

Abstract
Defective DNA damage response (DDR) signaling is a common mechanism that initiates and maintains the cellular senescence phenotype. Dysfunctional telomeres activate DDR signaling, genomic instability, and cellular senescence, but the links among these events remains unclear. Here, using an array of biochemical and imaging techniques, including a highly regulatable CRISPR/Cas9 strategy to induce DNA double strand breaks specifically in the telomeres, ChIP, telomere immunofluorescence, fluorescence in situ hybridization (FISH), micronuclei imaging, and the telomere shortest length assay (TeSLA), we show that chromosome mis-segregation due to imperfect DDR signaling in response to dysfunctional telomeres creates a preponderance of chromatin fragments in the cytosol, which leads to a premature senescence phenotype. We found that this phenomenon is caused not by telomere shortening, but by cyclic GMP–AMP synthase (cGAS) recognizing cytosolic chromatin fragments and then activating the stimulator of interferon genes (STING) cytosolic DNA-sensing pathway and downstream interferon signaling. Significantly, genetic and pharmacological manipulation of cGAS not only attenuated immune signaling, but also prevented premature cellular senescence in response to dysfunctional telomeres. The findings of our study uncover a cellular intrinsic mechanism involving the cGAS-mediated cytosolic self-DNA–sensing pathway that initiates premature senescence independently of telomere shortening.
Keywords: cyclic GMP-AMP synthase (cGAS), micronuclei, genome maintenance, DNA damage, senescence, telomere, CRISPR/Cas, DNA damage response, chromatin, cyclic GMP-AMP synthase (cGAS), genome maintenance, micronuclei
Senescence is an intrinsic cellular response that induces irreversible cell-cycle arrest. Although there are many triggers for cellular senescence, defective DNA damage response (DDR) signaling is thought to be one of the common mechanisms that induces and maintains the senescence phenotype (1). DDR signaling is often activated by telomere dysfunction due to loss of or defects in telomere-binding proteins, endonuclease (Isce1)-based introduction of double strand breaks (DSBs) in the subtelomeric regions (2), telomeric DSBs induced by TRF1 fusing with the Fokl enzyme (3), telomeric damage induced by laser microirradiation (4), targeted DNA damage at individual telomeres caused by KillerRed chromophore fused to TRF1 (5), and irreparable telomeric DSBs induced by ionizing radiation (6, 7). This DDR activation can result in chromosome fusions and subsequent progression through the cell cycle, causing breakage-bridge fusion cycles, which eventually lead to genome instability and cellular senescence (8–10). Cellular senescence due to replicative exhaustion, i.e. telomere shortening, has been studied extensively (11). However, the signaling cascade responsible for initiating cellular senescence independently of telomere shortening, but in response to telomere dysfunction, remains incompletely characterized.
Recent findings have provided mechanistic insights into how genomic instability due to defective DDR signaling triggers immune signaling. Cyclic GMP-AMP synthase (cGAS), a cytosolic DNA sensor, detects cytosolic DNA as a danger-associated molecular pattern and initiates immune responses (12). Several reports suggest that the accumulation, due to improper DDR signaling, of chromatin fragments in the form of micronuclei and the subsequent rupture of the nuclear envelope underlie cGAS activation (13–18). As a result of cGAS activation in response to cytosolic chromatin fragments, cells can undergo apoptosis, progress to cancer, or undergo irreversible cell cycle arrest, i.e. cellular senescence (19–23). However, it is not known whether DDR signaling activated in response to telomere damage can also activate the cytosolic DNA-sensing pathway and trigger cellular senescence.
In this study, we have not only developed a novel approach to induce DSBs specifically in telomeric DNA, but we also provide novel evidence that cGAS-mediated innate immune signaling is activated in response to cytosolic chromatin fragments generated by dysfunctional telomeres. Furthermore, we show that cells that harbor fused chromosomes in response to dysfunctional telomeres progress to mitosis, which leads to the accumulation of chromatin fragments in the cytoplasm. Within the cytoplasm, cGAS binds to chromatin fragments harboring DSBs and activates innate immune signaling, which often leads to premature senescence. Thus, cGAS recognizes cytosolic chromatin fragments generated in response to dysfunctional telomeres, and that triggers premature senescence independently of telomere shortening.
Results
Telomeric DNA DSBs cause micronuclei accumulation due to inefficient DNA damage response signaling
To induce DSBs specifically in telomeric DNA, we used a novel telomere DNA-directed CRISPR/Cas9 genome editing system (24). This two-component system consists of a single-guide RNA (sgRNA) and a Cas9 nuclease (Fig. 1A). The sgRNA (sgTelomere) contains a 22-nucleotide telomere targeting sequence with enhanced sgRNA-Cas9 assembly characteristics (25). This well-characterized sgRNA has been used previously to study telomere dynamics in living human cells (25). Cas9 is an endonuclease with two active DNA-cutting sites, one for each strand of the helix (24). The sgTelomere directs the Cas9 nuclease to telomeric DNA. To induce regulated DSBs within the telomeric DNA, we fused Cas9 to FK506-binding protein 12 (FKBP or DD-degradation domain) to produce dose-dependent, small-molecule–regulated post-translational stabilization. In the presence of Shield1, a small (750 Da), membrane-permeant, stabilizing ligand, the DD-tagged Cas9 (DD-Cas9) is stabilized (protected from proteasomal degradation) and accumulates in the cell. Ligand-dependent stabilization occurs very quickly (26). Additionally, Cas9 is controlled by a tetracycline-inducible (TetO) promoter. Hence, adding Shield1 to cells treated with doxycycline (DOX) stabilizes Cas9, and withdrawing Shield1 results in the rapid (within 12 h) degradation of Cas9 (Fig. 1, B, Ci, and Di). We noticed a clear co-localization of a major fraction of γH2AX foci with TRF2 foci and a telomere DNA-specific (TelC) PNA probe at 24 h after adding DOX + Shield1 (Fig. 1C, ii, top and middle panels). Under similar conditions, we observed a distinct co-localization between phosphorylated DNA-PKcs (pS2056) and TRF2 (Fig. 1C, ii, lower panel).
Figure 1.
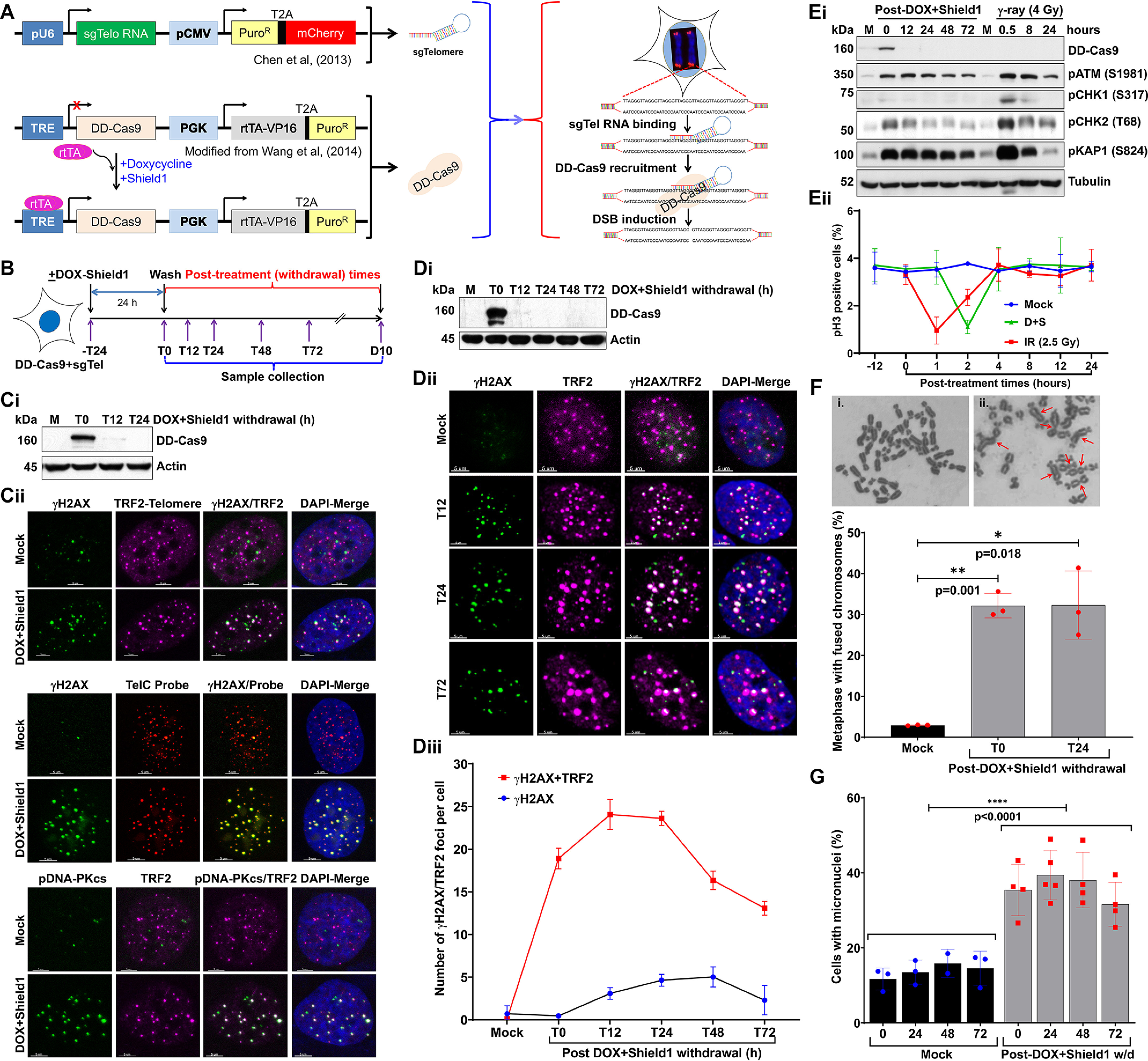
Defective repair of telomeric DSBs generates micronuclei. A, schematics showing sgTelomere RNA and DD-Cas9–expressing plasmid constructs used for inducing DSBs in the telomeric DNA region. Constitutively expressed sgTelomere RNA binds to telomeric DNA; adding DOX induces expression of DD-Cas9, and adding Shield1 stabilizes DD-Cas9. Subsequently, DD-Cas9 binds to sgTelomere RNA and induces DSBs in the telomeric DNA, TRE-tetracycline responsive element. B, schematics of the experimental design used in all panels of this figure (and Fig. 2), illustrating treatment of HT1080 cells stably expressing sgTelomere RNA and DD-Cas9 with 500 ng/ml of DOX and 1 mm Shield1 for 24 h, as well as the collection of samples at different post-DOX/Shield1 treatment times; T, hours; D, days. C, Western blotting shows DD-Cas9 stabilization at 24 h after DOX + Shield1 treatment (T0) and rapid disappearance of DD-Cas9 at 12 h after withdrawing DOX + Shield1 (T12, i). Representative images show co-localization of DSB markers γH2AX and phosphorylated DNA-PKCs (pDNA-PKcs; S2056) with telomere markers TRF2 and telomeric DNA probe (TelC PNA probe) at 24 h after DOX + Shield1 withdrawal (ii). D, Western blotting shows DD-Cas9 expression (i), representative images show co-localization of γH2AX and TRF2 foci (ii), and the line graph shows the appearance and disappearance of telomere-associated γH2AX foci (iii) in mock-treated cells and in treated cells at different post-DOX + Shield1 withdrawal times. Graph presents the mean ± S.D of 100-120 cells per condition from two independent experiments. E, Western blotting shows the extent of ATM (S1981), CHK1 (S317), and CHK2 (T68) kinases and KAP1 (S824) activation (i), and the graph shows the quantification of phosphorylated histone 3 (pH3)-positive mitotic phase cells (ii) at different times after mock (M), DOX + Shield1, and ionizing radiation (IR) treatments. Line graph presents the mean ± S.D. from two independent experiments. IR served as a positive control to check the activation of checkpoint proteins. F, representative images show fused chromosomes (red arrows) in (i) mock- and (ii) DOX + Shield1–treated cells (top). Bar graph shows the frequency of fused chromosomes in mock-treated cells and in treated cells at different post-DOX + Shield1 withdrawal times (bottom). Bar graph presents the mean ± S.D. from three independent experiments. G, bar graph shows the frequency of micronuclei at different post-DOX + Shield1 withdrawal times and in mock-treated HT1080 + sgTelomere + DD-Cas9 cells at corresponding time points. Bar graph presents the mean ± S.D. from 3 to 5 independent experiments. Statistical analyzes were performed using Student's t test (F) and two-way ANOVA (G).
Although the sgRNA that we have used for targeting telomeric repeat DNA in this study has been well characterized, we cannot rule out the recognition of interstitial telomeric repeats by this sgTelomere RNA. Therefore, we further verified the specificity of sgTelomere RNA by ChIP using anti-γH2AX antibodies and next-generation sequencing (NGS). For this purpose, we induced DSBs in the telomeric-DNA by treating U2OS-sgTelomere + DD-Cas9 cells with doxycycline and Shield1 for 24 h (Fig. S1, A–C), protein-DNA complex was cross-linked with 4% paraformaldehyde and the chromatin fraction was co-immunoprecipitated using anti-γH2AX antibodies (27). Subsequently, we purified genomic DNA and then conducted NGS. Our NGS data analysis using different bioinformatic tools revealed that about 88% of the total sequences (or peaks) were localized around the telomeric region, i.e. at the end of the chromosome (Fig. S1D). Thus, these results imply that a majority of DSBs induced by sgTel-DD-Cas9 are localized within the telomeric region.
Data indicate that telomeric DSBs (T-DSBs) are more refractory to repair than nontelomeric DSBs (6). To test telomeres' ability to repair DSBs induced by our sgTelomere RNA + DD-Cas9 system, we treated cells with DOX + Shield1 for 24 h and then immunostained the cells with TRF2 and γH2AX antibodies at different post–DOX + Shield1 withdrawal times (Fig. 1D, ii). Similar to a previous study with telomere damage induced by KillerRed-fused TRF1 (5), we found the most (20-28 TRF2 + γH2AX foci/cell) DSBs (γH2AX) in the telomeric region (TRF2) at 12-24 h (T12–T24) after removing DOX + Shield1 from the culture medium (Fig. 1D, iii). We observed that ∼55% (13 TRF2 + γH2AX foci per cell) of these telomeric DNA DSBs persisted even 72 h (T72) after the withdrawal of DOX + Shield1. Besides these γH2AX + TRF2 colocalized foci, we also detected some γH2AX foci that were not-colocalized with TRF2 (Fig. 1D, iii). These noncolocalized γH2AX foci could represent DSBs those occur as a result of endogenous metabolic reactions and replication stress (28). Thus, under our experimental conditions, a major fraction of telomeric DSBs were repairable, but some remained unrepairable.
Evidence suggests that the G2/M checkpoint allows cells enough time to repair DSBs before they enter mitosis. This checkpoint has an estimated threshold of 10-20 DSBs, below which cells can enter mitosis (29–31). We found that the number of unrepairable telomeric DSBs was below the threshold required for G2/M checkpoint activation. Therefore, we systematically examined the induction and maintenance of the G2/M checkpoint. We found that phosphorylation of ATM and CHK1/2, crucial kinases involved in G2/M checkpoint maintenance, was less activated in response to T-DSBs than to DSBs generated by ionizing radiation (Fig. 1E, i). Further examination of the G2/M checkpoint via the anti–phosphohistone 3 (pHis-3) antibody revealed that cells with T-DSBs were only transiently arrested in M phase and progressed through G2/M phase normally (Fig. 1E, ii). Overall, G2/M checkpoint activation in response to telomeric DSBs is transient and insufficient to permanently arrest cells that harbor T-DSBs in G2/M phase.
Evidence suggests that the repair of dysfunctional telomeres causes end-to-end fusion of chromosomes (32). Therefore, it is possible that the progression of cells from G2 to mitotic phase causes mis-segregation of end-to-end fused chromosomes in anaphase, which results in the generation of micronuclei. Conventional chromosome analysis of metaphase spreads revealed significantly higher levels of chromosome aberrations, specifically dicentric chromosomal (chromosome–chromosome fusion) aberrations, in cells with telomeric DSBs than in mock-treated cells (Fig. 1F). To determine the fate of fused chromosomes after cytokinesis, we examined the frequency of micronuclei formation after the induction of telomeric DSBs. We found that the frequency of micronuclei-positive cells was significantly higher in cells with telomeric DSBs than in mock-treated cells (Fig. 1G). Furthermore, we observed higher levels of micronuclei at T0–T72 h after removing DOX + Shield1 than in mock-treated samples at the corresponding time points (Fig. 1G). Overall, the DDR is more muted in response to telomeric DSBs, thus allowing cells to proceed to mitosis with residual damage or fused chromosomes, which results in the generation of micronuclei upon cytokinesis.
Telomeric DNA DSBs trigger cGAS accumulation in cytosolic chromatin fragments and cause cellular senescence
Besides serving as a marker for genomic instability and a binding substrate for DDR factors, micronuclei can recruit cGAS and activate the cytosolic DNA-sensing pathway (14–18). Therefore, we examined, first, cGAS recruitment to micronuclei in response to T-DSBs (Fig. 2A, i). As with previous reports, we noticed not only an increase (38–40%) in cGAS localization in the micronuclei generated in response to T-DSBs, but also that cGAS co-localized with γH2AX (Fig. 2A, ii). Second, we evaluated, by co-immunostaining, whether cGAS binding to micronuclei activates STING in cells harboring micronuclei whose nuclear envelope had ruptured (Lamin A/C-negative), that is, cytosolic chromatin fragments (CCFs) (Fig. 2B, i). As with previous findings, we found that most of the Lamin A/C-negative, CCF-positive cells had STING puncta in the cytosol (Fig. 2B, ii).
Figure 2.
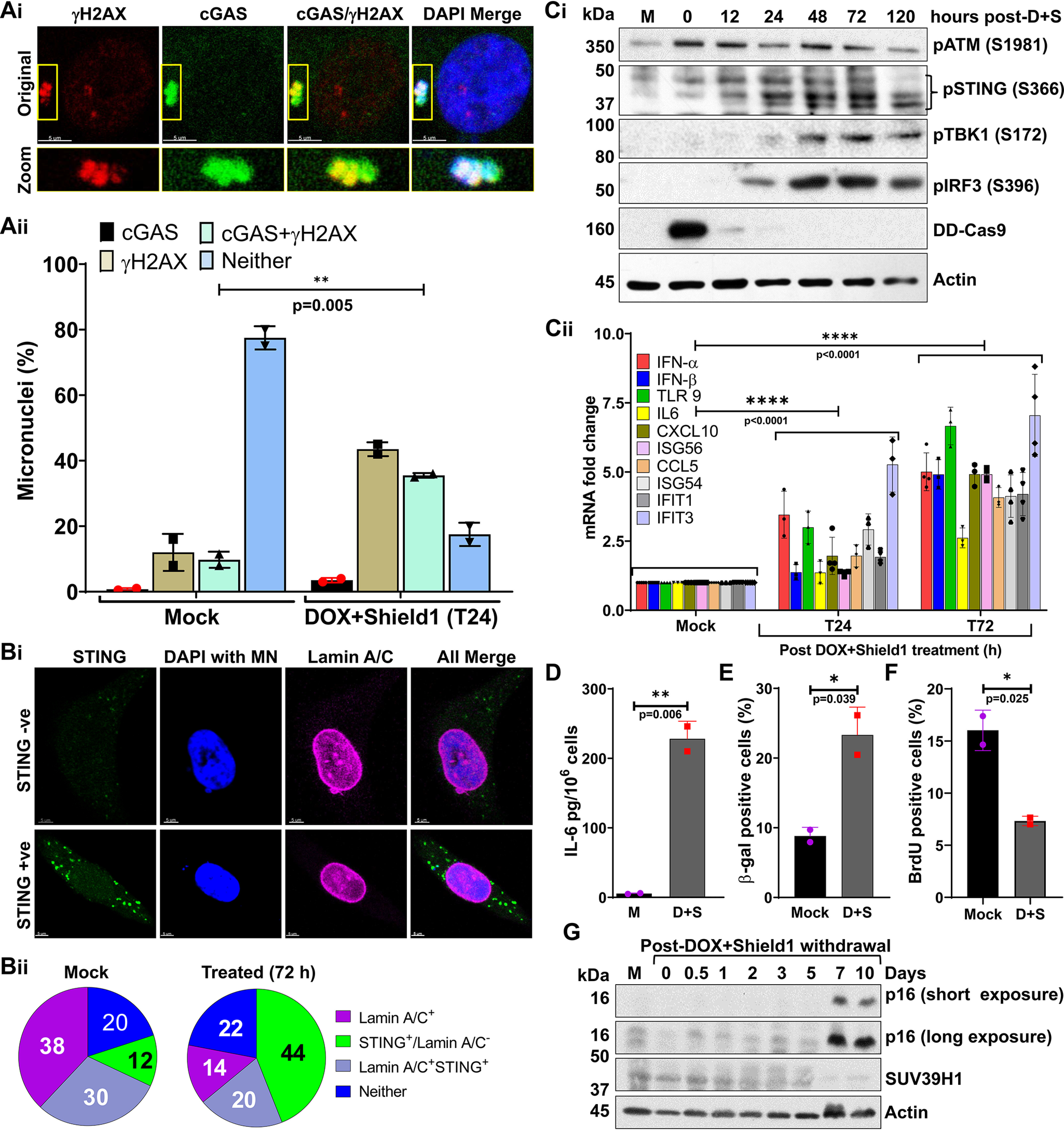
Micronuclei generated in response to defective telomeric DSB repair recruit cGAS and cause cellular senescence. A, representative images show co-localization of cGAS and γH2AX on micronuclei at 24 h after DOX + Shield1 withdrawal (i); the graph shows the frequency of micronuclei harboring γH2AX, cGAS, both or neither (ii) in mock-treated cells and in treated cells at 24 h after withdrawing DOX + Shield1. Bar graph presents the mean ± S.D. from two independent experiments. B, lack of nuclear envelope (Lamin A/C) around micronuclei correlates with STING activation in the cytosol. Representative images show the presence of STING speckles in the cytosol of cells harboring Lamin A/C-negative micronuclei (i). Pie charts show the frequency of micronuclei harboring Lamin A/C coating, STING, Lamin A/C and STING, or neither in mock-treated cells and in treated cells at 72 h after DOX + Shield1 withdrawal (ii). Pie charts represent the percentage of cells with the indicated markers from 50 different imaging fields of each experimental condition. MN, micronuclei. C and D, Western blots show increased activation of STING (S366), IRF3 (S396), and TBK1 (S172) (C, i); the graph shows increased levels of mRNA associated with immune signaling quantified by qRT-PCR (C, ii); and the graph shows IL-6 levels in cell culture supernatant measured by ELISA at the indicated times after DOX + Shield1 withdrawal (D). Bar graph presents the mean mean ± S.D. from 2 to 4 independent experiments. w/d, withdrawal; D + S, DOX + Shield1. E–G, telomeric DSBs trigger a senescence phenotype. Bar graphs show the frequency of cells positive for β-gal staining (E) and BrdU (F) at 10 days after DOX + Shield1 withdrawal. Bar graphs present the mean ± S.D. from two independent experiments. Western blots show p16 expression and SUV39H1 levels in mock-treated cells and in treated cells at different times after DOX + Shield1 withdrawal (G). Statistical analysis were performed using Student's t test (Aii, D, E, and F) and two-way ANOVA (C, ii).
Third, we evaluated whether cGAS recruitment to CCFs and STING aggregation activate downstream innate immune signaling in response to telomeric DSBs. We found that phosphorylation of STING, TBK1, and IRF3, accepted markers for the cytosolic DNA-mediated immune signaling pathway, increased after the induction of T-DSBs (Fig. 2C, i). Similarly, the expression of a panel of innate immune genes also increased after the generation of T-DSBs (Fig. 2C, ii), which correlated with the secretion of IL-6 in the culture medium (Fig. 2D).
We investigated whether the CCF-mediated activation of immune signaling we found is specific to one-cell type by testing our notion on U2OS cells, an established cell model for the alternative lengthening of telomeres pathway, which stably expressed SgTelomere RNA and DD-Cas9 (Fig. S1, A–C). Then, we assessed the generation of micronuclei and the activation of immune signaling in these cells. As with the HT1080 cells, inducing DSBs in the telomeric DNA of U2OS cells not only increased the number of micronuclei (Fig. S1A), but it also activated proteins involved in immune signaling and the expression of inflammatory cytokine genes (Fig. S1, B and C). Taking all of this evidence into account, we concluded that cGAS recruitment to CCFs correlates positively with the activation of immune signaling in response to telomeric DSBs and is independent of the nature of telomere length maintenance pathways.
Next, we examined whether cGAS-STING signaling activation in response to telomeric DSBs causes cellular senescence independently of telomere shortening. First, we counted the number of cells positive for β-gal, a well-accepted marker for the senescence phenotype. We found that the number of β-gal–positive cells was significantly higher 10 days after the induction of T-DSBs with DOX + Shield1 than in mock-treated cells (Fig. 2E). Second, we assessed the number of proliferating cells (S-Phase) by pulse labeling cultures with BrdU for 60 min before β-gal staining. We noticed that the number of BrdU-positive cells was significantly lower in DOX + Shield1–treated cells than in mock-treated cells (Fig. 2F). Third, we evaluated p16 expression and SUV39H1 levels, well-known markers of the senescence phenotype, by Western blotting (33, 34). We found elevated levels of p16 expression and reduced levels of SUV39H1 starting 7 days after the generation of T-DSBs (Fig. 2G).
Because evidence suggests that telomere shortening is a major driver of cellular senescence, we examined telomere length in HT1080-sgTelomere-DD-Cas9 cells with damaged telomeric DNA by the Telomere Shortest Length Assay (TeSLA), as described previously (35). We found that the average telomere lengths were not statistically significant between days 1 and 7 post-DOX + Shield1 treatment groups (Figs. S1Ei and Eii). Additionally, we noticed that the percentage of telomeres below 1.6-kb lengths was comparable between days 1 (20.88%) and 7 (20.11%) groups but it was relatively higher than the mock-treated (7.02%) group. We would like to point out that the TeSLA approach requires free telomeric ends for detection and the presence of large numbers of fused chromosomes in telomeric DNA DSBs groups (days 1-7) can have some impact on the detection of telomere lengths in days 1-7 groups. Nevertheless, these results imply that inducing DSBs in the telomeric DNA generates micronuclei, and this, in turn, activates cGAS-STING signaling, which culminates in cellular senescence.
Micronuclei generated in response to dysfunctional telomeres recruit cGAS and cause premature senescence
Next, we wanted to determine whether the cGAS recruitment to CCFs, the immune signaling activation, and the cellular senescence that we observed with telomeric DSBs induced by sgTelomere RNA + DD-Cas9 occurred in other models of telomere dysfunction. For this purpose, we stably expressed a tetracycline-inducible dominant-negative form of TRF2, TRF2ΔBΔM (DN-TRF2), in HT1080 cells (Fig. 3B, i). Similar to previous findings (32), inducing DN-TRF2 expression triggered telomere dysfunction-induced foci, known as TIF (Fig. 3B, ii). As with sgTelomere + DD-Cas9–induced DSBs, we observed significantly higher levels of dicentric chromosomes (Fig. 3C, i) and micronuclei generation (Fig. 3C, ii) in cells expressing DN-TRF2 than in mock-treated cells. Importantly, as with micronuclei induced by T-DSBs, we noticed that ∼30% of the micronuclei were positive for cGAS staining (Fig. 3D, i-ii). Additionally, we found that cGAS binding to CCFs of DN-TRF2–expressing cells correlated positively with the activation of STING, IRF3, and TBK1 (Fig. 3E, i), increased the percentage of micronuclei-positive cells with STING puncta (Fig. 3E, ii), elevated expression of innate immune genes (Fig. 3F), and secretion of the cytokine IL-6 (Fig. 3G). As expected, a significantly higher proportion of DN-TRF2–expressing cells than mock-treated cells exhibited β-gal staining (Fig. 3H). Furthermore, the greater number of β-gal cells correlated well with the elevated expression of p16 (Fig. 3I). Thus, cGAS binding to micronuclei triggers immune signaling and cellular senescence upon telomere dysfunction induced by DN-TRF2 expression.
Figure 3.
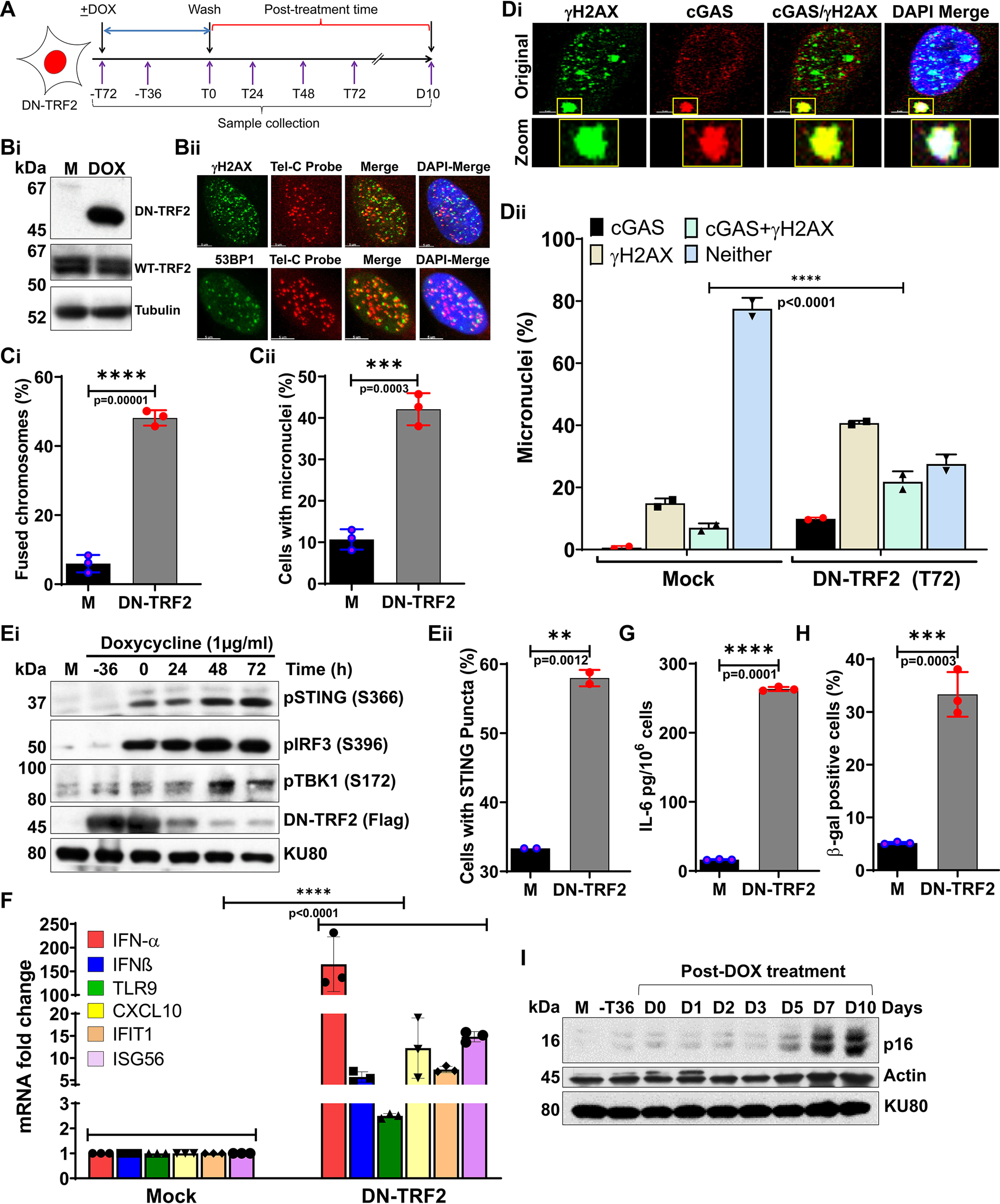
Micronuclei produced in response to dysfunctional telomeres recruit cGAS and trigger cellular senescence. A, schematics of the experimental design used in panels B-I, illustrating treatment of HT1080 cells stably expressing DN TRF2 with 500 ng/ml of DOX for 72 h and the collection of samples at different post-DOX treatment times; T, hours, D, days. B, Western blotting shows expression of DN-TRF2 cells (i), and representative indirect immunostaining images show co-localization of γH2AX and 53BP1 with telomeric DNA probe (telC PNA probe; ii) at 72 h (T72) post-DOX treatment; M, mock. C, bar graphs show quantification of percentages of chromosome–chromosome fusions (i) and micronuclei (ii) in mock-treated and 72 h (T72) post-DOX treatment samples. Bar graph presents the mean ± S.D. from three independent experiments. D, representative images show co-localization of cGAS and γH2AX in micronuclei (i), and the graph shows quantification of percentage of cGAS, γH2AX, cGAS/γH2AX, and neither positive micronuclei (ii) in DN-TRF2–expressing cells treated with either mock treatment or DOX for 72 h (T0). E–G, Western blots show phosphorylation status of STING (S366), IRF3 (S396), and TBK1 (S172) at the indicated times during and after DOX treatment (E, i); the graph shows the percentage of micronuclei-positive cells with STING puncta at 72 h after DOX treatment (E, ii); the graph shows increased levels of mRNAs associated with immune signaling quantified by qRT-PCR at 48 h post-DOX (T48) treatment time (F); and the graph shows IL-6 levels in cell culture supernatant at 48 h post-DOX (T48) treatment (G). Bar graph presents the mean ± S.D. from three independent experiments. H and I, dysfunctional telomeres cause premature senescence. Bar graph shows frequency of β-gal–stained HT1080-DN-TRF2 cells at 10 days post ± DOX treatment time (H). Bar graph presents the mean ± S.D. from 10-15 fields from three independent experiments. Representative Western blots show p16 expression at the indicated times during DOX treatment and after DOX removal (I). Statistical analysis were performed using Student's t test (Ci, Cii, Eii, G, and H) and two-way ANOVA (Dii and F).
Micronuclei generated by telomeric DNA replication stress recruit cGAS and cause premature senescence
To validate the above findings, we induced dysfunctional telomeres by using the newly-reported nucleoside analog, 6-thio-2′-deoxyguanosine (6-thio-dG) (36–38). 6-Thio-dG is recognized by telomerase and incorporated into de novo synthesized telomeres. This alters the overall chemistry, structure, and function of the shelterin complex (such as G-quadruplex–forming properties and protein recognition) (39, 40), which leads to their being recognized as TIF signals, but almost exclusively in cells expressing telomerase (36). As we reported previously (36, 37), treating cells with 6-thio-dG (3 μm) caused TIF (Fig. 4B). As with cells expressing sgTelomere + DD-Cas9 and DN-TRF2, we observed significantly higher levels of micronuclei formation in cells treated with 6-thio-dG than in mock-treated cells, with levels varying as a function of treatment time (Fig. 4C). Similarly, not only did we observe cGAS recruitment to ∼30–40% of micronuclei (Fig. 4D, i and ii), but we also noticed greater activation of STING, TBK1, and IRF3 (Fig. 4E, i), increased percentage of micronuclei positive cells with STING puncta (Fig. 4E, ii), and higher expression of innate immune genes (Fig. 4F) and IL-6 secretion (Fig. 4G) in 6-thio-dG–treated cells than in mock-treated cells. As seen in other experiments, a significantly higher proportion of 6-thio-dG (3 μm)-treated cells than mock-treated cells underwent senescence (Fig. 4H) and expressed p16 (Fig. 4I). So, cGAS recognizes a certain percentage of micronuclei, which triggers immune signaling and cellular senescence after 6-thio-dG–induced telomere dysfunction.
Figure 4.
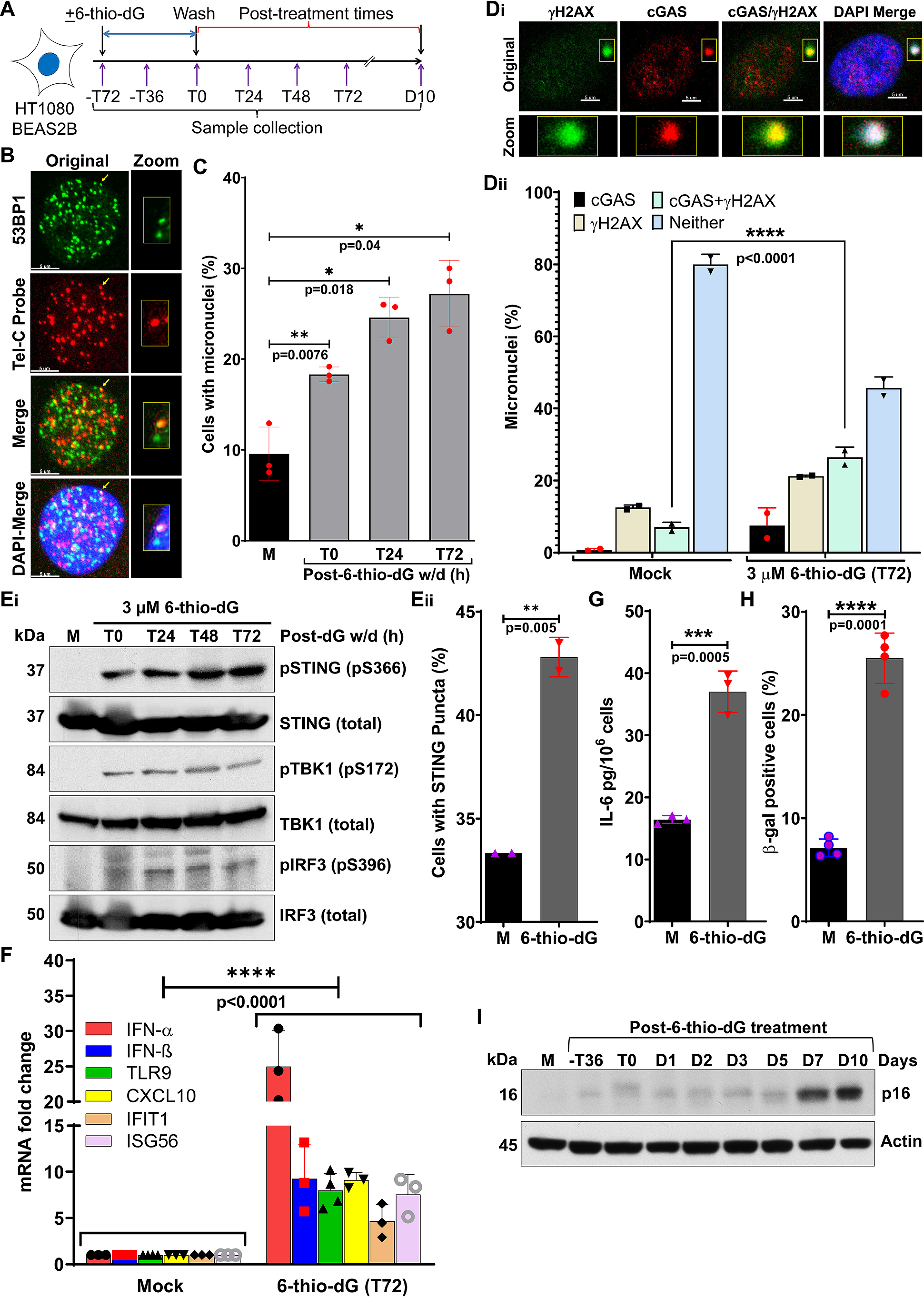
Micronuclei generated in response to telomeric DNA replication stress recruit cGAS and cause cellular senescence. A, schematics of the experimental strategy used in panels B–I, showing treatment of HT1080 cells with 3 μm 6-thio-dG for 72 h, recovery in drug-free medium, and collection of samples at different post-treatment times; T, hours; D, days. B, 6-thio-dG treatment induces dysfunctional telomeres. Representative images show co-localization of TIF marker 53BP1 with telomere DNA marker (TelC) at 48 h (T48) after 3 μm 6-thio-dG withdrawal. C, bar graph shows the quantification of micronuclei percentage in mock-treated cells and in treated cells at the indicated times after 3 μm 6-thio-dG withdrawal. Bar graph presents the mean ± S.D. from three independent experiments. D, cGAS is recruited to micronuclei induced by 6-thio-dG. Representative images show co-localization of cGAS and γH2AX in micronuclei (i), and the graph shows the quantification of percentage of micronuclei positive for cGAS, γH2AX, cGAS/γH2AX, and neither (ii) in mock-treated cells and in treated cells at 72 h (T72) after 6-thio-dG withdrawal. Bar graph presents the mean ± S.D. from two independent experiments. E–G, dysfunctional telomeres induced by 6-thio-dG (dG) activate innate immune signaling. Western blots show phosphorylation of STING (S366), TBK1 (S172), and IRF3 (S396) at the indicated times after 6-thio-dG withdrawal in BEAS-2B cells (E, i); the graph shows the percentage of micronuclei-positive cells with STING puncta at 72 h after 6-thio-dG removal (E, ii); the graph shows increased levels of mRNAs associated with immune signaling quantified by qRT-PCR at 72 h after 6-thio-dG withdrawal (F); and the graph shows IL-6 levels in cell culture supernatant at 72 h (T72) after 6-thio-dG withdrawal (G). Bar graphs present the mean ± S.D. from 2 to 4 experimental triplicates. H and I, graph shows elevated levels of β-gal staining at 10 days (D10) after 6-thio-dG withdrawal (H). Bar graph presents the mean ± S.D. of four independent experiments. Western blotting shows p16 expression at the indicated post–6-thio-dG treatment times (I). Statistical analysis were performed using Student's t test (C, Eii, G, and H) and two-way ANOVA (Dii and F).
To further demonstrate that this phenomenon is not limited to transformed cells nor to one cell type, we induced telomeric DNA damage in hTERT-immortalized human skin fibroblasts by 6-thio-dG treatment (Fig. S2A). As seen in HT1080 cells, dysfunctional telomeres not only increased the number of micronuclei but also activated immune signaling and premature senescence in hTERT-immortalized fibroblasts (Fig. S2, B–E). Overall, these results indicate that, irrespective of the mechanism that causes telomere dysfunction or the type of cells used, micronuclei resulting from dysfunctional telomeres in conjunction with cGAS recruitment initiate immune signaling, which culminates in cellular senescence.
To verify that this premature senescence phenotype caused by cytosolic chromatin fragments is distinct from senescence due to replicative exhaustion, we assessed micronuclei formation and immune signaling in primary fetal lung fibroblasts (IMR90) undergoing replicative senescence, a widely used model for replicative senescence caused by progressive telomere shortening (41). We noticed that the frequency of senescence increased significantly after we cultured the cells for 3 weeks (Fig. S2F). However, neither the number of micronuclei nor the expression of innate immune signaling pathway genes changed substantially between early (1 week) and late (2 week) passage IMR90 cells (Fig. S2, G and H). Thus, our results clearly indicate that micronuclei generated by dysfunctional telomeres activate cGAS-STING signaling and cause cellular senescence, which is distinct from replicative senescence caused by telomere shortening.
The progression from G2/M to G1 in response to dysfunctional telomeres is critical for micronuclei formation and premature senescence
The progression of G2/M phase cells with defective repair of telomeric DSBs to G1 results in the generation of micronuclei, which culminates in immune signaling-mediated premature senescence. Therefore, as we reported previously (16), we hypothesized that preventing the progression of G2/M cells with unrepaired or misrepaired T-DSBs to the G1 would prevent premature senescence. To test this notion, we used a selective small-molecule inhibitor of CDK1, RO-3306, that reversibly arrests cells at the G2/M border of the cell cycle (Fig. 5A) (42). Similar to a previous report (16), we found that blocking the cells at G2/M border did not affect the induction of telomeric DSBs (Fig. 5B), but it did prevent telomeric DSB-mediated micronuclei formation (Fig. 5C) and inflammatory signaling (Fig. 5, D and E) in HT1080 + sgTel RNA + DD-Cas9 cells upon induction of telomeric DSBs. Furthermore, blocking cells at the G2/M border also prevented the onset of premature senescence in cells with telomeric DNA damage (Fig. 5F). These results clearly demonstrate that insufficient G2/M checkpoint activation in response to telomeric DSBs is a major contributor to micronuclei generation, which results in immune signaling and premature senescence.
Figure 5.
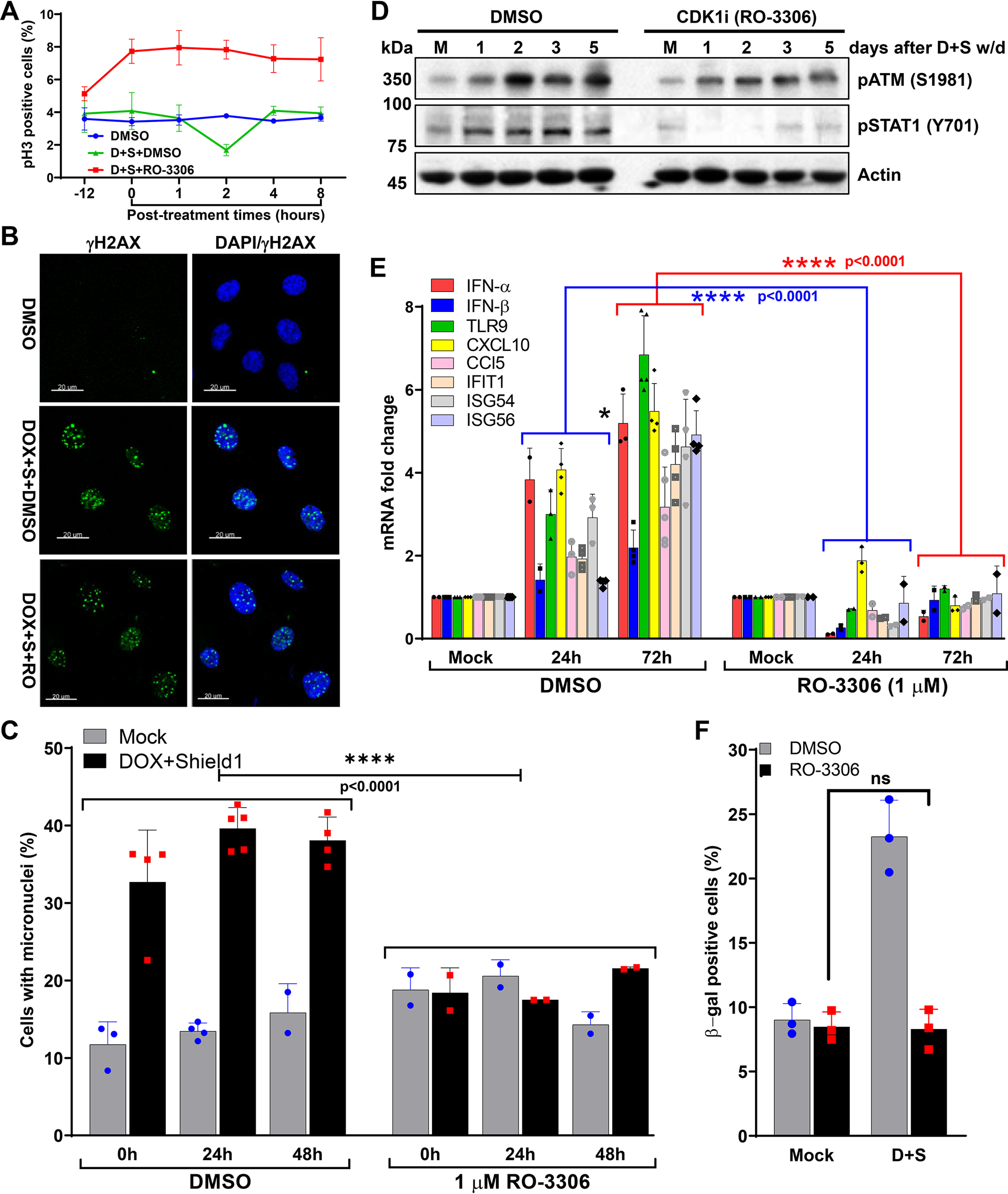
Progression from G2/M to G1 phase in response to dysfunctional telomeres is critical for micronuclei formation and premature senescence. A, the graph shows the quantification of phosphorylated histone 3 (pH3)-positive metaphase HT1080 + sgTel + DD-Cas9 cells at different times during (−12 and 0) and after (1 through 8) DMSO + DOX + Shield1 and 1 μm RO-3306 + DOX + Shield1 treatments. Line graph presents the mean ± S.D. from two independent experiments. −12, denotes 12 h after the drug exposure; 0, denotes 24 h after the drug exposure, and immediately after DOX + Shield1 removal. D + S-doxycycline + Shield1. B, representative images show the induction of telomeric DSBs, as assessed by γH2AX foci, in HT1080 cells co-expressing sgTelomere RNA and DD-Cas9 24 h after treatment with DMSO, DOX + Shield1 + DMSO or DOX + Shield1 + CDK1 inhibitor (1 μm, RO-3306). DOX + S-doxycycline + Shield1; RO-RO-3306. C, micronuclei formation is significantly reduced in response to CDK1 inhibition. Bar graph shows the frequency of micronuclei formation in HT1080 + sgTelomere RNA + DD-Cas9 cells with telomeric DSBs at different treatment conditions and times. Data presented here are the mean ± S.D. of 2 to 5 independent experiments. D–F, CDK1 inhibition attenuates immune signaling and senescence after telomeric DNA damage. Western blotting shows the attenuated activation of STAT1 (Y701) (D), which correlated well with the attenuated expression of innate immune genes in HT1080 + sgTelomere RNA + DD-Cas9 cells were treated with RO-3306 ± DOX + Shield1 (D+S) compared with cells treated with DMSO ± DOX + Shield1 (D+S) (E). Data presented here are the mean ± S.D. of 2-5 independent experiments. CDK1 inhibition prevents cells from undergoing senescence (F). Bar graphs represent the proportion of positively β-gal–stained cells, 5 days post-DOX and Shield1 (D+S) withdrawal in DMSO- and RO3306-treated cells. Data presented in the bar graphs are the mean ± S.D. of three independent experiments. D+S, doxycycline + Shield1; w/d, withdrawal; ns, not significant. Statistical analysis were performed using two-way ANOVA (C and E) and Student's t test (F).
Only a limited number of micronuclei harbor both cGAS and telomeric DNA in response to dysfunctional telomeres
To quantify the percentages of micronuclei positive for telomeric DNA and cGAS, both individually and co-localized, we treated HT1080-DN-TRF2 cells with doxycycline for 72 h, then subjected the cells to IF-FISH with Cy3-labeled telomeric DNA (TelC) probe, anti-γH2AX, and anti-cGAS antibodies (Fig. 6, A and B, images). First, we noticed that about 27–30% of the micronuclei were positive for telomeric DNA in response to dysfunctional telomeres, whereas only 8% of the micronuclei contained telomeric DNA in the mock-treated group (Fig. 6A, pie charts). These observations suggest that only a limited number of micronuclei, not all of them, harbor telomeric DNA. Second, we noticed that about 13% of the micronuclei were positive for both telomeric DNA and cGAS, and ∼43% of the telomeric DNA signal positive micronuclei harbor cGAS (Fig. 6B, pie charts). Thus, cGAS is recruited to a limited number of telomeric DNA-positive micronuclei.
Figure 6.

Telomeric DNA and cGAS are co-localized in a limited number of micronuclei. A, not all micronuclei harbor telomeric DNA. Representative images show the co-localization of γH2AX and telomeric DNA probe (TelC) (top), and the pie charts show the distribution of γH2AX, TelC, γH2AX + TelC, and neither in response to dysfunctional telomeres. HT1080-DN-TRF2 cells were treated with doxycycline for 72 h, and samples were collected at the times indicated. Cells were then subjected to IF-FISH with Cy3-labeled telomeric DNA (TelC) probe and anti-γH2AX antibodies. “Z” stack images of individual micronuclei-positive cells were imaged in each group and then examined for γH2AX alone, TelC alone, and γH2AX + TelC co-localizing micronuclei. Fifty to 60 micronuclei-positive cells were imaged in blinded fashion, and the presence or absence of TelC and γH2AX signals was quantified in blinded fashion. B, only a limited number of micronuclei harbor both cGAS and telomeric DNA. Representative images show the co-localization of cGAS and telomeric DNA probe (TelC) (top), and the pie charts show the distribution of cGAS, TelC, cGAS + TelC, and neither in response to dysfunctional telomeres. HT1080-DN-TRF2 cells were treated with doxycycline for 72 h, and samples were collected at the times indicated. Cells were then subjected to IF-FISH with Cy3-labeled telomeric DNA (TelC) probe and anti-cGAS antibodies. Z-stack images of individual micronuclei-positive cells were imaged in each group and then examined for cGAS alone, TelC alone, and cGAS + TelC co-localizing micronuclei. Fifty to 60 micronuclei-positive cells were imaged in blinded fashion, and the presence or absence of TelC and cGAS signals was quantified in blinded fashion.
cGAS is essential for causing premature senescence in response to telomere damage
To examine whether cGAS activation in response to CCFs induced by dysfunctional telomeres is responsible for cellular senescence, we depleted cGAS in BEAS-2B cells by using shcGAS RNA (Fig. 7A, inset) and caused dysfunctional telomeres by treating with 6-thio-dG. cGAS depletion did not affect the formation of micronuclei in response to telomere dysfunction (Fig. 7A, graph). However, cGAS-depleted cells exhibited lower activation of IRF3 than cGAS-proficient cells (Fig. 7B). Furthermore, cGAS-depleted cells failed to up-regulate the expression of immune signaling genes after telomere dysfunction (Fig. 7C). Ultimately, the reduced immune signaling resulting from cGAS depletion prevented cellular senescence in response to dysfunctional telomeres (Fig. 7D).
Figure 7.
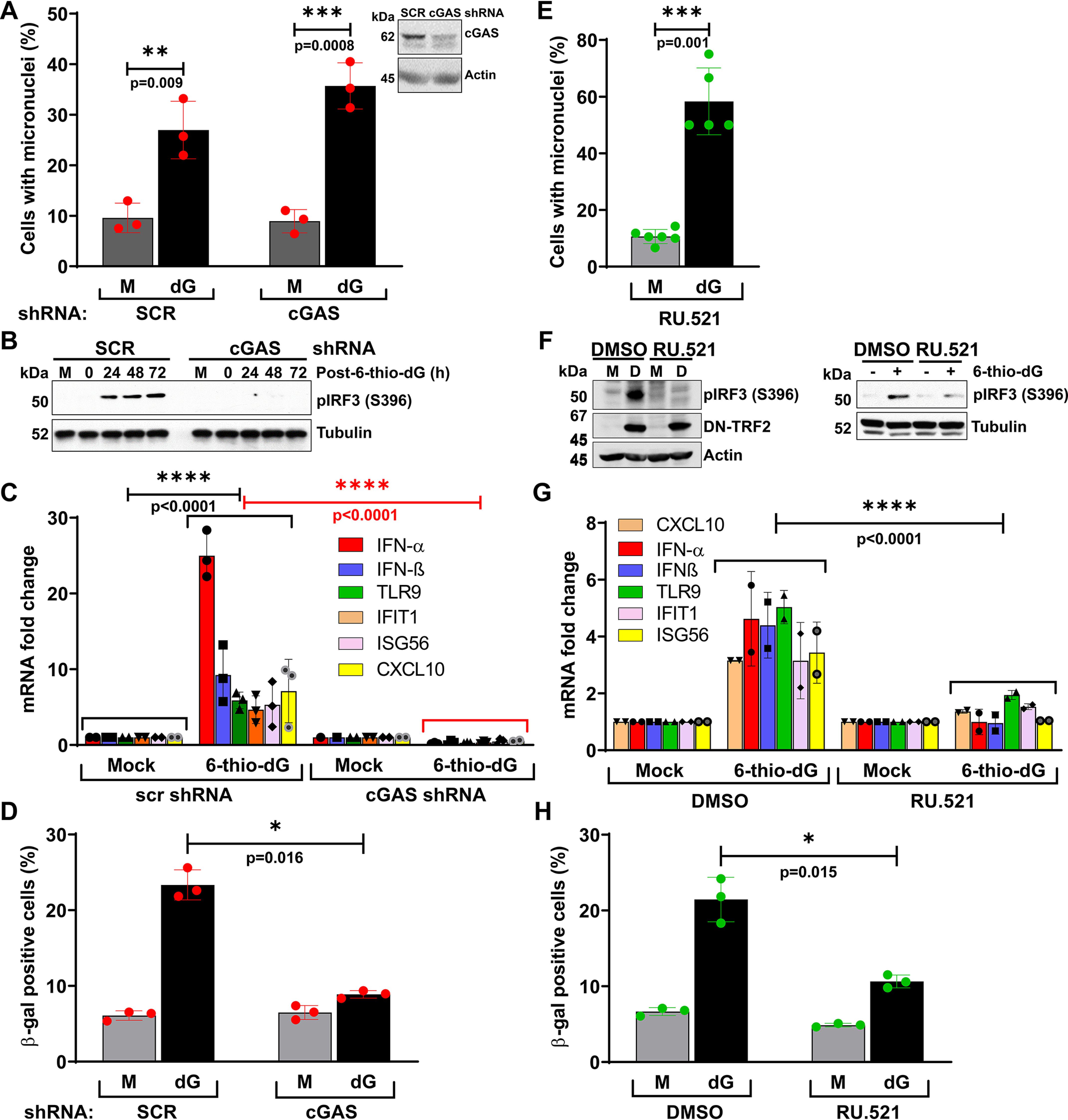
cGAS causes premature senescence phenotype in response to dysfunctional telomeres. A–D, cGAS is critical for causing the cellular senescence phenotype in response to dysfunctional telomeres. Bar graph shows frequency of micronuclei (A), Western blots show reduced activation of IRF3 (S396) (B), and graph shows reduced expression of immune pathway genes (C) in cGAS-depleted BEAS-2B cells relative to cGAS-proficient BEAS-2B cells in response to dysfunctional telomeres caused by 6-thio-dG (dG). BEAS-2B cells were treated with 6-thio-dG for 72 h, washed, recovered in drug-free medium for 72 h, and then used for different end point assessments. Bar graph shows percentage of β-gal–positive cGAS-proficient and -depleted cells at 10 days after 6-thio-dG (dG) withdrawal (D). Bar graphs present the mean ± S.D. of three independent experiments. E–H, inhibiting cGAS's enzymatic activity using a small molecule inhibitor, RU.521, reduces immune signaling and premature senescence phenotypes in response to dysfunctional telomeres. Bar graph shows the frequency of micronuclei formation in HT1080 cells treated with RU.521 with and without the induction of telomere dysfunction (dG). Bar graph presents the mean ± S.D. of 50 fields from each experimental group from 5 to 6 independent experiments (E). Western blots show decreased activation of IRF3 in HT1080-DN-TRF2 cells treated with cGAS inhibitor (RU.521) for 72 h after DOX withdrawal (F, i) and in HT1080 cells concomitantly treated with 6-thio-dG and cGAS inhibitor (RU.521) for 72 h (F, ii). Bar graphs show reduced expression of innate immune genes in HT1080-DN-TRF2 cells treated with cGAS inhibitor 72 h after the induction of DN-TRF2 expression with doxycycline (G). Bar graph presents the mean ± S.D. of two independent experiments. Bar graph shows the reduced level of β-gal staining in HT1080 cells treated with cGAS inhibitor 10 days after 6-thio-dG (I) withdrawal (H). Bar graphs present the mean ± S.D. of three independent experiments. M, mock; D, doxycycline. Statistical analysis were performed using Student's t test (A, D, E, and H) and two-way ANOVA (C and G).
To validate these results, we inhibited the enzymatic activities of cGAS using a recently identified small molecule inhibitor, RU.521 (43). RU.521 treatment did not affect micronuclei formation after 6-thio-dG treatment (Fig. 7E), but as with cGAS-depleted BEAS-2B cells, RU.521-treated HT1080-DN-TRF2 (Fig. 7F, i) and HT1080 + 6-thio-dG (Fig. 7F, ii) cells showed less IRF3 phosphorylation. Additionally, RU.521-treated HT1080-DN-TRF2 exhibited lower expression of immune signaling genes than mock-treated cells (Fig. 7G). Furthermore, the proportion of cells positive for β-gal staining was also significantly reduced when HT1080 cells were simultaneously exposed to RU.521 and 6-thio-dG (Fig. 7H). Taken together, these findings indicate that the cGAS-mediated cytosolic DNA sensing pathway is necessary to cause cellular senescence in response to dysfunctional telomeres.
Discussion
Cellular senescence restricts uncontrolled cell proliferation and plays critical roles in both aging and tumor suppression. We can distinguish at least two types of cellular senescence: replicative senescence, which depends on telomere shortening, and stress-induced premature senescence, which does not depend on telomere shortening. Although many stimuli may cause premature senescence, this study provides evidence that the accumulation of cytosolic chromatin fragments caused by dysfunctional telomeres initiates the cGAS-mediated cytosolic DNA-sensing pathway and the premature senescence phenotype. This study supports the idea that a mechanism that depends on telomere damage, but not telomere shortening, contributes to this phenotype.
It is generally accepted that most human cancers and tissue culture cells have overcome cellular senescence and achieved immortality. However, the present study shows that cancer cells and hTERT-immortalized cells can be induced to enter cellular senescence independently of telomere shortening when cGAS senses cytosolic chromatin fragments generated in response to dysfunctional telomeres. This is most likely because the cell's inability to accurately process dysfunctional telomeres, combined with an improperly functioning cell-cycle checkpoint mechanism, leads to the accumulation of cytosolic chromatin fragments and ultimately activates the cGAS-mediated cytosolic DNA sensing pathway-dependent premature senescence. Consequently, suppressing cell cycle progression reduces cellular senescence even in cells that harbor high levels of dysfunctional telomeres (Fig. 5). Similarly, it has been shown that genotoxic stress caused by ionizing radiation rapidly activates G2/M checkpoint arrest; however, release from G2 arrest occurs before DSB repair is complete, which results in micronuclei formation and activates cGAS-mediated cytosolic DNA sensing signaling (16, 31). Thus, dysfunctional telomeres together with inadequate DDR signaling contribute to micronuclei formation, cGAS-mediated immune signaling activation, and a telomere shortening-independent premature senescence phenotype.
A previous study reported that the activation of the alternative lengthening of telomere pathway causes an extra chromosomal telomere repeat DNA translocation to the cytoplasm that is recognized by the cGAS DNA sensor and thus activates the STING/TBK1 signaling cascade to trigger IRF3 phosphorylation, which results in IFNβ expression (44). However, we found that about 27–30% of the micronuclei were positive for telomeric DNA (Fig. 6A), and ∼43% of the telomeric DNA signal-positive micronuclei harbor cGAS (Fig. 6B). Furthermore, a recent report showed that dysfunctional telomeres generate cytosolic chromatin fragments and thus activate cGAS-STING signaling, which results in autophagic cell death and prevents genomic instability (45). However, we hardly detected any increase in the levels of cell death in our experimental models (Fig. S3). Nevertheless, defects in telomeres are associated with multiple cellular phenotypes, including senescence, cancer and autophagy, via the cGAS-STING signaling pathway. Specifically, our current study revealed that dysfunctional telomeres together with imperfect DDR signaling contribute to micronuclei formation, cGAS-mediated immune signaling activation, and a telomere shortening-independent premature senescence phenotype.
Together with previous findings (14, 15, 18), our study suggests that cGAS plays an essential role in initiating the signals required for telomere shortening-independent premature senescence. However, the question of how cGAS establishes the senescence phenotype remains unanswered (46). This is intriguing because cGAS recognition of cytosolic DNA fragments and the subsequent activation of innate immune signaling precede the appearance of this phenotype. In contrast, depleting cGAS or inhibiting its activity suffices to ameliorate premature senescence in the presence of cytosolic chromatin fragments due to dysfunctional telomeres. These results indicate that the initial activation of the cGAS-mediated cytosolic DNA sensing pathway suffices to establish a senescence phenotype. Reports suggest that IL-8 and IL-6 feed back to the secreting cells to reinforce senescence signaling (47, 48). Therefore, as suggested previously (46, 49), cGAS-mediated expression of pro-inflammatory factors, such as IFN-β, IL-6, and IL-8, in response to dysfunctional telomeres could serve as an autocrine signal that is required to establish a senescence phenotype. It is worth mentioning that the cell autonomous mechanism of cGAS-mediated premature senescence observed in vitro may be different from what occurs in an organism with an intact immune system.
In conclusion, this study provides evidence that micronuclei are generated when inefficient G2/M checkpoints resulting from telomere dysfunction allows cells with fused chromosomes to progress to mitosis. Upon nuclear envelope rupture, cGAS binds to these CCFs. This binding of cGAS to CCFs initiates innate immune signaling, which culminates in a premature senescence phenotype not caused by telomere shortening (Fig. 8). Further understanding how DDR factors help to regulate cGAS activation by self-DNA generated by dysfunctional telomeres will help in intervening in cases of excessive innate immune signaling activation in individuals with either loss of or dysfunction in telomere-binding factors.
Figure 8.

Model depicting the mechanism of cGAS-mediated premature senescence in response to dysfunctional telomeres.
Experimental procedures
Cell lines and culture conditions
HT1080, U2OS, BEAS-2B, and IMR90 cells were obtained from the American Type Culture Collection (ATCC, USA). Human telomerase reverse transcriptase (hTERT) immortalized 82-6 fibroblasts have been described previously (27). All cell lines were grown in standard tissue culture conditions at 5% CO2 and maintained either in Dulbecco's modified Eagle's medium (BEAS-2B) or α-modified Eagle's medium (HT1080, U2OS, IMR90, and 82-6) supplemented with 10% fetal bovine serum, 2 mm glutamine, and 0.1 mm nonessential amino acids. Mycoplasma contamination was frequently tested by PCR; only mycoplasma-free cells were used for our experiments.
Generation of stable cell lines
To make stable HT1080 + DD-Cas9/SgTelomere RNA and U2OS + DD-Cas9/SgTelomere RNA cell lines, we first infected HT1080 and U2OS cells with a lentivirus carrying tetracycline-inducible DD-Cas9 and then placed the cells under puromycin selection (0.5 and 2.5 µg/ml for HT1080 and U2OS cells, respectively). We isolated stable clones and evaluated DD-Cas9 expression by Western blotting and immunostaining using anti-FLAG antibody after treating cells with doxycycline (500 ng/ml) and Shield1 (1 μm) for 24 h. Second, we infected stable clones positive for DD-Cas9 with a lentivirus harboring sgTelomere (25) and mCherry fluorescent protein and then plated a single cell in each well of a 96-well–plate. Wells containing single colonies were selected, and clones positive for mCherry were further validated.
To make a stable HT1080 dominant-negative (DN)-TRF2 (45 to 453 amino acid regions of TRF2) cell line, we infected HT1080 cells with a lentivirus carrying tetracycline-inducible DN-TRF2, then placed the cells under puromycin selection (0.5 µg/ml). We isolated stable clones and treated them with doxycycline (1 µg/ml) for 72 h, then verified the expression of DN-TRF2 by both Western blotting and immunostaining using anti-FLAG antibodies.
To make stable BEAS-2B-shcGAS RNA cell lines, we infected BEAS-2B cells with pooled lentiviral particles carrying cGAS-specific shRNAs (Sigma) and then placed cells under puromycin selection (0.5 µg/ml). Stable clones were isolated, and the expression of cGAS was verified by Western blotting.
DNA manipulation and construction of the expression vectors
Standard molecular biology procedures were used to make all mammalian expression plasmids. A panel of human cGAS-specific shRNAs (TRCN0000128706, TRCN0000128310, TRCN0000149984, TRCN0000146282, TRCN0000150010) was purchased from Sigma. We added degradation domain (DD) to the N-terminal of Cas9 by PCR amplification, then ligated it into the NheI sites of pCW57.1-Cas9. The dominant-negative TRF2 fragment corresponding to the 45-453–amino acid region of TRF2 was PCR amplified using TRF2 cDNA and cloned into NheI/SalI sites of pCW-57.1 (Table S1).
Addgene plasmids used in this study
pSLQ1651-sgTelomere(F + E) was a gift from Bo Huang and Stanley Qi (Addgene plasmid No. 51024); pCW57.1 was a gift from David Root (Addgene plasmid No. 41393); pCW-Cas9 was a gift from Eric Lander and David Sabatini (Addgene plasmid No. 50661) (50).
Lentiviral production
293T cells were co-transfected with highly purified expression plasmids and pPLP2/pPLP1/pVSVG by using either Lipofectamine 2000 (Invitrogen), according to manufacturer's instructions, or the calcium phosphate method, as described in Kwon and Firestein (51). We collected cell culture supernatant containing viral particles 72 h after the transfection, filtered it through 0.22-μm polyvinylidene difluoride membrane filters (Millipore), and used the flow through to infect the cells.
Chemicals
We used CDK1 inhibitor RO3306 (Tocris, No. 4181), 6-thio-dG (Sigma, No. 1296), cGAS inhibitor RU.521 (AOBIOUS, No. 37877), Shield1 (Cheminpharma, No. S1-0005), and puromycin (AdipoGen).
Doxycycline and shield1 treatment
To induce DSBs in the telomeric DNA in HT1080 or U2OS-sgTelomere-DD-Cas9 cells, we co-treated cells with 500 ng/ml of doxycycline (Sigma) and 1 μm Shield1 (Cheminpharma) for 24 h. Cells were then washed three times with warm PBS and allowed to recover in regular growth media; samples were collected at different time points. To induce DN-TRF2 expression, we treated HT1080-DN-TRF2 cells with 1000 ng/ml of doxycycline (Sigma) for 72 h. Cells were then washed three times with warm PBS and allowed to recover in regular growth media; samples were collected at different post-DOX withdrawal time points.
6-Thio-2'-deoxyguanosine treatment
Twenty-four hours after plating, we treated cells with 2-10 μm 6-thio-dG for 72 h, washed three times in warm PBS, and then cultured in drug-free medium for an additional 0-15 days. Samples were collected at different post–6-thio-dG recovery times.
CDK1 inhibitor treatment
Exponentially growing HT1080-sgTelomere-DD-Cas9 cells were co-treated for 24 h with doxycycline (500 ng/ml), Shield1 (1 μm), and 1 μm RO3306 dissolved in DMSO. Cells were then washed three times with PBS and cultured in growth medium containing 1 μm CDK1 inhibitor. Samples were collected at different post-treatment times.
cGAS inhibitor (RU.521) treatment
Cells were treated with 1 μm RU.521 dissolved in DMSO for the entire duration of the experiment. Fresh drug was added to the medium every 48 h for continuous inhibition of cGAS.
Ionizing radiation (IR)
We exposed exponentially growing cells to 2-5 gray γ-rays by using a γ-irradiator (Mark 1 irradiator, JL Shepherd and Associates), as described previously (52).
Cell extract preparation and Western blotting
Whole-cell extracts were prepared by suspending cell pellets in RIPA buffer containing protease (PMSF, aprotinin, leupeptin, pepstatin A, DTT; all at 1:1000 dilutions) and phosphatase (Na3VO4 at 1:500 and NAF at 1:200 dilutions) inhibitors on ice for 30 min, then centrifuging at 14,000 rpm for 30 min at 4 °C to remove insoluble material. Whole-cell extracts (30 to 100 μg) were resolved by 6–12% SDS-PAGE, transferred onto polyvinylidene difluoride membranes, and incubated with antibodies of interest.
Antibodies used in this study
All of the primary antibodies used in this study are detailed in Table S2, including vendors' information, application, and dilution conditions. Manufacturers' validation criteria were used when applying all antibodies. For Western blotting, horseradish peroxidase-conjugated goat anti-rabbit and anti-mouse secondary antibodies were purchased from Bio-Rad and used at 1:1000 dilutions in 5% BSA or milk. For indirect immuno-fluorescence staining, fluorescent-conjugated secondary antibodies Alexa-488, Alexa-555, and Alexa-633 were purchased from Molecular Probes (Invitrogen) and used at 1:1000 dilutions.
Indirect immunostaining
Approximately 0.5-1 × 105 cells were plated in a 6-well–plate containing cover glasses and incubated for 36 h. Cells were treated with various chemicals for different time periods, as described above. Cells were fixed with 4% PFA for 20 min at room temperature at different post-treatment times and subjected to indirect immunofluorescence, as described previously (31). Briefly, cells were permeabilized in Triton X-100 (0.5% in PBS) on ice for 5 min, washed three times with PBS, incubated in blocking solution (5% goat serum in PBS) at room temperature for 60 min, and then incubated with primary antibodies (diluted in 5% goat serum) at room temperature for another 3 h or at 4 °C overnight. Then, cells were washed with wash buffer (1% BSA in PBS), incubated with appropriate secondary antibodies (1:1000 in 2.5% goat serum, 1% BSA, and PBS) at room temperature for 60 min, washed five times with 1% BSA, and mounted with mounting medium containing DAPI (Vectashield).
Telomere immunofluorescence fluorescence in situ hybridization (FISH)
Cells grown on coverslips were fixed in 4% PFA for 20 min at room temperature and then subjected to indirect immunostaining, as described above. Immediately after cells were incubated with a secondary antibody, coverslips were washed, re-fixed in 4% PFA for 10 min, washed in ethanol series solutions (70, 90, and 100%, 5 min each), and denatured with hybridization mixture (70% formamide, 1% blocking reagent, 10 mm Tris-HCl, pH 7.4, and 0.5 μm Cy3-labeled TelC PNA probe [PNA Bio; No. F1002]) for 6 min at 80 °C. Then, the samples were incubated in a 37 °C dark-humidified chamber for 4 h. Samples were then washed with Wash Solution I (70% formamide, 0.1% BSA, 10 mm Tris-HCl, pH 7.4, 2 times, 15 min apart) and Wash Solution II (water, 150 mm NaCl, 0.07% between 20 and 100 mm Tris-HCl, pH 7.4, 3 times at 10-min intervals). Samples were then mounted with mounting medium containing DAPI, and images were acquired using an LSM510 Meta confocal microscope (Zeiss).
Image acquisition and foci dissolution kinetics assay
Images were captured using an LSM 510 Meta laser scanning confocal microscope with a ×63 1.4 NA Plan-Apochromatic oil immersion objective. Images were taken of Z-sections (15-20 sections) at 0.35-µm intervals using 488-nm (EGFP and Alexa 488), 543-nm (Alexa 555), 633-nm (Alexa 633), and 405-nm (for DAPI) lasers. The tube current of the 488-nm argon laser was set at 6.1 A. The laser power was typically set to 3–5% transmission with the pinhole opened to 1-2 Airy units. To count foci, we assembled the Z-sections by using the Imaris software and analyzed them as described previously. For counting individual and co-localized TRF2 and γH2AX foci, we utilized the spot detection and co-localization functions of the Imaris software. We quantified individual and co-localized foci from images of 100-150 cells per time point from two to three independent experiments.
Micronuclei imaging and quantification
Cells fixed with 4% PFA were mounted with mounting solution containing DAPI (Vectashield). Images were acquired via Axio-Invert (Zeiss) microscope using the DAPI channel, and the exposure time ranged from 400 to 500 ms per frame. Micronuclei in 300-600 cells per experimental condition in 2-6 independent experiments were calculated in a blinded fashion via ImageJ (NIH).
Metaphase spread preparation
Chromosome aberrations were carried out as described previously (31). Twenty-four to 72 h after inducing dysfunctional telomeres, we prepared chromosomes by accumulating metaphases in the presence of 0.1 mg/ml of colcemid (Irvine Scientific) for 4 h. Cells were trypsinized and washed once with PBS and incubated in 10 ml of hypotonic solution (0.075 m KCl) at 37 °C for 15 min. Cells were pre-fixed with 1/10 volume of ice-cold methanol-acetic acid (3:1 ratio) in hypotonic solution and then centrifuged at 1000 rpm at 4 °C for 5 min. Then, the cells were fixed with methanol-acetic acid (3:1 ratio) on ice for 30 min and kept at −20°C until used. Cells were dropped onto warm (50 °C) pre-cleaned slides, stained with 5% Giemsa (KaryoMAX, GIBCO) at room temperature for 4 min, washed with dH2O, and then mounted using Vectashield mounting medium. Images were acquired via Olympus microscope (×100 objective) equipped with an Image Spot camera (Spot Imaging Solutions), and the presence or absence of fused chromosomes in 150-200 metaphase spreads were scored in a blinded fashion.
Senescence assay
Freshly fixed samples were subjected to β-gal staining using either the β-gal staining kit (Cell Signaling), following the manufacturer's instructions, or homemade β-gal staining solution, as described previously (53). Random images were acquired using the ×10 objective of a KEYENCE Microscope (BZ-X710) in a blinded manner. We counted in a blinded fashion the number of β-gal–positive cells in 20-30 random fields consisting of 1000-5000 total cells per experimental condition in two to three independent experiments.
Quantitative real-time PCR (qRT-PCR)
cDNA was synthesized from 1-3 μg of total RNA by using SuperScript III Reverse Transcriptase (18-080-051; Fisher Scientific) in a total volume of 20 µl, according to the manufacturer's instructions. We subjected the cDNA to qRT-PCR for several genes by using the primer sets (Table S3), CFX96 Touch Real-time PCR Detection System (Bio-Rad) and iTaq Universal SYBR Green Supermix (Bio-Rad, No. 1725121), according to the manufacturer's instructions. Relative gene expression was determined by the ΔΔCT method. The difference in cycle times, ΔCT, was determined as the difference between the tested gene of interest and the reference housekeeping β-actin gene. We then obtained ΔΔCT by finding the difference between the groups. The fold-change (FC) was calculated as FC = 2−ΔΔCT. All primers were purchased from Invitrogen. qRT-PCR assays were carried out in triplicate for each sample, and the mean value was used to calculate mRNA expression levels.
Bromodeoxyuridine (BrdU) labeling and detection
Cells with mock and induced dysfunctional telomeres at 10-15 days post-treatment times were labeled with 30 μm BrdU (Sigma) for 1 h in growth medium, washed three times with warm 1× PBS, and then fixed with ice-cold 70% ethanol at −20 °C overnight. Cells were then treated with ∼4 m HCl containing 0.5% Tween 20, incubated at 37 °C for 30 min, and then neutralized by washing twice with PBS. After neutralization, cells were incubated in 100 μl of PBS containing 0.1% Tween 20, 1% BSA, and anti-BrdU antibody conjugated with FITC (1:100; Molecular Probes) at room temperature for 2 h. After being washed with 1% BSA, cells were stained with DAPI (Vectashield), and images were acquired using the ×40 objective of a KEYONCE microscope. BrdU-positive cells in a total of 5000-10000 cells were counted manually in a blinded fashion.
ELISA
We collected cell culture supernatant at different post-treatment times and centrifuged at 800 × g for 5 min at room temperature to remove any cell debris. We then measured cytokine levels by ELISA using the Biolegend human IL-6 (catalog No. 430504) kit, as per the manufacturer's instructions and as described previously (54).
Measurement of telomere length by TeSLA
To analyze telomere length, we treated exponentially growing HT1080 + SgTelomere RNA + DD-Cas9 cells with doxycycline and Shield1 for 24 h, then purified the genomic DNA from mock-treated cells and from treated cells 1 and 7 days after DOX and Shield1 treatment was withdrawn. We then subjected 50 ng of genomic DNA from each group to the TeSLA technique developed by Lai et al. (35).
ChIP
To verify the specificity of sgTelomere RNA in recognizing telomeric DNA, we carried out ChIP using anti-γH2AX antibodies. Briefly, U2OS cells stably expressing sgTelomere + DD-Cas9 were treated with either DMSO or DOX + Shield1 for 24 h, washed three times in 1× PBS, and maintained in regular growth medium for 24 more hours. Cells were cross-linked with paraformaldehyde and the chromatin fraction was co-immunoprecipitated using anti-γH2AX antibodies. Subsequently, we used HiFi genome sequencing on the isolated DNA (UT Southwestern Medical Center, Genomic Sequencing Core) and obtained raw sequencing data files in the form of fastq files. We applied Bowtie2, Samtools, Bedtools, and MACS2 tools to verify the enrichment of sgTelomere RNA-telomeric region interactions. Please also see supporting information for details.
Statistics
Statistical analysis of data were performed using GraphPad Prism Software (version 8.4.2). Two tailed unpaired Student's t tests and two-way analysis of variance (ANOVA) were used for statistical analysis, and unless otherwise noted, all results are representative of at least two independent biological experiments and are reported as the mean ± S.D. GraphPad Prism (version 8.4.2) was used to create the graphs.
Data availability
All the data described in this study are contained within the article.
Supplementary Material
Acknowledgments
We thank Dr. Jonathan Feinberg for editing this manuscript.
This article contains supporting information.
Author contributions—S. A., S. B., S. M., and A. A. conceptualization; S. A. resources; S. A., S. B., S. M., D. S., K. S., M. Z., E. A. A., and A. A. data curation; S. A. software; S. A., S. B., S. M., D. S., K. S., M. Z., E. A. A., and A. A formal analysis; S. A. and A. A. supervision; S. A., S. B., S. M., D. S., and K. S. validation; S. A., S. B., S. M., K. S., H. A. S., J. W. S., and A. A. investigation; S. A., S. B., S. M., D. S., and K. S. visualization; S. A., S. B., S. M., and J. W. S. methodology; S. A., S. B., S. M., and A. A. writing-original draft; H. A. S. and A. A. funding acquisition; A. A. project administration.
Funding and additional information–This work was supported by National Institutes of Health Grants R01AG053341 (to A. A.) and R01HL115275 (to H. A. S.) and Cancer Prevention and Research Institute of Texas Grants RP190435 (to A. A. and H. A. S.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. J. W. S. is supported by NCI SPORE P50CA70907, NIH grant C06RR30414, and NCI P30CA142543. J.W.S. holds the Southland Financial Corporation Distinguished Chair in Geriatrics Research.
Conflict of interest—J. W. S. has an ownership interest (including patents) in and has an unpaid consultant/advisory board relationship with Maia Biotechnology.
- DDR
- DNA damage response
- DSB
- double strand breaks
- cGAS
- cyclic GMP-AMP synthase
- sgRNA
- single-guide RNA
- shRNA
- short hairpin RNA
- DAPI
- 4′,6-diamidino-2-phenylindole
- DOX
- doxycycline
- NGS
- next-generation sequencing
- T-DSB
- telomeric DSB
- CCF
- cytosolic chromatin fragment
- TeSLA
- telomere shortest length assay
- IL
- interleukin
- 6-thio-dG
- 6-thio-2′-deoxyguanosine
- hTERT
- human telomerase reverse transcriptase
- DN
- dominant-negative
- PFA
- paraformaldehyde
- FISH
- fluorescence in situ hybridization
- qRT
- quantitative RT
- IRF3
- interferon regulatory factor 3
- DD
- degradation domain
- FC
- fold-change
- STING
- stimulator of interferon genes.
References
- 1. Fumagalli M., and d'Adda di Fagagna F. (2009) SASPense and DDRama in cancer and ageing. Nat. Cell Biol. 11, 921–923 10.1038/ncb0809-921 [DOI] [PubMed] [Google Scholar]
- 2. Miller D., Reynolds G. E., Mejia R., Stark J. M., and Murnane J. P. (2011) Subtelomeric regions in mammalian cells are deficient in DNA double-strand break repair. DNA Repair 10, 536–544 10.1016/j.dnarep.2011.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cho N. W., Dilley R. L., Lampson M. A., and Greenberg R. A. (2014) Interchromosomal homology searches drive directional ALT telomere movement and synapsis. Cell 159, 108–121 10.1016/j.cell.2014.08.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Silva B. A., Stambaugh J. R., Yokomori K., Shah J. V., and Berns M. W. (2014) DNA damage to a single chromosome end delays anaphase onset. J. Biol. Chem. 289, 22771–22784 10.1074/jbc.M113.535955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sun L., Tan R., Xu J., LaFace J., Gao Y., Xiao Y., Attar M., Neumann C., Li G. M., Su B., Liu Y., Nakajima S., Levine A. S., and Lan L. (2015) Targeted DNA damage at individual telomeres disrupts their integrity and triggers cell death. Nucleic Acids Res. 43, 6334–6347 10.1093/nar/gkv598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fumagalli M., Rossiello F., Clerici M., Barozzi S., Cittaro D., Kaplunov J. M., Bucci G., Dobreva M., Matti V., Beausejour C. M., Herbig U., Longhese M. P., and d'Adda di Fagagna F. (2012) Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat. Cell Biol. 14, 355–365 10.1038/ncb2466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hewitt G., Jurk D., Marques F. D., Correia-Melo C., Hardy T., Gackowska A., Anderson R., Taschuk M., Mann J., and Passos J. F. (2012) Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nature Commun. 3, 708 10.1038/ncomms1708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. de Lange T. (2009) How telomeres solve the end-protection problem. Science 326, 948–952 10.1126/science.1170633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Denchi E. L. (2009) Give me a break: how telomeres suppress the DNA damage response. DNA Repair 8, 1118–1126 10.1016/j.dnarep.2009.04.013 [DOI] [PubMed] [Google Scholar]
- 10. Maciejowski J., Li Y., Bosco N., Campbell P. J., and de Lange T. (2015) Chromothripsis and kataegis induced by telomere crisis. Cell 163, 1641–1654 10.1016/j.cell.2015.11.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Harley C. B., Futcher A. B., and Greider C. W. (1990) Telomeres shorten during ageing of human fibroblasts. Nature 345, 458–460 10.1038/345458a0 [DOI] [PubMed] [Google Scholar]
- 12. Sun L., Wu J., Du F., Chen X., and Chen Z. J. (2013) Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791 10.1126/science.1232458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bartsch K., Knittler K., Borowski C., Rudnik S., Damme M., Aden K., Spehlmann M. E., Frey N., Saftig P., Chalaris A., and Rabe B. (2017) Absence of RNase H2 triggers generation of immunogenic micronuclei removed by autophagy. Human Mol. Genet. 26, 3960–3972 10.1093/hmg/ddx283 [DOI] [PubMed] [Google Scholar]
- 14. Dou Z., Ghosh K., Vizioli M. G., Zhu J., Sen P., Wangensteen K. J., Simithy J., Lan Y., Lin Y., Zhou Z., Capell B. C., Xu C., Xu M., Kieckhaefer J. E., Jiang T., et al. (2017) Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 550, 402–406 10.1038/nature24050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gluck S., Guey B., Gulen M. F., Wolter K., Kang T. W., Schmacke N. A., Bridgeman A., Rehwinkel J., Zender L., and Ablasser A. (2017) Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat. Cell Biol. 19, 1061–1070 10.1038/ncb3586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Harding S. M., Benci J. L., Irianto J., Discher D. E., Minn A. J., and Greenberg R. A. (2017) Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 548, 466–470 10.1038/nature23470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mackenzie K. J., Carroll P., Martin C. A., Murina O., Fluteau A., Simpson D. J., Olova N., Sutcliffe H., Rainger J. K., Leitch A., Osborn R. T., Wheeler A. P., Nowotny M., Gilbert N., Chandra T., et al. (2017) cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 548, 461–465 10.1038/nature23449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yang H., Wang H., Ren J., Chen Q., and Chen Z. J. (2017) cGAS is essential for cellular senescence. Proc. Natl. Acad. Sci. U.S.A. 114, E4612–E4620 10.1073/pnas.1705499114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kranzusch P. J., Lee A. S., Berger J. M., and Doudna J. A. (2013) Structure of human cGAS reveals a conserved family of second-messenger enzymes in innate immunity. Cell Rep. 3, 1362–1368 10.1016/j.celrep.2013.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kranzusch P. J., and Vance R. E. (2013) cGAS dimerization entangles DNA recognition. Immunity 39, 992–994 10.1016/j.immuni.2013.11.012 [DOI] [PubMed] [Google Scholar]
- 21. Civril F., Deimling T., de Oliveira Mann C. C., Ablasser A., Moldt M., Witte G., Hornung V., and Hopfner K. P. (2013) Structural mechanism of cytosolic DNA sensing by cGAS. Nature 498, 332–337 10.1038/nature12305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li X., Shu C., Yi G., Chaton C. T., Shelton C. L., Diao J., Zuo X., Kao C. C., Herr A. B., and Li P. (2013) Cyclic GMP-AMP synthase is activated by double-stranded DNA-induced oligomerization. Immunity 39, 1019–1031 10.1016/j.immuni.2013.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang X., Wu J., Du F., Xu H., Sun L., Chen Z., Brautigam C. A., Zhang X., and Chen Z. J. (2014) The cytosolic DNA sensor cGAS forms an oligomeric complex with DNA and undergoes switch-like conformational changes in the activation loop. Cell Rep. 6, 421–430 10.1016/j.celrep.2014.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J. A., and Charpentier E. (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 10.1126/science.1225829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen B., Gilbert L. A., Cimini B. A., Schnitzbauer J., Zhang W., Li G. W., Park J., Blackburn E. H., Weissman J. S., Qi L. S., and Huang B. (2013) Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell 155, 1479–1491 10.1016/j.cell.2013.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Banaszynski L. A., Chen L. C., Maynard-Smith L. A., Ooi A. G., and Wandless T. J. (2006) A rapid, reversible, and tunable method to regulate protein function in living cells using synthetic small molecules. Cell 126, 995–1004 10.1016/j.cell.2006.07.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Su F., Mukherjee S., Yang Y., Mori E., Bhattacharya S., Kobayashi J., Yannone S. M., Chen D. J., and Asaithamby A. (2014) Nonenzymatic role for WRN in preserving nascent DNA strands after replication stress. Cell Rep. 9, 1387–1401 10.1016/j.celrep.2014.10.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mehta A., and Haber J. E. (2014) Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harb. Perspect. Biol. 6, a016428 10.1101/cshperspect.a016428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Deckbar D., Birraux J., Krempler A., Tchouandong L., Beucher A., Walker S., Stiff T., Jeggo P., and Lobrich M. (2007) Chromosome breakage after G2 checkpoint release. J. Cell Biol. 176, 749–755 10.1083/jcb.200612047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Terzoudi G. I., Manola K. N., Pantelias G. E., and Iliakis G. (2005) Checkpoint abrogation in G2 compromises repair of chromosomal breaks in ataxia telangiectasia cells. Cancer Res. 65, 11292–11296 10.1158/0008-5472.CAN-05-2148 [DOI] [PubMed] [Google Scholar]
- 31. Asaithamby A., Hu B., and Chen D. J. (2011) Unrepaired clustered DNA lesions induce chromosome breakage in human cells. Proc. Natl. Acad. Sci. U.S.A. 108, 8293–8298 10.1073/pnas.1016045108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. van Steensel B., Smogorzewska A., and de Lange T. (1998) TRF2 protects human telomeres from end-to-end fusions. Cell 92, 401–413 10.1016/S0092-8674(00)80932-0 [DOI] [PubMed] [Google Scholar]
- 33. Brookes S., Rowe J., Gutierrez Del Arroyo A., Bond J., and Peters G. (2004) Contribution of p16(INK4a) to replicative senescence of human fibroblasts. Exp. Cell Res. 298, 549–559 10.1016/j.yexcr.2004.04.035 [DOI] [PubMed] [Google Scholar]
- 34. Zhang W., Li J., Suzuki K., Qu J., Wang P., Zhou J., Liu X., Ren R., Xu X., Ocampo A., Yuan T., Yang J., Li Y., Shi L., Guan D., et al. (2015) Aging stem cells: a Werner syndrome stem cell model unveils heterochromatin alterations as a driver of human aging. Science 348, 1160–1163 10.1126/science.aaa1356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lai T. P., Zhang N., Noh J., Mender I., Tedone E., Huang E., Wright W. E., Danuser G., and Shay J. W. (2017) A method for measuring the distribution of the shortest telomeres in cells and tissues. Nat. Commun. 8, 1356 10.1038/s41467-017-01291-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mender I., Gryaznov S., Dikmen Z. G., Wright W. E., and Shay J. W. (2015) Induction of telomere dysfunction mediated by the telomerase substrate precursor 6-thio-2'-deoxyguanosine. Cancer Discov. 5, 82–95 10.1158/2159-8290.CD-14-0609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mender I., LaRanger R., Luitel K., Peyton M., Girard L., Lai T. P., Batten K., Cornelius C., Dalvi M. P., Ramirez M., Du W., Wu L. F., Altschuler S. J., Brekken R., Martinez E. D., et al. (2018) Telomerase-mediated strategy for overcoming non-small cell lung cancer targeted therapy and chemotherapy resistance. Neoplasia 20, 826–837 10.1016/j.neo.2018.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sengupta S., Sobo M., Lee K., Senthil Kumar S., White A. R., Mender I., Fuller C., Chow L. M. L., Fouladi M., Shay J. W., and Drissi R. (2018) Induced telomere damage to treat telomerase expressing therapy-resistant pediatric brain tumors. Mol. Cancer Therap. 17, 1504–1514 10.1158/1535-7163.MCT-17-0792 [DOI] [PubMed] [Google Scholar]
- 39. Csaszar K., Spackova N., Stefl R., Sponer J., and Leontis N. B. (2001) Molecular dynamics of the frame-shifting pseudoknot from beet western yellows virus: the role of non-Watson-Crick base-pairing, ordered hydration, cation binding and base mutations on stability and unfolding. J. Mol. Biol. 313, 1073–1091 10.1006/jmbi.2001.5100 [DOI] [PubMed] [Google Scholar]
- 40. Štefl R., Špacková N., Berger I., Koča J., and Šponer J. (2001) Molecular dynamics of DNA quadruplex molecules containing inosine, 6-thioguanine and 6-thiopurine. Biophys. J. 80, 455–468 10.1016/S0006-3495(01)76028-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sherwood S. W., Rush D., Ellsworth J. L., and Schimke R. T. (1988) Defining cellular senescence in IMR-90 cells: a flow cytometric analysis. Proc. Natl. Acad. Sci. U.S.A. 85, 9086–9090 10.1073/pnas.85.23.9086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Vassilev L. T., Tovar C., Chen S., Knezevic D., Zhao X., Sun H., Heimbrook D. C., and Chen L. (2006) Selective small-molecule inhibitor reveals critical mitotic functions of human CDK1. Proc. Natl. Acad. Sci. U.S.A. 103, 10660–10665 10.1073/pnas.0600447103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vincent J., Adura C., Gao P., Luz A., Lama L., Asano Y., Okamoto R., Imaeda T., Aida J., Rothamel K., Gogakos T., Steinberg J., Reasoner S., Aso K., Tuschl T., et al. (2017) Small molecule inhibition of cGAS reduces interferon expression in primary macrophages from autoimmune mice. Nat. Commun. 8, 750 10.1038/s41467-017-01770-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chen Y. A., Shen Y. L., Hsia H. Y., Tiang Y. P., Sung T. L., and Chen L. Y. (2017) Extrachromosomal telomere repeat DNA is linked to ALT development via cGAS-STING DNA sensing pathway. Nat. Struct. Mol. Biol. 24, 1124–1131 10.1038/nsmb.3498 [DOI] [PubMed] [Google Scholar]
- 45. Nassour J., Radford R., Correia A., Fuste J. M., Schoell B., Jauch A., Shaw R. J., and Karlseder J. (2019) Autophagic cell death restricts chromosomal instability during replicative crisis. Nature 565, 659–663 10.1038/s41586-019-0885-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li T., and Chen Z. J. (2018) The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med. 215, 1287–1299 10.1084/jem.20180139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Acosta J. C., O'Loghlen A., Banito A., Guijarro M. V., Augert A., Raguz S., Fumagalli M., Da Costa M., Brown C., Popov N., Takatsu Y., Melamed J., d'Adda di Fagagna F., Bernard D., Hernando E., and Gil J. (2008) Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 133, 1006–1018 10.1016/j.cell.2008.03.038 [DOI] [PubMed] [Google Scholar]
- 48. Kuilman T., Michaloglou C., Vredeveld L. C., Douma S., van Doorn R., Desmet C. J., Aarden L. A., Mooi W. J., and Peeper D. S. (2008) Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 133, 1019–1031 10.1016/j.cell.2008.03.039 [DOI] [PubMed] [Google Scholar]
- 49. Umbreit N. T., and Pellman D. (2017) Genome jail-break triggers lockdown. Nature 550, 340–341 10.1038/nature24146 [DOI] [PubMed] [Google Scholar]
- 50. Wang T., Wei J. J., Sabatini D. M., and Lander E. S. (2014) Genetic screens in human cells using the CRISPR-Cas9 system. Science 343, 80–84 10.1126/science.1246981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kwon M., and Firestein B. L. (2013) DNA transfection: calcium phosphate method. Methods Mol. Biol. 1018, 107–110 10.1007/978-1-62703-444-9_10 [DOI] [PubMed] [Google Scholar]
- 52. Asaithamby A., and Chen D. J. (2009) Cellular responses to DNA double-strand breaks after low-dose γ-irradiation. Nucleic Acids Res. 37, 3912–3923 10.1093/nar/gkp237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Debacq-Chainiaux F., Erusalimsky J. D., Campisi J., and Toussaint O. (2009) Protocols to detect senescence-associated β-galactosidase (SA-βgal) activity, a biomarker of senescent cells in culture and in vivo. Nat. Protoc. 4, 1798–1806 10.1038/nprot.2009.191 [DOI] [PubMed] [Google Scholar]
- 54. Millrine D., Tei M., Gemechu Y., and Kishimoto T. (2016) Rabex-5 is a lenalidomide target molecule that negatively regulates TLR-induced type 1 IFN production. Proc. Natl. Acad. Sci. U.S.A. 113, 10625–10630 10.1073/pnas.1611751113 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the data described in this study are contained within the article.