

Abstract
Directionality of the hole transfer processes between DNA-backbone and -base was investigated using phosphorodithioate [P(S−)=S] components. ESR spectroscopy in homogeneous frozen aqueous solutions, pulse radiolysis in aqueous solution at ambient temperature confirmed initial formation of G•+-P(S−)=S. The ionization potential of G-P(S−)=S was calculated to be slightly lower than that of guanine in 5’-dGMP. Subsequent thermally-activated hole transfer from G•+ to P(S−)=S led to dithiyl radical (P-2S•) formation on μs timescale. In parallel, ESR spectroscopy and pulse radiolysis, and density functional theory (DFT) calculations confirmed P-2S• formation in abasic phosphorodithioate model compound. ESR investigations at low temperatures and higher G-P(S−)=S concentrations showed a bimolecular conversion of P-2S• to the σ2-σ*1-bonded dimer anion radical [-P-2S∸2S-P-]− [ΔG (150 K, DFT) = −7.2 kcal/mol]. However, [-P-2S∸2S-P-]− formation was not observed by pulse radiolysis [ΔG° (298 K, DFT) = −1.4 kcal/mol]. Neither P-2S• nor [-P-2S∸2S-P-]− oxidized guanine base; base-to-backbone hole transfer occurs in phosphorodithioate.
Keywords: Phosphorodithioate, backbone-to-base hole transfer, base-to-backbone hole transfer, guanine cation radical, thiyl radicals
Graphical Abstract
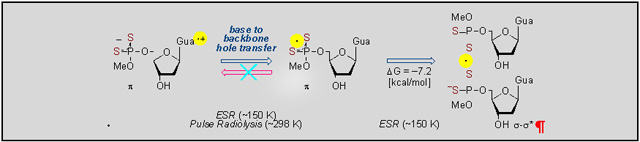
The nature and directionality of the hole transfer pathways in phosphorodithioate systems and identification of the radicals involved were achieved by employing a unique combination of synthesis, electron spin resonance spectroscopic studies in frozen aqueous solution at low temperature, pulse radiolysis in aqueous solution at room temperature, and ab initio DFT calculations.
Introduction
Investigation of hole transfer processes in DNA have been actively pusued owing to their relevance to development of nanowires, biosensors, electrochemical sequencing techniques, and tumor radiotherapy.[1–8] The pathways of hole transfer processes in DNA-components can be investigated in different temperature regimes using complementary tools. Continuous wave electron spin resonance (CW ESR) spectroscopy at low temperature detects the involvement of thermodynamically stable radicals, whereas pulse radiolysis (PR) at room temperature allows radical characterization through absorption spectra as well as determines radical reaction kinetics. ESR studies have established that two distinct pathways – tunneling and thermally-activated hopping are involved in these hole transfer processes.[5–11] Both base-to-base and backbone-to-base hole transfer processes have been investigated.[8–11] The pioneering work of Giese established photolytic hole injection method in defined DNA sequences via formation of C4•′ intermediate which resulted in a backbone-to-base hole transfer process.[7,12,13] Inspired by these works, we have already investigated modified DNA-components, such as phosphorothioate system (PO3S−). Application of ESR has aided to identify the latter type of hole transfer process.[9] ESR investigations of the oxidation of PO3S− incorporated oligomers (S-oligomers) with defined GC and AT sequences showed formation of the two-center three-electron σ2-σ*1-bonded adduct radical (-P-S∸Cl).[9] These studies evidenced that -P-S∸Cl formation takes place by Cl2•− addition to the >P=S moiety of phosphorothioate group followed by Cl− elimination. Subsequently, via thermally activated hopping process -P-S∸Cl oxidizes G (Gua) to form the DNA guanine base cation radical (G•+). On the other hand, Employing defined sequences of ds S-oligomers, ESR studies has established that -P-S∸Cl does not oxidize A, T, and C. Thus, the midpoint potential (E7) of -P-S∸Cl with respect to the standard hydrogen electrode should be bracketed between those of G (1.29 V) and A (1.42 V).[9] This work further established that ionization potential as well as one-electron oxidation potential of the PO3S− moiety in an S-oligomer is less than that of the phosphate moiety in the unmodified oligomer. As a result, the PO3S− moiety of a S-oligomer becomes an effective temporary trap of the hole prior to its transfer to G and, thus allow the backbone-to-base hole transfer process in DNA to be directly studied using ESR spectroscopy.[9] Additionally, ESR studies showed that -P-S∸Cl reacted bimolecularly with a proximate PO3S− moiety to generate the dimer disulfide anion radical ([-P-S∸S-P-]−) for ds S-oligomers of defined AT sequences.[9] Characterization of -P-S∸Cl and [-P-S∸S-P-]− were achieved by employing diisopropyl phosphorothioate, a model compound of PO3S− systems.[9]
Recently, pulse radiolysis has been employed to model the backbone phosphate-to-base hole transfer process.[10] Reactions between the phosphate radical (H2PO4•) and DNA bases G, A, T, and C in 6 M H3PO4 have been carried out using picosecond pulse radiolysis. The time-resolved observations show that H2PO4• efficiently oxidizes G, A and T to produce G•+, A•+ and T•+ via single electron (charge) transfer process; however, the reaction between H2PO4• and cytosine is found to be very slow. These experimental observations are supported by the calculations performed using B3LYP-PCM/6–31++G** DFT method,[10] which confirm phosphate (backbone)-to-base hole transfer.[9–11]
In order to elucidate the effect of the increased number of oxygen substitution by sulfur on the hole transfer processes from backbone-to-base or base-to-backbone taking place in modified nucleic acids, two distinct classes of phosphorodithioate compounds – an abasic system and a nucleotide, ammonium O,O’-diethyl dithiophosphate (DETP(S−)=S) and 5’-O-methoxyphosphorodithioyl-2’-deoxyguanosine (G-P(S−)=S) were selected as models (Figure 1).[14,15]
Figure 1.
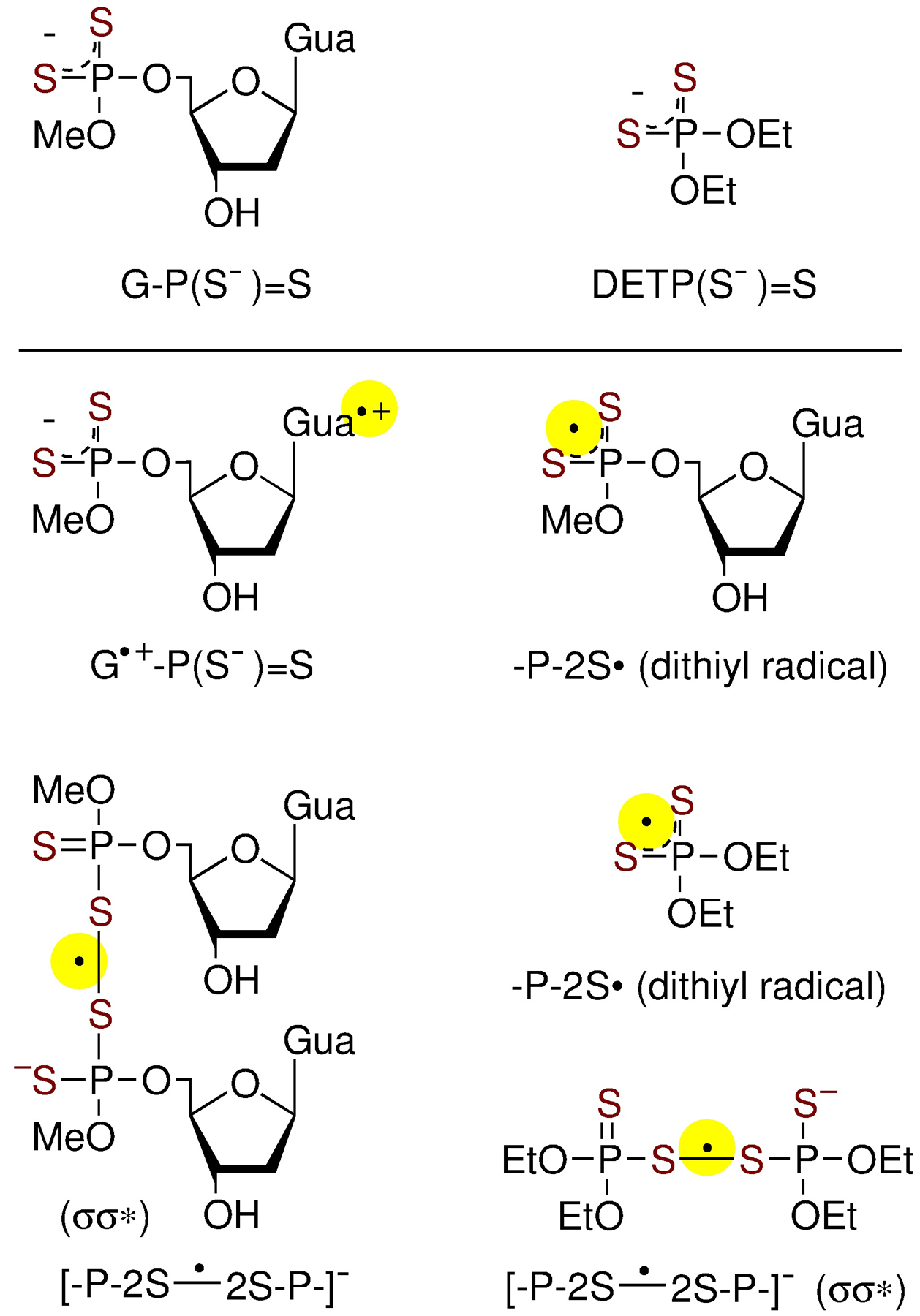
Phosphorodithioate nucleoside model, G-P(S−)=S (top left) and phosphorodithioate without base, DETP(S−)=S (top right), and radicals investigated in this work.
Employing these compounds, ESR spectroscopy, pulse radiolysis, and theoretical calculation (Figure 1 for the radicals investigated in this work) led to several important findings:
Reaction of the one-electron oxidant (e.g., Cl2•−) with DETP(S−)=S and G-P(S−)=S did not result in the formation of a two-center three-electron σ2-σ*1-bonded adduct radical, P(2S∸Cl), as found for phosphorothioates.[9] Instead, oxidation of DETP(S−)=S led to formation of a dithiyl radical, P-2S•, in which the unpaired spin is equally shared by both sulfurs. On the other hand, oxidation of G-P(S−)=S led to G•+-P(S−)=S formation. Subsequently, G•+-P(S−)=S converted unimolecularly to P-2S• by temperature activated hopping of the hole from the guanine base to the P(S−)=S moiety (backbone).
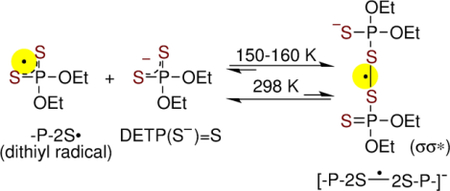
The formation of the dimer anion radical, [-P-2S∸2S-P-]−, via addition of P-2S• to a proximate parent molecule (DETP(S−)=S or G-P(S−)=S) was observed by ESR in homogeneous frozen aqueous solutions but not by pulse radiolysis in aqueous solution at ambient temperature. These results elucidate the strong effect of temperature on the [-P-2S∸2S-P-]− formation owing to the weak S-S bond.
P-2S• and [-P-2S∸2S-P-]− did not oxidize the guanine base. In contrast, for monothio S-oligomers, the two-center three-electron σ2-σ*1-bonded P(S∸Cl) oxidized the guanine base.[9]
This investigation constitutes the first study of transient radical species formed in models of 2S-oligomers. This work presents a unique combination of synthesis, ESR studies in homogeneous frozen aqueous solutions, pulse radiolysis studies in aqueous solution at ambient temperature, and ab initio quantum chemical calculations based on density functional theory (DFT). Studies on G-P(S−)=S, along with our previous work on monothio S-oligomers[9] elucidate the effect of incremental S-atom substitution at the phosphate moiety on the directionality of the hole transfer process between the phosphate and the base.
Results and Discussion
A. Formation of the dithiyl radical, -P-2S• by oxidation of the abasic phosphorodithioate model DETP(S−)=S:
A.1. ESR studies in homogeneous aqueous glassy solutions at low temperatures:
In Figure 2A, ESR spectrum (black) of a γ-irradiated homogeneous glassy (7.5 M LiCl/D2O) sample of DETP(S−)=S (2 mg/mL) is shown. Comparison of this spectrum (250 G scan) with the central part of the total 880 G wide multiplet Cl2•− spectrum[9,16,17] shows that only the expected two low field resonances from Cl2•− and a sharp singlet due to SO4•− are present at the center (Figure 2A). Previous works from our laboratory[9,16,17] have established that scavenging of radiation-produced holes by the matrix (7.5 M LiCl) leads to formation of the matrix radical, Cl2•− (reactions 1a,b).
Figure 2.
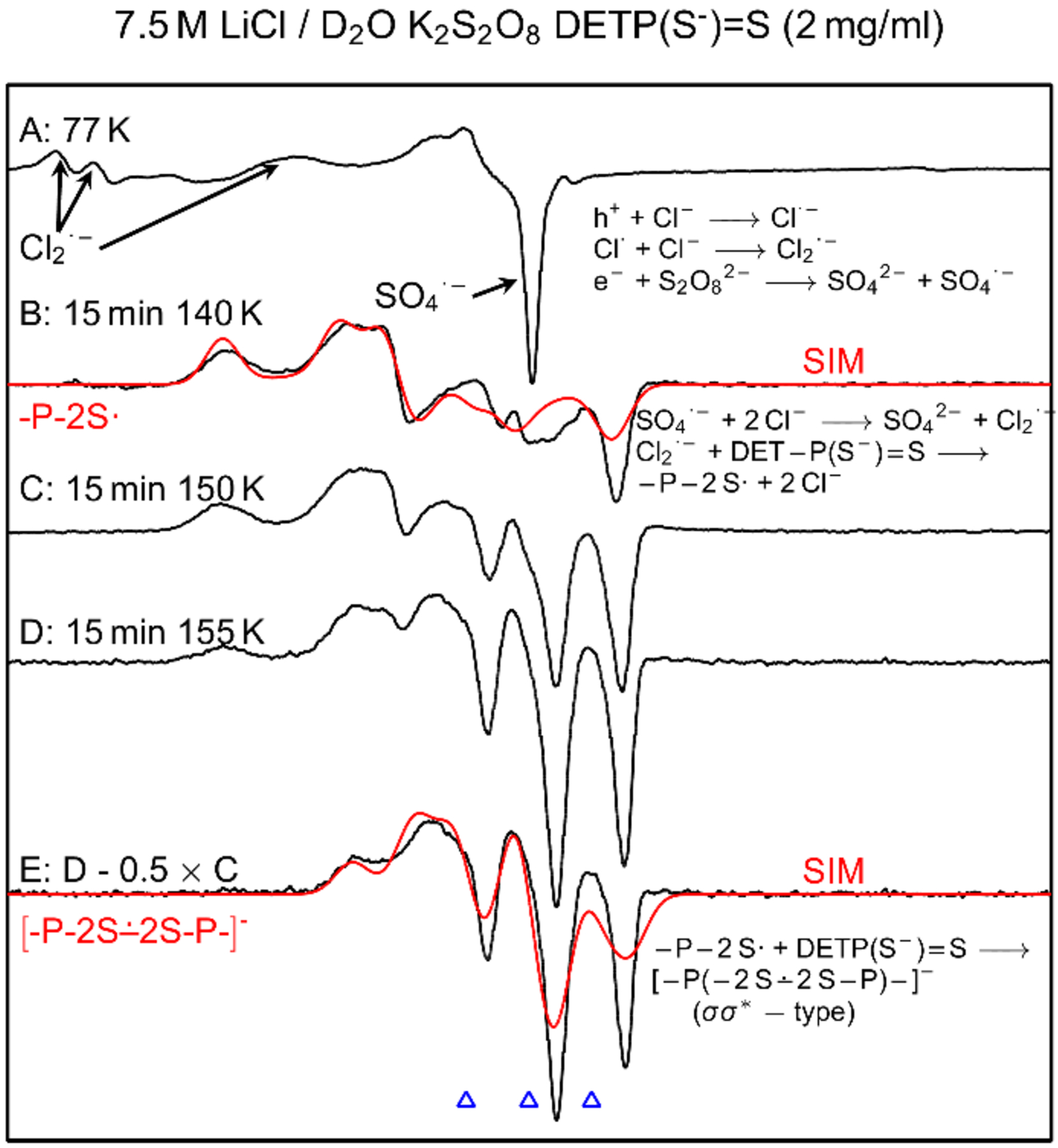
(A) ESR spectrum (black) showing SO4•− and Cl2•− formation in γ-irradiated (absorbed dose = 1.4 kGy at 77 K) homogeneous glassy sample of DETP(S−)=S (2 mg/mL) in the presence of electron scavenger K2S2O8 at native pD of 7.5 M LiCl/D2O (ca. 5). (B) Spectrum of the sample in (A) after annealing to ca. 140 K for 15 min. (C) After further annealing at ca. 150 K for 15 min. (D) After further annealing at ca. 155 K for 15 min. (E) Subtraction of 50% of spectrum C from spectrum D resulted in the black spectrum. Red spectra in (B) and (E) are the simulated spectra of -P-2S• and [-P-2S∸2S-P-]− respectively. See text for details of simulation. All experimentally obtained spectra were recorded at 77 K. The three reference markers (open triangles) in this Figure and in other Figures in the manuscript and in supporting information show the position of Fremy’s salt resonance with the central marker at g = 2.0056. Each of these markers is separated from each other by 13.09 G.
| (1a) |
| (1b) |
Reaction of radiation-produced electrons with S2O82− leads to the formation of SO4•− (reaction 2).[9,16–18]
| (2) |
Annealing the sample at and above ca. 125 K leads to gradual softening of the glass matrix thereby allowing to migration of radicals. Following our previous works,[9,16–18] annealing of the sample at ca. 140 K leads to formation of additional Cl2•− via reaction of SO4•− with Cl− (reaction 3).
| (3) |
Thus, Cl2•− becomes the only remaining radical species that reacts with the solute, i.e. DETP(S−)=S. Reaction of Cl2•− with DETP(S−)=S via annealing of this sample at ca. 140 K for 15 min in the dark produces a black multiplet spectrum (250 G scan) shown in Figure 2B. This spectrum is obtained via subtraction of line components of Cl2•− spectrum from the experimentally recorded spectrum. The black spectrum in Figure 2B does not match the reported spectrum of -P-S∸Cl.[9] Instead, it is an anisotropic spectrum consisting of a near isotropic doublet of ca. 24.2 G due to an α-P coupling (Table 1) and a high g anisotropy. The simulated spectrum, red in Figure 2B, is superimposed on the black spectrum for comparison and matches quite well. The parameters employed to obtain this simulated spectrum are the following: α-P couplings (24, 21, 27.5) G, (gxx, gyy, gzz) = (2.038, 2.0153, 2.0024), and a mixed Lorentzian/Gaussian (1/1) isotropic linewidth of 7 G. These parameters are consistent with those of the dithiyl radical, P-2S• (Table 1). We note here that for monothiyl radicals generated earlier from cysteine, glutathione, and pencillamine in the glassy system, anisotropic g values were poorly resolved and observed over a broad region from 2.01 to 2.13.[19,20] In contrast, the dithiyl radical spectrum shown in Figure 2B has well-defined g-values. Thus, these results demonstrate that the dithiyl radical (-P-2S•, Figure 2) is formed by one-electron oxidation of DETP(S−)=S by Cl2•− (reaction 4).
Table 1.
HFCCs values in Gauss (G) obtained by theory and experiment. ωB97XD/6–31G** method including solvation model (IEF-PCM) has been used for the calculation.
| Molecule | Radicala | Atoms | HFCC (G) | |||
|---|---|---|---|---|---|---|
| Theory | Expb | |||||
| AIso | AAniso | Total | ||||
| DETP(S−)=S | P-2S• | P | −19.26 | ca. 24.2 | ||
| 0.99 | −18.27 | |||||
| [−P-2S∸2S-P-]− | P1 | −11.89 | ca. 13.7 | |||
| 0.37 | −11.52 | |||||
| P2 | −11.90 | ca. 13.7 | ||||
| 0.37 | −11.53 | |||||
| G-P(S−)=S | P-2S• | P | −19.71 | ca. 24.7 | ||
| 1.03 | −18.68 | |||||
| [-P-2S∸2S-P-]−c | P1 | −11.89 | ca. 13.7 | |||
| 0.37 | −11.52 | |||||
| P2 | −11.90 | ca. 13.7 | ||||
| 0.37 | −11.53 | |||||
Geometry was fully optimized using the ωB97XD/6–31G** method including PCM.
The sign associated with HFCC values are provided not by experiment, only by calculations. Experiment gives only the magnitude.
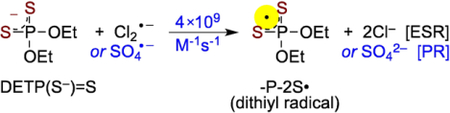 |
(4) |
Assignment of -P-2S• spectrum is supported by DFT calculated HFCC values and g-values which compare well with experiment (Table 1 and supporting information). This assignment is further supported by pulse radiolysis (section A.2), theoretically calculated spectrum of -P-2S• along with its spin density distributions (section C and supporting information).
The experimental spectra (black) found upon further annealing of this sample to 150 K and then to 155 K, are shown in Figures 2C and 2D respectively. Subtraction of 50% of spectrum C from spectrum D resulted in the black spectrum in Figure 2E. Comparison of this spectrum with that in Figure 2B shows the absence of line components due to -P-2S• in it. The spectrum obtained by subtraction isolates the line components assigned to the σσ* tetrasulfide anion radical ([-P-2S∸2S-P-]−, Figure 1) produced via bimolecular reaction of P-2S• with an unreacted DETP(S−)=S molecule (reaction 5).
 |
(5) |
Line components due to HFCC values from two near-isotropic α-P (ca. 13.7 G each) are found in the black spectrum (Figure 2E) with again a highly anisotropic g tensor. This spectrum has been simulated employing two near-isotropic α-P couplings of (13.0, 15.0, 13.0) G (Table 1), a mixed Lorentzian/Gaussian (1/1) isotropic linewidth of 7 G and g-values (2.0049, 2.0009, 2.0200). It is evident from Figure 2E that the simulated spectrum (red) matches well with experimentally obtained (black) spectrum. Assignment of the spectra in Figure 2E to [-P-2S∸2S-P-]− is based on the fact that two α-P couplings are found by experiment along with a good fit of experimental and theoretical hyperfine couplings for [-P-2S∸2S-P-]− (section C and supporting information). This assignment is further supported by our previous work with phosphorothioates showing that -P-S∸Cl reacted bimolecularly with a proximate PO3S− moiety to generate the σσ* dimer disulfide anion radical, [-P-S∸S-P-]−.[9]
Studies employing matched samples of DETP(S−)=S at different concentrations (0.3 mg/mL, 0.5 mg/mL, and 2 mg/mL) under same conditions (gain, modulation, and microwave power) showed that signal-to-noise ratios of ESR spectra increase substantially with increasing concentrations of DETP(S−)=S (supporting information Figures S1 to S3). Moreover, comparison of the black spectrum in Figure 2B with the corresponding ones in Figures S1 to S3 clearly establish that at low DETP(S−)=S concentrations (0.3 mg/mL and 0.5 mg/mL), formation of [-P-2S∸2S-P-]− (reaction 5) does not occur. However, at higher DETP(S−)=S concentration (2 mg/mL), [-P-2S∸2S-P-]− formation becomes favorable. Thus, the concentration dependent [-P-2S∸2S-P-]− production provides evidence that formation of [-P-2S∸2S-P-]− occurs via a bimolecular reaction of P-2S• with an unreacted proximate DETP(S−)=S molecule (reaction 5).
A.2. Pulse Radiolysis studies in aqueous solutions of DETP(S−)=S at room temperature:
Pulse radiolysis measurements employing aqueous solutions with various concentrations of DETP(S−)=S were performed to investigate the formation of P-2S• (reaction 4) and of [-P-2S∸2S-P-]− (reaction 5) at room temperature.
Aqueous solutions of native DETP(S−)=S was found to be quite acidic (pKa ca. 1) and pH of these solutions depended on the DETP(S−)=S concentrations. It is well-established in the literature that a part of the radiation-produced hydrated electrons in acidic solutions are scavenged by the protons to produce H-atoms.[21,22] Pulse radiolysis studies were carried out using either argon-saturated aqueous solutions with a variety of concentrations of DETP(S−)=S ([DETP(S−)=S]=0.25 to 10 mM), S2O82− (5 mM), t-butanol (0.5 M) or N2O-saturated aqueous solutions in the presence or absence of NaN3. The role of t-butanol is to scavenge H-atom and hydroxyl radical (•OH) that are produced via radiolysis of water and to generate the C-centered radical, (H2C(•)-C(CH3)2(OH)), by H-atom abstraction from the methyl group of t- butanol.[21–23] This radical has a weak absorption maximum in the UV at ca. 250 nm.[23]
The hydrated electron (observed at 600 nm within 0.1 μs, Figure 3A) reacts with S2O82− leading to the formation of the highly oxidizing SO4•− (reaction 2).[23] The hydrated electrons react with N2O yielding •OH.[21] In the presence of N3−, •OH is converted into the neutral azide radical, N3•, and this radical undergoes one-electron oxidation.[21]
Figure 3.
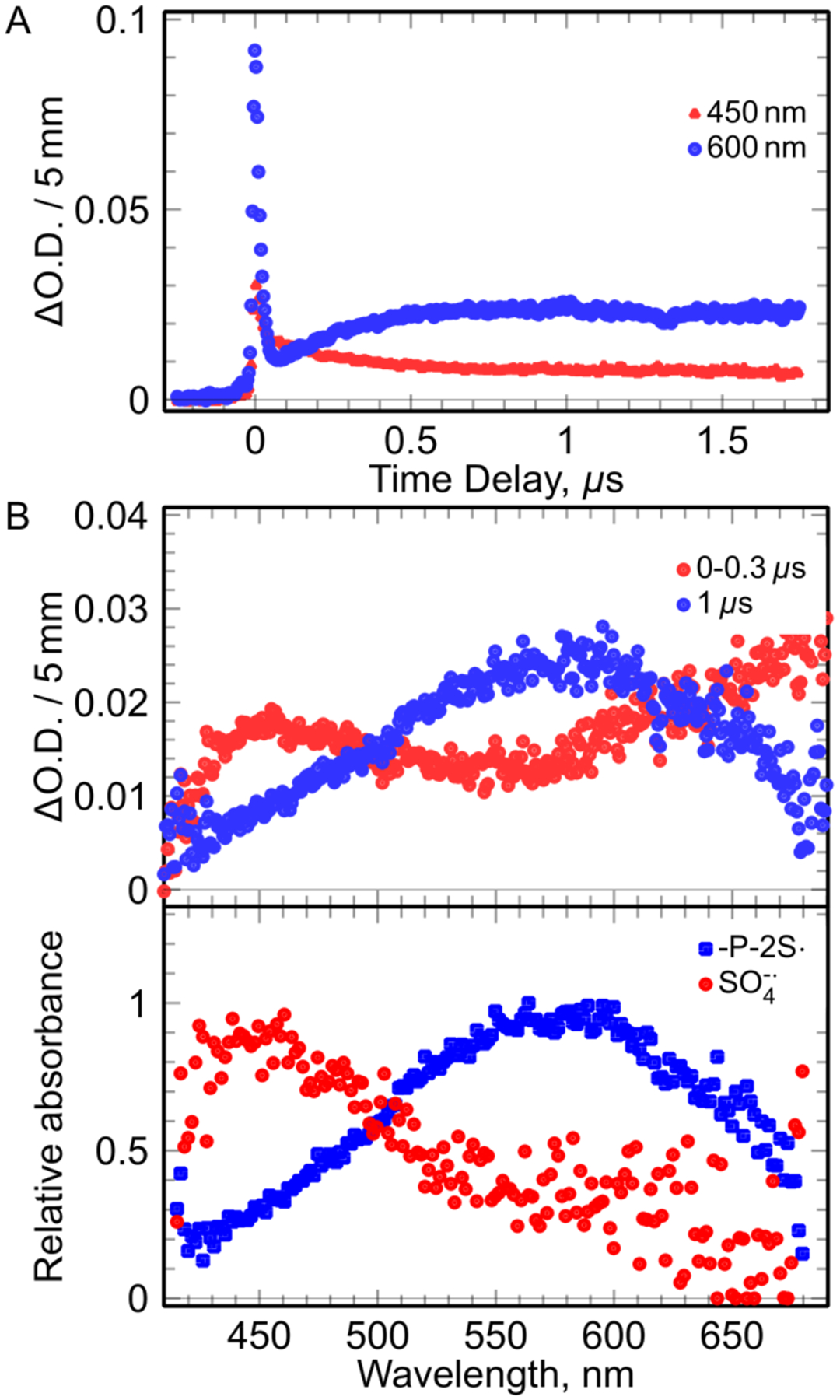
The kinetics and spectra observed in solution (pH 3) containing (DETP(S−)=S (1 mM), S2O82− (5 mM), t-butanol (0.5 M). (A) Kinetics at 450 nm and at 600 nm. (B): Spectra at different wavelength. (B, top): recorded spectra at different time. (B, bottom): The spectra of the species involved in the mechanism obtained by deconvolution of the all spectro-kinetic data.
Typically, for 1 mM DETP(S−)=S solution (pH = 3), the absorption recorded between 0 and 300 ns showed the presence of SO4•− and in less extend of the hydrated electron (Figure 3B, top). In the presence of DETP(S−)=S, the decay of SO4•− was observed clearly at 300 ns (Figure 3B, bottom). Analyses of the spectro-kinetic data show that after decay of the hydrated electron, there are only two absorbing species in solution, one absorbing at 450 nm (SO4•−) and the another one presenting a large absorption band with a maximum at 575 nm. Following the ESR spectral results, the radical species formed via oxidation of DETP(S−)=S was assigned to -P-2S• (reaction 4), absorbing at 575 nm (Figure 3B, bottom). These measurements alllowed us to obtain the value of the rate constant of the oxidation of DETP(S−)=S by SO4•− (reaction 4) as 4 × 109 M−1s−1. Corresponding pulse radiolysis studies of identically prepared argon-saturated DETP(S−)=S solutions containing (DETP(S−)=S (1 mM)), S2O82− (5 mM), and t-butanol (0.5 M) at pH 8.9 led to similar results (Figure S4).
By increasing the DETP(S−)=S concentrations up to 10 mM, same absorption spectra with a maximum at 575 nm were observed after the decay of SO4•−. Oxidation of DETP(S−)=S by SO4•−was found to occur at shorter times without any change in the shape and position of the absorption band recorded at 575 nm. However, the decay rate of -P-2S• was found unaltered.
As the decay rate of -P-2S• was found to be independent of DETP(S−)=S concentrations and it followed a second order (Figure 4, inset), this excludes the possibility of formation of [-P-2S∸2S-P-]− via reaction 5 at ambient temperature in aqueous solution.
Figure 4.
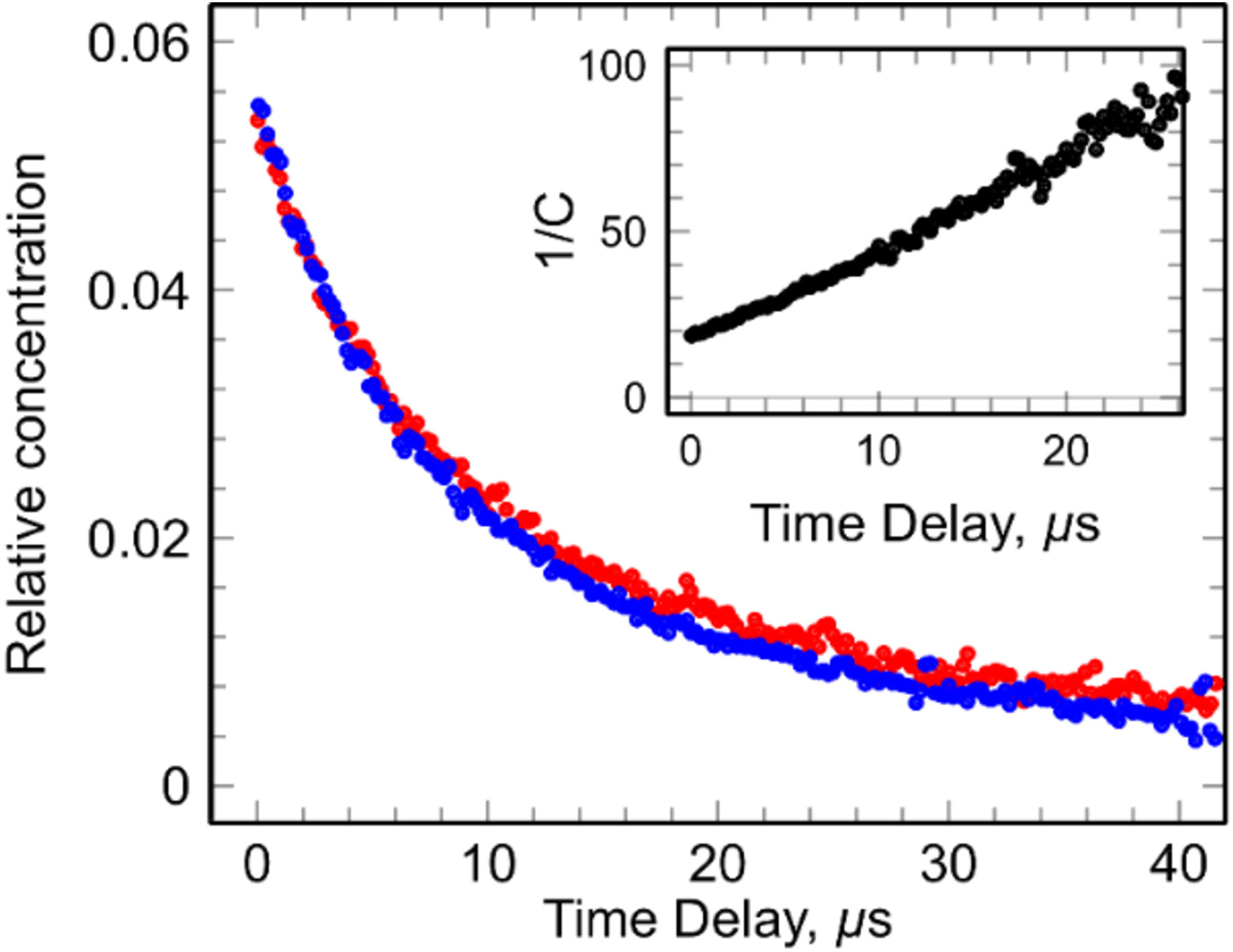
Kinetics observed in argon-saturated solutions containing (DETP(S−)=S 10 mM (red) and 1 mM (blue), with S2O82− ([S2O82-] = 5 mM) and t-butanol = 0.5 M) at pH 8.9. The inset shows second order treatment of the kinetics.
To support these results, additional pulse radiolysis using DETP(S−)=S solutions ([DETP(S−)=S] = 1 mM) under N2O-saturated conditions in the presence or absence of N3− were carried out. The same absorption spectra along with the same absorption band with a maximum at 575 nm was observed. Thus, the spectral assignment of -P-2S• was confirmed. Additional support for this assignment was obtained by theoretical calculations of spectra (section C and supporting information). -P-2S• was found to decay at a longer time than found for [DETP(S−)=S] = 10 mM. Half-life of this radical was found to be 20 μs (Figure 4).
Moreover, in these solutions without S2O82−, -P-2S• was found to undergo a second order decay over 20 μs and the decay did not depend on DETP(S−)=S concentration. In addition, the decay kinetics of -P-2S• was found to be very similar to those shown in Figures 4 and S4 (i.e., in solutions with S2O82−). Product of the decay of -P-2S• did not show any absorption between 400 and 700 nm. We note here that the absorption of neutral and oxidized glutathione dimer (GSSG) were found in the UV region.[24–26] Based on pulse radiolysis results, we conclude that (a) the same dithiyl radical species, P-2S•, is formed via oxidation of DETP(S−)=S by SO4•−, •OH, or by N3• (reaction 4), (b) P-2S• does not react with DETP(S−)=S to form [-P-2S∸2S-P-]− in aqueous solutions at room temperature, and (c) the neutral stable product [-P-2S-2S-P-] formed due to the decay of P-2S• via a second order reaction (reaction 6), did not absorb in the visible range.
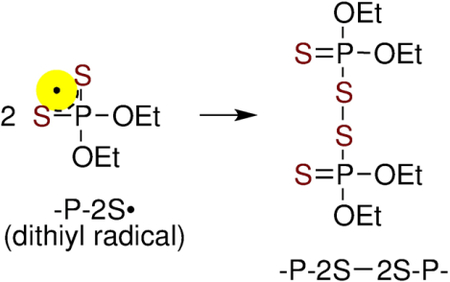 |
(6) |
B. Synthesis of G-P(S−)=S:
The investigated compounds were prepared using the oxathiaphospholane methodology, developed by Stec et al. for the stereocontrolled synthesis of phosphorothioate oligonucleotides.[27,28] The approach has also been successfully applied to the phosphorylation and thiophosphorylation of nucleosides and amino acids.[29–31] The synthesis starts from phosphitylation reaction of 3’-acyl-protected dGuo (1),[32] which was combined with chlorodithiaphospholane (2)[33] as illustrated in Scheme 1. The oxidation of tricoordinate phosphorus intermediates (not isolated and not illustrated) to the corresponding 2-thio-1,3,2-dithiaphospholane (3)[34] was accomplished with the use of elemental sulfur in pyridine. Key intermediate 3 reacted with O-nucleophile (methanol) in the presence of a strong base (DBU), followed by deprotection (aqueous ammonia). The resulting phosphorodithioate was converted into its sodium salt G-P(S−)=S Na+ (Dowex Na+; 86% yield).
Scheme 1.
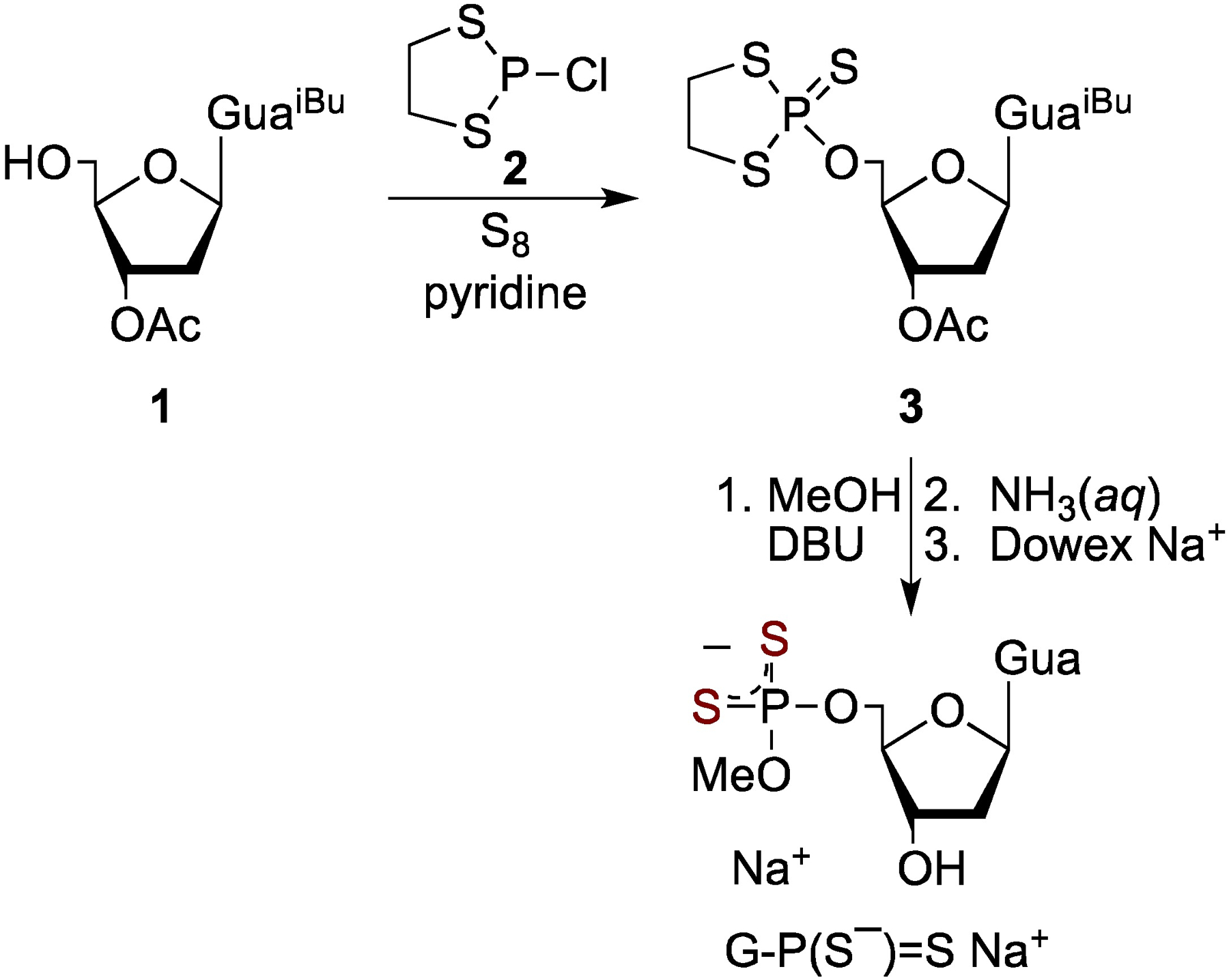
Synthesis of phosphorodithioate G-P(S−)=S Na+.
B.1. Oxidation of 5’-O-methoxyphosphorodithioyl-2’-deoxyguanosine (G-P(S−)=S):
In section A one-electron oxidation of DETP(S−)=S forming -P-2S• (reaction 4) and its bimolecular conversion to [-P-2S∸2S-P-]− (reaction 5) at low temperature (Figure 2) is described. However this reaction was not observed at room temperature by pulse radiolysis (Figure 4). Therefore, ESR spectroscopy (Section B.1.1) and pulse radiolysis (Section B.2) were performed employing in-house synthesized samples of G-P(S−)=S with various concentrations to investigate production of the nucleotides -P-2S• and [-P-2S∸2S-P-]−.
B.1.1. ESR studies:
In Figure 5A, ESR spectrum of a homogeneous glassy sample of G-P(S−)=S (1.5 mg/mL) that was γ-irradiated at 77 K is shown. This spectrum is identical to that shown in Figure 2A. Therefore, based on our assignment of spectrum in Figure 2A, the spectrum (black) in Figure 5A was assigned to a cohort of two radicals (Cl2•− and SO4•−, reactions 1 and 2).
Figure 5.
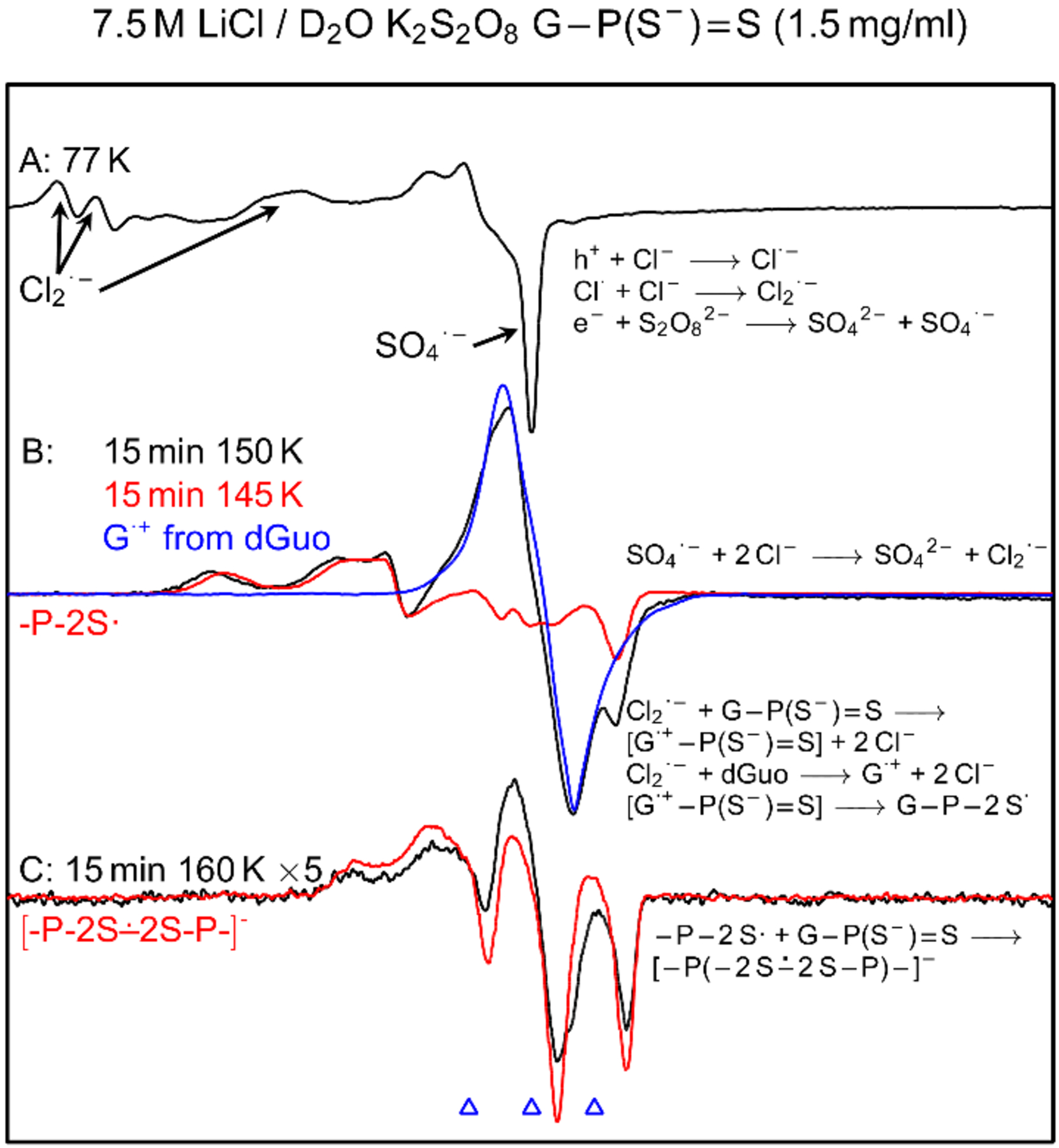
(A) ESR spectrum (black) showing SO4•− and Cl2•− formation in γ-irradiated (absorbed dose = 1.4 kGy at 77 K) homogeneous glassy sample of G-P(S−)=S (1.5 mg/mL) in the presence of electron scavenger K2S2O8 at native pD of 7.5 M LiCl/D2O (ca. 5). (B) Spectrum (black) of the sample in (A) after annealing to ca. 150 K for 15 min. The red spectrum in 5(B) is the black spectrum in Figure 2B that was assigned to -P-2S•. Blue spectrum is the reported spectrum of guanine cation radical (G•+) from homogeneous glassy samples of dGuo or guanine containing DNA-oligonucleotides.[35–44] These spectra are superimposed for comparison. (C) After further annealing at ca. 160 K for 15 min. This spectrum is expanded by digitally multiplying the original spectrum by 5. The red spectrum in 5(C) is the black spectrum in Figure 2E that is assigned to [-P-2S∸2S-P-]−. All experimentally obtained spectra were recorded at 77 K.
Subsequent annealing this sample to 150 K for 15 min resulted in the black spectrum in Figure 5B. This spectrum was recorded under the same conditions (gain, modulation, and microwave power) as employed to record the spectrum 5A. Experimentally recorded spectrum (red) of -P-2S• from DETP(S−)=S (black spectrum in Figure 2B) and the reported spectrum (blue) of guanine cation radical (G•+) from dGuo and from various DNA-model systems containing G[9,35–44] are superimposed on the black spectrum in Figure 5B. Using these spectra as benchmarks, analysis of the black spectrum in Figure 5B clearly show that this spectrum is a cohort of two radicals (-P-2S•, ca. 70%, and G•+-P(S−)=S, the singlet at the center, ca. 30%). These results further establish that -P-2S• from DETP(S−)=S and -P-2S• from G-P(S−)=S have very similar ESR spectrum, and hence, very similar ESR characteristics (i.e., α-P couplings, anisotropic g-values, centre of the spectrum, total hyper-fine splitting). Following our findings in the model compound DETP(S−)=S, ESR spectral studies of G-P(S−)=S showed no observable line component due to P(2S∸Cl) (Figure 5, and Figures S5 and S6).
However, ESR studies using matched samples of G-P(S−)=S at lower concentration, 0.5 mg/mL, showed formation of G•+-P(S−)=S only and no observable formation of -P-2S• (spectra B and C in Figure S5). On the other hand, using matched samples of G-P(S−)=S at higher concentration, [G-P(S−)=S] = 6 mg/mL, ESR studies showed predominant formation of -P-2S• along with small (ca. <10%) formation of G•+-P(S−)=S (spectrum B in Figure S6) under similar conditions. In addition, pulse radiolysis studies of oxidation of G-P(S−)=S Na+ at different concentrations (section B.2.1) showed formation of G•+-P(S−)=S only.
These results point out that oxidation of G-P(S−)=S involves only the oxidation of guanine base (reaction 7) and not of the P(S−)=S moiety (i.e., reaction 8). As a result, observation of the line components of -P-2S• in the experimental spectrum (black) in Figure 5B (also in spectra D and B in Figures S5 and S6 respectively) can be explained on the basis of a hole transfer process from the oxidized guanine base (G•+) to the backbone (P(S−)=S moiety, reaction 9).
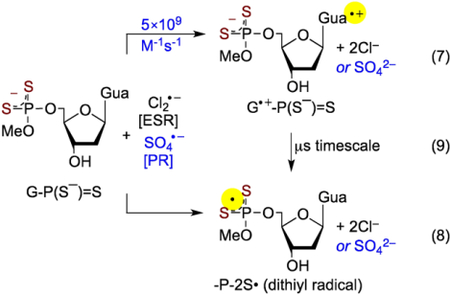 |
Note that, midpoint potential (E7) values of thiyl radicals (RS• (R= H-atom or alkyl group) and σσ* disulfide anion radical ([-S∸S-]−)) fall in the range of 0.5 to 0.9 V.[21,45–47] It is expected, therefore, that corresponding midpoint potentials of dithiyl radicals reported in this work (-P-2S•, [-P-2S∸2S-P-]−) should be similar. As midpoint potential of G•+ (E7, from dGuo/Guo) = 1.29 V)[9,21,22] being quite higher than corresponding expected midpoint potentials of -P-2S• and [-P-2S∸2S-P-]−, it seems very reasonable that neither -P-2S• nor [-P-2S∸2S-P-]− would oxidize dGuo/Guo. But, the reverse reaction, i.e., oxidation of -P(S−)=S moiety of G-P(S−)=S by G•+ via hole transfer (reaction 9) is clearly feasible and is observed in Figure 5B. Based on these ESR spectral results, we propose that -P-2S• formation happens via thermally activated hole hopping from G•+ to the -P(S−)=S moiety in G•+-P(S−)=S. Thus, ESR spectra in Figure 5B and spectra B and C in Figure S5 (0.5 mg/mL), and spectra B in Figure S6 (6 mg/mL) provide evidence of base-to-backbone hole transfer in G-P(S−)=S.
ESR spectrum (black) of this sample obtained after annealing at ca. 160 K for 15 min in the dark is shown in Figure 5C. This spectrum was also recorded under the same conditions (gain, modulation, and microwave power) as employed to record the spectra 5A and 5B. This spectrum was digitally multiplied by 5 to make its line components clearly visible. This spectrum matches very well with that (blue) of [-P-2S∸2S-P-]− observed (Figure 2E) from DETP(S−)=S along with a small extent of G•+-P(S−)=S line component found at the centre. Therefore, this black spectrum in Figure 5C is predominantly due to [-P-2S∸2S-P-]− from G-P(S−)=S. However, at [G-P(S−)=S] = 0.5 mg/mL, formation of [-P-2S∸2S-P-]− is not observed (Figure S5) albeit observed at higher concentrations (Figures 5, S6). Using matched samples of G-P(S−)=S with [G-P(S−)=S] = 6 mg/mL, the spectrum obtained after annealing at 160 K for 15 min was found to be only due to [-P-2S∸2S-P-]− along with no observable line component of G•+-P(S−)=S at the center (Figure S6). These results show that [-P-2S∸2S-P-]− production from G-P(S−)=S occurs via a bimolecular reaction between -P-2S• and a proximate G-P(S−)=S (reaction 10), similar to our findings in the model compound, DETP(S−)=S (reaction 5 and Figures S1 to S3).
ESR spectral studies in mixtures of DETP(S−)=S with dGuo at different ratios (Figures S7 and S8) support the evidence of reactions 9 and 10 as well. Especially, in the sample having a mixture of dGuo (5 mg/mL) and DETP(S−)=S (1 mg/mL), increase in the intensities of line components due to -P-2S• along with concomitant decrease of the intensity of G•+ spectrum was observed (black spectra in Figures S7C to S7E). ESR spectra of these samples were also compared with those shown in Figure 5. These observation established that the hole transfer does indeed occur from G•+ in dGuo to the proximate P(S−)=S moiety of DETP(S−)=S bimolecularly via thermally activated hopping. On the other hand, ESR spectra (Figure S8) obtained from the sample with a mixture of dGuo (1 mg/mL) and DETP(S−)=S (5 mg/mL), showed a very small G•+ line component at the centre of the spectrum similar to that in the spectrum S8B. These results indicate that the presence of a very small central G•+ line component is due to the remaining G•+ after the favourable hole transfer reaction 9. Formation of [-P-2S∸2S-P-]− found by ESR studies in these samples confirmed the bimolecular reaction of -P-2S• with a proximate P(S−)=S moiety (reaction 10).
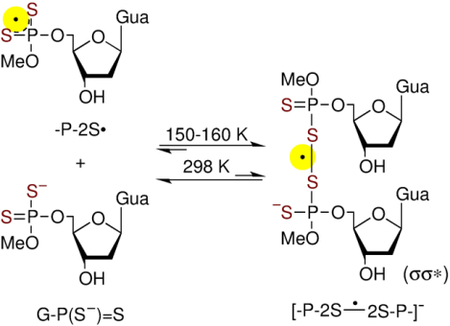 |
(10) |
All these ESR spectral results present no evidence of electron transfer from [-P-2S∸2S-P-]− to G•+-P(S−)=S unlike our previous observation of electron transfer from [-P-2S∸2S-P-]− to G•+-PO3S−.[9]
B.2. Pulse radiolysis:
Pulse radiolysis measurements were performed employing aqueous solutions at room temperature to establish the nature (intramolecular or intermolecular) of the hole transfer process occurring from G•+ to backbone (P(S−)=S moiety) in G•+- P(S−)=S (reaction 9). To achieve this goal, in the first step pulse radiolysis was performed to reproduce the data in the literature [21,22] on the formation of G•+ via oxidation of the guanine base of Guo by SO4•− (Figure S9) were reproduced by pulse radiolysis. Thereafter, pulse radiolysis were carried out in aqueous solution by employing a mixture of Guo (2 mM) and DETP(S−)=S (0.2 mM, Figure S10) and by using solutions of G-P(S−)=S at different concentrations (section B.2.1).
B.2.1. Pulse radiolysis of G-P(S−)=S:
The solution employed for pulse radiolysis studies contained G-P(S−)=S (1 mM), S2O82- (5 mM), t-butanol (0.5 M), and was thoroughly bubbled with argon to remove the dissolved oxygen. Similar to our results of pulse radiolysis studies of the oxidation of DETP(S−)=S by SO4•− (Figures 3,4), within 100 ns, decay of the hydrated electron observed at 650 nm was complete and SO4•− formation was observed at 450 nm (Figure 6). The spectra at 10 ns and 100 ns after the pulse confirmed the presence of SO4•−, which exhibited an absorption band at 450 nm. This radical decayed within 1 μs leading to the formation of G•+-P(S−)=S by SO4•− mediated oxidation of G-P(S−)=S (reaction 7) thereby confirming ESR spectral results (Figures 5, S5 to S8).
Figure 6.
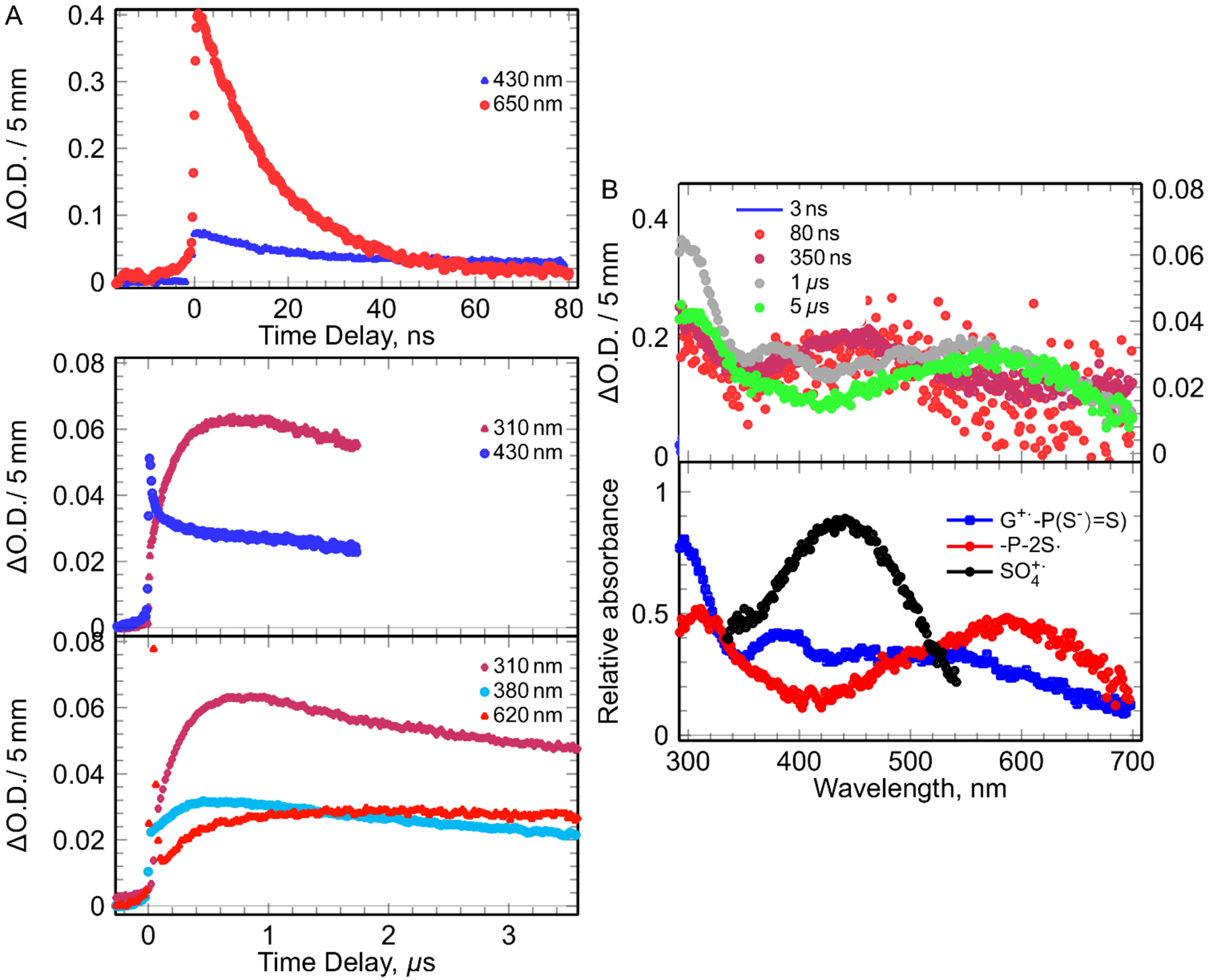
The kinetics and spectra observed in solution containing [G-P(S−)=S] (1 mM), S2O82- (5 mM), t-butanol (0.5 M) by pulse radiolysis. Left (A): kinetics at different times and wavelengths. Right (B): recorded spectra at different times (top) and the spectra of the species involved in the mechanism obtained by deconvolution of the all spectro-kinetic data with arbitrary unit (bottom).
The decay of SO4•− was correlated with the increase of three absorption bands: one broad band around 500 nm, one band at 360 nm, and an intense absorption band around 300 nm. These results agree nicely with the reported G•+ spectrum from dGuo or Guo by Candeias and Steenken,[22] and our independent experiments with Guo (Figures S9 and S10). These G•+ spectra match nicely with those of G•+-P(S−)=S from pulse radiolysis in aqueous solution at ambient temperature and with the reported spectrum of G•+ from homogeneous glassy samples of dGuo and DNA-oligomers containing guanine.[35–42] In addition, the absorption spectrum of G•+-P(S−)=S from G-P(S−)=S matches nicely with the corresponding spectrum of G•+ from dGuo in homogeneous glassy solution at 77 K.[35] These results unequivocally establish that only G•+-P(S−)=S is formed via oxidation of G-P(S−)=S (reaction 7) and oxidation of G-P(S−)=S does not lead to -P-2S• formation, i.e., oxidation of G-P(S−)=S does not proceed via reaction 8.
In agreement with pulse radiolysis studies of the mixture of Guo (2 mM) and DETP(S−)=S (0.2 mM) (Figure S10), G•+-P(S−)=S from G-P(S−)=S was found to decay and along with formation of a new species that absorbed mainly in the red wavelengths. The increase of absorption at 620 nm at longer time gave evidence of the formation of these new species (Figure 6). At longer timescale, the absorption due to G•+-P(S−)=S disappeared and a new and broad absorption maximum at 585 nm appeared that matched well with the absorption spectrum of -P-2S• from oxidation of DETP(S−)=S (Figure 3) and from oxidation of DETP(S−)=S by G•+ from Guo (Figure S10). These measurements led us to obtain the value of the rate constant of the oxidation of DETP(S−)=S by G•+ from Guo (i.e., of reaction 4, G•+ from Guo is the oxidant) as 4 × 109 M−1s−1. These results confirm the evidence of hole transfer in G•+-P(S−)=S leading to the formation of -P-2S•.(reaction 9). Most importantly, this reaction was found to be unimolecular as its kinetics was independent of G-P(S−)=S concentration. This base-to-backbone hole transfer rate is found to be several orders of magnitude higher than that of the 8-oxo-G formation via nucleophilic addition of water to the C-8 of guanine base of the one-electron oxidized G:C base pair followed by deprotonation and subsequent hole transfer in oligomers of defined sequences and in highly polymerized DNA.[43,44] These results indicate that 8-oxo-G formation through guanine cation radical[44] will be substantially hindered in phosphorodithioate-incorporated DNA. Similar to the results shown in Figure 4, the decay rate of -P-2S• (generated by reaction 9) was very similar to that in DETP(S−)=S, was observed to be independent of G-P(S−)=S concentrations and hence, was second order in nature.
ESR studies of G-P(S−)=S present the evidence of bimolecular conversion of -P-2S• from G-P(S−)=S to [-P-2S∸2S-P-]− at low temperature, similar to our observations in DETP(S−)=S (section A). However, this bimolecular conversion is not observed at room temperature by pulse radiolysis.
C. Theoretical Studies:
Theoretical studies were carried out to gain a more complete understanding of the mechanisms proposed by ESR and pulse radiolysis studies and to support the radical assignments of ESR and pulse radiolysis spectra due to various radicals formed by one-electron oxidation of G-P(S−)=S, of the phosphorodithioate model compound DETP(S−)=S, and subsequent reactions of these radicals in the aqueous phase.
Density functional theory (DFT) at the ωB97XD/6–31G** level was employed to calculate the radical properties, such as, spin density distribution and hyperfine coupling constant values (HFCCs), g values, and UV-Visible spectra. In addition, the free energy and enthalpy for reaction 5 were calculated to explain the formation of [-P-2S∸2S-P-]− at low temperature in glassy solutions (ESR studies) but not at room temperature (pulse radiolysis).
C.1. HFCC and spin density distributions:
The calculated HFCCs are presented in Table 1 along with experimental HFCCs. For -P-2S• generated from DETP(S−)=S, the calculated α-P isotropic (Aiso) and anisotropic (Aaniso) HFCCs are −19.3 G and (–1.59, 0.59, 0.99 G), respectively. Therefore, the total HFCCs (Aiso + Aaniso) are −20.9 G, −18.7 G and −18.3 G, respectively; this is in agreement with the experimental HFCCs ca. 24.2 G, Table 1. The calculated anisotropic total α-P HFCCs in -P-2S• formed by one-electron oxidation of G-P(S−)=S are (–21.5, −19.0, −18.7 G). These values are also in good agreement with the corresponding experimental value of α-P (ca. 25 G, Figure S6) in -P-2S•, Table 1. As an example of calculation of g-values, we calculated the g values of -P-2S• from G-P(S−)=S as (2.0588629, 2.0212272, 2.0042742) (see supporting information for details); the experimentally obtained g values for this radical were (2.038, 2.0153, 2.0024) and showed reasonable agreement between experiment and theory. Employing Time-Dependent Density Functional Theory (TDDFT) at the ωB97XD/6–31G** level, the theoretically calculated value of absorption maximum for -P-2S• from DETP(S−)=S and from G-P(S−)=S was found to be 1.9 eV (supporting information). For comparison, absorption maximum of -P-2S• from DETP(S−)=S and -P-2S• from G-P(S−)=S by pulse radiolysis were observed at 2.1 eV (Figures 3B and 6).
For [-P-2S∸2S-P-]− from DETP(S−)=S (reaction 5), the calculated HFCCs of each of the two α-P (P1 and P2, Table 1) are found to be as ca. −12 G which is in agreement with the experimental HFCCs of 13.7 G. From HFCCs of each of the two α-P in [-P-2S∸2S-P-]− and HFCCs of the α-P in -P-2S• it is apparent that the HFCC of an α-P in [-P-2S∸2S-P-]− is about half of that in -P-2S• as expected from our earlier investigation of phosphorothioates.[9]
The spin density distribution plots of -P-2S• and [-P-2S∸2S-P-]− are presented in the supporting information. In -P-2S•, the spin is delocalized on both the S atoms. This spin delocalization does not allow formation of a two-center three-electron σ2-σ*1-bonded adduct radical, P(2S∸Cl) from DETP(S−)=S or from G-P(S−)=S. On the other hand, in phosphorothioates (with one S atom), the spin localization on the S atom led to the formation of the two-center three-electron σ2-σ*1-bonded adduct radical, P(S∸Cl).[9]
In [-P-2S∸2S-P-]−, on the other hand, the spin is equally delocalized on only the two bonded S atoms (S-S). We note from Table 1 that HFCCs in P-2S• is not significantly affected by the substituent (diethyl or guanine). In addition, [-P-2S∸2S-P-]− from DETP(S−)=S or from G-P(S−)=S also have nearly identical couplings and support the ESR spectral results (Figures 5 and S3).
C.2. Calculation of the adiabatic ionization energy (AIE):
Calculation of the adiabatic ionization energy (AIE) of of the parent species (supporting information) are of interest as they might help to explain only the oxidation of guanine base (reaction 7). In Table 2, we have presented the ωB97XD-PCM/6–31++G** calculated AIE values for the G containing phosphorothioate, phosphorodithioate and phosphate as well as those of model compounds – diethyl ester phosphorothioate and DETP(S−)=S along with their spin density distributions on G, S and P, respectively. We note that for the one-electron oxidized G-P(S−)=S, spin is shared between the sulfur atoms whereas for the G-phosphorothioate the spin is solely on G (Table 2).
Table 2.
Adiabatic Ionization energy (AIE) in eV of systems substituted by sulfur atoms on phosphate. The calculations were carried out using the ωb97xd/6–31++G** method including PCM.
| System | ωb97xd-PCM/6–31++G** | Radical Site | |
|---|---|---|---|
| AIE [eV] | Spin Distribution | ||
| O2PS2-G (G-P(S−)=S) |
5.66 | S1 = 0.541; S2 = 0.541 P = −0.085 |
PS2 |
| O2PS2Et2 (DETP(S−)=S) | 5.65 | S1 = 0.552; S2 = 0.516 | PS2 |
| O3PS-G | 5.77 | G = 1.001; S = 0.003 P = −0.001 |
G |
| O4P-G | 5.76 | G = 1.0 | G |
| O3PSEt2 | 5.85 | S = 0.935 | S |
The abasic phosphorothioate O3PSEt2 and phosphorodithioate O2PS2Et2 (DETP(S−)=S) have AIE of 5.85 and 5.65 eV respectively, with the spin on the sulfur atoms. However, G-phosphate and G- phosphorothioate both have the spin on G and have AIEs 5.76 and 5.77 eV, respectively. These results predict that the ionization energy of G is intermediate between the phosphorothioate and phosphorodithioate species and for G-phosphorodithioate species, the oxidation should occur on sulfurs not on G and both G-P(S−)=S and DETP(S−)=S show nearly identical AIEs with spin on sulfurs. However, in ESR studies of glassy G-P(S−)=S samples at low concentrations the site of oxidation by Cl2•− is found to be on the G base (Figure S5). Employing various oxidants (e.g., SO4•−, N3•, •OH), pulse radiolysis studies in aqueous solution solutions of G-P(S−)=S ([G-P(S−)=S] = 1 to 10 mM, Figure 6) show that the site of oxidation is also on G (i.e., reaction 7 is favored). Based on these experimental and theoretical results, it is apparent that oxidation of G-P(S−)=S is driven by kinetics. This is not unexpected as the AIEs of the phosphorodithioates are very close (Table 2).
C.3. Formation of [-P-2S∸2S-P-]− becomes favorable at lower temperature:
For reaction 5, the standard free energy (ΔG°) for association of P-2S• with DETP(S−)=S was calculated as −1.4 kcal/ mole; the standard enthalpy (ΔH°) and standard entropy ΔS°) were calculated as −12.9 kcal/mol and 38.6 cal K−1 mol−1 respectively (see supporting information). However, at 150 K the free energy ΔG150K was calculated as −7.24 kcal/mol; the corresponding enthalpy ΔH150K and entropy ΔS150K were calculated as −13.3 kcal/mol and 40.7 cal K−1mol−1. These values predict that the formation of [-P-2S∸2S-P-]− becomes favorable at lower temperature thereby supporting its detection by ESR in the glassy samples at lower temperature (Figures 2, 5, S6–S8) albeit by pulse radiolysis at room temperature in aqueous solutions (Figures 3, 6, S4) due to a very weak S-S bond stemming from the positive entropy for the S-S bond dissociation.
Conclusion
The unique combination of synthesis of model systems, ESR studies at low temperatures, pulse radiolysis at ambient temperature and ab initio DFT calculations presented in this work has led to the following salient findings:
Unlike σ2-σ*1-bonded adduct radical (-P-S∸Cl) formation from Cl2•− addition to the >P=S moiety in phosphorothioates,[9] reaction of Cl2•− with abasic phosphorodithioate model compound - DETP(S−)=S leads to formation of -P2S• via one-electron oxidation and the unpaired spin is delocalized equally over both S atoms in -P2S•. However, in the nucleotide phosphorodithioate model (G-P(S−)=S), the site of oxidation is the guanine base, leading to the G•+-P(S−)=S formation.
Subsequent formation of the dithiyl radical, P-2S• was observed via a unimolecular and thermally activated hopping of the hole from the guanine base in G•+-P(S−)=S to the P(S−)=S moiety. These results are in contrast to our previous ESR and theoretical studies on phosphorothioate,[9] in which two-center three-electron σ2-σ*1-bonded adduct radical (-P-S∸Cl) resulted in hole transfer to G. Thus, our studies show that increasing the number of S-atoms as substituents in the phosphate moiety of sugar-phosphate backbone lowers the redox potential and allows for charge delocalization on the phosphorodithioate group which changes the directionality of the hole transfer process in DNA to base-to-backbone in phosphorodithioate from backbone-to-base in phosphorothioate[9].
Owing to a favorable free energy at low temperatures (ΔG150K = −7.2 kcal/mol), the addition of -P-2S• to a proximate DETP(S−)=S or to a proximate G-P(S−)=S leads to the formation of a stable σ2-σ*1-bonded adduct radical [-P-2S∸2S-P-]−. At room temperature, [-P-2S∸2S-P-]− formation is not observed by pulse radiolysis as the thermodynamic stability for [-P-2S∸2S-P-]− production is much less (ΔG° = −1.4 kcal/mol) owing to the positive entropy (40 cal K−1mol−1) on dissociation.
Neither -P2S• nor [-P-2S∸2S-P-]− was able to oxidize the guanine base in G-P(S−)=S, as well as in the mixture of dGuo and DETP(S−)=S, thereby showing that backbone-to-base hole transfer is not favored in DNA-phosphorodithioate.
A single phosphorodithioate in a long DNA strand can serve as a hole sink. However, this hole transfer process leads to the formation of backbone damage in the form of the dithiyl radical -P2S•. Recombination of -P2S• forms the neutral phosphorodithioate dimer [-P-2S-2S-P-] (reaction 6) with the disulfide (-S-S-) linkage. It is interesting to note that the plausible reaction of the thiyl radicals formed from amino acids containing thiol[21,45–47] with phosphorodithioate moiety could lead to S-S linkage which would hinder DNA replication or transcription processes. Such S-S linkage formation may constitute a key step involved in the mechanism of the phosphorodithioate-induced toxicity in human[48] and may interfere in electrochemical sequencing employing phosphorodithioate oligomers.
Experimental Section
Synthetic General methods for synthesis, ESR studies, pulse radiolysis, and theoretical studies are available in the Supporting Informations.
5’-O-Methoxyphosphorodithioyl-2’-deoxyguanosine sodium salt (G-P(S−)=S Na+):
A round bottom flask (25 mL) was charged with acyl-protected 2’-deoxyguanosine 1 (114 mg, 0.300 mmol (scheme 1, section B)), anhydrous pyridine (5 mL), and elemental sulfur (77 mg, 0.30 mmol). 2-Chloro-1,3,2-dithiaphospholane 2 (57 mg, 0.36 mmol, scheme 1) was added dropwise with stirring. The reaction mixture was stirred at room temperature for 12 h. The solvent was removed in vacuum and the residue was triturated with acetonitrile (10 mL). Undissolved sulfur was filtered off and the filtrate was concentrated in vacuum. The residue was dissolved in chloroform (2–3 mL) and subjected to a silica gel column chromatography (2.5 × 18 cm), methanol in chloroform (0 →10 %) to give 3 (121 mg, 0.228 mmol, 76%, scheme 1) as a white foam. A round bottom flask (25 mL) was charged with 3 (121 mg, 0.228 mmol) and anhydrous methanol (5 mL). DBU (52 μL, 0.34 mmol) was added and the reaction mixture was stirred for 6 h at room temperature. The solvent was removed by rotary evaporation, and the residue was dissolved in aqueous ammonia (5 mL, removal of the protecting groups). The solvent was removed by rotary evaporation. Silica gel column chromatography (2.5 × 18 cm), methanol in chloroform (2% →30%) gave nucleotide ammonium salt, which was converted into its sodium salt (Dowex 50 WX4 Na+) G-P(S−)=S Na+: (78.0 mg, 0.198 mmol, 86%). White foam. HRMS (ESI-TOF) [M+H]+ calculated for C11H16N5O5PS2 394.0403, found 394.0408. 1H NMR (DMSO-d6, δ): 10.64 (s, 1H, N-H), 8.14 (s, 1H, H-8), 6.47 (br s, 1H, H-1’), 6.15–6.10 (m, 1H, OH-3’), 4.38–4.35 (m, 1H, H-3’), 3.95–3.92 (m, 1H, H-4’), 3.91–3.88 (m, 1H, H-5’), 3.84–3.79 (m, 1H, H-5”), 3.37 and 3.34 (s, 3H, CH3), 2.58–2.52 (m, 1H, H-2’), 2.16–2.11 (m, 1H, H-2”); NMR (D2O, δ): 13C 158.03, 153.43, 150.83, 137.14, 114.74, 85.40, 83.71, 72.01, 65.24, 52.91, 38.96; 31P 114.89. RP-HPLC: Rt = 17.87 min.
Supplementary Material
Acknowledgements
AA, AK, and MDS thank NIH NCI (Grant R01CA045424), AA, MDS, and RD thank Research Excellence Fund (REF), and Center for Biomedical Research for support. RK, DK, and RD thank NIH NCI (CA111329) and Statutory Funds of CMMS PAS for support. We are also thankful to Dr. Hiroyuki Hayakawa (Yamasa Corporation, Biochemicals Division) for a generous supply of nucleosides. The National Science Foundation (NSF) instrumentation awards (CHE-1920110, CHE-0722547, and CHE-0821487) are also acknowledged.
References
- [1].(a) von Sonntag C, In Recent Trends in Radiation Chemistry edited by Rao BSM and Wishart J (World Scientific Publishing Co., Singapore, New Jersey, London, 2010, pp 543–562. [Google Scholar]; (b) Kanvah S, Joseph J, Schuster GB, Barnett RN, Cleveland CL, and Landman U, Acc. Chem. Res, 2010, 43, 280–287. [DOI] [PubMed] [Google Scholar]
- [2].(a) Peluso A, Caruso T, Landi A, and Capobianco A, Molecules, 2019, 24, 4044. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Fujitsuka M, and Majima T, Chem. Sci, 2017, 8, 1752–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Senthilkumar K, Grozema FC, Fonseca Guerra C, Bickelhaupt FM, Lewis FD, Berlin YA, Ratner MA, and Siebbeles LDA, J. Am. Chem. Soc, 2005, 127, 14894–14903. [DOI] [PubMed] [Google Scholar]
- [3].Sutherland BM, Bennett PV, Sidorkina O, and Laval J, Biochemistry, 2000, 39, 8026–8031. [DOI] [PubMed] [Google Scholar]
- [4].Burrows CJ, and Muller JG, Chem. Rev, 1998, 98, 1109–1151. [DOI] [PubMed] [Google Scholar]
- [5].Genereux JC, and Barton JK, Chem. Rev, 2010, 110, 1642–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Wagenknecht H–A, Ed. Charge Transfer in DNA: From Mechanism to Application. Wiley-VCH Verlag GmbH & Co. KGaA, Weiheim, 2005. [Google Scholar]
- [7].Shuster GB, Ed. Long Range Transfer in DNA I and II, Springer-Verlag, Berlin, Heidelberg, New York, 2004. [Google Scholar]
- [8].Sharma KKK, Tyagi R, Purkayastha S, and Bernhard WA, J. Phys. Chem. B, 2010, 114, 7672–7680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Adhikary A, Kumar A, Palmer BJ, Todd AD, and Sevilla MD, J. Am. Chem. Soc, 2013, 135, 12827–12838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ma J, Marignier J-L, Pernot P, Houée-Levin C, Kumar A, Sevilla MD, Adhikary A, and Mostafavi M, Phys. Chem. Chem. Phys, 2018, 20, 14927–14937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Adhikary A, Becker D, and Sevilla MD, In Applications of EPR in Radiation Research, edited by Lund A, A. and Shiotani M, Springer International Publishing: Heidelberg, New York, London, 2014, chapter 8, 299–352. [Google Scholar]
- [12].Giese B, and Wessely S, Chem. Commun, 2001, 2108–2109. [DOI] [PubMed] [Google Scholar]
- [13].Furrer E, and Giese B, Helvetica Chim. Acta, 2003, 86, 3623–3632. [Google Scholar]
- [14].Yang X,; Bassett SE, Li X, Luxon BA, Herzog NK, Shope RE, Aronsosn J, Prow TW, Leary JF, Kirby R, Ellington AD, and Gorenstein DG, Nucleic Acids Res, 2002, 30, e132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Yang X, Abeydeera ND, Liu F-W, and Egli M, Chem. Commun, 2017, 53, 10508–10511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Adhikary A, Kumar A, Heizer AN, Palmer BJ, Pottiboyina V, Liang Y, Wnuk SF, and Sevilla MD, J. Am. Chem. Soc, 2013, 135, 3121–3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Khanduri D, Adhikary A, and Sevilla MD, J. Am. Chem. Soc, 2011, 133, 4527–4537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Adhikary A, Malkhasian AYS, Collins S, Koppen J, Becker D, and Sevilla MD, Nucleic Acids Res, 2005, 33, 5553–5564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sevilla MD, Yan M, and Becker D, Biochem. Biophys. Res. Commun, 1988, 155, 405–410. [DOI] [PubMed] [Google Scholar]
- [20].Becker D, Swarts S, Champagne M, and Sevilla MD, Int. J. Radiat. Biol, 1988, 53, 767–786. [DOI] [PubMed] [Google Scholar]
- [21].von Sonntag C, Free-radical-induced DNA Damage and Its Repair; Springer-Verlag: Berlin, Heidelberg, 2006, 100–373. [Google Scholar]
- [22].Candeias LP, and Steenken S, J. Am. Chem. Soc, 1989, 111, 1094–1099. [Google Scholar]
- [23].Ma J, Denisov SA, Marignier J-L, Pernot P, Adhikary A, Seki S, and Mostafavi M, J. Phys. Chem. Lett, 2018, 9, 5105–5109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Abedinzadeh Z, Gardes-Albert M, and Ferradini C, Can. J. Chem, 1989, 67, 1247–1255. [Google Scholar]
- [25].Hofstetter D, Nauser T, and Koppenol WH, Chem. Res. Toxicol, 2010, 23, 1596–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Postal WS, Vogel EJ, Young CM, and Greenaway FT, J. Inorg. Biochem, 1985, 25, 25–33. [DOI] [PubMed] [Google Scholar]
- [27].Stec WJ, Grajkowski A, Karwowski B, Kobylańska A, Koziołkiewicz M, Misiura K, Okruszek A, Wilk A, Guga P, and Boczkowska M, J. Am. Chem. Soc, 1995, 117, 12019–12029. [Google Scholar]
- [28].Yang X, Curr. Protoc. Nucleic Acid Chem 2017, 70, 4.77.1–4.77.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Baraniak J, Kaczmarek R, and Stec WJ, Tetrahedron Lett, 2000, 41, 9139–9142. [Google Scholar]
- [30].Baraniak J, Kaczmarek R, Wasilewska E, and Stec WJ, Phosphorus Sulfur Silicon, 2002, 177, 1667–1670. [Google Scholar]
- [31].Baraniak J, Kaczmarek R, Korczyński D, and Wasilewska E, J. Org. Chem, 2002, 67, 7267–7274. [DOI] [PubMed] [Google Scholar]
- [32].Zhdanov RI, and Zhenodarova SM, Synthesis, 1975, 222–245. [Google Scholar]
- [33].Peake SC, Fild M, Schmutzler R, Harris RK, Nichols JM, and Rees RG, J. Chem. Soc., Perkin Trans 2, 1972, 380–385. [Google Scholar]
- [34].Okruszek A, Olesiak M, Krajewska D, Stec WJ, J. Org. Chem, 1997, 62, 2269–2272. [DOI] [PubMed] [Google Scholar]
- [35].Adhikary A, Kumar A, Becker D, and Sevilla MD, J. Phys. Chem. B, 2006, 110, 24171–24180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Adhikary A, Collins S, Khanduri D, and Sevilla MD, J. Phys. Chem. B, 2007, 111, 7415–7421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Adhikary A, Khanduri D, and Sevilla MD, J. Am. Chem. Soc, 2009, 131, 8614–8619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Adhikary A, Kumar A, Munafo SA, Khanduri D, and Sevilla MD, Phys. Chem. Chem. Phys, 2010, 12, 5353–5368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Adhikary A, Kumar A, and Sevilla MD, Radiat. Res, 2006, 165, 479–484. [DOI] [PubMed] [Google Scholar]
- [40].Close DM, In Radiation-induced Molecular Phenomena in Nucleic Acids: A Comprehensive Theoretical and Experimental Analysis, edited by Shukla MK, and Leszczynski J, Springer-Verlag, Berlin, Heidelberg, New York, 2008, 493–529. [Google Scholar]
- [41].Bernhard WA, In Radical and Radical Ion Reactivity in Nucleic Acid Chemistry, edited by Greenberg MM, John Wiley & Sons, Inc., New Jersey, 2009, chapter 2, 41–68. [Google Scholar]
- [42].Bernhard WA, and Close DM, DNA damage dictates the biological consequences of ionizing irradiation: the chemical pathways In Charged Particle and Photon Interactions with Matter Chemical, Physicochemical and Biological Consequences with Applications, edited by Mozumdar A, and Hatano Y, Marcel Dekker, Inc., New York, Basel, 2004, 431–470. [Google Scholar]
- [43].Rokhlenko Y, Cadet J, Geacintov NE, and Shafirovich V, J. Am. Chem. Soc, 2014, 136, 5956–5962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Shukla LI, Adhikary A, Pazdro R, Becker D, and Sevilla MD, Formation of 8-oxo-7,8-dihydroguanine-radicals in γ-irradiated DNA by multiple one-electron oxidations. Nucleic Acids Res, 2004, 32, 6565–6574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Surdhar PS, and Armstrong DA, J. Phys. Chem., 1986, 90, 5915–5917. [Google Scholar]
- [46].Surdhar PS, and Armstrong DA, J. Phys. Chem., 1987, 91, 6532–6537. [Google Scholar]
- [47].Madej E, and Wardman P, Arch. Biochem. Biophys., 2007, 462, 94–102. [DOI] [PubMed] [Google Scholar]
- [48]. https://cfpub.epa.gov/ncea/pprtv/documents/DiethylphosphorodithioateOO.pdf.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.