

Abstract
Here we demonstrate a technique to generate proteomic signatures of specific cell types within heterogeneous populations. While our method is broadly applicable across biological systems, we have limited the current work to study neural cell types isolated from human, post-mortem Alzheimer’s disease (AD) and aged-matched non-symptomatic (NS) brains. Motivating the need for this tool, we conducted an initial meta-analysis of current, human AD proteomics studies. While the results broadly corroborated major neurodegenerative disease hypotheses, cell type-specific predictions were limited. By adapting our Formaldehyde-fixed Intracellular Target-Sorted Antigen Retrieval (FITSAR) method for proteomics and applying this technique to characterize AD and NS brains, we generated enriched neuron and astrocyte proteomic profiles for a sample set of donors (available at www.fitsarpro.appspot.com). Results showed the feasibility for using FITSAR to evaluate cell-type specific hypotheses. Our overall methodological approach provides an accessible platform to determine protein presence in specific cell types and emphasizes the need for protein-compatible techniques to resolve systems complicated by cellular heterogeneity.
Keywords: Astrocytes, Alzheimer’s disease, brain, cellular heterogeneity, proteomics
INTRODUCTION
Resolving cell type-specific contributions is critical for understanding both normal, homeostatic processes and disease in biological systems complicated by cellular heterogeneity. In the field of neurodegeneration, debilitating neurological disorders have been primarily characterized by the presence of abnormal, aggregated proteins, like amyloid-beta (Aβ) or tau in Alzheimer’s disease (AD) or alpha-synuclein in Parkinson’s disease. While it was originally thought that these proteins provided toxic effects solely to neurons14,17,35, growing interest has turned to non-neuronal central nervous system cells and their potential role(s) in neurodegeneration. Given the inherent structural and functional interdependencies of cells in the brain, determining how specific neural cell types contribute to neurodegeneration is critical but remains logistically challenging.
Many techniques have been applied to resolve cell type-specific roles, including in the field of neurodegeneration. Highly accessible, low throughput assays, such as immunohistochemistry/immunofluorescence, have been used extensively to explore spatial localization of pathological indicators such as senile plaques and neurofibrillary tangles relative to cell types in AD human tissue5,25. On the opposite end of the technological spectrum, high-throughput assays, such as single-cell sequencing, provide the highest resolution to assess cellular heterogeneity. Recently, a flurry of new articles has been published applying single-cell sequencing to explore human neurodegenerative disease brain samples15,21,34,49. Although these studies are an important first step, sample sizes are often not large enough to confidently cluster many resident cell types. Furthermore, despite single-cell sequencing being well-suited for characterizing the cellular heterogeneity of tissues, the resulting gene expression data lack the finality of protein characterization. While mRNA abundance is commonly used as a proxy for protein abundance, this assumes that transcription is the primary factor determining the amount of protein made22. In practice, mRNA abundance has a relatively low correlation with protein synthesis (R2 = 0.09-0.46 in human systems) due to post-transcriptional modifications and regulation13,45. Therefore, we posit that cell type-specific protein profiling will provide additional, functionally relevant information not previously possible regarding a cell type’s role in disease pathology. While single-cell proteomics approaches (e.g., CyTOF28, CODEX multiplexed imaging20, SCoPE-MS7, NanoPOTS50, etc.) hold great promise, most are in their infancy, and the required equipment/processes remain impractical to implement broadly across the scientific community. The techniques described herein provide a bridge to this eventual goal, allowing researchers to immediately begin elucidating cell type-specific contributions in biological systems. For example, the majority of work characterizing protein in human AD samples has involved bulk brain regional isolates, which do not account for the heterogeneous composition of cell types. Instead, the entire sample is homogenized, effectively masking cell type-specific levels of common and unique proteins.
In this study, we developed and implemented a cell enrichment and processing method compatible with proteomics. The effectiveness and feasibility of this approach was demonstrated by investigating how a subset of human neural cell types contribute to AD pathology, a disease chosen because it is the most common neurodegenerative disorder and cause of dementia worldwide48. We began by conducting a meta-analysis to assess the few published proteomics datasets from human brain homogenates1,16,37,41 and predicted how different cell types contribute to AD dysfunctional mechanisms. We then adapted our Formaldehyde-fixed Intracellular Target-Sorted Antigen Retrieval (FITSAR) method39,40, a technique that allows for enrichment of specific cell subpopulations via intracellular antibody labeling and cell sorting prior to protein isolation, to allow for proteomic characterization of cell type-specific enriched subpopulations. We validated the technique by comparing similarly processed lysates from fresh and fixed rat brain cells, verifying that the generated proteomic signatures were highly similar. Final feasibility experiments included acquiring protein profiles for enriched neurons and astrocytes isolated from human, post-mortem, flash-frozen AD and aged-matched non-symptomatic (NS) brains. To demonstrate the potential impact of our enrichment/protein-characterization technique, resulting proteomics data were evaluated for AD-associated and cell type-specific proteins of interest. We conclude that the FITSAR method is a fast and cost-effective way to obtain cell type-specific proteomic datasets from post-mortem human samples.
MATERIALS AND METHODS
Meta-analysis
Proteomics studies characterizing AD and NS donor brains were included if they met the following criteria: 1) AD patients had similar diagnoses based on Braak scoring (VI/VI), and control patients were generally age-matched; 2) All samples were isolated from the prefrontal cortex; 3) Proteomics preparation was standardized (from lysis buffer composition and tissue homogenization to protein digestion, preparation, concentration, and mass spectrometry interrogation and analysis); and 4) Raw proteomic data were publicly available. Using these criteria, four separate datasets were identified1,16,37,41. All datasets were collected under a collaborative grant effort (U01AG046161) and were accessible through Synapse’s AMP-AD Knowledge Portal. In total, 206 AD and 156 NS patient datasets were analyzed. Within each study, differential proteins in AD versus NS samples were determined using a logistic generalized linear regression model19 and deemed statistically significant if their corrected p-values were less than 0.05. Functional signatures based on pooled protein classes were then derived by gene set enrichment analysis using Hallmark (50 gene sets), KEGG (186 gene sets), and Reactome (1499 gene sets) gene sets (Broad Institute’s Molecular Signature Database)44 for each dataset. Statistically significant pathways were identified after Bonferroni correction for testing multiple gene sets (Banner: 545; BLSA: 248; Emory: 232; UPenn: 84). Subsequent analyses only used pathways that were present in all four datasets. To predict how specific neural cell types (i.e., neurons, astrocytes, oligodendrocytes, and microglia) contribute to a dysfunctional mechanism, differential proteins were compared to a human cerebral cortex cell type-specific dataset12 within gene set variance analysis24 to generate probabilities that a specific cell type(s) caused mechanistic change. Resulting p-values were considered statistically significant if less than 0.05.
Antibodies
Antibodies used in the study are listed in Table 1. All antibodies were diluted in 1-5% wt/vol (1% for washes, 3% for sorts/immunofluorescence (IF), 5% for western blot (WB)) bovine serum albumin (BSA; Fisher Scientific) (Fraction IV) in 1X phosphate buffered saline (PBS, Fisher Scientific; for sorts/IF) or 1X tris-buffered saline tween (TBST; for WB).
Table 1.
Antibodies used for immunofluorescence (IF), fluorescence-activated cell sorting (FACS), and western blot (WB) experiments.
| Antibody | Vendor | Catalog # | [Ab] for IF | [Ab] for FACS | [Ab] for WB |
|---|---|---|---|---|---|
| mouse anti-beta-III tubulin | Biolegend | 801201 | 1:500 | ||
| rabbit anti-TUBB3 | Abcam | ab18207 | 1:50 | ||
| rabbit anti-glial fibrillary acidic protein | Dako | Z0334 | 1:1000 | ||
| rat anti-GFAP | Life Technologies | 130300 | 1:50 | ||
| mouse anti-oligodendrocyte marker 1 | R&D Systems | MAB2230 | 1:50 | 1:500 | |
| mouse anti-lysosomal associated membrane protein 2 | Abcam | 25631 | 1:100 | ||
| Alexa Fluor 555 mouse anti-TUBB3 | BD Biosciences | BD560339 | 1:20 | ||
| Alexa Fluor 647 mouse anti-GFAP | BD Biosciences | BD561470 | 1:100 | ||
| Brilliant Violet 421 rat anti-mouse IgG2a | BD Biosciences | BD 565817 | 1:100 | ||
| IRDye 800CW goat anti-mouse | LI-COR | 925-332210 | 1:15,000 | ||
| IRDye 680RD donkey anti-rabbit | LI-COR | 925-68073 | 1:15,000 | ||
| anti-mouse IgG H&L Alexa Fluor 488 | Abcam | 150113 | 1:500 | ||
| anti-rabbit IgG H&L Alexa Fluor 594 | Life Technologies | R37117 | 1:500 | ||
| anti-rat IgG Alexa Fluor 647 | Thermo Scientific | A21247 | 1:500 |
Abbreviations: Ab, antibody; FACS, fluorescence-activated cell sorting; IF, immunofluorescence; WB, western blot.
Primary rat brain dissociation
Brown University’s Animal Care Facility approved post-mortem harvesting of waste tissue from scheduled-to-be-euthanized sentinel rat dams. All methods were carried out in accordance with IACUC-approved protocols. After compressed carbon dioxide gas inhalation/asphyxiation and decapitation, rat brains were harvested and immediately dissociated into a single cell suspension by enzymatic digestion, mechanical trituration, filtration, and density gradient centrifugation (see Supplementary Materials and Methods for details). Isolated cells were then directly resuspended in either 1X PBS or fixed using 4% paraformaldehyde (PFA; Thermo Scientific) in PBS. Samples were incubated at room temperature (RT) for 10 minutes, centrifuged (700 x g for 3 minutes at 23°C), washed twice using cold 1% BSA/PBS buffer (wash buffer), and resuspended in buffer for use in subsequent protein isolation/characterization experiments.
Primary, flash frozen, post-mortem human brain dissociation
Six, primary, de-identified human prefrontal cortex samples (NS, N=3; AD, N=3) were obtained through Rhode Island Hospital’s Brain Tissue Resource Center (Title 45 CRF Part 46.102(f)). Donor information is listed in Table 2. Tissues were dissociated using the protocol described above for rat brain dissociation, with one additional step of thawing tissues in calcium- and magnesium-free PBS on ice until tissues sank (<3 minutes). Isolated cells were fixed with 4% PFA/PBS and stored in 1% BSA/PBS buffer at 4°C for up to 5 days, a time frame previously validated to have limited effect on the types/amounts of protein extracted from PFA-fixed samples40, before being used for subsequent FITSAR experiments.
Table 2. Human donor information and cell yields.
Rhode Island Hospital Pathology Core staff prepared/harvested/obtained all donor tissue, which was immediately flash frozen within a post-mortem interval of 12-24 hours and stored at −80°C to preserve sample integrity for downstream biochemical assays. AD-severe diagnosis indicates Braak staging of VI/VI. Abbreviations: F, female; M, male; y.o., years old.
| Donor# | Diagnosis | Gender | Age (y.o.) | Area of Brain | Tissue mass Processed (g) | Total Cell Yield | Cell Yield per g |
|---|---|---|---|---|---|---|---|
| 1 | NS | F | 74 | Prefrontal cortex | 12 | 70x106 | 5.8x106 |
| 2 | NS | M | 60 | Prefrontal cortex | 10 | 42x106 | 4.2x106 |
| 3 | NS | M | 39 | Prefrontal cortex | 9 | 61x10s | 6.8x106 |
| 4 | AD- severe | M | 80 | Prefrontal cortex | 10 | 60x106 | 6.0x106 |
| 5 | AD- severe | M | 90 | Prefrontal cortex | 12 | 68x106 | 5.7x106 |
| 6 | AD- severe | M | 56 | Prefrontal cortex | 10 | 53x106 | 5.3x106 |
Human neural cell type-specific enrichment using intracellular target-based fluorescence activated cell sorting (FACS)
To resolve cell type-specific contributions to AD pathology, neurons (identified by β-III tubulin (TUBB3) levels), astrocytes (identified by glial fibrillary acidic protein (GFAP) levels), and oligodendrocytes (identified by oligodendrocyte marker 1 (O1) levels) from AD and NS brains were enriched for by FACS. The FITSAR method was followed to prepare brain cell suspensions for said enrichment39. In brief, heterogeneous, PFA-fixed brain cell suspensions were permeabilized using 0.5% vol/vol Tween20 (Fisher Scientific) for 15 minutes in the dark, centrifuged (700 x g for 2.5 minutes at RT; standard settings), washed with wash buffer, and blocked in 3% BSA/PBS (blocking buffer) for 30 minutes at RT. Each donor sample was split into aliquots to prepare unstained controls, secondary antibody controls, single antibody controls, and triply-labeled samples for FACS. All diluted antibody solutions (listed in Table 1) were prepared in blocking buffer. 4’,6-diamindino-2-phenylindole (DAPI)-only controls (1:10,000, Thermo Scientific) were also prepared. Samples were counted and resuspended at 20x106 cells/mL in wash buffer. An aliquot was removed from each presorted sample for confocal imaging. Prior to sorting, each sample was filtered using a 40 μm mesh to minimize potential clogging.
An Influx high-speed cell sorter (BD Bioscience) was used to separate cell types by their characteristic markers. Lasers and associated filter sets used are listed in Table S1. Laser settings and gates were optimized for each donor. Gating strategy is explained in detail in Figs. S1 and S2. Populations were gated as follows: “neurons” were identified as TUBB3+/O1−/GFAP−; “oligodendrocytes” were identified as O1+/TUBB3−/GFAP−; and “astrocytes” were identified as GFAP+/TUBB3−/O1−. To measure the percentage of DAPI+ events in a sample, DAPI-labeled controls were assessed separately (Fig. S3). To maximize collected cell yields, all three populations of interest were collected simultaneously from a given sample. Additionally, triply antibody-labeled samples were also collected based on “R1” settings and are referred to as an “unsorted” condition (representing the average, mixed cell population that was still fully processed). Event and cell counts corresponding to each donor’s cell subpopulations are listed in Table S2. All results were analyzed and graphed using FlowJo v10.
Total protein isolation and quantification
All samples were processed using the protein isolation methods described in FITSAR method to allow further protein analysis (see Supplementary Materials and Methods for details). Rat samples were lysed within 6 hours of harvesting brain tissue. Human samples were lysed within a week of tissue dissociation. Resulting protein yields for rats were 48 ± 4 μg for fresh and 29 ± 1 μg for fixed. Higher total protein yields in fresh versus fixed samples was consistent with previous findings39,40. Resulting protein yields for all human sorted samples are listed in Table S2.
Western blot
Western blots determined protein levels of cell type-specific markers across all enriched donor samples, using previously described protocols39 (see Supplementary Materials and Methods for details). Specifically, TUBB3, O1, and GFAP levels were normalized to Total Protein Stain and were then assessed to determine how successfully neural cell types were enriched for based on our intracellular target-based sorting strategy.
Proteomics
All proteomics runs were prepared and analyses completed at Yale University’s Keck Mass Spectrometry & Proteomics Resource Core. Twenty-five μg of protein lysates from fresh (N = 3) and fixed (N = 3) rat brain cells and 2 μg of protein from human unsorted, neuron, and astrocyte conditions (N = 3 per condition) were sent to Yale. Details on sample preparation for proteomics are listed in Supplementary Materials and Methods. Digested sample was injected onto a Q-Exactive Plus (Thermo Fisher Scientific) LC-MS/MS system equipped with a Waters nanoAcquity UPLC system that uses a Waters Symmetry® C18 180 μm x 20mm trap column and a 1.7 μm, 75 μm x 250 mm nanoAcquity™ UPLC™ column (37°C) for peptide separation (additional details listed in Supplementary Materials and Methods). For rat samples, collected data were analyzed with Scaffold Viewer 4 (Proteome Software, LLC) using Mascot search algorithm (Matrix Science). Identified proteins were compared between fresh and fixed conditions, assessing both number and abundance (i.e., spectrum count) of proteins identified. Also, proteins common in both fresh and fixed conditions were compared to cell component datasets (Broad Institute’s Molecular Signature Database) to determine in which cellular compartments identified proteins were localized. For human samples, collected data were processed with Mascot Distiller, and protein identification was searched using the Mascot search algorithm. Cell type-specific and AD-associated markers were evaluated within generated datasets, and variance of scaled protein abundance (i.e., each protein is equally important regardless of total abundance, and variance differences represent standard deviation away from the average protein abundance) was visualized using the ClustVis heatmap webtool36.
Data Availability
All proteins identified from rat and human samples are available at www.fitsarpro.appspot.com, an open-source website created using the Data spectra package27. Raw spectra from proteomic runs are available through the MassIVE repository (rat: MSV000083746; human: MSV000083747).
Confocal imaging
To complement flow cytometry assessment, pre- and post-sorted, immunolabeled cells were imaged using a Nikon AIR Multiphoton microscope (settings listed in Table S3). A 20X objective (Nikon Plan APO 20X DIC M N2) was used to visualize overall enrichment simultaneously across many cells, and a 40X objective (Nikon APO 40X WI lambdas DIC N2) was used to visualize spatial organization of fluorescence intensity in single cells. To determine the localization of TUBB3 within GFAP+ astrocytes, formalin-fixed paraffin embedded superior frontal gyrus tissue sections (5 μm thick) from a NS patient (age 76, male) and severe AD patient (age 89, female) were provided through NYU’s AD Research Center. Tissue sections were labeled for DAPI, lysosomal-associated membrane protein 2, TUBB3, and GFAP (details on preparation listed in Supplementary Materials and Methods). Tissue sections were imaged using a 63X objective (Apochromat 63X/1.40) on a Zeiss LSM800 with Airyscan confocal microscope (settings listed in Table S3). Z-stack images were processed using Imaris (Bitplane). Setup was kept consistent across all samples for each respective confocal experiment. Presented images were enhanced by adjusting brightness and contrast uniformly across the entire image to highlight spatial localization.
Statistical Analysis
R revision 3.5.1, Sigma Plot version 12.5 (Systat Software Inc.) and Prism 7.0 (GraphPad) were used to perform all statistical analyses. All data described represented arithmetic means (± standard deviations, if applicable). Cell type proportion and TUBB3 quantification data passed Shapiro-Wilks normality and equal-variance tests. GFAP quantification data failed the Shapiro-Wilks normality test but passed the equal variance test. Two-way ANOVAs were used to detect significance for all experiments involving multiple comparisons (e.g., cell types and disease states). Significance levels were determined using Tukey post-hoc test (p < 0.05).
RESULTS
Meta-analysis of published neural proteomic datasets and post-hoc predictions of cell type-specific contributions to AD dysfunctional mechanisms
To evaluate the need for cell type-specific proteomic characterization, we performed a meta-analysis on published proteomic datasets collected from human AD and NS bulk prefrontal cortex samples1,16,37,41. Our goals were twofold: (1) identify protein targets common across multiple AD patients, and (2) predict the extent that each major neural cell type contributes to those dysfunctional mechanisms. Rather than assessing individual proteins, pooled protein classes were analyzed. Results corroborated well-established neurodegenerative hypotheses35, including changes in neurotrophic signaling, protein processing/trafficking, metabolism, cell stress/apoptosis, and inflammation. To predict the role of major neural cell types (e.g., neurons, astrocytes, oligodendrocytes, microglia) in identified dysfunctional pathways, associated pathway-specific genes were compared to a human cortical cell dataset12. Based on these comparisons, we were able to predict if specific cell type(s) were associated with AD mechanistic change (Fig. 1). While specific changes in neurons were not resolved using these combined datasets, changes in glia were apparent, including changes in reactive oxygen species generation (astrocytes), epithelial-to-mesenchymal transitions (astrocytes), calcium dysregulation (oligodendrocytes), and immune signaling (microglia). While these findings were encouraging, the vast majority of identifiable AD-related pathways (23 out of 34) were unassignable to specific cell types. Inability to predict cell type contributions are due in large part to our dependency on virtual, “post-hoc” assignment of cell types, which were defined by uniquely expressed genes12 (i.e., >10-fold difference in expression between cell type of interest and other cells). Based on our meta-analysis results, insufficient information existed to make robust cell type-specific predictions for most pathways, and therefore, real-world cell enrichment was required to accurately determine cell type-specific proportional contributions to AD pathology.
Figure 1. Meta-analysis of existing datasets corroborated major neurodegenerative disease hypotheses but revealed gaps in understanding how neural cell heterogeneity influences AD-associated dysfunctional mechanisms.
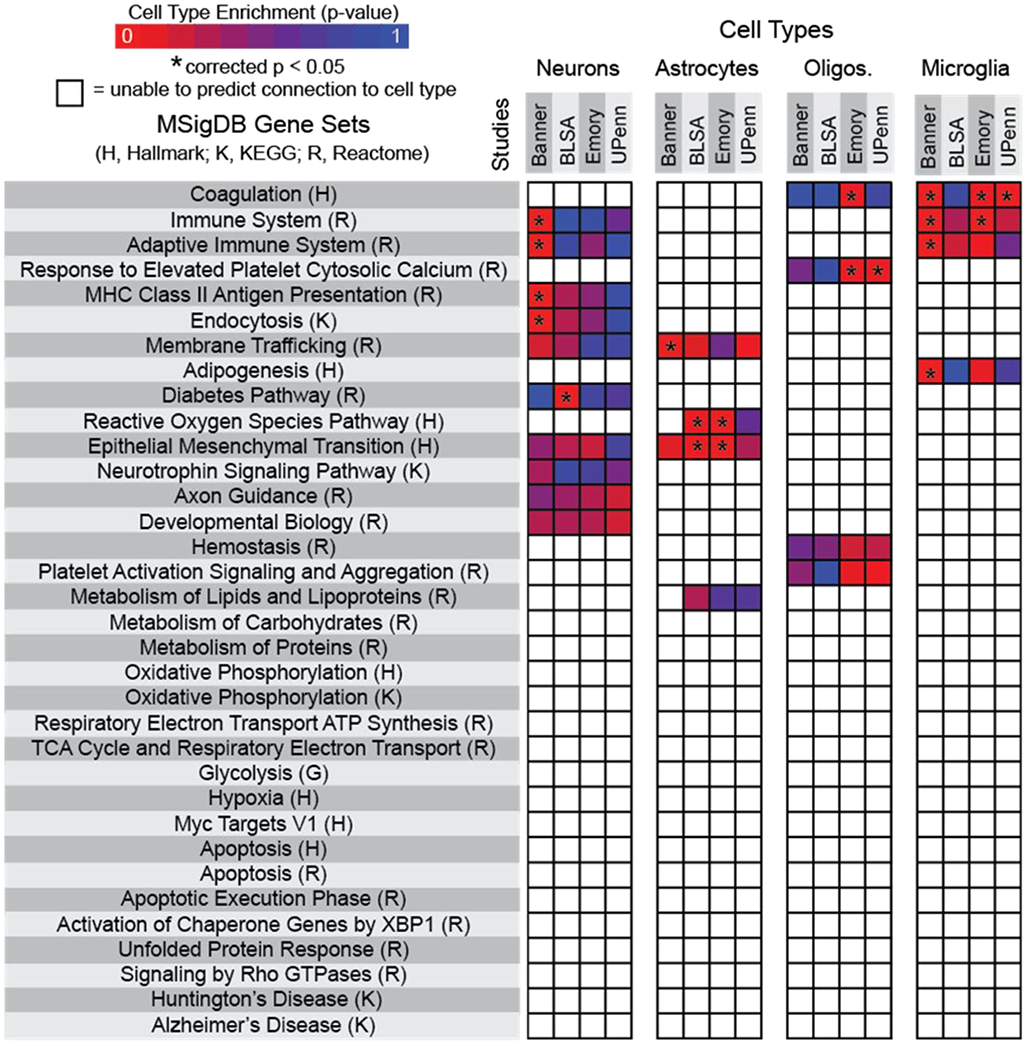
Four datasets (Banner Sun Health Research Institute Study (Banner), Baltimore Longitudinal Study on Aging Study (BLSA), Emory University Alzheimer’s Disease Research Center Brain Bank Study (Emory), and the Perelman School of Medicine University of Pennsylvania Proteomics Pilot Study (UPenn)) were assessed1,16,37,41. Listed gene sets include all pathways significantly different in Alzheimer’s disease versus non-symptomatic proteomic profiles. Heat maps represent specific cell type predictive contributions to dysfunctional pathways, with asterisks indicating statistical significance (corrected p < 0.05). Non-colored boxes indicate inconclusive tests in which a p-value was unable to be generated.
Comparison of proteomic profiles from fresh and paraformaldehyde-fixed cell samples
Previously, we established that FITSAR provides a platform in which proteins may be extracted from PFA-fixed, immunolabeled, and sorted cells without compromising protein yield or quality when using targeted protein assays, including western blot39 and multiplex enzyme-linked immunosorbent assays40. Given our goal to generate untargeted proteomic signatures from fixed, enriched cell populations, we first investigated whether cell fixation would irrevocably bias resulting protein signatures (i.e., differences in numbers, amounts, and/or localizations of proteins present in fixed samples when compared to fresh samples). To evaluate these parameters, rat brain cells were either fixed with 4% PFA or remained fresh, and isolated protein was assessed by quantitative tandem mass spectrometry (Fig. 2a). Overall protein signatures from fresh and fixed cells were highly similar. Eighty-three percent of all proteins identified were present in both fresh and fixed conditions, while a small minority of unique proteins were identified in fresh only (2%) and fixed only (15%) conditions (Fig. 2b). Of these overlapping proteins, fresh and fixed conditions had highly correlative (R2 = 0.80) spectrum count quantifications (Fig. 2c) and shared 72% of the most prevalent proteins present (Table S4). Although surprising that fixed samples had more proteins identified than fresh samples, this proportion of proteins was minor and may reflect protein extraction methods that were optimized for fixed samples, using detergents and boiling to adequately untangle proteins and induce methylene bridge thermal hydrolysis33. Finally, to determine if identified proteins exhibited any bias towards specific sub-cellular compartments, proteins were compared to gene ontology reference cell component datasets. Results showed that proteins were represented in all major compartments within eukaryotic cells regardless of fixation, and overlapping proteins within these compartments represented 86% ± 7% of total proteins across all categories. Additional proteins identified in fixed conditions were evenly distributed across all sub-cellular compartment categories. Ultimately, these data validate that protein signatures derived from PFA-fixed cells were highly similar to fresh counterparts using the same FITSAR-based protein isolation procedure.
Figure 2. After processing, fresh and paraformaldehyde (PFA)-fixed rat brain cells exhibit similar numbers, amounts, and cell component localization of proteins.
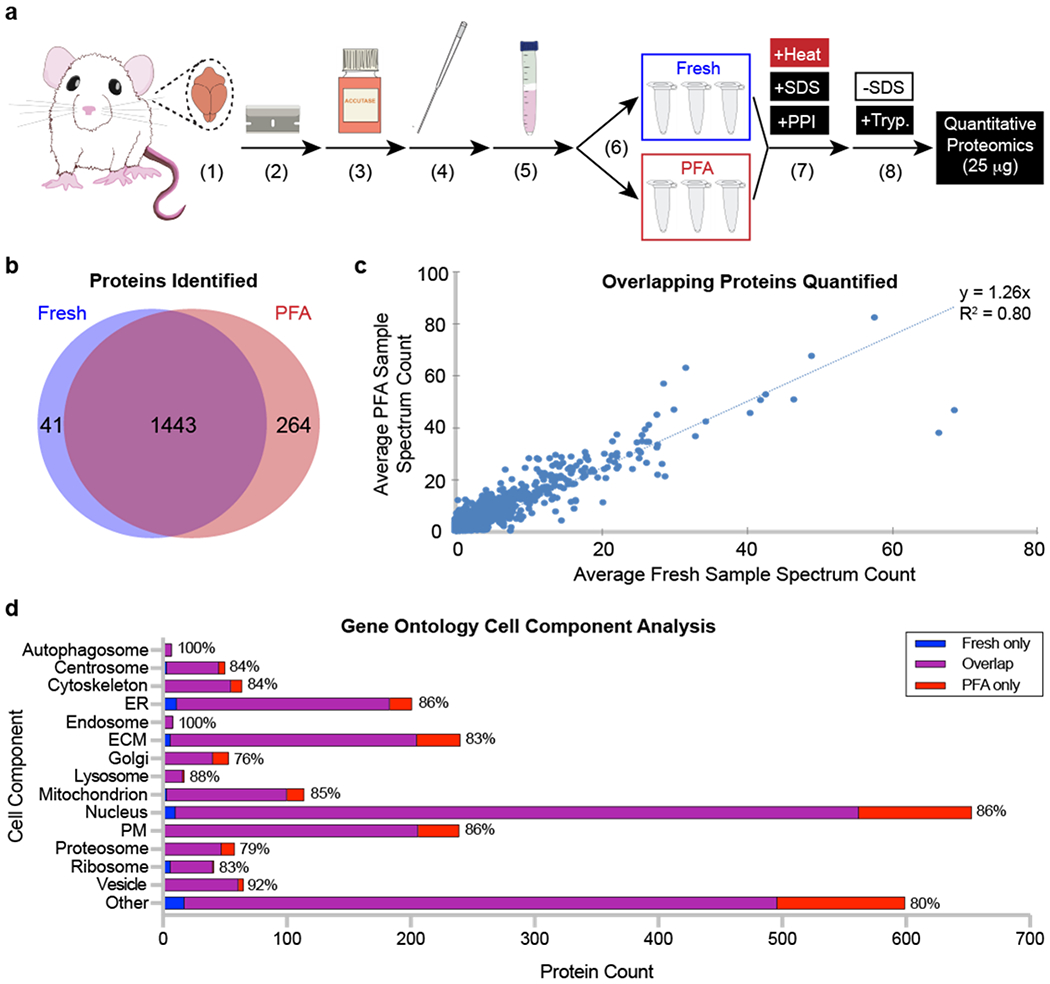
(a) Rat sample processing. A rat brain was (1) isolated, (2) diced into small pieces, and dissociated using (3) enzymatic digestion and (4) mechanical trituration. (5) Cells were isolated using a Percoll gradient before (6) being split into fresh and PFA samples (N = 3 per condition). (7) All samples were processed using detergent and high temperature. (8) Sodium dodecyl sulfate (SDS) was removed from protein lysates, and samples were digested using trypsin before being analyzed by tandem mass spectrometry. (b) Unique proteins identified in fresh and PFA samples. (c) Quantity of unique proteins identified compared between fresh and PFA samples. (d) Cell component localization from fresh only, overlapping, and PFA only identified proteins. Percentages at the end of each bar represent the proportion of overlapping proteins compared to the total number of proteins identified across all conditions. Abbreviations: ER, endoplasmic reticulum; ECM, extracellular matrix; PM, plasma membrane; PPI, protease and phosphatase inhibitor; SDS, sodium dodecyl sulfate; Tryp., trypsin.
Enrichment validation of neural cells isolated from AD and NS brain
As validation of the technique was successful using fresh and fixed primary rodent cells, we next applied FITSAR to human AD and NS tissue samples to better understand how resident cells contribute to AD pathology by characterizing a subset of neural cell types. While we opted to use flash frozen human post-mortem brains, our initial investigations in rodent suggested that researchers can use either fresh or frozen tissue, depending on availability. We did not evaluate whether the techniques would be compatible with non-dissociated, fixed tissues, although we assumed these samples would be problematic since they cannot be easily produce single cell suspensions due to extensive crosslinking between cells and the surrounding matrix. For the current study, flash-frozen cerebral cortex samples from three severe AD patients and three aged-matched NS patients were prepared identically to freshly harvested rat brain with the addition of immunolabeling and sorting for neural cell subpopulations (Fig. 3a). Tissue-mass normalized cell yields were similar among donors regardless of disease pathology (Fig. 3b). We obtained appreciable numbers of neurons and astrocytes through this process but had substantially more difficulty acquiring oligodendrocytes. As such, downstream proteomic analyses were limited to only two of the three major cell types that we originally targeted. Also, given the flash-frozen state of accessible human brain tissue and the need for significant mechanical/shear force to process it, the majority of collected events were neural cell bodies that lacked large extensions. On average, each donor cell suspension contained 95 ± 4% DAPI+ events (Fig. S3), verifying successful isolation of cell bodies and exclusion of the substantial myelin debris. Under these sorting parameters, identifiable cell type proportions (Fig. 3c; Fig. S4) and collected cell type-specific yields (Fig. 3d) were not statistically different between AD and NS brains. Across all donors, ~30% of sorted cells associated with a single, cell-type population, while the remaining cells were unresolvable due to auto-fluorescent noise, attributed to lipofuscin accumulation in aged tissues6 and/or limitations of antigen detection/antibody efficiency in flash frozen tissues.
Figure 3. Using FITSAR, protein from neurons and astrocytes could be characterized from human post-mortem AD and age-matched NS brain.
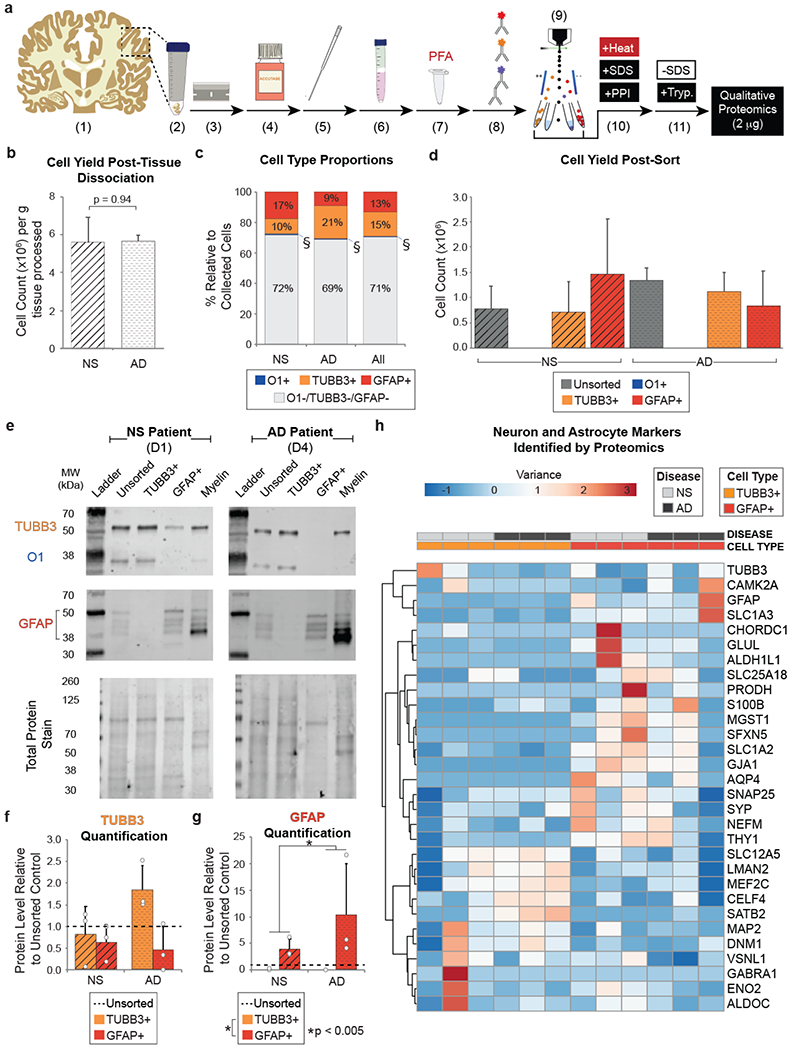
(a) Human sample processing. (1) Frozen tissue (~10 g) was (2) thawed on ice, (3) diced, and dissociated using (4) enzymatic digestion and (5) mechanical trituration. (6) Cells were isolated using a Percoll gradient before (7) being fixed with PFA. (8) Samples were antibody labeled for oligodendrocyte marker 1 (O1), β-III tubulin (TUBB3), and glial fibrillary acidic protein (GFAP) and (9) sorted using fluorescence-activated cell sorting (FACS). (10) Collected TUBB3+ and GFAP+ cells were processed using detergent and high temperature. (11) SDS was removed from protein lysates, and samples were digested using trypsin before analysis by tandem mass spectrometry. (b) Cell yield collected per gram of tissue processed from AD and NS brain. A Student’s t-test was performed to determine significance between disease states (p = 0.94). (c) Average cell type proportions of oligodendrocytes (O1+), neurons (TUBB3+), and astrocytes (GFAP+) relative to the total number of cells analyzed by FACS from AD and NS brain. O1 cell populations (§) made up <1 % of cell total in all donors assessed. A 2-way ANOVA was performed to determine significance among disease states (p = 1), cell types (p < 0.001), and interactions between factors (p = 0.082). (d) Cell type-specific yields post-sort. A 2-way ANOVA was performed to determine significance between disease states (p = 0.72) cell type-specific cell yields (p < 0.01), and interactions between factors (p = 0.29). (e) Example western blots of post-FACS, collected cell populations and myelin debris protein lysates for a NS patient (Donor 1) and an AD patient (Donor 4), assessing TUBB3 (55 kDa), O1 (35 kDa), GFAP (38-50 kDa), and Total Protein Stain. Densitometry-based quantification of (f) TUBB3 and (g) GFAP (50 kDa isoform). 2-way ANOVAs were performed to determine significance between disease states (TUBB3 p = 0.66; GFAP p < 0.005), cell conditions (TUBB3 p = 0.13; GFAP p < 0.005), and interactions between factors (TUBB3 p = 0.40; GFAP p = 0.16). (h) Common neuron and astrocyte markers identified by proteomics supported successful enrichment. Heatmap generated using ClustVis web tool. Abbreviations: D, donor; PPI, protease and phosphatase inhibitor; SDS, sodium dodecyl sulfate; Tryp, trypsin.
Protein lysates were initially evaluated by western blot to verify successful FACS-based enrichment of each cell type. Given insufficient yield of oligodendrocytes for protein analysis, protein isolated from myelin debris was run alongside FACS-collected populations, as we hypothesized that additional O1+ oligodendrocytes may have remained in this debris layer during Percoll density gradient clean-up (Fig. 3e; Fig. S5). However, O1 protein level was variable across donor samples and was not enriched in myelin debris conditions, suggesting that these cells were lost before density gradient separation and may be more sensitive to enzymatic, mechanical, and/or filtering steps required to isolate single cells from flash-frozen tissue. Therefore, only TUBB3 and GFAP protein levels were quantified and normalized to their respective unsorted controls to allow for comparisons within and across donors. TUBB3 protein levels were 0.3-1.5 fold higher in TUBB3+ neurons than their corresponding GFAP+ astrocytes (with the exception of Donor 2) but did not reach statistical significance (p = 0.13) (Fig. 3f). In contrast, GFAP (50 kDa isoform) protein levels were 3-21 fold higher in GFAP+ astrocytes relative to unsorted and TUBB3+ neurons (p < 0.005). Additionally, AD donors had significantly higher GFAP than NS donors (p < 0.005) (Fig. 3g), which has been previously established as a common, albeit non-ideal, identifier of reactive astrogliosis in neurodegeneration and neuroinflammation31.
In addition to predetermined proteins of interest assessed by western blot, enriched protein lysates were evaluated by proteomics to determine if clusters existed based on a variety of cell type-specific markers8,12,23,48 (Fig. 3h). Indeed, results further validated the feasibility of our method, while also demonstrating its potential for broader biological hypothesis testing. When assessing neuron- and astrocyte-specific markers, two major clusters were identified: one neuronal (e.g., SLC12A5, LMAN2, MEF2C, CELF4, SATB2, MAP2, DNM1, and VSNL1) and the other astroglial (e.g., GFAP, SLC1A3, CHORDC1, GLUL, ALDH1L1, SLC25A18, PRODH, S100β, MGST1, SFXN5, SLC1A1, GJA1, and AQP4). Although the majority of markers associated with the astrocyte cluster were astrocyte-specific, there was a small number of neuron-specific markers captured within this group, which corresponded with neuronal proteins that localize to synapses (e.g., SNAP25, SYP) and the cell surface (e.g., THY1).
To investigate why neuron-specific markers were observed in sorted GFAP+ astrocytes, we imaged both TUBB3+ and GFAP+ sorted cells to determine the presence and localization of these primary cell type-specific markers. Low-magnification images revealed overall successful enrichment, as defined by the majority of cells having their respective, single cell type-specific marker of interest (Fig. S6). On closer inspection some “contamination” was observed between TUBB3+ neurons and GFAP+ astrocytes. Specifically, GFAP+ astrocytes often had TUBB3 protein present on the cell, whereas TUBB3+ neurons rarely had GFAP (Fig. 4; consistent with Fig. 3F and 3G). Was this cell type-contamination just an artifact from mechanical dissociation of flash frozen brain tissue, or was TUBB3 also in GFAP+ astrocytes, which are known to phagocytose synapses9 and clear protein aggregates accumulated during AD progression2, engulfing neuronal debris? Our preliminary imaging experiments localized TUBB3 protein to lysosomes in GFAP+ astrocytes, suggesting the latter is at least possible (Fig. S7). Future investigation is required to dissect this disease-relevant biological phenomenon. Ultimately, we demonstrated significant enrichment for astrocytes and neurons from a complex mixture of heterogeneous cells isolated from flash-frozen human tissue (Fig. 3h).
Figure 4. Confocal imaging revealed differences in degree of contamination by neighboring cell processes between sorted TUBB3+ and GFAP+ cells in suspension.
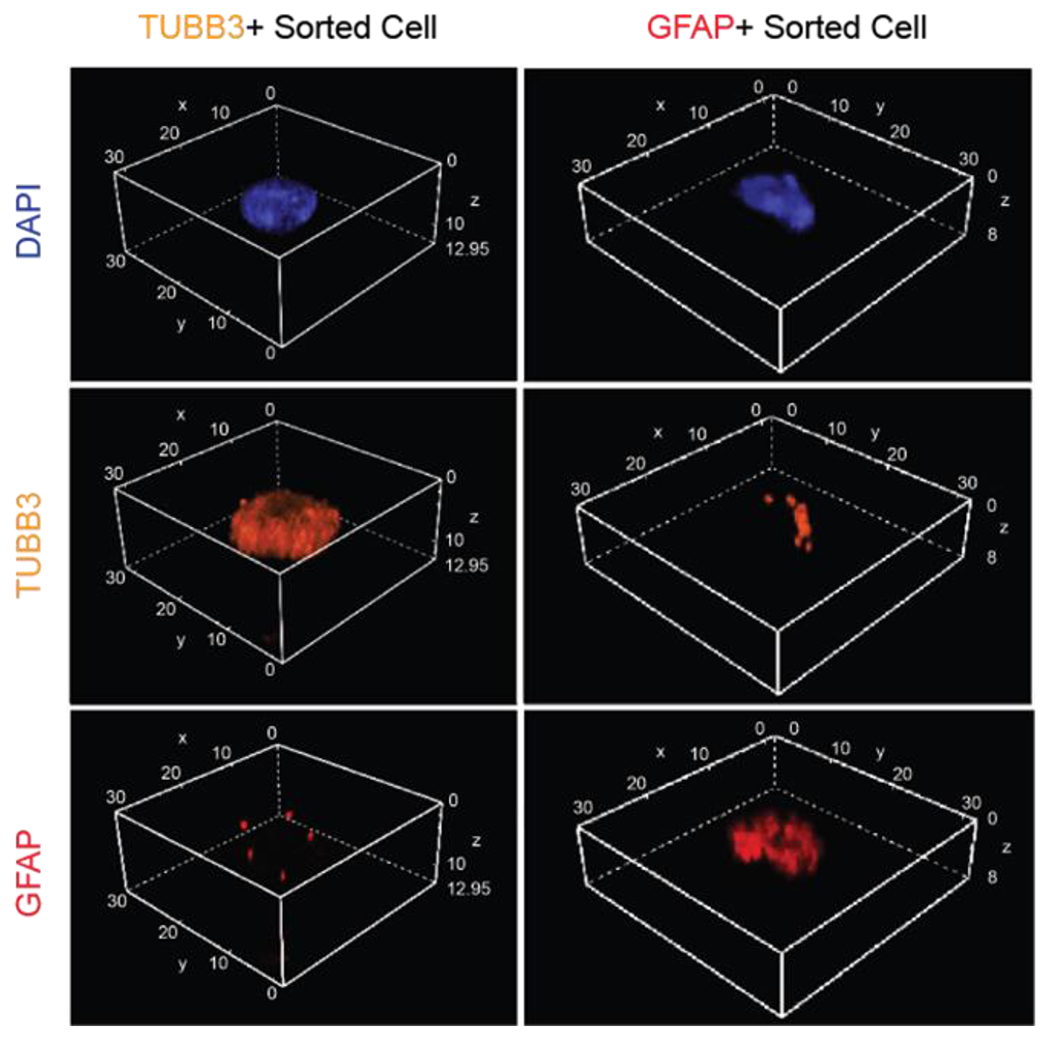
Volumetric stacked images of single, TUBB3+ and GFAP+ sorted cells, respectively. All dimensions listed in μm. Blue, DAPI; Orange, TUBB3; Red, GFAP.
Identifying cell type-specific contributions to disease using proteomics
After successful generation of neuron and astrocyte proteomic profiles from AD and NS brains, we next investigated how these cell types potentially contribute to AD pathology. This analysis was only conducted as an exercise to demonstrate the feasibility for pursuing these types of cell type-specific assessments. Statistically valid conclusions would require substantially larger sample sizes than those used here (N=3 for NS and AD). Mass spectrometry identified ~4,300 proteins across donor samples, and we evaluated this dataset for known genetic risk factors, markers of inflammation, and aging, as identified by previously published transcriptomic characterizations. First, we identified AD risk factors from recent AD genome-wide association studies and reviews3,11,18,26,30,42 (Fig. 5a). We found that TUBB3+ neurons from AD patients were enriched for PICALM, APBA2, LRP1, APP, BCR, CELF1, and PTK2B compared to NS TUBB3+ neurons and all GFAP+ astrocytes. This is not surprising given the strong evidence for pathological roles of APP and Aβ in AD14,17,35 We also saw that GFAP+ astrocytes had higher levels of CLU and APOE, highlighting the important role for apolipoprotein metabolism/regulation in astrocytes for disease initiation/progression43. Next, we examined our data for reactive astrocyte markers (Fig. 5b), given growing evidence for major pathological changes associated with astrocyte function in neurodegenerative diseases32,38,47,48, including AD32,43. Surprisingly, GFAP+ astrocytes had increased levels of classical reactive astrocyte markers regardless of disease pathology. Given the similarities of reactive astrocyte markers between AD and NS GFAP+ astrocytes, we hypothesized that age may be a more significant driving factor than AD-specific disease pathology in our samples. To explore this hypothesis, we mined recent aging astrocyte transcriptomic datasets4,10 and examined these markers in our proteomics dataset (Fig. 5c). Under these parameters, many age-associated astrocyte markers at the gene level were also enriched for in our proteomics analyses (Fig. 5c). Our proteomic dataset highlighted several proteins that would be interesting to investigate in more detail, including glutamate transporter GLT-1 (SLC1A2), vimentin (VIM), aquaporin 4 (AQP4), and complement component C1Q (C1Q), which have roles in neurodegeneration hypotheses ranging from neuronal excitotoxicity to glymphatic dysfunction29,46 and inflammation32 in astrocytes. While the current sample sizes are too small for explicit conclusions, the above exercise demonstrates how FITSAR is an easily accessible and powerful platform to facilitate protein profile characterization of specific cell subpopulations in primary tissues.
Figure 5. Proteomic profiling demonstrated the feasibility for examining cell type-specific hypotheses associated with AD pathology.
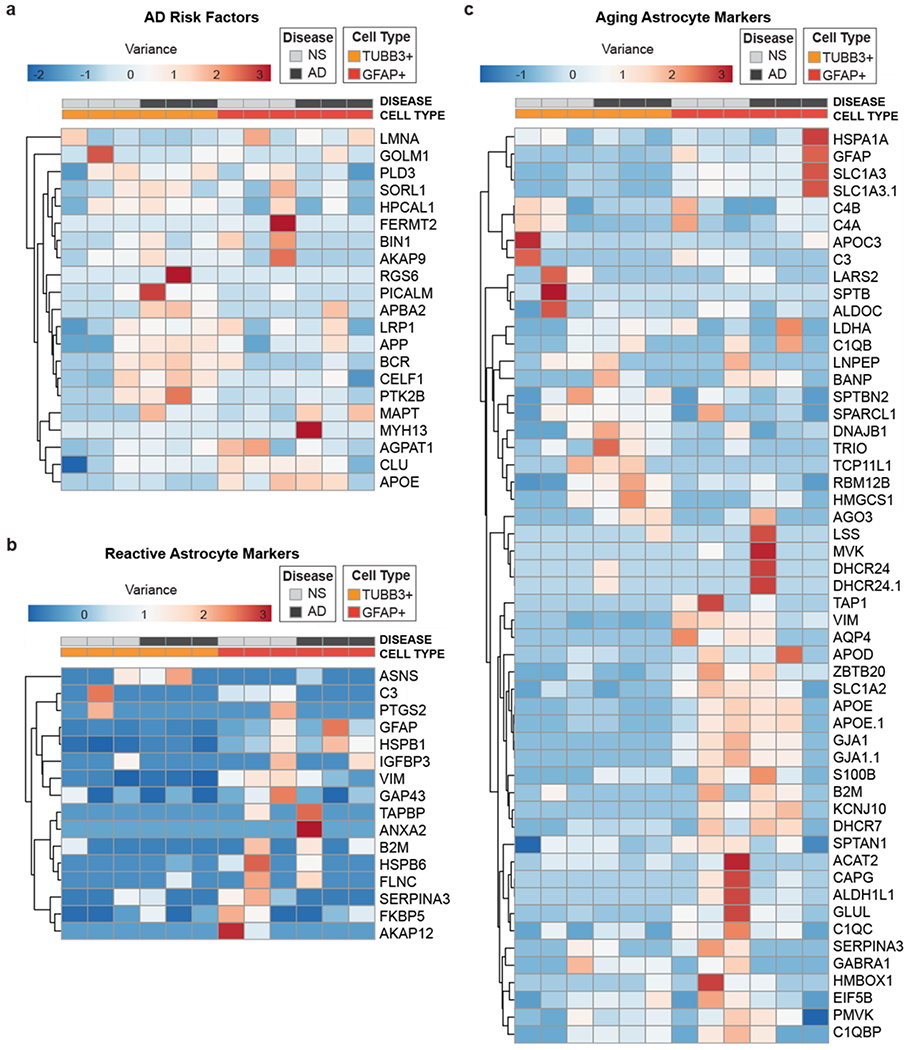
(a) Common AD risk factors. (b) Common reactive astrocyte markers. (c) Aged astrocyte markers. Unit variance scaling was applied to all rows. Rows and columns are clustered using correlation distance and average linkage. Heatmap generated using ClustVis web tool.
DISCUSSION
Here we present a transformative methodological approach that facilitates the investigation of protein signatures for targeted cell types in heterogeneous samples. As a practical demonstration, we conducted the first proteomic study comparing neurons and astrocytes isolated from human, post-mortem AD and aged-matched NS brain to gain a better understanding of their respective contributions to AD. Initially, we attempted to predict these cell type-specific contributions to AD through bioinformatic analysis of published bulk proteomic datasets. However, these predictions were limited due to the few available proteomics datasets for AD human samples and was entirely dependent on virtual assignment of cell types, which gave us greater impetus to empirically generate proteomic signatures from enriched cell types. In this pursuit, we adapted the FITSAR method for proteomics and found that it allowed for detection of individual proteins equally well in either fresh or fixed cell samples, providing a means to use fixation to access intracellular targets. In our application of this technique to human tissues, we found that no detectable differences in sample preparation requirements or the depth of proteomic signatures obtained, although there was substantial variability among human donor samples as is evident from enrichment validation western blots. Additionally, we demonstrated that multiple cell populations could be simultaneously isolated from the same piece of tissue, allowing for efficient, direct comparison within a single donor sample. FITSAR is therefore a fast, cost-effective, and simple method to generate protein signatures from specific, intracellularly targeted cell populations. Finally, we demonstrated how the resulting data could be used to test existing and new hypotheses (e.g., AD risk factors, astrocyte reactivity, aging, etc.), which could spur additional, follow-up studies.
Our AD FITSAR-generated proteomic datasets, currently available on an open access, interactive website (www.fitsarpro.appspot.com), provide a unique resource for the field to continue to mine and compare to new findings. However, most importantly, our study supports the application of FITSAR as a platform to study targeted cell subpopulations in any tissue, organoid, cell culture, or biological system that is complicated by heterogeneity. The same methods described here can also be used to examine other cell types (e.g., microglia, pericytes, endothelial cells), subtypes (e.g., glutamatergic, GABAergic, or dopaminergic neurons), or phenotypes (e.g., C3-positive cells indicating ‘A1’ reactive astrocytes32), provided well-defined markers and antibodies are available. In addition to incorporating robust markers for sorting a cell population of interest, subsequent cell and protein yields are also critical for successful application of this technique. In our study, a minimum of 250,000 cells per cell type was necessary to extract enough protein (~5 μg) for western blot and qualitative proteomic analyses. Therefore, to apply FITSAR to highly rare cell populations, multiple biological samples may need to be pooled together until significant improvements to sorting efficiencies and/or protein detection by mass spectrometry are developed. Although these practical limitations exist, FITSAR provides a significant advancement for characterizing the proteomes of specific cell populations using widely available reagents and instrumentation.
Overall, our study establishes the feasibility and necessity of characterizing cell type-specific protein profiles, using post-mortem human brain as a test case, to more accurately understand proportional cell contributions to healthy and diseased tissue states. Enriching for specific cell populations prior to characterization increases experimental reproducibility and bolsters subsequent interpretations. By better understanding how different cell subpopulations contribute to mechanisms driving pathologies such as AD, we anticipate increased identification of novel therapeutic targets to treat these diseases.
Supplementary Material
ACKNOWLEDGEMENTS:
The authors would like to thank Rebecca Hamelin and Brown’s Animal Care Facility for providing sentinel rats for method optimization, Dr. Edward Stopa, Terra D. Velilla, and the Brain Tissue Resource Center for providing primary human brain samples, Mark Dooner and the COBRE Flow Cytometry Core at Rhode Island Hospital for his assistance with FACS runs, Dr. TuKiet Lam, Jean Kanyo, Wei Wei Wang, and Keck Mass Spectrometry & Proteomics Resource Core at Yale University for the mass spectrometry sample preparation and data analysis, Dr. Thomas Neubert for his feedback on the proteomics analyses, Dr. Thomas Wisniewski, Dr. Arline Faustin, and the New York University Alzheimer’s Disease Center (funded in part by PHS Grant P30 AG08051) for providing primary human brain tissue sections, and Dr. Ryosuke Kita for his help on using data spectra.
FUNDING:
This work was supported by the National Institutes of Health (R01 AR063642 to EMD, T32 to JSS via T32AG052909 (Wisniewski, Scharfman)), the National Science Foundation (CAREER CBET 1253189 and EAGER CBET 1547819 to EMD, GRFP 2014183678 to JSS), the Cure Alzheimer’s Fund (SAL), and the Alzheimer’s Disease Resource Center at NYU Langone (SAL and JSS).
Abbreviations and symbols used:
- Aβ
amyloid-beta
- AD
Alzheimer’s disease
- D
donor
- DAPI
4’,6-diamindino-2-phenylindole
- ECM
extracellular matrix
- ER
endoplasmic reticulum
- F
female
- FACS
fluorescence-activated cell sorting
- FSC
forward scatter
- FITSAR
Formaldehyde-fixed Intracellular Target-Sorted Antigen Retrieval
- GFAP
glial fibrillary acidic protein
- H
Hallmark gene sets
- K
KEGG gene sets
- M
male
- NS
non-symptomatic
- O1
oligodendrocyte marker 1
- PBS
phosphate saline buffer
- PFA
paraformaldehyde
- PM
plasma membrane
- PPI
protease and phosphatase inhibitor
- R
Reactome gene sets
- R#
region #
- SDS
sodium dodecyl sulfate
- SSC
side scatter
- Tryp
trypsin
- TUBB3
β-III tubulin
- y.o.
years old
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
CONFLICT OF INTEREST:
No benefits in any form have been or will be received from a commercial party related directly or indirectly to the subject of this manuscript.
REFERENCES
- 1.The perelman school of medicine upenn proteomics pilot (upppilot) study. <https://www.synapse.org/#!Synapse:syn5477237> 2015.
- 2.Bard F et al. Peripherally administered antibodies against amyloid β-peptide enter the central nervous system and reduce pathology in a mouse model of alzheimer disease. Nat Med. 6: 916–919, 2000. [DOI] [PubMed] [Google Scholar]
- 3.Bertram L, and Tanzi RE. Genome-wide association studies in alzheimer’s disease. Hum Mol Genet. 18: R137–145, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boisvert MM, Erikson GA, Shokhirev MN, and Allen NJ. The aging astrocyte transcriptome from multiple regions of the mouse brain. Cell Rep. 22: 269–285, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Braak Η, Alafuzoff I, Arzberger T, Kretzschmar H, and Del Tredici K. Staging of alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 112: 389–404, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brunk UT, and Terman A. Lipofuscin: Mechanisms of age-related accumulation and influence on cell function. Free Radic Biol Med. 33: 611–619, 2002. [DOI] [PubMed] [Google Scholar]
- 7.Budnik B, Levy E, Harmange G, and Slavov N. Scope-ms: Mass spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation. Genome Biol. 19: 161, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cahoy JD et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: A new resource for understanding brain development and function. J Neurosci. 28: 264–278, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chung WS et al. Astrocytes mediate synapse elimination through megf10 and mertk pathways. Nature. 504: 394–400, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clarke LE et al. Normal aging induces a1-like astrocyte reactivity. Proc Natl Acad Sci U S A. 115: E1896–E1905, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cuyvers E, and Sleegers K. Genetic variations underlying alzheimer’s disease: Evidence from genome-wide association studies and beyond. The Lancet Neurol. 15: 857–868, 2016. [DOI] [PubMed] [Google Scholar]
- 12.Darmanis S et al. A survey of human brain transcriptome diversity at the single cell level. Proc Natl Acad Sci U S A. 112: 7285–7290, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Sousa Abreu R, Penalva LO, Marcotte EM, and Vogel C. Global signatures of protein and mrna expression levels. Mol Biosyst. 5: 1512–1526, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Strooper B, and Karran E. The cellular phase of alzheimer’s disease. Cell. 164: 603–615, 2016. [DOI] [PubMed] [Google Scholar]
- 15.Del-Aguila JL et al. A single-nuclei rna sequencing study of mendelian and sporadic ad in the human brain. Alzheimers Res Ther. 11: 71,2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duong D The baltimore longitudinal study on aging (blsa) study. <https://www.synapse.org/#!Synapse:syn3606086> 2015.
- 17.Ginsberg SD, Che S, Counts SE, and Mufson EJ. Single cell gene expression profiling alzheimer’s disease. NeuroRx. 3: 302–318, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Giri M, Zhang M, and Lu Y. Genes associated with alzheimer’s disease: An overview and current status. Clin Interv Aging. 11: 665–681, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goeman JJ, van de Geer SA, and van Houwelingen HC. Testing against a high dimensional alternativ. J. R. Statist. Soc. B 68: 477–493, 2006. [Google Scholar]
- 20.Goltsev Y et al. Deep profiling of mouse splenic architecture with codex multiplexed imaging. Cell. 174: 968–981 e915, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grubman A et al. A single-cell atlas of entorhinal cortex from individuals with alzheimer’s disease reveals cell-type-specific gene expression regulation. Nat Neurosci. 22: 2087– 2097, 2019. [DOI] [PubMed] [Google Scholar]
- 22.Gry M et al. Correlations between rna and protein expression profiles in 23 human cell lines. BMC Genomics. 10, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guttenplan KA, and Liddelow SA. Astrocytes and microglia: Models and tools. J Exp Med. 216: 71–83, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hanzelmann S, Castelo R, and Guinney J. Gsva: Gene set variation analysis for microarray and rna-seq data. BMC Bioinformatics. 14, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hyman BT et al. National institute on aging–alzheimer’s association guidelines for the neuropathologic assessment of alzheimer’s disease. Alzheimers Dement. 8: 1–13, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jansen IE et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing alzheimer’s disease risk. Nat Genet, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kita R Dataspectra2. <https://github.com/rkita/dataspectra2> 2018.
- 28.Korin B et al. High-dimensional, single-cell characterization of the brain’s immune compartment. Nat Neurosci. 20: 1300–1309, 2017. [DOI] [PubMed] [Google Scholar]
- 29.Kress BT et al. Impairment of paravascular clearance pathways in the aging brain. Ann Neurol. 76: 845–861,2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lambert JC et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for alzheimer’s disease. Nat Genet. 45: 1452–1458, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liddelow SA, and Barres BA. Reactive astrocytes: Production, function, and therapeutic potential. Immunity. 46: 957–967, 2017. [DOI] [PubMed] [Google Scholar]
- 32.Liddelow SA et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 541: 481–487, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Magdeldin S, and Yamamoto T. Toward deciphering proteomes of formalin-fixed paraffin-embedded (ffpe) tissues. Proteomics. 12: 1045–1058, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mathys H et al. Single-cell transcriptomic analysis of alzheimer’s disease. Nature, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mattsson N, Schott JM, Hardy J, Turner MR, and Zetterberg H. Selective vulnerability in neurodegeneration: Insights from clinical variants of alzheimer’s disease. J Neurol Neurosurg Psychiatry. 87: 1000–1004, 2016. [DOI] [PubMed] [Google Scholar]
- 36.Metsalu T, and Vilo J. Clustvis: A web tool for visualizing clustering of multivariate data using principal component analysis and heatmap. Nucleic Acids Res. 43: W566–570, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peters M The banner sun health research institute (banner) study. <https://www.synapse.org/#!Synapse:syn7170616> 2016.
- 38.Rothhammer V et al. Microglial control of astrocytes in response to microbial metabolites. Nature. 557: 724–728, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sadick JS, Boutin ME, Hoffman-Kim D, and Darling EM. Protein characterization of intracellular target-sorted, formalin-fixed cell subpopulations. Sci Rep. 6, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sadick JS, and Darling EM. Processing fixed and stored adipose-derived stem cells for quantitative protein array assays. Biotechniques. 63: 275–280, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seyfried NT et al. A multi-network approach identifies protein-specific co-expression in asymptomatic and symptomatic alzheimer’s disease. Cell Syst. 4: 60–72 e64, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shen L, and Jia J. An overview of genome-wide association studies in alzheimer’s disease. Neurosci Bull. 32: 183–190, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shi Y et al. Apoe4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature. 549: 523–527, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Subramanian A et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 102: 15545–15550, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vogel C, and Marcotte EM. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat Rev Genet. 13: 227–232, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weller RO, Subash M, Preston SD, Mazanti I, and Carare RO. Perivascular drainage of amyloid-beta peptides from the brain and its failure in cerebral amyloid angiopathy and alzheimer’s disease. Brain Pathol. 18: 253–266, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yun SP et al. Block of a1 astrocyte conversion by microglia is neuroprotective in models of parkinson’s disease. Nat Med. 24: 931–938, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zamanian JL et al. Genomic analysis of reactive astrogliosis. J Neurosci. 32: 6391–6410, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou Y et al. Human and mouse single-nucleus transcriptomics reveal trem2-dependent and trem2-independent cellular responses in alzheimer’s disease. Nat Med. 26: 131–142, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhu Y et al. Nanodroplet processing platform for deep and quantitative proteome profiling of 10-100 mammalian cells. Nat Commun. 9: 882, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All proteins identified from rat and human samples are available at www.fitsarpro.appspot.com, an open-source website created using the Data spectra package27. Raw spectra from proteomic runs are available through the MassIVE repository (rat: MSV000083746; human: MSV000083747).