

Abstract
Infection with the Gram-negative, microaerophilic bacterium Helicobacter pylori induces an inflammatory response and oxidative DNA damage in gastric epithelial cells that can lead to gastric cancer (GC). However, the underlying pathogenic mechanism is largely unclear. Here, we report that the suppression of Nei-like DNA glycosylase 2 (NEIL2), a mammalian DNA glycosylase that specifically removes oxidized bases, is one mechanism through which H. pylori infection may fuel the accumulation of DNA damage leading to GC. Using cultured cell lines, gastric biopsy specimens, primary cells, and human enteroid-derived monolayers from healthy human stomach, we show that H. pylori infection greatly reduces NEIL2 expression. The H. pylori infection-induced downregulation of NEIL2 was specific, as Campylobacter jejuni had no such effect. Using gastric organoids isolated from the murine stomach in coculture experiments with live bacteria mimicking the infected stomach lining, we found that H. pylori infection is associated with the production of various inflammatory cytokines. This response was more pronounced in Neil2 knockout (KO) mouse cells than in WT cells, suggesting that NEIL2 suppresses inflammation under physiological conditions. Notably, the H. pylori-infected Neil2-KO murine stomach exhibited more DNA damage than the WT. Furthermore, H. pylori-infected Neil2-KO mice had greater inflammation and more epithelial cell damage. Computational analysis of gene expression profiles of DNA glycosylases in gastric specimens linked the reduced Neil2 level to GC progression. Our results suggest that NEIL2 downregulation is a plausible mechanism by which H. pylori infection impairs DNA damage repair, amplifies the inflammatory response, and initiates GC.
Keywords: Helicobacter pylori, DNA damage, base excision repair, gastric cancer, bacterial infection, inflammation, Nei-like DNA glycosylase 2 (NEIL2), cytokine response, oxidative stress, gastric organoid, infection, DNA repair, stem cells, enteroids, Nei-like DNA glycosylase
The World Health Organization and the American Cancer Society (ACS) estimate that gastric cancer (GC) is the fourth most prevalent cancer worldwide and the third leading cause of death in the United States (1). Recognized risk factors for developing GCs include genetic predisposition (variants in DNA repair genes have been reported as key factors in GC susceptibility and survival) (2, 3), dietary factors (for example, high salt intake), obesity, smoking, and Helicobacter pylori infection (4, 5). H. pylori is a Gram-negative, microaerophilic bacterium that infects the gastric mucosa, causing gastritis and often leading to devastating complications, such as peptic ulcers and GC. H. pylori infection increases the risk of developing GCs by 5–6-fold (6). Treatment and eventual eradication of the infection could reduce the risk of developing such complications but cannot completely erase the possible threat. Additionally, an increasing number of studies report antibiotic resistance among H. pylori strains (7, 8). This makes complete eradication of H. pylori infection challenging, and recurrent infections are common without extreme or noticeable symptoms (5, 9, 10).
As for the pathogenesis of GCs, infection with H. pylori of both gastric cell lines and primary gastric epithelial cells has been shown to trigger a unique DNA damage pattern that preferentially targets transcribed regions and regions proximal to telomeres (6, 11). However, how H. pylori infection results in the accumulation of DNA damage remains unknown. Generally, bacterial infection-induced inflammation leads to the generation of reactive oxygen species (ROS) that cause oxidative genome damage in the host cells. Without timely repair, mutagenic DNA lesions can cause several pathologies, including cancer (12–15). Oxidized DNA bases are primarily repaired via the DNA base excision repair (BER) pathway (16, 17). BER is a multistep process that is initiated with recognition and excision of the lesion by a family of repair proteins called DNA glycosylases (DGs). All four DNA bases are subjected to oxidative modifications leading to approximately two dozen lesions; only 5 oxidized base-specific DNA glycosylases with overlapping substrate specificities have been characterized so far in mammalian cells. Among them, OGG1 and NTH1, which were identified and characterized earlier, primarily remove oxidized purines and pyrimidines, respectively, and were thought to be the major mammalian DNA glycosylases. However, null mice of these two DGs did not show any visible phenotype (16, 18–24). We and several other groups then found that mammalian cells possess a new family of DNA glycosylases and named them E. coli Nei-like (NEIL1–3) proteins. NEILs are capable of removing both purines and pyrimidines, but they are unique with respect to DNA damage recognition. Whereas OGG1 and NTH1 remove the base lesions from duplex DNA, NEILs prefer to initiate excision of lesions that are located in a single-stranded region, a replication fork, or transcription bubble mimic (15, 25, 26). Indeed, it was found that NEIL1 and NEIL3 are mostly involved in repairing the damage in replicating cells, and NEIL2 primarily removes the oxidized DNA bases from transcribed regions (26). Recently, copy number variation analysis of the COSMIC and TCGA databases has clearly shown that, among all DNA repair proteins, NEIL2 is the most affected in various cancers, and its level is generally low (27). A mutant or functional variant of NEIL2 was also reported as a risk factor for lung cancer and squamous cell carcinoma in the oral cavity (28–31), and the loss of NEIL2 is associated with poor survival in patients with estrogen receptor-positive breast cancer (32).
Several reports indicate that H. pylori-induced ROS can cause oxidative DNA damage leading to the accumulation of mutations (33, 34). Hence, it is expected that aberrant expression and/or compromised activities of the DGs will be contributing factors for various pathologies, including carcinogenesis. Surprisingly, despite the accumulation of significantly larger amounts of mutagenic 8-oxoG in Ogg1−/− mice, these animals did not develop tumors when exposed to KBrO3, an oxygen radical-forming agent (35). Moreover, Ogg1-null mice are resistant to H. pylori-induced inflammation. These data suggest that oxidized DNA base lesions alone are not sufficient for developing GCs; some other factor(s) is necessary for developing tumors in the affected tissues.
Based on the link between NEIL2 and various types of cancers, we hypothesized that H. pylori infection triggers the accumulation of mutations via dysregulation of NEIL2. Using the stem cell-based ex vivo technique in which gastric organoids were isolated from the stomach of WT and Neil2 knockout (KO) mice and an in vivo infection model, we report here that the level of NEIL2, but not that of other DGs, is significantly decreased upon H. pylori infection, leading to increased DNA damage accumulation and inflammatory responses. Because ROS, DNA damage, and inflammation are closely intertwined, and because NEIL2 is an anti-inflammatory protein in addition to its role in DNA repair (26), we conclude that the combinatorial effect of the accumulation of oxidized DNA lesions along with chronic inflammation likely plays a critical role in the initiation and progression of H. pylori-associated GCs.
Results
H. pylori infection is associated with altered expression of NEIL2 in a dose- and time-dependent manner
To determine the expression of BER proteins in gastric epithelial cells after H. pylori infection, AGS cells were transfected with the C-terminal FLAG-tagged expression plasmids of OGG1, NEIL1, NEIL2 and NTH1, followed by infection with H. pylori. Surprisingly, the level of NEIL2-FLAG (with a 5-fold higher expression than endogenous NEIL2 [Fig. S1]), but not that of the other DNA glycosylases, was significantly decreased in the AGS cell lysate at the protein level (Fig. 1A). To determine whether the stably expressed NEIL2-FLAG is downregulated in transcript level after infection, we specifically detected the NEIL2-FLAG transcript level (Fig. S2A and B). We indeed found its downregulation at 24 h and 48 h postinfection. The empty vector-expressing control cells did not show any amplification of the NEIL2-FLAG transcript, indicating specific amplification in NEIL2-FLAG-expressing cells (Fig. S3A). However, the NEIL2 transcript level was decreased significantly following H. pylori infection in control cells (Fig. S3A and B), indicating the downregulation of the endogenous NEIL2 as well.
Figure 1.

The downregulation of NEIL2 in gastric epithelial cells after H. pylori infection. A, NEIL2-FLAG, NEIL1-FLAG, OGG1-FLAG, and NTHI-FLAG plasmids were transfected in AGS cells, followed by infection with H. pylori for 24 h. FLAG expression was determined in the whole-cell extract by Western blotting (WB). B, endogenous levels of NEIL-2 and OGG1 proteins were determined in the whole-cell extract by WB in uninfected and H. pylori-infected AGS cells. C, time-dependent changes in the NEIL2 expression were determined in AGS cells after infection with H. pylori for the indicated times. D, the effect of H. pylori MOI on NEIL2 expression was evaluated in AGS cells by using H. pylori (MOI, 10:1 and 100:1). A–D, in all experiments, α-tubulin was used as a loading control. B–D, densitometry was performed to quantify the level of NEIL2 expression after normalization with the loading control.
To further confirm this, AGS cells were infected with H. pylori, and it was found that the endogenous expression of NEIL2, but not OGG1, was significantly decreased (Fig. 1B). Furthermore, the expression of NEIL2 following H. pylori infection was decreased in a time-dependent manner. NEIL2 protein and its transcript level started to decrease at 6 h postinfection and became undetectable at 24 h and 48 h (Fig. 1C). To test the effect of H. pylori inoculum on NEIL2 expression, AGS cells were infected with H. pylori at a multiplicity of infection (MOI) of 10 and 100, and the expression of NEIL2 was determined by Western blot analysis. The reduction of NEIL2 expression was more pronounced in AGS lysates at higher bacterial load (Fig. 1D), indicating that H. pylori infection is linked to the downregulation of NEIL2.
H. pylori infection in primary and transformed gastric cells downregulates NEIL2 at the transcript level
To understand the impact of H. pylori infection on NEIL2 expression in primary and transformed gastric cells (Fig. 2A), we used the epithelial cell lines AGS, NCI-N87 from gastric carcinoma, and normal human fetal gastric/intestinal cells, Hs738 (36). Figure 2B demonstrates the time-dependent changes in the NEIL2 transcript level at 0, 1, 3, 6, and 12 h post-H. pylori infection in AGS cells. The NEIL2 mRNA expression was increased at 1 h postinfection, followed by a significant decrease in the transcript level in a time-dependent manner (Fig. 2B). Similarly, the level of NEIL2 transcription was also downregulated in Hs738, the nontransformed gastric cells from the normal human fetus (Fig. 2C), and in NCI-N87, the transformed epithelial cell line (Fig. 2D), after 12 h of H. pylori infection. Next, we evaluated the physiological relevance of this finding in human gastric biopsy specimens (Fig. 2E, i) that were either H. pylori positive or H. pylori negative. Interestingly, H. pylori-positive specimens had lower NEIL2 expression than H. pylori-negative specimens (Fig. 2E, ii), indicating a link between H. pylori infection and the level of NEIL2 in the stomach. To understand further the effect of H. pylori infection on primary gastric epithelial cells, organoids or enteroids were isolated from healthy human gastric biopsy specimens. The enteroid-derived monolayers (EDMs) showed a significant reduction in the level of NEIL2 following H. pylori infection (Fig. 2E, iii).
Figure 2.

The downregulation of NEIL2 transcript in primary and transformed gastric epithelial cells after H. pylori infection. A, the schematic diagram shows the infection of the primary (Hs738) and transformed (AGS, NCI-N87) cells with H. pylori. B, the expression of NEIL2 transcript was determined by qRT-PCR (qPCR) in AGS cells after H. pylori infection at the indicated time points. C and D, NEIL2 transcript expression was tested in primary Hs738 cells (C) and NCI-N87 cells (D) after H. pylori infection. B–D, the expression of the uninfected control was taken as 1. E, i, the schematic shows the gastric biopsies used in step ii and H. pylori-infected enteroid-derived monolayers (EDMs) used in step iii. ii, NEIL2 expression was detected by qPCR in H. pylori-positive and -negative human gastric biopsy specimens (6 negative and 4 positive). The expression of the H. pylori-negative sample was taken as 1. iii, EDMs from gastric specimens of healthy subjects were infected with H. pylori. Data were generated from the means ± S.D. from three independent experiments. *, p < 0.05.
The downregulation of NEIL2 is H. pylori-specific and independent of cag pathogenicity island (CagA)
To investigate whether the reduced expression of NEIL2 was H. pylori infection specific, the NEIL2 expression level was measured in AGS cells infected with either H. pylori or Campylobacter jejuni, a gastrointestinal pathogen similar to H. pylori (37). We found that NEIL2 expression was downregulated with H. pylori infection but not with C. jejuni infection (Fig. 3A). To investigate if NEIL2 expression depended on specific genes or the virulence factor of H. pylori, we selected two mutant H. pylori strains: 1) alkyl hydroperoxide reductase (AhpC) (an enzyme crucial for oxidative stress resistance and the survival of the bacterium in the host that generates higher amounts of 8-oxo-guanine associated with their DNA [38]), and 2) NapA (proteins are known to regulate oxidative stress and ROS generation [39]). We also tested the effect of H. pylori mutant strains for Cag pathogenicity island or CagA (H. pylori virulence factors) on NEIL2 expression and compared it to the effect of the WT H. pylori 26695 strain. We found that NEIL2 expression was downregulated in all of the mutant strains to a level similar to that of WT H. pylori (Fig. 3B–D), indicating that the downregulation of NEIL2 was independent of bacterial virulence factors. To understand the effect of bacterially secreted factors, bacterial supernatant and pellet were added to the cells, and the expression was compared with that of the whole bacteria. The decrease in NEIL2 expression in epithelial cells was mediated only by bacterial pellets (Fig. 3E, second to last lane), and boiling the pellet abolished the effect (Fig. 3E, last lane), indicating that the bacterial factor responsible for NEIL2 downregulation is heat sensitive.
Figure 3.

The downregulation of NEIL2 is H. pylori specific and does not depend on Cag pathogenicity island. NEIL2 expression was determined by WB of whole-cell extracts of AGS cells after H. pylori and C. jejuni infection (A), 43504 WT and AhpC and NapA mutant strain infection (B), and WT and Cag and CagA mutant strain infection (C). D, the relative expression of NEIL2 transcript in WT and Cag and CagA mutant-infected AGS cells was determined by qRT-PCR. The expression of the uninfected control was taken as 1. E, NEIL2 expression was determined by WB after treatment of AGS cells with whole H. pylori, supernatant from infected cells, boiled supernatant, bacterial pellet, and boiled bacterial pellet. In all Western blots (A–C and E), α-tubulin was used as a loading control. One representative blot from 3 independent experiments is shown.
The inflammatory responses of gastric epithelial cells following H. pylori infection is NEIL2 dependent
To understand the effect of infection on gastric epithelial cells under conditions similar to those in vivo, EDMs were prepared from the stomachs of WT and Neil2 KO mice (Fig. S4) and healthy human subjects (Fig. S6). We established EDMs (Fig. S4B) from gastric spheroids (Fig. S4A) of WT and Neil2 KO mice infected with H. pylori. The EDMs were collected for functional assays as shown in Fig. 4A. To understand the mechanistic basis of NEIL2-mediated inflammation, we conducted a cytokine proteome profiler array on the supernatant collected from the H. pylori-infected EDMs of WT and Neil2 KO mice (Fig. 4B). The cytokines/chemokines at low and high exposures are listed in Table 1. The expression of KC, IL1β, IL-6, TNFα, MCP-1, and IFN-γ was measured in the gastric EDMs of WT and Neil2 KO organoids. Interestingly, the level of proinflammatory cytokines was significantly higher in the KO EDMs after 48 h of H. pylori infection (Fig. 4C). To verify the findings, we also conducted KC, MCP-1, and IL-6 ELISA on the mock-treated and H. pylori-infected EDMs. H. pylori infection showed more inflammatory cytokines that were further upregulated in Neil2 KO EDMs (Fig. 4D–F). A similar result was observed with KO EDMs for TNFα and IL-1β (Fig. S6A and B). We also confirmed that EDMs generated from healthy human subjects infected with H. pylori had higher levels of the proinflammatory cytokines MCP-1, IL-8, and IL-6 (Fig. S5).
Figure 4.
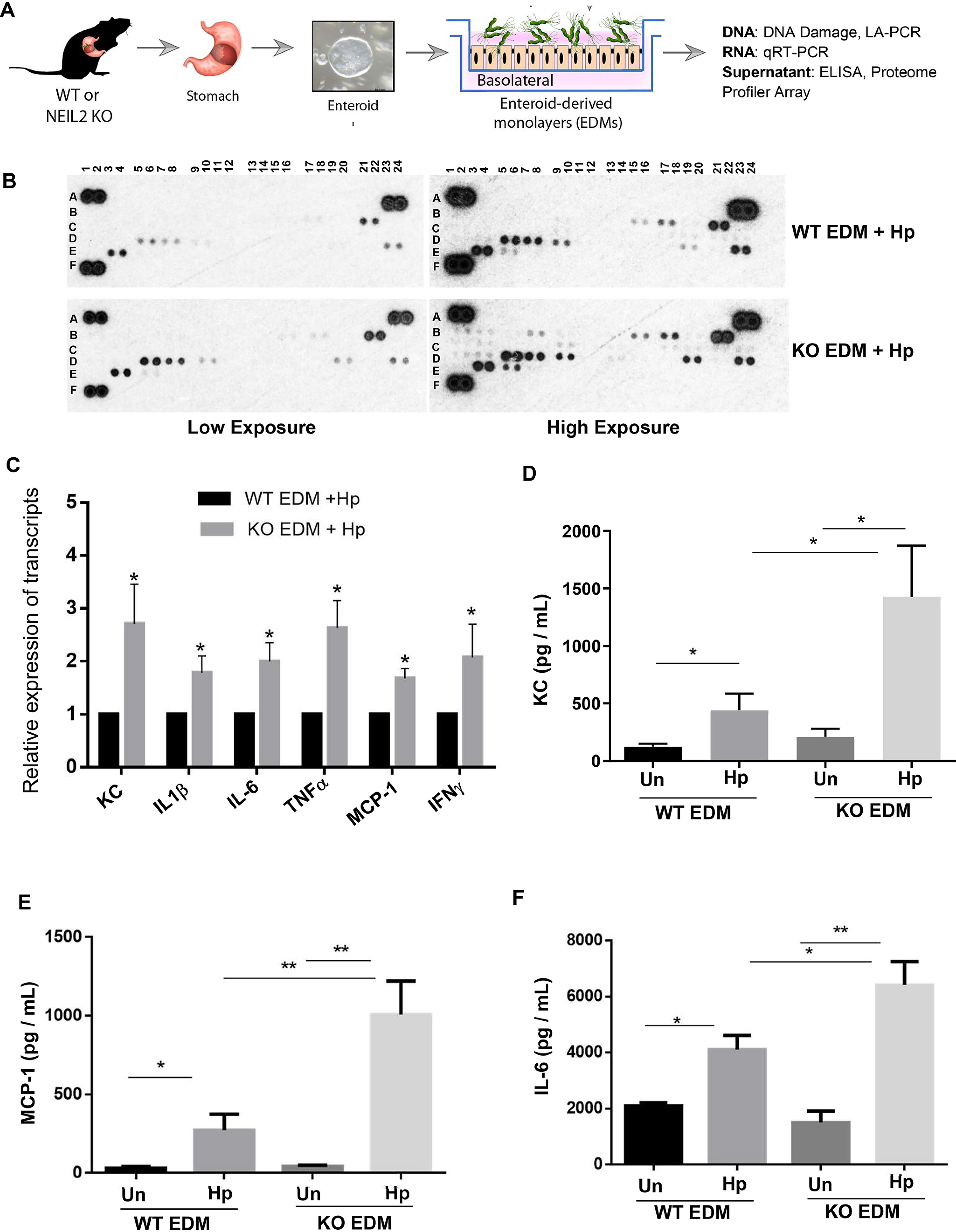
The proinflammatory responses in H. pylori-infected EDMs derived from murine gastric tissue depend on NEIL2. A, the schematic diagram shows the plan for functional assays of enteroids isolated from WT or Neil2 KO mice. B, the proteome profiler array was conducted with the supernatant collected from the H. pylori-infected WT and Neil2 KO EDMs. Two blots from low exposures and high exposures were analyzed, and the cytokines that appeared in the array are listed in the table. C, the differential expression of selected cytokines was verified by qRT-PCR of RNA samples isolated from EDMs used in panel B. D–F, the supernatants from the EDMs of WT or Neil2 KO mice, either untreated (Un) or infected with H. pylori (Hp), were further verified by ELISA for KC, MCP-1, and IL-6 expression. Data were generated from the mean ± S.D. from three independent experiments. *, p < 0.05; **, p < 0.01.
Table 1.
Analysis of proteome profiler array
| Well no./location | Detail | Cytokine appeared | Cytokine appeared |
|---|---|---|---|
| A1, A2; A23, A24; F1, F2 | Reference spot | Low exposure | High exposure |
| B1, B2 | CXCL13 | Yes | |
| B3, B4 | C5/C5a | Yes | |
| B7, B8 | GM-CSF | Yes | |
| B15, B16 | IFN− | Yes | Yes |
| B 17, B18 | IL1-a | Yes | Yes |
| B21, B22 | IL-1Ra | Yes | Yes |
| C3, C4 | IL-4 | Yes | |
| C9, C10 | IL-7 | Yes | |
| C17, C18 | IL-16 | Yes | |
| C23, C24 | Il-27 | Yes | |
| D1, D2 | IP-10 | Yes | |
| D3, D4 | I-TAC | Yes | |
| D5, D6 | KC | Yes | Yes |
| D7, D8 | M-CSF | Yes | Yes |
| D9, D10 | CCL2/MCP-1 | Yes | Yes |
| D19, D20 | MIP-2 | Yes | Yes |
| D23, D24 | CXCL12/S.D.F-1 | Yes | Yes |
| E3, E4 | TIMP-1 | Yes | Yes |
| E5, E6 | TNF-α | Yes | Yes |
To understand the link between H. pylori-mediated inflammatory responses and the role of NEIL2 therein, we transfected AGS cells with vector control or NEIL2-overexpressing plasmids and infected both with H. pylori for 12 h and 24 h. The multiplex cytokine assay showed IL-8 as the prevalent proinflammatory cytokine after H. pylori infection (not shown). Therefore, IL-8 ELISA was performed with control versus NEIL2-overexpressing AGS cells. NEIL2 overexpression resulted in lower levels of the inflammatory cytokine IL-8 (Fig. S6C).
NEIL2 KO mouse stomach showed higher levels of inflammation
To examine the in vivo significance of H. pylori infection, WT and Neil2 KO mice were infected/inoculated with H. pylori via oral gavage (as shown in Fig. 5A). KC (mouse IL-8 analog), TNFα, and IFN-γ mRNA expression levels were measured in the gastric tissue by qRT-PCR (Fig. 5B–D). The level of KC expression was ∼12-fold higher, whereas the TNFα and IFN-γ levels were 1.5- and 2-fold higher, respectively, in gastric tissue collected from the infected Neil2 KO mice compared with those of WT-infected mice (Fig. 5B–D). Similarly, the levels of IL-1β and IL-6 expression were higher in KO-infected tissue than in the WT tissue (Fig. S6D–E). To evaluate the role of NEIL2 in preventing gastric lesions following H. pylori infection, we performed hematoxylin and eosin (H&E) staining of the gastric specimens. Histology score was measured on the basis of inflammation, as stated in Materials and methods (Fig. 5E). Representative images indicate the differences in the extent of the infiltration of immune cells (Fig. 5F). All the data in Fig. 4 and 5 with KO mouse tissue and EDMs suggest that NEIL2 plays an important role in mediating the inflammatory response against H. pylori infection.
Figure 5.
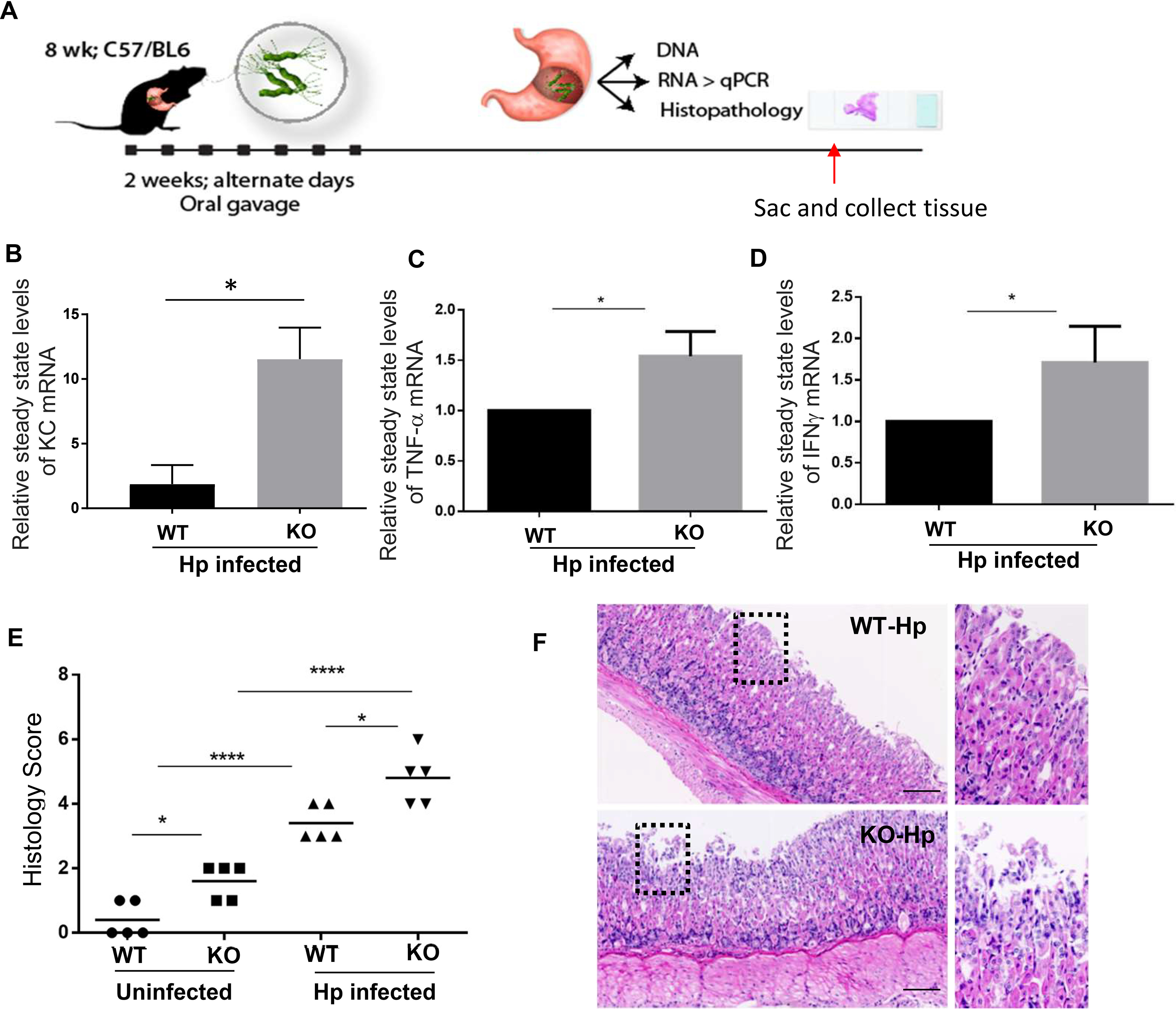
The proinflammatory responses in gastric tissue following H. pylori infection depend on NEIL2. A, schematics show the planning of animal experiments with C57-BL/6 mice to isolate RNA and histopathology. B–D, the relative mRNA expressions of IL-8 (KC), TNF-α, and IFN-γ were determined by qRT-PCR of gastric tissues from H. pylori-infected WT and Neil2 KO mice. E, histology scores of stomach specimens of WT mice and Neil2 KO mice before and after H. pylori infection. *, p < 0.05; ***, p < 0.001. F, representative images from the stomach specimens of H. pylori-infected WT and Neil2 KO mouse groups.
NEIL2 KO mouse stomach showed increased DNA damage after H. pylori infection
To understand the effect of NEIL2 depletion on the accumulation of oxidative DNA base lesions, the supernatants from the coculture of EDMs of WT and Neil2 KO mice with H. pylori infection were measured for oxidative DNA adducts. Neil2 KO stomach EDMs accumulated greater oxidative DNA damage at 48 h of H. pylori infection than the WT (Fig. 6A). To determine whether ROS-mediated DNA damage can induce NEIL2 expression, we treated the AGS cells with glucose oxidase (GO) for 45 min, followed by recovery of oxidative DNA damage for 3, 6, 12, and 24 h. After GO treatment, we could detect the lower intensity of the long amplicon for HPRT and Polβ genes (Fig. 6B), indicating the accumulation of oxidative base lesions. Interestingly, after GO treatment, we detected no significant changes in the NEIL2 level (Fig. 6C). Thus, the depletion of NEIL2 is independent of ROS generation, and it is possible that another factor(s) from H. pylori can affect the expression of NEIL2. Nonetheless, DNA damage was assessed by long amplicon quantitative-PCR (LA-qPCR) in representative Polβ and β-globin genes in the genomic DNA extracted from mouse stomach epithelial enteroids upon H. pylori infection (Fig. 6D). We observed a significantly larger amount of oxidative damage accumulation in both the Polβ and β-globin genes, particularly after 48 h of H. pylori infection in Neil2 KO enteroids compared with that of WT enteroids.
Figure 6.

Evaluation of the role of NEIL2 in H. pylori infection-induced accumulation of oxidized DNA bases in stomach epithelial cells. A, the stomach epithelial EDM of WT and Neil2 KO mice were either untreated or infected with H. pylori for 48 h. The supernatant at each condition was used to measure the oxidative damage of DNA as mentioned in Materials and methods. B, AGS cells were either untreated or treated with glucose oxidase (GO) for 45 min. The genomic DNA was used for long amplicon (LA) and short amplicon (SA) qPCR for HPRT and POLB. The histogram represents the accumulation of oxidative DNA base lesions after densitometry from 3 independent sets of experiment. One representative gel figure is shown. The band intensity in the control cell was arbitrarily set as 100. The error bars represent the standard deviation of the mean band intensity. ***, p < 0.001. C, the NEIL2 expression level was measured by Western blotting from nuclear extract of AGS cells following GO treatment for the indicated time. Con, untreated control. HDAC2, histone deacetylase 2, used as a nuclear loading control. D, stomach epithelial enteroids of WT and Neil2 KO mice were either mock infected or infected with H. pylori for 24 h, 48 h, or 72 h and then harvested for genomic DNA isolation. LA-qPCR was performed to evaluate the level of oxidized DNA base lesions after H. pylori infection. Representative gels show the amplification of each long fragment (∼7-8 kB) (upper) normalized to that of a short fragment (∼250 bp) (lower) of the corresponding (Pol β and β-Globin) genes. The histogram represents the accumulation of oxidative DNA base lesions after densitometry from 4 different sets of WT versus Neil2 KO mice. The band intensity in the WT mice was arbitrarily set as unity (n = 1). The error bars represent the standard deviation from the mean band intensity.
Elevated NEIL2 gene expression in GCs is correlated to a higher probability of disease-free survival, and NEIL2 is lowered in H. pylori-positive atrophic gastritis
NEIL2 mRNA expression levels in GCs were analyzed using publicly available RNA-Seq datasets at RRID:SCR_007834. NEIL2 mRNA expression levels were higher in both intestinal and diffuse-type gastric adenocarcinomas than in noncancerous gastric mucosa (Fig. 7A and B). However, analysis of the disease-free survival data from 631 patients diagnosed with GCs of various clinical stages (40) showed that lower NEIL2 expression correlated with lower probability of disease-free survival for stage I alone, stage II alone, and all stages of GC (Fig. 7C–E).
Figure 7.
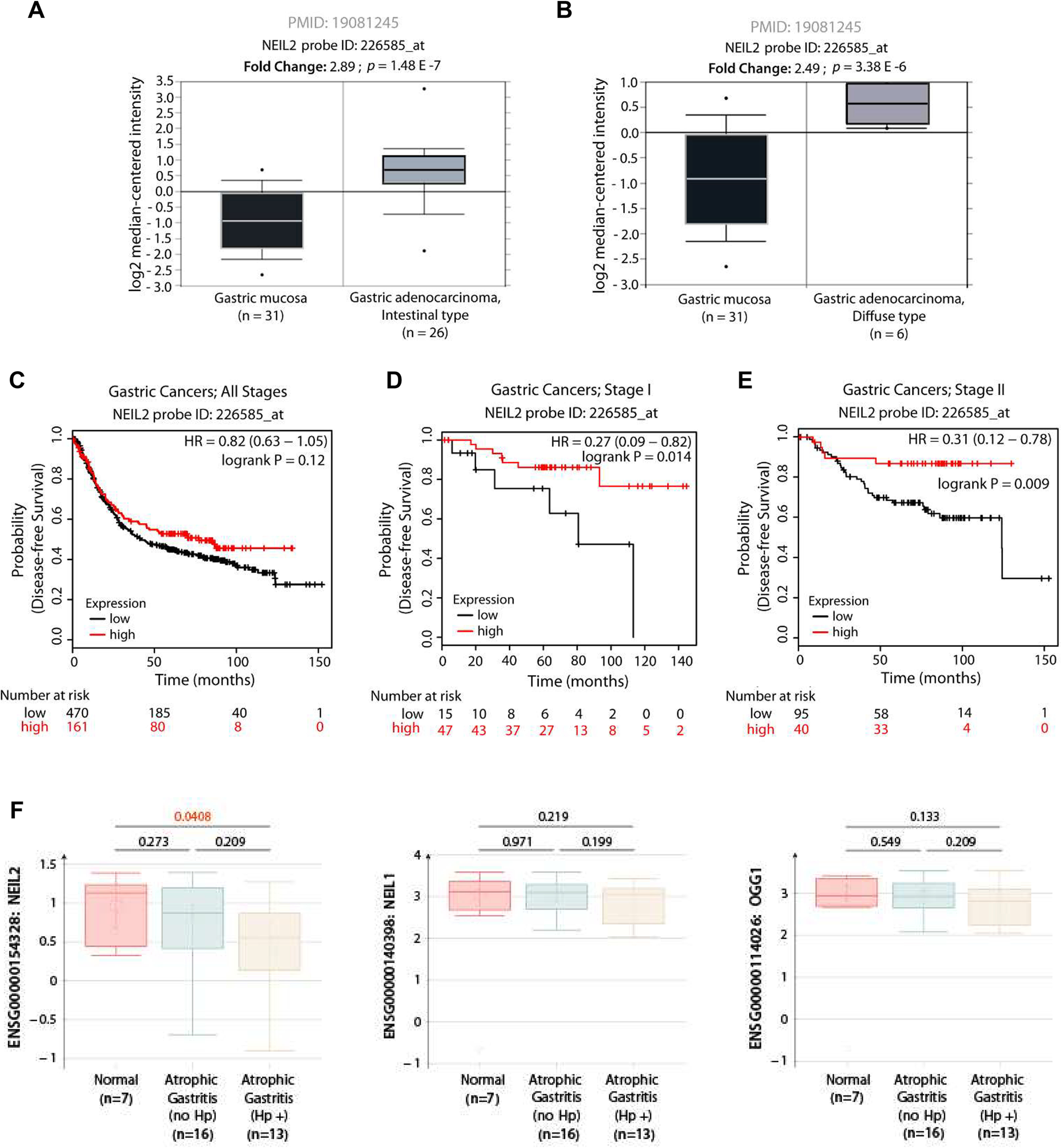
NEIL2 level is specifically downregulated in H. pylori-positive atrophic gastritis, and low levels of expression in early GCs lead to a poor prognosis. A–B, NEIL2 mRNA expression is elevated in both the intestinal (A) and diffuse (B) subtypes of GCs. Expression levels of NEIL2 mRNA in normal versus cancer subjects were analyzed in publicly available RNA-Seq datasets (53) using RRID:SCR_007834. C–E, the Kaplan-Meier plotter [K-M plotter; RRID:SCR_018753] program was used to analyze disease-free survival among 631 patients diagnosed with GCs of various clinical stages (40), stratified into high (red) versus low (black) NEIL2 expression. All stages, C; stage I alone, D; stage II alone, E. F, The levels of NEIL2, NEIL1, and OGG1 were analyzed in the publicly available data set with GDS5338 (41) from the biopsy specimens of stomachs of H. pylori-positive and -negative subjects.
The analysis of publicly available data identified that NEIL2 is specifically decreased with GC progression compared with other repair proteins
The publicly available data set with accession no. GDS5338 (41) from the biopsy specimens of the stomach from H. pylori-positive and -negative human subjects was analyzed, and the level of NEIL2 was found to be significantly decreased in H. pylori-positive gastric atrophy compared with that in the negative specimens. Interestingly, the downregulation of BER proteins in gastric atrophy was prominent only for NEIL2 but not for the other two DNA glycosylases, NEIL1 and OGG1 (Fig. 7F).
Next, we performed computational analysis of gene expression profiles of publicly available datasets (GSE78523) (42) with incomplete metaplasia (IIM-C) and incomplete metaplasia that progressed to GCs (IIM-GC). We chose the incomplete metaplasia group, as it has a higher chance of progression to GCs than the complete metaplasia group (42). The expression data of the BER enzymes showed that, specifically, the level of Neil2 is reduced compared with those of other DNA glycosylases (Neil1, Neil3, NTH1, and Ogg1) in gastric specimens collected from incomplete metaplasia controls (Fig. 8A and B). The significant reduction of NEIL2 in incomplete metaplasia controls correlated with the progression of GCs and suggests that the reduced level of Neil2 can accelerate the accumulation of mutations that can initiate cancer progression.
Figure 8.
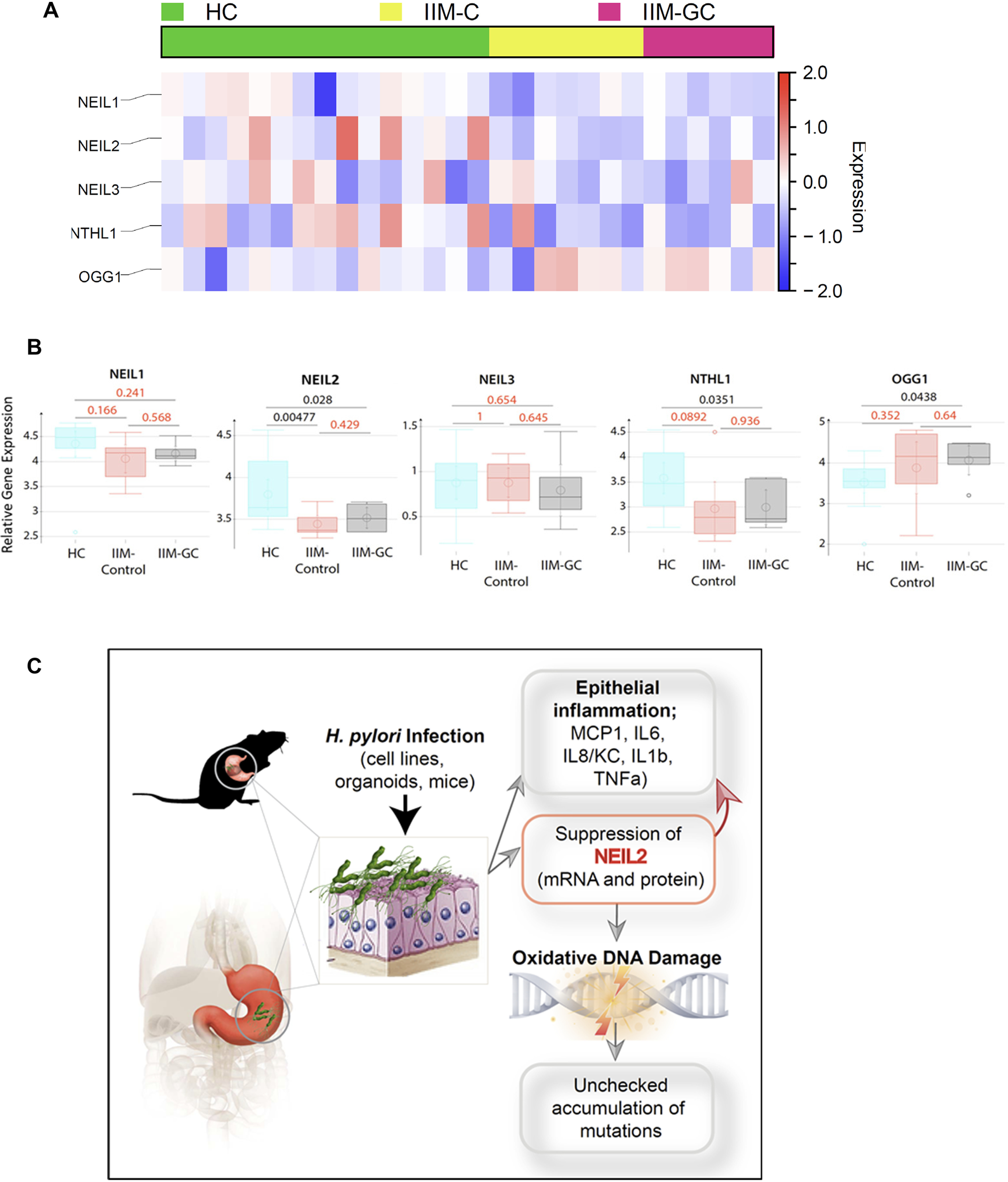
The heat map and the schematic summarize the role of NEIL2 in the initiation of GC. A, a heatmap was generated after analyzing the expression profile of base excision repair genes in the microarray of the publicly available data set (GSE78523) (42). The microarray was performed with the RNA extracted from FFPE cuts of gastric biopsies from a total of 45 human subjects from different disease groups: healthy controls (HC), incomplete metaplasia (IIM-C), and complete metaplasia (IIM-GC). Incomplete metaplasia had a higher progression rate to GC than complete metaplasia. The level of NEIL2 was significantly reduced compared with those of all the genes analyzed in the heatmap. B, NEIL1, NEIL2, NEIL3, OGG1, and NTH1 expression were detected in this cohort. C, H. pylori infection has serious negative effects on the gastric mucosa because of chronic mucosal inflammation leading to DNA damage and downregulation of the base-excision repair protein NEIL2. NEIL2, which actively repairs the damage that occurs during transcription, has impaired repair activity following H. pylori infection and, therefore, accumulates mutations that can lead to GC. D, the schematic diagram summarizes the findings of the current study.
Discussion
Chronic infections and infection-induced proinflammatory cytokines can lead to the production of ROS and reactive nitrogen intermediates. These reactive species can induce DNA damage and thereby propel genomic instability during tumor initiation and progression (14, 43). The vicious feed-forward cycle of inflammation and oxidative stress continuously targets cellular macromolecules, including the genomic DNA (gDNA), inducing mostly oxidized base damage. These oxidized DNA base lesions are primarily repaired via the BER pathways (44, 45). Defects in BER will adversely affect cellular function and allow DNA damage to accumulate and may trigger the development of cancers. Many carcinopathogens utilize this paradigm to induce inflammation-induced genomic instability (46, 47) via mechanisms that remain poorly understood. The major discovery we report here is a mechanism by which the carcinopathogen H. pylori derails a host defense mechanism that is geared to protect cells from oxidized DNA base lesions by repairing them.
H. pylori is a major human pathogen causing chronic, progressive gastric mucosal damage that is linked to atrophic gastritis (AG) (48). AG is linked to an increased risk for intestinal-type adenocarcinoma and type 1 gastric carcinoid, specifically in the presence of extensive intestinal metaplasia (49). Studies have shown that eradication of the bacteria can reduce the risk of cancer development (5, 50). Since H. pylori infection generally develops into a chronic infection, the accompanying persistent inflammatory response, ROS production, and accumulation of DNA damage could be a mechanism for the development of GC (51, 52). The in silico data analysis (Fig. 8, A and B) showed that, among all the BER proteins (NEIL1, NEIL2, NEIL3, OGG1, and NTH1), the level of NEIL2 is significantly reduced in the incomplete metaplasia of gastric specimens that progress to GCs. No study to date has identified the link between NEIL2 or any other BER deficiency with accumulated mutation and their involvement in infection-mediated cancer. Therefore, we studied the BER protein NEIL2 using Neil2-null mice that accumulate oxidative genomic damage, mostly in the transcribed regions, and are more responsive than WT mice to inflammatory agents (26). Interestingly, in gastric epithelial cells, the H. pylori-induced accumulation of a unique DNA damage pattern occurs preferentially in transcribed regions (6).
To establish a link between H. pylori infection and the functional role of DNA glycosylases, we assessed the expression level of BER proteins following H. pylori infection in human gastric epithelial cells (cultured cells as well as in enteroids isolated from gastric biopsies of healthy human subjects). H. pylori downregulated NEIL2 expression in a time- and dose-dependent manner but was Cag independent; consequently, DNA damage was accumulated that could lead to GC.
In our study, we showed the link between NEIL2 expression and inflammatory response (IL-8/KC, MCP-1, IL-6, IL-1β, and TNFα); overexpression of NEIL2 led to a lower inflammatory response, whereas Neil2 KO mice had a greater inflammatory response. Therefore, we hypothesized that NEIL2 expression is linked to both indirect DNA damage caused by the unchecked inflammatory response as well as to a direct build-up of oxidative DNA damage by inhibition of BER. This process implicates the involvement of NEIL2 in gastric carcinogenesis after H. pylori infection and could represent a potential biomarker for GC progression and prevention. From a database search, we found that NEIL2 is expressed at higher levels in various gastric adenocarcinomas than in normal gastric tissue. However, higher NEIL2 expression within each stage of cancer led to a higher probability of survival than that of patients with lower NEIL2 expression (53). In addition to being a potential biomarker for risk assessment and the diagnosis of carcinogenesis, NEIL2 expression may act as a prognostic biomarker to aid in determining the course of the disease. In future studies, it will be important to determine the NEIL2 expression after H. pylori infection and correlate expression levels to gastric carcinogenesis to identify a more significant and clinically relevant link between H. pylori infection, NEIL2 expression, and gastric carcinogenesis.
Besides NEIL2 downregulation, we found that H. pylori infection increased the inflammatory response (IL-8 production) in vitro (AGS cells, Hs738, and enteroid-derived monolayers) and in vivo (human biopsy samples and mice). Since inflammation is a risk factor for cancer development (47, 54), we hypothesized that H. pylori could promote cancer progression by increasing inflammatory signaling. To link IL-8 expression with the function of NEIL2, Neil2 KO mice and WT mice were tested for several inflammatory cytokines (IL-8/KC, MCP-1, IL-6, IL-1β, and TNFα) upon infection with H. pylori. We found that Neil2 KO mice had a greater inflammatory response than the WT. We confirmed our findings by infecting EDM prepared from WT and Neil2 KO mice with H. pylori. IL-8 levels decreased in the NEIL2 overexpressed cells. All the inflammatory cytokines are increased in Neil2 KO mice or enteroid-derived cells, further confirming the regulatory role of NEIL2 on the resolution of inflammation. This reveals an additional immune-homeostatic role of NEIL2 apart from its DNA glycosylase activity, and this is in conjunction with recent findings that show that OGG1 (another dedicated DNA glycosylase) facilitates NF-κB's binding to its consensus sequence and promotes inflammatory responses (24, 55–57).
Inflammatory cytokines can induce DNA damage that, in turn, promotes senescence (58, 59). This is further established as shown in Fig. S8, where an increase in IL-8 (KC) and IL-6 levels in KO EDMs started at 24 h and increased further at 48 h. However, the DNA damage is significantly increased in NEIL2 KO EDMs only after 48 h of infection (Fig. 6D), suggesting that the inflammatory cytokines induce ROS generation, followed by DNA damage. We provide a perspective on DNA repair inhibition and inflammatory response and propose that both phenomena are regulated by NEIL2 and exacerbate one another, leading to the accumulation of tumorigenic DNA damage (Fig. 8C). Moreover, we found that the regulation of NEIL2 expression was independent of the presence of ROS and that the expression of NEIL2 is H. pylori dose and time dependent. It was demonstrated earlier that the bacterial effector EspF from enteropathogenic Escherichia coli (EPEC) abrogates host cell mismatch repair (MMR) (60). Thus, we postulate that such bacterial effectors are also involved in downregulating NEIL2 expression in H. pylori-mediated infection. Hence, how the factors from H. pylori can downregulate NEIL2 expression will be an exciting area of future research. The knowledge gained from such studies should help in understanding the mechanistic link between infection-mediated inflammation, DNA damage, and GC, which will ultimately have a great translational impact to benefit human health.
Materials and methods
Spheroid isolation and maintenance
Gastric stem cells were isolated from WT and Neil2 KO mice, as described previously (61, 62). The pylorus section of the stomach was removed, washed with PBS, and then incubated with collagenase type I solution containing gentamicin (50 µg/ml) at 37 °C for 30 min with mixing at 10-min intervals. The cells were filtered through a 70-µm cell strainer, and the density and viability of the gastric stem cells were checked using trypan blue and a hemocytometer. The cells were suspended in Matrigel, and 50% conditioned medium (CM) was added to each well (61). Spheroid cultures were passaged every 3 to 4 days using 0.25% trypsin in PBS-EDTA. EDM was prepared as previously described (22, 63). The spheroids were briefly trypsinized, and the isolated cells were filtered, counted, suspended in 5% CM, and pelleted with Matrigel (1:30 dilution) in the apical part of the transwell at a density of 5 × 105 cells/well. The 5% CM was added to the base of the transwell. A similar isolation and culture protocol was used to generate spheroids from human stomach tissue, as previously described (64, 65).
Cell line maintenance
AGS cells were cultured in ATCC-formulated F-12K medium (catalog no. 30-2004) containing 10% fetal bovine serum (FBS), penicillin (1 units/ml), and streptomycin (100 µg/ml) and then kept in an incubator at 37 °C and 5% CO2.
H. pylori infection
H. pylori strain ATCC-26695 was incubated with Brucella broth supplemented with 10% FBS 1 day before infection and left on a shaker at 37 °C in 10% CO2. EDM and gastric epithelial cell lines were infected with the bacteria at a multiplicity of infection (MOI) of 100:1 for 24 h and 48 h. The Cag and CagA mutants were described in our previous study (66). The AhpC and NapA mutants were grown as published previously (38, 39). For the supernatant and bacterial pellet, the bacterial culture was centrifuged down at 10,000 rpm and the supernatant was separated from the bacterial pellet. The same number of bacteria was used for preparing the whole bacteria, pellet, and supernatant.
RNA isolation and RT-PCR
RNA was isolated from uninfected and H. pylori-infected cells using the RNeasy Micro kit (Qiagen, Valencia, CA) according to the manufacturer's instructions. cDNA was synthesized using the superscript kit (Invitrogen). qPCR was carried out using the SYBR Green master mix for target genes and normalized to the housekeeping gene encoding GAPDH or β-actin using the ΔΔCT method (30). Primers were designed using the Roche Universal Probe Library Assay Design software (Table 2).
Table 2.
List of primers used in the study
| Gene | Species | Forward primer (5′→3′) | Reverse primer (5′→3′) |
|---|---|---|---|
| NEIL2 | Mouse | CTGCCGCCTTTCAGTCTCT | TCTGGATCAAACCGAAGGAA |
| Human | GGGGCAGCAGTAAGAAGCTA | GGAATAATTTCTTTCCATGGACCT | |
| IL-8/KC | Mouse | CGCTTCTCTGTGCAGCGCTGCTGCT | AAGCCTCGCGACCATTCTTGAGTC |
| Human | GAGCACTCCATAAGGCACAAA | ATGGTTCCTTCCGGTGGT | |
| IL-6 | Mouse | CCCCAATTTCCAATGCTCTCC | CGCACTAGGTTTGCCGAGTA |
| Human | TCAATATTAGAGTCTCAACCCCCA | TTCTCTTTCGTTCCCGGTGG | |
| IFN-γ | Mouse | CGGCACAGTCATTGAAAGCC | TGTCACCATCCTTTTGCCAGT |
| Human | GGCTTTTCAGCTCTGCATCG | TCTGTCACTCTCCTCTTTCCA | |
| MCP-1 | Mouse | AAGTGCAGAGAGCCAGACG | TCAGTGAGAGTTGGCTGGTG |
| Human | AGTCTCTGCCGCCCTTCT | GTGACTGGGGCATTGATTG | |
| TNF-α | Mouse | CCACCACGCTCTTCTGTCTA | AGGGTCTGGGCCATAGAACT |
| Human | CGCTCCCCAAGAAGACAG | AGAGGCTGAGGAACAAGCAC | |
| IL-1β | Mouse | GAAATGCCACCTTTTGACAGT | CTGGATGCTCTCATCAGGACA |
| Β-actin | Mouse | GACGGCCAGGTCATCACTAT | ACATCTGCTGGAAGGTGGAC |
| Human | AGAGCTACGAGCTGCCTGAC | AGCACTGTGTTGGCGTACAG | |
| NEIL2 FLAG | ATGCCAGAAGGGCCGTTG | TTGTCATCGTCGTCCTTGTAGT | |
| Endogenous NEIL-2 | ATGCCAGAAGGGCCGTTG | GGAGAACTGGCACTGCTCTG |
Western blotting
Cell lysates, collected from uninfected and H. pylori-infected monolayers and cell lines, were tested for NEIL2 protein expression using a purified anti-NEIL-2 antibody (dilution, 1:500) as described previously (67).
Quantification of NEIL2 transcript level
AGS cells stably expressing NEIL2-FLAG or empty FLAG vector, as a control, were infected with H. pylori at different time points (0 h, 1 h, 6 h, 12 h, 24 h, and 48 h), total RNA was isolated, and the levels of both NEIL2-FLAG and endogenous NEIL2 mRNA were determined by RT-PCR using the primers listed in Table 2.
ELISA
Supernatants collected from apical and basolateral parts of uninfected and H. pylori-infected monolayers on transwells were tested for IL-8 using the mouse CXCL1/KC DuoSet ELISA kit (R&D Systems) according to the manufacturer's instructions. The level of IL-8 was compared in H. pylori-infected EDMs versus the uninfected EDMs with the same cell number.
Infection of Neil2-KO mice with H. pylori
Mice used in the study were WT and Neil2 KO C57BL/6 mice and were generated and characterized as previously described (26). The maintenance, breeding, and infection study protocols were approved by the Animal Care and Ethics Committee of UTMB, Galveston, TX. Mice were inoculated with H. pylori strain SS1 (108 bacterial counts/dose) via oral gavage for 3 days/week as described previously (68). Animals were sacrificed after 3 months, and stomach specimens were collected for RNA and DNA isolation.
Histopathology of Neil2 KO mice following H. pylori infection
Gastric tissues from H. pylori-infected WT and NEIL2 KO mice were stained with H&E. The slides were scored using the criteria used in the published literature (69, 70). The scoring system was graded as 1, mild multifocal; 2, mild widespread or moderate multifocal; 3, mild widespread and moderate multifocal or severe multifocal; 4, moderate widespread; 5, moderate widespread and severe multifocal; and 6, severe widespread.
Induction of ROS by GO for NEIL2 expression
Human gastric epithelial (AGS) cells with GO (final concentration, 200 ng/ml) were treated for 1 h, a period known to generate ROS, and the cells were allowed to recover for 24 h following induction of ROS to measure the NEIL2 level.
Induction of ROS by GO to determine its effect on NEIL2 expression
AGS cells were treated with GO (final concentration, 200 ng/ml; 45-min incubation in reduced serum medium), known to generate ROS, and cells were allowed to recover in complete medium for 24 h following induction of ROS. Cells were collected at various time points for isolation of nuclear extract (NE) to assess the NEIL2 level by Western blotting.
Preparation of Nuclear Extract NE
NEs were prepared as described previously (26). Briefly, cells were lysed in buffer A (10 mm Tris-HCl [pH 7.9], 0.34 M sucrose, 3 mm CaCl2, 2 mm magnesium acetate, 0.1 mm EDTA, 1 mm DTT, 0.5% Nonidet P-40 [NP-40], and 1× protease inhibitor mixture [Roche]) and centrifuged at 3,500 × g for 15 min. Nuclear pellets were washed with buffer A without NP-40 and then lysed in buffer B (20 mm HEPES [pH 7.9], 3 mm EDTA, 10% glycerol, 150 mm potassium acetate, 1.5 mm MgCl2, 1 mm DTT, 0.1% NP-40, 1 mm sodium orthovanadate [vanadate], and 1× protease inhibitors) by homogenization. Supernatants were collected after centrifugation at 15,000 × g for 30 min, and DNA/RNA in the suspension was digested with 0.15 units/μl Benzonase (Novagen) at 37 °C for 1 h. The samples were centrifuged at 20,000 × g for 30 min and the supernatants collected as NEs.
Biopsy samples
Gastric biopsy samples were collected from gastro-esophageal-duodenoscopy patients according to Institutional Review Board-approved human subject protocols. All human studies abide by the Declaration of Helsinki principles. Histopathology and rapid urease testing were used to detect the H. pylori infection. NEIL2 transcript expression was determined using qRT-PCR.
DNA damage analysis
WT and Neil2 KO mice were infected with H. pylori, and gDNA was isolated from stomach epithelial enteroids of uninfected control and H. pylori-infected mice using a genomic-tip 20/G kit (Qiagen, catalog no. 10223, with corresponding buffer sets) per the manufacturer's protocol. This kit has the advantage of minimizing DNA oxidation during the isolation steps; thus, it can be used reliably for isolation of high-molecular-weight DNA with excellent template integrity for detecting endogenous DNA damage using LA-qPCR. The extracted gDNA was quantified by Pico Green (Molecular Probes) in a 96-well black-bottomed plate, and the presence of H. pylori was confirmed by PCR. To induce strand breaks at the sites of unrepaired oxidized DNA base lesions, 150 ng of gDNA was treated with 4 units of E. coli enzyme Fpg (New England Biolabs) for 1 h at 37 °C. The enzyme was inactivated by incubating the reaction mixture at 65 °C for 15 min. The gene-specific DNA damage was measured by LA-qPCR using 15 ng of Fpg-treated gDNA per PCR reaction by following the protocol reported earlier by Chakraborty et al. (20). LA-qPCR was carried out to amplify a 6.5-kb region of polβ and the 8.7-kb region of globin genes, as done previously (26). To maintain linearity, the final PCR condition was optimized at 94 °C for 30 s (94 °C for 30 s, 55–60°C for 30 s depending on the oligo annealing temperature, 65 °C for 10 min) for 25 cycles and 65 °C for 10 min. Since amplification of a small region would be independent of DNA damage, a small DNA fragment for each gene was also amplified to normalize the amplification of large fragments, as done before (20). The amplified products then were visualized on gels and quantified with an ImageJ automated digitizing system (National Institutes of Health) based on two independent replicate PCRs. The extent of damage was calculated in terms of the relative band intensity with the WT mice considered as one. A similar method was followed to measure the oxidative DNA damage following GO treatment of AGS cells.
Measurement of DNA/RNA oxidative damage
The level of DNA damage in WT stomach EDM and NEIL2 KO EDM was challenged with H. pylori infection and quantified with the DNA/RNA oxidative damage ELISA kit (Cayman Chemical, USA) according to the manufacturer's instructions and previous papers (71, 72). This immunoassay detects oxidized guanine species: 8-hydroxy-2′-deoxaguanosine from DNA and 8-hydroxyguanine from either DNA or RNA. Briefly, this assay is based on competition between oxidatively damaged guanosine species and 8-hydroxy deoxaguanosine acetylcholinesterase conjugate (DNA/RNA oxidative damage tracer) for a limited amount of DNA/RNA oxidative damage mAb. The amount of tracer, which is held constant, that will bind to the mAb is inversely proportional to the concentration of oxidatively damaged guanine species in the well. The secondary antibody (goat polyclonal anti-mouse IgG) has been previously attached to the well and will react with antibody oxidatively damaged guanine complex. The reaction is developed by the addition of Ellman's reagent that has substrate to acetylcholinesterase. The intensity of the color is determined spectrophotometrically at a wavelength of 405–420 nm and is proportional to the amount of oxidative damage tracer bound to the well, which is inversely proportional to the amount of free 8-hydroxy guanosine present in the well. According to the manufacturer's instructions, standard curves were developed using standards of 3000 pg/ml to 10.3 pg/ml and were prepared using the same culture media of the infection experiment to ensure that the matrix of standards is comparable to the samples.
Statistics
Results are expressed as mean ± S.D. and were compared by using a two-tailed Student t test. Differences were considered significant if P values were lower than 0.05.
Data availability
All data are contained within the manuscript.
Supplementary Material
Acknowledgments
We are thankful to HUMANOID CoRE for organoid-media and biobanking of organoids. We thank Sarah Toombs Smith, Ph.D., ELS, for critically editing this manuscript.
This article contains supporting information.
Author contributions—I. M. S., A. Z. S., T. V., V. E. R., I. P., D. S., P. G., T. K. H., and S. D. formal analysis; I. M. S., A. Z. S., T. V., A. C., D. M., D. B., E. J. B., V. E. R., I. P., D. S., P. G., T. K. H., and S. D. methodology; I. M. S., A. Z. S., A. C., I. P., D. S., P. G., T. K. H., and S. D. writing-review and editing; D. M. and S. D. data curation; D. B., E. J. B., V. E. R., I. P., D. S., P. G., T. K. H., and S. D. resources; I. P., D. S., P. G., and T. K. H. software; P. G., T. K. H., and S. D. conceptualization; P. G., T. K. H., and S. D. supervision; P. G., T. K. H., and S. D. funding acquisition; P. G., T. K. H., and S. D. investigation; P. G., T. K. H., and S. D. project administration; T. K. H. and S. D. validation; T. K. H. and S. D. writing-original draft.
Funding and additional information—This work was supported, in whole or in part, by National Institutes of Health grants DK107585 and DK099275, C3 Padre Pedal MCC pilot grant (to S. D.), R01 NS073976 (to T. H.), R01HL145477 (T. K. H.), W81XWH-18-1-0743 (to T. K. H.), R01 AI141630 (to P. G.), R01-GM138385 (to D. S.), and DoD CA150375 (to V. E. R.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—All authors declare that they have no conflict of interest.
- GC
- gastric cancer
- EDMs
- enteroid-derived monolayers
- MOI
- multiplicity of infection
- BER
- base excision repair
- DGs
- DNA glycosylases
- LA-qPCR
- long amplicon quantitative PCR
- AG
- gastric atrophy
- ROS
- reactive oxygen species
- qRT-PCR
- quantitative reverse transcription-PCR
- GO
- glucose oxidase
- CM
- conditioned medium
- NE
- nuclear extract
- gDNA
- genomic DNA.
References
- 1. World Health Organization. (2018) The global cancer observatory. International Agency for Research on Cancer, World Health Organization, Geneva, Switzerland. [Google Scholar]
- 2. Mou X., Li T., Wang J., Ali Z., Zhang Y., Chen Z., Deng Y., Li S., Su E., Jia Q., He N., Ni J., and Cui D. (2015) Genetic variation of BCL2 (rs2279115), NEIL2 (rs804270), LTA (rs909253), PSCA (rs2294008) and PLCE1 (rs3765524, rs10509670) genes and their correlation to gastric cancer risk based on universal tagged arrays and Fe3O4 magnetic nanoparticles. J. Biomed. Nanotechnol. 11, 2057–2066 10.1166/jbn.2015.2113 [DOI] [PubMed] [Google Scholar]
- 3. Ronchetti L., Melucci E., De Nicola F., Goeman F., Casini B., Sperati F., Pallocca M., Terrenato I., Pizzuti L., Vici P., Sergi D., Di Lauro L., Amoreo C. A., Gallo E., Diodoro M. G., et al. (2017) DNA damage repair and survival outcomes in advanced gastric cancer patients treated with first-line chemotherapy. Int. J. Cancer 140, 2587–2595 10.1002/ijc.30668 [DOI] [PubMed] [Google Scholar]
- 4. Crew K. D., and Neugut A. I. (2006) Epidemiology of gastric cancer. World J. Gastroenterol. 12, 354–362 10.3748/wjg.v12.i3.354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Malfertheiner P., Megraud F., O'Morain C. A., Atherton J., Axon A. T. R., Bazzoli F., Gensini G. F., Gisbert J. P., Graham D. Y., Rokkas T., El-Omar E. M., and Kuipers E. J. and European Helicobacter Study Group. (2012) Management of Helicobacter pylori infection–the Maastricht IV/Florence Consensus Report. Gut 61, 646–664 10.1136/gutjnl-2012-302084 [DOI] [PubMed] [Google Scholar]
- 6. Koeppel M., Garcia-Alcalde F., Glowinski F., Schlaermann P., and Meyer T. F. (2015) Helicobacter pylori infection causes characteristic DNA damage patterns in human cells. Cell Rep. 11, 1703–1713 10.1016/j.celrep.2015.05.030 [DOI] [PubMed] [Google Scholar]
- 7. Abadi A. T. B. (2017) Resistance to clarithromycin and gastroenterologist's persistence roles in nomination for Helicobacter pylori as high priority pathogen by World Health Organization. World J. Gastroenterol. 23, 6379–6384 10.3748/wjg.v23.i35.6379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Savoldi A., Carrara E., Graham D. Y., Conti M., and Tacconelli E. (2018) Prevalence of antibiotic resistance in Helicobacter pylori: a systematic review and meta-analysis in World Health Organization regions. Gastroenterology 155, 1372–1382 10.1053/j.gastro.2018.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Flores-Trevino S., Mendoza-Olazaran S., Bocanegra-Ibarias P., Maldonado-Garza H. J., and Garza-Gonzalez E. (2018) Helicobacter pylori drug resistance: therapy changes and challenges. Expert Rev. Gastroenterol. Hepatol. 12:819–827 10.1080/17474124.2018.1496017 [DOI] [PubMed] [Google Scholar]
- 10. Liou J. M., Chen P. Y., Luo J. C., Lee J. Y., Chen C. C., Fang Y. J., Yang T. H., Chang C. Y., Bair M. J., Chen M. J., Hsu Y. C., Hsu W. F., Chang C. C., Lin J. T., Shun C. T., et al. (2018) Efficacies of genotypic resistance-guided vs empirical therapy for refractory Helicobacter pylori infection. Gastroenterology 155, 1109–1119 10.1053/j.gastro.2018.06.047 [DOI] [PubMed] [Google Scholar]
- 11. Alzahrani S., Lina T. T., Gonzalez J., Pinchuk I. V., Beswick E. J., and Reyes V. E. (2014) Effect of Helicobacter pylori on gastric epithelial cells. World J. Gastroenterol. 20, 12767–12780 10.3748/wjg.v20.i36.12767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Valko M., Leibfritz D., Moncol J., Cronin M. T., Mazur M., and Telser J. (2007) Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 39, 44–84 10.1016/j.biocel.2006.07.001 [DOI] [PubMed] [Google Scholar]
- 13. Valko M., Rhodes C. J., Moncol J., Izakovic M., and Mazur M. (2006) Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chemico-Biol. Interactions 160, 1–40 10.1016/j.cbi.2005.12.009 [DOI] [PubMed] [Google Scholar]
- 14. Wiseman H., and Halliwell B. (1996) Damage to DNA by reactive oxygen and nitrogen species: role in inflammatory disease and progression to cancer. Biochem. J. 313, 17–29 10.1042/bj3130017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sahan A. Z., Hazra T. K., and Das S. (2018) The pivotal role of DNA repair in infection mediated-inflammation and cancer. Front. Microbiol. 9, 663 10.3389/fmicb.2018.00663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang L. J., Ma F., Tang B., and Zhang C. Y. (2016) Base-excision-repair-induced construction of a single quantum-dot-based sensor for sensitive detection of DNA glycosylase activity. Anal. Chem. 88, 7523–7529 10.1021/acs.analchem.6b00664 [DOI] [PubMed] [Google Scholar]
- 17. Maynard S., Schurman S. H., Harboe C., de Souza-Pinto N. C., and Bohr V. A. (2009) Base excision repair of oxidative DNA damage and association with cancer and aging. Carcinogenesis 30, 2–10 10.1093/carcin/bgn250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Scharer O. D., and Jiricny J. (2001) Recent progress in the biology, chemistry and structural biology of DNA glycosylases. BioEssays 23, 270–281 [DOI] [PubMed] [Google Scholar]
- 19. Hegde M. L., Hazra T. K., and Mitra S. (2008) Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res. 18, 27–47 10.1038/cr.2008.8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sancar A., Lindsey-Boltz L. A., Unsal-Kacmaz K., and Linn S. (2004) Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 73, 39–85 10.1146/annurev.biochem.73.011303.073723 [DOI] [PubMed] [Google Scholar]
- 21. Hazra T. K., Das A., Das S., Choudhury S., Kow Y. W., and Roy R. (2007) Oxidative DNA damage repair in mammalian cells: a new perspective. DNA Rep. 6, 470–480 10.1016/j.dnarep.2006.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. den Hartog G., Chattopadhyay R., Ablack A., Hall E. H., Butcher L. D., Bhattacharyya A., Eckmann L., Harris P. R., Das S., Ernst P. B., and Crowe S. E. (2016) Regulation of Rac1 and reactive oxygen species production in response to infection of gastrointestinal epithelia. PLoS Pathog. 12, e1005382 10.1371/journal.ppat.1005382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bhattacharyya A., Chattopadhyay R., Hall E. H., Mebrahtu S. T., Ernst P. B., and Crowe S. E. (2010) Mechanism of hypoxia-inducible factor 1 alpha-mediated Mcl1 regulation in Helicobacter pylori-infected human gastric epithelium. Am. J. Physiol. Gastrointest. Liver Physiol. 299, G1177–G1186 10.1152/ajpgi.00372.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ba X., Aguilera-Aguirre L., Rashid Q. T., Bacsi A., Radak Z., Sur S., Hosoki K., Hegde M. L., and Boldogh I. (2014) The role of 8-oxoguanine DNA glycosylase-1 in inflammation. Int. J. Mol. Sci. 15, 16975–16997 10.3390/ijms150916975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Banerjee D., Mandal S. M., Das A., Hegde M. L., Das S., Bhakat K. K., Boldogh I., Sarkar P. S., Mitra S., and Hazra T. K. (2011) Preferential repair of oxidized base damage in the transcribed genes of mammalian cells. J. Biol. Chem. 286, 6006–6016 10.1074/jbc.M110.198796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chakraborty A., Wakamiya M., Venkova-Canova T., Pandita R. K., Aguilera-Aguirre L., Sarker A. H., Singh D. K., Hosoki K., Wood T. G., Sharma G., Cardenas V., Sarkar P. S., Sur S., Pandita T. K., Boldogh I., et al. (2015) Neil2-null mice accumulate oxidized DNA bases in the transcriptionally active sequences of the genome and are susceptible to innate inflammation. J. Biol. Chem. 290, 24636–24648 10.1074/jbc.M115.658146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shinmura K., Kato H., Kawanishi Y., Igarashi H., Goto M., Tao H., Inoue Y., Nakamura S., Misawa K., Mineta H., and Sugimura H. (2016) Abnormal expressions of DNA glycosylase genes NEIL1, NEIL2, and NEIL3 are associated with somatic mutation loads in human cancer. Oxidative Med. Cell. Longevity 2016, 1546392 10.1155/2016/1546392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dou H., Mitra S., and Hazra T. K. (2003) Repair of oxidized bases in DNA bubble structures by human DNA glycosylases NEIL1 and NEIL2. J. Biol. Chem. 278, 49679–49684 10.1074/jbc.M308658200 [DOI] [PubMed] [Google Scholar]
- 29. Dey S., Maiti A. K., Hegde M. L., Hegde P. M., Boldogh I., Sarkar P. S., Abdel-Rahman S. Z., Sarker A. H., Hang B., Xie J., Tomkinson A. E., Zhou M., Shen B., Wang G., Wu C., et al. (2012) Increased risk of lung cancer associated with a functionally impaired polymorphic variant of the human DNA glycosylase NEIL2. DNA Rep. 11, 570–578 10.1016/j.dnarep.2012.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sarker A. H., Chatterjee A., Williams M., Lin S., Havel C., Jacob P. 3rd, Boldogh I., Hazra T. K., Talbot P., and Hang B. (2014) NEIL2 protects against oxidative DNA damage induced by sidestream smoke in human cells. PLoS One 9, e90261 10.1371/journal.pone.0090261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhai X., Zhao H., Liu Z., Wang L. E., El-Naggar A. K., Sturgis E. M., and Wei Q. (2008) Functional variants of the NEIL1 and NEIL2 genes and risk and progression of squamous cell carcinoma of the oral cavity and oropharynx. Clin. Cancer Res. 14, 4345–4352 10.1158/1078-0432.CCR-07-5282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Anurag M., Punturi N., Hoog J., Bainbridge M. N., Ellis M. J., and Haricharan S. (2018) Comprehensive Profiling of DNA Repair Defects in Breast Cancer Identifies a Novel Class of Endocrine Therapy Resistance Drivers. Clin. Cancer Res. 24, 4887–4899 10.1158/1078-0432.CCR-17-3702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Handa O., Naito Y., and Yoshikawa T. (2010) Helicobacter pylori: a ROS-inducing bacterial species in the stomach. Inflamm. Res. 59, 997–1003 10.1007/s00011-010-0245-x [DOI] [PubMed] [Google Scholar]
- 34. Davies G. R., Simmonds N. J., Stevens T. R., Sheaff M. T., Banatvala N., Laurenson I. F., Blake D. R., and Rampton D. S. (1994) Helicobacter pylori stimulates antral mucosal reactive oxygen metabolite production in vivo. Gut 35, 179–185 10.1136/gut.35.2.179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Touati E., Michel V., Thiberge J. M., Ave P., Huerre M., Bourgade F., Klungland A., and Labigne A. (2006) Deficiency in OGG1 protects against inflammation and mutagenic effects associated with H. pylori infection in mouse. Helicobacter 11, 494–505 10.1111/j.1523-5378.2006.00442.x [DOI] [PubMed] [Google Scholar]
- 36. Beswick E. J., Das S., Pinchuk I. V., Adegboyega P., Suarez G., Yamaoka Y., and Reyes V. E. (2005) Helicobacter pylori-induced IL-8 production by gastric epithelial cells up-regulates CD74 expression. J. Immunol. 175, 171–176 10.4049/jimmunol.175.1.171 [DOI] [PubMed] [Google Scholar]
- 37. Moran A. P. (2010) The role of endotoxin in infection: Helicobacter pylori and Campylobacter jejuni. Subcell. Biochem. 53, 209–240 10.1007/978-90-481-9078-2_10 [DOI] [PubMed] [Google Scholar]
- 38. Wang G., Conover R. C., Olczak A. A., Alamuri P., Johnson M. K., and Maier R. J. (2005) Oxidative stress defense mechanisms to counter iron-promoted DNA damage in Helicobacter pylori. Free Radic. Res. 39, 1183–1191 10.1080/10715760500194018 [DOI] [PubMed] [Google Scholar]
- 39. Wang G., Hong Y., Olczak A., Maier S. E., and Maier R. J. (2006) Dual roles of Helicobacter pylori NapA in inducing and combating oxidative stress. Infect. Immun. 74, 6839–6846 10.1128/IAI.00991-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Szendrői A., Szász A. M., Kardos M., Tőkés A.-M., Idan R., Szűcs M., Kulka J., Nyirády P., Szendrői M., Szállási Z., Győrffy B., and Tímár J. (2016) Opposite prognostic roles of HIF1alpha and HIF2alpha expressions in bone metastatic clear cell renal cell cancer. Oncotarget 7, 42086–42098 10.18632/oncotarget.9669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nookaew I., Thorell K., Worah K., Wang S., Hibberd M. L., Sjovall H., Pettersson S., Nielsen J., and Lundin S. B. (2013) Transcriptome signatures in Helicobacter pylori-infected mucosa identifies acidic mammalian chitinase loss as a corpus atrophy marker. BMC Med. Genomics 6, 41 10.1186/1755-8794-6-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Companioni O., Sanz-Anquela J. M., Pardo M. L., Puigdecanet E., Nonell L., Garcia N., Parra Blanco V., Lopez C., Andreu V., Cuatrecasas M., Garmendia M., Gisbert J. P., Gonzalez C. A., and Sala N. (2017) Gene expression study and pathway analysis of histological subtypes of intestinal metaplasia that progress to gastric cancer. PLoS One 12, e0176043 10.1371/journal.pone.0176043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mantovani A., Allavena P., Sica A., and Balkwill F. (2008) Cancer-related inflammation. Nature 454, 436–444 10.1038/nature07205 [DOI] [PubMed] [Google Scholar]
- 44. Jackson S. P., and Bartek J. (2009) The DNA-damage response in human biology and disease. Nature 461, 1071–1078 10.1038/nature08467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hoeijmakers J. H. (2001) Genome maintenance mechanisms for preventing cancer. Nature 411, 366–374 10.1038/35077232 [DOI] [PubMed] [Google Scholar]
- 46. Kadaja M., Isok-Paas H., Laos T., Ustav E., and Ustav M. (2009) Mechanism of genomic instability in cells infected with the high-risk human papillomaviruses. PLoS Pathog. 5, e1000397 10.1371/journal.ppat.1000397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Colotta F., Allavena P., Sica A., Garlanda C., and Mantovani A. (2009) Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis 30, 1073–1081 10.1093/carcin/bgp127 [DOI] [PubMed] [Google Scholar]
- 48. Ebule I. A., Longdoh A. N., and Paloheimo I. L. (2013) Helicobacter pylori infection and atrophic gastritis. Afr. Health Sci. 13, 112–117 10.4314/ahs.v13i1.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. de Vries A. C., van Grieken N. C., Looman C. W., Casparie M. K., de Vries E., Meijer G. A., and Kuipers E. J. (2008) Gastric cancer risk in patients with premalignant gastric lesions: a nationwide cohort study in the Netherlands. Gastroenterology 134, 945–952 10.1053/j.gastro.2008.01.071 [DOI] [PubMed] [Google Scholar]
- 50. Wong B. C., Lam S. K., Wong W. M., Chen J. S., Zheng T. T., Feng R. E., Lai K. C., Hu W. H., Yuen S. T., Leung S. Y., Fong D. Y., Ho J., Ching C. K., Chen J. S., and China Gastric Cancer Study Group. (2004) Helicobacter pylori eradication to prevent gastric cancer in a high-risk region of China: a randomized controlled trial. JAMA 291, 187–194 10.1001/jama.291.2.187 [DOI] [PubMed] [Google Scholar]
- 51. Miftahussurur M., Yamaoka Y., and Graham D. Y. (2017) Helicobacter pylori as an oncogenic pathogen, revisited. Exp. Rev. Mol. Med. 19, e4 10.1017/erm.2017.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Butcher L. D., den Hartog G., Ernst P. B., and Crowe S. E. (2017) Oxidative stress resulting from Helicobacter pylori infection contributes to gastric carcinogenesis. Cell Mol. Gastroenterol. Hepatol. 3, 316–322 10.1016/j.jcmgh.2017.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. D'Errico M., de Rinaldis E., Blasi M. F., Viti V., Falchetti M., Calcagnile A., Sera F., Saieva C., Ottini L., Palli D., Palombo F., Giuliani A., and Dogliotti E. (2009) Genome-wide expression profile of sporadic gastric cancers with microsatellite instability. Eur J. Cancer 45, 461–469 10.1016/j.ejca.2008.10.032 [DOI] [PubMed] [Google Scholar]
- 54. Kiraly O., Gong G., Olipitz W., Muthupalani S., and Engelward B. P. (2015) Inflammation-induced cell proliferation potentiates DNA damage-induced mutations in vivo. PLoS Genet. 11, e1004901 10.1371/journal.pgen.1004901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Aguilera-Aguirre L., Hosoki K., Bacsi A., Radak Z., Sur S., Hegde M. L., Tian B., Saavedra-Molina A., Brasier A. R., Ba X., and Boldogh I. (2015) Whole transcriptome analysis reveals a role for OGG1-initiated DNA repair signaling in airway remodeling. Free Radic Biol. Med. 89, 20–33 10.1016/j.freeradbiomed.2015.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ba X., and Boldogh I. (2018) 8-Oxoguanine DNA glycosylase 1: beyond repair of the oxidatively modified base lesions. Redox Biol. 14, 669–678 10.1016/j.redox.2017.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pan L., Zhu B., Hao W., Zeng X., Vlahopoulos S. A., Hazra T. K., Hegde M. L., Radak Z., Bacsi A., Brasier A. R., Ba X., and Boldogh I. (2016) Oxidized guanine base lesions function in 8-oxoguanine DNA glycosylase-1-mediated epigenetic regulation of nuclear factor kappaB-driven gene expression. J. Biol. Chem. 291, 25553–25566 10.1074/jbc.M116.751453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Fumagalli M., Rossiello F., Mondello C., and d'Adda di Fagagna F. (2014) Stable cellular senescence is associated with persistent DDR activation. PLoS ONE 9, e110969 10.1371/journal.pone.0110969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Orjalo A. V., Bhaumik D., Gengler B. K., Scott G. K., and Campisi J. (2009) Cell surface-bound IL-1alpha is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc. Natl. Acad. Sci. U S A 106, 17031–17036 10.1073/pnas.0905299106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Maddocks O. D., Scanlon K. M., and Donnenberg M. S. (2013) An Escherichia coli effector protein promotes host mutation via depletion of DNA mismatch repair proteins. mBio 4, e00152–13 10.1128/mBio.00152-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Miyoshi H., and Stappenbeck T. S. (2013) In vitro expansion and genetic modification of gastrointestinal stem cells in spheroid culture. Nat. Protoc. 8, 2471–2482 10.1038/nprot.2013.153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sato T., Vries R. G., Snippert H. J., van de Wetering M., Barker N., Stange D. E., van Es J. H., Abo A., Kujala P., Peters P. J., and Clevers H. (2009) Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459, 262–265 10.1038/nature07935 [DOI] [PubMed] [Google Scholar]
- 63. Suarez K., Lim E., Singh S., Pereira M., Joosen L. P., Ibeawuchi S.-R., Dunkel Y., Mittal Y., Ho S. B., Chattopadhyay R., Guma M., Boland B. S., Dulai P. S., Sandborn W. J., Ghosh P., et al. (2018) Dysregulation of the engulfment pathway in the gut fuels inflammatory bowel disease. bioRxiv 280172 https://www.biorxiv.org/node/86078.full
- 64. Sebrell T. A., Hashimi M., Sidar B., Wilkinson R. A., Kirpotina L., Quinn M. T., Malkoc Z., Taylor P. J., Wilking J. N., and Bimczok D. (2019) A novel gastric spheroid co-culture model reveals chemokine-dependent recruitment of human dendritic cells to the gastric epithelium. Cell Mol. Gastroenterol. Hepatol. 8, 157–171 10.1016/j.jcmgh.2019.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sebrell T. A., Sidar B., Bruns R., Wilkinson R. A., Wiedenheft B., Taylor P. J., Perrino B. A., Samuelson L. C., Wilking J. N., and Bimczok D. (2018) Live imaging analysis of human gastric epithelial spheroids reveals spontaneous rupture, rotation and fusion events. Cell Tissue Res. 371, 293–307 10.1007/s00441-017-2726-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Beswick E. J., Pinchuk I. V., Suarez G., Sierra J. C., and Reyes V. E. (2006) Helicobacter pylori CagA-dependent macrophage migration inhibitory factor produced by gastric epithelial cells binds to CD74 and stimulates procarcinogenic events. J. Immunol. 176, 6794–6801 10.4049/jimmunol.176.11.6794 [DOI] [PubMed] [Google Scholar]
- 67. Das S., Chattopadhyay R., Bhakat K. K., Boldogh I., Kohno K., Prasad R., Wilson S. H., and Hazra T. K. (2007) Stimulation of NEIL2-mediated oxidized base excision repair via YB-1 interaction during oxidative stress. J. Biol. Chem. 282, 28474–28484 10.1074/jbc.M704672200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lina T. T., Pinchuk I. V., House J., Yamaoka Y., Graham D. Y., Beswick E. J., and Reyes V. E. (2013) CagA-dependent downregulation of B7-H2 expression on gastric mucosa and inhibition of Th17 responses during Helicobacter pylori infection. J. Immunol. 191, 3838–3846 10.4049/jimmunol.1300524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sutton P., Danon S. J., Walker M., Thompson L. J., Wilson J., Kosaka T., and Lee A. (2001) Post-immunisation gastritis and Helicobacter infection in the mouse: a long term study. Gut 49, 467–473 10.1136/gut.49.4.467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Yamaoka Y., Kita M., Kodama T., Imamura S., Ohno T., Sawai N., Ishimaru A., Imanishi J., and Graham D. Y. (2002) Helicobacter pylori infection in mice: role of outer membrane proteins in colonization and inflammation. Gastroenterology 123, 1992–2004 10.1053/gast.2002.37074 [DOI] [PubMed] [Google Scholar]
- 71. Gao X., Holleczek B., Cuk K., Zhang Y., Anusruti A., Xuan Y., Xu Y., Brenner H., and Schottker B. (2019) Investigation on potential associations of oxidatively generated DNA/RNA damage with lung, colorectal, breast, prostate and total cancer incidence. Sci. Rep. 9, 7109 10.1038/s41598-019-42596-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Rodrigues D. G. B., de Moura Coelho D., Sitta A., Jacques C. E. D., Hauschild T., Manfredini V., Bakkali A., Struys E. A., Jakobs C., Wajner M., and Vargas C. R. (2017) Experimental evidence of oxidative stress in patients with l-2-hydroxyglutaric aciduria and that l-carnitine attenuates in vitro DNA damage caused by d-2-hydroxyglutaric and l-2-hydroxyglutaric acids. Toxicol. In Vitro 42, 47–53 10.1016/j.tiv.2017.04.006 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are contained within the manuscript.