

Abstract
Background
Glioblastoma (GBM) is an aggressive malignant brain tumor where median survival is approximately 15 months after best available multimodal treatment. Recurrence is inevitable, largely due to O6 methylguanine DNA methyltransferase (MGMT) that renders the tumors resistant to temozolomide (TMZ). We hypothesized that pretreatment with bortezomib (BTZ) 48 hours prior to TMZ to deplete MGMT levels would be safe and tolerated by patients with recurrent GBM harboring unmethylated MGMT promoter. The secondary objective was to investigate whether 26S proteasome blockade may enhance differentiation of cytotoxic immune subsets to impact treatment responses measured by radiological criteria and clinical outcomes.
Methods
Ten patients received intravenous BTZ 1.3 mg/m2 on days 1, 4, and 7 during each 4th weekly TMZ‐chemotherapy starting on day 3 and escalated from 150 mg/m2 per oral 5 days/wk via 175 to 200 mg/m2 in cycles 1, 2, and 3, respectively. Adverse events and quality of life were evaluated by CTCAE and EQ‐5D‐5L questionnaire, and immunological biomarkers evaluated by flow cytometry and Luminex enzyme‐linked immunosorbent assay.
Results
Sequential BTZ + TMZ therapy was safe and well tolerated. Pain and performance of daily activities had greatest impact on patients' self‐reported quality of life and were inversely correlated with Karnofsky performance status. Patients segregated a priori into three groups, where group 1 displayed stable clinical symptoms and/or slower magnetic resonance imaging radiological progression, expanded CD4+ effector T‐cells that attenuated cytotoxic T‐lymphocyte associated protein‐4 and PD‐1 expression and secreted interferon γ and tumor necrosis factor α in situ and ex vivo upon stimulation with PMA/ionomycin. In contrast, rapidly progressing group 2 patients exhibited tolerised T‐cell phenotypes characterized by fourfold to sixfold higher interleukin 4 (IL‐4) and IL‐10 Th‐2 cytokines after BTZ + TMZ treatment, where group 3 patients exhibited intermediate clinical/radiological responses.
Conclusion
Sequential BTZ + TMZ treatment is safe and promotes Th1‐driven immunological responses in selected patients with improved clinical outcomes (Clinicaltrial.gov (NCT03643549)).
Keywords: bortezomib, immune checkpoint, MGMT, recurrent GBM, temozolomide, Th1/Th2 cytokine ratios
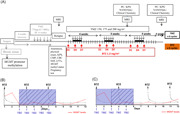
Schematic of trial schedule. A, Timeline showing BORTEM‐17 treatment regimen. Bortezomib administered intravenous at days 1, 4, and 7 (48 hours pretreatment to deplete MGMT protein) before target TMZ 200 mg/m2 dose for 5 days (from and including days 3‐7), repeated in six cycles. In n = 3 patients per each dose 150 vs 175 vs 200 mg/m2 TMZ in dose pathfinding, safety evaluation. Clinical chemistry for renal, hepatic, and bone marrow monitoring for toxicity based on CTCAE v. 4.03. MRI tumor monitoring radiological response assessment based on RANO criteria. Rationale for sequential treatment schedule based on preclinical data. B, sequential administration in BORTEM‐17 clinical trial vs (C) previous studies where BTZ on days 1, 4, 8, and 11 was given concomitantly with TMZ from day 1 to 5, when MGMT levels were high. Dashed boxes mark days when TMZ doses might be more effective, (B) all five doses vs (C) three doses every month. BTZ, bortezomib; CBC Diff, complete blood count with differential test; CMP, comprehensive metabolic panel; KPS, Karnofsky performance score; LFT, liver function test; MGMT, O6‐methyl guanine DNA methyltransferase; MRI, magnetic resonance imaging; NANO, neurologic assessment in neuro‐oncology; QoL, quality of life; TMZ, temozolomide.
1. INTRODUCTION
Glioblastoma (GBM) is the most prevalent and aggressive brain tumor in humans. Standard first‐line treatment for fit patients consists of surgery aiming for maximal safe tumor resection, 1 , 2 followed by radiochemotherapy with daily temozolomide (TMZ) administered concomitantly with external beam fractionated ionizing radiation for 6 weeks to a total dose of 60 Gy in 30 fractions. Adjuvant TMZ is administered for 5 days every 4th week for up to six cycles. 3 Despite this multimodal treatment, median overall survival is only approximately 15 months. 3 , 4 , 5 For patients harboring GBM with unmethylated O6 methylguanine DNA methyltransferase (MGMT) promoter, 2‐year survival rate is only 14% compared to 46% for those with methylated MGMT promoter 6 and thus, MGMT promoter status is both prognostic and predictive of treatment response to TMZ chemotherapy. Tumor recurrence is inevitable and median time to neoplastic progression is approximately 6.9 months. 3 There is no standard treatment after recurrence, therefore, options are dependent on physician's choice or practice at the given institution. The European society for medical oncology guidelines recommend that patients be treated within investigational protocols. 7
Profound cellular and molecular heterogeneity typifies GBM 8 , 9 thus, combination therapy to mitigate drug resistance has become the benchmark of neuro‐oncology. Nevertheless, the alkylating chemotherapeutic agents, temozolomide and lomustine are still the backbone of systemic glioma therapy. 10 Several clinical studies have explored blocking of MGMT to improve TMZ efficacy in treatment resistant GBM. 11 , 12 , 13 , 14 Transcription factors and coactivators, including nuclear factor κB (NF‐κB), 15 tumor protein 53 (TP53), 16 , 17 transcription factor Sp1, 16 hypoxia inducible factor‐1α (HIF‐1α), 18 cyclic adenosine monophosphate response element‐binding protein, 19 MGMT enhancer binding protein 20 and signal transducer and activator of transcription 3 (STAT3) 21 have also been investigated for their potential to regulate MGMT protein expression. It has been established that NF‐κB binding sites are present within the MGMT promoter region, that MGMT messenger RNA (mRNA) is induced by NF‐κB/p65, and that MGMT expression correlates with NF‐κB activation regardless of promoter methylation status. 15 , 22 NF‐κB activation requires 26S proteasomal processing. 23
Bortezomib (BTZ) is a proteasome inhibitor that has been approved for treatment of multiple myeloma, mantle cell lymphoma and trialed in early phase trials for treatment of GBM. It blocks the chymotryptic activity of the 26S proteasome and prevents degradation of misfolded proteins or abundant short‐lived proteins such as transcription factors that may be important in regulation of tumor and immune cell differentiation, proliferation, and apoptosis. We recently showed that GBM pretreatment with BTZ for 48 hours prohibited phosphorylation of IkBα, resulting in reduced nuclear translocation of the activated phosphorylated NF‐κB p65/RelA subunit. This correlated with reduced MGMT protein and mRNA expression by 70%‐80% and sensitized the GBM cells to TMZ chemotherapy. 22 Two other studies also investigated MGMT depletion via mechanisms associated with activation of NFκB, MAPK, STAT3, and HIF‐1α pathways after BTZ treatment. 16 , 18 As well as depleting the tumor's cytoprotective mechanisms, agents that simultaneously promote tumor recognition by cells of the immune system may be attractive anticancer candidates. Indeed, we demonstrated that natural killer (NK) cells treated with BTZ exhibited more mature, activated and cytotoxic CD57+CD16dimCD69+ phenotype, and that efficacy of combination treatment of GBM cells with BTZ + NK cells in vitro and in vivo was augmented by tumor necrosis factor related ligand (TRAIL)‐receptor interactions, as well as tumor expression of stress‐ligands recognized by activating NKG2D receptor. 24 Sequential combination of BTZ 48 hours before TMZ 164 mg/m2 treatment, depleted MGMT mRNA in vivo, attenuated tumor growth and significantly prolonged animal survival. 22 Thus, this phase IB of our BORTEM‐17 clinical trial (NCT03643549) was launched to investigate whether pretreatment of recurrent GBM patients with BTZ 1.3 mg/m2 on days 1, 4, and 7, to deplete MGMT levels 48 hours before TMZ, commencing on days 3 to 7 every 4th week, might sensitize MGMT unmethylated GBM to TMZ chemotherapy. We assessed the maximal safe escalated TMZ dose administered in sensitization schedule with BTZ and whether treatment enhanced differentiation of cytotoxic immune subsets to impact treatment responses.
2. MATERIALS AND METHODS
2.1. Treatment plan
The study protocol (Figure 1A) was approved by the regional ethical board for Western Norway (2017/2084) and the Norwegian Medicines Agency (17/17445‐17). All eligible patients signed the approved consent form for study participation before undergoing any study related procedures. Inclusion/exclusion criteria are summarized in the Supporting Information and further details available on: https://clinicaltrials.gov/ct2/show/NCT03643549?term=Bortezomib+and+temozolomide&cond=GBM&draw=2&rank=1
Figure 1.
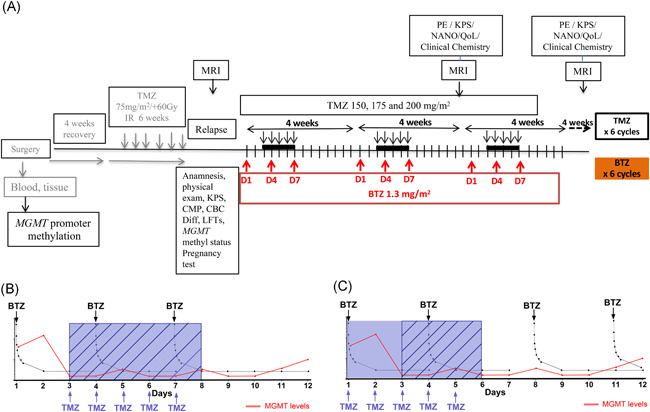
Schematic of trial schedule. A, Timeline showing BORTEM‐17 treatment regimen. Bortezomib administered intravenous at days 1, 4, and 7 (48 hours pretreatment to deplete MGMT protein) before target TMZ 200 mg/m2 dose for 5 days (from and including days 3‐7), repeated in six cycles. In n = 3 patients per each dose 150 vs 175 vs 200 mg/m2 TMZ in dose pathfinding, safety evaluation. Clinical chemistry for renal, hepatic, and bone marrow monitoring for toxicity based on CTCAE v. 4.03. MRI tumor monitoring radiological response assessment based on RANO criteria. Rationale for sequential treatment schedule based on preclinical data. B, sequential administration in BORTEM‐17 clinical trial vs (C) previous studies where BTZ on days 1, 4, 8, and 11 was given concomitantly with TMZ from day 1 to 5, when MGMT levels were high. Dashed boxes mark days when TMZ doses might be more effective, (B) all five doses vs (C) three doses every month. BTZ, bortezomib; CBC Diff, complete blood count with differential test; CMP, comprehensive metabolic panel; KPS, Karnofsky performance score; LFT, liver function test; MGMT, O6‐methyl guanine DNA methyltransferase; MRI, magnetic resonance imaging; NANO, neurologic assessment in neuro‐oncology; QoL, quality of life; TMZ, temozolomide
Ten patients (n = 8 males and n = 2 females) were enrolled between September 2018 and October 2019 and their characteristics are summarized in Table 1. BTZ 1.3 mg/m2 was administered as 30 seconds bolus intravenous injection on days 1, 4, and 7 during each 4th week chemotherapy cycle. The first three patients received 150 mg/m2 TMZ per oral 5 days/wk starting on day 3 after BTZ administration in the first cycle and thereafter 175 and 200 mg/m2 every 4 weeks in cycles 2 and 3, respectively (Figure 1A). If all three patients at a given TMZ dose did not develop a dose limiting toxicity (ie, grade 3 or 4 on Common Terminology Criteria for Adverse Events [CTCAE]), the next cohort of three patients would be treated at the next TMZ dose level. The ultimate aim was to attain combination treatment with BTZ and TMZ at 200 mg/m2 which was closest to the most effective dose in the preclinical study. 22 Total treatment duration per patient was estimated for 6 to 12 months unless disease progression.
Table 1.
BORTEM‐17 patient characteristics
| Patient ID | Age | Sex | MGMT status | % of methylation (pyrosequence) mean ± SEM | KPS | Molecular pathology | No. of recurrence before recruitment | Treatment after recurrence before recruitment | Treatment after withdrawn | No. of BORTEM cycle after recruitment | OS, mo | Survival after first recurrence | Survival after 2nd recurrence | Survival after recruitment | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| KIR2DS4 | IDH1 | ATRX | TP53 | ||||||||||||||
| P01 | 45 | M | UM | 3 ± 1.1 | 90 | Func/Del | Mut | Mut | Mut | 2 | SRS + TMZ | LAVA | 3 | 27.3 | 13.4 | 7.1 | 6.6 |
| P02 | 53 | F | UM | 2 ± 0.6 | 80 | Func/Func | Wt | Wt | n/a | 2 | S + SRS | No antineoplastic treatment | 2 | 24.0 | 9.6 | 6.7 | 6.2 |
| P03 | 54 | M | UM | 2 ± 0.6 | 80 | Del/Del | Wt | n/a | n/a | 2 | TMZ | Lomustin + RT | 6 | 33.8 | 18.6 | 15.2 | 14.5 |
| P04 | 40 | F | UM | 3 ± 1.7 | 90 | Del/Del | Mut | Wt | Wt | 1 | No antineoplastic treatment | No antineoplastic treatment | 2 | 13.8 | 4.7 | 2.1 | 3.8 |
| P05 a | 35 | M | UM | 2 ± 0.3 | 90 | Del/Del | Mut | Mut | Mut | 2 | S | LAVA | 2 | 30.6 | 25.0 | 14.7 | 14.4 |
| P06 | 35 | F | UM | 3 ± 0.7 | 90 | Func/Func | Wt | Wt | Wt | 2 | SRS | SRS | 2 | 15.2 | 6.1 | 4.9 | 3.2 |
| P07 a | 26 | M | UM | 1 ± 0.2 | 100 | Del/Del | Mut | Mut | Wt | 1 | No antineoplastic treatment | LAVA | 2 | 28.0 | 11.3 | 8.9 | 10.6 |
| P08 | 57 | M | UM | 2 ± 0.4 | 80 | Func/Del | Wt | Wt | n/a | 1 | No antineoplastic treatment | LAVA | 2 | 18.7 | 4.6 | 2.3 | 4.2 |
| P09 | 58 | M | UM | 2 ± 0.8 | 90 | Del/Del | Wt | n/a | n/a | 2 | SRS | No antineoplastic treatment | 2 | 12.8 | 3.9 | 2.6 | 2.2 |
| P10 | 52 | M | UM | 4 ± 1.3 | 80 | Func/Del | Wt | n/a | n/a | 2 | S | No antineoplastic treatment | 2 | 11.2 | 6.4 | 3.3 | 2.4 |
| Median survival, mo | 21.4 | 8.0 | 5.8 | 5.2 | |||||||||||||
Abbreviations: ATRX, α‐thalassemia/mental retardation, X‐linked; BORTEM‐17, bortezomib and temozolomide trial 2017; Del, KIR2DS4*00101 deleted (Del/Del homozygous); F, female; Func, KIR2DS4*00101 full length (Func/Func homozygous, Func/Del heterozygous); IDH1, isocitrate dehydrogenase‐1; KIR2DS4, killer cell immunoglobulin like receptor, two Ig domains and short cytoplasmic tail 4; KPS, Karnofsky performance status; LAVA, combination of lomustine, avastin and vincristine; M, male; Mut, mutated; n/a, not available; OS, (overall survival in months) from date of first operation; RT, radiotherapy; S, surgery; SRS, stereotactic radio surgery; Survival after first recurrence, survival after date of MRI showing 1st progression; Survival after recruitment, survival after date of recruitment to last date of update; Survival after second recurrence, survival after date of MRI showing 2nd progression; TMZ, temozolomide; TP53, tumour protein 53; UM MGMT, unmethylated O6 methylguanine DNA methyltransferase; Wt, wild type.
Alive.
2.2. NeuroImaging
Brain magnetic resonance imaging (MRI) was performed using the 3T Siemens MAGNETOM Prisma (Siemens Healthineers, Munich, Germany) at the Radiology Department at Haukeland University Hospital (details of sequences and quantification of radiologic tumor growth are reported in the Supporting Information). MRI was undertaken no earlier than 2 weeks before enrollment to confirm disease progression using T2 and T1 MRI sequences with and without gadolinium contrast agent (Clariscan; GE Healthcare) and reported by the study neuroradiologist based on RANO criteria. 25 MRI was also undertaken at day 56, after two cycles of BTZ and TMZ treatment, and thereafter every 8th week until endpoint evaluation at 6 months.
2.3. Clinical evaluation and self‐reported quality of life
Clinical evaluation including neurological assessment according to Neuro‐Oncology (NANO) scale, 26 EQ‐5D‐5L quality of life questionnaire, 27 and Karnofsky performance status (KPS) were recorded during consultations every 4th week and before MRI evaluations. Hematological tests were obtained within 1 week of study enrollment and subsequently within each treatment cycle on days 1, 4, 7, 11, 15, 22, and 28 before commencing the next cycle. Molecular pathology including MGMT promoter methylation, IDH1, TP53, and ATRX mutation analysis was undertaken at the pathology department. Adverse events were graded using the National Cancer Institute CTCAE version 4.03.
2.4. Immune monitoring
Immune monitoring was performed on peripheral blood mononuclear cells (PBMCs) before, during and after BTZ + TMZ treatment in time course analyses on day 1, 4, 7, 11, 15, and 22 of the first treatment cycle. PBMCs were stained with fluorescent conjugated primary antibody mix for NK and T cells (Table S1 and Figure S1), data acquired on BD LSR FORTESSA (BD Biosciences, Trondheim, Norway) and analysed with FlowJo software version 10 (Tree Star Inc, Ashland, OR) as detailed in the Supporting Information.
2.5. Statistical analysis
Linear mixed effects model regression analysis was used to analyse tumor volumes, EQ‐5D‐5L health scores and their correlation with NANO scores and ability to predict KPS throughout the course of treatment using Stata version 15.1 (StataCorp LLC, Texas). One‐ and two‐way analysis of variance (ANOVA) with Bonferroni correction for multiple testing was used to analyse immunomonitoring and enzyme‐linked immunosorbent assay data using Graphpad PRISM 8.0 software (La Jolla, CA). Two‐sided P values less than .05 were considered significant.
3. RESULTS
3.1. BORTEM‐17 trial schedule
Our treatment regimen was based on a previous preclinical study where we demonstrated that BTZ pretreatment of GBM cells for 48 hours depleted MGMT levels and sensitized to TMZ. Thus, the sequential schedule was adopted, administering intravenous bolus 1.3 mg/m2 BTZ on days 1, 4, and 7 before target TMZ 200 mg/m2 dose for 5 days (starting from and including days 3‐7), repeated in six cycles (Figure 1A). Previous studies had administered BTZ on days 1, 4, 8, and 11, where this was concomitantly administered with TMZ from day 1 to 5 (when MGMT levels would be high the first 2 days in MGMT unmethylated patients). Our rationale was thus to maximize the therapeutic potential of all five TMZ doses by only administering the chemotherapy 48 hours after BTZ when MGMT levels would be lowest (Figure 1B,C).
3.2. BORTEM‐17 patient characteristics
The basic clinical and molecular characteristics of the patients are indicated in Table 1. Our patient cohort included eight males and two females with a median age of 49 years (range, 26‐58) and median KPS score 80 (range, 80‐100) at enrollment. In accordance with the inclusion criteria, all patients had unmethylated MGMT promoter (quantitatively <4% methylation), while n = 4/10 (40%) were IDH1 R132H mutant with accompanying TP53 and ATRX mutations. Five patients possessed the KIR2DS4*00101 allele as homozygous or heterozygous alleles that we previously demonstrated to be an independent good prognostic factor. 28 At the time of primary diagnosis, five patients had undergone gross total resection while the remaining five had subtotal resection or biopsy. Primary tumor location was temporal (2/10), frontal (3/10), parietal lobes (3/10), and in two patients multifocal. Seven patients (70%) were enrolled onto the study protocol after their second recurrence and three of them had had secondary surgery. Three patients were treated at first tumor recurrence.
3.3. BTZ administered in combination with TMZ is appropriately metabolized
Next, we investigated time course pharmacokinetic distribution of BTZ after administration to establish that in these heavily pretreated GBM patients, BTZ was metabolized as previously described. 29 The plasma concentration of BTZ showed a biphasic decline, with a fast distribution phase and a slower elimination phase consistent with a two‐compartment model (Figure 2A). There was also an increase in plasma concentrations at all time‐points on day 7 where estimated C max was twice as high, (P < .001; Figure 2A and Table 2) compared to day 1. Both the increased C max and area under the curve values (ng/mL) were consistent with previous findings in adult patients receiving BTZ on days 1, 4, 8, and 11. 30 Taken together, the data indicate that BTZ was metabolized similarly to chemotherapy naïve patients.
Figure 2.
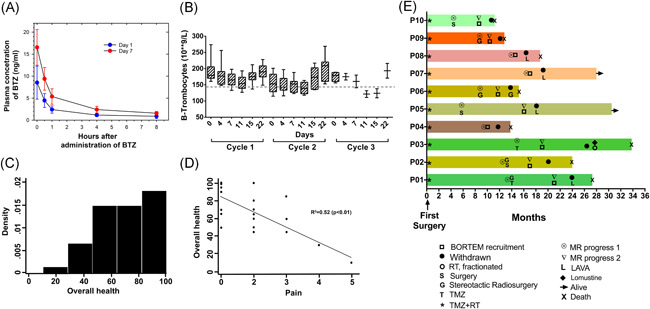
Clinical, radiological and quality of life response. A, Pharmacokinetic analysis of bortezomib concentration (ng/mL) in plasma of patients following five timepoints on day 1 and 7 of treatment. B, Thrombocyte counts during the first three treatment cycles. Data represent the mean ± SEM from n = 10 patients. C, Density scores for perception of overall health on EQ‐5D‐5L questionnaire. D, Correlation of self‐reported overall health and levels of pain on EQ‐5D‐5L. E, Swimmer plot of individual patients depicting treatment start/stop times of all trial patients, aligned according to their first surgery. Bars represent survival in months from first surgery, first and second MRI progression. Rightward arrow indicates that the patient was still alive at the time of final data collection
Table 2.
Estimated bortezomib concentration in plasma at day 1 and 7 in patients receiving BTZ at day 1, 4, and 7
| Day 1 | Day 7 | |||
|---|---|---|---|---|
| Patient ID | C max | AUC | C max | AUC |
| P01 | 6.10 | 460.2 | 19.35 | 2403.7 |
| P02 | 5.56 | 1178.2 | 16.17 | 2845.3 |
| P03 | 15.47 | 2344.6 | 15.14 | 2071.0 |
| P04 | 6.37 | 889.8 | 14.52 | 1883.7 |
| P05 | 7.76 | 1068.3 | 16.05 | 2538.4 |
| P06 | 4.43 | 1096.9 | 10.97 | 2189.9 |
| P07 | 5.51 | 1125.2 | 14.19 | 1818.6 |
| P08 | 9.42 | 1403.1 | 14.39 | 2738.8 |
| P09 | 13.37 | 1513.8 | 18.80 | 2985.6 |
| P10 | 11.37 | 2159.9 | 25.90 | 4015.3 |
| Average | 8.54 | 1324.0 | 16.55*** | 2549.0*** |
| STD | 3.748 | 701.90 | 4.059 | 380.43 |
Note: Plasma concentration at t = 0 hour (Cmax) and AUC at day 1 and 7 in 10 patients receiving BTZ at day 1, 4, and 7.
Abbreviation: AUC, area under the curve; STD, standard deviation
Denotes significance at P < .001, respectively, using two‐sided Student's t test to compare mean C max or AUC from day 1 and 7.
3.4. Combination BTZ and TMZ is well tolerated
Sequential therapy with BTZ and TMZ was safe, well tolerated and thrombocyte levels consistently normalized by day 22 of each cycle (Figure 2B and Table 3). There was no dose limiting toxicity and most adverse events were mild to moderate in severity (grades 1 or 2). Only 6/10 patients reported toxicities that included grade 2 fatigue (3/10 patients: 30%) where in one patient it was worsening of pre‐existing fatigue. Grade 2 pruritus and urticaria was experienced by 1/10 patient (10%), while grade 1 paresthesia, nausea, gastrointestinal infection, intracranial hemorrhage, and diarrhea/constipation were recorded in 5/10 (50%) patients, respectively (Table 3).
Table 3.
Observed adverse events
| Any grade | Grade 1 | Grade 2 | ||||
|---|---|---|---|---|---|---|
| Adverse events | n | % | n | % | n | % |
| Fatigue | 3/10 | 30 | 0 | 0 | 3/10 | 30 |
| Nausea | 1/10 | 10 | 1/10 | 10 | 0 | 0 |
| Constipation | 1/10 | 10 | 1/10 | 10 | 0 | 0 |
| Pruritus | 1/10 | 10 | 0 | 0 | 1/10 | 10 |
| Paresthesia | 1/10 | 10 | 1/10 | 10 | 0 | 0 |
| Urticaria | 1/10 | 10 | 0 | 0 | 1/10 | 10 |
| Vomiting | 1/10 | 10 | 1/10 | 0 | 0 | 0 |
| Gastrointestinal infection (suspected) | 1/10 | 10 | 1/10 | 0 | 0 | 0 |
| Intracranial hemorrhage | 1/10 | 10 | 1/10 | 0 | 0 | 0 |
| Diarrhea | 1/10 | 10 | 1/10 | 0 | 0 | 0 |
| Thrombocytes | 8/10 | 80 | 8/10 | 80 | 0 | 0 |
| Total number of patient with adverse events | 6/10 | 60 | 5/10 | 50 | 4/10 | 40 |
| Adverse events due to BTZ treatment | 3/10 | 30 | 2/10 | 20 | 1/10 | 10 |
| Adverse events due to TMZ treatment | 5/10 | 50 | 1/10 | 10 | 4/10 | 40 |
| Worsening of a pre‐existing condition | 2/10 | 20 | 1/10 | 10 | 1/10 | 10 |
Note: Bold values indicate percentage.
3.5. Pain had greatest impact on self‐reported quality of life
We used the EQ‐5D‐5L questionnaire to detect clinically relevant differences in health‐related quality of life and sought to identify variables that most predicted the patients' general quality of life. The patients' overall health evaluations were skewed towards higher values indicating generally good perceived health (Figure 2C). However, unadjusted analyses showed a significant relation between overall health perception and usual activities of daily living (b‐coefficient, −18.26; 95% confidence interval [CI] [−30.904 to −5.020]; P = .005). An increase of pain perception by one point on the EQ‐5D‐5L was associated with 17 times reduced overall health assessment (b‐coefficient, −17.592; 95% CI [−24.909 to −10.276]; P < .001). Likewise, perceived increased anxiety or depression was greatly correlated with diminished overall health assessment (b‐coefficient, −13.2048; 95% CI [−22.842 to −3.254]; P = .009). We next performed a stepwise analysis to determine which variable had the most significant impact on patients' overall health perception and found that pain (b‐coefficient, −13.293; 95% CI [−18.567 to −8.019]; P < .001) and ability to perform usual activities (b‐coefficient, −11.309; 95% CI [−15.090 to −7.528]; P < .001) had the most impact (Figure 2D). However, the time point of the patients' pain experience was not significant (b‐coefficient, 1.628; 95% CI [0.002‐3.255]; P = .050) indicating that this was not associated with their study participation. Moreover, analysis of individual effects of mobility (P = .001), self‐care (P = .003), ability to perform usual daily activities (P = .001), pain (P < .0001), depression/anxiety (P = .012) all showed inverse association with KPS. Median KPS after second evaluation was 80 (range, 70‐100) and 80 (range, 60‐90) at the third evaluation. In stepwise adjusted analyses, self‐care (b‐coefficient, −4.893; 95% CI [−9.654 to −0.132]; P = .044) and ability to perform usual daily activities (b‐coefficient, −4.39; 95% CI [−6.967 to −1.826]; P = .001) were most correlated with KPS. Furthermore, stepwise adjusted correlation analyses of NANO scale variables with KPS, revealed inverted association of vision (P = .020), strength (P < .0001), ataxia (P < .0001), and language (P = .010) with KPS, however, ultimately, the ability to perform usual daily activities (b‐coefficient, −3.587; 95% CI [−5.534 to −1.640]; P < .0001), ataxia (b‐coefficient, −14.03; 95% CI [−21.554 to −6.514]; P < .0001), and strength (b‐coefficient, −8.587; 95% CI [−14.896 to −2.278]; P = .008) had the greatest effect on KPS.
3.6. Clinical and radiological responses
As of April 2020, 10 patients met the RANO radiological or clinical progression criteria, eight were withdrawn after two cycles. Two patients completed three and six cycles of treatment. Of these 10 patients, eight died of tumor progression while on second line treatment or after study medication was discontinued (Figure 2E and Table 1). After study treatment withdrawal five patients (P01, P03, P05, P07, P08) were transitioned to lomustine or the combination lomustine, avastin and vincristine (LAVA), whereas four patients did not receive further anti‐neoplastic treatment, (P02, P04, P09, P10; Table 1). Notwithstanding, patient‐02 was alive for 4.5 months after study withdrawal without further anti‐neoplastic treatment, before his death. Patient‐03 had radiological stable disease according to RANO criteria after the second cycle and completed the six cycles of treatment (Figures 2E and 3A). After a treatment break, he had a third MRI progression, and received reirradiation and lomustin and continued lomustine every 6th week. He died 14.5 months after inclusion in the BORTEM‐17 study and had overall survival of 33.8 months. Patient‐05 and ‐07 are still alive and have survived 14.4 and 10.6 months, respectively, post recruitment. No problems of noncompliance were recorded. After recruitment, median survival of the 10 patients was 5.2 months and median overall survival was 21.4 months (Table 1).
Figure 3.
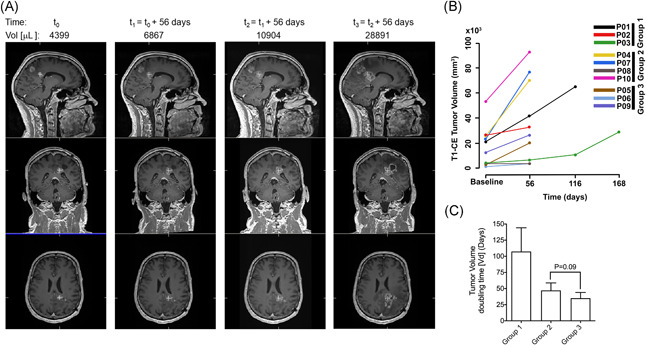
Tumour growth and population doubling time. A, Coregistered 3D T1 weighted gadolinium contrast enhanced serial MR image of patient‐03 (IDHwt; MGMT UM, 54 years male) treated for 6 months, time indicated in days and tumour volume in µL. B, Mean of 3D measured tumour volumes in mm3 from T1‐weighted MR images with contrast of all patients, and (C) tumor population doubling time (in days) for group 1, 2, and 3 patients
3.7. Slower tumour growth kinetics in group 1 patients with positive MRI and clinical responses
The patients were grouped a priori based on clinical or MRI progression, where group 1 patients (01, 02, and 03) were characterized by long survival, stable clinical symptoms and/or MRI confirmed stable disease during BORTEM‐17 treatment. Group 2 patients (04, 07, 08, and 10) exhibited rapid clinical deterioration and MRI confirmed progressive disease, while group 3 patients (05, 06, and 09) exhibited mixed positive and negative clinical and radiological effects that did not fit the profiles of neither group 1 nor group 2. The median TMZ dose received for group 1, 2, and 3 patients was 150, 175, and 175 mg/m2, respectively. Patient‐03 from group 1 had a left splenium contrast enhancing lesion and achieved clinical and radiological stable disease (20% progression from baseline according to RANO criteria) 56 days after commencing treatment (Figure 3A), subsequently completing all six therapy cycles. However, volumetric quantification of contrast enhancing tumour growth on 3D T1 weighted MRI revealed overall change of growth over time in all patients (b‐coefficient, 12 403.7; 95% CI [4880.8‐19 926.7]; P = .001; Figure 3B). Mean‐ tumour volumes between the groups were not statistically different, although compared to baseline volumes, there was significant change after 112 days (b‐coefficient, 24 220; 95% CI [−6435.8‐42 004.2]; P = .008) and 168 days (b‐coefficient, 30 828.4; 95% CI [7512.3‐54 144.6]; P = .01) in group 1 patients. Likewise, group 2 patients' MRI‐tumour volumes after 56 days were significantly different from group 1 baseline volumes, (b‐coefficient, 43 825.6; 95% CI [14 670.8‐72 980.4]; P = .003). Group 1 patient tumours exhibited the slowest growth rate indicated by longest population doubling time (approximately 107.4 days) compared to group 2 and 3 patient tumours (47.1 and 35.1 days, respectively; Figure 3C).
3.8. Immunological mechanisms of patients with positive clinical responses
BTZ‐induced tumour death generates immunogenic antigens and sensitizes to dendritic cell immunotherapy. 24 , 31 , 32 Thus, we investigated the patients' T cell activation and maturation status in blood and cytokines circulating in their plasma at baseline, after exposure to BTZ only (within the first 8 hours after administration), BTZ + TMZ (on days 4‐7) and during the 3 weeks recovery period (days 11, 15, and 22) in cycle 1. We also investigated responses to stimulation with PMA/ionomycin or autologous tumour cells ex vivo of patients' PBMCs obtained at baseline (BTZ naïve) versus those obtained during recovery (day 22‐56) after initial exposure to BTZ + TMZ.
Remarkably, group 2 patients had substantially higher levels of multiple immune tolerising Th2 cytokines in plasma compared to group 1 and 3 patients. During BTZ + TMZ treatment fourfold to sevenfold higher concentrations of interleukin 4 (IL‐4) was observed in group 2 compared to group 3 patients (P < .05; Figure 4A). Group 2 patients also had consistently higher IL‐5/IFNγ and IL‐4/IFNγ ratios (P < .05) from baseline throughout treatment compared to group 3 patients. In contrast, group 3 patients had threefold higher IL‐10 plasma levels compared to group 1 and group 2 patients during BTZ and TMZ treatment (two way ANOVA, P < .0001 and P < .001, respectively; Figure 4A), implying a tolerising effect of TMZ chemotherapy through IL‐10, in particular for group 3 patients.
Figure 4.
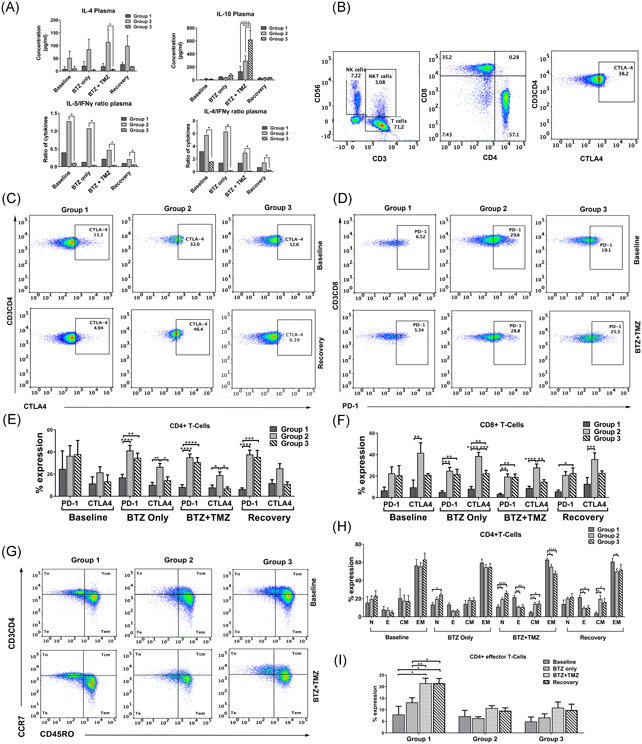
Immune mechanisms and augmented differentiation of CD4+ and CD8+ T cell subsets in patients with positive clinical responses. A, Cytokine concentrations in pg/mL in patients' plasma, (top panels) IL‐4 and IL‐10, and (bottom panels) the ratio between the average concentrations of IL‐5 and IL‐4 against IFNγ during treatment. B, Representative dot plots showing (from left to right) patient NK and T cell lymphocytes in CD56 vs CD3 gates, CD8+ vs CD4+ T cell subsets and CD3+CD4+ T cells expressing CTLA‐4. C, Representative dot plots showing CD3+CD4+ T cell subsets expressing CTLA‐4 and (D) Representative dot plots showing CD3+CD8+ T cell subsets expressing PD‐1. E, Mean ± SEM % of CD3+CD4+ T cells expression of PD‐1 and CTLA‐4 in group 1, 2, and 3 patients before, during and after treatment with BTZ and TMZ. F, Mean ± SEM % of CD3+CD8+ expressing PD‐1 and CTLA‐4 before and after treatment with BTZ and TMZ. G, Dot plots showing CCR7 vs CD45RO within CD3+CD4+ T cell subsets in group 1, 2, and 3 patients, before and during treatment with BTZ and TMZ. H, Mean ± SEM % expression of naïve (N), effector (E), central memory (CM) and effector memory (EM) CD4+ T‐cells, and (I) CD4+ T‐effector cells before, during and after treatment with BTZ and TMZ in group 1, 2, and 3 patients. *P < .05, **P < .01, ***P < .001, ****P < .0001, n = 10 patients. BTZ, bortezomib; IFNγ, interferon γ; IL, interleukin; NK, natural killer; TMZ, temozolomide
3.9. Rapidly progressing patients are characterized by Th2 immune responses and tolerised CD8+ T cell phenotype
Given their profound Th2‐cytokine driven anti‐inflammatory responses, we investigated their relative expression of cytotoxic T‐lymphocyte–associated antigen 4 (CTLA‐4) and programmed death 1 (PD‐1) immune checkpoints on CD4+ and CD8+ T cell subsets (Figure 4B‐F) as an indication of negative immune regulation and T cell exhaustion. Although there were no significant differences in numbers of CD4+CTLA‐4+ T cells at baseline (Figures 4C and 4E), CD4+CTLA‐4+ T cells were increased by twofold to threefold after BTZ and during combination with BTZ and TMZ in the rapidly progressing group 2 patients compared to group 1, (Figures 4C and 4E; one way ANOVA, P < .01, P < .05) respectively, and compared to group 3 patients, (P < .05, respectively). PD‐1+ CD4+ T cells were increased by twofold to fourfold in group 2 compared to group 1 patients during combination BTZ with TMZ and later, during recovery (one way ANOVA, P < .0001, both, Figures 4C and 4E). Group 3 patients also upregulated numbers of PD‐1+ CD4+ T cells by twofold to fourfold compared to group 1 patients (one way ANOVA, P < .0001, Figures 4C and 4E) during combination BTZ with TMZ when IL‐10 levels were elevated in their plasma (Figure 4A) and in the recovery period, respectively.
Larger fractions of cytotoxic CD8+ T cells from group 2 patients expressed CTLA‐4 compared to group 1 patients at baseline (one way ANOVA, P < .01), during BTZ (P < .0001), also after combination BTZ with TMZ treatment (one way ANOVA, P < .0001) and during recovery, (P < .001; Figure 4F). Group 2 patients also had higher levels of CD8+ CTLA‐4+ T cells compared to group 3 patients during BTZ (one way ANOVA, P < .01), at combination BTZ with TMZ treatment (P < .01; Figure 4F). Collectively, these data might indicate that CD4+ T cells from the rapidly progressing patients are biased towards a Th2 phenotype and tolerised at earlier stages (days 1‐7 of BTZ, as well as BTZ + TMZ treatment). This is consistent with the profound Th2 cytokine profiles in their plasma. On the other hand, CD8+ effector T cells from rapidly progressive and mixed benefit patients from group 2 and 3, respectively, upregulated the inhibitory receptor PD‐1 (Figures 4D and 4F) only at later stages during recovery (days 4‐15.) Importantly, this phenomenon was not evident in group 1 patients.
3.10. Augmented differentiation of CD4+ and CD8+ T cell subsets in patients with positive clinical responses
Given the potential tolerization and exhaustion of T cells denoted by differential Th2/Th1 cytokine ratios and expression of PD‐1 and CTLA‐4 checkpoint molecules in the patients undergoing treatment, we sought to investigate their maturation phenotypes. We used CCR7, CD45RO, CD28 surface expression to discriminate distinct differentiation stages from naïve (TN), effector (TE), central memory (TCM), effector memory (TEM), and NK‐cells (Figures 4G and S2). While there was no difference in differentiated CD4+ T cell subsets between patients at baseline, group 1 patients with positive clinical and immunological responses had increased CD4+ TE subsets after BTZ + TMZ combination treatment compared to rapidly progressive group 2 and 3 patients (two way ANOVA, P < .001, respectively (Figure 4G,H) and during the recovery period (two way ANOVA P < .05; Figure 4G,H). Importantly, CD4+ TE subsets were not only increased after BTZ + TMZ treatment within group 1 patients, but also during recovery compared to baseline (two way ANOVA, P < .05, both), and BTZ only treatment, (Figure 4I; two way ANOVA P < .01 and P < .05, respectively). Correspondingly, these patients had fewer CD4+ TCM during combination BTZ + TMZ treatment and recovery periods (one way ANOVA, P < .05, and P < .01), respectively (Figure 4H). These data indicate that the group 1 patients with positive clinical and Th1 driven immune responses possibly mobilized and expanded CD4+ TE subsets in response to novel antigens produced as a result of BTZ + TMZ induced tumor cell death.
3.11. Patients with positive clinical responses diminish CTLA‐4+ CD4+T cells and attenuate IL‐10 secretion after treatment
To investigate whether the Th2‐biased and tolerised T cell phenotype before and after BTZ treatment in some patients could be reversed, we used PMA/ionomycin to stimulate PBMCs isolated from patients' blood at baseline and on day 22 during recovery after exposure to BTZ and TMZ therapy. Stimulation of baseline PBMCs with PMA/ionomycin increased the level of CTLA‐4+CD4+ Th cells in group 2 and 3 patients by sixfold compared to group 1 patients (Figure 5A,B, two way ANOVA, P < .0001, respectively), and CTLA‐4+CD8+ T cells by threefold (group 2 vs 1, P < .001; Figure 5C). However, stimulation of PBMCs that had been exposed to BTZ + TMZ treatment in situ induced the greatest numbers of CTLA‐4+ expressing CD4+ Th cells in group 3, compared to both group 1 and 2 patients (Figure 5B, two way ANOVA P < .0001), respectively. Notably, stimulation of PBMCs from rapidly progressing group 2 patients exposed to BTZ + TMZ treatment diminished the fractions of CTLA‐4+ CD4+ Th cells by ca. 15% (Figure 5B) compared to stimulation at baseline, implying positive effects of the treatment, also indicated by diminishing immune suppressive Th2/Th1 cytokine ratios in plasma after treatment (Figure 4A). CD8+ T cell subsets from group 1 patients had moderate levels of intracellular interferon γ (IFNγ) compared to group 3 patients stimulated at baseline or during treatment recovery (two way ANOVA P < .05 respectively, Figure 5D,E). Correspondingly, group 3 patients with greatest fractions of CTLA‐4+CD4+ Th cells also exhibited greater immunosuppressive IL‐10/IFNγ ratios at recovery compared to both group 1 and group 2 patients (P < .0001, respectively; Figure 5F). Confirming the profound immune suppressive and Th2‐driven responses, also observed in plasma cytokines, unstimulated PBMCs from both group 2 and 3 patients exhibited greater IL‐10/TNF‐α at recovery compared to group 1 patients (P < .05, respectively; Figure 5G), and IL4/TNF‐α ratios in unstimulated PBMCs at baseline compared to group 1 patients (P < .001, respectively; Figure 5H). In patient‐02 from group 1 whose tumour was available for coculture with autologous PBMCs, the CD8+T cells were not tolerised by coculture with the tumour, compared to PBMCs not cocultured with tumour cells, or PBMCs stimulated with PMA/ionomycin (one way ANOVA, P < .0001, Figure 5I). Robust stimulation with PMA/ionomycin increased CD107a degranulation in CD8+ T cells compared to PBMCs exposed or not to BTZ in situ or cocultured with patient‐02' autologous tumour cells (P < .05). Nevertheless, release of cytolytic granzyme B by CD8+ T cells was least potent upon coculture with autologous tumour cells (P < .0001) compared to BTZ naïve PBMCs (Figure 5I). IL‐2 secretion was greatest after stimulation with PMA/ionomycin of PBMCs at baseline and after BTZ exposure (P < .001; Figure 5J). Although attenuated, cytotoxic CD8+ T cells increased tumour necrosis factor α (TNF‐α) secretion when exposed to autologous patient‐02' tumour cells and when compared to unstimulated PBMCs from baseline (P < .01), at day 22 during recovery after BTZ + TMZ treatment (P < .05) and at day 56 post treatment (P < .001; Figure 5K). Collectively, these data might indicate that this patient's killer T cells were able to launch appropriate responses against his tumour ex vivo.
Figure 5.
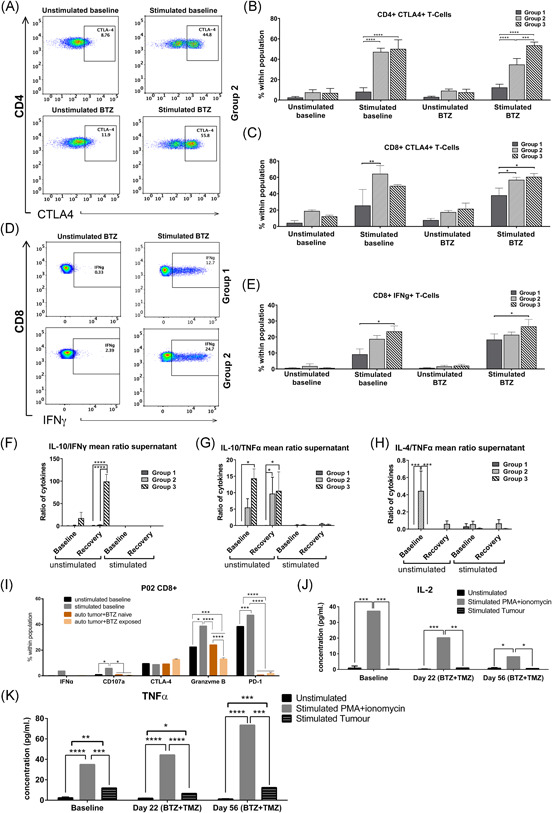
PBMCs from patients with positive clinical responses diminish CTLA‐4+ CD4+Th cells, attenuate IL‐10 secretion and have moderate intracellular IFNγ after bortezomib + temozolomide treatment. A, Representative dot plots showing CD4 vs CTLA‐4 in patient PBMCs unstimulated and stimulated, with PMA/ionomycin before or after exposure to BTZ or BTZ + TMZ treatment in situ. B, Mean ± SEM % of CD4+ CTLA‐4+ T cells at baseline or after treatment/stimulation conditions in group 1, 2, and 3 patients. C, Mean ± SEM % of CD8+ CTLA‐4+ at baseline or after treatment/stimulation conditions in group 1, 2, and 3 patients. D Representative dot plots showing intracellular IFNγ in CD8+T cells in patients' PBMCs unstimulated and stimulated with PMA/ionomycin in group 1 vs 2 patients. E, Mean ± SEM % of CD8+ IFNγ+ T cells at baseline or after treatment/stimulation conditions in group 1, 2, and 3 patients. Cytokines present in supernatants from patients' PBMCs after stimulation with PMA/ionomycin represented as mean ± SEM of Th2/Th1 ratio between (F) IL‐10 and IFNγ, (G) IL‐10 and TNF‐α, and (H) IL‐4 and TNF‐α in group 1, 2, and 3 patients at baseline or during treatment recovery. I, Mean ± SEM % expression of markers in patient‐02 tumour or PBMCs unstimulated and stimulated with PMA/ionomycin or P02‐tumour before or after exposure to bortezomib + temozolomide in situ. Cytokine concentrations in pg/mL from supernatants from patient‐02 tumour or PBMCs unstimulated or stimulated ex vivo with PMA + ionomycin or patient‐02 tumour or before and after exposure to bortezomib + temozolomide in situ of (J) IL‐2 and (K) TNF‐α. *P < .05; **P < .01; ***P < .001: ****P < .0001, n = 10 patients. BTZ, bortezomib; IFNγ, interferon γ; IL, interleukin; TMZ, temozolomide; TNF‐α, tumor necrosis factor α
3.12. BTZ induced NK cell maturation phenotype and expression of inhibitory checkpoints
Group 1 patients possessed more mature CD16+CD57+ NK cells at baseline (P < .01), during BTZ (P < .05), BTZ + TMZ treatment (P < .01) and during recovery (P < .05) compared to group 3 patients, and during BTZ + TMZ (P < .05), and recovery (P < .0001) compared to group 2 patients (Figure 2A‐2E). Surprisingly, these mature subsets in the group 1 patients upregulated the inhibitory checkpoint receptors NKG2A and PD‐1 (Figure S2F,S2G), and diminished activating DNAM‐1 and CD69 receptors (Figure S2H,S2I). In contrast, during BTZ + TMZ treatment group 2 and 3 patients who secreted high levels of IL‐10 and had less profound T cell responses also decreased CD16+CD57+ mature NK cell fractions compared to group 1 patients (CD16: group 2 vs 1, P < .05; group 3 vs 1, P < .01) and CD57: group 2 vs 1, P < 0.01; group 3 vs 1, P < .05). Within the reduced CD16+ CD57+ NK cell fractions in group 2 and 3 patients (Figure S2D,S2E) inhibitory NK cells expressing NKG2A and PD‐1 were also diminished compared to group 1 patients (Figure S2F,S2G), whereas subsets expressing DNAM‐1 and CD69 were increased after BTZ and TMZ treatment (Figure S2H,S2I), potentially indicating increased activation.
Taken together, copious IL‐10 levels in plasma may have contributed to greater activation of their NK cells but attenuated T cell responses after BTZ and TMZ treatment. Unfortunately, due to limited PBMCs and lack of access to fresh biopsy tissue for all patients, we could not perform functional assays with purified NK cells to mechanistically interrogate these findings.
4. DISCUSSION
We sought herein to investigate as primary objective whether sequential pretreatment with BTZ to sensitize recurrent GBM patients with unmethylated MGMT promoter to TMZ chemotherapy was safe and tolerated. This is because previous analyses utilized a concurrent schedule with monotherapy BTZ, metronomic doses of TMZ, or were conducted in newly diagnosed patients. 33 , 34 , 35 , 36 , 37 In this phase IB study, we successfully dose escalated TMZ to the target 200 mg/m2 dose for combination with 1.3 mg/m2 BTZ and no grade 3 or 4 toxicities were registered in our study. This is consistent with previous trials using concurrent schedules 22 with intensive and prolonged treatment, as well as with various drug combinations. 33 , 34 , 35 , 36 , 37 , 38 , 39
Our patients scored relatively high on the quality of life EQ‐5D‐5L questionnaire that was previously validated for the Norwegian population. 40 The ability to perform usual daily activity, ataxia, and changes in strength were the strongest predictors of KPS and this finding is corroborated by a previous study of Norwegian patients after surgery. 41 BTZ crosses the blood brain barrier in human and mice 22 , 39 and higher concentrations were found in brain tissue than in plasma. 39 Our pharmacokinetic analysis showed similar rapid redistribution of the drug from plasma to tissue as was reported for newly diagnosed patients. 29 , 30
Nevertheless, the BORTEM‐17 patients segregated a priori into three groups based on clinical and/or radiological progression. The grouping was concordant with tumor growth characteristics where group 2 patients with the most rapid progression had the greatest tumour burden (ca. 34 × 103 mm3) at baseline. Moreover, their tumour population doubling time from baseline to first MRI evaluation at day 56 was 47.1 days, nearly twofold faster than the group 1 patients whose baseline tumour volume was ca. 17.2 × 103 mm3 and had a prolonged doubling time of 107.4 days. Although two‐thirds patients in groups 1 and 3 had prior treatment with stereotactic radiosurgery (SRS) after their second recurrence and before study recruitment, these patients had differential clinical responses. To minimize the challenge of differentiating tumour recurrence from radiation necrosis, study recruitment was based on MR contrast enhancing lesion greater than or equal to 12 weeks after completed radiotherapy. Furthermore, the SRS treated lesions were not considered as target lesions in the RANO evaluation of tumour progression.
IDH1 mutant genotype is associated with improved outcome 10 , 42 but 2/3 (66%) of best performing group 1 patients had wt IDH1 compared to 2/4 (50%) and 2/3 (66%) patients from group 2 and 3, respectively. Since the IDH‐status had similar heterogeneous frequency within the groups, this was therefore not considered the underlying cause for differential responses of the patients. Furthermore, nine of the 10 patients had de novo GBM, with no evidence of a less malignant lesion before GBM diagnosis. Patient 07's IDH1‐mutant GBM tumor had progressed from an initial grade III anaplastic astrocytoma, thus representing a secondary GBM. Thus, the primary vs secondary GBM status also could not explain the differential prognosis between the patient groups. We did not however, classify the tumors based on previously reported gene expression profiles 43 , 44 , 45 , 46 because we only had access to formalin fixed paraffin embedded tumor tissue. Three molecular GBM subtypes termed proneural, proliferative, and mesenchymal 44 were identified and reported to have prognostic value that was independent of the World Health Organization histological grading and/or presence of necrosis. Further subclassifications identified four subtypes–classic, mesenchymal, proneural, and neural signatures 45 , 46 and now, more recently, greater heterogeneity at the cellular level was confirmed. 43 However, the proneural GBM subtype with assumed good prognosis in the Phillip et al's 44 study was associated with poor prognosis in the Verhaak study. 45 Furthermore, the subtypes coexist and vary spatially within the individual tumors as shown by multiregional sampling. Longitudinal analyses also indicate that the subtype signatures may change over time, affected, for example, by therapy. Thus, although the molecular subtypes provide great insight and opportunities for targeting therapies to the aberrant gene pathways, they fail to demonstrate robust differences in survival between them and are thus, not currently utilized clinically for diagnosis or prognostication. For this reason, we did not classify the tumors according to these molecular subtypes.
Remarkably, group 2 patients who also exhibited profound tolerance within their peripheral immune cell compartments demonstrated by dominant Th2 vs. Th1 suppressive cytokine ratios in plasma, consistently elevated PD‐1 and CTLA‐4 immune checkpoints on both CD4+ Th and CD8+ cytotoxic T cells throughout the treatment. They also had fewer fractions of CD16+CD57+ mature NK cells and subsets expressing inhibitory NKG2A and PD‐1 checkpoint molecules. In contrast, NK‐ cell populations expressing CD69 and DNAM‐1 were increased after BTZ and TMZ treatment, potentially indicating increased activation. Indeed, when BTZ naïve PBMCs from group 2 patients were stimulated with PMA/ionomycin ex vivo, they responded by secreting suppressive Th2 cytokines such as IL‐4, IL‐10 and upregulated CTLA‐4+CD4+ and CTLA‐4+CD8+ T cell fractions. Under the influence of IL‐4, tumor cells secrete IL‐10 which hinders activation and recruitment of adaptive immune mediators to the tumor microenvironment, by for example, downregulating CD28 expression, inhibiting proliferation and secretion of Th1 cytokines (IFNγ, TNF‐α, IL‐2) by T cells, as well as counteracting antigen presentation by dendritic cells. 47 , 48 IL‐10 also diminishes tumor cell expression of class I major histocompatibility molecules (MHC), 49 and provides opportunity for NK cell mediated cytotoxicity. IL‐10 thus also regulates NK cell cytotoxicity and is established to have a dual pro‐ and anti‐inflammatory role in different cell types and under different conditions. 50 IL‐10 gene polymorphisms that reduce IL‐10 levels correlate with increased melanoma incidence, while high IL‐10 levels are observed at sites of spontaneous rejection in primary melanoma, 51 potentially NK cell mediated. Intravenous administration of IL‐10 to healthy volunteers induced increased IFN‐γ and granzyme release. 52 IL‐10 conditioning of NK cells in vitro upregulated mRNA transcripts for type I interferons, high mobility group 1, CD69 and secretogranin 1 (TIA‐1) molecules and increased NK cytotoxicity against resistant Daudi cells. 53 Thus, while mediating tolerance and escape from adaptive immunity, IL‐10 cytokine may promote NK cells' antitumor activity in early stages. Thus, the rapid progression and high levels of IL‐10 secreted by group 2 and group 3 patients after BTZ + TMZ treatment may have acutely induced their activated NK cell phenotype.
However, such a substantial Th2 immune response is likely to down‐regulate tumor‐specific Th1 and CD8+T cell responses 54 in the long run and contribute to tumor immune evasion and cancer progression in these patients. CTLA‐4 expression would further decrease T cell activation, proliferation and effector functions. 55 , 56 In contrast, group 1 patients with lower disease burden and slower tumor growth kinetics had less suppressive Th2 driven cytokine responses and specifically IL‐10, and expanded CD4+ effector T cells in response to treatment. We speculate that this reflects beneficial Th1 and CD8+ T cell mediated antitumor responses enhanced by BTZ + TMZ combination therapy, possibly due to enhanced tumor antigen presentation.
Surprisingly, group 3 patients with mixed clinical/radiological responses had the lowest tumor burden (ca. 5.5 × 103 mm3) yet exhibited the most rapid population doubling time of 35.1 days. These patients also had multiple neoplastic lesions. In response to BTZ + TMZ treatment, group 3 patients secreted the highest concentrations of immunosuppressive IL‐10 cytokine in plasma and exhibited a tolerogenic phenotype (partially overlapping with the group 2 patients) denoted by upregulated PD‐1+CD4+ and CTLA‐4 +CD8+ T cell subsets compared to group 1 patients. TMZ can suppress immune responses in some glioma patients as it has been shown to downregulate activation of the JAK/STAT pathway and induce PD‐L1 expression on tumor cells. 57
Although group 2 and 3 patients responded to PMA/ionomycin stimulation by secreting high levels of the IFNγ Th1 cytokine, their levels of IL‐10 were many magnitudes higher, culminating in an overall immune suppressive phenotype. These data might indicate that the tolerant phenotype is not simply a feature of high tumor burden but may reflect tumor cell intrinsic features as the tumor leverages multipronged strategies to escape immune detection and destruction. 58 The caveat to this study is the small sample size and lack of randomized controls as it was powered primarily for evaluation of safety and tolerance of sequentially combined BTZ with dose escalated TMZ, as well as radiological and immunological biomarker analyses. The latter was challenged by the unavailability of sufficient ex vivo PBMCs for NK cell isolation and fresh autologous tumor cells from all patients for functional assessment.
Nevertheless, cytokine signaling locally in the brain cross‐talks with that in the systemic circulation, and aberrant function is often reflected in unison with phenotypic defects. In conclusion, 3 of 4 patients in group 2 were recruited into the BORTEM‐17 trial after their first tumor recurrence and this distinguished them from both group 1 and group 3 patients where all patients were recruited after their second recruitment. Group 1 patients further distinguish from both group 2 and group3 by diminished IL‐10 levels. Thus, levels of IL‐10 after BTZ + TMZ treatment could be monitored prospectively as a potential biomarker for probability of immunological responses defining the patients as group 1‐, 2‐, or 3‐type. Combination BTZ + TMZ treatment was safe, tolerated and immunological and MRI biomarkers provided robust indication for best responder patients exhibiting Th1 driven immune responses.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
AUTHOR CONTRIBUTIONS
Conducted experiments/analysed data: MAR, JB, AGN, VA, MH, AW, HM, JH, LO, LH, SAL, and AL. Treated and followed‐up patients: JB, DG, and PB. Writing the manuscript: MAR, VA, and MC. Revised manuscript: all authors. Designed research: MC and DG. Acquired funding: MC
Supporting information
Supplementary Figure 1. Fluorescence‐minus‐one (FMO) controls for the selected antibodies form the panel of antibody used in this study. Dot plots of multicolor flow cytometry showing the fluorescence spread into the channels shown by the FMO control compared to an unstained control, right most graphs show fully stained PBMC samples. The graphs show the FMO for (A) BV570 CD4, (B) PE Cy7 CD8, (C) PE PD‐1, (D) BV786 CTLA4, (E) APC CD45RO and (F) V450 CD56. Blue line represents FMO gating boundary
Supplementary Figure 2. Bortezomib induces NK cell maturation phenotypes and expression of inhibitory checkpoints. Representative dot plots showing CD56 vs. CD16 expression within the CD56+ CD3‐ NK cell gate. (B) Mean ± S.E.M. % of CD56 bright NK cell subsets in group 1, 2 and 3 patients before, during and after treatment with BTZ and TMZ. (C) Representative dot plots showing CD57 expression within the CD56 gate. (D) Mean ± S.E.M. % of CD16+ NK cell subsets in group 1, 2 and 3 patients before, during and after treatment with BTZ and TMZ. Mean ± S.E.M % of (E) CD57, (F) NKG2A, (G) PD‐1, (H) DNAM‐1, and (I) CD69 expression in NK cells from group 1, 2 and 3 patients, before, during and after treatment with BTZ and TMZ. Data represents the mean ± S.E.M. of n = 10 patients. Two‐way ANOVA, Bonferroni's multiple comparison test, * P < 0.05; ** P < 0.01; *** P < 0.001: **** P < 0.0001
Supporting information
Supporting information
ACKNOWLEDGMENTS
Flow cytometry was performed at the Flow Cytometry Core Facility, University of Bergen. We thank Line Ellila Lorentzen, Hedda Christensen and NORCrin for study administration, The Norwegian Brain Tumour Society, Rolf Ledal, and Monica Solberg Hansen for study representation and dissemination. The Norwegian Cancer Society (grant #190170 and #206965 to Martha Chekenya). All authors read and consented to publication of the final version of the manuscript.
Rahman MA, Brekke J, Arnesen V, et al. Sequential bortezomib and temozolomide treatment promotes immunological responses in glioblastoma patients with positive clinical outcomes: A phase 1B study. Immun Inflamm Dis. 2020;8:342–359. 10.1002/iid3.315
Mohummad A. Rahman and Jorunn Brekke contributed equally to this study.
Dorota Goplen and Martha Chekenya are co‐senior authors.
REFERENCES
- 1. Sanai N, Polley MY, McDermott MW, Parsa AT, Berger MS. An extent of resection threshold for newly diagnosed glioblastomas. J Neurosurg. 2011;115:3‐8. 10.3171/2011.2.JNS10998 [DOI] [PubMed] [Google Scholar]
- 2. Stummer W, Pichlmeier U, Meinel T, et al. Fluorescence‐guided surgery with 5‐aminolevulinic acid for resection of malignant glioma: a randomised controlled multicentre phase III trial. Lancet Oncol. 2006;7:392‐401. 10.1016/S1470-2045(06)70665-9 [DOI] [PubMed] [Google Scholar]
- 3. Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987‐996. 10.1056/NEJMoa043330 [DOI] [PubMed] [Google Scholar]
- 4. Chinot OL, Wick W, Mason W, et al. Bevacizumab plus radiotherapy‐temozolomide for newly diagnosed glioblastoma. N Engl J Med. 2014;370:709‐722. 10.1056/NEJMoa1308345 [DOI] [PubMed] [Google Scholar]
- 5. Gilbert MR, Dignam JJ, Armstrong TS, et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med. 2014;370:699‐708. 10.1056/NEJMoa1308573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997‐1003. 10.1056/NEJMoa043331 [DOI] [PubMed] [Google Scholar]
- 7. Popescu RA, Schäfer R, Califano R, et al. The current and future role of the medical oncologist in the professional care for cancer patients: a position paper by the European Society for Medical Oncology (ESMO). Ann Oncol. 2014;25:9‐15. 10.1093/annonc/mdt522 [DOI] [PubMed] [Google Scholar]
- 8. Louis DN, Perry A, Reifenberger G, et al. The 2016 world health organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016;131:803‐820. 10.1007/s00401-016-1545-1 [DOI] [PubMed] [Google Scholar]
- 9. Ohgaki H, Kleihues P. The definition of primary and secondary glioblastoma. Clin Cancer Res. 2013;19:764‐772. 10.1158/1078-0432.CCR-12-3002 [DOI] [PubMed] [Google Scholar]
- 10. Wick W, Hartmann C, Engel C, et al. NOA‐04 randomized phase III trial of sequential radiochemotherapy of anaplastic glioma with procarbazine, lomustine, and vincristine or temozolomide. J Clin Oncol. 2009;27:5874‐5880. 10.1200/JCO.2009.23.6497 [DOI] [PubMed] [Google Scholar]
- 11. Quinn JA, Desjardins A, Weingart J, et al. Phase I trial of temozolomide plus O6‐benzylguanine for patients with recurrent or progressive malignant glioma. J Clin Oncol. 2005;23:7178‐7187. 10.1200/JCO.2005.06.502 [DOI] [PubMed] [Google Scholar]
- 12. Quinn JA, Pluda J, Dolan ME, et al. Phase II trial of carmustine plus O(6)‐benzylguanine for patients with nitrosourea‐resistant recurrent or progressive malignant glioma. J Clin Oncol. 2002;20:2277‐2283. [DOI] [PubMed] [Google Scholar]
- 13. Viel T, Monfared P, Schelhaas S, et al. Optimizing glioblastoma temozolomide chemotherapy employing lentiviral‐based anti‐MGMT shRNA technology. Mol Ther. 2013;21:570‐579. 10.1038/mt.2012.278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Weingart J, Grossman SA, Carson KA, et al. Phase I trial of polifeprosan 20 with carmustine implant plus continuous infusion of intravenous O6‐benzylguanine in adults with recurrent malignant glioma: new approaches to brain tumor therapy CNS consortium trial. J Clin Oncol. 2007;25:399‐404. 10.1200/JCO.2006.06.6290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lavon I, Fuchs D, Zrihan D, et al. Novel mechanism whereby nuclear factor kappaB mediates DNA damage repair through regulation of O(6)‐methylguanine‐DNA‐methyltransferase. Cancer Res. 2007;67:8952‐8959. 10.1158/0008-5472.CAN-06-3820 [DOI] [PubMed] [Google Scholar]
- 16. Bocangel D, Sengupta S, Mitra S, Bhakat KK. p53‐Mediated down‐regulation of the human DNA repair gene O6‐methylguanine‐DNA methyltransferase (MGMT) via interaction with Sp1 transcription factor. Anticancer Res. 2009;29:3741‐3750. [PMC free article] [PubMed] [Google Scholar]
- 17. Harris LC, Remack JS, Houghton PJ, Brent TP. Wild‐type p53 suppresses transcription of the human O6‐methylguanine‐DNA methyltransferase gene. Cancer Res. 1996;56:2029‐2032. [PubMed] [Google Scholar]
- 18. Persano L, Pistollato F, Rampazzo E, et al. BMP2 sensitizes glioblastoma stem‐like cells to Temozolomide by affecting HIF‐1alpha stability and MGMT expression. Cell Death Dis. 2012;3:e412 10.1038/cddis.2012.153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bhakat KK, Mitra S. Regulation of the human O(6)‐methylguanine‐DNA methyltransferase gene by transcriptional coactivators cAMP response element‐binding protein‐binding protein and p300. J Biol Chem. 2000;275:34197‐34204. 10.1074/jbc.M005447200 [DOI] [PubMed] [Google Scholar]
- 20. Chen FY, Harris LC, Remack JS, Brent TP. Cytoplasmic sequestration of an O6‐methylguanine‐DNA methyltransferase enhancer binding protein in DNA repair‐deficient human cells. Proc Natl Acad Sci USA. 1997;94:4348‐4353. 10.1073/pnas.94.9.4348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kohsaka S, Wang L, Yachi K, et al. STAT3 inhibition overcomes temozolomide resistance in glioblastoma by downregulating MGMT expression. Mol Cancer Ther. 2012;11:1289‐1299. 10.1158/1535-7163.MCT-11-0801 [DOI] [PubMed] [Google Scholar]
- 22. Rahman MA, Gras Navarro A, Brekke J, et al. Bortezomib administered prior to temozolomide depletes MGMT, chemosensitizes glioblastoma with unmethylated MGMT promoter and prolongs animal survival. Br J Cancer. 2019;10:545‐555. 10.1038/s41416-019-0551-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sun SC. The non‐canonical NF‐kappaB pathway in immunity and inflammation. Nat Rev Immunol. 2017;17:545‐558. 10.1038/nri.2017.52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gras Navarro A, Espedal H, Joseph JV, et al. Pretreatment of glioblastoma with bortezomib potentiates natural killer cell cytotoxicity through TRAIL/DR5 mediated apoptosis and prolongs animal survival Cancers, 2019:11 10.3390/cancers11070996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wen PY, Macdonald DR, Reardon DA, et al. Updated response assessment criteria for high‐grade gliomas: response assessment in neuro‐oncology working group. J Clin Oncol. 2010;28:1963‐1972. 10.1200/JCO.2009.26.3541 [DOI] [PubMed] [Google Scholar]
- 26. Nayak L, DeAngelis LM, Brandes AA, et al. The neurologic assessment in neuro‐oncology (NANO) scale: a tool to assess neurologic function for integration into the response assessment in neuro‐oncology (RANO) criteria. Neuro Oncol. 2017;19:625‐635. 10.1093/neuonc/nox029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Brooks R. EuroQol: the current state of play. Health Policy. 1996;37:53‐72. [DOI] [PubMed] [Google Scholar]
- 28. Dominguez–Valentin M, Gras Navarro A, Rahman AM, et al. Identification of a natural killer cell receptor allele that prolongs survival of cytomegalovirus‐positive glioblastoma patients. Cancer Res. 2016;76:5326‐5336. 10.1158/0008-5472.CAN-16-1162 [DOI] [PubMed] [Google Scholar]
- 29. Reece DE, Sullivan D, Lonial S, et al. Pharmacokinetic and pharmacodynamic study of two doses of bortezomib in patients with relapsed multiple myeloma. Cancer Chemother Pharmacol. 2011;67:57‐67. 10.1007/s00280-010-1283-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hanley MJ, Mould DR, Taylor TJ, et al. Population pharmacokinetic analysis of bortezomib in pediatric leukemia patients: model‐based support for body surface area‐based dosing over the 2‐ to 16‐year age range. J Clin Pharmacol. 2017;57:1183‐1193. 10.1002/jcph.906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chang CL, Hsu YT, Wu CC, et al. Immune mechanism of the antitumor effects generated by bortezomib. J Immunol. 2012;189:3209‐3220. 10.4049/jimmunol.1103826 [DOI] [PubMed] [Google Scholar]
- 32. Schumacher LY, Vo DD, Garban HJ, et al. Immunosensitization of tumor cells to dendritic cell‐activated immune responses with the proteasome inhibitor bortezomib (PS‐341, Velcade). J Immunol. 2006;176:4757‐4765. 10.4049/jimmunol.176.8.4757 [DOI] [PubMed] [Google Scholar]
- 33. Kong XT, Nguyen NT, Choi YJ, et al. Phase 2 study of bortezomib combined with temozolomide and regional radiation therapy for upfront treatment of patients with newly diagnosed glioblastoma multiforme: safety and efficacy assessment. Int J Radiat Oncol Biol Phys. 2018;100:1195‐1203. 10.1016/j.ijrobp.2018.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kubicek GJ, Werner‐Wasik M, Machtay M, et al. Phase I trial using proteasome inhibitor bortezomib and concurrent temozolomide and radiotherapy for central nervous system malignancies. Int J Radiat Oncol Biol Phys. 2009;74:433‐439. 10.1016/j.ijrobp.2008.08.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. McCracken DJ, Celano EC, Voloschin AD, Read WL, Olson JJ. Phase I trial of dose‐escalating metronomic temozolomide plus bevacizumab and bortezomib for patients with recurrent glioblastoma. J Neurooncol. 2016;130:193‐201. 10.1007/s11060-016-2234-6 [DOI] [PubMed] [Google Scholar]
- 36. Phuphanich S, Supko JG, Carson KA, et al. Phase 1 clinical trial of bortezomib in adults with recurrent malignant glioma. J Neurooncol. 2010;100:95‐103. 10.1007/s11060-010-0143-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Portnow J, Frankel P, Koehler S, et al. A phase I study of bortezomib and temozolomide in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2012;69:505‐514. 10.1007/s00280-011-1721-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Odia Y, Kreisl TN, Aregawi D, Innis EK, Fine HA. A phase II trial of tamoxifen and bortezomib in patients with recurrent malignant gliomas. J Neurooncol. 2015;125:191‐195. 10.1007/s11060-015-1894-y [DOI] [PubMed] [Google Scholar]
- 39. Raizer JJ, Chandler JP, Ferrarese R, et al. A phase II trial evaluating the effects and intra‐tumoral penetration of bortezomib in patients with recurrent malignant gliomas. J Neurooncol. 2016;129:139‐146. 10.1007/s11060-016-2156-3 [DOI] [PubMed] [Google Scholar]
- 40. Nord E. EuroQol: health‐related quality of life measurement. Valuations of health states by the general public in Norway. Health Policy. 1991;18:25‐36. 10.1016/0168-8510(91)90141-j [DOI] [PubMed] [Google Scholar]
- 41. Jakola AS, Unsgard G, Solheim O. Quality of life in patients with intracranial gliomas: the impact of modern image‐guided surgery. J Neurosurg. 2011;114:1622‐1630. 10.3171/2011.1.JNS101657 [DOI] [PubMed] [Google Scholar]
- 42. Sanson M, Marie Y, Paris S, et al. Isocitrate dehydrogenase 1 codon 132 mutation is an important prognostic biomarker in gliomas. J Clin Oncol. 2009;27:4150‐4154. 10.1200/JCO.2009.21.9832 [DOI] [PubMed] [Google Scholar]
- 43. Neftel C, Laffy J, Filbin MG, et al. An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell. 2019;178:835‐849 e821. 10.1016/j.cell.2019.06.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Phillips HS, Kharbanda S, Chen R, et al. Molecular subclasses of high‐grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9:157‐173. 10.1016/j.ccr.2006.02.019 [DOI] [PubMed] [Google Scholar]
- 45. Verhaak RGW, Hoadley KA, Purdom E, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98‐NF110. 10.1016/j.ccr.2009.12.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang Q, Hu B, Hu X, et al. Tumor evolution of glioma‐intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell. 2017;32:42‐56 e46. 10.1016/j.ccell.2017.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Filella X, Alcover J, Zarco MA, Beardo P, Molina R, Ballesta AM. Analysis of type T1 and T2 cytokines in patients with prostate cancer. Prostate. 2000;44:271‐274. [DOI] [PubMed] [Google Scholar]
- 48. Huang M, Wang J, Lee P, et al. Human non‐small cell lung cancer cells express a type 2 cytokine pattern. Cancer Res. 1995;55:3847‐3853. [PubMed] [Google Scholar]
- 49. Petersson M, Charo J, Salazar‐Onfray F, et al. Constitutive IL‐10 production accounts for the high NK sensitivity, low MHC class I expression, and poor transporter associated with antigen processing (TAP)‐1/2 function in the prototype NK target YAC‐1. J Immunol. 1998;161:2099‐2105. [PubMed] [Google Scholar]
- 50. Mocellin S, Panelli MC, Wang E, Nagorsen D, Marincola FM. The dual role of IL‐10. Trends Immunol. 2003;24:36‐43. 10.1016/s1471-4906(02)00009-1 [DOI] [PubMed] [Google Scholar]
- 51. Conrad CT, Ernst NR, Dummer W, Brocker EB, Becker JC. Differential expression of transforming growth factor beta 1 and interleukin 10 in progressing and regressing areas of primary melanoma. J Exp Clin Cancer Res. 1999;18:225‐232. [PubMed] [Google Scholar]
- 52. Lauw FN, Pajkrt D, Hack CE, Kurimoto M, van Deventer SJ, van der Poll T. Proinflammatory effects of IL‐10 during human endotoxemia. J Immunol. 2000;165:2783‐2789. 10.4049/jimmunol.165.5.2783 [DOI] [PubMed] [Google Scholar]
- 53. Mocellin S, Panelli M, Wang E, et al. IL‐10 stimulatory effects on human NK cells explored by gene profile analysis. Genes Immun. 2004;5:621‐630. 10.1038/sj.gene.6364135 [DOI] [PubMed] [Google Scholar]
- 54. Kidd P. Th1/Th2 balance: the hypothesis, its limitations, and implications for health and disease. Altern Med Rev. 2003;8:223‐246. [PubMed] [Google Scholar]
- 55. Buchbinder EI, Desai A. CTLA‐4 and PD‐1 pathways: similarities, differences, and implications of their inhibition. Am J Clin Oncol. 2016;39:98‐106. 10.1097/COC.0000000000000239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chambers CA, Sullivan TJ, Truong T, Allison JP. Secondary but not primary T cell responses are enhanced in CTLA‐4‐deficient CD8+ T cells. Eur J Immunol. 1998;28:3137‐3143. [DOI] [PubMed] [Google Scholar]
- 57. Sengupta S, Marrinan J, Frishman C, Sampath P. Impact of temozolomide on immune response during malignant glioma chemotherapy. Clin Dev Immunol. 2012;2012:831090‐831097. 10.1155/2012/831090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kmiecik J, Poli A, Brons NH, et al. Elevated CD3+ and CD8+ tumor‐infiltrating immune cells correlate with prolonged survival in glioblastoma patients despite integrated immunosuppressive mechanisms in the tumor microenvironment and at the systemic level. J Neuroimmunol. 2013;264:71‐83. 10.1016/j.jneuroim.2013.08.013 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Fluorescence‐minus‐one (FMO) controls for the selected antibodies form the panel of antibody used in this study. Dot plots of multicolor flow cytometry showing the fluorescence spread into the channels shown by the FMO control compared to an unstained control, right most graphs show fully stained PBMC samples. The graphs show the FMO for (A) BV570 CD4, (B) PE Cy7 CD8, (C) PE PD‐1, (D) BV786 CTLA4, (E) APC CD45RO and (F) V450 CD56. Blue line represents FMO gating boundary
Supplementary Figure 2. Bortezomib induces NK cell maturation phenotypes and expression of inhibitory checkpoints. Representative dot plots showing CD56 vs. CD16 expression within the CD56+ CD3‐ NK cell gate. (B) Mean ± S.E.M. % of CD56 bright NK cell subsets in group 1, 2 and 3 patients before, during and after treatment with BTZ and TMZ. (C) Representative dot plots showing CD57 expression within the CD56 gate. (D) Mean ± S.E.M. % of CD16+ NK cell subsets in group 1, 2 and 3 patients before, during and after treatment with BTZ and TMZ. Mean ± S.E.M % of (E) CD57, (F) NKG2A, (G) PD‐1, (H) DNAM‐1, and (I) CD69 expression in NK cells from group 1, 2 and 3 patients, before, during and after treatment with BTZ and TMZ. Data represents the mean ± S.E.M. of n = 10 patients. Two‐way ANOVA, Bonferroni's multiple comparison test, * P < 0.05; ** P < 0.01; *** P < 0.001: **** P < 0.0001
Supporting information
Supporting information