively charged methylsulfonium group (CH3–S+) (Fig. 1) (56, 57). In flowering plants, the biosynthesized l-methionine can be methylated to S-methylmethionine by a SAM-dependent reaction, catalyzed by methionine S-MTase (20). Although the biochemistry of methionine methylation is best characterized in the context of small molecules, the significance of protein methionine methylation is less clear. Reports of protein methionine methylation are limited to two examples in single-cell organisms involving a partially purified enzyme activity active on cytochrome c in the flagellate Euglena gracilis (21) and an automethylation of LaeA in the filamentous fungus Aspergillus nidulans (22).
Figure 1.

Thiomethylation. In plants, free amino acid methionine (Met) is methylated to become S-methylmethionine (SMM), which itself serves as a methyl donor for homocysteine (Hcy) methylation, generating two molecules of methionine. Homocysteine is a nonproteinogenic amino acid, differing from methionine and cysteine by lacking a terminal methyl group (CH3) or an additional methylene bridge (CH2), respectively. Like cysteine, homocysteine carries a reactive thiol nucleophile. In humans, the level of homocysteine, an increase of which is considered a risk factor for cardiovascular diseases, is regulated by at least three pathways to regenerate methionine: (i) methionine synthase; (ii) betaine homocysteine S-methyltransferase (BHMT-1) (56), using the methyl donor betaine (also known as trimethylglycine); and (iii) betaine-homocysteine S-methyltransferase 2 (BHMT-2), which uses S-methylmethionine, but not betaine, as a methyl donor (57). In addition, a pathogenic bacterial secreted effector protein NleE methylates cysteine-S in a human zinc finger protein.
SETD3 was identified as the first metazoan histidine MTase that works on an actin histidine residue (23–26). We have noted that SETD3 shares a high degree of global similarity with two characterized protein lysine MTases (human SETD6 and plant rubisco LSMT) and detected that SETD3 has weak lysine methylation activity on an actin variant in which the target His73 is substituted by a lysine (H73K) (27, 28). Prompted by the inhibitory effect of oncohistone mutations of lysine-to-methionine and similar endogenous MTases inhibiting sequences, here we report that unexpectedly, SETD3 has methylation activity on an actin variant in which the target His73 is substituted by a methionine (H73M). We investigate the structural and molecular determinants of generating S-methylmethionine in the context of a protein peptide.
Results
SETD3 methylation of methionine
Our previous characterizations of SETD3 indicated that SETD3 exhibited in vitro activity on full-length recombinant actin-Lys73, albeit much weaker than actin-His73 activity (27) (Fig. 2A). Unexpectedly we observed SETD3 activity on actin-Met73, in which the target His73 was substituted by a methionine (Fig. 2A). As a negative control, no activity was observed on actin-Ala73, in which His73 was replaced by an alanine. Consistent with the full-length recombinant protein, SETD3 exhibited activities on peptide substrates bearing either histidine, lysine, or methionine at position 73 (Fig. 2B). Under the saturating conditions of overnight reactions in which SETD3 completed reactions on its native substrate His73, we observed ∼50% reduced activity on Met73 and much reduced activity on Lys73.
Figure 2.
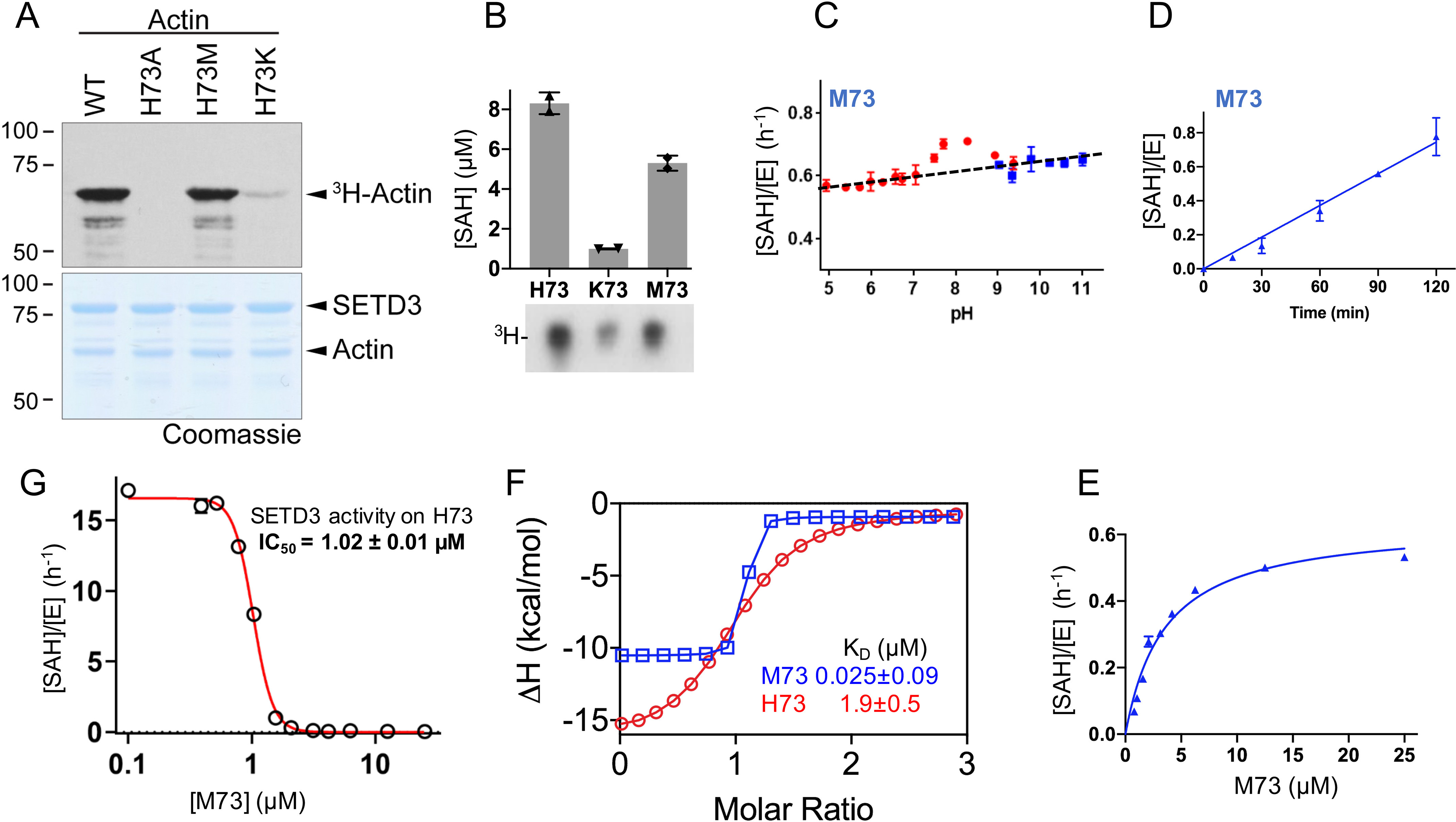
SETD3 is active on Met73. A, in vitro methylation reactions with SETD3, indicated full-length actin proteins, and radiolabeled methyl donor ([3H]SAM). The reactions were resolved by SDS–PAGE (bottom panel), and activity was visualized by autoradiography (top panel). B, overnight reaction of SETD3 activity on actin peptides containing His73, Lys73, or Met73, by two assays: Promega MTase-GloTM assay measures the reaction by-product (SAH) concentration (y axis) and autoradiography (3H peptides). C, SETD3 activity on Met73 as a function of pH (red points for below pH 9 and blue points for above pH 9). D, SETD3 activity on Met73 is linear as a function of time. E, single-turnover kinetics of SETD3 on Met73 under pH 8 for 1-h reaction. F, ITC measurements of SETD3 against Met73 and His73 (for original graphs, see Fig. S1, B and C). G, inhibition of SETD3 activity on His73 methylation by Met73-containing peptide.
As aforementioned, the methyl donor SAM carries a positively charged methylsulfonium group (CH3–S+), and SAM-dependent MTases require the target atom be in a deprotonated state. In agreement with this mechanism, SETD3 has varied, substrate-dependent optimum pH values: an optimum pH of 7 and above for histidine where the imidazole ring (with a typical pKa value near 6) is uncharged and a pH of ∼10.5 for lysine (mainly because of its typical pKa value of ∼10) (27). In comparison, methionine has a nonpolar S-methyl thioether side chain, and SETD3 has a relatively constant activity on Met73 over a pH range of 5–11 with a small increase of activity at pH 8.0 (Fig. 2C) at the linear time point of 1 h (Fig. 2D). Although SETD3 has a catalytic efficiency kcat/Km of 1.8 h−1 μm−1 on His73 methylation (28), SETD3 is a slow enzyme on Met73 without turnover (i.e. the product over the enzyme concentration is less than 1; [SAH]/[E] < 1). Under the first-order kinetics conditions, we estimated that SETD3 showed an activity on Met73 with an apparent kcat/Km value of ∼0.12 h−1 μm−1 (Fig. 2E).
Next, we measured the dissociation constant between SETD3 and Met73 peptide by isothermal titration calorimetry (Fig. 2F). We observed a KD value of 25 nm, an increase of binding by a factor of 76 from the SETD3 dissociation of His73 peptide, which might partially explain the low turnover rate of Met73 from SETD3. This result of enhanced binding (reduced KD value) by the methionine-substituted peptide agrees with previous studies on LSD1 (∼30-fold) (14) and PRC2 (∼20-fold) (12). In addition, the 25 nm dissociation constant matches well with the KD value of 32 nm observed between G9a and H3 peptide with the Lys9 replacement with norleucine in the presence of added SAM (11). Under the conditions that activity of SETD3 on Met73 is negligible (Fig. S1A), the Met73 peptide inhibits SETD3 activity on His73 with an IC50 of 1 μm (Fig. 2G), consistent with G9a inhibition by H3 peptide K9M with IC50 of 2.3 μm (11).
Structure of SETD3 in complex with Met73-containing peptide
SETD3 was readily crystallized with the Met73 (residues 66–88) peptide in the presence of SAH, and we observed electron density for peptide residues 66–84 (Fig. 3A). The ternary structure was determined to resolution of 1.9 Å (Table S1). The SETD3-Met73 structure is virtually identical to the previously determined structures of SETD3 in complex with His73 (PDB code 6MBJ), methylated His73 (PDB code 6OX2), or Lys73 (PDB code 6OX3) with pairwise comparison of less than 0.2 Å of root-mean-square deviation across two complexes of each crystal structure in the crystallographic asymmetric unit in space group P21 (Fig. 3B). In the methionine-occupied active site (Fig. 3C), the sulfur atom (Sδ) of Met73 is 5.3 Å away from the sulfur atom of SAH, in which a transferable methyl group would be attached (Fig. 3D). The observed conformations of the Met73 side chain and the methionine moiety of SAH in the active site is nearly symmetric to each other by a rotation of 180°. The nonpolar S-methyl thioether side chain is packed in between the aromatic rings of Tyr312 (via the Cγ atom) and Trp273. The terminal Cε carbon of Met73 is in van der Waals contact with the main chain Cα atom of Asp274 (3.4 Å), the indole ring of Trp273 (3.5 Å), and the side chain of Ile310 (4.0 Å) (Fig. 3E). In addition, the main chain Cα carbon of Met73 of actin forms a C–H···O type of hydrogen bond (29) with side chain of Asn255 (Fig. 3F). The dominant nonpolar interactions give rise to favorable binding of Met73 in an active site constructed mainly of aromatic and hydrophobic residues; a greasy environment helps to deprotonate (lower the pKa value) lysine, as evident by the observed pKa value of 6.5 for a lysine fully buried inside the hydrophobic core in a mutant T4 lysozyme (30). Thus, the mutations of the polar residue Asn255 required for hydrogen bonding with His73 during catalysis (27) to hydrophobic residues alanine and valine improved activity for Met73 methylation by ∼3-fold (Fig. 3, G and H).
Figure 3.
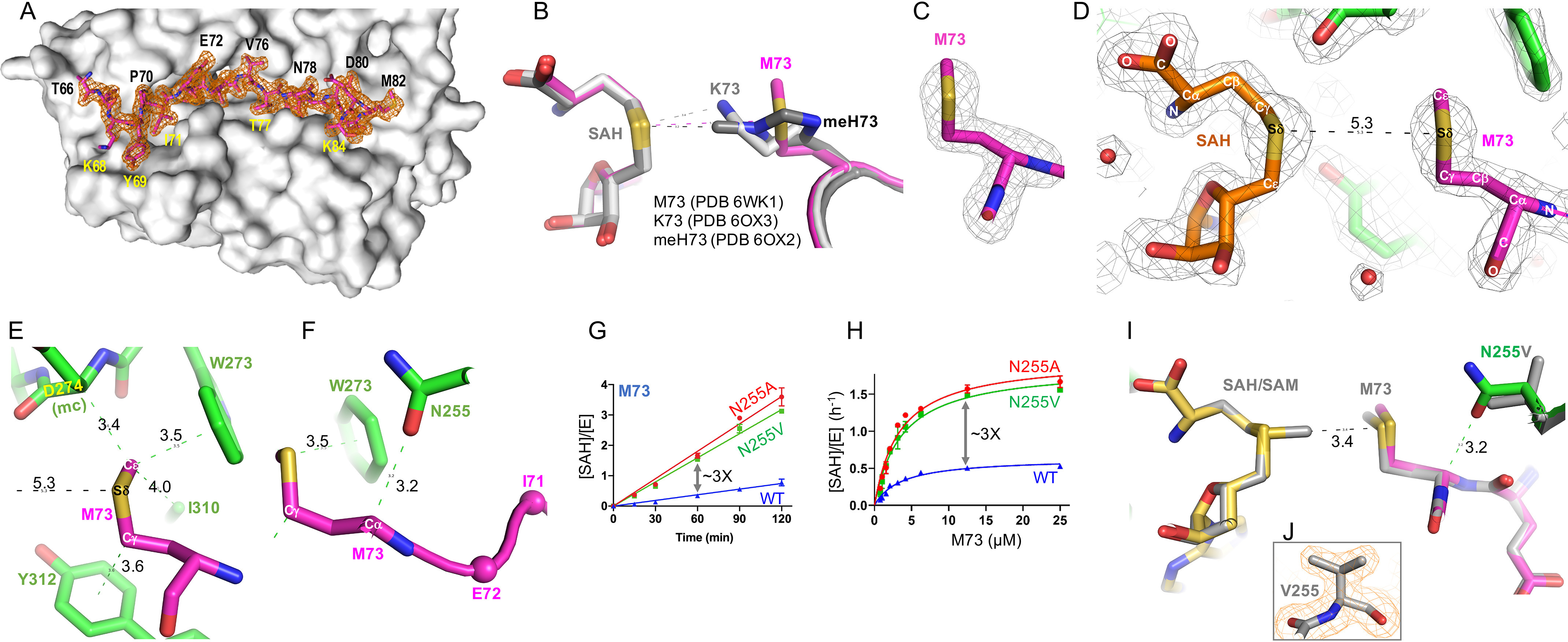
Structure of SETD3 in complex with Met73 in the active site. A, a surface representation of SETD3 (colored gray) accommodation of Met73 peptide in a long surface groove. Omit electron density for ordered residues 66–84 (in stick model) is contoured at 3σ above the mean. B, superimposition of SETD3 in complex with Met73, Lys73, and methylated His73. C, omit electron density (Fo − Fc) for Met73 contoured at 4σ above the mean. D, the Met73 side chain and the methionine moiety of SAH is nearly symmetric to each other by a rotation of 180°. E, the nonpolar S-methyl thioether side chain is in contact with aromatic and hydrophobic residues. F, a C–H···O hydrogen bond is formed between the main chain Cα atom of Met73 of actin and side chain of Asn255 of SETD3. G and H, SETD3 N255V and N255A mutants have high activity on Met73. I, superimposition of SETD3 (N255) (PDB code 6WK1) and its mutant N255V (PDB code 6WK2) in complex with actin Met73 peptide. J, electron density (2Fo − Fc) for Val255 contoured at 2.0σ above the mean.
Next, we determined the structure of mutant N255V in complex with the Met73 peptide to resolution of 1.76 Å (Table S1). The mutant structure is nearly identical to that of the WT enzyme, with a root-mean-square deviation of 0.16 Å across 946 pairs of Cα atoms (Fig. 3, I–J). The increased hydrophobicity of Val255, accompanied with the loss of polar interaction associated with Asn255, allows the side chain of Met73 of actin to move slightly toward to the methyl donor (Fig. 3I).
Comparison of SETD3 with other MTases in binding of methionine
We compared the structures of the SETD3-Met73 complex with that of three histone H3 MTases (G9a, PRC2, and SETD2) in complex with peptides containing methionine mutations at lysine 9, 27, and 36, respectively (10, 12, 13). By superimposing the respective cofactor SAM or SAH, we observed two major differences. First, the histone H3 peptides and actin peptide run in opposite directions (Fig. 4A). Second, the methionine residues in the context of histone H3 are further away from the transferrable methyl group of SAM, whereas in the actin peptide, the methyl acceptor (sulfur Sδ) is located within a shorter distance of and in line with the incoming methyl group of the methylsulfonium of SAM (Fig. 4B). Such a linear arrangement comprising the nucleophile (Met-S:), the methyl group, and the leaving thioester group of SAM in the transition state is required for the SN2 reaction mechanism used by SAM-dependent MTases (Fig. 4C).
Figure 4.

Comparison of active-site configuration for histone SET domain proteins and SETD3. A, superimposition of SETD3, G9a, PRC2, and SETD2 in complex with methionine-substituted peptide substrates. B, a linear arrangement comprising the Sδ atom of Met73, the methyl group, and the leaving thioester group of SAM. C, the Sn2 reaction mechanism by SAM-dependent methylation. D, SET7, PRDM9, and GLP are not active on methionine-substituted histone H3 peptides. E, PRDM9 is automethylated (see Fig. S2). F, superimposition of SETD3 in complex with methylated histidine, SET7 with monomethylated lysine, and PRDM9 with dimethylated lysine substrates.
Next, we tested three additional SET domain proteins, SET7 (31), PR/SET domain 9 (PRDM9) (32), and G9a-like protein (GLP) (33), on histone peptides containing methionine substitution at either lysine position 4 or 9. Each enzyme is active on its respective lysine-containing peptide substrate (Fig. 4D) but not the peptides with the corresponding methionine substitution. The activity observed with PRDM9 on H3M4 peptide is probably due to the automethylation of PRDM9 (Fig. 4E), because intramolecular automethylation was observed previously on lysine residues by PRDM9 (34) (Fig. S2).
The difference of activity on methionine by SETD3 as compared with other SET domain proteins on histone H3 probably reflects the difference of their natural substrates: the smaller histidine residue needs to enter deeper into the active site of SETD3 and positions the target nitrogen atom pointing toward, and within a short distance of, the incoming methyl group, whereas the terminal nitrogen atom of the larger lysine residue in an extended side chain conformation can be positioned in the same target position even with the backbone of peptide being further away (Fig. 4F). The inactivity on methionine by histone MTases presumably reflects the fact that the methionine side chain is not juxtaposed in close enough proximity to the donor methyl group of SAM for nucleophilic attack (Fig. 4B). We note that in the case of SETD3 bound with Lys73 peptide, the side chain of Lys73 is in a bent conformation (Fig. 3B) (27, 28). We also note that, unlike G9a (11), we did observe stable interactions between GLP and histone H3K9M in the presence of either SAM or SAH (Fig. S3), which forms heterodimer with G9a (33). However, the affinity for GLP-H3K9M-SAM (KD = 120 nm) is less than four times weaker than that of G9a.
SETD3 generates S-methylmethionine
HPLC–MS/MS confirmed and localized the methylation of actin peptide on Met73 by SETD3 resulting in S-methylmethionine. For the unmodified peptide, the only observable signal is from the expected mass of the peptide (2855.378 Da, 0.35 ppm error) and oxidized forms (Fig. 5A). A major precursor with a mass shift consistent with methylation (2869.390 Da, 0.94 ppm error) was identified when actin was reacted with SETD3 (Fig. 5B). The +4 charge state precursor of the modified peptide was isolated and fragmented by method of electron transfer dissociation (ETD).
Figure 5.
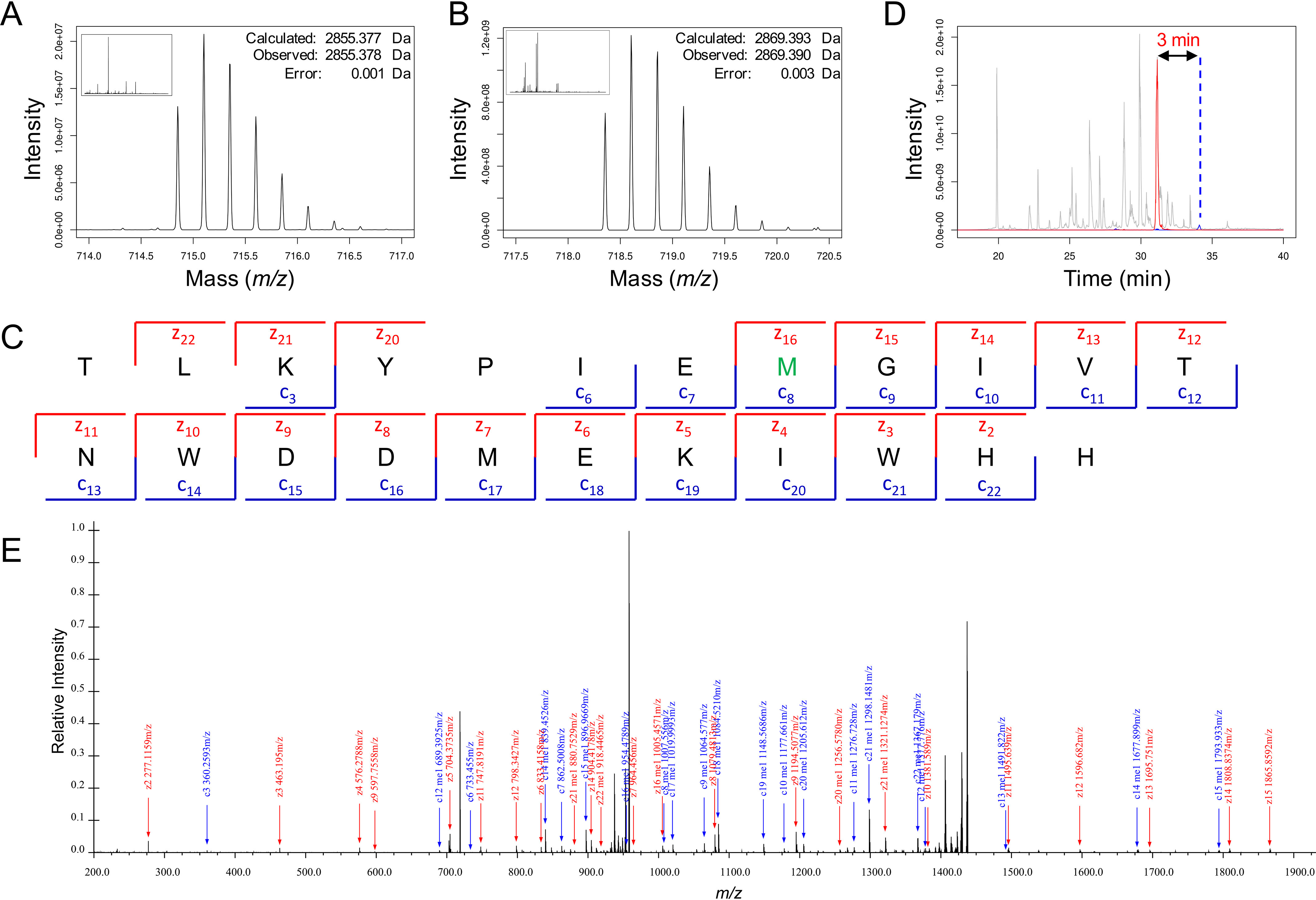
Identification and localization of methionine methylation by MS. A, MS1 spectra at 34-min retention time matches unmodified actin (<1 ppm error). B, MS1 spectra at 31-min retention time matches actin +14.0157 Da, which is consistent with methylation (<1 ppm error). C, matched ions for MS2 generated from fragmenting the methylated actin precursor. The methylation is unambiguously localized to Met73 (green). D, extracted ion chromatogram of TIC (gray), unmodified actin (blue), and methylated actin (red). A 3-min retention time shift is observed consistent with an increase in hydrophilicity. E, annotated spectra from averaged MS2 scans for the methylated actin.
ETD of the modified peptide resulted in an unambiguous localization of a +14.012 Da (which equals 2869.390–2855.378) mass shift to Met73 (Fig. 5C). This evidence suggests two possible additions. The first is that CH2 is added and results in a mass change of +14.016 Da (calculated). The second possibility is that the addition of a CH3 to the sulfur results in an addition of +15.0235 Da (calculated), and the formation of a positively charged sulfonium ion that results from the loss of an electron, when the valence shell is expanded to accommodate the additional bond; however, this fixed positive charge would normally be accounted for by a proton, which therefore must be subtracted from the uncharged mass, also resulting in a final mass difference of 14.016 Da (which equals 15.0235–1.0079). Thus, the mass of the modification alone does not distinguish between these two modes of methylation.
Changes in retention time support the assignment of S-methylmethionine (Fig. 5D). The methyl modified peptide elutes 3 min earlier than the unmodified peak on our reversed phase C3 column. Such a shift is consistent with a decrease in hydrophobicity, consistent with the addition of a strongly hydrophilic fixed positive charge. Monomethylation of carbon, which seems unlikely, increases hydrophobicity and would result in the opposite: a shift to longer retention times. Thus, the shift in retention time is consistent with the S-methylmethionine and the formation of a fixed positive charge and highly hydrophilic sulfonium.
In addition, we also note unusual charge state distributions and mass-to-charge ratios in the ETD-based fragmentation that could be explained by the unusual peptide chemistry from the fixed charged sulfonium ion (Fig. 5E). Usually during fragmentation, fragment ions are almost exclusively observed in an m/z range of 400–1200 because of correlation between the mass and the number of basic sites. Here, we observed +1 fragment ions extending to 2000 m/z. This unusual distribution of ions is likely due to the fixed positive charge of the S-methylmethionine.
There are multiple lines of evidence for assigning the product as S-methylmethionine based on intact mass, fragment sequence coverage, chromatographic shifts toward more hydrophilic retention times, and unusual behavior in fragmentation. The precedent for this type of reaction and sulfonium ions in general is readily apparent in the production of SAM, which is itself a sulfonium ion carrying a fixed positive charge. Thus, the proposed reaction that produces S-methylmethionine would start and end with a sulfonium ion (Fig. 4C).
Likely because of the unique chemical nature of the sulfonium moiety of the S-methylmethionine, we did not observe fragments that include modified Met73, using collision-induced dissociation (CID) or high-energy collision dissociation (HCD). It is not clear why the proposed sulfonium structure significantly changes the characteristic of the peptide. However, it is our conjecture that it interferes with the mobile proton associated with HCD/CID dissociation method (35).
Discussion
A wide variety of SAM-dependent protein methylation occurs on nitrogen atoms of the protein N terminus and the side chains of arginine, lysine, glutamine, and histidine and the carboxylate oxygen atoms of the side chains of glutamate and aspartate, as well as the protein C terminus (36). In addition to two previous reports (21, 22), we show here that the sulfur of methionine, in the context of an actin variant, can be methylated by the SET-domain MTase SETD3 in vitro. We note that previous examples of protein methionine methylation are generated by either an unidentified methyltransferase in E. gracilis (21) or LaeA of A. nidulans, a member of the seven-strand MTase family (22) that is structurally distinct from the SET-family MTases (37).
Identification and localization of methylation on methionine required use of ETD, a technique that is infrequently used in bottom-up proteomics. Thus, it is unlikely that previous proteomic analyses would have localized methylation to methionine residues. Future whole proteome profiling with ETD targeting methionine methylation may yield a host of novel modifications and better explain previously obfuscated mechanisms. Further, by including methionine methylation as a potential post-translational modification, one may prevent incorrect assignments. The unusual fragmentation characteristics observed may be due to the nature of the peptide, and thus further efforts are required for assigning a pattern for methionine methylation.
In addition to methionine–S-methylation, protein cysteine–S-methylation has been reported in a zinc-coordinating cysteine in a human zinc finger protein, methylation of which is catalyzed by a pathogenic bacterial secreted effector protein NleE and results in loss of zinc binding (38). The Escherichia coli DNA repair protein Ada automethylates by transferring the aberrant methyl group from damaged DNA (methyl phosphotriester lesion or mutagenic O6-methylguanine lesion) to one of its own cysteine residues (39). Methionine and cysteine are the two common sulfur-containing amino acids, the former being hydrophobic and the latter being highly nucleophilic. Thiomethylation switches these properties of the two amino acids (Fig. 1): S-methylmethionine contains a highly reactive and charged methylsulfonium group, whereas S-methylcysteine mostly results in elimination of cysteine's intended function (39–42). The charged methylmethionine had an effect on the pI of cytochrome c from E. gracilis (21). We note that a human S-methyltransferase (Thiopurine methyltransferase) discovered several decades ago (43, 44) that is an important drug-metabolizing enzyme active on thiopurine drugs (45, 46) has no known natural substrates even now. Two human enzymes have been reported to have automethylation on cysteine residues (42, 47).
The hydrophobicity of methionine within substrate peptides is well-accommodated in aromatic-rich active sites, and methionine replacement of the target His73 leads to an impressive increase in binding affinity for SETD3, as well as for the peptides with methionine substitution of target lysine of histone modifying enzymes (LSD1 and PRC2), effectively inhibiting the enzymatic activities on their native substrates. We show that placement of methionine properly in the active site—close distance to and in line with the incoming methyl group (Fig. 4B)—allows SETD3 and its mutant variants to methylate methionine. In humans, many of the 55 SET domain–containing proteins (48) have not been fully characterized, and further study could potentially reveal that some of them can selectively methylate methionine in a protein context.
Materials and methods
Purification of SETD3
Recombinant human SETD3 (pXC2003) and its mutants, N255F (pXC2092), N255A (pXC2093), and N255V (pXC2094), expressed in E. coli BL21(DE3) CodonPlusTM cells (Stratagene) as GST fusions, were purified as described (27, 28). Briefly, a Bio-Rad NGCTM system was used to conduct three-column chromatography of GSH-Sepharose, HiTrap Q-HP, and Superdex 200. PreScisson Protease (purified in-house) was used to remove the GST tag, leaving five additional N-terminal residues (GPLGS). The concentrated SETD3 proteins at ∼25–35 mg/ml were kept at −80 °C in storage buffer of 20 mm Tris (pH 8.0), 200 mm NaCl, 5% glycerol, and 0.5 mm tris (2-carboxyethyl) phosphine. The actin peptides His73 (residues 66–80), Lys73 (residues 66–88), and Met73 (residues 66–88) containing varied amino acid at residue 73 (histidine, lysine, or methionine) were synthesized by GenScript.
Crystallography
A ternary complex of SETD3, Met73 peptide (residues 66–88), and SAH was formed by mixing the three components in a molar ratio of 1:4:5 (protein:Met73:SAH) and incubated on ice for 1 h. For the ternary complex of SETD3 mutant N255V, Met73 (residues 66–88) peptide, and SAM, the components were mixed at a molar ratio of 1:5:10 (protein:Met73:SAM) and incubated on ice overnight. Sitting drops (0.4-μl) at ∼20 °C were set up by an Art Robbins Gryphon Crystallization Robot containing the ternary complexes (∼14 mg/ml or ∼0.2 mm) plus the well solutions of 0.2 m ammonium acetate, 0.1 m sodium citrate tribasic dihydrate, pH 5.6, and 30% (w/v) PEG 4000.
Cryoprotectant buffer containing 25% (v/v) ethylene glycol and the crystallization solution was used to flash-freeze single crystals. The SER-CAT Beamline 22ID of the Advanced Photon Source at Argonne National Laboratory was used remotely to collect X-ray diffraction data, processed with HKL2000 (49). The previously solved structure of human SETD3 (PDB code 6MBJ) was used as the search model in the molecular replacement of PHENIX PHASER module (50, 51). Randomly chosen 5% reflections were used for the validation of the Rfree value throughout the process of structure refinement with PHENIX REFINE (52). Manual building and corrections of structural models between refinement rounds were carried out using COOT (53, 54). The PDB validation server was used to ratify the quality of the final refined structure. The molecular graphic program PyMOL (Schrödinger, LLC) was used to generate figure panels.
Methylation assay
Methylation assays on full-length actin were performed as previously described (27). Briefly, batch-purified GST-SETD3 (2 μm) and GST–β-actin (1 μm) were reacted with 2 μCi of [3H]SAM (American Radiolabeled Chemicals) in 50 mm Tris (pH 8.0), 20 mm KCl, 5 mm MgCl2, and 10% glycerol (v/v). The reactions were incubated overnight at 30 °C, resolved by SDS-PAGE, and analyzed by autoradiography and Coomassie Blue staining.
For peptide substrates, the Promega bioluminescence assay (MTase-GloTM) (55) was used to measure the methylation reaction by-product SAH, which is converted into ATP in a two-step reaction, and ATP is detected through a luciferase reaction. A low-volume 384-well plate with each well containing an aliquot of 5 μl of reaction mixture was used to measure luminescence signal by a Synergy 4 multimode microplate reader (BioTek).
Optimal pH analysis for actin M73 peptide
A reaction mixture contained 3.6 μm SETD3, 50 μm Met73 peptide, 40 μm SAM, 50 mm NaCl, 0.1 mg/ml BSA, 1 mm DTT, and varied pH with 20 mm citric acid, bis-tris propane (below pH 9) or 20 mm glycine/NaOH (above pH 9). The 20 mm citric acid, bis-tris propane is a mixture of 10 mm citric acid and 10 mm Bis-Tris propane. The reaction was performed at 37 °C for 1 h, terminated by the addition of TFA to a final concentration of 0.4% (v/v), and then diluted 4-fold with 20 mm Tris-HCl, pH 8.0, 50 mm NaCl, 0.1 mg/ml BSA, and 1 mm DTT (the reaction buffer).
Time-course measurement
The reaction mixture contained 0.36 μm enzyme (SETD3, N255A, and N255V), 20 μm Met73 peptide (residues 66–88), and 40 μm SAM in the reaction buffer. The reaction was performed at 37 °C and was terminated at the indicated time with the addition of 0.1% (v/v) TFA to 10-μl reaction aliquot.
Single-turnover kinetics
The reaction mixture of 20 μl contained SETD3 ([WT] = 1.44 μm, [N255A/V] = 0.72 μm), 40 μm SAM, and varying concentration of Met73 peptide in the reaction buffer. The reactions were carried out at 37 °C for 1 h. The apparent kcat/Km value was estimated from the slope of low substrate concentration data point where the reaction rate varies linearly with substrate concentration.
Activity of SET proteins on methionine-containing peptides
The reaction mixture contained 2 μm enzyme of each methyltransferase (SETD3, SET7, PRDM9, and GLP), 40 μm SAM, and 10 μm respective peptide in the reaction buffer and left overnight at room temperature. Actin peptide Met73 (residues 66–88), histone H3 peptides Lys4 (residues 1–24), Met4 (residues 1–18), Lys9 (residues 1–24), and Met9 (residues 1–21) were used in the assays. In addition, reaction products using 15 μm [methyl-3H]SAM (PerkinElmer NET155V001MC) were visualized by fluorography. An aliquot of 5 μl of the reaction samples were run on an 18% SDS-PAGE gel. The gel was soaked in EN3HANCE solution (PerkinElmer) for 30 min, dried, and exposed to Hyperfilm (GE Healthcare) at −80 °C for 72 h.
Inhibition of SETD3 activity on His73 by Met73
A reaction mixture contained 0.18 μm SETD3, 40 μm SAM, and incubated with increasing concentration of Met73 peptide (0–100 μm) in the reaction buffer for 30 min. The addition of 20 μm His73 peptide started the reaction for 20 min at room temperature.
Isothermal titration calorimetry
A MicroCal PEAQ-ITC automated system (Malvern Instrument Ltd.) was operated at 25 °C. SETD3 (20 μm in 20 mm Tris-HCl, pH 8.0, 50 mm NaCl, 5% glycerol, and 0.5 mm tris (2-carboxyethyl) phosphine) was maintained in the sample cell. The actin peptides (400 μm) in the identical buffer were injected into the cell by a syringe under continuous stirring (750 rpm) with the reference power set as 8 µcal/s. The volume of each injection was 2 µl with a fixed duration time of 4 s, and the spacing time between the injections was 250 s to achieve equilibrium. Binding constants were calculated by fitting the data using a single–binding site model by the ITC data analysis program supplied by the manufacturer. We also measured GLP (20 μm in the reaction buffer) binding to histone H3 peptide Met9 (residues 1–21) (200 μm) in the presence of SAH or SAM (50 μm).
HPLC–MS/MS
Actin peptides were desalted, dried, and resuspended with 2% acetonitrile and 0.1% formic acid, resulting in a final concentration of 10 μm. HPLC was performed on a Dionex U-3000 Pro-flow system. A 20-min gradient was performed from 1 to 35% of 98% acetonitrile and 0.1% formic acid. An aliquot of sample (1 μl) was loaded onto a C-18 column with a 100-μm-diameter and 10-cm-long (Waters XBridgeTM C18 3.5 μm 130 Å).
Acquisition was performed by a Thermo Scientific Orbitrap Fusion Lumos. A static spray voltage of 2000 V and an ion-transfer tube temperature of 320 °C were set for the source. MS1 was performed by the Orbitrap in positive mode, with the ETD parameters listed in Table S2.
Two m/z peaks were targeted for MS2 fragmentation: 714.8508 (for the unmodified actin peptide) and 718.3551 (for the modified actin peptide) both are +4 ions. Two different ETD parameters were used (Table S2), resulting in two different fragment spectra per duty cycle per precursor.
Multiple other MS settings were tested, most notably CID and HCD for MS2 fragmentation and MS3 using ETD and CID/HCD. These attempts did not yield sufficient coverage or interpretable data and are not presented. Raw files were initially analyzed by our in-house software and confirmed with ProSight 4.0 (Thermo Scientific). Final analysis for presented spectra was performed manually, and all annotations were manually validated.
Data availability
The X-ray structures (coordinates and structure factor files) have been submitted to the PDB under accession numbers 6WK1 (SETD3-Met73-SAH) and 6WK2 (N225V-Met73-SAM). The MS proteomics data have been submitted to the ProteomeXchange via the PRIDE database (accession PXD019203).
Supplementary Material
Acknowledgments
We thank Da Jia for purification of SET7 used for activity assays, members of the Cheng laboratory for discussion, and Dr. Or Gozani (Stanford University) for support under National Institutes of Health Grant R01 GM133051.
This article contains supporting information.
Author contributions—S. D., M. V. H., A. P., and A. W. W. data curation; S.D., J. R. H., C. B.W., X. Z., and N. L. Y. formal analysis; S.D. and X. Z. investigation; X. Z. and X. C. supervision; N. L. Y. methodology; N. L. Y., A. W. W., and X. C. writing-review and editing; A. W. W. and X. C. conceptualization; X. C. funding acquisition; X. C. writing-original draft.
Funding and additional information—This work was supported by National Institutes of Health Grants R35GM134744 and CPRIT RR160029 (to X. C.) and R01GM133051 (to A. W. W.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare no competing interests with the contents of this article.
- MTase
- methyltransferase
- SAM
- S-adenosyl-l-methionine
- SAH
- S-adenosyl-l-homocysteine
- PDB
- Protein Data Bank
- ETD
- electron transfer dissociation
- CID
- collision-induced dissociation
- HCD
- high-energy collision dissociation
- GST
- glutathione S-transferase
- ITC
- isothermal titration calorimetry.
References
- 1. Paik W. K., Paik D. C., and Kim S. (2007) Historical review: the field of protein methylation. Trends Biochem. Sci. 32, 146–152 10.1016/j.tibs.2007.01.006 [DOI] [PubMed] [Google Scholar]
- 2. Shechter D. (2019) Introduction to the multi-author review on methylation in cellular physiology. Cell. Mol. Life Sci. 76, 2871–2872 10.1007/s00018-019-03141-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tolsma T. O., and Hansen J. C. (2019) Post-translational modifications and chromatin dynamics. Essays Biochem. 63, 89–96 10.1042/EBC20180067 [DOI] [PubMed] [Google Scholar]
- 4. Zhao Z., and Shilatifard A. (2019) Epigenetic modifications of histones in cancer. Genome Biol. 20, 245 10.1186/s13059-019-1870-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Allis C. D., and Jenuwein T. (2016) The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 17, 487–500 10.1038/nrg.2016.59 [DOI] [PubMed] [Google Scholar]
- 6. Butler J. S., Koutelou E., Schibler A. C., and Dent S. Y. (2012) Histone-modifying enzymes: regulators of developmental decisions and drivers of human disease. Epigenomics 4, 163–177 10.2217/epi.12.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schwartzentruber J., Korshunov A., Liu X. Y., Jones D. T., Pfaff E., Jacob K., Sturm D., Fontebasso A. M., Quang D. A., Tönjes M., Hovestadt V., Albrecht S., Kool M., Nantel A., Konermann C., et al. (2012) Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482, 226–231 10.1038/nature10833 [DOI] [PubMed] [Google Scholar]
- 8. Wu G., Broniscer A., McEachron T. A., Lu C., Paugh B. S., Becksfort J., Qu C., Ding L., Huether R., Parker M., Zhang J., Gajjar A., Dyer M. A., Mullighan C. G., Gilbertson R. J., et al. (2012) Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 44, 251–253 10.1038/ng.1102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lowe B. R., Maxham L. A., Hamey J. J., Wilkins M. R., and Partridge J. F. (2019) Histone H3 mutations: an updated view of their role in chromatin deregulation and cancer. Cancers (Basel) 11, 660 10.3390/cancers11050660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shan C. M., Wang J., Xu K., Chen H., Yue J. X., Andrews S., Moresco J. J., Yates J. R., Nagy P. L., Tong L., and Jia S. (2016) A histone H3K9M mutation traps histone methyltransferase Clr4 to prevent heterochromatin spreading. eLife 5, e17903 10.7554/eLife.17903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jayaram H., Hoelper D., Jain S. U., Cantone N., Lundgren S. M., Poy F., Allis C. D., Cummings R., Bellon S., and Lewis P. W. (2016) S-Adenosyl methionine is necessary for inhibition of the methyltransferase G9a by the lysine 9 to methionine mutation on histone H3. Proc. Natl. Acad. Sci. U.S.A. 113, 6182–6187 10.1073/pnas.1605523113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Justin N., Zhang Y., Tarricone C., Martin S. R., Chen S., Underwood E., De Marco V., Haire L. F., Walker P. A., Reinberg D., Wilson J. R., and Gamblin S. J. (2016) Structural basis of oncogenic histone H3K27M inhibition of human polycomb repressive complex 2. Nat. Commun. 7, 11316 10.1038/ncomms11316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yang S., Zheng X., Lu C., Li G. M., Allis C. D., and Li H. (2016) Molecular basis for oncohistone H3 recognition by SETD2 methyltransferase. Genes Dev. 30, 1611–1616 10.1101/gad.284323.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Forneris F., Binda C., Adamo A., Battaglioli E., and Mattevi A. (2007) Structural basis of LSD1-CoREST selectivity in histone H3 recognition. J. Biol. Chem. 282, 20070–20074 10.1074/jbc.C700100200 [DOI] [PubMed] [Google Scholar]
- 15. Hübner J. M., Müller T., Papageorgiou D. N., Mauermann M., Krijgsveld J., Russell R. B., Ellison D. W., Pfister S. M., Pajtler K. W., and Kool M. (2019) EZHIP/CXorf67 mimics K27M mutated oncohistones and functions as an intrinsic inhibitor of PRC2 function in aggressive posterior fossa ependymoma. Neuro. Oncol. 21, 878–889 10.1093/neuonc/noz058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jain S. U., Do T. J., Lund P. J., Rashoff A. Q., Diehl K. L., Cieslik M., Bajic A., Juretic N., Deshmukh S., Venneti S., Muir T. W., Garcia B. A., Jabado N., and Lewis P. W. (2019) PFA ependymoma-associated protein EZHIP inhibits PRC2 activity through a H3 K27M-like mechanism. Nat. Commun. 10, 2146 10.1038/s41467-019-09981-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Piunti A., Smith E. R., Morgan M. A. J., Ugarenko M., Khaltyan N., Helmin K. A., Ryan C. A., Murray D. C., Rickels R. A., Yilmaz B. D., Rendleman E. J., Savas J. N., Singer B. D., Bulun S. E., and Shilatifard A. (2019) CATACOMB: An endogenous inducible gene that antagonizes H3K27 methylation activity of Polycomb repressive complex 2 via an H3K27M-like mechanism. Sci. Adv. 5, eaax2887 10.1126/sciadv.aax2887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ragazzini R., Pérez-Palacios R., Baymaz I. H., Diop S., Ancelin K., Zielinski D., Michaud A., Givelet M., Borsos M., Aflaki S., Legoix P., Jansen P., Servant N., Torres-Padilla M. E., Bourc'his D., et al. (2019) EZHIP constrains polycomb repressive complex 2 activity in germ cells. Nat. Commun. 10, 3858 10.1038/s41467-019-11800-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cantoni G. L. (1952) The nature of the active methyl donor formed enzymatically from l-methionine and adenosinetriphosphate. J. Am. Chem. Soc. 74, 2942–2943 10.1021/ja01131a519 [DOI] [Google Scholar]
- 20. Grunau J. A., and Swiader J. M. (1991) Chromatographic quantitation of free amino acids: S-methylmethionine, methionine and lysine in corn. J. Plant Nutr. 14, 653–662 10.1080/01904169109364232 [DOI] [Google Scholar]
- 21. Farooqui J. Z., Tuck M., and Paik W. K. (1985) Purification and characterization of enzymes from Euglena gracilis that methylate methionine and arginine residues of cytochrome c. J. Biol. Chem. 260, 537–545 [PubMed] [Google Scholar]
- 22. Patananan A. N., Palmer J. M., Garvey G. S., Keller N. P., and Clarke S. G. (2013) A novel automethylation reaction in the Aspergillus nidulans LaeA protein generates S-methylmethionine. J. Biol. Chem. 288, 14032–14045 10.1074/jbc.M113.465765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kwiatkowski S., Seliga A. K., Vertommen D., Terreri M., Ishikawa T., Grabowska I., Tiebe M., Teleman A. A., Jagielski A. K., Veiga-da-Cunha M., and Drozak J. (2018) SETD3 protein is the actin-specific histidine N-methyltransferase. eLife 7, e37921 10.7554/eLife.37921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wilkinson A. W., Diep J., Dai S., Liu S., Ooi Y. S., Song D., Li T. M., Horton J. R., Zhang X., Liu C., Trivedi D. V., Ruppel K. M., Vilches-Moure J. G., Casey K. M., Mak J., et al. (2019) SETD3 is an actin histidine methyltransferase that prevents primary dystocia. Nature 565, 372–376 10.1038/s41586-018-0821-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Diep J., Ooi Y. S., Wilkinson A. W., Peters C. E., Foy E., Johnson J. R., Zengel J., Ding S., Weng K. F., Laufman O., Jang G., Xu J., Young T., Verschueren E., Kobluk K. J., et al. (2019) Enterovirus pathogenesis requires the host methyltransferase SETD3. Nat. Microbiol. 4, 2523–2537 10.1038/s41564-019-0551-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Guo Q., Liao S., Kwiatkowski S., Tomaka W., Yu H., Wu G., Tu X., Min J., Drozak J., and Xu C. (2019) Structural insights into SETD3-mediated histidine methylation on beta-actin. eLife 8, e43676 10.7554/eLife.43676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dai S., Horton J. R., Woodcock C. B., Wilkinson A. W., Zhang X., Gozani O., and Cheng X. (2019) Structural basis for the target specificity of actin histidine methyltransferase SETD3. Nat. Commun. 10, 3541 10.1038/s41467-019-11554-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dai S., Horton J. R., Wilkinson A. W., Gozani O., Zhang X., and Cheng X. (2020) An engineered variant of SETD3 methyltransferase alters target specificity from histidine to lysine methylation. J. Biol. Chem. 295, 2582–2589 10.1074/jbc.RA119.012319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Horowitz S., and Trievel R. C. (2012) Carbon-oxygen hydrogen bonding in biological structure and function. J. Biol. Chem. 287, 41576–41582 10.1074/jbc.R112.418574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dao-Pin S., Anderson D. E., Baase W. A., Dahlquist F. W., and Matthews B. W. (1991) Structural and thermodynamic consequences of burying a charged residue within the hydrophobic core of T4 lysozyme. Biochemistry 30, 11521–11529 10.1021/bi00113a006 [DOI] [PubMed] [Google Scholar]
- 31. Wang H., Cao R., Xia L., Erdjument-Bromage H., Borchers C., Tempst P., and Zhang Y. (2001) Purification and functional characterization of a histone H3-lysine 4-specific methyltransferase. Mol. Cell 8, 1207–1217 10.1016/S1097-2765(01)00405-1 [DOI] [PubMed] [Google Scholar]
- 32. Mihola O., Trachtulec Z., Vlcek C., Schimenti J. C., and Forejt J. (2009) A mouse speciation gene encodes a meiotic histone H3 methyltransferase. Science 323, 373–375 10.1126/science.1163601 [DOI] [PubMed] [Google Scholar]
- 33. Tachibana M., Ueda J., Fukuda M., Takeda N., Ohta T., Iwanari H., Sakihama T., Kodama T., Hamakubo T., and Shinkai Y. (2005) Histone methyltransferases G9a and GLP form heteromeric complexes and are both crucial for methylation of euchromatin at H3-K9. Genes Dev. 19, 815–826 10.1101/gad.1284005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Koh-Stenta X., Poulsen A., Li R., Wee J. L., Kwek P. Z., Chew S. Y., Peng J., Wu L., Guccione E., Joy J., and Hill J. (2017) Discovery and characterisation of the automethylation properties of PRDM9. Biochem. J. 474, 971–982 10.1042/BCJ20161067 [DOI] [PubMed] [Google Scholar]
- 35. Wysocki V. H., Tsaprailis G., Smith L. L., and Breci L. A. (2000) Mobile and localized protons: a framework for understanding peptide dissociation. J. Mass Spectrom. 35, 1399–1406 [DOI] [PubMed] [Google Scholar]
- 36. Clarke S. (1993) Protein methylation. Curr. Opin. Cell Biol. 5, 977–983 10.1016/0955-0674(93)90080-a [DOI] [PubMed] [Google Scholar]
- 37. Schubert H. L., Blumenthal R. M., and Cheng X. (2003) Many paths to methyltransfer: a chronicle of convergence. Trends Biochem. Sci. 28, 329–335 10.1016/S0968-0004(03)00090-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang L., Ding X., Cui J., Xu H., Chen J., Gong Y. N., Hu L., Zhou Y., Ge J., Lu Q., Liu L., Chen S., and Shao F. (2011) Cysteine methylation disrupts ubiquitin-chain sensing in NF-κB activation. Nature 481, 204–208 10.1038/nature10690 [DOI] [PubMed] [Google Scholar]
- 39. He C., Hus J. C., Sun L. J., Zhou P., Norman D. P., Dötsch V., Wei H., Gross J. D., Lane W. S., Wagner G., and Verdine G. L. (2005) A methylation-dependent electrostatic switch controls DNA repair and transcriptional activation by E. coli ada. Mol. Cell 20, 117–129 10.1016/j.molcel.2005.08.013 [DOI] [PubMed] [Google Scholar]
- 40. Szilák L., Finta C., Patthy A., Venetianer P., and Kiss A. (1994) Self-methylation of BspRI DNA-methyltransferase. Nucleic Acids Res. 22, 2876–2881 10.1093/nar/22.15.2876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hanck T., Schmidt S., and Fritz H. J. (1993) Sequence-specific and mechanism-based crosslinking of Dcm DNA cytosine-C5 methyltransferase of E. coli K-12 to synthetic oligonucleotides containing 5-fluoro-2′-deoxycytidine. Nucleic Acids Res. 21, 303–309 10.1093/nar/21.2.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Siddique A. N., Jurkowska R. Z., Jurkowski T. P., and Jeltsch A. (2011) Auto-methylation of the mouse DNA-(cytosine C5)-methyltransferase Dnmt3a at its active site cysteine residue. FEBS J. 278, 2055–2063 10.1111/j.1742-4658.2011.08121.x [DOI] [PubMed] [Google Scholar]
- 43. Weinshilboum R. M. (2006) Pharmacogenomics: catechol O-methyltransferase to thiopurine S-methyltransferase. Cell Mol. Neurobiol. 26, 539–561 10.1007/s10571-006-9095-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Weinshilboum R. M. (1992) Methylation pharmacogenetics: thiopurine methyltransferase as a model system. Xenobiotica 22, 1055–1071 10.3109/00498259209051860 [DOI] [PubMed] [Google Scholar]
- 45. Marinaki A. M., and Arenas-Hernandez M. (2020) Reducing risk in thiopurine therapy. Xenobiotica 50, 101–109 10.1080/00498254.2019.1688424 [DOI] [PubMed] [Google Scholar]
- 46. Asadov C., Aliyeva G., and Mustafayeva K. (2017) Thiopurine S-methyltransferase as a pharmacogenetic biomarker: significance of testing and review of major methods. Cardiovasc. Hematol. Agents Med. Chem. 15, 23–30 10.2174/1871525715666170529091921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Patel A., Vought V. E., Swatkoski S., Viggiano S., Howard B., Dharmarajan V., Monteith K. E., Kupakuwana G., Namitz K. E., Shinsky S. A., Cotter R. J., and Cosgrove M. S. (2014) Automethylation activities within the mixed lineage leukemia-1 (MLL1) core complex reveal evidence supporting a “two-active site” model for multiple histone H3 lysine 4 methylation. J. Biol. Chem. 289, 868–884 10.1074/jbc.M113.501064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Carlson S. M., and Gozani O. (2016) Nonhistone lysine methylation in the regulation of cancer pathways. Cold Spring Harb. Perspect. Med. 6, a026435 10.1101/cshperspect.a026435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Otwinowski Z., Borek D., Majewski W., and Minor W. (2003) Multiparametric scaling of diffraction intensities. Acta Crystallogr. A 59, 228–234 10.1107/s0108767303005488 [DOI] [PubMed] [Google Scholar]
- 50. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Storoni L. C., McCoy A. J., and Read R. J. (2004) Likelihood-enhanced fast rotation functions. Acta Crystallogr. D Biol. Crystallogr. 60, 432–438 10.1107/S0907444903028956 [DOI] [PubMed] [Google Scholar]
- 52. Headd J. J., Echols N., Afonine P. V., Grosse-Kunstleve R. W., Chen V. B., Moriarty N. W., Richardson D. C., Richardson J. S., and Adams P. D. (2012) Use of knowledge-based restraints in phenix.refine to improve macromolecular refinement at low resolution. Acta Crystallogr. D Biol. Crystallogr. 68, 381–390 10.1107/S0907444911047834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 54. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hsiao K., Zegzouti H., and Goueli S. A. (2016) Methyltransferase-Glo: a universal, bioluminescent and homogenous assay for monitoring all classes of methyltransferases. Epigenomics 8, 321–339 10.2217/epi.15.113 [DOI] [PubMed] [Google Scholar]
- 56. Pajares M. A., and Pérez-Sala D. (2006) Betaine homocysteine S-methyltransferase: just a regulator of homocysteine metabolism? Cell. Mol. Life Sci. 63, 2792–2803 10.1007/s00018-006-6249-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Szegedi S. S., Castro C. C., Koutmos M., and Garrow T. A. (2008) Betaine-homocysteine S-methyltransferase-2 is an S-methylmethionine-homocysteine methyltransferase. J. Biol. Chem. 283, 8939–8945 10.1074/jbc.M710449200 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The X-ray structures (coordinates and structure factor files) have been submitted to the PDB under accession numbers 6WK1 (SETD3-Met73-SAH) and 6WK2 (N225V-Met73-SAM). The MS proteomics data have been submitted to the ProteomeXchange via the PRIDE database (accession PXD019203).