

Abstract
The mechanisms underlying atrial-selective prolongation of effective refractory period (ERP) and suppression of atrial fibrillation (AF) by NS8593 and UCL1684, small conductance calcium-activated potassium (SK) channel blockers, are poorly defined. The purpose of the study was to confirm the effectiveness of these agents to suppress AF and to probe the underlying mechanisms. Transmembrane action potentials and pseudo-ECGs were recorded from canine isolated coronary-perfused canine atrial and ventricular wedge preparations. Patch clamp techniques were used to record sodium channel current (INa) in atrial and ventricular myocytes and human embryonic kidney (HEK) cells. In both atria and ventricles, NS8593 (3–10 μM) and UCL1684 (0.5 μM) did not significantly alter action potential duration (APD), suggesting little to no SK channel inhibition. Both agents caused atrial-selective: 1) prolongation of ERP secondary to development of post-repolarization refractoriness [PRR], 2) reduction of Vmax, and 3) increase of diastolic threshold of excitation (all are sodium-mediated parameters). NS8593 and UCL1684 significantly reduced INa density in HEK cells as well as in atrial but not in ventricular myocytes at physiologically relevant holding potentials. NS8593 caused a shift of steady-state inactivation to negative potentials in atrial but not ventricular cells. NS8593 and UCL1684 prevented induction of acetylcholine-mediated AF in 6/6 and 8/8 preparations, respectively. This anti-AF effect was associated with strong rate-dependent depression of excitability. The SK channel blockers, NS8593 and UCL1684, are effective in preventing the development of AF due to potent atrial-selective inhibition of INa, causing atrial-selective prolongation of ERP secondary to induction of PRR.
Keywords: Pharmacology, Electrophysiology, Atrial fibrillation, Antiarrhythmic drugs
INTRODUCTION
A major limitation of pharmacological approaches to suppression of atrial fibrillation (AF) is the risk of induction of ventricular pro-arrhythmia. This liability can be reduced or eliminated with the use of agents that produce atrial-selective electrophysiological effects.1, 2 The small conductance calcium-activated potassium (SK) channels have been suggested as prime targets for development of drugs for AF suppression because of atrial-selective expression of small-conductance Ca2+-activated K+ channel current carried by these channels.3, 4 The relative contribution of the SK channel to atrial electrophysiology, however, remains poorly defined.4, 5 Agents that block the SK channels can cause atrial-selective prolongation of the effective refractory period (ERP) and effectively suppress AF.6–9 However, the mechanism underlying atrial selective ERP prolongation by SK channel blockers is poorly understood. The present study was designed to evaluate the electrophysiological effects of the SK channel blockers NS8593 and UCL16846, 7, 10–12 in canine coronary-perfused atrial and ventricular preparations and to assess their effectiveness in preventing the development of AF in an acetylcholine-mediated AF model. We also determined their ability to inhibit sodium channel activity in HEK cells as well as in native canine atrial and ventricular myocytes.
METHODS
Adult mongrel dogs of either sex were used. This investigation conforms to the Guide for Care and Use of Laboratory Animals. Preparations consisting of right atrium with a rim of right ventricle (1.0–2.0 cm) were isolated from the canine hearts. The ostium of the right coronary artery was cannulated with polyethylene tubing (i.d., 1.75 mm; o.d., 2.1 mm) and the preparations were perfused with cold Tyrode’s solution (12–15°C) containing 8.5 [K+]0. With continuous coronary perfusion, all ventricular branches of the right coronary artery were immediately clamped with metal clips (1.5×10 mm, Kent Scientific Corp., Torrington, CT, USA). The total time from excision of the heart to cannulation and perfusion of the artery was <4 min. Ventricular and atrial coronary branches were ligated using silk thread. The preparations were placed in a temperature-controlled bath (8 × 6 × 4 cm) and perfused at a rate of 8–10 ml/min using Tyrode’s solution. The composition of the Tyrode’s solution was (in mM): NaCl 129, KCl 4, NaH2PO4 0.9, NaHCO3 20, CaCl2 1.8, MgSO4 0.5, and D-glucose 5.5, bubbled with 95% O2 and 5% CO2 (37±0.5 °C, pH=7.35). The perfusate was delivered to the artery by a roller pump. An air trap was used to avoid bubbles in the perfusion line.
Transmembrane action potential (AP) recordings (sampling rate 41 kHz) were obtained using floating glass microelectrodes (2.7 M KCl, 10–25 MΩ DC resistance). A pseudo-electrocardiogram (ECG) was recorded using two electrodes consisting of Ag/AgCl half cells placed in the Tyrode’s solution bathing the preparation, 1.0 to 1.2 cm from opposite ends of the coronary-perfused preparations (Fig. 1). Effective refractory period (ERP) was measured by delivering premature stimuli after every 10th basic beat at a pacing cycle length (CL) of 500 ms. The diastolic threshold of excitation (DTE) was determined by increasing stimulus intensity in 0.01 mA steps starting from 0.1 mA, until a steady 1:1 activation was achieved. The shortest S1-S1 pacing interval permitting a 1:1 activation was measured at a 2xDTE, with DTE being determined at a CL of 500 ms. Post-repolarization refractoriness (PRR) was recognized when ERP exceeded APD90 in the ventricle and APD70 in atria. Under normal conditions, ventricular ERP coincided with APD90, whereas atrial ERP generally coincided with APD70–75.13, 14 In the presence of acetylcholine, atrial ERP corresponds to APD90. Maximum rate of rise of the AP upstroke (Vmax): Stable AP recordings and Vmax measurements are difficult to obtain in vigorously contracting perfused cardiac preparations.15 The effect of NS8593 and UCL1684 on Vmax was therefore determined by selecting the largest Vmax in the absence and presence of these agents at a CL of 500 ms in each preparation. The effect of NS8593 and UCL1684 on Vmax was also evaluated as Vmax changes upon acceleration from a CL of 500 to 300 ms. In vigorously beating cardiac preparations, resting membrane potential (RMP) can only be approximated. The change in RMP was approximated by comparing the largest amplitude of phase 0 of AP recorded in control and following drug administration. Significant reduction of phase 0 AP amplitude and depolarization of RMP are generally correlated.16, 17
Figure 1. NS8593 produces atrial-selective prolongation of effective refractory period (ERP) due to development of post-repolarization refractoriness (PRR). Little change is observed in action potential duration (APD) in either atria or ventricles.
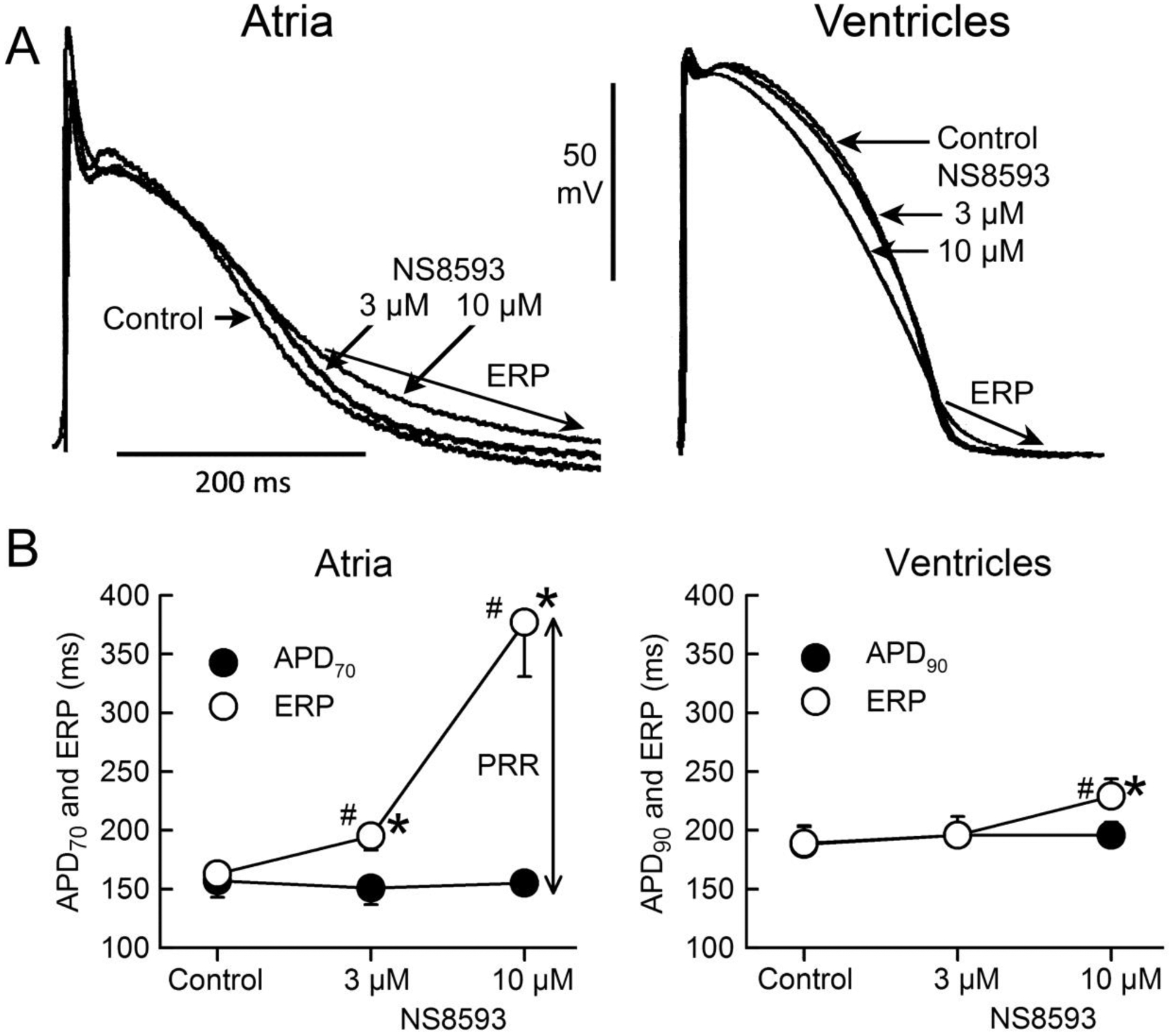
A: Atrial and ventricular superimposed action potentials recorded under control conditions and following addition of 3 and 10 μM NS8593 to the coronary perfusate.
B: Summary data of the effect of NS8593 to alter APD and ERP in atria and ventricles. Note that ERP corresponds to APD70–75 in atria and to APD90 in ventricles. N=6 for atria and 8 for ventricles because 2 out of the 8 atria were either very depressed or inexcitable in the presence of 10 μM NS8593. CL = 500 ms. * p<0.05 vs. control. # - p<0.05 vs. respective APD70 value.
Isolation of atrial and ventricular myocytes were prepared from canine hearts using techniques described previously.13 Briefly, left atrial and ventricular preparations were dissected and coronary perfused first with cardioplegic solution and then with a nominally Ca2+-free solution (mM): NaCl 129, KCl 5.4, MgSO4 2.0, NaH2PO4 0.9, glucose 5.5, and NaHCO3 20, bubbled with 95% O2/5% CO2 containing 0.1% BSA for a period of about 5 min. The preparations were then subjected to enzyme digestion with the nominally Ca2+-free solution supplemented with 0.5 mg/ml collagenase, 0.1 mg/ml protease, and 1 mg/ml BSA for 8–12 min. Then the tissues were minced and incubated in fresh buffer containing 0.5 mg/ml collagenase, 1 mg/mL BSA and agitated. The specific activities of the enzymes were as follows: Protease (Sigma P5147, 3.5 U/mg) and collagenase (Worthington, Type 2, 290U/mg). The supernatant was filtered, centrifuged at 200 rpm for 2 min, and the myocyte-containing pellet was stored in 0.5 mM Ca2+ HEPES buffer at room temperature.
Cell transfection.
HEK cells were grown in DMEM with Glutamax supplemented with 10% FBS in 35 mm culture dishes and placed in a 5% CO2 incubator at 37 °C. To assess the effect on INa, HEK cells were co-transfected using FuGeneHD (Active Motif, Carlsbad, CA,) with a 1:1 molar ratio of WT human SCN5A, and WT SCN1B ion channel genes cloned into the pcDNA3.1. In addition, enhanced green fluorescent protein cDNA-Nav1.5 was tagged for identification of transfected cells for electrophysiologic study 12–24 hours after transfection.
Experimental Protocols.
The equilibration period was 30 min. The electrophysiological parameters were recorded before (control) and after addition of NS8593 or UCL1684 to the perfusion solution. The concentration of NS8593 (3 and 10 μM) was increased in a step-wise manner, with at least 30 minutes at each concentration before the start of recording. Reported data with UCL1684 (0.5 μM) were obtained following >1 hour of exposure to the drug.
The anti-AF effect of NS8593 and UCL1684 was evaluated using an acetylcholine (ACh) -mediated AF model (1 μM). ACh permits electrical induction of persistent AF in 100% atrial preparations (by premature electrical stimulation or rapid pacing). After completion of the main protocol, ACh (1 μM) was added to the coronary perfusate in the presence of 10 μM of NS8593 and 0.5 μM of UCL1684 and induction of AF was attempted using premature electrical stimulation and rapid pacing.
Voltage clamp recordings of peak INa.
Peak INa, was measured as previously described with minor modification.13 Experiments were performed using a MultiClamp 200B (Molecular Devices, Foster City, CA). Command voltages were delivered, and data acquired via a DigiData 1550B computer interface using pClamp9 (Molecular Devices) with data stored on a computer hard disk. Patch pipettes were pulled from borosilicate glass (1.5 mm o.d. and 1.1 mm i.d.) on a Model DMZ-Universal-Electrode-puller (Zeitz-Instruments, Germany). The electrode resistance was 1.5–2.5 MΩ when filled with the internal solution (see below). The membrane was ruptured by applying negative pressure and series resistance compensated by 75 to 80%. Whole cell current data was acquired at 20–50 kHz and filtered at 5 kHz. Cell capacitance was measured by integrating the transient charge following application of a −5mV voltage clamp step. Currents were normalized to cell capacitance and expressed as density (pA/pF). External solution contained the following (in mM): choline Cl 120, NaCl 10, Na+ acetate 2.8, CaCl2 0.5, KCl 4, MgCl2 1.5, CoCl2 1, glucose10, HEPES10, NaOH5, and BaCl2 0.1, pH adjusted to 7.4 with NaOH/HCl. The pipette solution contained the following (mM): NaCl 15, CsF 120, MgCl2 1, KCl 5, HEPES 10, Na2ATP4, and EGTA 10, pH adjusted to 7.2 with CsOH. Recordings of INa were made at least 5 min after cell membrane rupture to minimize the effects of time-dependent negative shift of steady-state (SS) inactivation that occurs in conventional voltage clamp experiments. Whole cell currents were analyzed using the Clampfit analysis program pClamp 9 (Molecular Devices).
Drugs:
NS8593 and UCL1684 (NeuroSearch, Denmark) were dissolved in 100% DMSO as a stock of 10 and 1 mM, respectively.
Statistics:
Statistical analysis was performed using paired or unpaired t test and one-way repeated measures analysis of variance (ANOVA) followed by Bonferroni’s or Student-Newman-Keuls test, as appropriate. All tissue data are expressed as mean ± SD and single cell data as mean ± SEM.
RESULTS:
Electrophysiological effects of NS8593 and UCL1684 in atria and ventricles
NS8593 (3–10 μM) caused marked depression of excitability in atria, but not in ventricles. At 10 μM and a CL of 500 ms, 2 of 8 atria but 0 of 5 ventricles were markedly depressed, failing to respond to stimulation in a 1:1 fashion, so that the electrophysiological parameter could not be reasonably measured in these two atria. NS8593 (3–10 μM) produced little to no change in APD70–90 in either atria or ventricles (Fig. 1). At a concentration of 3 μM, NS8593 prolonged ERP only in atria whereas at a concentration of 10 μM, ERP was prolonged in both atria and ventricles, but to a much greater extent in the atria (Fig. 1). The NS8593-mediated ERP prolongation was due to induction of PRR, which was much greater in atria vs. ventricles (Fig. 1). NS8593 significantly decreased Vmax in an atrial-selective manner. However, a significant reduction of Vmax was evident in the ventricles as well (Fig. 2). DTE and the shortest S1 – S1 pacing interval permitting 1:1 activation was also affected by NS8593 in an atrial-selective manner (Fig. 3). Of note, at an NS8593 concentration of 10 μM and CL of 300 ms, 0/8 atria but 5/5 ventricles displayed 1:1 activation (with a DTEx10 determined at a CL of 500 ms), demonstrating potent rate-dependent atrial-selective effect of NS8593 on excitability. Electrophysiological effects of UCL1684 in the cardiac preparations were similar to those observed with NS8593. APD70–90 was not significantly altered in both atrial and ventricular preparations and ERP was prolonged in an atrial-selective manner due to development of PRR (Fig. 4). Vmax, DTE, and the shortest S1-S1 were also affected exclusively or primarily in atria vs. ventricles (Fig. 4).
Figure 2. Atrial-selective rate-dependent reduction of maximum rate of rise of the action potential (AP) upstroke (Vmax) and depression of excitability by NS8593.
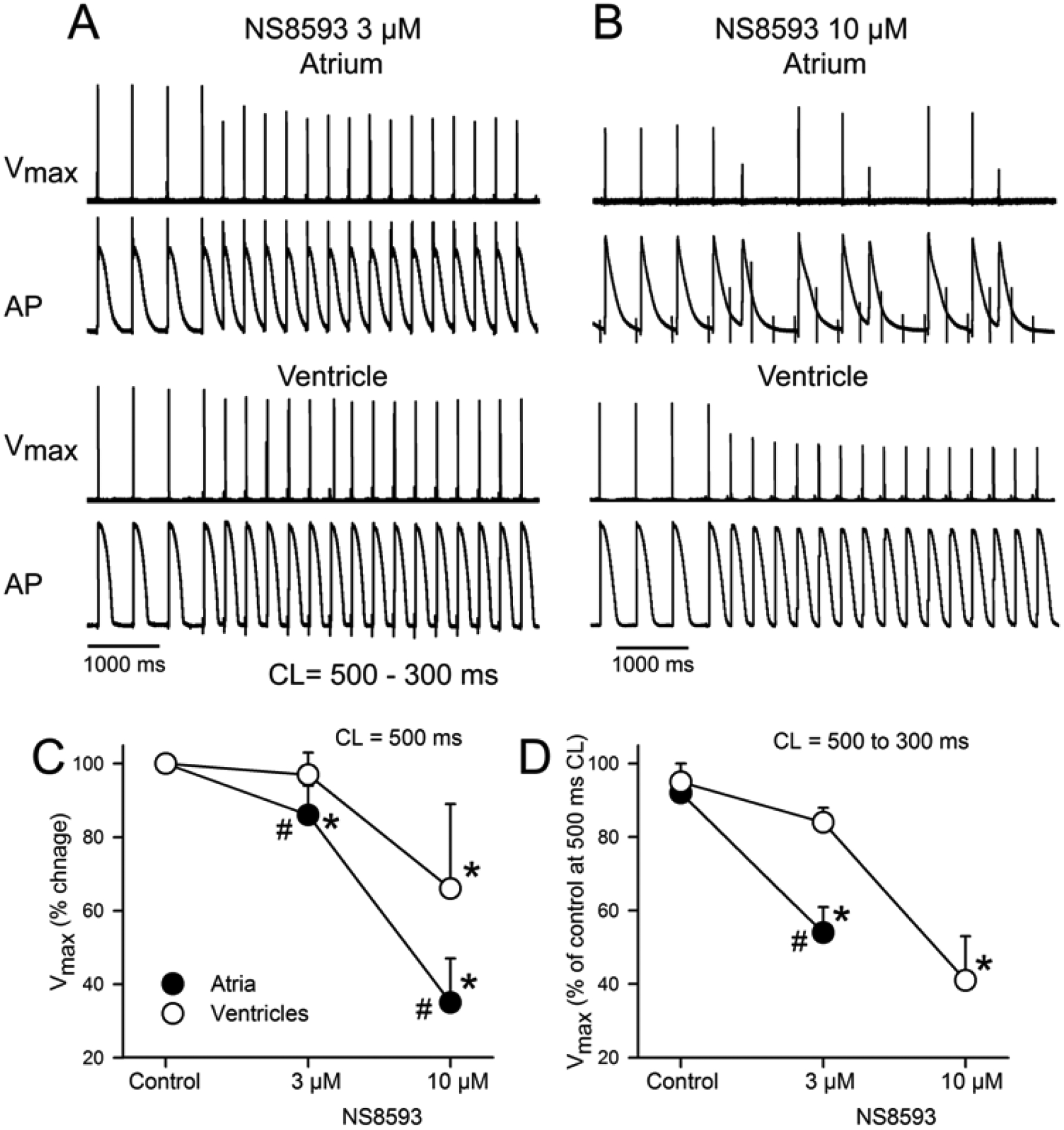
A and B: Action potentials and corresponding Vmax values recorded from atria and ventricles during acceleration of pacing rate from a CL of 500 to 300 ms following exposure to 3 or 10 μM of NS8593. Failure of 1:1 activation was observed in 100% of atrial preparations at a CL of 300 ms in the presence of 10 μM NS8593. C: Composite data of Vmax of atria and ventricles at a CL of 500 ms expressed as % of control. D: Composite data of Vmax of atrial and ventricular APs following acceleration from a CL of 500 to 300 ms expressed as % of Vmax value at a CL of 500 ms in Controls. N=6–8. * - p<0.05 vs. respective control. # - p<0.05 vs. respective ventricular values.
Figure 3.
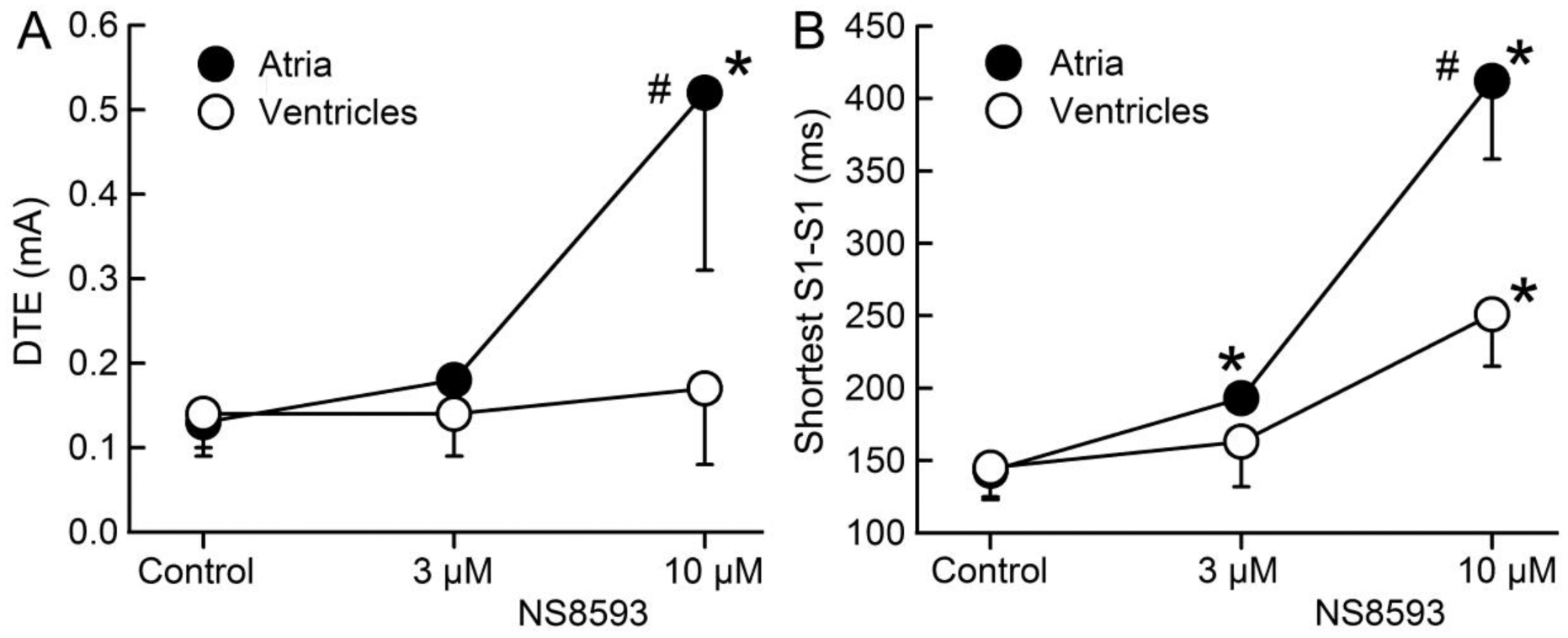
NS8593 increases diastolic threshold of excitation (DTE) and prolongs the shortest S1 – S1 interval permitting 1:1 activation much more in atria than in ventricles. * p <0.05 vs. control. # - p<0.05 vs. respective ventricular value. N=6–8. DTE was measured at a CL of 500 ms.
Figure 4.
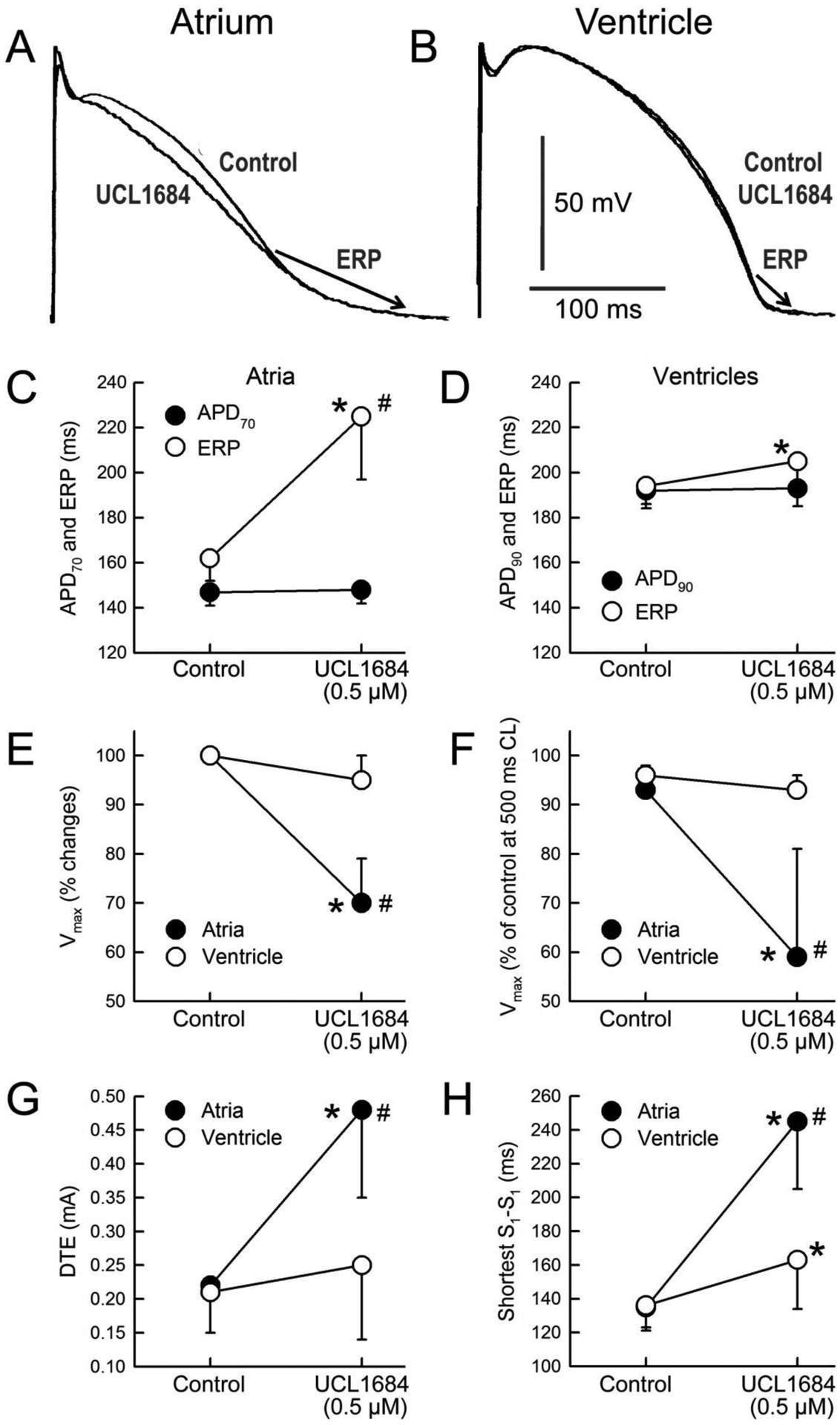
Atrial selective electrophysiological effects of UCL1684 in the induction of PRR (A-D), reduction of Vmax (E-F), increase in DTE (G), and prolongation of the shortest S1-S1 interval permitting 1:1 activation (H). Composite data of Vmax of atrial and ventricular APs following acceleration from a CL of 500 to 300 ms expressed as % of Vmax value recorded at a CL of 500 ms in Controls (F).* p <0.05 vs. control. # - p<0.05 vs. respective ventricular value. N=7–10. Data presented in C, D, E, and G were obtained at a pacing CL = 500 ms.
The maximum amplitude of atrial APs recorded in the presence of 3 and 10 μM of NS8593 and 0.5 μM of UCL1684 was 96±7, 73±13, and 94±6%, respectively, of those recorded under control conditions (p=0.14, p<0.001, and p=0.09, respectively; CL = 500 ms; n=6–8), consistent with the effect of these agents to block INa and suggesting the possibility that 10 μM NS8593 depolarized RMP. The maximum amplitude of APs recorded from the ventricles was not statistically significantly affected by 3 or 10 μM NS8583 or 0.5 μM UCL1684 (98±6, 94±10 and 98±5% of controls, respectively n=8–10, CL 500 ms).
Effect of NS8593 and UCL1684 to suppress atrial fibrillation
ACh alone (1 μM) significantly abbreviated atrial APD and ERP, permitting induction of persistent AF in 100% of atria (Fig. 5; Table 1). Addition of ACh in the presence of NS8593 (10 μM) or UCL1684 (0.5 μM) resulted in a much smaller abbreviation of APD and ERP than observed with ACh alone (Table 1). Both drugs maintained their ability to induce PRR in the presence of ACh. Rate-dependent excitability was also significantly depressed, as evident by prolongation of the shortest S1-S1 interval, so that ERP at rapid activation rates was far longer than APD (Table 1). AF could not be induced in the presence of these agents in any of the preparations tested (Table 1). Potent anti-AF action of the drugs was associated with strong rate-dependent depression of excitability, which did not permit rapid activation of the atrium (Fig. 5; Table 1).
Figure 5. NS8593 (10 μM) prevents the induction of acetylcholine (ACh) - mediated atrial fibrillation largely by depressing excitability.
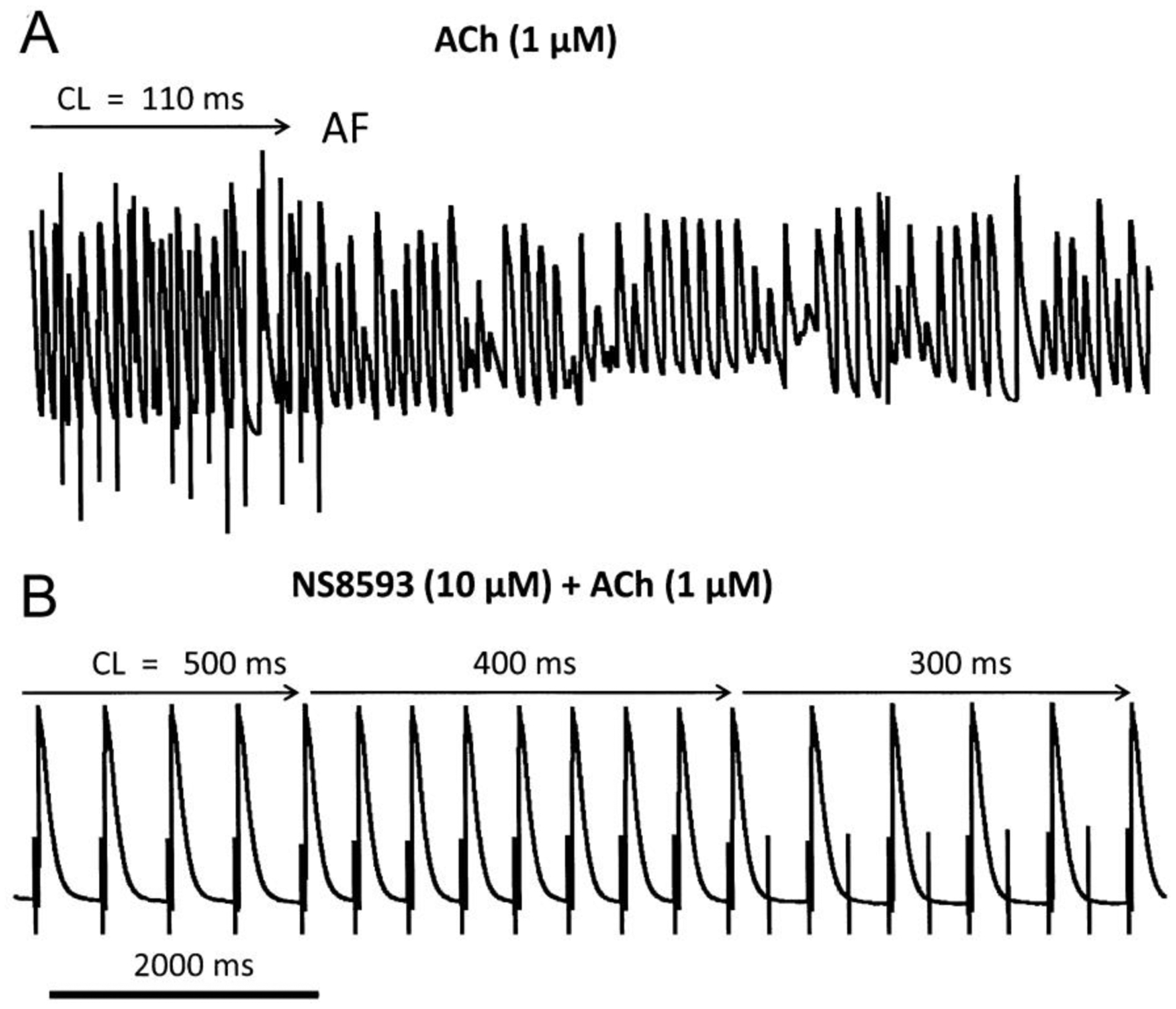
Shown are action potential tracings. A: Rapid pacing induced AF in the presence of ACh alone. B: Rapid pacing fails to induce AF in the presence of NS8593 and ACh due to significant rate-dependent depression of excitability, manifested as a failure of 1:1 activation.
Table 1.
Effects of ACh on atrial electrophysiological parameters in the presence of NS8593 and UCL1684 and on induction of persistent AF in isolated canine coronary-perfused right atria.
| APD90 (ms) | APD70 (ms) | ERP (ms) | PRR (ms) | Shortest S1-S1 (ms) | Induction of AF | |
|---|---|---|---|---|---|---|
| Control | 204±11 | 153±9 | 158±17 | 4±7 | 129±8 | 0% (0/10) |
| ACh (1 μM) | 51±8 | 48±7 | 61±8 | 10±8 | 65±14 | 100% (10/10) |
| NS8593 (10 μM) + ACh (1 μM) | 115±11* | 104±10* | 332±56†# | 217±56† | 371±76† | 0% (0/6) |
| UCL1684 (0.5 μM) + ACh (1 μM) | 94±12* | 87±11* | 177±64†# | 83±51† | 205±52† | 0% (0/8) |
Data were obtained at a cycle length (CL) of 500 ms (n=6 for each).
<0.05 vs. acetylcholine alone (ACh, 1.0 μM).
p<0.001 vs. acetylcholine alone.
p<0.001 vs. APD90.
Post-repolarization refractoriness (PRR) was defined as the difference between ERP and APD90 in the presence of ACh, and between ERP and APD70 in control. ACh was added in the presence of the SK channel blockers. The APD and ERP values for ACh alone are historical data collected in similar preparations in previous studies.
Effects of NS8593 and UCL1684 on INa
Both agents significantly reduced INa in HEK cells, measured at HP of −80 mV (Fig. 6A). To test the hypothesis that the effect of SK channel blockers to produce atrial-selective depression of excitability is the result of inhibition of INa, we studied the direct effect of NS8593 on inhibition of INa in isolated canine atrial and ventricular myocytes at two HPs (−120 and −90 mV) using patch clamp techniques. NS8593 (10 μM) caused a minor non-significant reduction of INa in both atrial and ventricular myocytes at a HP of −120 mV but a potent atrial- predominant decrease of INa at a HP of – 90 mV (Fig. 6B). Steady-state inactivation of the sodium channels (h-curve) was not affected by NS8583 (10 μM) in the ventricular myocytes, but was shifted to negative potentials in the atrial myocytes (Fig. 6C). As we reported previously,13 the h-curve of atrial cells is typically more negative than that of ventricular cells in the absence of drug (Fig. 6C).
Figure 6. UCL1684 and NS8593 produce direct atrial-selective inhibition of sodium channel current (INa) and shift steady-state inactivation of the cardiac sodium channels.
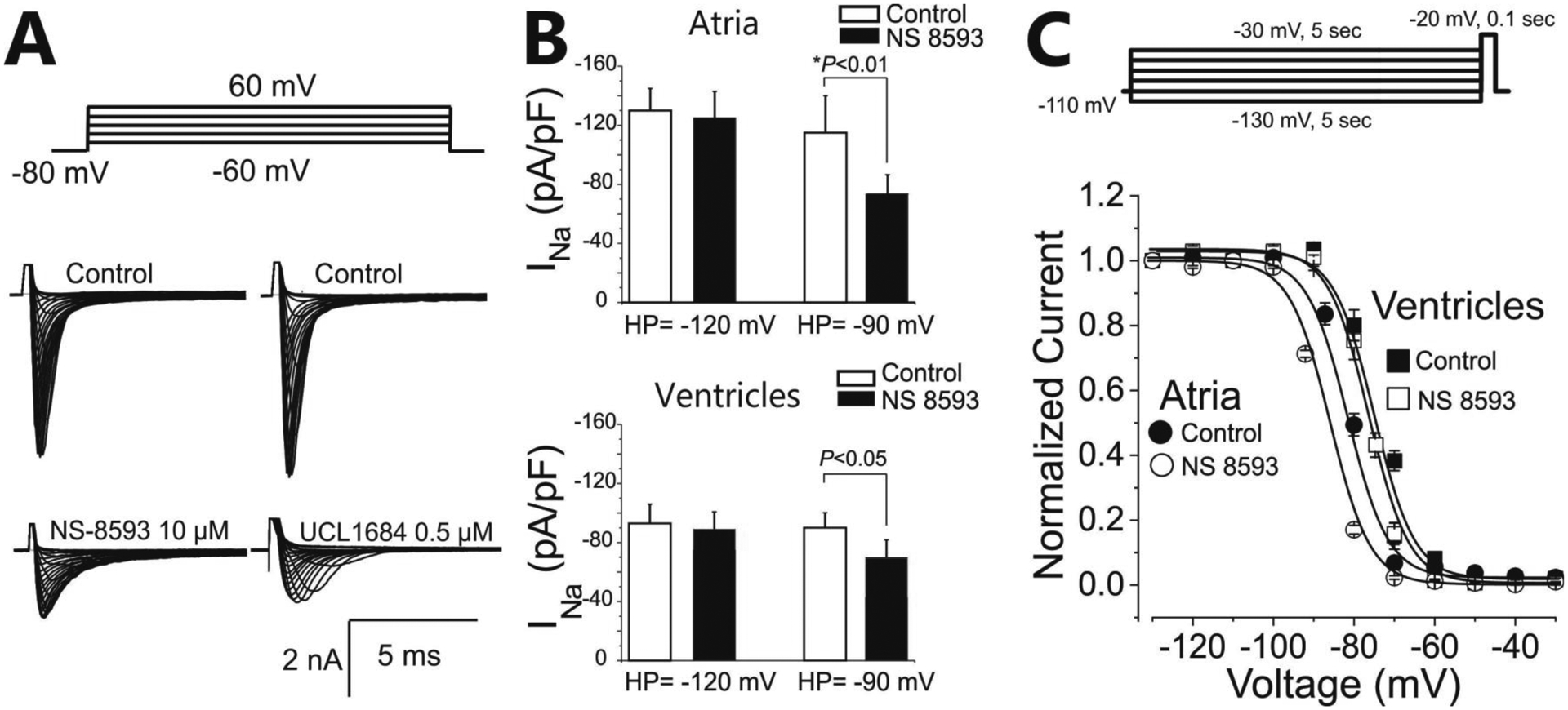
A: Representative INa recordings from a NaV1.5-WT plus NavB1-WT expressed in HEK cells in presence of UCL1684. B: Effect of NS8593 (10 μM) effect on sodium channel current density at test potential of −20 mV in canine atrial and ventricular myocytes recorded at holding potentials (HP) of −120 mV and −90 mV. C. Effect of NS8593 (10 μM) on steady-state inactivation of the sodium channels in atrial and ventricular cardiac myocytes.
DISCUSSION:
The principal findings of the present study are that both SK channel inhibitors studied cause electrophysiological alterations in canine cardiac single cells and multicellular preparations (i.e., reduction of INa density, negative shift of SS-inactivation, PRR induction, reduced Vmax, and increased DTE) consistent with potent atrial-selective inhibition of sodium channel activity. Both drugs have previously been shown to inhibit SK channels11, 12 and these channels have previously been shown to be expressed in canine atria.11 However, the absence of APD prolongation in our atrial preparations with either NS8593 or UCL1684 suggests little to no functional contribution of SK channel inhibition in canine atria under these experimental conditions. Our data demonstrate an effect of both NS8593 and UCL1684 to prevent induction of vagally-mediated AF, due to potent depression of excitability secondary to block of sodium channel activity. Collectively, our findings suggest that SK channel inhibition by NS8593 and UCL1684 does not contribute meaningfully to their potent anti-AF effects.
SK channels have been identified in the hearts of mouse, rat, rabbit, dog, and human and the Ca+2-activated potassium current (ISK) carried by these channels is exclusively or largely expressed in the atrium.6, 8, 10, 18 The selective expression of ISK in the atria is thought to make these channels potential targets for effecting atrial-selective APD and ERP prolongation, thus contributing to the management of AF without adverse consequences in the ventricles.3, 4, 8 Some agents that block the SK channels (such as NS8553, UCL1684, ICA, ICAGEN, AP14145, and AP30663) have been shown to prolong atrial ERP and suppress AF without prolongation of QT in experimental settings.6, 7, 9, 19 There are however reports demonstrating a pro-AF effect induced by genetic ablation of SK channels3 or SK channel inhibition with apamin.20 The mechanism of atrial-selective ERP prolongation by SK channel inhibitors is not well defined. These inhibitors may or may not prolong atrial APD.5, 8 Similar to the results of the present study (Figs. 1–4), previous studies have reported that NS8593, UCL1684, and ICA produce atrial-selective alterations in sodium-channel mediated parameters, i.e., reducing Vmax, slowing conduction, and inducing PRR,10, 21 indicating atrial-selective inhibition of the sodium channel. However, the effect of these agents to depress sodium channel parameters were attributed to an effect of ISK inhibition to effect depolarization of RMP. A significant NS8593-induced reduction of Vmax in the ventricles (where SK channel are not or poorly expressed) clearly points to a direct effect of NS8593 on INa (Fig. 2). Of note, the concentration of UCL1684 used in the current study was relatively low, resulting in minor depression of sodium-channel-mediated parameters in the ventricle (Fig. 4). In the present study, we also demonstrate a direct effect of NS8593 and UCL1684 to inhibit INa using both isolated canine myocytes and HEK cells expressing the alpha and beta1 subunits of the cardiac sodium channel. Moreover, we demonstrate that INa inhibition and its atrial-selectivity are critically dependent on holding potential (HP). Inhibition of INa is clearly evident at a HP of −90 mV, but not at a HP of −120 mV (Fig. 6).
It is important to recognize that the ability of the other SK channel blockers (i.e., ICA, ICAGEN, AP14145, AP30663, etc.) to inhibit the sodium channel have either not been tested or have been tested at a HP of −120 mV. Our data emphasize the need to test such agents under physiologically relevant conditions (i.e., HPs and pacing rates) in order to avoid missing their atrial-selective actions to inhibit INa. Our observation with regard to the SK channel inhibitors is reminiscent of the recent experience with IKur blockers, including vernakalant, AVE0118, AZD1305, which upon close examination proved to exert their actions to prolong ERP solely or largely via atrial-selective inhibition of sodium channel current and not as a result of inhibition of IKur.15, 22–24 Our findings provide yet another example of potassium channel blockers exerting their antiarrhythmic effect principally via inhibition of sodium channel activity.
INa blockers are known to abbreviate atrial APD, via inhibition of late INa25 and it is possible that a similar action on the part of NS8593 and UCL1684 might have negated the effect of SK channel inhibition to prolong atrial APD.
As discussed above, the depression of INa produced by SK channel blockers has in previous studies been attributed to drug-induced depolarization of RMP in atria.21 This suggestion is based largely on the observation that RMP is slightly depolarized in the presence of high concentrations of NS8553, ICAGEN, and ICA in human and rat atrial cells and tissues (by 2–4 mV).21 The contribution of such RMP depolarization is unlikely to provide an explanation for the strong reduction in sodium channel activity because a 2–4 mV depolarization cannot account for the 40–50% reduction of Vmax recorded.17, 21 Our demonstration using patch clamp techniques of a direct effect of NS8593 or UCL1684 to significantly produce an atrial-selective inhibition of INa (Fig. 6) can account for the atrial-selective induction of PRR and reduction of Vmax. It stands to reason that concomitant depolarization of RMP by whatever means is likely to accentuate the effect of the SK channel blockers to inhibit INa. Several studies have reported that inhibition of the SK channels alone does not alter RMP.3, 5, 8, 26 Also relevant to this issue is the observation that relatively high concentrations of INa blockers are associated with depolarization of RMP, suggesting that depolarization of RMP observed in some settings may be secondary to INa inhibition rather than SK channel inhibition.16, 27, 28
Moreover, ICA at10 μM has been shown to be effective against AF but to only depolarize atrial RMP by 1–2 mV (non-significantly) in rat cardiac preparations.21, 29 Further studies are needed to assess the contribution of SK channel inhibitors to indirect inhibition of INa via depolarization of RMP as well as the contribution of this mechanism to their anti-arrhythmic efficacy.
Atrial selective peak INa block is a typical feature of many INa blockers displaying relatively rapid unbinding kinetics, including ranolazine, amiodarone, vernakalant, dronedarone and AZD1305.13, 22–24, 30–32 Previous work from our group has shown that atrial selectivity of INa blockers is due to several factors including: 1) the presence of a more depolarized resting membrane potential in atria vs. ventricles; 2) a more negative half-inactivation voltage for the sodium channel in atrial vs. ventricular cells33–35 (both of these factors reduce the availability of sodium channels in atria more than ventricles) and 3) a more gradual phase 3 repolarization in atrial cells; this characteristic of the atrial action potential leads to progressive diminution or disappearance of the diastolic interval at rapid rates of activation, which reduces the ability of sodium channel blockers to dissociate from the sodium channel, thus leading to accumulation of sodium channel block. These distinctions between atrial and ventricular cell electrophysiology led us to suggest a novel strategy for AF suppression, namely atrial-selective sodium channel block.13, 33, 34 The ability of NS8593 to preferentially shift atrial V0.5 to more negative potentials contributes to further accentuation of the atrial-selective inhibition of INa (Fig 6).
Both NS8593 and UCL1684 stunted the effect of ACh to abbreviate atrial APD. This would appear to be consistent with SK channel inhibition, but because ACh dramatically reduces intracellular calcium activity,36 it is expected to reduce SK channel activity (assuming that calcium activity is reduced in the vicinity of the SK channels). Accordingly, inhibition of SK channel is unlikely to prolong APD owing to the dramatically reduced levels of intracellular calcium. The effect NS8593 and UCL1684 to blunt the ability of ACh to abbreviate APD is similar to that reported with other sodium channel blockers. Previous studies have shown that all sodium channel blockers tested at therapeutically-relevant concentrations in the coronary perfused canine right atrial preparations, including ranolazine, lidocaine, propafenone, amiodarone, AZD1305, Wenxin Keli, blunt the effect ACh to abbreviate atrial APD.13, 23, 30, 37, 38 Similar data have been reported with regard to the effect of flecainide and ICA to blunt the ability of carbachol to abbreviate repolarization in rat atrial tissues.29 These effects may be due to the ability of INa blockers to directly inhibit IK-ACh as previously reported for flecainide, propafenone, disopyramide, and pilsicainide.39, 40 Because the extent of PRR prolongation greatly exceeded that of APD prolongation in the presence of ACh (Table 1), it can be argued that sodium channel block by NS8593 and UCL1684 is the most important driver underlying the observed anti-arrhythmic effect of these agents.
Study limitations:
The electrophysiological actions of NS8593 and UCL1684 were studied acutely in isolated normal non-remodeled Tyrode’s-perfused atrial preparations and acutely dissociated atrial and ventricular myocytes. SK channel expression is reported to be augmented in diseased ventricles (e.g., in heart failure),41 thus reducing the predominance of SK channels in atria. This however is unlikely to alter the principal conclusion of our study, i.e., that the electrophysiological and anti-AF effects of NS8593 and UCL1684 are due to atrial selective INa inhibition. We studied the efficacy of NS8593 and UCL1684 to prevent but not to terminate AF. It is possible that SK channel inhibition by these agents may play more of a role in suppressing AF since AF is known to increase intracellular Ca2+ activity (including during ACh-mediated AF36), which may increase the influence of SK channels on atrial repolarization and thus the efficacy of SK channel blockers to prolong atrial ERP during AF. This hypothesis remains to be tested with specific SK channel blockers. The presence of autonomic influences and other factors present in vivo may modulate the effect of the drugs, resulting in outcomes different from those observed in the present study.
CONCLUSION
NS8593 and UCL1684 are very effective in preventing the development of AF in our ACh models of AF. The electrophysiological alterations induced by NS8593 and UCL1684 in canine cardiac preparations and isolated cells suggest that these compounds exert their electrophysiological and anti-AF effects predominantly or exclusively via atrial-selective inhibition of the sodium channel. These conclusions pertain to the specific drugs and conditions studied and cannot be directly applied to other conditions or other SK channel blockers. Our findings, coupled with available data, suggest that NS8593 and UCL1684 are effective for the management of AF and lack any pro-arrhythmic liability in the ventricle because of their atrial-selectivity.
Acknowledgements
We are grateful to Robert Goodrow for expert technical support.
Funding sources:
Grants We acknowledge grant support from NHLBI (HL47678, HL138103 and HL152201, CA and AB), W.W. Smith Charitable Trust (CA and AB) and Wistar and Martha Morris Fund (CA)
Footnotes
Open access was paid by the Acesion Pharma
Disclosures:
Morten Grunnet is an employee of Acesion Pharma. Charles Antzelevitch is a consultant to Novartis. The other authors have no conflicts of interest.
REFERENCES
- 1.Ehrlich JR and Nattel S. Novel approaches for pharmacological management of atrial fibrillation. Drugs. 2009;69:757–774. [DOI] [PubMed] [Google Scholar]
- 2.Burashnikov A and Antzelevitch C. Novel pharmacological targets for the rhythm control management of atrial fibrillation. PharmacolTher. 2011;132:300–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li N, Timofeyev V, Tuteja D, Xu D, Lu L, Zhang Q, Zhang Z, Singapuri A, Albert TR, Rajagopal AV, Bond CT, Periasamy M, Adelman J and Chiamvimonvat N. Ablation of a Ca2+-activated K+ channel (SK2 channel) results in action potential prolongation in atrial myocytes and atrial fibrillation. J Physiol. 2009;587:1087–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grunnet M, Bentzen BH, Sorensen US and Diness JG. Cardiac ion channels and mechanisms for protection against atrial fibrillation. RevPhysiol Biochem Pharmacol. 2012;162:1–58. [DOI] [PubMed] [Google Scholar]
- 5.Nagy N, Marton Z, Kiss L, Varro A, Nanasi PP and Toth A. Role of Ca(2)+-sensitive K+ currents in controlling ventricular repolarization: possible implications for future antiarrhytmic drug therapy. Curr Med Chem. 2011;18:3622–3639. [DOI] [PubMed] [Google Scholar]
- 6.Diness JG, Sorensen US, Nissen JD, Al-Shahib B, Jespersen T, Grunnet M and Hansen RS. Inhibition of small-conductance Ca2+-activated K+ channels terminates and protects against atrial fibrillation. Circ Arrhythm Electrophysiol. 2010;3:380–390. [DOI] [PubMed] [Google Scholar]
- 7.Skibsbye L, Diness JG, Sorensen US, Hansen RS and Grunnet M. The duration of pacing-induced atrial fibrillation is reduced in vivo by inhibition of small conductance Ca(2+)-activated K(+) channels. J Cardiovasc Pharmacol. 2011;57:672–681. [DOI] [PubMed] [Google Scholar]
- 8.Qi XY, Diness JG, Brundel BJ, Zhou XB, Naud P, Wu CT, Huang H, Harada M, Aflaki M, Dobrev D, Grunnet M and Nattel S. Role of small-conductance calcium-activated potassium channels in atrial electrophysiology and fibrillation in the dog. Circulation. 2014;129:430–40. [DOI] [PubMed] [Google Scholar]
- 9.Diness JG, Skibsbye L, Simo-Vicens R, Santos JL, Lundegaard P, Citerni C, Sauter DRP, Bomholtz SH, Svendsen JH, Olesen SP, Sorensen US, Jespersen T, Grunnet M and Bentzen BH. Termination of Vernakalant-Resistant Atrial Fibrillation by Inhibition of Small-Conductance Ca(2+)-Activated K(+) Channels in Pigs. Circ Arrhythm Electrophysiol. 2017;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Skibsbye L, Poulet C, Diness JG, Bentzen BH, Yuan L, Kappert U, Matschke K, Wettwer E, Ravens U, Grunnet M, Christ T and Jespersen T. Small-conductance calcium-activated potassium (SK) channels contribute to action potential repolarization in human atria. Cardiovasc Res. 2014;103:156–67. [DOI] [PubMed] [Google Scholar]
- 11.Qi XY, Diness JG, Brundel B, Zhou XB, Naud P, Wu CT, Huang H, Harada M, Aflaki M, Dobrev D, Grunnet M and Nattel S. Role of small conductance calcium-activated potassium channels in atrial electrophysiology and fibrillation in the dog. Circulation. 2014;129:430–440. [DOI] [PubMed] [Google Scholar]
- 12.Rosa JC, Galanakis D, Ganellin CR, Dunn PM and Jenkinson DH. Bis-quinolinium cyclophanes: 6,10-diaza-3(1,3),8(1,4)-dibenzena-1,5(1,4)- diquinolinacyclodecaphane (UCL 1684), the first nanomolar, non-peptidic blocker of the apamin-sensitive Ca(2+)-activated K+ channel. J Med Chem. 1998;41:2–5. [DOI] [PubMed] [Google Scholar]
- 13.Burashnikov A, Di Diego JM, Zygmunt AC, Belardinelli L and Antzelevitch C. Atrium-selective sodium channel block as a strategy for suppression of atrial fibrillation: differences in sodium channel inactivation between atria and ventricles and the role of ranolazine. Circulation. 2007;116:1449–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bode F, Kilborn M, Karasik P and Franz MR. The repolarization-excitability relationship in the human right atrium is unaffected by cycle length, recording site and prior arrhythmias. J Am Coll Cardiol. 2001;37:920–925. [DOI] [PubMed] [Google Scholar]
- 15.Burashnikov A, Barajas-Martinez H, Hu D, Nof E, Blazek J and Antzelevitch C. Atrial-selective prolongation of refractory period with AVE0118 is due principally to inhibition of sodium channel activity. J Cardiovasc Pharmacol. 2012;59:539–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bigger JT, Jr. and Mandel WJ. Effect of lidocaine on the electrophysiological properties of ventricular muscle and purkinje fibers. J Clin Invest. 1970;49:63–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miyata A, Dowell JD, Zipes DP and Rubart M. Rate-dependent [K+](o) accumulation in canine right atria in vivo: electrophysiological consequences. Am J Physiol Heart Circ Physiol. 2002;283:H506–H517. [DOI] [PubMed] [Google Scholar]
- 18.Tuteja D, Xu D, Timofeyev V, Lu L, Sharma D, Zhang Z, Xu Y, Nie L, Vazquez AE, Young JN, Glatter KA and Chiamvimonvat N. Differential expression of small-conductance Ca2+-activated K+ channels SK1, SK2, and SK3 in mouse atrial and ventricular myocytes. Am J Physiol Heart Circ Physiol. 2005;289:H2714–H2723. [DOI] [PubMed] [Google Scholar]
- 19.Diness JG, Kirchhoff JE, Speerschneider T, Abildgaard L, Edvardsson N, Sorensen US, Grunnet M and Bentzen BH. The KCa2 Channel Inhibitor AP30663 Selectively Increases Atrial Refractoriness, Converts Vernakalant-Resistant Atrial Fibrillation and Prevents Its Reinduction in Conscious Pigs. Front Pharmacol. 2020;11:159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hsueh CH, Chang PC, Hsieh YC, Reher T, Chen PS and Lin SF. Pro-arrhythmic Effect of Blocking the Small Conductance Calcium Activated Potassium Channel in Isolated Canine Left Atrium. Heart Rhythm. 2013;10:891–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Skibsbye L, Wang X, Axelsen LN, Bomholtz SH, Nielsen MS, Grunnet M, Bentzen BH and Jespersen T. Antiarrhythmic Mechanisms of SK Channel Inhibition in the Rat Atrium. J Cardiovasc Pharmacol. 2015;66:165–76. [DOI] [PubMed] [Google Scholar]
- 22.Burashnikov A, Pourrier M, Gibson JK, Lynch JJ and Antzelevitch C. Rate-dependent effects of vernakalant in the isolated non-remodeled canine left atria are primarily due to block of the sodium channel. Comparison with ranolazine and dl-sotaol. Circ Arrhythm Electrophysiol. 2012;5:400–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burashnikov A, Zygmunt AC, Di Diego JM, Linhardt G, Carlsson L and Antzelevitch C. AZD1305 exerts atrial-predominant electrophysiological actions and is effective in suppressing atrial fibrillation and preventing its re-induction in the dog. J Cardiovasc Pharmacol. 2010;56:80–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wettwer E, Christ T, Endig S, Rozmaritsa N, Matschke K, Lynch JJ, Pourrier M, Gibson JK, Fedida D, Knaut M and Ravens U. The new antiarrhythmic drug Vernakalant: ex-vivo study of human atrial tissue from sinus rhythm and chronic atrial fibrillation. Cardiovasc Res. 2013;98:145–154. [DOI] [PubMed] [Google Scholar]
- 25.Burashnikov A, Di Diego JM, Goodrow RJ, Jr., Belardinelli L and Antzelevitch C. Atria are More Sensitive Than Ventricles to GS-458967-Induced Inhibition of Late Sodium Current. J Cardiovasc Pharmacol Ther. 2015;20:501–8. [DOI] [PubMed] [Google Scholar]
- 26.Ko JS, Guo S, Hassel J, Celestino-Soper P, Lynnes TC, Tisdale JE, Zheng JJ, Taylor SE, Foroud T, Murray MD, Kovacs RJ, Li X, Lin SF, Chen Z, Vatta M, Chen PS and Rubart M. Ondansetron blocks wild-type and p.F503L variant small-conductance Ca(2+)-activated K(+) channels. Am J Physiol Heart Circ Physiol. 2018;315:H375–h388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Satoh H, Ishii M and Hashimoto K. An electrophysiological comparison of a novel class Ic antiarrhythmic agent, NIK-244 (ethacizin) and flecainide in canine ventricular muscle. Br J Pharmacol. 1989;98:827–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schmidt C, Wiedmann F, Schweizer PA, Becker R, Katus HA and Thomas D. Class I antiarrhythmic drugs inhibit human cardiac two-pore-domain K(+) (K2 (2)p) channels. European journal of pharmacology. 2013;721:237–48. [DOI] [PubMed] [Google Scholar]
- 29.Skibsbye L, Bengaard AK, Uldum-Nielsen AM, Boddum K, Christ T and Jespersen T. Inhibition of Small Conductance Calcium-Activated Potassium (SK) Channels Prevents Arrhythmias in Rat Atria During beta-Adrenergic and Muscarinic Receptor Activation. Front Physiol. 2018;9:510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burashnikov A, Di Diego JM, Sicouri S, Ferreiro M, Carlsson L and Antzelevitch C. Atrial-selective effects of chronic amiodarone in the management of atrial fibrillation. Heart Rhythm. 2008;5:1735–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Suzuki T, Morishima M, Kato S, Ueda N, Honjo H and Kamiya K. Atrial selectivity in Na+ channel blockade by acute amiodarone. Cardiovasc Res. 2013;98:136–144. [DOI] [PubMed] [Google Scholar]
- 32.Bogdan R, Goegelein H and Ruetten H. Effect of dronedarone on Na(+), Ca (2+) and HCN channels. Naunyn Schmiedebergs ArchPharmacol. 2011;383:347–356. [DOI] [PubMed] [Google Scholar]
- 33.Burashnikov A and Antzelevitch C. Atrial-selective sodium channel block for the treatment of atrial fibrillation. ExpertOpinEmergDrugs. 2009;14:233–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Antzelevitch C and Burashnikov A. Atrial-selective sodium channel block as a novel strategy for the management of atrial fibrillation. J Electrocardiol. 2009;42:543–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Burashnikov A and Antzelevitch C. Atrial-selective sodium channel blockers: do they exist? JCardiovasc Pharmacol. 2008;52:121–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Burashnikov A and Antzelevitch C. Reinduction of atrial fibrillation immediately after termination of the arrhythmia is mediated by late phase 3 early afterdepolarization-induced triggered activity. Circulation. 2003;107:2355–2360. [DOI] [PubMed] [Google Scholar]
- 37.Burashnikov A, Petroski A, Hu D, Barajas-Martinez H and Antzelevitch C. Atrial-selective inhibition of sodium channel current by Wenxin Keli is effective in suppressing atrial fibrillation. Heart Rhythm. 2012;9:125–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Burashnikov A, Belardinelli L and Antzelevitch C. Atrial-selective sodium channel block strategy to suppress atrial fibrillation: ranolazine versus propafenone. J PharmacolExpTher. 2012;340:161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Voigt N, Rozmaritsa N, Trausch A, Zimniak T, Christ T, Wettwer E, Matschke K, Dobrev D and Ravens U. Inhibition of IK,ACh current may contribute to clinical efficacy of class I and class III antiarrhythmic drugs in patients with atrial fibrillation. Naunyn Schmiedebergs Arch Pharmacol. 2010;381:251–9. [DOI] [PubMed] [Google Scholar]
- 40.Inomata N, Ohno T, Ishihara T and Akaike N. Antiarrhythmic agents act differently on the activation phase of the ACh-response in guinea-pig atrial myocytes. Br J Pharmacol. 1993;108:111–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chua SK, Chang PC, Maruyama M, Turker I, Shinohara T, Shen MJ, Chen Z, Shen C, Rubart-von der LM, Lopshire JC, Ogawa M, Weiss JN, Lin SF, Ai T and Chen PS. Small-conductance calcium-activated potassium channel and recurrent ventricular fibrillation in failing rabbit ventricles. Circ Res. 2011;108:971–979. [DOI] [PMC free article] [PubMed] [Google Scholar]