

Abstract
The development of the dendritic arbor in pyramidal neurons is critical for neural circuit function. Here, we uncovered a pathway in which δ-catenin, a component of the cadherin–catenin cell adhesion complex, promotes coordination of growth among individual dendrites and engages the autophagy mechanism to sculpt the developing dendritic arbor. Using a rat primary neuron model, time-lapse imaging, immunohistochemistry, and confocal microscopy, we found that apical and basolateral dendrites are coordinately sculpted during development. Loss or knockdown of δ-catenin uncoupled this coordination, leading to retraction of the apical dendrite without altering basolateral dendrite dynamics. Autophagy is a key cellular pathway that allows degradation of cellular components. We observed that the impairment of the dendritic arbor resulting from δ-catenin knockdown could be reversed by knockdown of autophagy-related 7 (ATG7), a component of the autophagy machinery. We propose that δ-catenin regulates the dendritic arbor by coordinating the dynamics of individual dendrites and that the autophagy mechanism may be leveraged by δ-catenin and other effectors to sculpt the developing dendritic arbor. Our findings have implications for the management of neurological disorders, such as autism and intellectual disability, that are characterized by dendritic aberrations.
Keywords: δ-catenin, hippocampal neurons, dendrite, autophagy, dendritic arbor, catenin delta 2 (CTNND2), autism, neuronal development, ATG7, neuron, development, hippocampus
In pyramidal neurons of the hippocampus and cortex, dendrites are the major sites of information input (1, 2). Pyramidal neurons have a distinct architecture with multiple dendrites and a single axon. In addition to axon-dendrite polarity, pyramidal neurons have dendritic polarity with distinct apical and basolateral dendrites, a feature that is critical for appropriate synaptic input (3–6). Dendrites have both active and passive properties that contribute to neuronal computation (7–14). The development of the dendritic arbor, with distinct apical and basolateral dendrites, is tightly orchestrated (1, 15) to allow the appropriate formation of neuronal circuits and networks. In primary neurons, apical-basolateral dendritic polarity is maintained and correlates with dendritic length (16–19), with the longest dendrite (primary) correlating with the apical and the second longest (secondary) correlating with the basolateral dendrite. Our knowledge of the mechanisms that guide the formation and maintenance of individual dendrites and the overall dendritic architecture is far from complete.
The dendritic arbor in pyramidal neurons is complex, both in vitro and in vivo, with multiple dendrites that originate in the cell body, each bearing multiple higher-order branches with distinct regions for synaptic input (1, 4,5, 20). For a developing neuron, it is critical that every single dendrite that originates in the cell body is correctly sculpted and generates an appropriate number of higher-order branches while maintaining overall arbor shape and connectivity (21). Given this, it is likely that molecular mechanisms exist that allow for interdendritic communication that drive overall dendritic patterning in a coordinated manner. However, the identity of such mechanisms remains unclear. Determining the mechanisms that regulate coordinated growth of multiple dendrites within the same neuron has implications for our ability to understand the underpinnings of neural circuit wiring in the healthy and diseased brain.
Mutations in CTNND2 have been identified in autism (22, 23), and loss of CTNND2 is associated with intellectual disability (24, 25). CTNND2 encodes δ-catenin, a component of the cadherin-catenin cell adhesion complex that binds to cadherin at its juxtamembrane region. Components of the cadherin-catenin cell adhesion complex, including δ-catenin, have been implicated in a variety of neuronal functions associated with dendritic and synaptic structure, function, and plasticity, indicating that they are key components of the molecular machinery underlying cognition (5, 26–28). Others and we have previously demonstrated a critical role for δ-catenin in sculpting the developing dendritic arbor (29–33). Whereas some molecular components that cooperate with δ-catenin in regulating the dendritic arbor have been identified, our knowledge of the mechanisms that allow δ-catenin to sculpt the developing dendritic arbor remains far from complete.
Autophagy is a key cellular pathway that allows removal and degradation of cellular components (34, 35), thus promoting normal cellular homeostasis (36). Macroautophagy allows for sequestration of substrates within an autophagosome that ultimately fuses with the lysosome within which protein degradation occurs. The majority of the autophagy proteins associate within a macro complex that include kinases and several ATG proteins that function in an orderly assembly mechanism. Dysregulation of autophagy has been identified and characterized in the context of neurodegenerative disorders (37–43). However, our understanding of the role of autophagy underlying normal neuronal developmental processes is incomplete (44–48). Autophagy is a critical regulator of synaptic density and architecture with roles in both presynaptic (49) and postsynaptic compartments and axons (50–56). Little is known, however, about the role of autophagy in dendritic development in pyramidal neurons. Here, we provide evidence to demonstrate that δ-catenin promotes coordinated development of the apical and basolateral dendrites in primary neurons and engages the autophagy mechanism to sculpt the developing dendritic arbor.
Results
Dendrite dynamics during development
During development, the dendritic arbor is sculpted by extension and retraction of the individual dendritic arbors. We took advantage of a primary rat neuronal cell culture system (57) to examine dendritic arbor dynamics during development. Rat primary hippocampal neurons were maintained in culture as described previously (57). Neurons were transfected with EGFP and imaged by live time lapse imaging at DIV 11, 14, and 19 to allow visualization of the dendritic tree (Fig. 1A). For these studies, we define the primary (apical) dendrite as the longest and the secondary (basolateral) dendrite as the second longest dendrite that originate in the cell body (16, 17, 19). We quantitated dendritic lengths of the primary and secondary dendrites separately as measures of dendritic extension. During the period between DIV 11 and DIV 14, both primary and secondary dendrites predominantly showed extension with little retraction (Fig. 1B). However, with advancing age, DIV 14–19, there was more of a balance between extension and retraction (Fig. 1C). Overall, during the time period between DIV 11 and 19, both primary and secondary dendrites had net extension (Fig. 1D) and similar levels of average extension (Fig. 1E). These data suggest that in this model system, DIV 11–14 represents a period of dynamic dendrite extension, whereas both extension and retraction are balanced at DIV 14–19, leading to overall dendrite extension. Further, primary and secondary dendrites show similar dynamics of extension and retraction, suggesting that they are coordinately sculpted.
Figure 1.
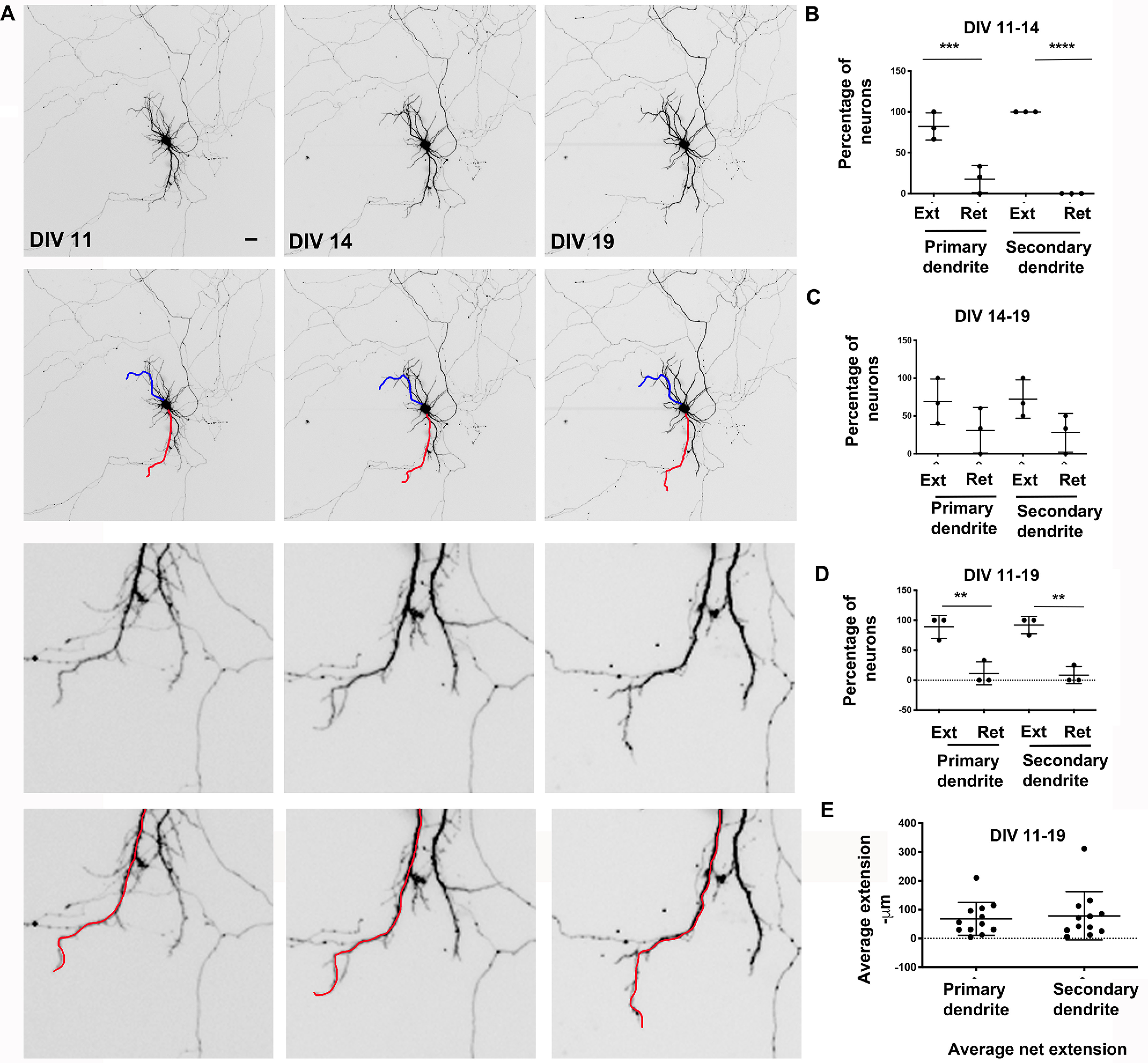
Dendrite dynamics during development in hippocampal neurons. A, representative images from time lapse imaging of rat hippocampal primary neurons in culture at the indicated time points. The red and blue lines overlay the primary and secondary dendrites, respectively. Bottom panels, magnified regions of the top panels. Scale bar, 12.5 μm. B, percentage of neurons with extension or retraction of primary and secondary dendrites (DIV 11–14). C, percentage of neurons with extension or retraction of primary and secondary dendrites (DIV 14–19). D, percentage of neurons with extension or retraction of primary and secondary dendrites (DIV 11–19). Data analysis by one-way ANOVA with Tukey's multiple-comparison test for B–D. *, p < 0.05; **, p < 0.005; ***, p < 0.0005. Data represent mean ± S.D. (error bars). E, average extension in microns of primary and secondary dendrites (DIV 11–19). Data analysis by unpaired t test assuming equal variances. *, p < 0.05; **, p < 0.005; ***, p < 0.0005 for C and D. Data represent mean ± S.D.
δ-Catenin-N-term mice have altered dendrite dynamics
The δ-catenin-N-term mouse (also referred to as the δ-catenin knockout mouse (58)) retains the N-terminal fragment of δ-catenin and is the only δ-catenin mouse model that is currently available. We have previously demonstrated that δ-catenin-N-term mice have reduced dendritic arbors (32). We examined the length of primary, secondary, tertiary, and quaternary dendrites in primary neurons from control (WT/WT) and mutant (δ-catenin-N-term/δ-catenin-N-term) mice at DIV 8 and DIV 16–17. These dendrites are all dendrites that originate in the cell body and were classified as primary, secondary, tertiary, or quaternary based on their lengths with the primary being the longest. These classifications only refer to the length of the main dendritic shaft and do not reflect dendritic branches that arise from the main shaft. At DIV 8, the length of the primary dendrites in mutant neurons was not significantly different from the control neurons. Surprisingly, the lengths of secondary, tertiary, and quaternary dendrites were all increased in the mutants compared with the control (Fig. 2, A and B). However, at DIV 16–17, this scenario was significantly different in that the length of the primary dendrite was significantly reduced in the mutant compared with the control, whereas none of the other dendrites showed any significant alterations in length (Fig. 2, C and D). Thus, the loss of δ-catenin leads to an uncoupling of the coordination between primary and secondary dendritic dynamics.
Figure 2.

Loss of δ-catenin in a mouse model leads to dendritic deficits during development. A, representative confocal images from primary hippocampal neurons from WT/WT and δ-catenin-N-term/δ-catenin-N-term mice at DIV 8 in vitro. Scale bar, 25 μm. B, dendrite lengths of primary, secondary, tertiary, and quarternary dendrites from primary hippocampal neurons from WT/WT and δ-catenin-N-term/δ-catenin-N-term mice at DIV 8 in vitro (data analysis by Student's t test; *, p < 0.05; **, p < 0.005; ***, p < 0.0005; data represent mean ± S.E. (error bars)). C, representative confocal images from primary hippocampal neurons from WT/WT and δ-catenin-N-term mice at DIV 16–17 in vitro. Scale bar, 25 μm. D, dendrite lengths of primary, secondary, tertiary, and quarternary dendrites from primary hippocampal neurons from WT/WT and δ-catenin-N-term/δ-catenin-N-term mice at DIV 16–17 in vitro. Data analysis was by Student's t test; *, p < 0.05; **, p < 0.005; ***, p < 0.0005. Data represent mean ± S.E.
Knockdown of δ-catenin leads to retraction of primary but not secondary dendrites
The reduction in the dendritic arbor with loss or knockdown of δ-catenin could be mediated by either a reduction in the extension or an increase in the retraction of dendrites. To examine which of these mechanisms is responsible, we examined the development of the dendritic arbor in primary neurons expressing vector or a previously validated (32, 59) shRNA to δ-catenin by time-lapse analysis (Fig. 3A). These studies were done in primary neurons from DIV 13 to DIV 17 with neurons imaged at DIV 13, 15, and 17. We examined the percentage of neurons with net extension or retraction of primary and secondary dendrites. Between DIV 13 and 15, the neurons expressing vector were balanced in terms of extension and retraction of primary dendrites (Fig. 3D). However, neurons expressing shRNA to δ-catenin had an increased level of retraction and reduced extension (Fig. 3D). Similar results were observed at DIV 15–17 (Fig. 3E). Consolidation of DIV 13–17 (Fig. 3F) data indicated that in neurons expressing δ-catenin shRNA, the percentage of neurons retracting primary dendrites was higher than control, whereas the percentage demonstrating extension was lower than neurons expressing vector (Fig. 3F). In these neurons, the average length of dendrite extended or retracted was not significantly different between neurons expressing vector or shRNA to δ-catenin (Fig. 3B). A similar analysis of secondary dendrites (Fig. 3, C, G, H, and I) indicated that knockdown of δ-catenin did not alter the extension or retraction of secondary dendrites compared with neurons expressing vector only. These studies indicate that the knockdown of δ-catenin leads to an increase in the percentage of neurons that retract the primary dendrite accompanied by a decrease in the percentage of neurons that extend primary dendrites. Interestingly, these effects are confined to primary dendrites, because secondary dendrites in neurons expressing vector or shRNA were not significantly different in extension and retraction. These data are consistent with the data in Fig. 2 (C and D) demonstrating that, at a comparable stage of development, the primary dendrites in δ-catenin-N-term mice are shorter compared with control, whereas the secondary dendrites are not affected. Taken together, these results indicate that the loss or knockdown of δ-catenin predominantly leads to a reduction in the length of the primary, but not secondary, dendrite by an active retraction mechanism. Further, these results are in agreement with data from the δ-catenin-N-term mice and suggest that loss or knockdown of δ-catenin uncouples the dynamics between the primary and secondary dendrites in developing neurons.
Figure 3.

Knockdown of δ-catenin leads to uncoupling of primary and secondary dendrite dynamics. A, representative images of individual neurons expressing vector or shRNA to δ-catenin at DIV 13 and DIV 17. Scale bar, 12.5 μm. B, average length of extension and retraction of primary dendrites in neurons expressing vector or shRNA to δ-catenin. C, average length of extension and retraction of secondary dendrites in neurons expressing vector or shRNA to δ-catenin. D, percentage of neurons with net extension and retraction of primary dendrites in neurons expressing vector or shRNA to δ-catenin at DIV 13–15. E, percentage of neurons with net extension and retraction of primary dendrites in neurons expressing vector or shRNA to δ-catenin at DIV 15–17. F, percentage of neurons with net extension and retraction of primary dendrites in neurons expressing vector or shRNA to δ-catenin at DIV 13–17; cumulative data from D (e.g. percentage of neurons with net extension and retraction of secondary dendrites in neurons expressing vector or shRNA to δ-catenin at DIV 13–15). H, percentage of neurons with net extension and retraction of secondary dendrites in neurons expressing vector or shRNA to δ-catenin at DIV 15–17. I, percentage of neurons with net extension and retraction of secondary dendrites in neurons expressing vector or shRNA to δ-catenin at DIV 13–17; cumulative data from G and H. Data analysis was by one-way ANOVA with Sidak's multiple-comparison test; *, p < 0.05; **, p < 0.005; ***, p < 0.0005. Data represent mean ± S.D. (error bars).
Expression of markers for autophagy during development in the hippocampus and cortex
The autophagy mechanism has been implicated in multiple aspects of neuronal development; however, little is known about its role in dendritic development. To begin to evaluate whether the autophagy pathway might be involved in dendritic arborization during development, we examined whether major mediators of the autophagy pathway were expressed in the hippocampus and cortex during development. We examined the expression of some components of the autophagy pathway, namely LC3-I/II, beclin, and ATG5. In the rat hippocampus, we could detect expression of LC3-I/II, beclin, and ATG5 at P5, P14, and P21 (Fig. 4A) coinciding with the periods of dendritic and synaptic development. These markers were also expressed in the adult as expected. We also examined the expression of these autophagy markers in primary rat hippocampal and cortical neurons in cultures at stages coinciding with dendritic and spine development. LC3-I was expressed at DIV 7, 14, and 21 in both cortical and hippocampal neurons (Fig. 4A), whereas the expression of LC3-II was more robust at DIV 14 and 21 in both groups of neurons. Whereas beclin was expressed at all three stages examined in both groups of neurons, we could detect little expression of ATG5 in DIV 7 neurons, but ATG5 was well-expressed at DIV 14 and 21. We also took advantage of GFP-LC3 as a marker for autophagosomes (60) to examine autophagy in primary rat neurons. Primary rat hippocampal neurons were transfected with GFP-LC3 and examined at DIV 7, 14, and 21 by confocal microscopy. The number of autophagosomes in the cell body was evaluated. Autophagosomes were detected in the cell body at all stages examined (Fig. 4, B and C). These data indicate that markers of autophagy are expressed in hippocampal and cortical neurons at stages coinciding with dendrite development.
Figure 4.

Expression of components of the autophagy pathway during time periods coinciding with dendrite and spine development. A, Western blots for LC3-I/II, beclin, and ATG5 in lysates from rat hippocampus or primary rat hippocampal or primary rat cortical neurons at the indicated time points. B, representative images of cell bodies from primary hippocampal neurons expressing GFP LC3 at the indicated ages. Scale bars, 5 μm (top) and 1 μm (bottom). C, quantitation of the number of GFP LC3 puncta in cell bodies from primary hippocampal neurons at the indicated time points. Data represent mean ± S.D. (error bars).
Knockdown of ATG7 leads to an increase in dendrite complexity
We chose to examine whether the autophagy pathway has a role in dendrite development. To this end, we examined the effects of knockdown of ATG7 on the dendritic arbor at time points coinciding with early dendrite development. Primary rat hippocampal neurons in culture were transfected with GFP-expressing plasmids encoding vector or ATG7 shRNA that has been previously validated (60) at DIV 4. The neurons were fixed at DIV 12, and neuronal morphology was examined by confocal microscopy (Fig. 5A). Neurons expressing shRNA to ATG7 had a significant increase in the total length of dendrites (Fig. 5B). This was accompanied by no change in the number of primary dendrites (Fig. 5C). Further, examination of the dendritic arbor by Sholl analysis (Fig. 5D) indicates a trend toward an increase in the number of intersections proximal to the cell body, but not distally, in neurons expressing shRNA to ATG7. Taken together, these data indicate that knockdown of ATG7 during the early phases of dendrite development enhances the dendritic arbor. We used a similar strategy to examine effects of knockdown of ATG7 at later time periods of dendrite development. Similar to the results obtained with knockdown of ATG7 at early phases of dendrite development, loss of ATG7 at later time points (DIV 12–18) (Fig. 5E) of development led to an increase in the total dendritic length (Fig. 5F) with no change in the number of primary dendrites (Fig. 5G). Sholl analysis demonstrated significant increases in the number of intersections at distal sites, likely reflecting reduced pruning at dendritic tips (Fig. 5H). We also examined densities of spines on primary dendrites within 60 μm of the cell body. In neurons expressing ATG7 shRNA, there was a trend toward increased spine density, although this did not reach significance (Fig. 5, I and J). Thus, knockdown of ATG7 leads to an increase in the dendritic arbor, both at early and late stages of dendrite development, with no significant effects on spine densities.
Figure 5.

Knockdown of ATG7 enhances the dendritic arbor. A, representative images from primary hippocampal neurons expressing vector or shRNA to ATG7 (DIV 4–12). Scale bar, 25 μm. B, total dendritic length from primary hippocampal neurons expressing vector or shRNA to ATG7 (DIV 4–12). C, total number of primary dendrites in primary hippocampal neurons expressing vector or shRNA to ATG7 (DIV 4–12). Data analysis by unpaired t test assuming equal variances. *, p < 0.05; **, p < 0.005; ***, p < 0.0005 for C and D. Data represent mean ± S.D. (error bars). D, Sholl analysis in primary hippocampal neurons expressing vector or shRNA to ATG7 (DIV 4–12). Data analysis by two-way ANOVA with Sidak's multiple-comparison test. *, p < 0.05; **, p < 0.005; ***, p < 0.0005. Data represent mean ± S.D. E, representative images from primary hippocampal neurons expressing vector or shRNA to ATG7 (DIV 11–18). Scale bar, 25 μm. F, total dendritic length from primary hippocampal neurons expressing vector or shRNA to ATG7 (DIV 11–18). G, total number of primary dendrites in primary hippocampal neurons expressing vector or shRNA to ATG7 (DIV 11–18). Data analysis was by unpaired t test assuming equal variances. *, p < 0.05; **, p < 0.005; ***, p < 0.0005 for C and D. Data represent mean ± S.D. H, Sholl analysis in primary hippocampal neurons expressing vector or shRNA to ATG7 (DIV 11–18). Data analysis by two-way ANOVA with Sidak's multiple-comparison test. *, p < 0.05; **, p < 0.005; ***, p < 0.0005. Data represent mean ± S.D. I, representative images of dendritic segments from primary hippocampal neurons expressing vector or ATG7 shRNA. Scale bar, 2 μm. J, average spine density from primary hippocampal neurons expressing vector or ATG7 shRNA. Data analysis was by unpaired t test assuming equal variances. NS, not significant.
Knockdown of ATG16L1 leads to increased dendrite complexity
We also examined the effects of knockdown of ATG16L1, another component of the autophagy pathway, on dendrite arborization. Primary rat neurons in culture were transfected with plasmids encoding vector or ATG16L1 shRNA (61) at DIV 12, and their morphology was examined by confocal microscopy at DIV 18. Similar to the results obtained with knockdown of ATG7, knockdown of ATG16L1 (Fig. 6A) led to an increase in total dendritic length (Fig. 6B), with no change in the number of primary dendrites (Fig. 6C). Sholl analysis indicates a trend toward increase at more distal dendritic terminals in neurons expressing the shRNA to Atg16L1 (Fig. 6D), similar to the data observed with ATG7 knockdown. We also examined the densities of spines as above in neurons expressing vector or shRNA to ATG16L1. In neurons expressing ATG16L1 shRNA, there was a trend toward a decrease in spine densities, although this did not reach significance. These results suggest that loss of ATG16L1 enhances dendrite arborization, similar to loss of ATG7, without affecting spine densities.
Figure 6.

Knockdown of ATG16L1 during later development enhances the dendritic arbor. A, representative images from primary hippocampal neurons expressing vector or shRNA to ATG16L1 (DIV 11–18). Scale bar, 25 μm. B, total dendritic length from primary hippocampal neurons expressing vector or shRNA to ATG16L1 (DIV 11–18). C, total number of primary dendrites in primary hippocampal neurons expressing vector or shRNA to ATG16L1 (DIV 11–18). Data analysis was by unpaired t test assuming equal variances. *, p < 0.05; **, p < 0.005; ***, p < 0.0005 for C and D. Data represent mean ± S.D. (error bars). D, Sholl analysis in primary hippocampal neurons expressing vector or shRNA to ATG16L1 (DIV 11–18). Data analysis by two-way ANOVA with Sidak's multiple-comparison test. *, p < 0.05; **, p < 0.005; ***, p < 0.0005. Data represent mean ± S.D. E, representative images of dendritic segments from primary hippocampal neurons expressing vector or ATG16L1 shRNA. Scale bar, 2 μm. F, average spine density in from primary hippocampal neurons expressing vector or ATG16L1 shRNA. Data analysis by unpaired t test assuming equal variances. NS, not significant.
Rescue of δ-catenin knockdown phenotype by concomitant knockdown of ATG7
We examined the ability of ATG7 knockdown to rescue the dendritic phenotype observed with knockdown of δ-catenin. Primary rat neurons in culture were transfected with vector or δ-catenin shRNA with or without vector and ATG7 shRNA. In neurons expressing the δ-catenin shRNA, the total dendritic length and number of dendritic end points were significantly lower than in neurons expressing vector (Fig. 7, A–C). Concurrent expression of the ATG7 shRNA with the δ-catenin shRNA significantly restored both the total dendritic length and number of dendritic end points. Under all of these conditions, the number of primary dendrites was not significantly affected (Fig. 7D). Similar results are observed by Sholl analysis (Fig. 7E). These data indicate that the compromise in the dendritic arbor observed with the knockdown of δ-catenin can be partially rescued by concomitant knockdown of ATG7, implicating a role for δ-catenin in engaging the autophagy pathway to sculpt the developing dendritic arbor.
Figure 7.

Knockdown of ATG7 partially rescues the δ-catenin dendrite knockdown phenotype. A, representative confocal images of neurons expressing the indicated constructs. Scale bar, 25 μm. B, total dendrite length from primary hippocampal neurons expressing the indicated constructs (DIV 11–18). C, total number of dendritic end points in primary hippocampal neurons expressing the indicated constructs (DIV 11–18). Data analysis was by one-way ANOVA with Sidak's multiple-comparison test. *, p < 0.05; **, p < 0.005; ***, p < 0.0005. Data represent mean ± S.D. (error bars). D, total number of primary dendrites in primary hippocampal neurons expressing the indicated constructs (DIV 11–18). Data analysis was by one-way ANOVA with Sidak's multiple-comparison test. *, p < 0.05; **, p < 0.005; ***, p < 0.0005. Data represent mean ± S.D. G, Sholl analysis in primary hippocampal neurons expressing the indicated constructs (DIV 11–18). Data analysis was by two-way ANOVA with Dunnet's multiple-comparison test; comparison with δ-cat shRNA + ATG7 vector. *, p < 0.05; **, p < 0.005; ***, p < 0.0005. Data represent mean ± S.D.
Discussion
Our studies provide evidence to make several significant conclusions on dendrite development including the following: 1) developing dendrites are dynamic; however, these dynamics are similar for both primary (apical) and secondary (basolateral) dendrites, suggesting that these two dendrites are coordinately sculpted; 2) loss or knockdown of δ-catenin leads to an uncoupling of the developmental dynamics between the primary and secondary dendrites; 3) there is a role for the autophagy mechanism in regulating dendrite arborization; and 4) there is a role for δ-catenin in engaging the autophagy mechanism to sculpt the developing dendritic arbor.
The development of the appropriate dendritic arbor is a key requirement for appropriate neural circuit formation and function. Developing dendrites are dynamic and are sculpted by both extension and retraction. Our previous studies that suggest that individual dendrites have some level of autonomous control over their growth (16). Our data suggest that there are additional cellular mechanisms in place to promote coordination of growth of individual dendritic arbors, and δ-catenin is a component of the underlying machinery. To our knowledge, our studies provide the first evidence that the apical (primary) and basolateral (secondary) dendrites show coordinated dynamics that can be uncoupled via a molecular mechanism. We do note that the significant reduction in the total dendritic length (Fig. 7) with loss or knockdown of δ-catenin is unlikely to be mediated solely by an effect of on the primary dendrite because the overall number of dendritic end points is also affected by loss or knockdown of δ-catenin. Published data (62, 63) suggest that there are cellular mechanisms that regulate dendrite branching at different levels, so it is possible that δ-catenin additionally regulates dendrite extension or retraction of the some of the higher-order dendritic branches during development. We recognize that the rescue of the δ-catenin dendrite phenotype by concurrent knockdown of ATG7 is not complete. These results are completely expected, because we anticipate that this is not the sole mechanism engaged by δ-catenin to regulate the dendritic arbor. Published studies indicate that δ-catenin also engages the small GTPases and the actin cytoskeleton to regulate the dendritic arbor (29, 33, 64), so we anticipate that there is some cross-talk between these pathways in sculpting the developing dendritic arbor. We also note that the gross levels of LC3 are not altered in the δ-catenin-N-term/δ-catenin-N-term mouse cortical lysates (data not shown), suggesting that loss of δ-catenin does not lead to a global alteration in the levels of components of the autophagy pathway, but the impacts are at the functional level.
Our data demonstrate a novel role for the macroautophagy pathway in regulating the sculpting of the dendritic arbor in hippocampal pyramidal neurons during development. Components of the autophagy pathway are expressed in the hippocampus and cortex at time points coincident with development. Knockdown of ATG7 or ATG16L1, key components of the pathway, promote an increase in the dendritic arbor. Taken together, these data provide the first evidence that the autophagy pathway is a key mechanism that contributes to the sculpting of the dendritic arbor during development, thus likely making critical contributions to neural circuit wiring in the developing brain. The role of autophagy in neurons has been predominantly characterized in the context of neurodegeneration (40, 65–68), although its roles in axonal morphogenesis and synaptic function are emerging (56, 69, 70). By providing evidence for a role for autophagy in dendrite sculpting, our data suggest that autophagy mechanisms in neurons may be highly relevant to the etiology of neurodevelopmental disorders. Several neurodevelopmental disorders associated with autism, intellectual disability, and related disorders are associated with dendritic aberrations (71), and our studies imply that autophagic mechanisms may be of interest in these devastating disorders.
Several components of the molecular machinery that underlie dendrite sculpting during development have been identified. Our data implicate the autophagy pathway as a novel mechanism for dendrite sculpting during development. We propose that inhibition of the autophagy pathway inhibits dendrite pruning rather than promoting dendrite extension. The sculpting of the dendritic arbor is governed by several extrinsic and intrinsic cues (15, 72), including growth factors, contact-mediated mechanisms, and neuronal activity–mediated mechanisms. It is tempting to speculate that at least some of these signaling pathways converge on the autophagy pathway to regulate the dendritic arbor during development. The autophagy pathway regulates multiple aspects of cellular homeostasis. To allow autophagy to specifically regulate dendrite arborization, we propose the existence of specific upstream effectors, in addition to δ-catenin, which transduce signals that allow the engagement of the autophagy pathway specifically for dendrite morphogenesis. Such mechanisms would allow specific control of dendrite sculpting in response to specific extracellular and intracellular cues and could be either positive or negative. For example, we have previously demonstrated that morphine-mediated regulation of spine and synapse density in the hippocampus is mediated via an autophagic mechanism (60) through a series of intermediate effectors, including reactive oxygen species and endoplasmic reticulum stress pathways. Future efforts aimed at identifying upstream effectors might provide key insights into the key pathways that take advantage of the autophagy machinery to elicit sculpting of the developing dendritic arbor.
In neurons, the autophagy pathway has been predominantly studied in the context of neurodegeneration (39). Autophagic aberrations have been linked to neurodegeneration in a variety of human disorders, including ALS (73–75), polyglutamine disorders (38), and Lafora's disease (76). Further, mutations in genes with links to autophagy mechanisms have been identified or implicated in neurodegenerative disorders, including SENDA (77). Several of these disorders are accompanied by behavioral deficits. By identifying a role for autophagy in modulating the dendritic architecture, our studies suggest that some of the behavioral impairments observed in these disorders may have their roots in aberrant dendritogenesis and consequently disrupted neural connectivity.
Our data indicate that knockdown of ATG7 (DIV 12–18) does not significantly alter spine densities, although there is a trend toward an increase in spine densities that does not reach statistical significance. These results are in contrast to published reports that indicate a role for ATG7 in spine pruning in the cortex and hippocampus (78). Specifically, in vivo data indicated that synaptic densities are higher in the cortex of the ATG7 null animals at P29–30 and in primary hippocampal neurons (DIV 21). However, our studies are in agreement with our previous studies that demonstrate that knockdown of ATG7 at DIV 7–21 does not alter excitatory synaptic density significantly but does show a trend toward increase in neurons expressing ATG7 shRNA (60). The basis of these differences is not entirely clear; however, there are several possibilities. One possibility is that there is a small window during which spine pruning is highly regulated and our time points of analysis encompass a much larger window. Another possibility is that there may be non-cell-autonomous roles for autophagy in spine pruning. Our studies induce knockdown of ATG7 in individual neurons that network with WT neurons, whereas published studies were performed in neurons from the ATG7 flox/CamK2 Cre or in ATG7 floxed neurons in which Cre was expressed using a lentivirus, allowing for significantly higher levels of infection than our technique. In this case, the ATG7 knockdown neurons are in a network with other ATG7 knockdown neurons. A third possibility is that, because shRNA knockdowns are rarely complete, there is sufficient ATG7 in the knockdown neurons to function in spine morphogenesis. Further studies are necessary to tease apart these possibilities. Our data also indicate that knockdown of ATG16L1 does not alter spine density, but in contrast to the ATG7 knockdown, knockdown of ATG16L1 shows a trend toward decrease in spine density that does not reach statistical significance. These results suggest that perhaps different components of the autophagy pathway may function differently in spine morphogenesis in the developing hippocampus. We have previously observed that loss or knockdown of δ-catenin leads to an increase in the density of spines (79, 80). Based on the data demonstrating that knockdown of ATG7 or 16L1 does not alter the spine density, it is unlikely that δ-catenin cooperates with the autophagy pathway to regulate the density of spines.
Our results support the idea that δ-catenin and autophagy are critical components of the molecular machinery that regulates the dendritic arbor during development. Several neurodevelopmental disorders are associated with aberrations in dendritic and spine architecture (32, 81, 82). In addition to δ-catenin, we anticipate that other upstream factors that engage autophagy to promote dendrite development are likely to be of significance to neurodevelopmental disorders. Identifying these upstream factors and elucidating the mechanisms by which they link to the autophagy pathway to regulate dendrite development in the developing hippocampus will have major implications for our understanding of neural circuit development in health and disease.
Several questions remain, including the molecular mechanisms by which δ-catenin engages the autophagy mechanism to sculpt dendrites and whether the autophagy mechanism is engaged by other known effectors of dendrite development to sculpt the dendritic arbor, making it a more universal mechanism. Whereas answering these questions will require considerable effort and will form the focus of our future investigation, we anticipate that our studies can more acutely lead to clinical translational studies that examine the beneficial effects of knockdown of autophagy components on restoring the dendritic arbor in mouse models of disease that have a compromise in the dendritic arbor. Given the toxicity associated with complete loss of autophagy, these studies need to be performed during a short developmental time window in a neuron type–specific manner. If such studies are successful, they hold great promise for our ability to target neurological disorders with dendritic aberrations.
Experimental procedures
Rat tissue collection and primary rat neuron cultures
Hippocampal tissue was collected from rats at indicated ages. Primary rat hippocampal and cortical neurons were isolated from embryonic day 18 rats and maintained in culture as described previously (57, 83). All manipulations were performed in accordance with animal protocols approved by the institutional animal care and use committee at the University of Nebraska Medical Center. Primary neurons were maintained in media without araC treatment for the indicated times as described previously. Transfections were performed at DIV indicated using Lipofectamine 2000 (Thermo Fisher Scientific). Embryos obtained from a single mother (8–12 embryos) were pooled to obtain neurons for one experiment (n = 1).
Plasmids
Vector and shRNA to ATG7 (60) and ATG 16L1 have been described and validated previously. The hairpin sequence for the ATG16L1 is CCGGCAATGTGTAATGAGTGGACATCTCGAGATGTCCACTCATTACACATTGTTTTTTG. The plasmids were kind gifts from Dr. Ana Maria Cuervo (Albert Einstein College of Medicine). GFP LC3 plasmid was a kind gift from Dr. Howard Fox (University of Nebraska Medical Center).
Antibodies
The following primary antibodies were used: LC3 (1:3000), beclin (1:1000), ATG5 (1:2000), and β-tubulin (1:100).
Time lapse imaging
These studies were performed at the Eppley Cancer Center Molecular Biology Core/High-throughput screening facility at the University of Nebraska Medical Center using a ×20 objective.
Immunocytochemistry
Primary neurons in culture on coverslips were transfected as above and fixed in 4% paraformaldehyde, 4% sucrose. Neurons were permeabilized in a 0.1% Triton X-100/PBS solution for 10 min, washed, blocked with 10% BSA in PBS, incubated with primary antibody overnight, washed, incubated with fluorophore-conjugated secondary antibody for 1–2 h at room temperature, washed, treated with 4′,6-diamidino-2-phenylindole, and mounted using AntiFade.
Western blotting
Tissue samples were collected and placed in radioimmune precipitation assay buffer containing 1% Triton X-100, 1% SDS, 50 mm Tris, pH 7.4, 100 mm NaCl, 5 mm EDTA, 5 mm EGTA, 5 mm protease inhibitors, and 5 mm phosphatase inhibitors. Tissue samples were sonicated for 5 s until solid matter was dissociated. Insoluble material was pelleted at 15,000 rpm for 15 min at 4 °C in a tabletop centrifuge. The supernatant was collected and boiled at 95 °C for 5 min in Laemmli buffer.
Cells were collected by adding the radioimmune precipitation assay lysis buffer solution and by using a cell scraper and syringe to dissociate whole cells. Lysates were spun at 15,000 rpm for 15 min at 4 °C in a tabletop centrifuge. The supernatant was collected, and Laemmli buffer was added.
The samples were run on a 15% SDS-polyacrylamide gel and transferred to a polyvinylidene difluoride membrane for 2.5 h at 100 V. The membrane was blocked with 5% milk with TBS and incubated with primary antibody overnight at 4 °C. After washing, the membrane was incubated with secondary antibody conjugated with horseradish peroxidase for 1 h at room temperature. The blots were washed again and then imaged using ECL or Dura substrates.
Confocal microscopy
Confocal images were obtained using a Zeiss LSM 700 inverted scope. Z-stacks of neurons were used to create two-dimensional maximum projection images used to visualize GFP-LC3 puncta within the cell body. Images were taken using the ×40 objective at ×3 zoom. Neurons imaged to measure dendritic length were taken using the ×40 objective.
Data collection/analysis
Quantitative data were gathered on the LC3 puncta counts by visually observing max projections of Z-stacks taken from the confocal ×40 objective with ×3 zoom and counting only within the region of the cell body. 10 neurons per coverslip were counted for each time point: DIV 7, DIV 15, and DIV 21 from three independent cultures.
Data on dendritic length for transfected neurons was measured using ImageJ and tracing branching. Axons were excluded. 10–15 neurons per coverslip were analyzed from 3–5 independent experiments. Statistical analysis was performed as indicated in the figure legends.
Sholl analysis
Sholl analysis was performed using an ImageJ plugin to obtain parameters described in the figures.
Spine analysis
Images were taken on an inverted Zeiss LSM 700 confocal microscope. Z-stacks were obtained using the ×63 objective at ×3 zoom, 1024 × 1024 pixels. Dendrite length was measured using ImageJ line tool. Spines were counted manually by a nonblinded observer. Spine density was calculated as spines/μm.
Numbers for experimental analysis
All data were obtained from three or more independent experiments unless otherwise indicated: Fig. 1, total of 12–13 neurons from three independent experiments (for B–D, data from an individual experiment were averaged to obtain n = 1, and averaged data from three independent experiments are shown); Fig. 2, 145–150 total neurons for each condition from three animals of each genotype; Fig. 3, 19–26 neurons for each condition (for D, G, E, H, F, and I, data from an individual experiment were averaged to obtain n = 1, and averaged data from three independent experiments are shown); Fig. 4 (for A, tissue from three animals for each time point/age and primary neurons from 3–4 independent cultures for each DIV; for B and C, n = 31–34 neurons for each condition); Fig. 5 (for A–D, 39–42 neurons for each condition; E–H, 44–45 neurons for each condition; I and J, 31–38 neurons); Fig. 6 (for A–D, 31 neurons for each condition; for E and F, 37–41 neurons for each condition); Fig. 7 (for A, B, D, and E, 48–56 neurons; for C, 36–44 neurons for each condition).
Statistical analysis
For two-group comparisons, Student's t test was used, and p < 0.05 was considered significant. For multiple groups, data were analyzed using one-way or two-way ANOVA as indicated in the figure legends. No statistical method was used to predetermine sample size. Sample size was based on the literature.
Data availability
All data are contained within the article. Original data sets are available upon request to Jyothi Arikkath (Jyothi.arikkath@howard.edu).
Author contributions—C. L., E. S., E. J. S., L. Y., T. R. C., Y. C., and J. A. investigation; C. L. and J. A. methodology; C. L., E. J. S., S. B., S. J. B., and J. A. writing-review and editing; E. S., E. J. S., N. W. D., L. Y., T. R. C., Y. C., and J. A. formal analysis; N. W. D., L. Y., T. R. C., Y. C., and J. A. data curation; S. B., S. J. B., and J. A. supervision; S. B., S. J. B., and J. A. funding acquisition; J. A. conceptualization; J. A. resources; J. A. validation; J. A. visualization; J. A. writing-original draft; J. A. project administration.
Funding and additional information—This work was supported by Startup funds from the Munroe-Meyer Institute and grants from the Alzheimer's association, the Nebraska Research Initiative, an Institutional Development Award (IDeA) under NIGMS, National Institutes of Health, Grant 5P20GM103471-10 (to C. L., E. S., and J. A.), Nebraska EPSCoR Grant EPS-1004094 (to J. A.), RO3 from National Institutes of Health Grant 1R03MH110726-01 (to J. A.), and the Edna Ittner Pediatric Research Fund. L. Y. and E. S. were supported in part by a graduate student fellowship from the University of Nebraska Medical Center. This work was also supported by NIA, National Institutes of Health, Grant AG031158 (to N. W. D., T. R. C., and S. J. B.) and NIMH, National Institutes of Health, Grant MH106425 (to S. J. B.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- DIV
- days in vitro
- P
- postnatal day
- ANOVA
- analysis of variance.
References
- 1. Spruston N. (2008) Pyramidal neurons: dendritic structure and synaptic integration. Nat. Rev. Neurosci. 9, 206–221 10.1038/nrn2286 [DOI] [PubMed] [Google Scholar]
- 2. Harnett M. T., Makara J. K., Spruston N., Kath W. L., and Magee J. C. (2012) Synaptic amplification by dendritic spines enhances input cooperativity. Nature 491, 599–602 10.1038/nature11554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bloss E. B., Cembrowski M. S., Karsh B., Colonell J., Fetter R. D., and Spruston N. (2016) Structured dendritic inhibition supports branch-selective integration in CA1 pyramidal cells. Neuron 89, 1016–1030 10.1016/j.neuron.2016.01.029 [DOI] [PubMed] [Google Scholar]
- 4. Kupferman J. V., Basu J., Russo M. J., Guevarra J., Cheung S. K., and Siegelbaum S. A. (2014) Reelin signaling specifies the molecular identity of the pyramidal neuron distal dendritic compartment. Cell 158, 1335–1347 10.1016/j.cell.2014.07.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Basu R., Duan X., Taylor M. R., Martin E. A., Muralidhar S., Wang Y., Gangi-Wellman L., Das S. C., Yamagata M., West P. J., Sanes J. R., and Williams M. E. (2017) Heterophilic type II cadherins are required for high-magnitude synaptic potentiation in the hippocampus. Neuron 96, 160–176.e8 10.1016/j.neuron.2017.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yamawaki N., Li X., Lambot L., Ren L. Y., Radulovic J., and Shepherd G. M. G. (2019) Long-range inhibitory intersection of a retrosplenial thalamocortical circuit by apical tuft-targeting CA1 neurons. Nat. Neurosci. 22, 618–626 10.1038/s41593-019-0355-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Branco T., and Häusser M. (2011) Synaptic integration gradients in single cortical pyramidal cell dendrites. Neuron 69, 885–892 10.1016/j.neuron.2011.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schmidt-Hieber C., Toleikyte G., Aitchison L., Roth A., Clark B. A., Branco T., and Häusser M. (2017) Active dendritic integration as a mechanism for robust and precise grid cell firing. Nat. Neurosci. 20, 1114–1121 10.1038/nn.4582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Makara J. K., and Magee J. C. (2013) Variable dendritic integration in hippocampal CA3 pyramidal neurons. Neuron 80, 1438–1450 10.1016/j.neuron.2013.10.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Branco T., and Häusser M. (2010) The single dendritic branch as a fundamental functional unit in the nervous system. Curr. Opin. Neurobiol. 20, 494–502 10.1016/j.conb.2010.07.009 [DOI] [PubMed] [Google Scholar]
- 11. Stuart G. J., and Spruston N. (2015) Dendritic integration: 60 years of progress. Nat. Neurosci. 18, 1713–1721 10.1038/nn.4157 [DOI] [PubMed] [Google Scholar]
- 12. Shimell J. J., Shah B. S., Cain S. M., Thouta S., Kuhlmann N., Tatarnikov I., Jovellar D. B., Brigidi G. S., Kass J., Milnerwood A. J., Snutch T. P., and Bamji S. X. (2019) The X-linked intellectual disability gene Zdhhc9 is essential for dendrite outgrowth and inhibitory synapse formation. Cell Rep. 29, 2422–2437e2428 10.1016/j.celrep.2019.10.065 [DOI] [PubMed] [Google Scholar]
- 13. Sheffield M. E., and Dombeck D. A. (2019) Dendritic mechanisms of hippocampal place field formation. Curr. Opin. Neurobiol. 54, 1–11 10.1016/j.conb.2018.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Payeur A., Béïque J. C., and Naud R. (2019) Classes of dendritic information processing. Curr. Opin. Neurobiol. 58, 78–85 10.1016/j.conb.2019.07.006 [DOI] [PubMed] [Google Scholar]
- 15. Arikkath J. (2012) Molecular mechanisms of dendrite morphogenesis. Front. Cell Neurosci. 6, 61 10.3389/fncel.2012.00061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yuan Y., Seong E., Yuan L., Singh D., and Arikkath J. (2015) Differential regulation of apical-basolateral dendrite outgrowth by activity in hippocampal neurons. Front. Cell Neurosci. 9, 314 10.3389/fncel.2015.00314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wu Y. K., Fujishima K., and Kengaku M. (2015) Differentiation of apical and basal dendrites in pyramidal cells and granule cells in dissociated hippocampal cultures. PLoS ONE 10, e0118482 10.1371/journal.pone.0118482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Horton A. C., Yi J. J., and Ehlers M. D. (2006) Cell type-specific dendritic polarity in the absence of spatially organized external cues. Brain Cell Biol. 35, 29–38 10.1007/s11068-006-9003-y [DOI] [PubMed] [Google Scholar]
- 19. Horton A. C., Rácz B., Monson E. E., Lin A. L., Weinberg R. J., and Ehlers M. D. (2005) Polarized secretory trafficking directs cargo for asymmetric dendrite growth and morphogenesis. Neuron 48, 757–771 10.1016/j.neuron.2005.11.005 [DOI] [PubMed] [Google Scholar]
- 20. Biever A., Donlin-Asp P. G., and Schuman E. M. (2019) Local translation in neuronal processes. Curr. Opin. Neurobiol. 57, 141–148 10.1016/j.conb.2019.02.008 [DOI] [PubMed] [Google Scholar]
- 21. Duman J. G., Mulherkar S., Tu Y. K., Erikson K. C., Tzeng C. P., Mavratsas V. C., Ho T. S., and Tolias K. F. (2019) The adhesion-GPCR BAI1 shapes dendritic arbors via Bcr-mediated RhoA activation causing late growth arrest. Elife 8, 10.7554/eLife.47566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Turner T. N., Sharma K., Oh E. C., Liu Y. P., Collins R. L., Sosa M. X., Auer D. R., Brand H., Sanders S. J., Moreno-De-Luca D., Pihur V., Plona T., Pike K., Soppet D. R., Smith M. W., et al. (2015) Loss of δ-catenin function in severe autism. Nature 520, 51–56 10.1038/nature14186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lu Q., Aguilar B. J., Li M., Jiang Y., and Chen Y. H. (2016) Genetic alterations of δ-catenin/NPRAP/Neurojungin (CTNND2): functional implications in complex human diseases. Hum. Genet. 135, 1107–1116 10.1007/s00439-016-1705-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Medina M., Marinescu R. C., Overhauser J., and Kosik K. S. (2000) Hemizygosity of δ-catenin (CTNND2) is associated with severe mental retardation in cri-du-chat syndrome. Genomics 63, 157–164 10.1006/geno.1999.6090 [DOI] [PubMed] [Google Scholar]
- 25. Belcaro C., Dipresa S., Morini G., Pecile V., Skabar A., and Fabretto A. (2015) CTNND2 deletion and intellectual disability. Gene 565, 146–149 10.1016/j.gene.2015.03.054 [DOI] [PubMed] [Google Scholar]
- 26. Bian W. J., Miao W. Y., He S. J., Qiu Z., and Yu X. (2015) Coordinated spine pruning and maturation mediated by inter-spine competition for cadherin/catenin complexes. Cell 162, 808–822 10.1016/j.cell.2015.07.018 [DOI] [PubMed] [Google Scholar]
- 27. Li M. Y., Miao W. Y., Wu Q. Z., He S. J., Yan G., Yang Y., Liu J. J., Taketo M. M., and Yu X. (2017) A critical role of presynaptic cadherin/catenin/p140Cap complexes in stabilizing spines and functional synapses in the neocortex. Neuron 94, 1155–1172.e8 10.1016/j.neuron.2017.05.022 [DOI] [PubMed] [Google Scholar]
- 28. Elia L. P., Yamamoto M., Zang K., and Reichardt L. F. (2006) p120 catenin regulates dendritic spine and synapse development through Rho-family GTPases and cadherins. Neuron 51, 43–56 10.1016/j.neuron.2006.05.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gilbert J., and Man H. Y. (2016) The X-linked autism protein KIAA2022/KIDLIA regulates neurite outgrowth via N-cadherin and δ-catenin signaling. eNeuro 3, ENEURO.0238-16.2016 10.1523/ENEURO.0238-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kim H., Han J. R., Park J., Oh M., James S. E., Chang S., Lu Q., Lee K. Y., Ki H., Song W. J., and Kim K. (2008) δ-Catenin-induced dendritic morphogenesis: an essential role of p190RhoGEF interaction through Akt1-mediated phosphorylation. J. Biol. Chem. 283, 977–987 10.1074/jbc.M707158200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Matter C., Pribadi M., Liu X., and Trachtenberg J. T. (2009) δ-Catenin is required for the maintenance of neural structure and function in mature cortex in vivo. Neuron 64, 320–327 10.1016/j.neuron.2009.09.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Arikkath J., Israely I., Tao Y., Mei L., Liu X., and Reichardt L. F. (2008) Erbin controls dendritic morphogenesis by regulating localization of δ-catenin. J. Neurosci. 28, 7047–7056 10.1523/JNEUROSCI.0451-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Martinez M. C., Ochiishi T., Majewski M., and Kosik K. S. (2003) Dual regulation of neuronal morphogenesis by a δ-catenin-cortactin complex and Rho. J. Cell Biol. 162, 99–111 10.1083/jcb.200211025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bento C. F., Renna M., Ghislat G., Puri C., Ashkenazi A., Vicinanza M., Menzies F. M., and Rubinsztein D. C. (2016) Mammalian autophagy: how does it work? Annu. Rev. Biochem. 85, 685–713 10.1146/annurev-biochem-060815-014556 [DOI] [PubMed] [Google Scholar]
- 35. Mizushima N., Levine B., Cuervo A. M., and Klionsky D. J. (2008) Autophagy fights disease through cellular self-digestion. Nature 451, 1069–1075 10.1038/nature06639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tasset I., and Cuervo A. M. (2016) Role of chaperone-mediated autophagy in metabolism. FEBS J 283, 2403–2413 10.1111/febs.13677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wong E., and Cuervo A. M. (2010) Autophagy gone awry in neurodegenerative diseases. Nat. Neurosci. 13, 805–811 10.1038/nn.2575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ashkenazi A., Bento C. F., Ricketts T., Vicinanza M., Siddiqi F., Pavel M., Squitieri F., Hardenberg M. C., Imarisio S., Menzies F. M., and Rubinsztein D. C. (2017) Polyglutamine tracts regulate beclin 1-dependent autophagy. Nature 545, 108–111 10.1038/nature22078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Menzies F. M., Fleming A., and Rubinsztein D. C. (2015) Compromised autophagy and neurodegenerative diseases. Nat. Rev. Neurosci. 16, 345–357 10.1038/nrn3961 [DOI] [PubMed] [Google Scholar]
- 40. Menzies F. M., Fleming A., Caricasole A., Bento C. F., Andrews S. P., Ashkenazi A., Füllgrabe J., Jackson A., Jimenez Sanchez M., Karabiyik C., Licitra F., Lopez Ramirez A., Pavel M., Puri C., Renna M., et al. (2017) Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron 93, 1015–1034 10.1016/j.neuron.2017.01.022 [DOI] [PubMed] [Google Scholar]
- 41. Wong Y. C., and Holzbaur E. L. (2015) Autophagosome dynamics in neurodegeneration at a glance. J. Cell Sci. 128, 1259–1267 10.1242/jcs.161216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yang Y., Coleman M., Zhang L., Zheng X., and Yue Z. (2013) Autophagy in axonal and dendritic degeneration. Trends Neurosci. 36, 418–428 10.1016/j.tins.2013.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Martinez-Vicente M., Talloczy Z., Wong E., Tang G., Koga H., Kaushik S., de Vries R., Arias E., Harris S., Sulzer D., and Cuervo A. M. (2010) Cargo recognition failure is responsible for inefficient autophagy in Huntington's disease. Nat. Neurosci. 13, 567–576 10.1038/nn.2528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Maday S., and Holzbaur E. L. (2016) Compartment-specific regulation of autophagy in primary neurons. J. Neurosci. 36, 5933–5945 10.1523/JNEUROSCI.4401-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Maday S., Wallace K. E., and Holzbaur E. L. (2012) Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J. Cell Biol. 196, 407–417 10.1083/jcb.201106120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Maday S., and Holzbaur E. L. (2012) Autophagosome assembly and cargo capture in the distal axon. Autophagy 8, 858–860 10.4161/auto.20055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kononenko N. L., Claßen G. A., Kuijpers M., Puchkov D., Maritzen T., Tempes A., Malik A. R., Skalecka A., Bera S., Jaworski J., and Haucke V. (2017) Retrograde transport of TrkB-containing autophagosomes via the adaptor AP-2 mediates neuronal complexity and prevents neurodegeneration. Nat. Commun. 8, 14819 10.1038/ncomms14819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Okerlund N. D., Schneider K., Leal-Ortiz S., Montenegro-Venegas C., Kim S. A., Garner L. C., Waites C. L., Gundelfinger E. D., Reimer R. J., and Garner C. C. (2018) Bassoon controls presynaptic autophagy through Atg5. Neuron 97, 727 10.1016/j.neuron.2018.01.010 [DOI] [PubMed] [Google Scholar]
- 49. Hernandez D., Torres C. A., Setlik W., Cebrián C., Mosharov E. V., Tang G., Cheng H. C., Kholodilov N., Yarygina O., Burke R. E., Gershon M., and Sulzer D. (2012) Regulation of presynaptic neurotransmission by macroautophagy. Neuron 74, 277–284 10.1016/j.neuron.2012.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Maday S., and Holzbaur E. L. (2014) Autophagosome biogenesis in primary neurons follows an ordered and spatially regulated pathway. Dev. Cell 30, 71–85 10.1016/j.devcel.2014.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang T., Martin S., Papadopulos A., Harper C. B., Mavlyutov T. A., Niranjan D., Glass N. R., Cooper-White J. J., Sibarita J. B., Choquet D., Davletov B., and Meunier F. A. (2015) Control of autophagosome axonal retrograde flux by presynaptic activity unveiled using botulinum neurotoxin type A. J. Neurosci. 35, 6179–6194 10.1523/JNEUROSCI.3757-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Vijayan V., and Verstreken P. (2017) Autophagy in the presynaptic compartment in health and disease. J. Cell Biol. 216, 1895–1906 10.1083/jcb.201611113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Okerlund N. D., Schneider K., Leal-Ortiz S., Montenegro-Venegas C., Kim S. A., Garner L. C., Waites C. L., Gundelfinger E. D., Reimer R. J., and Garner C. C. (2017) Bassoon controls presynaptic autophagy through Atg5. Neuron 93, 897–913.e7 10.1016/j.neuron.2017.01.026 [DOI] [PubMed] [Google Scholar]
- 54. Wakatsuki S., Tokunaga S., Shibata M., and Araki T. (2017) GSK3B-mediated phosphorylation of MCL1 regulates axonal autophagy to promote Wallerian degeneration. J. Cell Biol. 216, 477–493 10.1083/jcb.201606020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Soukup S. F., Kuenen S., Vanhauwaert R., Manetsberger J., Hernández-Díaz S., Swerts J., Schoovaerts N., Vilain S., Gounko N. V., Vints K., Geens A., De Strooper B., and Verstreken P. (2016) A LRRK2-dependent EndophilinA phosphoswitch is critical for macroautophagy at presynaptic terminals. Neuron 92, 829–844 10.1016/j.neuron.2016.09.037 [DOI] [PubMed] [Google Scholar]
- 56. Ban B. K., Jun M. H., Ryu H. H., Jang D. J., Ahmad S. T., and Lee J. A. (2013) Autophagy negatively regulates early axon growth in cortical neurons. Mol. Cell Biol. 33, 3907–3919 10.1128/MCB.00627-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Beaudoin G. M. 3rd, Lee S. H., Singh D., Yuan Y., Ng Y. G., Reichardt L. F., and Arikkath J. (2012) Culturing pyramidal neurons from the early postnatal mouse hippocampus and cortex. Nat. Protoc. 7, 1741–1754 10.1038/nprot.2012.099 [DOI] [PubMed] [Google Scholar]
- 58. Israely I., Costa R. M., Xie C. W., Silva A. J., Kosik K. S., and Liu X. (2004) Deletion of the neuron-specific protein δ-catenin leads to severe cognitive and synaptic dysfunction. Curr. Biol. 14, 1657–1663 10.1016/j.cub.2004.08.065 [DOI] [PubMed] [Google Scholar]
- 59. Brigidi G. S., Sun Y., Beccano-Kelly D., Pitman K., Mobasser M., Borgland S. L., Milnerwood A. J., and Bamji S. X. (2014) Palmitoylation of δ-catenin by DHHC5 mediates activity-induced synapse plasticity. Nat. Neurosci. 17, 522–532 10.1038/nn.3657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Cai Y., Yang L., Hu G., Chen X., Niu F., Yuan L., Liu H., Xiong H., Arikkath J., and Buch S. (2016) Regulation of morphine-induced synaptic alterations: role of oxidative stress, ER stress, and autophagy. J. Cell Biol. 215, 245–258 10.1083/jcb.201605065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cai Y., Arikkath J., Yang L., Guo M. L., Periyasamy P., and Buch S. (2016) Interplay of endoplasmic reticulum stress and autophagy in neurodegenerative disorders. Autophagy 12, 225–244 10.1080/15548627.2015.1121360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Patel M. V., Swiatkowski P., Kwon M., Rodriguez A. R., Campagno K., and Firestein B. L. (2018) A novel short isoform of cytosolic PSD-95 interactor (cypin) regulates neuronal development. Mol. Neurobiol. 55, 6269–6281 10.1007/s12035-017-0849-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. O'Neill K. M., Kwon M., Donohue K. E., and Firestein B. L. (2017) Distinct effects on the dendritic arbor occur by microbead versus bath administration of brain-derived neurotrophic factor. Cell Mol. Life Sci. 74, 4369–4385 10.1007/s00018-017-2589-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Abu-Elneel K., Ochiishi T., Medina M., Remedi M., Gastaldi L., Caceres A., and Kosik K. S. (2008) A δ-catenin signaling pathway leading to dendritic protrusions. J. Biol. Chem. 283, 32781–32791 10.1074/jbc.M804688200 [DOI] [PubMed] [Google Scholar]
- 65. Yue Z., Horton A., Bravin M., DeJager P. L., Selimi F., and Heintz N. (2002) A novel protein complex linking the δ2 glutamate receptor and autophagy: implications for neurodegeneration in lurcher mice. Neuron 35, 921–933 10.1016/S0896-6273(02)00861-9 [DOI] [PubMed] [Google Scholar]
- 66. Komatsu M., Waguri S., Chiba T., Murata S., Iwata J., Tanida I., Ueno T., Koike M., Uchiyama Y., Kominami E., and Tanaka K. (2006) Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441, 880–884 10.1038/nature04723 [DOI] [PubMed] [Google Scholar]
- 67. Pandey U. B., Nie Z., Batlevi Y., McCray B. A., Ritson G. P., Nedelsky N. B., Schwartz S. L., DiProspero N. A., Knight M. A., Schuldiner O., Padmanabhan R., Hild M., Berry D. L., Garza D., Hubbert C. C., et al. (2007) HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 447, 859–863 10.1038/nature05853 [DOI] [PubMed] [Google Scholar]
- 68. Ivankovic D., Drew J., Lesept F., White I. J., Lopez Domenech G., Tooze S. A., and Kittler J. T. (2020) Axonal autophagosome maturation defect through failure of ATG9A sorting underpins pathology in AP-4 deficiency syndrome. Autophagy 16, 391–407 10.1080/15548627.2019.1615302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Tomoda T., Yang K., and Sawa A. (2019) Neuronal autophagy in synaptic functions and psychiatric disorders. Biol. Psychiatry 87, 787–796 10.1016/j.biopsych.2019.07.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Hoffmann S., Orlando M., Andrzejak E., Bruns C., Trimbuch T., Rosenmund C., Garner C. C., and Ackermann F. (2019) Light-activated ROS production induces synaptic autophagy. J. Neurosci. 39, 2163–2183 10.1523/JNEUROSCI.1317-18.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kulkarni V. A., and Firestein B. L. (2012) The dendritic tree and brain disorders. Mol. Cell Neurosci. 50, 10–20 10.1016/j.mcn.2012.03.005 [DOI] [PubMed] [Google Scholar]
- 72. Lefebvre J. L., Sanes J. R., and Kay J. N. (2015) Development of dendritic form and function. Annu. Rev. Cell Dev. Biol. 31, 741–777 10.1146/annurev-cellbio-100913-013020 [DOI] [PubMed] [Google Scholar]
- 73. Gao F. B., Almeida S., and Lopez-Gonzalez R. (2017) Dysregulated molecular pathways in amyotrophic lateral sclerosis-frontotemporal dementia spectrum disorder. EMBO J. 36, 2931–2950 10.15252/embj.201797568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ramesh N., and Pandey U. B. (2017) Autophagy dysregulation in ALS: when protein aggregates get out of hand. Front. Mol. Neurosci. 10, 263 10.3389/fnmol.2017.00263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Nassif M., Woehlbier U., and Manque P. A. (2017) The enigmatic role of C9ORF72 in autophagy. Front. Neurosci. 11, 442 10.3389/fnins.2017.00442, 10.3389/fnhum.2017.00442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Knecht E., Aguado C., Sarkar S., Korolchuk V. I., Criado-García O., Vernia S., Boya P., Sanz P., Rodríguez de Córdoba S., and Rubinsztein D. C. (2010) Impaired autophagy in Lafora disease. Autophagy 6, 991–993 10.4161/auto.6.7.13308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Saitsu H., Nishimura T., Muramatsu K., Kodera H., Kumada S., Sugai K., Kasai-Yoshida E., Sawaura N., Nishida H., Hoshino A., Ryujin F., Yoshioka S., Nishiyama K., Kondo Y., Tsurusaki Y., et al. (2013) De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat. Genet. 45, 445–449449e441 10.1038/ng.2562 [DOI] [PubMed] [Google Scholar]
- 78. Tang G., Gudsnuk K., Kuo S. H., Cotrina M. L., Rosoklija G., Sosunov A., Sonders M. S., Kanter E., Castagna C., Yamamoto A., Yue Z., Arancio O., Peterson B. S., Champagne F., Dwork A. J., et al. (2014) Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 83, 1131–1143 10.1016/j.neuron.2014.07.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Yuan L., Seong E., Beuscher J. L., and Arikkath J. (2015) δ-Catenin regulates spine architecture via cadherin and PDZ-dependent interactions. J. Biol. Chem. 290, 10947–10957 10.1074/jbc.M114.632679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Arikkath J., Peng I. F., Ng Y. G., Israely I., Liu X., Ullian E. M., and Reichardt L. F. (2009) δ-Catenin regulates spine and synapse morphogenesis and function in hippocampal neurons during development. J. Neurosci. 29, 5435–5442 10.1523/JNEUROSCI.0835-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Yadav S., Oses-Prieto J. A., Peters C. J., Zhou J., Pleasure S. J., Burlingame A. L., Jan L. Y., and Jan Y. N. (2017) TAOK2 kinase mediates PSD95 stability and dendritic spine maturation through Septin7 phosphorylation. Neuron 93, 379–393 10.1016/j.neuron.2016.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Dang T., Duan W. Y., Yu B., Tong D. L., Cheng C., Zhang Y. F., Wu W., Ye K., Zhang W. X., Wu M., Wu B. B., An Y., Qiu Z. L., and Wu B. L. (2018) Autism-associated Dyrk1a truncation mutants impair neuronal dendritic and spine growth and interfere with postnatal cortical development. Mol. Psychiatry 23, 747–758 10.1038/mp.2016.253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Yuan L., Singh D., Buescher J. L., and Arikkath J. (2018) A role for proteolytic regulation of δ-catenin in remodeling a subpopulation of dendritic spines in the rodent brain. J. Biol. Chem. 293, 11625–11638 10.1074/jbc.RA118.001966 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data are contained within the article. Original data sets are available upon request to Jyothi Arikkath (Jyothi.arikkath@howard.edu).