

Abstract
Cyclooxygenase-2 (COX-2) overexpression is prominent in inflammatory diseases, neurodegenerative disorders, and cancer. Directly monitoring COX-2 activity within its native environment poses an exciting approach to account for and illuminate the effect of the local environments on protein activity. Herein, we report the development of CoxFluor, the first activity-based sensing approach for monitoring COX-2 within live cells with confocal microscopy and flow cytometry. CoxFluor strategically links a natural substrate with a dye precursor to engage both the cyclooxygenase and peroxidase activities of COX-2. This catalyzes the release of resorufin and the natural product, as supported by molecular dynamics and ensemble docking. CoxFluor enabled the detection of oxygen-dependent changes in COX-2 that are independent of protein expression within live macrophage cells.
Keywords: activity-based sensing, bioimaging, fluorescent probes, isoform selectivity, molecular dynamics and ensemble docking
Graphical Abstract
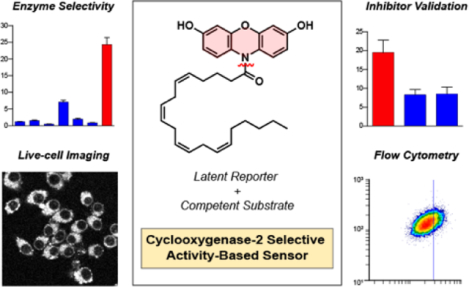
Activity-based sensing of Cyclooxygnease-2: An isoform-selective fluorogenic probe reports on enzymatic activity within live cells. Two-step activation facilitates the release of the natural product and resorufin for confocal imaging and flow cytometry. CoxFluor indicates that COX-2 activity can be regulated by oxygen without a change in protein expression level.
Introduction
Cyclooxygenase (COX, E.C. 1.14.99.1) is the key biosynthetic enzyme that initiates the synthesis of prostaglandins from their linear lipid precursor, arachidonic acid (AA).[1–3] After production, the prostaglandins function as important lipid-based mediators that regulate physiological processes, such as gastric epithelial protection, hemostasis, and sodium metabolism.[4,5] At higher concentrations the prostaglandins act as potent pro-inflammatory compounds through the production of cytokines.[6,7] These prostaglandins can be further elaborated by a variety of tissue-specific isomerases and synthases to afford the entire gambit of prostanoids (AA-derived prostaglandins).[5]
Two isoforms of human COX have been identified (COX-1 and −2), which display a high level of structural homology (~60% sequence identity).[8] Both isoforms catalyze the rate-limiting step for production of prostaglandin H2 (PGH2) through distinct cyclooxygenase and peroxidase active sites. The major difference between the two isoforms is their expression profile: COX-1 is constitutively expressed in most cell types, whereas COX-2 expression can either be constitutive or induced for rapid prostaglandin production depending on the tissue type.[2,4,9–12] COX overexpression represents a prominent phenotype in inflammation,[13,14] neurodegenerative disorders,[15] and cancer.[10,16,17] For example, high expression levels and cross-talk with inducible nitric oxide synthase in cancer can correlate with poor clinical outcomes through increased angiogenesis, proliferation, and cell migration.[18–22] It is important to note that a variety of factors beyond concentration can influence COX-2 activity (e.g., substrate concentration, allosteric regulators, and post-translational modifications), especially in vivo.[23–27]
To date, only a limited number of strategies have been introduced for detecting COX-2 in living systems. For example, selective inhibitors of COX-2 have been radiolabeled[28–34] or appended to dyes[35,36] for PET/SPECT or fluorescence imaging, respectively. These imaging agents report on relative COX-2 expression profiles; however, they remain in a constant ‘on’ state, regardless of whether they are bound to COX-2. More recently, activatable fluorescent inhibitors have been reported that afford an “off-on” response, where protein binding sequesters the inhibitor away from the fluorophore, disrupts the inhibitor-mediated fluorescence quenching, and renders the protein-small-molecule adduct fluorescent.[37,38] Despite these notable improvements, these technologies only report on whether COX-2 is present, but not whether the enzyme is catalytically active.
Activity-based sensing (ABS) represents a powerful detection strategy that relies on selective chemical reactivity. In contrast to binding-based methods, ABS provides a direct readout for the analyte or protein’s activity, rather than its concentration, enabling a more complete depiction of its contribution within living systems.[39,40] A variety of fluorescent probes have been reported for the detection of enzyme activity. However most display broad reactivity across a class of enzymes.[41] The development of isoform-selective activity-based sensors remains challenging due to the generally high structural similarities among isoforms. To date, some examples exist; however, they have largely targeted promiscuous enzymes involved in xenobiotic metabolism (e.g., cytochrome P450, monoamine oxidase, aldehyde dehydrogenase).[42–46] To address these limitations, we developed CoxFluor, the first isoform-selective, activity-based fluorescent probe for COX-2. Isoform selectivity was achieved by leveraging the subtle differences in the size and dynamics of the cyclooxygenase active sites.[2,47] CoxFluor enabled the validation of COX-2 inhibitors within in vitro assays, was successfully applied for live-cell imaging and flow cytometry, and illustrated oxygen-dependent COX-2 activity within live macrophage.
Results and Discussion
CoxFluor consists of 3,7-dihydroxyphenoxazine (reduced form of resorufin) linked to AA through a cleavable amide bond. We proposed this design with the hypothesis that the lipid tail could serve as a substrate for COX-2 in a manner similar to the AA (Scheme 1a). After binding within the cyclooxygenase active site, the probe could undergo hydrogen atom abstraction by the enzyme’s Tyr385, followed by dioxygenation and cyclization to afford the CoxFluor-PGG2 intermediate. Subsequent translocation to the peroxidase active site and oxidation by Compound I or II could yield an unstable oxygen-centered resorufin radical followed by dismutation and amide hydrolysis to release the fluorescent product and either PGG2 or PGH2 (after reduction, Scheme 1b).[1,48–50] When proposing this structure, we hypothesized that we could utilize the bulky dye to prevent COX-1 binding and catalysis. This was based on previous reports that indicate COX-1 is unable to accommodate large groups at the carboxylate[2] and that this moiety provides critical interactions with Arg120 for binding.[51–53] On the other hand, COX-2’s larger substrate pocket and expanded substrate scope, including amides, suggested that CoxFluor should be competent for catalysis.[54]
Scheme 1.
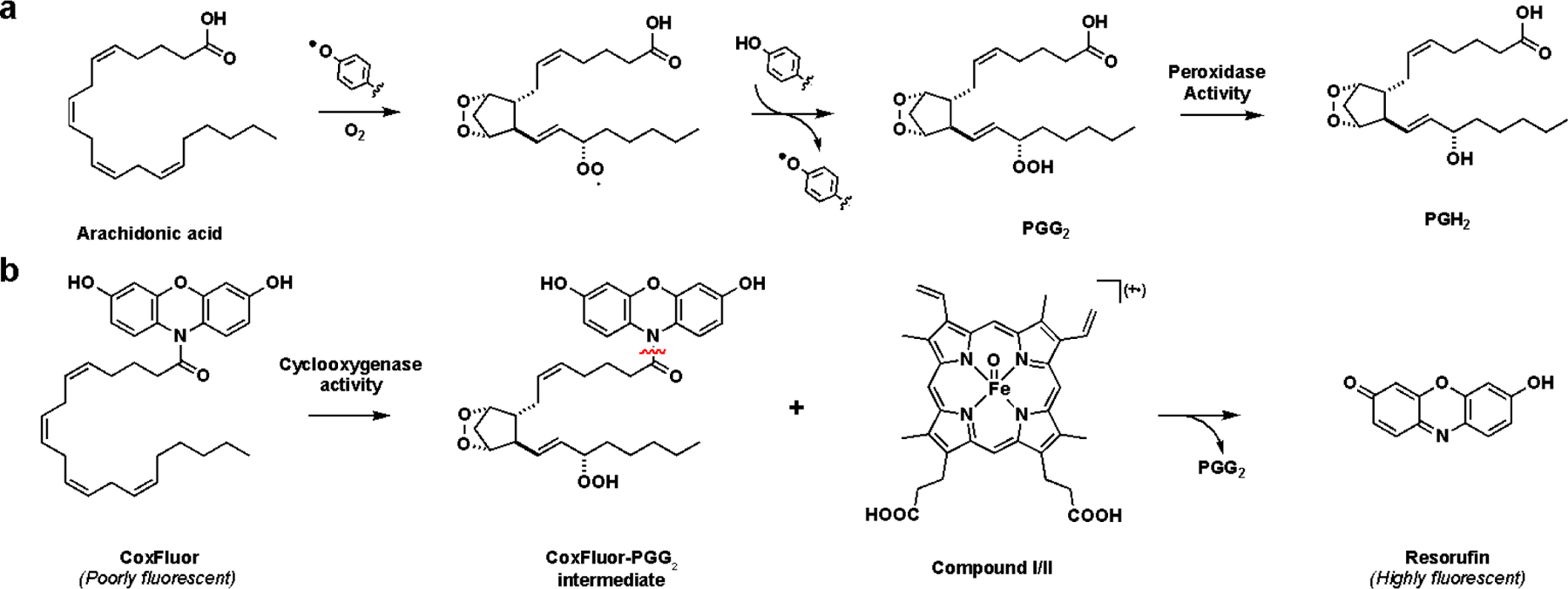
(a) Biosynthesis of PGH2 from AA by COX. (b) Proposed mechanism of enzyme-catalyzed resorufin production by COX via a CoxFluor-PGG2 intermediate via either Compound I (π cationic porphyrin radical) or Compound II (oxyferryl). The reaction between two CoxFluor-PGG2 intermediate radicals, dismutation, and hydrolysis are omitted for simplicity.
The synthesis of CoxFluor began with the Zn-mediated reduction of resazurin, followed by acetylation with acetic anhydride to afford 1 in 63% yield over two steps.[55] This intermediate was then coupled to arachidonyl chloride in the presence of potassium iodide to afford 2 in 49% yield. Finally, treatment of 2 with sodium methoxide (generated in situ) facilitated acetyl deprotection and provided CoxFluor in 68% yield (overall 21% over 4 steps, Scheme 2).
Scheme 2.
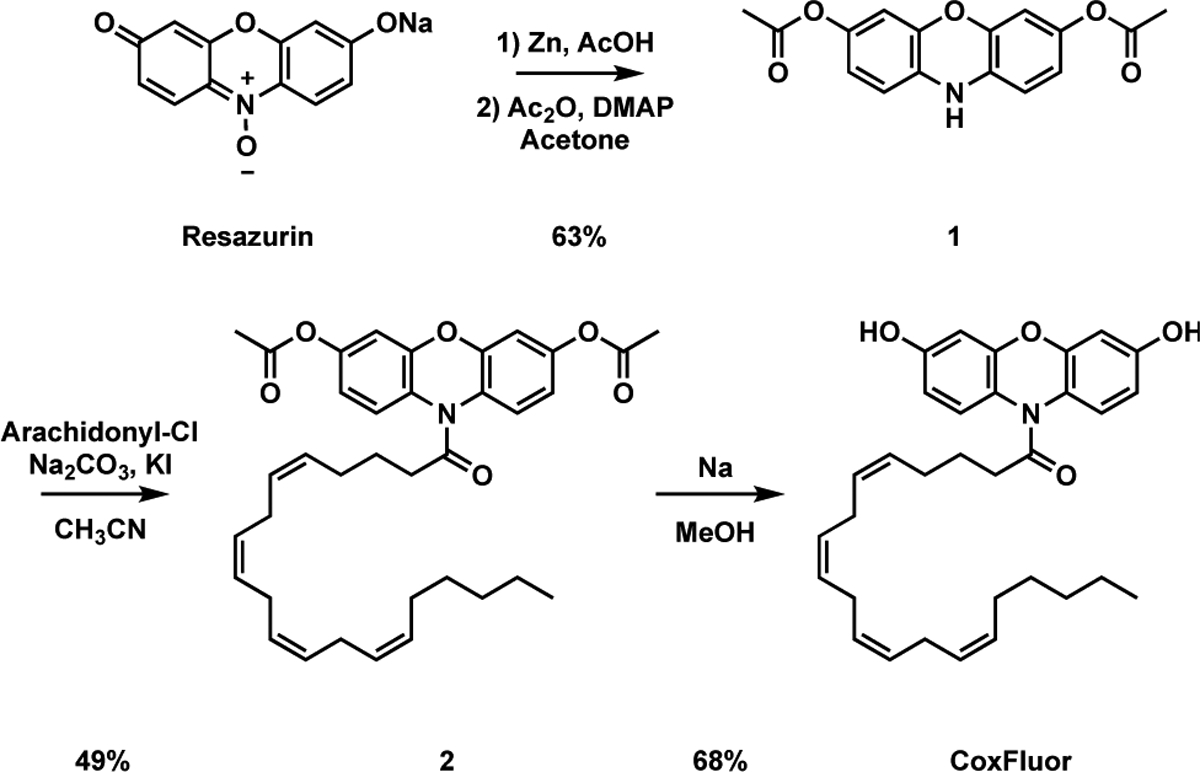
Synthesis of CoxFluor from resazurin.
With CoxFluor in hand, we first evaluated its response to recombinant human COX-2. Treatment of CoxFluor (ΦF = 0.29 , ε = 820 M−1cm−1 at 572 nm) with COX-2 resulted in the production of resorufin (ΦF = 0.55, ε = 73,000 M−1cm−1 at 573 nm)[56] and PGG2
(confirmed by LC-HRMS, Figure S1) with a 41-fold fluorescent turn-on response (Figure 1a).
Figure 1.
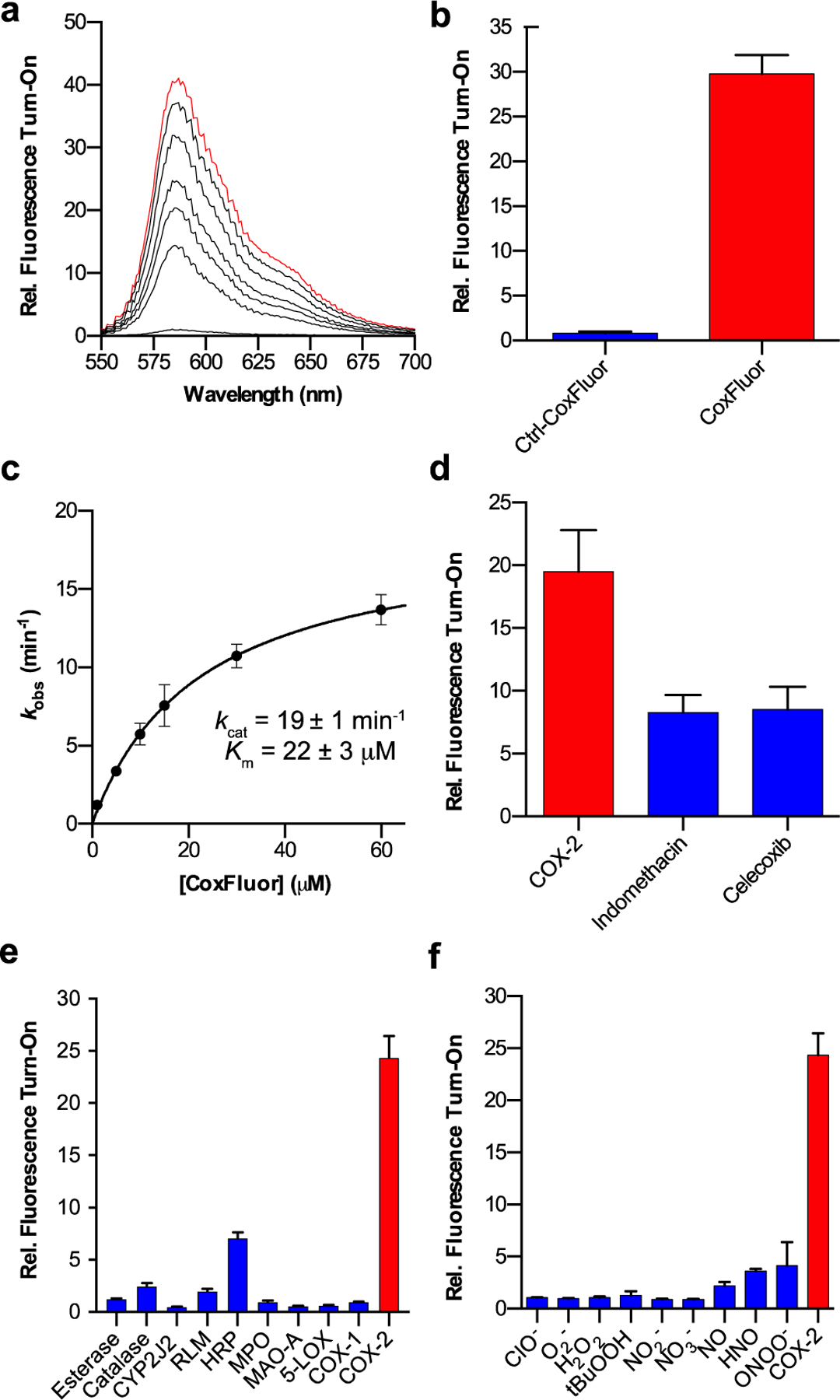
(a) Relative fluorescence for CoxFluor (10 μM) incubated with COX-2 (250 nM) and hemin (1 μM) over the course of 4 h. (b) Relative fluorescent intensity of CoxFluor and Ctrl-CoxFluor after incubation with COX-2 (250 nM) and hemin (1 μM) for 4 h. (c) Michaelis-Menten kinetics for COX-2-catalyzed (50 nM) release of resorufin in the presence of hemin (200 nM). (d) Inhibition of COX-2 (250 nM) and hemin (1 μM) by indomethacin (10 μM) and celecoxib (10 μM). (e) Relative fluorescence for CoxFluor incubated COX-2 (0.101 U), a panel of enzymes (10-fold excess) or (f) panel of reactive oxygen/nitrogen species (500 μM, 50 equiv.). All experiments were conducted at room temperature in 100 mM Tris HCl buffer (pH 8.0) except MPO, and 5-LOX (additional information in the Supporting Information). All data is reported as the mean ± standard deviation (n = 3) for parts b, d, e and f or the error from fitting for part c. Parts a/c and b/d-f were performed according to the fluorimeter and plate reader assays, respectively.
To confirm that activation was a consequence of enzyme-catalyzed oxidation rather than unbiased peroxidase activity, we prepared a control compound, Ctrl-CoxFluor, where the AA lipid was replaced with the saturated lipid tail (arachidic acid, Scheme S1). Ctrl-CoxFluor proved to be unreactive under the same reaction conditions (Figure 1b), strongly suggesting that the signal enhancement observed for CoxFluor was due to enzymatic activity. Under steady state conditions, CoxFluor underwent oxidation to resorufin by COX-2 with a kcat of 19 min−1 and Km of 22 μM (Figure 1c). The Km values are similar to those obtained for arachidonyl ethanolamide (Km = 24 μM) and were within an order of magnitude of AA (Km = 6 μM) and the measured rate for turn-over is within 100-fold of the natural substrate cyclooxygenase activity.[54] Moreover, CoxFluor’s fluorescence response was inhibited by both indomethacin and celecoxib (Figure 1d). This suggests that CoxFluor can be utilized to identify and/or evaluate COX-2 inhibitors, overcoming key drawbacks in existing methodologies that require radiolabeled compounds, purification of intermediates or products, or coupled-enzyme systems.[57,58] This also provides further support for the formation of a CoxFluor-PGG2 intermediate because both inhibitors bind within the cyclooxygenase active site of the protein.[59,60]
Isoform selectivity was evaluated by incubating CoxFluor with COX-1 isolated from bovine vesicles, where a minimal change in fluorescence was observed over the course of 4 h (1.3-fold fluorescence enhancement, 5% of COX-2 response, Figure S2). Off-target activation was also assessed against a panel of enzymes that possess closely related activities (lipoxygenase, peroxidase, catalase), could potentially cleave the amide (esterase), or are capable of metabolizing AA analogs (cytochrome P450s).[61] Even in the presence of 10-fold excess enzyme, CoxFluor displayed good selectivity against all of the tested enzymes (Figure 1e). Likewise, no undesirable activation was observed when CoxFluor was incubated with various biologically relevant reactive oxygen species, reactive nitrogen species, or other cellular oxidants/reductants (Figure 1f and S3). Moreover, no bleaching was observed when resorufin was treated with the same panel of reactive oxygen and nitrogen species (Figure S3). Importantly, incubation of CoxFluor with COX-2 in the presence of glutathione (GSH, 1 mM) maintained significant COX-2-specific fluorescence enhancement, indicating the potential for use for live-cell imaging (Figure S4). This is in contrast to Amplex® Red-based assays, which display cross-reactivity with GSH in the presence of peroxidases[62] or direct quenching of the radical intermediate.[50]
To further investigate the mechanism of CoxFluor activation by COX-2, we employed molecular dynamics (MD) and ensemble docking. Previous structures of COX-1 (PDB 5U6X)[63] and COX-2 (PDB 5KIR)[64] were selected, ligands were removed and the holoenzyme was simulated in explicit water over a period of 200 ns. Importantly, no global differences were observed for the peptide backbone across a panel of COX-1 and COX-2 structures, even in the presence of ligands and substrate (www.rcsb.org, all root-mean-standard-deviation < 2.5 Å, Table S1–2 and Figure S5).[65] Next, CoxFluor was docked into each pose along the simulation trajectory to probe the most probable binding modes (based on the predicted binding score),[66] where clear differences in the number and area available for binding were observed both overall and within the cyclooxygenase active site of COX-2 as compared to COX-1 (Figure 2a–c and S6). This finding is consistent with the difference in solvent accessible surfaces harbored within each cyclooxygenase active site.[2] CoxFluor also docks in a similar location to the reported indomethacin fluorescent inhibitors (Figure S7),[36,67] further supporting the proposed cyclooxyenase-dependent release of resorufin (Scheme 1b).
Figure 2.
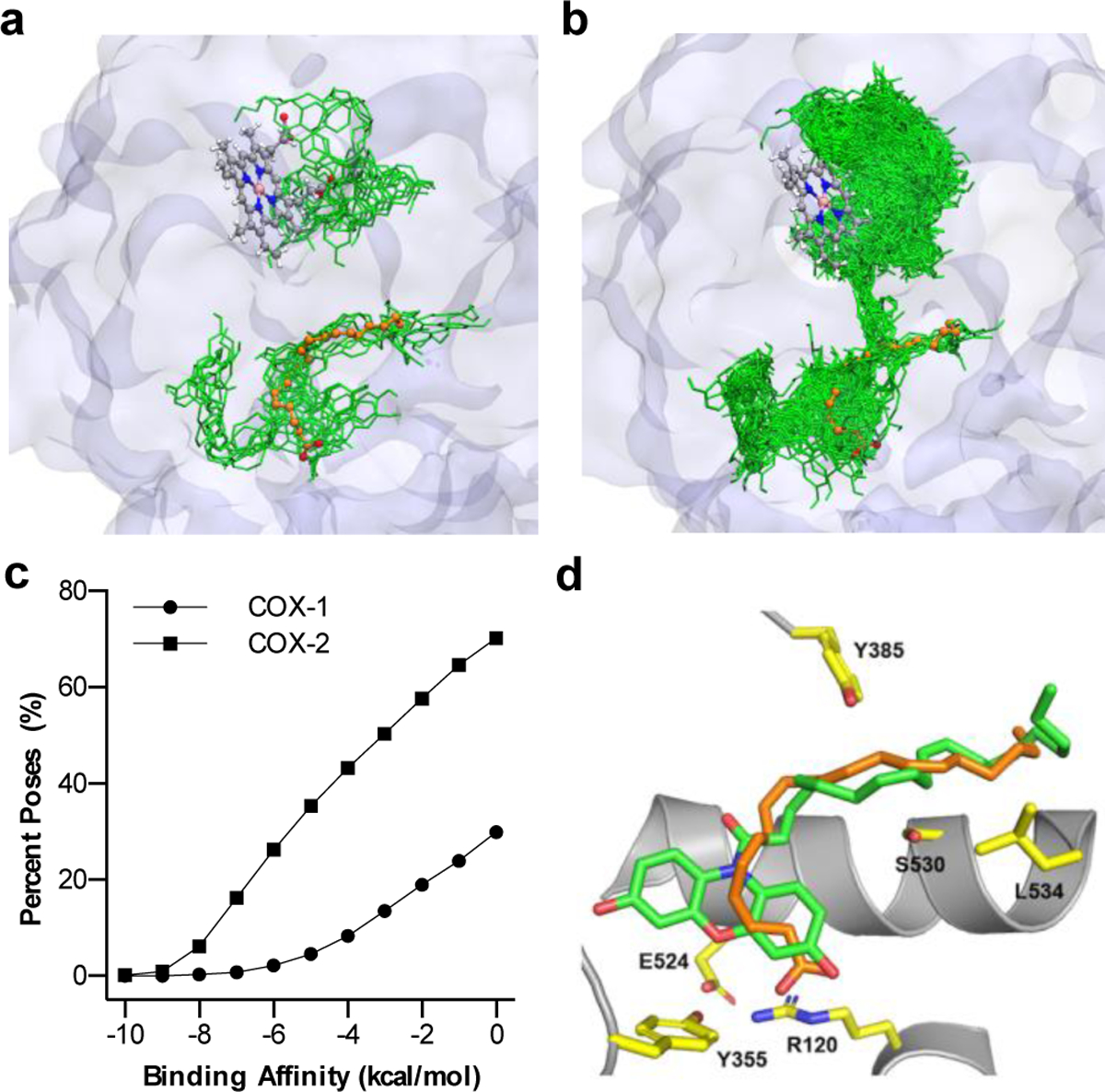
Molecular dynamics and ensemble docking studies of (a) COX-1 and (b) COX-2 with CoxFluor (green). Initial protein structures were prepared from PDB 5U6X and 5KIR and crystal bound AA (orange) is superimposed for comparison from PDB 1DIY and 3HS5, respectively. Structures display an overlay of all CoxFluor docked poses with scoring less than or equal to −7 kcal/mol. The heme from the simulation is represented as a ball and stick model to distinguish the cyclooxygenase and peroxidase active sites. (c) Percent of CoxFluor poses within the docking score binding affinities. (d) Competent binding of CoxFluor within the cyclooxygenase active site. Tyr385 (yellow) is oriented for hydrogen atom extraction from either CoxFluor (green) or AA (orange, PDB 3HS5).
To begin to interrogate the sequence of events, CoxFluor and the CoxFluor-PGG2 intermediate were docked within COX-2. Consistent with the proposed mechanism, CoxFluor binds significantly at the cyclooxygenase active site (66/597 poses at −7 kcal/mol cutoff). On the other hand, the CoxFluor-PGG2 intermediate displayed increased binding at the peroxidase active site for oxidation by either Compound I or Compound II (356/368 poses at −7 kcal/mol cutoff). This increased selectivity for the CoxFluor-PGG2 intermediate at the peroxidase active site is consistent over all spontaneous interactions (Figure S8). It is important to note that these results are consistent with the peroxidase-based oxidation[68] of CoxFluor in a similar manner to Amplex® Red[50] and 2,2′-azinobis(3-ethylbenzothiazolinesulfonic acid by horseradish peroxidase.[49,69] Finally, to confirm that CoxFluor can adopt the requisite conformation for oxidation by the active site Tyr385, CoxFluor was docked within the cyclooxygenase active site and compared to crystal bound AA (PDB 3HS5).[70] Structural comparisons indicate that CoxFluor can bind within the cyclooxygenase active site with proper orientation for oxidation (Figure 2d). Similar poses are also observed within the larger box that contains both the cyclooxygenase and peroxidase active sites. Together, these results suggest that CoxFluor binds within the substrate binding pocket or adjacent solvent-exposed sites to undergo oxidation and formation of the CoxFluor-PGG2 intermediate. After dissociation from the active site, the intermediate displays increased binding to the peroxidase active site for oxidation by Compound I or Compound II and subsequent release of resorufin.
Prior to performing cellular experiments, we evaluated CoxFluor’s stability and biocompatibility. Negligible fluorescence response was observed after 8 h of incubation at room temperature or 37 °C (Figure S9) and typical staining conditions yielded minimal toxicity in HEK 293T and RAW 264.7 macrophage cells (Figure S10). Next, we generated a transiently transfected HEK 293T cell line over-expressing human COX-2. GSH depletion was performed with N-ethylmaleimide (NEM)[71] prior to staining because GSH can generate[62] or quench radical intermediates of other peroxidase-based probes complicating the interpretation of the results.[50,72] Moreover, past work has demonstrated that COX-2 inhibitors (e.g., celecoxib analogs and indomethacin) can alter GSH levels,[73–76] which is consistent with our initial experimental observations (Figure S11). After incubation with CoxFluor for 3 h we observed a 1.2-fold fluorescence increase for transfected cells compared to the control, and no fluorescence enhancement was observed upon treatment with indomethacin (Figure S12).
Next, we applied CoxFluor for the detection and imaging of endogenous COX-2 activity within RAW 264.7 murine macrophage. As a key player in inflammatory and immune responses, macrophages undergo phenotypic changes to pro-inflammatory or anti-inflammatory states upon stimulation.[77] Because COX-2 activity is the rate-limiting step in prostaglandin biosynthesis (pro-inflammatory mediators), COX-2 expression and subsequent prostaglandin production can be measured in well-established lipopolysaccharide (LPS)-induced inflammation models as a biomarker for inflammation.[3,78–83] To date, these studies typically rely on mRNA quantification, western blot analysis, or downstream product quantification (e.g., ELISA assays) rather than direct quantification of COX-2 activity. This is due to the lack of isoform selectivity in current cyclooxygenase assays. First, we measured the effect of LPS stimulation time on COX-2’s specific activity in RAW 264.7 macrophage lysates. Variations due to GSH fluctuations were again minimized using NEM, which we and others have shown does not affect COX activity (Figure S13).[84] Within the first 4 h of activation only a modest 2.4-fold increase in COX-2 activity was observed. Over the next several hours, the activity increased in a linear fashion to a maximum of 17.6-fold at 19 h (Figure S14). These results are similar to previous reports of prostaglandin biosynthesis within both murine[82] and human[81] macrophage cells under LPS-stimulated conditions. The activity differences were then confirmed within live cells using both confocal microscopy (Figure 3) and flow cytometry (Figure S15). Rather than using NEM, we pre-treated with buthionine sulfoximine (BSO), a γ-glutamylcysteine synthase inhibitor because it is a less cytotoxic alternative.[85,86] Under these conditions we observed a 1.2-fold fluorescent enhancement for LPS-stimulated cells relative to the control using confocal microscopy and a 1.6-fold increase in median fluorescence for cells activated for 19 h as compared to 4 h with flow cytometry (Figure 3 and S15). Moreover, monitoring the ratio of fluorescence from CoxFluor stained cells to CoxFluor stained cells treated with indomethacin clearly identifies COX-2 activity across activation states (M0 versus M1, Figure S15). Finally, the ability of CoxFluor to detect activity changes without GSH depletion was confirmed with flow cytometry under similar conditions where a 1.2-fold increase in activity was observed after 19 h LPS-activation relative to 4 h LPS-stimulated cells (Figure S16).
Figure 3.
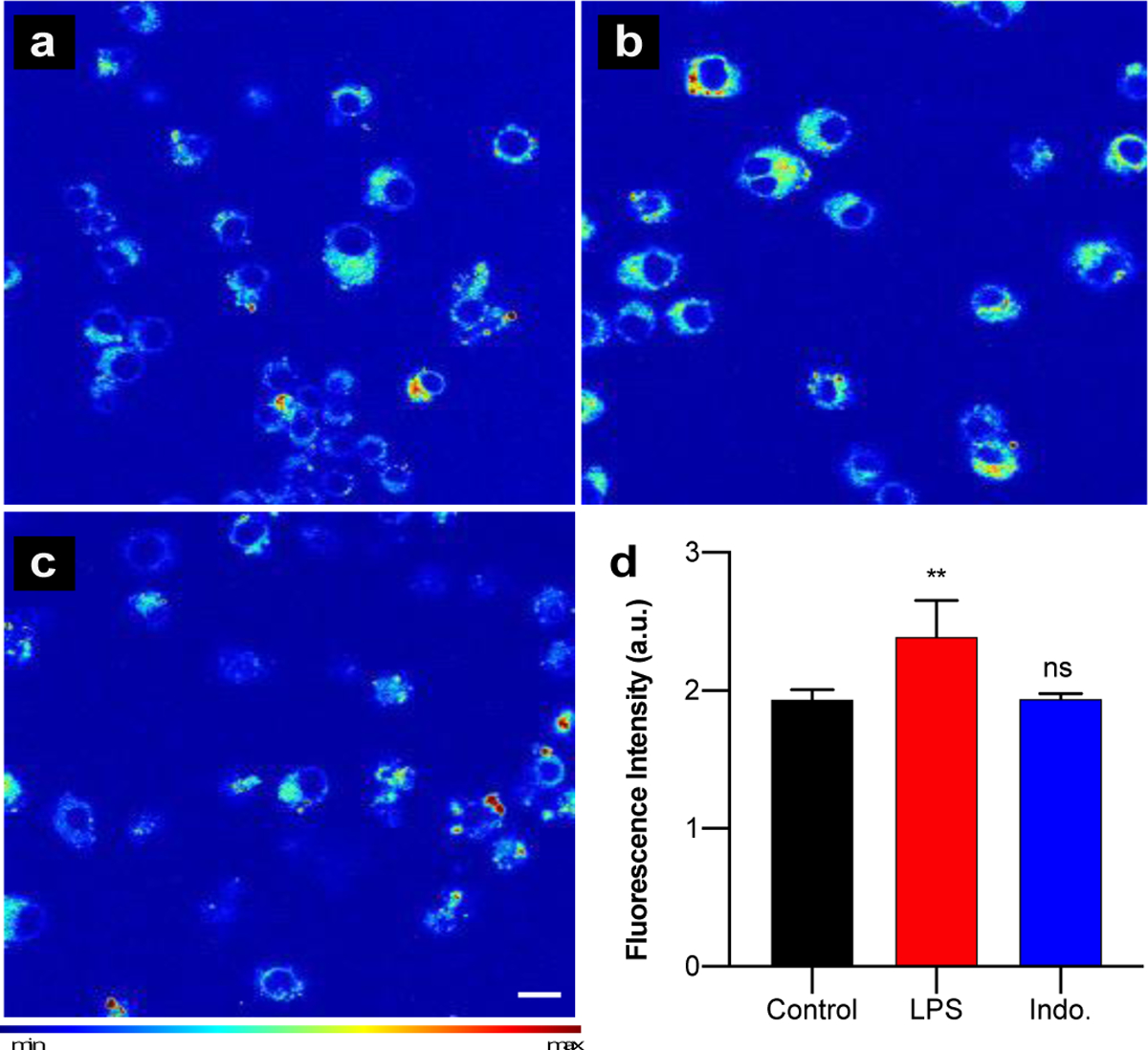
Confocal imaging of COX-2 activity in (a) control, (b) LPS-stimulated (1 μg/mL), and (c) indomethacin-treated (10 μM), LPS stimulated RAW 264.7 cells after 4 h incubation with CoxFluor (10 μM) at 37 °C following treatment with BSO (began 2 h prior to staining, 200 μM). Scale bar (white) represents 10 μm. (d) Quantified data. Values are reported as the mean ± standard deviation (n = 3). Statistical analysis was performed using one-way ANOVA (α = 0.05). **, p < 0.01.
Finally, we were interested in determining whether COX-2 activity can be regulated beyond the protein level within the native cellular environment. We hypothesized that oxygen concentrations may influence COX-2 activity because oxygen acts as a substrate but can also bind the heme prosthetic groups in the ferric state, yielding an off-cycle resting state.[87] The effect of oxygen on COX-2 activity was evaluated in RAW 264.7 macrophage cells after a 19 h activation with LPS. The cells were then stained under normoxic (~21%) or hypoxic conditions (prepared using AneroPack® for < 0.1% oxygen in a sealed container) and the fluorescent response was monitored with flow cytometry. Interestingly, higher activity was observed under hypoxic conditions, as indicated by a substantial shift in the fluorescence (Figure 4a–c). Moreover, this increase in fluorescence was COX-2-dependent, where treatment with indomethacin resulted in a 12% decrease in the ratio of median fluorescence for hypoxic to normoxic M1 macrophages (Figure 4d). To confirm the result and evaluate the dose-dependency of oxygen, we repeated the experiment under a 1 or 0.1% oxygen atmosphere. Here we observe an oxygen-dependent decrease in COX-2 activity as a function of oxygen (Figure 4d–e) that is independent of protein expression levels (Figure 4f). This result provides strong evidence that the local cellular environment plays a critical role in the activity of COX-2. This finding emphasizes the importance of measuring enzymatic activity in addition to measuring transcription and translation levels. To the best of our knowledge, this represents the first example in which an ABS approach has revealed activity differences that result from regulation beyond protein expression levels.
Figure 4.
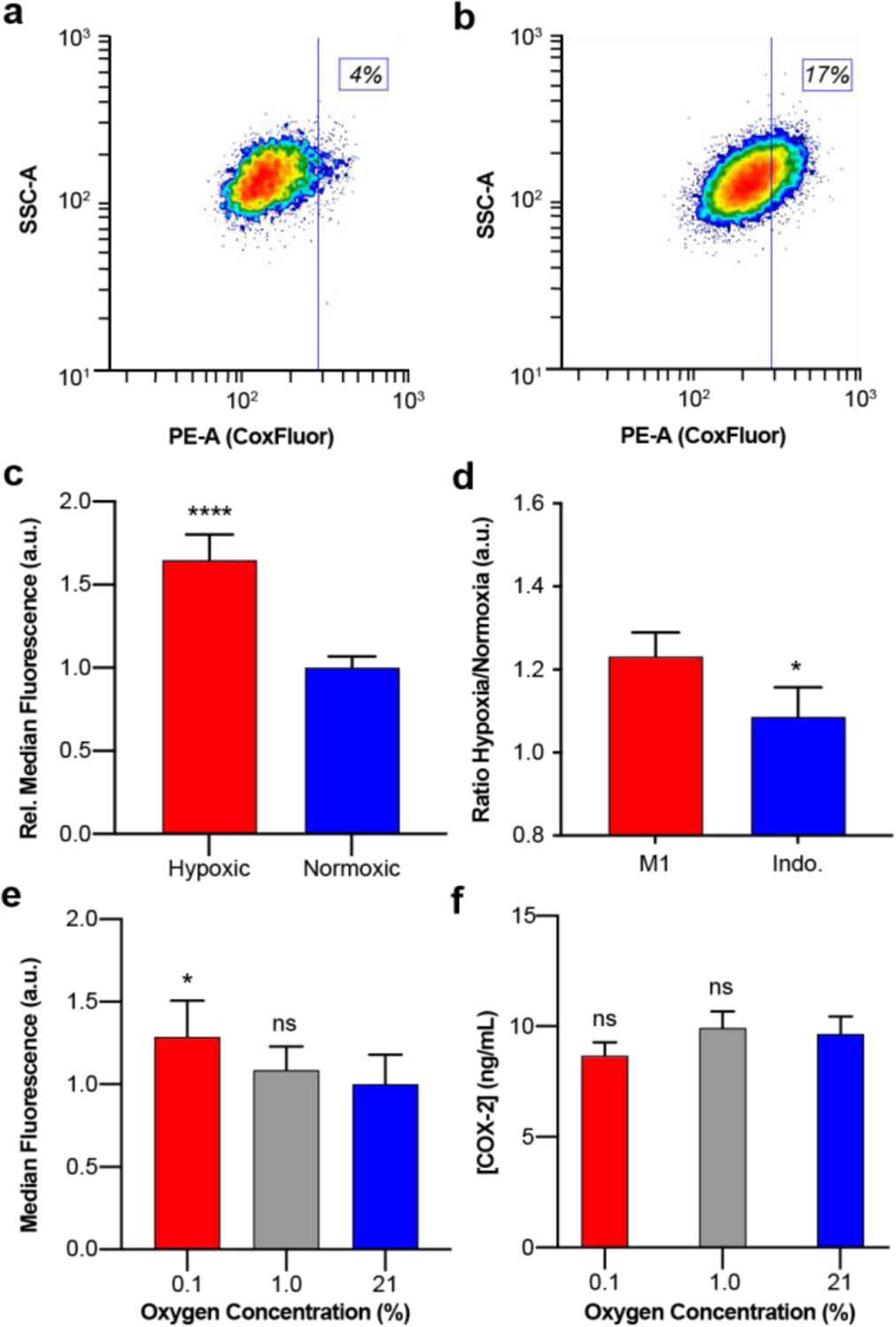
Contour plots of flow cytometry data from live LPS-activated RAW 264.7 macrophage cells stained with CoxFluor (10 μM) under (a) normoxic and (b) hypoxic conditions. (c) Quantified median fluorescence for hypoxic and normoxic cells. (d) Effect of indomethacin (20 μM) treatment on the ratio of hypoxic to normoxic median fluorescence. (e) Median fluorescence of RAW 264.7 macrophage cells stained under 0.1% (red), 1.0% (grey), or 21% (blue) oxygen concentrations. (f) ELISA quantification of COX-2 protein expression levels in 25 μg/mL protein lysates. Values are reported as the mean ± propagated standard deviation (n = 6 for flow cytometry data; n = 3 indomethacin inhibition and ELISA data). All staining was performed following treatment with BSO (began 2 h prior to staining, 200 μM) and oxygen dependent flow cytometry data was replicated on two separate days. Statistical analysis was performed with two-tail Student’s t test for c, one-tail Student’s t test for d, or one-way ANOVA for e (α = 0.05). *, p < 0.05, ****, p < 0.0001.
Conclusion
It is critical to develop methods for studying the influence of the local tissue on COX-2’s enzymatic activity. While it is reasonable to postulate that substrate availability should affect reaction rates, the outcome is particularly difficult to predict within living systems. We turned to an ABS approach to overcome this key challenge. CoxFluor represents the first isoform-selective sensor for measuring COX-2 activity. Strategic utilization of the bulky resorufin precursor and AA tail provided CoxFluor with the requisite selectively for interrogating COX-2 biology in complex mixtures (e.g., in the presence of COX-1), where current inhibitors display imperfect selectivity.[88] This isoform selectivity enables researchers to directly study COX-2 within its native environment, accounting for factors beyond transcription and translation that influence enzymatic activity.[89] Along these lines, we demonstrate that COX-2 activity can be regulated by oxygen availability within live cells. Taken together, these results suggest that CoxFluor will be a powerful tool for evaluating new therapeutics and for studying COX-2 in live cells.
Supplementary Material
Acknowledgements
The authors acknowledge the National Institute of Health for support (R35GM133581 to J.C.; R01GM115386 to M.G.M.; P41GM104601 and R01GM123455 to E.T.; R01GM1155884 and R03DA04236502 to A.D.). This work was supported in part by the Alfred P. Sloan Fellowship (FG-2017-8964 to J.C.) and by the American Heart Association Scientist Development Grant (15SDG25760064 to A.D). C.J.R. acknowledges the Chemistry-Biology Interface Training Grant (T32GM070421) and Seemon Pines Graduate Fellowship for support. Simulations were performed using the National Science Foundation computing resources through an XSEDE grant (TMGCA06N060 to E.T.) and PRAC allocation grant (ACI1713784 to E.T.). Major funding for the 500 MHz Bruker CryoProbeTM was provided by the Roy J. Carver Charitable Trust (Muscatine, Iowa; Grant #15-4521) to the School of Chemical Sciences NMR Lab. The Q-Tof Ultima mass spectrometer was purchased in part with a grant from the National Science Foundation, Division of Biological Infrastructure (DBI-0100085). We acknowledge Prof. Elvira de Mejia (Food Science and Human Nutrition, UIUC) for providing RAW 264.7 macrophage cells. We acknowledge the Core Facilities at the Carl R. Woese Institute for Genomic Biology for access to the Zeiss LSM 700 Confocal Microscope. Additionally, we acknowledge the Roy J. Carver Biotechnology Center at UIUC and Dr. Barbara Pilas for access to the BD LSR Fortessa Flow Cytometry Analyzer and for constructive discussions about instrumentation. We also thank Prof. Robert Gennis (Department of Biochemistry, UIUC) for access to the Strathkelvin oxygen electrode, Prof. H. Rex Gaskins (Department of Animal Sciences and Pathobiology, UIUC) and Emily (Jee-Wei) Chen (Department of Chemical and Biomolecular Engineering, UIUC) for access and assistance with the BioSpherix C-Chamber hypoxic incubator equipped with the corresponding OxyCycler C42 sub-chamber controller, and Daryl Meling and Amogh Kambalyal for the procurement and dissection of the bovine vesicles.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Supporting information for this article is given via a link at the end of the document.
References
- [1].Rouzer CA, Marnett LJ, J. Lipid Res 2009, 50, S29–S34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Blobaum AL, Marnett LJ, J. Med. Chem 2007, 50, 1425–1441. [DOI] [PubMed] [Google Scholar]
- [3].DeWitt DL, Biochim. Biophys. Acta 1991, 1083, 121–134. [DOI] [PubMed] [Google Scholar]
- [4].Kargman S, Charleson S, Cartwright M, Frank J, Riendeau D, Mancini J, Evans J, O’Neill G, Gastroenterology 1996, 111, 445–454. [DOI] [PubMed] [Google Scholar]
- [5].Smyth EM, Grosser T, Wang M, Yu Y, FitzGerald GA, J. Lipid Res 2009, 50, S423–S428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].St-Jacques B, Ma W, J. Neurochem 2011, 118, 841–854. [DOI] [PubMed] [Google Scholar]
- [7].Liebert MA, Williams JOYA, Pontzer CH, Shacter E, J. Interf. Cytokine Res 2000, 298, 291–298. [DOI] [PubMed] [Google Scholar]
- [8].Appleby SB, Ristimaki A, Neilson K, Narko K, Hla T, Biochem. J 1994, 302, 723–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Dubois RN, Abramson SB, Crofford L, Gupta RA, Simon LS, Van De Putte LBA, Lipsky PE, FASEB J. 1998, 12, 1063–1073. [PubMed] [Google Scholar]
- [10].Williams CS, Mann M, DuBois RN, Oncogene 1999, 18, 7908–7916. [DOI] [PubMed] [Google Scholar]
- [11].Fagerberg L, Hallström BM, Oksvold P, Kampf C, Djureinovic D, Odeberg J, Habuka M, Tahmasebpoor S, Danielsson A, Edlund K, et al. , Mol. Cell. Proteomics 2014, 13, 397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kirkby NS, Chan MV, Zaiss AK, Garcia-Vaz E, Jiao J, Berglund LM, Verdu EF, Ahmetaj-Shala B, Wallace JL, Herschman HR, et al. , Proc. Natl. Acad. Sci 2015, 113, 434–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kapoor S, Burke A, Mardini IA, McAdam BF, FitzGerald GA, Habib A, Lawson JA, J. Clin. Invest 2000, 105, 1473–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Samad TA, Moore KA, Sapirstein A, Billet S, Allchorne A, Poole S, Bonventre JV, Woolf CJ, Nature 2001, 410, 471–475. [DOI] [PubMed] [Google Scholar]
- [15].Teismann P, Tieu K, Choi D-K, Wu D-C, Naini A, Hunot S, Vila M, Jackson-Lewis V, Przedborski S, Proc. Natl. Acad. Sci 2003, 100, 5473–5478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Reddy BS, Hirose Y, Lubet R, Steele V, Kelloff G, Paulson S, Seibert K, Rao CV, Cancer Res. 2000, 60, 293–297. [PubMed] [Google Scholar]
- [17].Chen H, Cai W, Chu ESH, Tang J, Wong CC, Wong SH, Sun W, Liang Q, Fang J, Sun Z, et al. , Oncogene 2017, 36, 4415–4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kargman SL, O’Neill GP, Vickers PJ, Evans JF, Mancini JA, Jothy S, Cancer Res. 1995, 55, 2556–2559. [PubMed] [Google Scholar]
- [19].Basudhar D, Glynn SA, Greer M, Somasundaram V, No JH, Scheiblin DA, Garrido P, Heinz WF, Ryan AE, Weiss JM, et al. , Proc. Natl. Acad. Sci 2017, 114, 201709119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Mazhar D, Ang R, Waxman J, Br. J. Cancer 2006, 94, 346–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Patel NL, Stevens J, Haines DC, Kalen J, Seaman S, Zudaire E, Hilton MB, Conrads TP, Logsdon D, Croix B.St. , et al. , Sci. Transl. Med 2014, 6, 242ra84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Howe LR, Chang SH, Tolle KC, Dillon R, Young LJT, Cardiff RD, Newman RA, Yang P, Thaler HT, Muller WJ, et al. , Cancer Res. 2005, 65, 10113–10119. [DOI] [PubMed] [Google Scholar]
- [23].Smith WL, Urade Y, Jakobsson P-J, Chem. Rev 2011, 111, 5821–5865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Yuan C, Rieke CJ, Rimon G, Wingerd BA, Smith WL, Proc. Natl. Acad. Sci 2006, 103, 6142–6147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Yuan C, Sidhu RS, Kuklev DV, Kado Y, Wada M, Song I, Smith WL, J. Biol. Chem 2009, 284, 10046–10055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Dong L, Yuan C, Orlando BJ, Malkowski MG, Smith WL, J. Biol. Chem 2016, 291, 25641–25655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Dong L, Zou H, Yuan C, Hong YH, Kuklev DV, Smith WL, J. Biol. Chem 2016, 291, 4069–4078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Mccarthy TJ, Sheriff AU, Graneto MJ, Talley JJ, Welch MJ, J. Nucl. Med 2002, 43, 117–124. [PubMed] [Google Scholar]
- [29].de Vries EFJ, van Waarde A, Buursma AR, Vaalburg W, J. Nucl. Med 2003, 44, 1700–6. [PubMed] [Google Scholar]
- [30].Prabhakaran J, Underwood MD, Parsey RV, Arango V, Majo VJ, Simpson NR, Van Heertum R, Mann JJ, Kumar JSD, Bioorg. Med. Chem 2007, 15, 1802–1807. [DOI] [PubMed] [Google Scholar]
- [31].Schuller HM, Kabalka G, Smith G, Mereddy A, Akula M, Cekanova M, ChemMedChem 2006, 1, 603–610. [DOI] [PubMed] [Google Scholar]
- [32].Wuest F, Kniess T, Bergmann R, Pietzsch J, Bioorg. Med. Chem 2008, 16, 7662–7670. [DOI] [PubMed] [Google Scholar]
- [33].de Vries EFJ, Doorduin J, Dierckx RA, van Waarde A, Nucl. Med. Biol 2008, 35, 35–42. [DOI] [PubMed] [Google Scholar]
- [34].Dileep Kumar JS, Bai B, Zanderigo F, DeLorenzo C, Prabhakaran J, Parsey R, Mann JJ, Molecules 2018, 23, 1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Uddin MJ, Crews BC, Blobaum AL, Kingsley PJ, Gorden DL, McIntyre JO, Matrisian LM, Subbaramaiah K, Dannenberg AJ, Piston DW, et al. , Cancer Res. 2010, 70, 3618–3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Uddin MJ, Crews BC, Ghebreselasie K, Marnett LJ, Bioconjug. Chem 2013, 24, 712–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Zhang H, Fan J, Wang J, Dou B, Zhou F, Cao J, Qu J, Cao Z, Zhao W, Peng X, J. Am. Chem. Soc 2013, 135, 17469–17475. [DOI] [PubMed] [Google Scholar]
- [38].Zhang H, Fan J, Wang J, Zhang S, Dou B, Peng X, J. Am. Chem. Soc 2013, 135, 11663–11669. [DOI] [PubMed] [Google Scholar]
- [39].Aron AT, Reeves AG, Chang CJ, Curr. Opin. Chem. Biol 2018, 43, 113–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Su TA, Bruemmer KJ, Chang CJ, Curr. Opin. Biotechnol 2019, 60, 198–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Chyan W, Raines RT, ACS Chem. Biol 2018, 13, 1810–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Dai ZR, Ge GB, Feng L, Ning J, Hu LH, Jin Q, Wang DD, Lv X, Dou TY, Cui JN, et al. , J. Am. Chem. Soc 2015, 137, 14488–14495. [DOI] [PubMed] [Google Scholar]
- [43].Ning J, Liu T, Dong P, Wang W, Ge G, Wang B, Yu Z, Shi L, Tian X, Huo X, et al. , J. Am. Chem. Soc 2019, 141, 1126–1134. [DOI] [PubMed] [Google Scholar]
- [44].Li L, Zhang CW, Chen GYJ, Zhu B, Chai C, Xu QH, Tan EK, Zhu Q, Lim KL, Yao SQ, Nat. Commun 2014, 5, 3276. [DOI] [PubMed] [Google Scholar]
- [45].Li L, Zhang CW, Ge J, Qian L, Chai BH, Zhu Q, Lee JS, Lim KL, Yao SQ, Angew. Chemie - Int. Ed 2015, 54, 10821–10825. [DOI] [PubMed] [Google Scholar]
- [46].Anorma C, Hedhli J, Bearrood TE, Pino NW, Gardner SH, Inaba H, Zhang P, Li Y, Feng D, Dibrell SE, et al. , ACS Cent. Sci 2018, 4, 1045–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Furse KE, Pratt DA, Porter NA, Lybrand TP, Biochemistry 2006, 45, 3189–3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Kulmacz RJ, van der Donk WA, Tsai AL, Prog. Lipid Res 2003, 42, 377–404. [DOI] [PubMed] [Google Scholar]
- [49].Gorris HH, Walt DR, J. Am. Chem. Soc 2009, 131, 6277–6282. [DOI] [PubMed] [Google Scholar]
- [50].Debski D, Smulik R, Zielonka J, Michałowski B, Jakubowska M, Debowska K, Adamus J, Marcinek A, Kalyanaraman B, Sikora A, Free Radic. Biol. Med 2016, 95, 323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Picot D, Loll PJ, Garavito RM, Nature 1994, 367, 243–249. [DOI] [PubMed] [Google Scholar]
- [52].Rieke CJ, Mulichak AM, Garavito RM, Smith WL, J. Biol. Chem 1999, 274, 17109–17114. [DOI] [PubMed] [Google Scholar]
- [53].Bhattacharyya DK, Lecomte M, Rieke CJ, Garavito RM, Smith WL, J. Biol. Chem 1996, 271, 2179–2184. [DOI] [PubMed] [Google Scholar]
- [54].Yu M, Ives D, Ramesha CS, Biochemistry 1997, 272, 21181–21186. [DOI] [PubMed] [Google Scholar]
- [55].Hitomi Y, Takeyasu T, Funabiki T, Kodera M, Anal. Chem 2011, 83, 9213–9216. [DOI] [PubMed] [Google Scholar]
- [56].Klotz AV, Stegeman JJ, Walsh C, Anal. Biochem 1984, 140, 138–145. [DOI] [PubMed] [Google Scholar]
- [57].Ayoub SS, Flower RJ, Seed MP, Cyclooxygenases. Methods in Molecular Biology (Methods and Protocols), 2010. [Google Scholar]
- [58].Jiang J, Borisenko GG, Osipov A, Martin I, Chen R, Shvedova AA, Sorokin A, Tyurina YY, Potapovich A, Tyurin VA, et al. , J. Neurochem 2004, 90, 1036–1049. [DOI] [PubMed] [Google Scholar]
- [59].Mancini JA, Riendeau D, Falgueyret J-PP, Vickers PJ, O’Neill GP, Falgueyret J-PP, Vickers PJ, O’Neill GP, J. Biol. Chem 1995, 270, 29372–29377. [DOI] [PubMed] [Google Scholar]
- [60].Hood WF, Gierse JK, Isakson PC, Kiefer JR, Kurumbail RG, Seibert K, Monahan JB, Mol. Pharmacol 2003, 63, 870–877. [DOI] [PubMed] [Google Scholar]
- [61].McDougle DR, Watson JE, Abdeen AA, Adili R, Caputo MP, Krapf JE, Johnson RW, Kilian KA, Holinstat M, Das A, Proc. Natl. Acad. Sci 2017, 114, E6034–E6043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Votyakova TV, Reynolds IJ, Arch. Biochem. Biophys 2004, 431, 138–144. [DOI] [PubMed] [Google Scholar]
- [63].Cingolani G, Panella A, Perrone MG, Vitale P, Di Mauro G, Fortuna CG, Armen RS, Ferorelli S, Smith WL, Scilimati A, Eur. J. Med. Chem 2017, 138, 661–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Orlando BJ, Malkowski MG, Acta Crystallogr. Sect. F Struct. Biol. Commun 2016, 72, 772–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE, Nucleic Acids Res 2000, 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Trott O, Olson AJ, J. Comput. Chem 2010, 31, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Xu S, Uddin MJJ, Banerjee S, Marnett LJLJ, Jashim Uddin M, Banerjee S, Duggan K, Musee J, Kiefer JR, Ghebreselasie K, et al. , J. Biol. Chem 2019, 294, 8690–8689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Chance B, Arch. Biochem. Biophys 1952, 41, 416–424. [DOI] [PubMed] [Google Scholar]
- [69].Rodriguez-Lopez JN, Gilabert MA, Tudela J, Thorneley RNF, Garcia-Canovas F, Biochemistry 2000, 39, 13201–13209. [DOI] [PubMed] [Google Scholar]
- [70].Vecchio AJ, Simmons DM, Malkowski MG, J. Biol. Chem 2010, 285, 22152–22163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Yuan L, Lin W, Zhao S, Gao W, Chen B, He L, Zhu S, J. Am. Chem. Soc 2012, 134, 13510–23. [DOI] [PubMed] [Google Scholar]
- [72].Tsikas D, Suchy MT, Niemann J, Tossios P, Schneider Y, Rothmann S, Gutzki FM, Frölich JC, Stichtenoth DO, FEBS Lett. 2012, 586, 3723–3730. [DOI] [PubMed] [Google Scholar]
- [73].Ryan EP, Bushnell TP, Friedman AE, Rahman I, Phipps RP, Cancer Immunol. Immunother 2008, 57, 347–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Dengiz GO, Odabasoglu F, Halici Z, Suleyman H, Cadirci E, Bayir Y, Arch. Pharm. Res 2007, 30, 1426–1434. [DOI] [PubMed] [Google Scholar]
- [75].Pastoris O, Verri M, Boschi F, Kastsiuchenka O, Balestra B, Pace F, Tonini M, Natale G, Naunyn. Schmiedebergs. Arch. Pharmacol 2008, 378, 421–429. [DOI] [PubMed] [Google Scholar]
- [76].Abdel-Raheem IT, Basic Clin. Pharmacol. Toxicol 2010, 107, 742–750. [DOI] [PubMed] [Google Scholar]
- [77].Martinez FO, Gordon S, F1000Prime Rep. 2014, 6, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Bienkowskis MJ, Petro MA, Robinson J, Biochemistry 1989, 264, 6536–6544. [PubMed] [Google Scholar]
- [79].Giroux M, Descoteaux A, J. Immunol 2000, 165, 3985–3991. [DOI] [PubMed] [Google Scholar]
- [80].Eliopoulos AG, Dumitru CD, Wang CC, Cho J, Tsichlis PN, EMBO J. 2002, 21, 4831–4840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Grkovich A, Johnson CA, Buczynski MW, Dennis EA, J. Biol. Chem 2006, 281, 32978–32987. [DOI] [PubMed] [Google Scholar]
- [82].Rouzer CA, Jacobs AT, Nirodi CS, Kingsley PJ, Morrow JD, Marnett LJ, J. Lipid Res 2005, 46, 1027–1037. [DOI] [PubMed] [Google Scholar]
- [83].Font-Nieves M, Sans-Fons MG, Gorina R, Bonfill-Teixidor E, Salas-Peŕdomo A, Maŕquez-Kisinousky L, Santalucia T, Planas AM, J. Biol. Chem 2012, 287, 6454–6468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Smith CJ, Marnett LJ, Arch. Biochem. Biophys 1996, 335, 342–350. [DOI] [PubMed] [Google Scholar]
- [85].Buchmuller-Rouiller Y, Corradin SB, Smith J, Schneider P, Ransijn A, Jongeneel CV, Mauël J, Cell. Immunol 1995, 164, 73–80. [DOI] [PubMed] [Google Scholar]
- [86].Butzer U, Weidenbach H, Gansauge S, Gansauge F, Beger HG, Nussler AK, FEBS Lett. 1999, 445, 274–278. [DOI] [PubMed] [Google Scholar]
- [87].Trushkin NA, Filimonov IS, Vrzheshch PV, Biochem. 2010, 75, 1368–1373. [DOI] [PubMed] [Google Scholar]
- [88].Warner TD, Giuliano F, Vojnovic I, Bukasa A, Mitchell JA, Vane JR, Proc. Natl. Acad. Sci 1999, 96, 7563–7568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Mitchell JA, Akarasereenont P, Thiemermann C, Flower RJ, Vane JR, Proc. Natl. Acad. Sci. USA 1993, 90, 11693–11697. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.