

Abstract
Background:
Parkinson’s disease (PD) and schizophrenia (SCZ) are heritable brain disorders that both involve dysregulation of the dopaminergic system. Epidemiological studies have reported potential comorbidity between the disorders, and movement disturbances are common in SCZ patients before treatment with antipsychotic drugs. Despite this, little is known about shared genetic etiology between the disorders.
Methods:
We analyzed recent large genome-wide associations studies (GWAS) on SCZ (n=77,096) and PD (n=417,508) using a conditional/conjunctional false discovery rate (FDR) approach to evaluate overlap in common genetic variants and improve statistical power for genetic discovery. Using a variety of biological resources, we functionally characterized the identified genomic loci.
Results:
We observed genetic enrichment in PD conditional on associations with SCZ, and vice versa, indicating polygenic overlap. We then leveraged this cross-trait enrichment using conditional FDR analysis and identified nine novel PD risk loci and one novel SCZ locus at conditional FDR<0.01. Further, we identified nine genomic loci jointly associated with PD and SCZ at conjunctional FDR<0.05. There was an even distribution of antagonistic and agonistic effect directions among the shared loci, in line with the insignificant genetic correlation between the disorders. 65 out of 67 genes mapped to the shared loci are expressed in the human brain and show cell-type specific expression profiles.
Conclusions:
Altogether, the study increases the understanding of the genetic architectures underlying SCZ and PD, indicating that common molecular genetic mechanisms may contribute to overlapping pathophysiological and clinical features between the disorders.
Keywords: Parkinson’s disease, schizophrenia, GWAS, genetic overlap, RERE, ZDHHC2
Introduction
Parkinson’s disease (PD) and schizophrenia (SCZ) are severe brain disorders with complex etiologies that involve multiple genetic and environmental factors. While SCZ is considered a neurodevelopmental disorder with typical onset in early adulthood, PD is a neurodegenerative disorder with most cases occurring after the age of 60. PD is characterized by degeneration of dopaminergic neurons in the substantia nigra, manifesting clinically with motor symptoms such as bradykinesia, tremor and rigidity (1). Current treatment strategies in PD largely aim to increase dopaminergic activity (1). SCZ, on the other hand, is characterized by psychotic symptoms such as hallucinations, delusions and thought distortions, in addition to mood symptoms, loss of motivation and cognitive dysfunction (2). Despite their distinguishable clinical manifestations and different ages at onset, SCZ and PD share some disease characteristics, most notably abnormalities of the dopaminergic system (1, 2). Whereas PD is associated with dopaminergic deficiency (1), the psychotic symptoms of SCZ are associated with increased dopaminergic signaling in the striatum (2, 3). In fact, almost all effective antipsychotic drugs are antagonists of dopamine D2 receptors, with parkinsonian symptoms representing one of the most common adverse effects (4). Moreover, the non-psychotic symptoms of SCZ have been linked to lower dopaminergic transmission in the mesocortical pathway (3). Further, symptoms associated with SCZ, like hallucinations, depression and cognitive dysfunction, are common in patients with PD (5, 6), and may even occur in drug-naïve PD patients (5, 7). Intriguingly, meta-analyses have reported an increased prevalence of movement disturbances such as dyskinesia and parkinsonism among antipsychotic-naïve patients with SCZ and their healthy first-degree relatives (8, 9), suggesting that genetic risk may play a role. In line with this, a recent large population-based family cohort study found that first-degree relatives of patients with PD were more likely to develop SCZ (10). Additionally, two epidemiological studies reported higher rates of PD among patients with SCZ (11, 12), although the findings from these two studies may have been biased from drug-induced parkinsonism. However, despite the clinical and epidemiological associations between SCZ and PD, little is known about shared genetic etiology between the disorders.
The genetic architectures of both PD (13–15) and SCZ (16) are highly polygenic, with many genetic variants with small effects contributing to disease risk. Recent large GWAS on PD (13–15, 17) have identified 92 genome-wide significant loci, and more than hundred genome-wide significant loci are linked to SCZ (16, 18). Intriguingly, five genome-wide significant loci have been jointly associated with SCZ (16) and PD in recent GWAS (13, 15–17). Moreover, two systematic investigations of GWAS summary data implicated loci shared between SCZ and PD within NT5C2 (19) and SLC39A8 (20). Notably, both PD (21) and SCZ (22) are also associated with the 22q11.2 deletion syndrome. Despite the overlapping genetic associations between SCZ and PD, genetic studies have reported no genome-wide correlation between the disorders (23). A significant genome-wide correlation, however, requires consistent effect directions among the overlapping variants between two phenotypes (24), which is not necessarily the case despite polygenic overlap. Indeed, increasing evidence indicates that there is a mixed pattern of allelic effect directions among variants shared between a number of complex human traits and disorders (25–33), warranting more detailed analyses to disentangle the genetic relationship between complex phenotypes. In the present study we aimed to further investigate the shared genetic basis between SCZ and PD, and applied the conditional false discovery rate (condFDR) approach (34) to analyze recent large non-overlapping GWAS on PD (n=417,508) (13, 15) and SCZ (n=77,096) (16). The condFDR approach builds on an empirical Bayesian statistical framework, and increases power to detect shared loci by leveraging the combined power of two GWAS (34). We have in recent years used this approach to identify shared genetic influences between a number of complex human traits and disorders (25, 27, 30, 35–42).
Methods and Materials
GWAS data
We obtained GWAS results in the form of summary statistics (p-values and effect sizes) (13, 15, 16). All participants were of European ancestry. First, we meta-analyzed two independent GWAS on PD (13, 15): a 23andMe cohort on 6,476 cases and 302,042 controls described in Chang et al (2017) (13) and a PDGene cohort on 13,708 cases and 95,282 controls described in Nalls et al (2014) (15). A similar meta-analysis was conducted in the Chang et al (2017) study (13), but was restricted to the 10,000 most significant single-nucleotide polymorphisms (SNPs) from the PDGene study (15). For our meta-analysis, however, we utilized the complete summary statistics data from both the PDGene study (7,799,579 variants) (15) and the 23andMe cohort (12,896,220 variants) (13). We combined the association statistics for the meta-analysis of the 23andMe (13) and PDGene (15) cohorts using an inverse-variance weighted method as implemented in METAL (43). In the 23andMe cohort, cases and controls were designated on the basis of web-based self-report surveys on 23andMe research participants (13). The PDGene GWAS meta-analyzed 15 studies using different criteria for case/control ascertainment (15). Data on SCZ were acquired from the Psychiatric Genomics Consortium (PGC) (16), consisting of 46 case control samples (33,640 cases with SCZ or schizoaffective disorder and 43,456 controls) and 3 family-based association studies (1,235 parent affected-offspring trios) (16) (15,358,497 variants). We also included GWAS summary statistics on four disorders as negative controls, namely attention-deficit hyperactivity disorder (ADHD; 19,099 cases and 34,194 controls) (44), autism spectrum disorder (ASD; 8,381 cases and 27,969 controls) (45), haemorrhoids (12,102 cases, 349,092 controls) and prostate cancer (25,074 cases and 24,272 controls) (46). All GWAS investigated in the present study were approved by the relevant ethics committees, and informed consent was obtained from all participants. The Norwegian Institutional Review Board for the South-East Norway Region has evaluated the current protocol and found that no additional institutional review board approval was needed because no individual data were used. For details, see Supplemental Methods.
condFDR analysis
The condFDR method builds on an empirical Bayesian statistical framework (47) and capitalizes on the inherent power in combining two GWAS to improve discovery of genetic variants (34). First, we constructed conditional Q-Q plots to provide a visual pattern of overlap in SNP associations, i.e. “cross-trait enrichment”. Note that cross-trait enrichment indicates shared genomic loci between the phenotypes, which could reflect both shared or separate causal variants (48). The conditional Q-Q plots compare the association with a primary trait across all SNPs and within SNPs strata determined by their association with the secondary trait (34, 49). To improve SNP discovery in SCZ and PD, the condFDR statistical framework (34, 49) re-ranks the test-statistics of a primary phenotype based on the strength of the association with a secondary phenotype (34, 49). Inverting the roles of primary and secondary phenotypes gives the inverse condFDR value. The conjunctional FDR (conjFDR), defined in turn as the maximum of the two condFDR values, provides a conservative estimate of the FDR for association with both phenotypes. In line with the prior literature (25, 30, 35, 37–42), the significance threshold for shared loci was conjFDR<0.05 and for conditional loci condFDR<0.01. We excluded SNPs around the extended MHC region, chromosome 8p23.1 and the gene MAPT (genome build 19 locations chr6:25119106–33854733, chr8:7200000–12500000 and chr17:40000000–47000000, respectively) before fitting the FDR model, given that their intricate regional LD may bias FDR estimation (50). All p-values were corrected for inflation using a genomic inflation control procedure (40). For methodological details of the condFDR approach, see Supplemental Methods or the review by Smeland and colleagues (2019) (34).
Genomic loci definition
We defined independent genomic loci according to the FUMA (51) protocol. FUMA is an online platform for functional mapping of genetic variants (http://fuma.ctglab.nl/) (51). First, we identified independent significant SNPs as SNPs that were independent from each other at r2<0.6 and that reached a condFDR<0.01 or conjFDR<0.05, in the respective condFDR and conjFDR analyses. A subset of these in approximate linkage equilibrium with each other at r2<0.1 were then selected as lead SNPs. To define distinct genomic loci, we merged all loci which were less than 250kb apart selecting a SNP with the most significant p-value as a lead SNP of the merged locus. The borders of the genomic loci were defined by identifying all SNPs (candidate SNPs) in LD (r2≥0.6) with one of the independent significant SNPs in the locus. Overlapping signals within complex LD regions were represented by one independent lead SNP only. All LD information was calculated from the 1000 Genomes Project European-ancestry haplotype reference panel (52). We evaluated the directional effects of the loci shared between SCZ and PD by comparing their z-scores and odds ratios.
Functional annotation
Using FUMA (51), we functionally annotated all candidate SNPs in the genomic loci having an LD r2≧0.6 with one of the independent significant SNPs and having a condFDR or conjFDR value <0.10. SNPs were annotated with Combined Annotation Dependent Depletion (CADD) (53) scores, which predict how deleterious the SNP effect is on protein structure/function, RegulomeDB (54) scores, which predict likelihood of regulatory functionality, and chromatin states, which predict transcription/regulatory effects from chromatin states at the SNP locus (55, 56). Using FUMA (51), we linked candidate SNPs to genes using either of three gene-mapping strategies: 1) positional mapping to align SNPs to genes based on their physical proximity, 2) expression quantitative trait locus (eQTL) mapping to match cis-eQTL SNPs to genes whose expression is associated with allelic variation at the SNP level, and 3) chromatin interaction mapping to link SNPs to genes based on three-dimensional DNA–DNA interactions between each SNP’s genomic region and nearby or distant genes. Next, we determined the expression in the adult human brain of the identified genes using Genotype-Tissue Expression (GTEx) (57) data and we performed a gene-set analysis using FUMA. Finally, we used a publicly available RNA-sequencing transcriptome and splicing database (58) to determine the cell-specific expression within the human cerebral cortex of the genes implicated by the shared loci. The six cell types surveyed were neurons, fetal and mature astrocytes, oligodendrocytes, microglia/macrophages, and endothelial cells (58). For details, see Supplemental Methods.
Results
We observed enrichment in SCZ SNPs as a function of the significance of associations with PD (Figure 1A). This indicates polygenic overlap between the disorders. The reverse conditional Q-Q plots demonstrate consistent enrichment in PD given associations with SCZ (Figure 1B). We also observed cross-trait enrichment between SCZ and the psychiatric disorders ADHD and ASD, but not with haemorrhoids or prostate cancer (Supplemental Figures 1–4). In contrast, we did not observe cross-trait enrichment between PD and any of the four control disorders, despite polygenic signal. The results indicate specificity for the cross-trait enrichment between PD and SCZ. However, we cannot exclude the possibility that the negative findings are due to inadequate statistical power, and that future larger GWAS may show some overlap.
Figure 1.
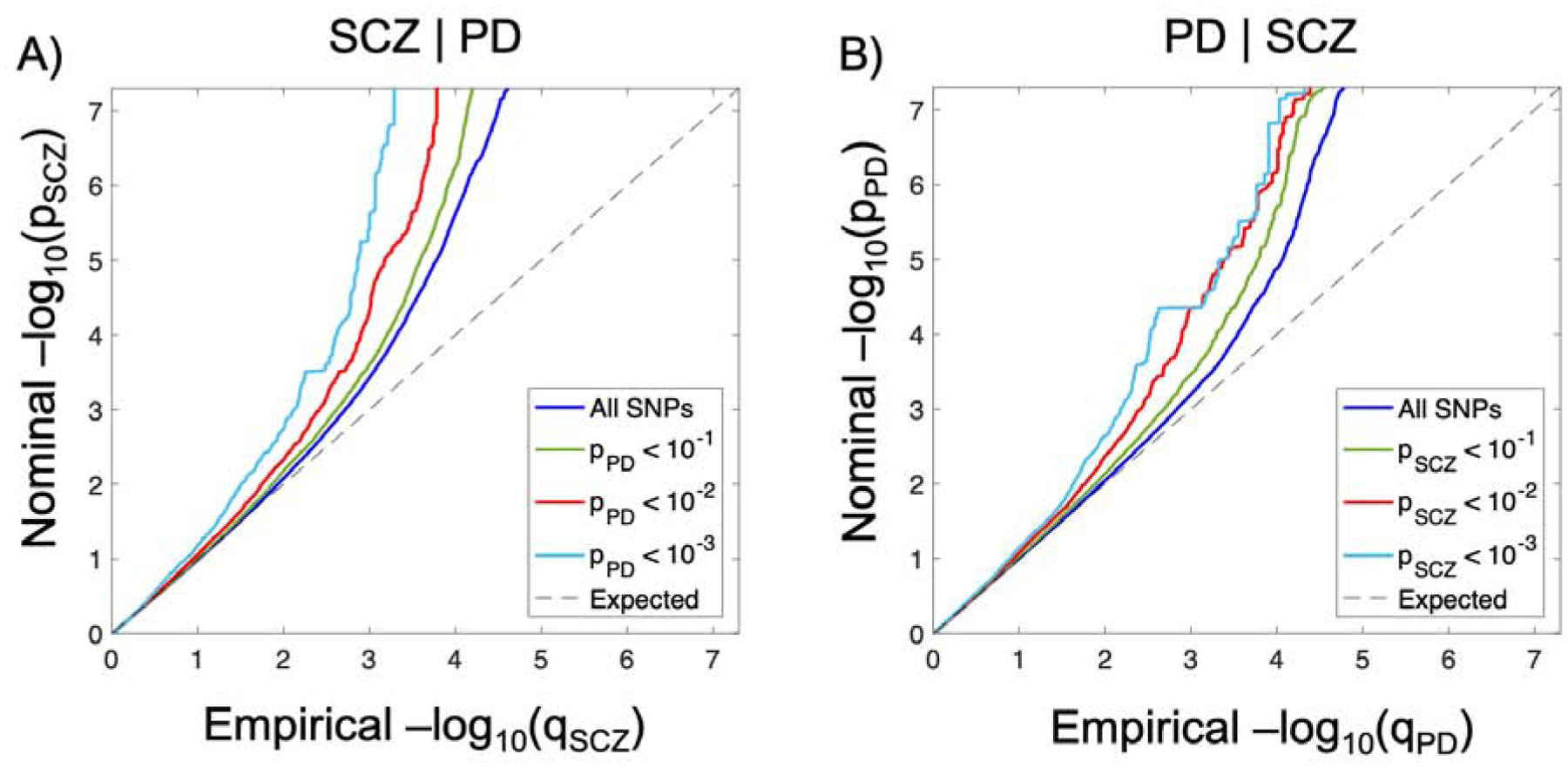
Polygenic overlap between schizophrenia (SCZ) and Parkinson’s disease (PD). Conditional Q-Q plots of nominal versus empirical −log10 p-values (corrected for inflation) in A) SCZ below the standard GWAS threshold of p<5×10−8 as a function of significance of association with PD, and B) vice versa, at the level of p ⩽ 0.1, p ⩽ 0.01, p ⩽ 0.001, respectively. The blue lines indicate all SNPs. The dashed lines indicate the null hypothesis.
To increase statistical power for genetic discovery, we leveraged the cross-trait enrichment between SCZ and PD using condFDR analysis (34) and re-ranked PD SNPs conditionally on their association with SCZ, and vice versa. At condFDR<0.01, we identified 53 loci associated with PD conditional on SCZ (Supplemental Table 1). Of these, 33 loci had been identified in the original PD GWAS (13, 15), two were identified in subsequent GWAS analyses (the NUCKS1 (41) and SLC39A8 (20) loci), while nine reached genome-wide significance in a recent larger PD GWAS (17). In total, we identified nine novel PD risk loci (Table 1), implicating 54 genes (Supplemental Table 3). Two of the lead SNPs in the novel PD risk loci, rs56379273 and rs181870458, were only present in the 23andMe dataset (13), but not the PDGene dataset (15), indicating that these associations may have lower confidence. Furthermore, we identified 119 loci associated with SCZ at condFDR<0.01 (Supplemental Table 4). Twenty-three of these loci were not identified in the original SCZ GWAS (16). However, all but one have been associated with SCZ in subsequent GWAS analyses (18, 25, 27, 30, 59, 60). The sole novel SCZ locus was detected near pseudogene RNU6–183P with rs9293740 as lead SNP (chromosome 5:77612246; p-value SCZ = 5.81 × 10−6, p-value PD = 9.27 × 10−4, condFDR = 8.16 × 10−3; Table 2). Five protein-coding genes were also mapped to this locus, with AP3B1 as nearest gene (Supplemental Table 6). Supplemental Figures 5–6 present the condFDR Manhattan plots for PD and SCZ, showing all SNPs with a condFDR<0.01 within an LD block in relation to their chromosomal location.
Table 1.
Novel genomic loci associated with Parkinson’s disease (PD) at conditional FDR (condFDR) < 0.01 given association with schizophrenia (SCZ)
| Locus | Chr | Lead SNP | A1/A2 | Nearest Gene | Functional category | P-value PD | Z-score PD | P-value SCZ | Odds ratio SCZ | condFDR |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1 | rs302714 | A/C | RERE | ncRNA_intronic | 4.23E-06 | 4.60 | 2.37E-08 | 0.94 | 3.16E-03 |
| 10 | 2 | rs113732002 | A/C | NGEF | intronic | 3.02E-07 | −5.12 | 1.29E-02 | 1.05 | 1.41E-03 |
| 13 | 3 | rs181870458 | A/G | GM2AP1 | intergenic | 1.55E-07 | 5.25 | 3.27E-01 | 0.96 | 2.52E-03 |
| 30 | 8 | rs4921739 | T/C | ZDHHC2 | intronic | 6.20E-06 | −4.52 | 1.24E-06 | 0.93 | 4.27E-03 |
| 32 | 9 | rs2278973 | T/G | CACNA1B | exonic | 2.70E-07 | 5.14 | 3.16E-02 | 0.96 | 1.82E-03 |
| 37 | 12 | rs77669894 | A/G | BICD1 | intergenic | 3.67E-08 | 5.51 | 9.64E-02 | 1.07 | 4.94E-04 |
| 45 | 15 | rs1027647 | A/C | TPM1 | intergenic | 2.14E-06 | 4.74 | 1.60E-02 | 0.97 | 8.12E-03 |
| 51 | 20 | rs6060983 | T/C | MYLK2 | intronic | 6.21E-07 | 4.98 | 5.01E-01 | 1.01 | 9.12E-03 |
| 53 | 21 | rs56379273 | A/G | TRPM2 | exonic | 8.59E-08 | 5.35 | 2.26E-02 | 1.11 | 5.86E-04 |
The most strongly associated SNPs in novel genomic loci associated with PD at condFDR<0.01 given association with SCZ after merging regions < 250 kb apart into a single locus. The table presents chromosomal position (Chr), nearest gene and functional category, as well as p-values and effect sizes (odds ratios or z-scores) from the original summary statistics on SCZ and PD. The effect sizes are given with reference to allele 1 (A1). For more details and the full list of all loci associated with PD at condFDR<0.01, see Supplemental Table 1. See Supplemental Table 3 for all genes mapped to these loci.
Table 2.
The novel genomic locus associated with schizophrenia (SCZ) at conditional FDR (condFDR) < 0.01 given association with Parkinson’s disease (PD)
| Locus | Chr | Lead SNP | A1/A2 | Nearest Gene | Functional category | P-value SCZ | Odds ratio SCZ | P-value PD | Z-score PD | condFDR |
|---|---|---|---|---|---|---|---|---|---|---|
| 42 | 5 | rs9293740 | A/G | RNU6–183P | upstream | 5.81E-06 | 0.95 | 9.27E-04 | −3.31 | 8.16E-03 |
The most strongly associated SNP in the novel genomic locus associated with SCZ at condFDR<0.01 given association with PD after merging regions < 250 kb apart into a single locus. The table presents chromosomal position (Chr), nearest gene and functional category, as well as p-values and effect sizes (odds ratios or z-scores) from the original summary statistics on SCZ and PD. The effect sizes are given with reference to allele 1 (A1). For more details and the full list of all loci associated with SCZ at condFDR<0.01, see Supplemental Table 4. See Supplemental Table 6 for all genes mapped to this locus.
Nine distinct genomic loci were jointly associated with SCZ and PD at conjFDR<0.05 (Table 3). All of these loci have previously been associated with SCZ (16, 18). Five of the shared loci already reached genome-wide significance in the PD GWAS presently analyzed (13, 15), while the CLCN3 locus was identified in the recent larger PD GWAS (17), and the NT5C2 (19) and SLC39A8 (20) loci have been associated with PD in other GWAS investigations. Altogether, we identified two novel PD risk loci among the shared loci, at RERE (lead SNP rs302714, chromosome 1:8486131, p-value SCZ = 2.37 × 10−8, p-value PD = 4.23 × 10−6, conjFDR = 3.16 × 10−3), and at ZDHHC2 (lead SNP rs4921739, chromosome 8:17029657, p-value SCZ = 1.24 × 10−6, p-value PD = 6.20 × 10−6, conjFDR=4.27 × 10−3). As denoted by the sign of the effect sizes, five of the shared loci have consistent effect directions in SCZ and PD, indicating that the respective risk alleles are linked to higher susceptibility to both SCZ and PD (Table 3). Two of the nine shared loci were located in regions with complex LD, specifically the extended MHC region (locus 4, lead SNP rs9468195) and chromosomal region 8p23.1 (locus 5, lead SNP rs2979160). Both of these loci have previously been associated with SCZ and PD at the genome-wide significance level (13, 14, 16). Due to their extensive LD, these regions span large numbers of genes (61, 62), and we consider these conjunctional hits as to reflect the involvement of the extended MHC and 8p23.1 regions in both disorders rather than any specific gene. Note that we excluded SNPs in these LD regions before fitting the condFDR model to avoid inflation of the FDR estimation.
Table 3.
Genomic loci jointly associated with Parkinson’s disease (PD) and schizophrenia (SCZ) at conjunctional FDR<0.05
| Locus | Chr | Lead SNP | A1/A2 | Nearest Gene | Functional category | P-value PD | Z-score PD | P-value SCZ | Odds ratio SCZ | conjFDR |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1 | rs302714# | A/C | RERE | ncRNA_intronic | 4.23E-06 | 4.60 | 2.37E-08 | 0.94 | 3.16E-03 |
| 2 | 4 | rs13107325 | T/C | SLC39A8 | exonic | 6.96E-08 | −5.39 | 1.54E-12 | 1.16 | 7.43E-05 |
| 3 | 4 | rs62333164 | A/G | CLCN3 | intronic | 1.09E-04 | −3.87 | 2.85E-05 | 1.05 | 3.20E-02 |
| 4* | 6 | rs9468195 | A/G | RSL24D1P1 | intergenic | 1.51E-09 | −6.04 | 5.88E-10 | 1.08 | 4.49E-06 |
| 5* | 8 | rs2979160 | A/G | SGK223 | intergenic | 2.96E-06 | −4.67 | 2.39E-06 | 0.95 | 4.11E-03 |
| 6 | 8 | rs4921739# | T/C | ZDHHC2 | intronic | 6.20E-06 | −4.52 | 1.24E-06 | 0.93 | 4.27E-03 |
| 7 | 10 | rs79780963 | T/C | NT5C2 | intronic | 2.12E-04 | −3.70 | 2.79E-16 | 0.85 | 4.97E-02 |
| 8 | 11 | rs3802921 | T/C | IGSF9B | UTR3 | 6.88E-09 | −5.79 | 2.40E-06 | 0.94 | 4.13E-03 |
| 9 | 12 | rs73228032 | A/C | HIP1R | intronic | 4.95E-05 | −4.06 | 6.51E-05 | 0.93 | 3.90E-02 |
The most strongly associated SNPs in independent genomic loci shared between SCZ and PD at conjFDR<0.05 after merging regions < 250 kb apart into a single locus. The table presents chromosomal position (Chr), nearest gene and functional category, as well as p-values and effect sizes (odds ratios or z-scores) from the original summary statistics on SCZ and PD. The effect sizes are given with reference to allele 1 (A1). Novel PD risk loci are labeled with#.
The significant shared signals in the major histocompatibility complex region (chr6:25119106–33854733; locus 4) and chromosomal region 8p23.1 (chr8:7200000–12500000; locus 5) are represented by only one independent lead SNP each given their extended LD. For more details and a list of all candidate variants in these loci, see Supplemental Table 7. See Supplemental Table 8 for genes mapped to these loci.
Functional annotation (51) of all SNPs having a conjFDR value<0.10 in the loci shared between SCZ and PD (n=551) demonstrated that these were mostly intronic and intergenic, while 1.3% were exonic (Supplemental Table 7). To visualize the genomic distribution of the shared variants and their nearest genes, we constructed a ‘conjFDR Manhattan plot’ where all SNPs without pruning are shown, and the independent significant lead SNPs are encircled in black (Figure 2). After excluding the MHC and 8p23.1 loci, we applied FUMA (51) to link the 97 candidate SNPs in the seven shared loci to 67 protein-coding genes (Supplemental Table 8).Positional mapping aligned the SNPs to 11 genes, cis-eQTL mapping implicated 51 genes, and chromatin interaction mapping implicated 27 genes. Three genes were implicated by all three gene-mapping procedures, specifically RERE and SLC45A1 at chromosome 1p36.23 and IGSF9B at chromosome 11q25, giving a higher credibility for the link between these genes and loci. Notably, seven genes were mapped to SNPs with cis-eQTL associations in separate eQTL databases on human brain tissue (57, 63–65), specifically RERE, SLC45A1, IGSF9B, VPS37A, MTMR7, CNNM2 and NT5C2. The gene mapping strategy only implicated 29 genes when restricting the eQTL associations and chromatin interactions to those detected in human brain tissue. Using GTEx data (57), we show that 65 of the genes mapped to the shared loci, but not CA6 and SPATA19, are expressed in the human brain (Supplemental Figure 7). Moreover, we demonstrate cell-type specific expression profiles in the major cell types in the human cerebral cortex for the 65 brain-expressed genes (Supplemental Figure 8). A gene-set analysis identified no biological process significantly associated with the 67 genes.
Figure 2.
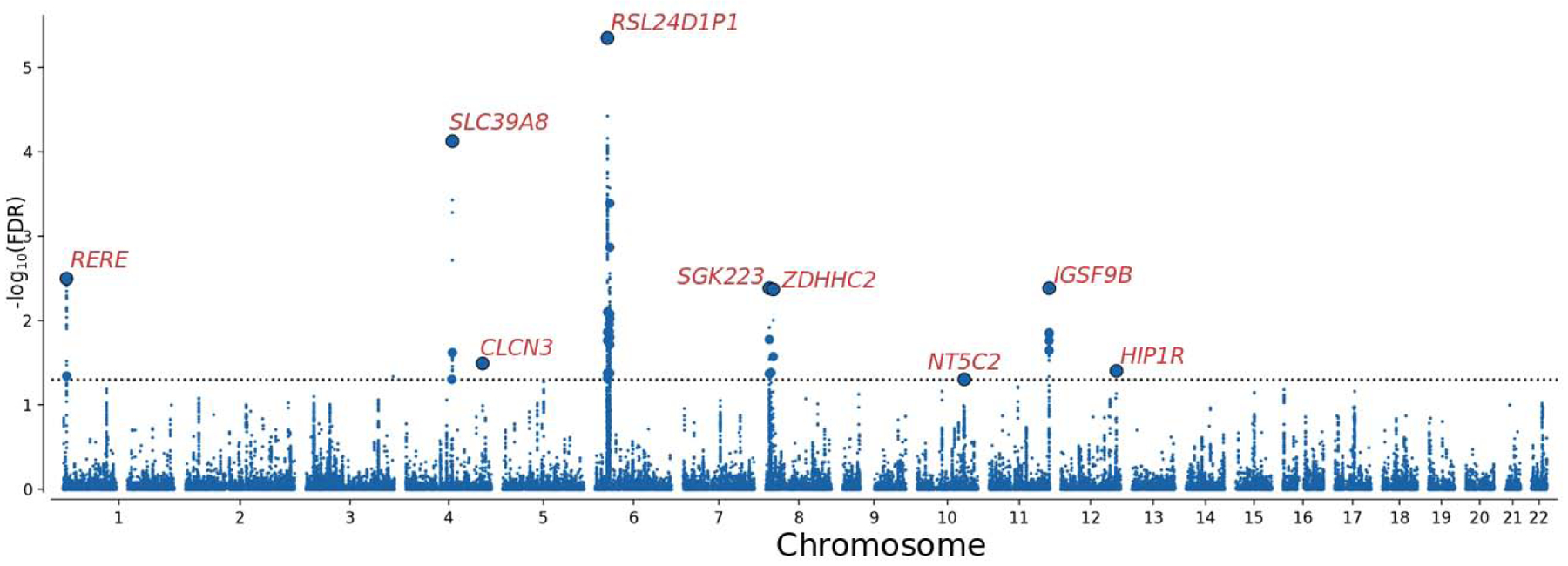
Common genetic variants jointly associated with schizophrenia (n=77,096) and Parkinson’s disease (n=417,508) at conjunctional false discovery rate (conjFDR) < 0.05. Manhattan plot showing the –log10 transformed conjFDR values for each SNP on the y-axis and chromosomal positions along the x-axis. The dotted horizontal line represents the threshold for significant shared associations (conjFDR<0.05, i.e. −log10(conjFDR)>1.3). Independent lead SNPs are encircled in black, and their nearest gene is displayed. The significant shared signals in the major histocompatibility complex region (chr6:25119106–33854733) and region 8p23.1 (chr8:8091701–11835712) are represented by one lead SNP only. Further details are provided in Table 3, and Supplemental Tables 7 and 8.
Discussion
In the present study, we investigated non-overlapping GWAS summary statistics on PD (13, 15) and SCZ (16) to increase insights into their shared genetic etiology. First, we constructed conditional Q-Q plots, which indicate polygenic overlap between SCZ and PD (Figure 1), supporting prior genetic findings (13, 15–17, 19, 20). Next, we leveraged the cross-trait enrichment using condFDR analysis and identified nine novel PD loci (Table 1), one novel SCZ locus (Table 2), and nine genomic loci jointly associated with SCZ and PD (Table 3), two of which are novel shared loci. The demonstration of polygenic overlap between SCZ and PD, including both agonistic and antagonistic SNP effects, is likely to generate novel hypotheses into their relationship and may inform clinical practices. The findings indicate that in some patients, genetic variants shared by SCZ and PD may possibly contribute to overlapping clinical and pathophysiological features between the disorders, for example dyskinesia and parkinsonism in antipsychotic-naïve SCZ patients (8, 9). However, the even distribution of directional effects among the shared loci does not explain the potential comorbidity between SCZ and PD as suggested by epidemiological studies (10–12), indicating that environmental factors or rare genetic variants may play a role. Furthermore, the results suggest that in order to develop valid genetic prediction tools identifying patients at higher risk of comorbid SCZ and PD symptomatology, these tools would need to focus on the subset of the overlapping variants with agonistic effects, in addition to the risk variants not shared by the disorders. Such tools are currently lacking in the clinic. The findings may also have implications for clinical trials in PD and SCZ, by supporting the collection and sequencing of DNA for better risk stratification.
Although much of the genetic architectures underlying SCZ and PD remain to be identified, seven genomic loci are already found to be shared between the disorders (13, 15–17, 19, 20). Here, we provide additional support for these overlapping associations and identify two novel shared loci, yielding a total number of nine loci shared between SCZ and PD (Table 3; Figure 2). Notably, a recent large genetic study (23) examined shared heritability between multiple brain disorders using LD score regression and reported no genetic correlation between SCZ and PD (23). This result mirrored the non-significant correlations between other psychiatric and neurological disorders (23), which has been interpreted to signify that psychiatric and neurological disorders have distinct non-overlapping genetic etiologies (23). However, genetic correlations can only capture consistent allelic effect directions among shared variants, and are agnostic about mixed patterns of directional effects (24). Among the nine shared loci identified here, the allelic effect directions were consistent at five loci (Table 3). This balanced mixture of directional effects complies with the non-significant genetic correlation between SCZ and PD (23), and indicates that agonistic and antagonistic overlapping molecular genetic mechanisms may be involved in SCZ and PD pathophysiology. Note that it is possible that our finding of polygenic overlap between SCZ and PD may have been biased from misdiagnosis in the separate GWAS investigated (13, 15, 16), i.e., that parkinsonian side effects in SCZ patients were misclassified as symptoms of PD, or that drug-induced psychosis in PD patients was misdiagnosed as SCZ. Although we are unable to control for this possible bias, the number of such cases is likely to have been very small, and this potential occurrence would be unable to explain the identification of shared variants with opposite effect directions in SCZ and PD.
Using the condFDR approach to improve genetic discovery, we identified 23 SCZ loci and 20 PD loci at condFDR<0.01 not identified in the original GWAS analyzed (13, 15, 16) (Supplemental Tables 1 and 4). However, among these, as many as 22 of the SCZ loci (18, 25, 27, 30, 59, 60) and eleven of the PD loci (17, 20, 41) have been associated with the disorders in subsequent GWAS analyses, supporting their role in SCZ and PD etiology. These results demonstrate the validity of the condFDR approach to increase the yield of existing GWAS. Among the candidate SNPs in the nine novel PD risk loci, two of the SNPs, rs2278973 (chromosome 9:141016262) and rs56379273 (chromosome 21:45826486), are nonsynonymous exonic variants within genes CACNA1B and TRPM2, respectively (Supplemental Table 2). These SNPs were also the most strongly associated SNPs in these loci. Nonsynonymous exonic variants are more likely to disrupt protein function by altering the amino acid sequence of the protein. CACNA1B encodes a voltage-dependent calcium channel subunit widely expressed in the nervous system, which regulates neurotransmission (66). A rare CACNA1B mutation has been implicated in SCZ (67). Another CACNA1B mutation has been associated with a myoclonus-dystonia syndrome (68), but the evidence is inconsistent (69). The TRPM2 variant had a CADD-score of 17.7, further suggesting deleteriousness (53) (Supplemental Table 2). TRPM2 encodes a member of the melastin-related transient receptor potential (TRPM) family of cation channels, which regulates ion homeostasis and membrane potential in various cell types (70). TRPM2 has been linked to the regulation of GABAergic activity in the substantia nigra (71), and apoptosis in dopaminergic neurons after exposure to neurotoxins (72). Note that the locus at TRPM2 was also mapped to two other protein-coding genes, PFKL and AP001579.1, through eQTL associations (Supplemental Table 3).
The novel PD risk loci at RERE and ZDHHC2 were also found to be shared between PD and SCZ at conjFDR<0.05 (Table 3). The RERE locus reached genome-wide significance in the original SCZ GWAS (16), while the ZDHHC2 locus reached genome-wide significance in a recent transancestry SCZ GWAS (18) that also included the original PGC SCZ GWAS dataset (16). At the RERE locus, the SCZ risk allele was associated with lower PD risk (Table 3). In addition to being the nearest gene to most of the candidate SNPs in this locus, RERE was implicated through eQTL associations in multiple tissues including the brain, and through chromatin interactions (Supplemental Table 8). Nine other genes were also mapped to this locus, but with lower confidence. RERE encodes a nuclear receptor coregulator that regulates retinoic acid signaling in several tissues during development (73). Deleterious variants within RERE have been linked to neurodevelopmental disease (74), and common RERE variants have been implicated in major depression (75). The SCZ and PD risk alleles were agonistic at the ZDHHC2 locus (Table 3). ZDHHC2 was mapped to this locus as both the nearest gene and through eQTL functionality in different tissues, including the cerebellum (Supplemental Table 8). Five other genes were also linked to this locus, but with lower confidence. ZDHHC2 encodes a palmitoyl transferase, which regulates diverse protein activities (76), including organization of the postsynaptic density (77). ZDHHC2 was identified as a key protective gene regulator in a mouse model of PD, which prevented dopaminergic cell death in the substantia nigra pars compacta (78). Additionally, transcriptional activity of ZDHHC2 was decreased in the substantia nigra of patients with incipient PD (78). Of note, as previously shown (20), there are strong antagonistic effects on SCZ and PD associated with rs13107325 (Table 3), a highly pleiotropic missense variant within SLC39A8, which encodes a metal ion transporter (79). The minor T allele of rs13107325, which is linked to higher SCZ risk and lower PD risk (20), is also associated with poorer cognitive function (30) and larger gray matter volume in the putamen (80), a brain structure part of the nigrostriatal dopaminergic pathway (81). Our functional analysis further demonstrated that 65 out of the 67 protein-coding genes mapped to the shared loci are expressed in the human brain (Supplemental Figure 7), in which they present distinct expression profiles across the major cell types (58) (Supplemental Figure 8).
In conclusion, we corroborate prior genetic findings (13, 15–17, 19, 20) by demonstrating polygenic overlap between SCZ and PD and highlighting nine shared loci with a mixed pattern of directional effects (Table 3). Further studies are needed to clarify the distinct causal variants underlying the shared genomic associations detected here, which could arise from both shared or separate causal variants (48), and to determine whether the variants influence SCZ and PD pathobiology through common intermediary phenotypes or more basic neurobiological mechanisms, such as interfering with dopaminergic function. The study demonstrates the utility of the condFDR statistical framework to increase genetic discovery and uncover polygenic overlap between complex phenotypes despite the absence of genetic correlation (34, 49), which may be useful for elucidating genetic overlap between other psychiatric and neurological disorders.
Supplementary Material
Acknowledgments
We thank the Psychiatric Genomics Consortium, UK Biobank, 23andMe and PDGene for access to data, and the many people who provided DNA samples. The work was supported by the Research Council of Norway (262656, 249711, 248980, 248778, 226971, 223273), South-East Norway Regional Health Authority (2016–064) and KG Jebsen Stiftelsen (SKGJ-MED-008). A.M.D. was supported by NIH grant U24DA041123.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
O.A.A. has received speaker’s honorarium from Lundbeck and is a consultant for Healthlytix. A.M.D. is a founder of and holds equity interest in CorTechs Labs and serves on its scientific advisory board. He is also a member of the Scientific Advisory Board of Healthlytix and receives research funding from General Electric Healthcare (GEHC). The terms of these arrangements have been reviewed and approved by the University of California, San Diego in accordance with its conflict of interest policies. Remaining authors report no biomedical financial interests or potential conflicts of interest.
References
- 1.Lees AJ, Hardy J, Revesz T (2009): Parkinson’s disease. Lancet. 373:2055–2066. [DOI] [PubMed] [Google Scholar]
- 2.Owen MJ, Sawa A, Mortensen PB (2016): Schizophrenia. Lancet. 388:86–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Howes OD, Kapur S (2009): The dopamine hypothesis of schizophrenia: version III--the final common pathway. Schizophr Bull. 35:549–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miyamoto S, Miyake N, Jarskog LF, Fleischhacker WW, Lieberman JA (2012): Pharmacological treatment of schizophrenia: a critical review of the pharmacology and clinical effects of current and future therapeutic agents. Mol Psychiatry. 17:1206–1227. [DOI] [PubMed] [Google Scholar]
- 5.Schapira AHV, Chaudhuri KR, Jenner P (2017): Non-motor features of Parkinson disease. Nat Rev Neurosci. 18:435–450. [DOI] [PubMed] [Google Scholar]
- 6.Forsaa EB, Larsen JP, Wentzel-Larsen T, Goetz CG, Stebbins GT, Aarsland D, et al. (2010): A 12-year population-based study of psychosis in Parkinson disease. Arch Neurol. 67:996–1001. [DOI] [PubMed] [Google Scholar]
- 7.Pagonabarraga J, Martinez-Horta S, Fernandez de Bobadilla R, Perez J, Ribosa-Nogue R, Marin J, et al. (2016): Minor hallucinations occur in drug-naive Parkinson’s disease patients, even from the premotor phase. Mov Disord. 31:45–52. [DOI] [PubMed] [Google Scholar]
- 8.Koning JP, Tenback DE, van Os J, Aleman A, Kahn RS, van Harten PN (2010): Dyskinesia and parkinsonism in antipsychotic-naive patients with schizophrenia, first-degree relatives and healthy controls: a meta-analysis. Schizophr Bull. 36:723–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peralta V, Cuesta MJ (2017): Motor Abnormalities: From Neurodevelopmental to Neurodegenerative Through “Functional” (Neuro)Psychiatric Disorders. Schizophr Bull. 43:956–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu FC, Lin HT, Kuo CF, Hsieh MY, See LC, Yu HP (2018): Familial aggregation of Parkinson’s disease and coaggregation with neuropsychiatric diseases: a population-based cohort study. Clin Epidemiol. 10:631–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smith DJ, Langan J, McLean G, Guthrie B, Mercer SW (2013): Schizophrenia is associated with excess multiple physical-health comorbidities but low levels of recorded cardiovascular disease in primary care: cross-sectional study. BMJ Open. 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin HL, Lin HC, Chen YH (2014): Psychiatric diseases predated the occurrence of Parkinson disease: a retrospective cohort study. Ann Epidemiol. 24:206–213. [DOI] [PubMed] [Google Scholar]
- 13.Chang D, Nalls MA, Hallgrimsdottir IB, Hunkapiller J, van der Brug M, Cai F, et al. (2017): A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat Genet. 49:1511–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.International Parkinson Disease Genomics C, Nalls MA, Plagnol V, Hernandez DG, Sharma M, Sheerin UM, et al. (2011): Imputation of sequence variants for identification of genetic risks for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet. 377:641–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nalls MA, Pankratz N, Lill CM, Do CB, Hernandez DG, Saad M, et al. (2014): Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat Genet. 46:989–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schizophrenia Working Group of the Psychiatric Genomics C (2014): Biological insights from 108 schizophrenia-associated genetic loci. Nature. 511:421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nalls MA, Blauwendraat C, Vallerga CL, Heilbron K, Bandres-Ciga S, Chang D, et al. (2019): Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 18:1091–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Z, Chen J, Yu H, He L, Xu Y, Zhang D, et al. (2017): Genome-wide association analysis identifies 30 new susceptibility loci for schizophrenia. Nat Genet. 49:1576–1583. [DOI] [PubMed] [Google Scholar]
- 19.Nalls MA, Saad M, Noyce AJ, Keller MF, Schrag A, Bestwick JP, et al. (2014): Genetic comorbidities in Parkinson’s disease. Hum Mol Genet. 23:831–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pickrell JK, Berisa T, Liu JZ, Segurel L, Tung JY, Hinds DA (2016): Detection and interpretation of shared genetic influences on 42 human traits. Nat Genet. 48:709–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Butcher NJ, Kiehl TR, Hazrati LN, Chow EW, Rogaeva E, Lang AE, et al. (2013): Association between early-onset Parkinson disease and 22q11.2 deletion syndrome: identification of a novel genetic form of Parkinson disease and its clinical implications. JAMA Neurol. 70:1359–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bassett AS, Chow EW, AbdelMalik P, Gheorghiu M, Husted J, Weksberg R (2003): The schizophrenia phenotype in 22q11 deletion syndrome. Am J Psychiatry. 160:1580–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brainstorm C, Anttila V, Bulik-Sullivan B, Finucane HK, Walters RK, Bras J, et al. (2018): Analysis of shared heritability in common disorders of the brain. Science. 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bulik-Sullivan B, Finucane HK, Anttila V, Gusev A, Day FR, Loh PR, et al. (2015): An atlas of genetic correlations across human diseases and traits. Nat Genet. 47:1236–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smeland OB, Wang Y, Frei O, Li W, Hibar DP, Franke B, et al. (2018): Genetic Overlap Between Schizophrenia and Volumes of Hippocampus, Putamen, and Intracranial Volume Indicates Shared Molecular Genetic Mechanisms. Schizophr Bull. 44:854–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee PH, Baker JT, Holmes AJ, Jahanshad N, Ge T, Jung JY, et al. (2016): Partitioning heritability analysis reveals a shared genetic basis of brain anatomy and schizophrenia. Mol Psychiatry. 21:1680–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smeland OB, Bahrami S, Frei O, Shadrin A, O’Connell K, Savage J, et al. (2019): Genome-wide analysis reveals extensive genetic overlap between schizophrenia, bipolar disorder, and intelligence. Mol Psychiatry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schmitt J, Schwarz K, Baurecht H, Hotze M, Folster-Holst R, Rodriguez E, et al. (2016): Atopic dermatitis is associated with an increased risk for rheumatoid arthritis and inflammatory bowel disease, and a decreased risk for type 1 diabetes. J Allergy Clin Immunol. 137:130–136. [DOI] [PubMed] [Google Scholar]
- 29.Baurecht H, Hotze M, Brand S, Buning C, Cormican P, Corvin A, et al. (2015): Genome-wide comparative analysis of atopic dermatitis and psoriasis gives insight into opposing genetic mechanisms. Am J Hum Genet. 96:104–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smeland OB, Frei O, Kauppi K, Hill WD, Li W, Wang Y, et al. (2017): Identification of Genetic Loci Jointly Influencing Schizophrenia Risk and the Cognitive Traits of Verbal-Numerical Reasoning, Reaction Time, and General Cognitive Function. JAMA Psychiatry. 74:1065–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bansal V, Mitjans M, Burik CAP, Linner RK, Okbay A, Rietveld CA, et al. (2018): Genome-wide association study results for educational attainment aid in identifying genetic heterogeneity of schizophrenia. Nat Commun. 9:3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bipolar Disorder and Schizophrenia Working Group of the Psychiatric Genomics Consortium (2018): Genomic Dissection of Bipolar Disorder and Schizophrenia, Including 28 Subphenotypes. Cell. 173:1705–1715 e1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frei O, Holland D, Smeland OB, Shadrin AA, Fan CC, Maeland S, et al. (2019): Bivariate causal mixture model quantifies polygenic overlap between complex traits beyond genetic correlation. Nat Commun. 10:2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smeland OB, Frei O, Shadrin A, O’Connell K, Fan CC, Bahrami S, et al. (2019): Discovery of shared genomic loci using the conditional false discovery rate approach. Hum Genet. [DOI] [PubMed] [Google Scholar]
- 35.Broce I, Karch CM, Wen N, Fan CC, Wang Y, Tan CH, et al. (2018): Immune-related genetic enrichment in frontotemporal dementia: An analysis of genome-wide association studies. PLoS Med. 15:e1002487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Desikan RS, Schork AJ, Wang Y, Thompson WK, Dehghan A, Ridker PM, et al. (2015): Polygenic Overlap Between C-Reactive Protein, Plasma Lipids, and Alzheimer Disease. Circulation. 131:2061–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Andreassen OA, Harbo HF, Wang Y, Thompson WK, Schork AJ, Mattingsdal M, et al. (2014): Genetic pleiotropy between multiple sclerosis and schizophrenia but not bipolar disorder: differential involvement of immune-related gene loci. Mol Psychiatry. 20:207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Karch CM, Wen N, Fan CC, Yokoyama JS, Kouri N, Ross OA, et al. (2018): Selective Genetic Overlap Between Amyotrophic Lateral Sclerosis and Diseases of the Frontotemporal Dementia Spectrum. JAMA Neurol. 75:860–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu JZ, Hov JR, Folseraas T, Ellinghaus E, Rushbrook SM, Doncheva NT, et al. (2013): Dense genotyping of immune-related disease regions identifies nine new risk loci for primary sclerosing cholangitis. Nat Genet. 45:670–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Andreassen OA, Djurovic S, Thompson WK, Schork AJ, Kendler KS, O’Donovan MC, et al. (2013): Improved detection of common variants associated with schizophrenia by leveraging pleiotropy with cardiovascular-disease risk factors. Am J Hum Genet. 92:197–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Witoelar A, Jansen IE, Wang Y, Desikan RS, Gibbs JR, Blauwendraat C, et al. (2017): Genome-wide Pleiotropy Between Parkinson Disease and Autoimmune Diseases. JAMA Neurol. 74:780–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ferrari R, Wang Y, Vandrovcova J, Guelfi S, Witeolar A, Karch CM, et al. (2017): Genetic architecture of sporadic frontotemporal dementia and overlap with Alzheimer’s and Parkinson’s diseases. J Neurol Neurosurg Psychiatry. 88:152–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Willer CJ, Li Y, Abecasis GR (2010): METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 26:2190–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Demontis D, Walters RK, Martin J, Mattheisen M, Als TD, Agerbo E, et al. (2019): Discovery of the first genome-wide significant risk loci for attention deficit/hyperactivity disorder. Nat Genet. 51:63–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grove J, Ripke S, Als TD, Mattheisen M, Walters RK, Won H, et al. (2019): Identification of common genetic risk variants for autism spectrum disorder. Nat Genet. 51:431–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Eeles RA, Olama AA, Benlloch S, Saunders EJ, Leongamornlert DA, Tymrakiewicz M, et al. (2013): Identification of 23 new prostate cancer susceptibility loci using the iCOGS custom genotyping array. Nat Genet. 45:385–391, 391e381–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Efron B (2010): Large-scale inference : empirical Bayes methods for estimation, testing, and prediction. Cambridge ; New York: Cambridge University Press. [Google Scholar]
- 48.Solovieff N, Cotsapas C, Lee PH, Purcell SM, Smoller JW (2013): Pleiotropy in complex traits: challenges and strategies. Nat Rev Genet. 14:483–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smeland OB, Frei O, Shadrin A, O’Connell K, Fan CC, Bahrami S, et al. (2019): Discovery of shared genomic loci using the conditional false discovery rate approach. Hum Genet. [DOI] [PubMed] [Google Scholar]
- 50.Schwartzman A, Lin X (2011): The effect of correlation in false discovery rate estimation. Biometrika. 98:199–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Watanabe K, Taskesen E, van Bochoven A, Posthuma D (2017): Functional mapping and annotation of genetic associations with FUMA. Nat Commun. 8:1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.The 1000 Genomes Project Consortium (2015): A global reference for human genetic variation. Nature. 526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J (2014): A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 46:310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Boyle AP, Hong EL, Hariharan M, Cheng Y, Schaub MA, Kasowski M, et al. (2012): Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 22:1790–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Roadmap Epigenomics C, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, et al. (2015): Integrative analysis of 111 reference human epigenomes. Nature. 518:317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhu Z, Zhang F, Hu H, Bakshi A, Robinson MR, Powell JE, et al. (2016): Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet. 48:481–487. [DOI] [PubMed] [Google Scholar]
- 57.Consortium GTEx (2017): Genetic effects on gene expression across human tissues. Nature. 550:204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD, et al. (2016): Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron. 89:37–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Goes FS, McGrath J, Avramopoulos D, Wolyniec P, Pirooznia M, Ruczinski I, et al. (2015): Genome-wide association study of schizophrenia in Ashkenazi Jews. Am J Med Genet B Neuropsychiatr Genet. 168:649–659. [DOI] [PubMed] [Google Scholar]
- 60.Wang Y, Thompson WK, Schork AJ, Holland D, Chen CH, Bettella F, et al. (2016): Leveraging Genomic Annotations and Pleiotropic Enrichment for Improved Replication Rates in Schizophrenia GWAS. PLoS Genet. 12:e1005803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Horton R, Wilming L, Rand V, Lovering RC, Bruford EA, Khodiyar VK, et al. (2004): Gene map of the extended human MHC. Nat Rev Genet. 5:889–899. [DOI] [PubMed] [Google Scholar]
- 62.Tabares-Seisdedos R, Rubenstein JL (2009): Chromosome 8p as a potential hub for developmental neuropsychiatric disorders: implications for schizophrenia, autism and cancer. Mol Psychiatry. 14:563–589. [DOI] [PubMed] [Google Scholar]
- 63.Fromer M, Roussos P, Sieberts SK, Johnson JS, Kavanagh DH, Perumal TM, et al. (2016): Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat Neurosci. 19:1442–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ng B, White CC, Klein HU, Sieberts SK, McCabe C, Patrick E, et al. (2017): An xQTL map integrates the genetic architecture of the human brain’s transcriptome and epigenome. Nat Neurosci. 20:1418–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang D, Liu S, Warrell J, Won H, Shi X, Navarro FCP, et al. (2018): Comprehensive functional genomic resource and integrative model for the human brain. Science. 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Heyes S, Pratt WS, Rees E, Dahimene S, Ferron L, Owen MJ, et al. (2015): Genetic disruption of voltage-gated calcium channels in psychiatric and neurological disorders. Prog Neurobiol. 134:36–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Purcell SM, Moran JL, Fromer M, Ruderfer D, Solovieff N, Roussos P, et al. (2014): A polygenic burden of rare disruptive mutations in schizophrenia. Nature. 506:185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Groen JL, Andrade A, Ritz K, Jalalzadeh H, Haagmans M, Bradley TE, et al. (2015): CACNA1B mutation is linked to unique myoclonus-dystonia syndrome. Hum Mol Genet. 24:987–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mencacci NE, R’Bibo L, Bandres-Ciga S, Carecchio M, Zorzi G, Nardocci N, et al. (2015): The CACNA1B R1389H variant is not associated with myoclonus-dystonia in a large European multicentric cohort. Hum Mol Genet. 24:5326–5329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fleig A, Penner R (2004): The TRPM ion channel subfamily: molecular, biophysical and functional features. Trends Pharmacol Sci. 25:633–639. [DOI] [PubMed] [Google Scholar]
- 71.Lee CR, Machold RP, Witkovsky P, Rice ME (2013): TRPM2 channels are required for NMDA-induced burst firing and contribute to H(2)O(2)-dependent modulation in substantia nigra pars reticulata GABAergic neurons. J Neurosci. 33:1157–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sun Y, Sukumaran P, Selvaraj S, Cilz NI, Schaar A, Lei S, et al. (2018): TRPM2 Promotes Neurotoxin MPP(+)/MPTP-Induced Cell Death. Mol Neurobiol. 55:409–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fregeau B, Kim BJ, Hernandez-Garcia A, Jordan VK, Cho MT, Schnur RE, et al. (2016): De Novo Mutations of RERE Cause a Genetic Syndrome with Features that Overlap Those Associated with Proximal 1p36 Deletions. Am J Hum Genet. 98:963–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jordan VK, Fregeau B, Ge X, Giordano J, Wapner RJ, Balci TB, et al. (2018): Genotype-phenotype correlations in individuals with pathogenic RERE variants. Hum Mutat. 39:666–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wray NR, Ripke S, Mattheisen M, Trzaskowski M, Byrne EM, Abdellaoui A, et al. (2018): Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat Genet. 50:668–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Greaves J, Chamberlain LH (2011): DHHC palmitoyl transferases: substrate interactions and (patho)physiology. Trends Biochem Sci. 36:245–253. [DOI] [PubMed] [Google Scholar]
- 77.Fukata Y, Dimitrov A, Boncompain G, Vielemeyer O, Perez F, Fukata M (2013): Local palmitoylation cycles define activity-regulated postsynaptic subdomains. J Cell Biol. 202:145–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Brichta L, Shin W, Jackson-Lewis V, Blesa J, Yap EL, Walker Z, et al. (2015): Identification of neurodegenerative factors using translatome-regulatory network analysis. Nat Neurosci. 18:1325–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.He L, Girijashanker K, Dalton TP, Reed J, Li H, Soleimani M, et al. (2006): ZIP8, member of the solute-carrier-39 (SLC39) metal-transporter family: characterization of transporter properties. Mol Pharmacol. 70:171–180. [DOI] [PubMed] [Google Scholar]
- 80.Luo Q, Chen Q, Wang W, Desrivieres S, Quinlan EB, Jia T, et al. (2019): Association of a Schizophrenia-Risk Nonsynonymous Variant With Putamen Volume in Adolescents: A Voxelwise and Genome-Wide Association Study. JAMA Psychiatry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Beaulieu JM, Gainetdinov RR (2011): The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev. 63:182–217. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.