

Abstract
Gene expression programs change over time, differentiation and development and in response to stimuli. However, nearly all techniques for profiling gene expression in single cells do not directly capture transcriptional dynamics. Here, we present a method for combined single-cell combinatorial indexing and mRNA labelling (sci-fate), which uses combinatorial cell indexing and 4sU labeling of newly synthesized mRNA to concurrently profile the whole and newly synthesized transcriptome in each of many single cells. We used sci-fate to study the cortisol response in >6,000 single cultured cells. From these data, we quantified the dynamics of the cell cycle and of glucocorticoid receptor activation, and explored their intersection. Finally, we developed software to infer and analyze cell state transitions. We anticipate that sci-fate will be broadly applicable to quantitatively characterize transcriptional dynamics in diverse systems.
Editorial summary
Gene expression dynamics in single cells are tracked by labeling newly synthesized RNA in indexed cells.
During organismal development, as well as during myriad physiological and pathophysiological processes, individual cells traverse a manifold of molecularly and functionally distinct states. However, although experimental methods for profiling various aspects of single cell biology have recently proliferated, nearly all such methods deliver only a static snapshot of each cell. To address this in part, “pseudotime” methods computationally place individual cells along a continuous trajectory based on their transcriptomes1-6. However, pseudotime infers rather than directly measures transcriptional dynamics, is dependent on sufficient representation along the trajectory, and may fail to capture the detailed dynamics of individual cells (e.g. directionality, multiple superimposed potentials, etc.)7. In contrast, time-lapse microscopy can experimentally measure transcriptional dynamics, but is limited to visualization of a few marker genes in a few cells, and as such may be insufficient to decipher the complexity of many biological systems.
Here we describe a technique, sci-fate, to measure the dynamics of gene expression in large numbers of single cells and at the level of the whole transcriptome. In brief, we integrated protocols for labeling newly synthesized mRNA with 4-thiouridine (4sU)8,9 with single cell combinatorial indexing RNA-seq (sci-RNA-seq10). As a proof-of-concept, we applied sci-fate to a model system of cortisol response, characterizing expression dynamics in over 6,000 single cells. From these data, we quantify the dynamics of the transcription factor (TF) modules that underpin the cell cycle, glucocorticoid receptor activation, and other processes, and develop a framework for inferring the distribution of cell state transitions. The methods described here may be broadly applicable to quantitatively characterize transcriptional dynamics in diverse systems.
Overview of sci-fate
Briefly, sci-fate relies on the following steps (Fig. 1a): (i) Cells are incubated with 4-thiouridine (4sU), a thymidine analog, to label newly synthesized RNA11-17. (ii) Cells are harvested, fixed with 4% paraformaldehyde, and then subjected to a thiol(SH)-linked alkylation reaction which covalently attaches a carboxyamidomethyl group to 4sU by nucleophilic substitution8. (iii) Cells are distributed by dilution to 4 x 96 well plates. The first sci-RNA-seq molecular index is introduced via in situ reverse transcription (RT) with a poly(T) primer bearing both a well-specific barcode and a degenerate unique molecular identifier (UMI). During first strand cDNA synthesis, modified 4sU templates guanine rather than adenine incorporations. (iv) Cells from all wells are pooled and then redistributed by fluorescence-activated cell sorting (FACS) to multiple 96-well plates. (v) Double-stranded cDNA is synthesized. After Tn5 transposition, cDNA is PCR amplified via primers recognizing the Tn5 adaptor on the 5’ end and the RT primer on the 3’ end. These primers also bear a well-specific barcode that introduces the second sci-RNA-seq molecular index. (vi) PCR amplicons are subjected to massively parallel DNA sequencing. As with other sci- methods18-26, most cells pass through a unique combination of wells, such that their contents are marked by a unique combination of barcodes that can be used to group reads derived from the same cell. (vii) The subset of each cell’s transcriptome corresponding to newly synthesized transcripts is distinguished by T→ C conversions in reads mapping to mRNAs (Methods).
Fig. 1. Sci-fate enables joint profiling of whole and newly synthesized transcriptomes.
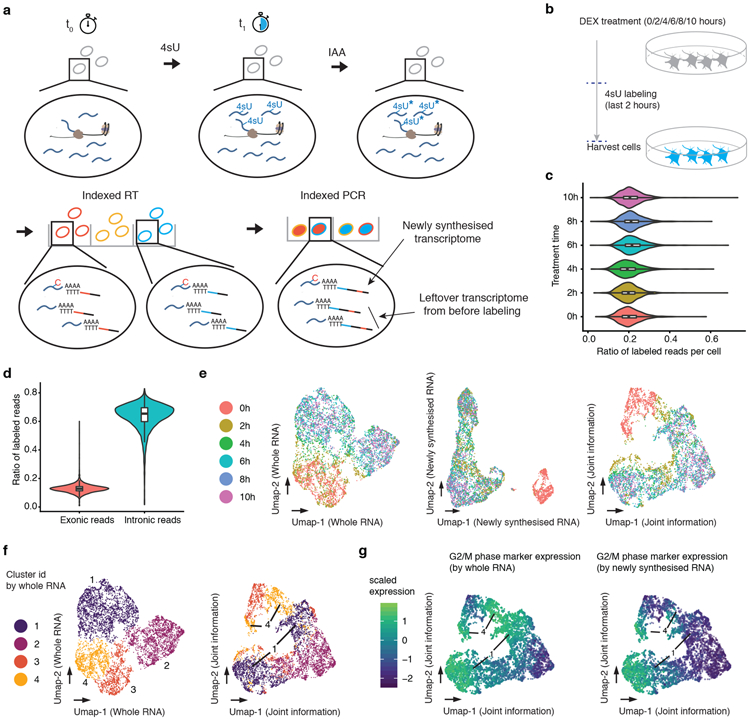
(a) The sci-fate workflow. Key steps are outlined in text. (b) Experimental scheme. A549 cells were treated with dexamethasone for varying amounts of time ranging from 0 to 10 hrs. Cells from all treatment conditions were labeled with 4sU two hours before harvest for sci-fate. (c) Violin plot showing the fraction of 4sU labeled reads per cell for each of the six treatment conditions. Cell number n = 1,054 (0h), 1,049 (2h), 949 (4h), 1,262 (6h), 1,041 (8h), and 1,325 (10h). For all violin plots in this figure: thick lines in the middle, medians; upper and lower box edges, first and third quartiles, respectively; whiskers, 1.5 times the interquartile range; circles, outliers. (d) Violin plot showing the fraction of 4sU labeled reads per cell (n = 6,680), split out by the subsets that map to exons vs. introns. (e) UMAP visualization of A549 cells (n = 6,680) based on their whole transcriptomes (left), newly synthesized transcriptomes (middle) or with joint analysis, i.e. combining the top PCs from each (right). (f) Same as left and right of panel e, respectively, but colored by cluster id from UMAP based on whole transcriptomes. (g) Same as right of panel e, but colored by normalized expression of G2/M marker genes by their overall expression levels (left) or their levels of newly synthesized transcripts (right). UMI counts for these genes are scaled by library size, log-transformed, aggregated and then mapped to Z-scores.
For quality control, we first tested sci-fate with a mixture of HEK293T (human) and NIH/3T3 (mouse) cells under four conditions: with vs. without 4sU labeling (200 nM, 6 hrs), and with vs. without the thiol(SH)-linked alkylation reaction. The resulting transcriptomes were overwhelmingly species-coherent (>99% purity for both human and mouse cells, 2.7% collisions; Supplementary Fig. 1ab) with similar mRNA recovery rates (overall median 21,342 unique molecular identifiers (UMIs) per cell; Supplementary Fig. 1c). However, only with 4sU labeling and thiol(SH)-linked alkylation did we observe a substantial proportion of reads bearing T → C conversions, i.e. newly synthesized transcripts (46% and 31% for treated human and mouse cells, respectively, vs. 0.8% for untreated cells; Supplementary Fig. 1d). The aggregated transcriptomes of cells derived from sci-fate and conventional sci-RNA-seq were highly correlated (Spearman’s correlation r = 0.99; Supplementary Fig. 1ef), suggesting that short-term labeling and conversion do not substantially bias transcript counts.
Profiling of transcriptome dynamics in cortisol response
To investigate the transcriptional dynamics of cortisol response27, we applied sci-fate to an in vitro model wherein dexamethasone (DEX), a synthetic mimic of cortisol, activates glucocorticoid receptor (GR), which binds to thousands of locations across the genome and rapidly alters gene expression28-31. Specifically, we treated lung adenocarcinoma-derived A549 cells for 0, 2, 4, 6, 8 or 10 hrs with 100 nM DEX. In each condition, cells were incubated with 4sU (200 nM) for the two hours immediately preceding harvest. We then performed a 384 x 192 sci-fate experiment (Fig. 1b). Each of the six conditions was represented by 64 wells during the first round of indexing, such that all samples could be processed in a single sci-RNA-seq experiment to minimize batch effects.
After filtering out low quality cells, potential doublets and a small subgroup of differentiated cells (Methods), we obtained single cell profiles for 6,680 cells (median 26,176 UMIs corresponding to mRNAs detected per cell). A median of 20% of mRNA UMIs were labeled per cell (Fig. 1c; Supplementary Fig. 2a-c). The proportion of newly synthesized mRNAs was markedly higher in reads mapping to intronic (65%) vs. exonic (13%) regions (p-value < 2.2e-16, two-sided Wilcoxon signed-rank test; Fig. 1d; Supplementary Fig. 2de), consistent with the expectation that the intronic reads are more likely to have been recently synthesized. We also compared intronic reads and newly synthesised mRNA for RNA velocity analysis32 and observed a subjectively consistent picture, suggesting they capture similar information (Supplementary Fig. 2f).
In exploring these data, we first asked whether the newly synthesized vs. whole transcriptome data convey identical or distinct information with respect to cell state. Performing dimensionality reduction with Uniform Manifold Approximation and Projection (UMAP)33 on whole transcriptomes failed to separate DEX untreated (0 hrs) vs. treated (2+ hrs) cells (Fig. 1e, left, Supplementary Fig. 2g). In contrast, applying UMAP to the newly synthesized subset of the single cell transcriptomes readily separated DEX untreated vs. treated cells (Fig. 1e, center). These patterns are likely a consequence of the fact that in DEX treated cells, the newly synthesized transcriptome more faithfully reflects the GR response itself. Illustrative of this, the classic markers for GR response, FGD428 and FKBP534, exhibited the highest fold induction in comparisons of the newly synthesized transcriptome at 0 hrs vs. 2 hrs, but the magnitude of their induction was dampened in comparisons of the whole transcriptome between the same time points (Supplementary Fig. 2hi; Supplementary Table 1).
To jointly make use of the information conveyed by the whole and newly synthesized transcriptomes, we combined their top principal components (PCs) for UMAP analysis. This approach separates cells that had experienced no (0 hrs), recent (2 hrs) or extended (4+ hrs) DEX treatment (Fig. 1e, right). With this joint approach, cells corresponding to two clusters defined by analysis of whole transcriptomes (clusters 1 & 4; Fig. 1f, left) each split into two groups (Fig. 1f, right). Examining the levels of newly synthesized mRNAs corresponding to cell cycle markers35, one pair of these new groups corresponds G2/M phase cells (high levels of both overall and newly synthesized G2/M markers), and the other to early G0/G1 phase cells (high levels of overall but low levels of newly synthesized G2/M markers) (Fig. 1g; Supplementary Fig. 2jk). Of note, cells from the 2 hr time point exhibited a distribution of cell cycle states according to this joint information (Fig. 1eg). Overall, these analyses illustrate how joint analysis of the newly synthesized and whole components of single cell transcriptomes can recover cell state information that is not easily obtained from the whole transcriptomes alone.
TF module activity decomposes cellular processes
Multiple dynamic gene regulatory processes are concurrently underway in this in vitro system -- minimally, the GR response and the cell cycle. We speculated that these might be disentangled, and their intersection probed, by first identifying the TF modules driving new mRNA synthesis in relation to each process.
TF modules, comprising candidate links between TFs and their regulated genes, were identified as follows. For each gene, across the 6,680 cells, we computed correlations between the levels of newly synthesized mRNA for that gene and the overall expression level of each of 859 expressed TFs, using LASSO (least absolute shrinkage and selection operator) regression. Out of 1,086 links involving TFs characterized by ENCODE36, 807 were validated by TF binding sites near the genes’ promoters36, a 4.3-fold enrichment relative to background expectation (odds ratio for validation = 2.89 for LASSO-identified links vs. 0.67 for background, p-value < 2.2e-16, two-sided Fisher’s Exact test). These covariance links were further filtered by ChIP-seq binding37, and supplemented with additional covariance links validated by motif38 enrichment analysis (Fig. 2a; Methods). Altogether, we identified 986 links between 29 TFs and 532 genes (Supplementary Fig. 3ab; Supplementary Table 2). As a control, we permuted the cell order of the cell x TF expression matrix (Ti) (Methods), and then repeated the analysis. No links were identified after permutation. Some of the identified TF-gene regulatory relationships are supported by a manually curated database of TF networks (TRRUST39), e.g. E2F1 (top enriched TF of E2F1-linked genes = E2F1, adjusted p-value = 8e-7)40, NFE2L2 (top enriched TF of NFE2L2-linked genes = NFE2L2, adjusted p-value = 0.003)40, and SREBF2 (top enriched TF of SREBF2-linked genes: SREBF2, adjusted p-value = 0.0006)40.
Fig. 2. Characterizing TF modules driving concurrent, dynamic gene regulatory processes in populations of single cells.
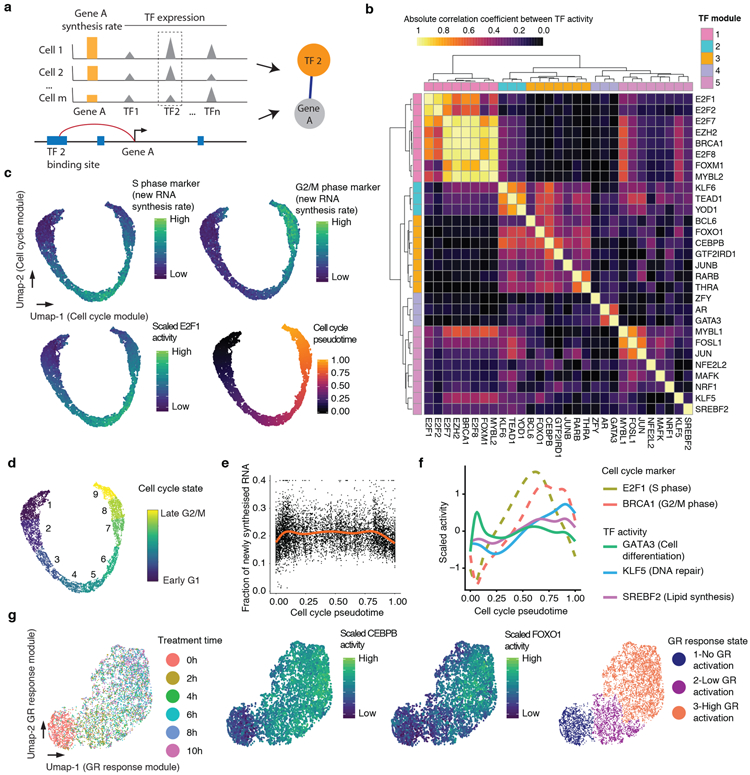
(a) Schematic of approach used to identify links between TFs and their regulated genes. (b) Heatmap showing the absolute Pearson’s correlation coefficient between the activities of pairs of TFs (Cell number n = 6,680). (c) UMAP visualization of A549 cells (n = 6,680) based on the activity of cell cycle-related TF module, colored by levels of newly synthesized mRNA corresponding to S phase markers (top left), G2/M phase markers (top right), and E2F1 activity (bottom left). Bottom right panel is colored by pseudotime based on point position on the principal curve estimated by princurve package65. (d) Same as panel c, but colored according to nine cell cycle states defined by unsupervised clustering analysis. In broad terms, cell cycle states 1-3 correspond to G1 phase, 4-6 to S phase, and 7-9 to G2/M phase. (e) Scatter plot showing the changes in the fraction of newly synthesized mRNA in each cell (n = 6,680) along cell cycle progression. The red line is the smoothed curve estimated by the geom_smooth function66. (f) Similar to panel e, but showing smoothed activity of selected TF modules as a function of cell cycle pseudotime. (g) UMAP visualization of A549 cells (n = 6,680) based on the activity of GR response-related TF module, colored by DEX treatment time (left), CEBPB or FOXO1 activity (middle panels), or cluster id from unsupervised clustering (right). Throughout figure, to calculate TF module activity, newly synthesized UMI counts for genes linked to module-assigned TFs are scaled by library size, log-transformed, aggregated and then mapped to Z-scores.
The 29 TFs with one or more gene links included well-established GR response effectors such as CEBPB41, FOXO142, and JUNB43 (Supplementary Fig. 3cd). This group also included several TFs not previously implicated in GR response, including YOD1 and GTF2IRD1, both of which exhibited greater expression and activity in DEX-treated cells (Supplementary Fig. 3ef). The main TFs driving cell cycle progression were also identified, e.g. E2F1, E2F2, E2F7, BRCA1, MYBL244. Notably, the expression levels of TFs such as E2F1 were more highly correlated with the levels of newly synthesized than overall target gene mRNAs (Supplementary Fig. 3g). We also observed regulatory links corresponding to TFs involved in cell differentiation such as GATA345, mostly expressed in a subset of quiescent cells, as well as TFs involved in oxidative stress response such as NRF146 and NFE2L2 (NRF2)47.
We calculated a measure of each of these 29 TFs’ activities in each cell, based on the normalized aggregation of the levels of newly synthesized mRNA for all of its target genes. We then computed the absolute correlation coefficient between each pair of TFs with respect to their activity across the 6,680 cells. Hierarchical clustering of these pairwise correlations identified several major TF modules, i.e. sets of TFs that appear to be regulating the same process (Fig. 2b). A first large TF module corresponds to all cell cycle-related TFs in the set, e.g. E2F1, FOXM144. A second large TF module corresponds to GR response-related TFs, e.g. FOXO1, CEBPB, JUNB, RARB41-43. The other modules include one corresponding to GR-activated G1/G2/M phase cells (KLF6, TEAD1, YOD1; Supplementary Fig. 3h), and another corresponding to likely-differentiating GR-activated G1 phase cells (GATA3, AR; Supplementary Fig. 3h)45,48. Additional TFs or TF modules appear to capture other processes that are heterogeneous in this population of cells, including NRF1 and NFE2L2 for stress response/apoptosis (top enriched pathway of NFE2L2-linked genes: ferroptosis, adjusted p-value = 1e-5)40,46,47,49; KLF5 for DNA damage repair (top pathway: ATM signaling, adjusted p-value = 0.018)40,50; and SREBF2 for cholesterol homeostasis (top pathway: “SREBF and miR33 in cholesterol and lipid homeostasis”, adjusted p-value = 9e-6)40,51.
To assign cell cycle states to individual cells, we first ordered cells by their cell cycle-linked TF module activity. This resulted in a smooth, nearly circular trajectory, in which the levels of newly synthesized mRNA corresponding to known cell cycle markers was dynamic (Fig. 2c)35. We observed a gap between late G2/M phase and early G1 phase, consistent with the dramatic cell state change during cell division. By unsupervised clustering of the activities of individual TFs within the cell cycle-linked TF module, we identified 9 cell cycle states spanning the early, middle and late cell cycle phases (Fig. 2d). Early G1 and late G2/M phase cells exhibited decreased synthesis of new RNA relative to other parts of the cell cycle, possibly due to chromosomal condensation during mitosis (Fig. 2e)52-54. Other (i.e. non-cell-cycle) TF modules exhibited different dynamics in relation to cell cycle progression (Fig. 2f). For example, GATA3 activity peaks in early G1 phase, potentially reflecting a cell differentiation pathway distinct from cell cycle reentry45. In contrast, the modules of KLF5 and SREBF2, associated with DNA repair and lipid homeostasis, respectively, exhibited greater activity from S to G2 phase, possibly related to roles in DNA replication and cell division, respectively55.
Similarly, the cells can also be ordered into a smooth trajectory based on GR response-linked TF module activity. As expected, this trajectory correlates well with DEX treatment time, as well as the activity of GR response-related TFs (Fig. 2g). By unsupervised clustering of the activities of individual TFs within the GR response-linked TF module, we identified GR response states corresponding to no, low and high levels of activation (Fig. 2g).
We next sought to explore the intersection of the 9 cell cycle states (Fig. 2d) and the 3 GR response states (Fig. 2g). Each of 27 possible state combinations were represented by some cells, with the smallest group corresponding to 1.1% of the overall dataset (n = 74 cells, intersection of “early G2/M” cell cycle state and “no GR activation” state; Supplementary Fig. 4ab). Although we observe several TF modules that appear specific to certain intersections of the cell cycle and GR response (KLF6/TEAD1/YOD1 and GATA3/AR, discussed above), several observations support the conclusion that the dynamics of the cell cycle and GR response operate largely independently. First, we observe minimal correlation between the activities of the primary TF modules for cell cycle and GR response across the 6,680 cells (Pearson’s correlation r = 0.004; Fig. 2b). Second, the relative proportions of each of the 27 possible state combinations are readily predicted by proportions of cell cycle and GR response states, i.e. with no interaction term (Supplementary Fig. 4b).
Inferring single cell transcriptome dynamics with sci-fate
We next sought to develop a strategy to use sci-fate data to infer the past transcriptional state of each cell, i.e. at the onset of 4sU labeling, which might in turn allow us to relate cells derived from different time points. The inference of past transcriptional state requires knowledge of two parameters -- first, the detection rate of newly synthesized transcripts (i.e. the proportion of newly synthesised transcripts containing one or more detected T > C mutations), and second, the degradation rate of each mRNA species. Below, we discuss how each of these parameters can be estimated directly from the sci-fate data generated for this experiment. A more detailed consideration is provided in the Methods.
Under the assumption that mRNA degradation rates are not affected by DEX treatment (this assumption is validated further below), it is relatively straightforward to estimate sci-fate’s detection rate for newly synthesized transcripts. Each sci-fate transcriptome in this dataset consists of two components -- the newly synthesized transcriptome, whose detection rate we hope to estimate, and the ‘leftover’ transcriptome, i.e. transcripts that were present at the onset of 4sU labeling, minus any degradation over the course of the two hours. Comparing the 0 hr (untreated) and 2 hr DEX treatment groups, we expect that their leftover bulk transcriptomes (at the onset of 2 hr 4sU labeling) should be identical, as should sci-fate’s detection rate for newly synthesized transcripts. As such, an equation can be constructed relating the transcriptomes of these treatment groups to one another (Methods). For each of 186 genes exhibiting the largest differences in new transcription between the two conditions, we solved this equation to estimate sci-fate’s detection rate. As these estimates were largely consistent across genes and robust to sequencing depth (Supplementary Fig. 5a-e), we used their median value (82%) as sci-fate’s estimated detection rate for all subsequent analyses.
We next sought to estimate the degradation rate of each mRNA species. As noted above, the bulk transcriptome at each time point in our experiment can be decomposed into the newly synthesized transcriptome and the leftover transcriptome. Furthermore, the leftover transcriptome should equal the bulk transcriptome from the time point two hours earlier, provided that we correct for mRNA degradation over that interval. From these assumptions, an equation can be constructed and solved to estimate the mRNA half-life of each gene, which we did independently for each 2 hr interval of the experiment (Methods; Supplementary Table 3). As a first quality check, we simply compared these estimated mRNA degradation rates between time points, and found them to be both consistent and robust to sequencing depth (Supplementary Fig. 5fg; median Pearson’s r = 0.92). As a second quality check, we compared them to orthogonally generated estimates of mRNA half-lives from the literature9. Despite the fact that different technologies were used on different cell lines (A549 vs. K562), the estimates of mRNA half-lives were reasonably consistent (Supplementary Fig. 5h; Pearson’s r = 0.76). Of note, the absolute differences in estimated mRNA half-lives between sci-fate and previous techniques could be due to the use of different cell lines or systematic differences between the techniques.
With these parameters in hand, we next estimated the past transcriptional state of each cell in our dataset (Methods; Supplementary Fig. 6ab), and sought to use these estimated states to link individual cells to one another across time points (Fig. 3a). Specifically, for each cell B (e.g. a cell from the 2 hr time point), we used a recently developed alignment method35 to identify a cell A profiled at an earlier time point (e.g. a cell from the 0 hr time point), wherein A’s current state was closest to B’s estimated past state. In this framework, A can be regarded as the parent state of B. Applying this strategy to each of the five intervals comprising our experiment, we constructed a set of linkages spanning the entire dataset and time course (Fig. 3b).
Fig. 3. Inferring single cell transcriptional dynamics with sci-fate.
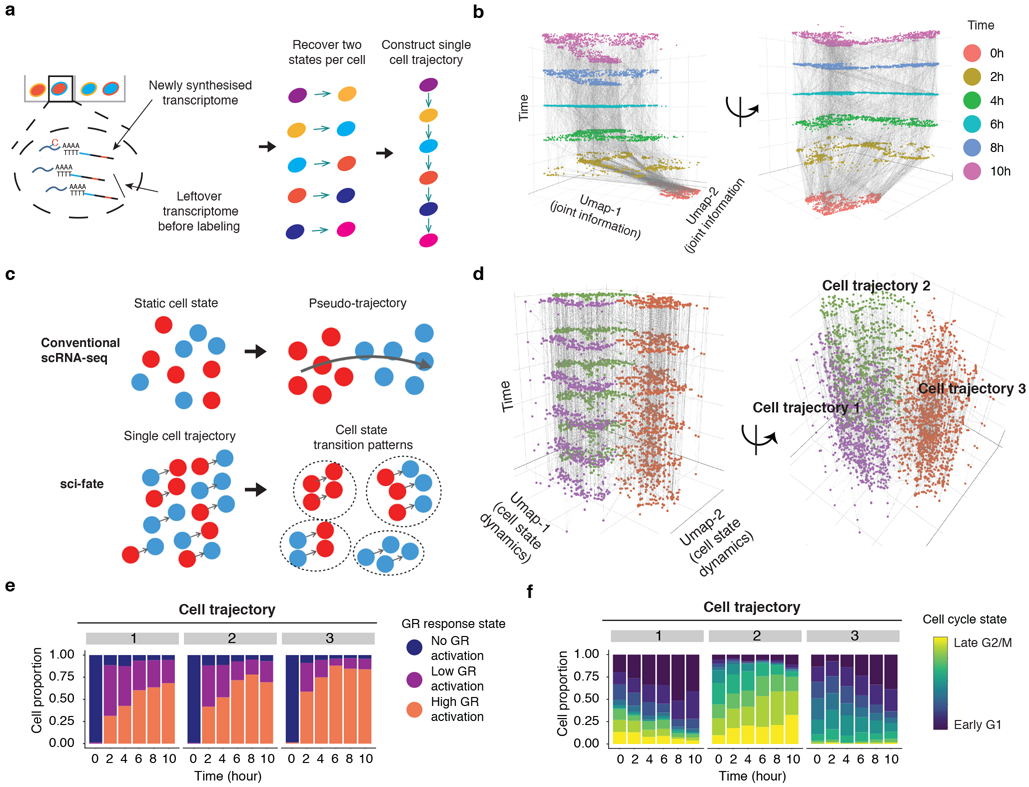
(a) Schematic of approach for linking cells based on estimated past transcriptional states to reconstruct single cell transition trajectories. (b) 3D plot of all cells (cell number n = 6,680). The x and y coordinates correspond to the joint information UMAP space shown in the rightmost panel of Fig. 1e. The z coordinate as well as colors correspond to DEX treatment time. Linked parent and child cells are connected with grey lines. (c) Schematic comparing conventional scRNA-seq and sci-fate for cell trajectory analysis. (d) Similar to panel b, except the x and y coordinates correspond to the UMAP space based on the single cell transition trajectories across the six time points (cell number n = 6,680). (e-f) Barplots showing the contributions of the 3 GR response states (e) and the 9 different cell cycle states (f) to each of three cell trajectory clusters.
A key contrast with conventional pseudotime is that with sci-fate, each cell is now characterized not only by its present state, but also by specific linkages to a series of distinct cells matching its predicted past and/or future states (Fig. 3c). To evaluate whether these mini-trajectories contain structure, we applied UMAP and unsupervised clustering, which resulted in three distinct trajectory clusters (Fig. 3d). To annotate these, we checked the proportions of each of the aforementioned three GR response states and nine cell cycle states in each of them, as a function of time. As expected, all three trajectories exhibited a rapid transition from no GR activation to low/high GR activation (Fig. 3e). However, each trajectory appears to correspond to a different starting point with respect to the cell cycle (Fig. 3f). Trajectory 1 corresponds to cells that transition from G2/M to G1 phase over the course of the 10 hr experiment. Trajectory 2 corresponds to cells that transition from late S phase to G2/M phase over the course of the experiment. Trajectory 3 corresponds to cells that transition from G1 to either S phase or G1 arrest over the course of the experiment. The inference of G1 arrest subsequent to DEX treatment is consistent with the dynamics of cell state proportions in this experiment as well as with previous research56,57. As a control, we clustered the cell state transition trajectories by simply aligning neighboring time points without knowledge of newly synthesized mRNA; this failed to recover expected cell cycle dynamics (Supplementary Fig. 6c).
Inferred cell transitions recapitulate expected dynamics
We next sought to evaluate whether the distribution of cell state transitions inferred by sci-fate are consistent with the expected dynamics. We assigned each cell into one of the 27 states (3 GR response x 9 cell cycle states) and computed a cell state transition network (Fig. 4a), with the assumption that the cell state transitions in this experiment follow a Markov process with transition probabilities that do not change over time. This assumption is validated in part by the observation that the distribution of predicted cell state transitions estimated from the last three time intervals (4-6 hrs, 6-8 hrs, 8-10 hrs) are highly correlated with each other (Supplementary Fig. 7a) despite varied cell state proportions at 4 hrs vs. the later time-points (Supplementary Fig. 7b). Consistent with DEX treatment, transitions are highly biased from G1 to S, S to G2/M, and G2/M to G1 phase of the cell cycle (Fig. 4a). As a control analysis, cell state transition networks were similarly derived, but based either on randomly permuted cell state transition links, or on links derived from mature mRNAs only; these both failed to recapitulate the expected pattern of cell cycle transitions (Supplementary Fig. 7c).
Fig. 4. Constructing a state transition network for GR response and cell cycle.
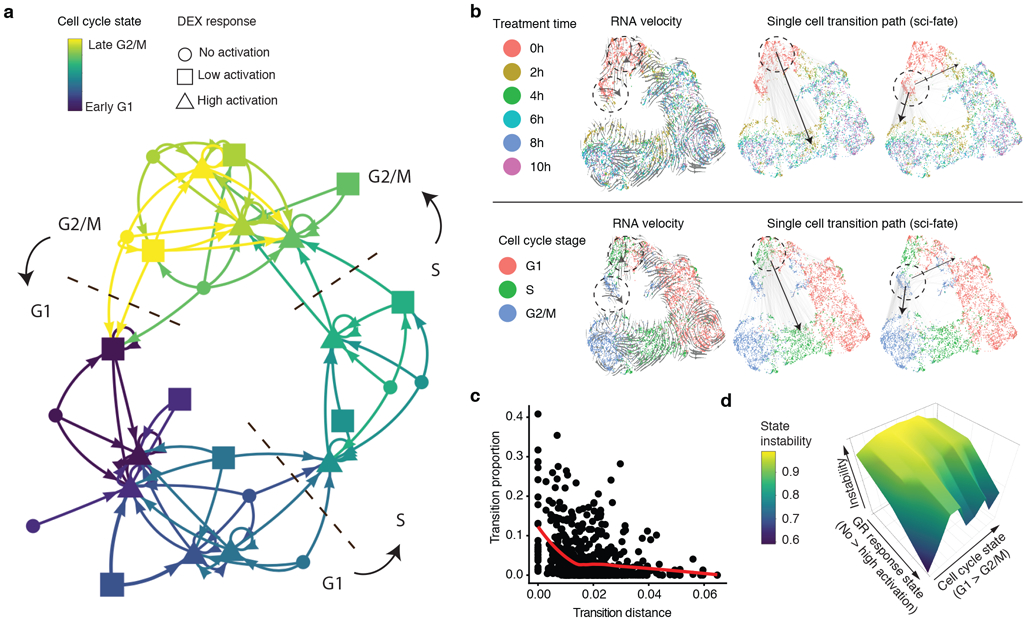
(a) Cell state transition network. The nodes are 27 cell states characterized by combinations of cell cycle and GR activation states. The links represents frequent cell state transition trajectories (transition proportion > 10%) between cell states. This threshold for defining a link corresponds to approximately two standard deviations from the mean transition proportion calculated after permuting cell transition links (n = 729). (b) The x and y coordinates correspond to the joint information UMAP space shown in the rightmost panel of Fig. 1e, colored by DEX treatment time (top) or inferred cell cycle state (bottom). Grey lines represent inferred cell state transition links between parent and child cells (left: cell state transition links starting from cells at the S phase and no GR activation stage (Link number n = 433); right: cell state transition links starting from cells at G2/M phase and no GR activation stage (Link number n = 365)). Black arrows show main cell state transition directions. (c) Scatter plot showing the relationship between transition distance (Pearson’s distance) and transition proportion (n = 729), together with the red LOESS smoothed line by ggplot266. (d) 3D plot showing the cell state stability landscape. X-axis represents GR response states (from no to low to high activation state). Y-axis represents the cell cycle states ordered from G1 to G2/M. Z-axis represents cell state instability, defined as the proportion of cells inferred to be moving out of a given state between time points.
The 27 states shown in Fig. 4a each correspond to subsets of cells whose transcriptomes are similar, making use of the joint information provided by distinguishing between old (> 2 hrs) vs. new (< 2 hrs) transcripts. Importantly, the distribution of transitions are inferred, rather than explicitly known, but supported by the fact that they correspond to expected phenomena, e.g. irreversible progression through GR response, as well as irreversible progression through the cell cycle. As examples, S phase cells without GR activation (0 hrs treatment) mostly transit into S phase cells with GR activation (2 hrs treatment), while G2/M phase cells with no GR activation (0 hrs treatment) mostly transit to G2/M or G1 phase cells with GR activation (2 hrs treatment) (Fig. 4b). For comparison, overlaying the same UMAP coordinates with RNA velocity vectors32 recovered similar patterns, but only when treatment time information was incorporated into the RNA velocity analysis (Supplementary Fig. 7d).
Can we use this framework to better understand the characteristics of transcriptional states that govern their dynamics? As a first approach, we calculated the pairwise Pearson’s distance between the aggregated transcriptomes of each of the 27 states. As expected, the greater the distance between any pair of states, the lower the proportional representation of that transition in the network (Spearman’s correlation coefficient = −0.38; Fig. 4c). As a second approach, we computed “instability” as the proportion of cells inferred to be moving out of a given state between time points (Fig. 4d). As expected, states corresponding to no GR activation were the least stable by this metric. Furthermore, amongst high GR activation states, states corresponding to early G1 were the most stable. These representations of the data are consistent with the transition network, wherein the states corresponding to high GR activation and early G1 are a frequent “destination” of all nearby states (purple triangles in Fig. 4a).
Discussion
Sci-fate captures information analogous to RNA velocity32, which distinguishes ‘older’ vs. ‘newer’ transcripts based on their splicing status. On one hand, RNA velocity is more straightforward than sci-fate, as it makes use of information that is indirectly captured by many single cell profiling technologies, while sci-fate requires 4sU labeling steps that cannot necessarily be used in all contexts. On the other hand, sci-fate lends itself to experimental control in a way that RNA velocity does not, as the timing and length of 4sU labeling can be specified whereas with RNA velocity it is a product of endogenous splicing dynamics. Furthermore, as we show, an experimental design that couples the labeling of newly synthesized mRNA to a time series enables the quantitative analysis of cells with complex transcriptional histories and futures.
While our manuscript was under review, two methods directed at the same goal, scSLAM-seq and NASC-seq, were reported58,59. Although there are similarities including the labeling strategy, we note major differences with respect to performance, accuracy and scalability: (1) Because sci-fate uses combinatorial indexing, we successfully measured newly synthesized mRNA in >6,000 cells in one experiment, compared with <200 cells for scSLAM-seq or NASC-seq. Given that sci-fate is easily adaptable to three level combinatorial indexing26, it should already be possible to profile newly synthesized mRNA is >1 million cells per experiment. (2) sci-fate costs <$0.20 per cell for library preparation with two-level indexing, and <$0.01 per cell with three-level indexing26. By comparison, both scSLAM-seq and NASC-seq utilize smart-seq which costs ~$11 per cell for library preparation60. On a related point, sci-fate required an order of magnitude fewer raw reads per cell (~200,000 sci-fate vs. ~2 million with scSLAM-seq), but achieved a greater number of genes detected per cell. (3) A key feature of sci-fate is that we performed in situ 4sU chemical conversion in bulk, fixed cells, resulting in a high reaction efficiency and low mRNA loss. In contrast, scSLAM-seq and NASC-seq require extracting mRNA from each cell followed by bead-based purification and chemical conversion. As a result, sci-fate exhibits higher efficiency to detect low abundance transcripts (median 6,500 genes detected per cell with sci-fate vs. ~4,000 with scSLAM-seq, despite of 1/10 of the raw sequencing depth). Furthermore, sci-fate exhibits a higher detection rate of newly synthesised mRNA (82% in sci-fate vs. <50% in scSLAM-seq). (4) The signal-to-noise ratio (labeled vs. unlabeled cells) of sci-fate is 38- to 58-fold, compared with only ~10-fold for scSLAM-seq or NASC-seq. This is partly due to the fact that the sci-fate library preparation is strand-specific, whereas smart-seq is not. (5) sci-fate enables direct counting of newly synthesised vs. pre-existing mRNA via 3’ tagged unique molecular identifiers (UMIs)61, which are used by neither scSLAM-seq nor NASC-seq. Additional advantages of sci-fate include compatibility with fixed cells and the ability to concurrently process multiple independent biological samples within a single experiment. Finally, it is notable that in situ 4sU chemical conversion requires cell permeabilization and, at least in our experience, PFA fixation, neither of which is straightforward to introduce on droplet-based scRNA-seq platforms such as 10x Genomics.
We note that while sci-fate enables quantification of mRNA synthesis in single cells, we remain in need of methods for measuring mRNA degradation rates in single cells. Related to that, our simplifying assumption that gene-specific degradation rates are constant across our DEX time course might not be a good choice in other systems. Specifically, in systems where the gene-specific degradation rates are expected or observed to substantially vary over time, these should be estimated for each time interval separately.
Sci-fate can be broadly applied to most in vitro systems to quantitatively characterize cell state dynamics within short time windows (e.g. one to several hours). For even shorter time frames, a concern is that signal-to-noise will drop as the rate of labeling falls towards the background rate of 0.8%. For longer time frames, a time series approach can be adopted as in the main experiment described here.
A major limitation of sci-fate is that 4sU labeling experiments are generally performed within in vitro cell culture models. However, recent studies have shown that 4sU can be used in conjunction with transgenic UPRT-expressing mice to stably label cell type-specific nascent RNA transcription in vivo62-64, suggesting that sci-fate, with further optimizations to enhance 4sU incorporation and detection rate, can potentially be used to profile single cell transcriptional dynamics in vivo and at scale.
Online Methods:
Mammalian cell culture
All mammalian cells were cultured at 37°C with 5% CO2, and were maintained in high glucose DMEM (Gibco cat. no. 11965) for HEK293T and NIH/3T3 cells or DMEM/F12 medium for A549 cells, both supplemented with 10% FBS and 1X Pen/Strep (Gibco cat. no. 15140122; 100U/ml penicillin, 100 μg/ml streptomycin). Cells were trypsinized with 0.25% typsin-EDTA (Gibco cat. no. 25200-056) and split 1:10 three times per week.
Sample processing for sci-fate
For HEK293T and NIH/3T3 cells, cells were incubated with 200uM 4sU for 6 hours before cell harvest. A549 cells were treated with 100 nM DEX for 0 hrs, 2 hrs, 4 hrs, 6 hrs, 8 hrs or 10 hrs. Cells in all treatment conditions were incubated with 200uM 4sU for the last two hours before cell harvest. Of note, an excessively long labeling time (the extreme of which results in all transcripts being labeled) may result in information loss. Through a test experiment on HEK293T cells, we found that the transcriptome degradation rate is around 10% per hour for each cell. We then selected two hours as the labeling time window, such that about 80% of detected transcriptomes per cell would be previously synthesized and usable to infer previous cell state. In theory, a shorter labeling time enables more accurate cell past state inference but also requires sampling more time points to cover a continuous process with a given time interval (i.e. 10 hours in our DEX treatment experiment). A short labeling time would also potentially be affected by greater noise. In our data, the background labeling rate (“labeled” reads ratio in non-labeled cells) is 0.8%. Given that 2 hours of labeling results in detection of ~20% of transcripts as newly synthesized, to keep at least a 10:1 signal-to-noise ratio, the minimum labeling period should be at least 50 minutes.
All cell lines (A549, HEK293T and NIH/3T3 cells) were trypsinized, spun down at 300xg for 5 min (4°C) and washed once in 1X ice-cold PBS. All cells were fixed with 4ml ice cold 4% paraformaldehyde (EMS) for 15 min on ice. After fixation, cells were pelleted at 500xg for 3 min (4°C) and washed once with 1ml PBSR (1 x PBS, pH 7.4, 1% BSA, 1% SuperRnaseIn, 1% 10mM DTT). After wash, cells were resuspended in PBSR at 10 million cells per ml, and flash frozen and stored in liquid nitrogen. Paraformaldehyde fixed cells were thawed on 37°C water bath, spun down at 500xg for 5 min, and incubated with 500ul PBSR including 0.2% Triton X-100 for 3min on ice. Cells were pelleted and resuspended in 500ul nuclease free water including 1% SuperRnaseIn. 3ml 0.1N HCl were added into the cells for 5min incubation on ice 24. 3.5ml Tris-HCl (pH = 8.0) and 35ul 10% Triton X-100 were added into cells to neutralize HCl. Cells were pelleted and washed with 1ml PBSR. Cells were resuspended in 100ul PBSR. 100ul PBSR with fixed cells were incubated with mixture including 40ul Iodoacetamide (IAA, 100mM), 40ul sodium phosphate buffer (500mM, pH = 8.0), 200ul DMSO and 20ul H2O, at 50°C for 15min. The reaction was quenched by 8ul DTT (1M) and 8.5ml PBS67. Of note, the cell lost rate is high (> 95%) in the chemical conversion and centrifugation step. Cells were pelleted and resuspended in 100ul PBSI (1 x PBS, pH 7.4, 1% BSA, 1% SuperRnaseIn). For all later washes, cells were pelleted by centrifugation at 500xg for 5 min (4°C).
The following steps are similar with sci-RNA-seq protocol with paraformaldehyde fixed nuclei19,20. Briefly, cells were distributed into four 96-well plates. For each well, 500 to 5,000 cells (2 μL) were mixed with 1 μl of 25 μM anchored oligo-dT primer (5′-ACGACGCTCTTCCGATCTNNNNNNNN[10bp index]TTTTTTTTTTTTTTTTTTTTTTTTTTTTTTVN-3′, where “N” is any base and “V” is either “A”, “C” or “G”; IDT) and 0.25 μL 10 mM dNTP mix (Thermo), denatured at 55°C for 5 min and immediately placed on ice. 1.75 μL of first-strand reaction mix, containing 1 μL 5X Superscript IV First-Strand Buffer (Invitrogen), 0.25 μl 100 mM DTT (Invitrogen), 0.25 μl SuperScript IV reverse transcriptase (200 U/μl, Invitrogen), 0.25 μL RNaseOUT Recombinant Ribonuclease Inhibitor (Invitrogen), was then added to each well. Reverse transcription was carried out by incubating plates at the following temperature gradient: 4°C 2 minutes, 10°C 2 minutes, 20°C 2 minutes, 30°C 2 minutes, 40°C 2 minutes, 50°C 2 minutes and 55°C 10 minutes. All cells were then pooled, stained with 4’,6-diamidino-2-phenylindole (DAPI, Invitrogen) at a final concentration of 3 μM, and sorted at 50 cells per well into 5 μL EB buffer. Cells were gated based on DAPI stain such that singlets were discriminated from doublets and sorted into each well. 0.66 μl mRNA Second Strand Synthesis buffer (NEB) and 0.34 μl mRNA Second Strand Synthesis enzyme (NEB) were then added to each well, and second strand synthesis was carried out at 16°C for 180 min. Each well was then mixed with 5 μL Nextera™ TD buffer (Illumina) and 1 μL i7 only TDE1 enzyme (25 nM, Illumina, diluted in Nextera™ TD buffer), and then incubated at 55°C for 5 min to carry out tagmentation. The reaction was stopped by adding 12 μL DNA binding buffer (Zymo) and incubating at room temperature for 5 min. Each well was then purified using 36 uL AMPure XP beads (Beckman Coulter), eluted in 16 μL of buffer EB (Qiagen), then transferred to a fresh multi-well plate.
For PCR reactions, each well was mixed with 2μL of 10 μM P5 primer (5′-AATGATACGGCGACCACCGAGATCTACAC[i5]ACACTCTTTCCCTACACGACGCTCTTCCGATCT-3′; IDT), 2 μL of 10 μM P7 primer (5′-CAAGCAGAAGACGGCATACGAGAT[i7]GTCTCGTGGGCTCGG-3′; IDT), and 20 μL NEBNext High-Fidelity 2X PCR Master Mix (NEB). Amplification was carried out using the following program: 72°C for 5 min, 98°C for 30 sec, 18-22 cycles of (98°C for 10 sec, 66°C for 30 sec, 72°C for 1 min) and a final 72°C for 5 min. After PCR, samples were pooled and purified using 0.8 volumes of AMPure XP beads. Library concentrations were determined by Qubit (Invitrogen) and the libraries were visualized by electrophoresis on a 6% TBE-PAGE gel. Libraries were sequenced on the NextSeq™ 500 platform (Illumina) using a V2 150 cycle kit (Read 1: 18 cycles, Read 2: 130 cycles, Index 1: 10 cycles, Index 2: 10 cycles).
Read alignment and downstream processing
Read alignment and gene count matrix generation for the single cell RNA-seq was performed using the pipeline that we developed for sci-RNA-seq10 with minor modifications. Reads were first mapped to a reference genome with STAR/v2.5.2b68, with gene annotations from GENCODE V19 for human, and GENCODE VM11 for mouse. For experiments with HEK293T and NIH/3T3 cells, we used an index combining chromosomes from both human (hg19) and mouse (mm10). For the A549 experiment, we used human genome build hg19.
The single cell sam files were first converted into alignment tsv file using sam2tsv function in jvarkit69. Next, for each single cell alignment file, mutations matching the background SNPs were filtered out. For background SNP reference of A549 cells, we downloaded the paired-end bulk RNA-seq data for A549 cells from ENCODE37 (sampled name: ENCFF542FVG, ENCFF538ZTA, ENCFF214JEZ, ENCFF629LOL, ENCFF149CJD, ENCFF006WNO, ENCFF828WTU, ENCFF380VGD). Each paired-end fastq files were first adaptor-clipped using trim_galore/0.4.170 with default settings, aligned to human hg19 genome build with STAR/v2.5.2b68. Unmapped and multiple mapped reads were removed by samtools/v1.371. Duplicated reads were filtered out by MarkDuplicates function in picard/1.10572. De-duplicated reads from all samples were combined and sorted with samtools/v1.371. Background SNPs were called by mpileup function in samtools/v1.371 and mpileup2snp function in VarScan/2.3.9 73. For HEK293T and NIH/3T3 test experiment, a background SNP reference was generated in a similar pipeline above, with the aggregated single cell sam data from control condition (no 4sU labeling and no IAA treatment condition).
For each single cell alignment file, all mutations with quality score <= 13 were removed. Mutations at both ends of each read were mostly due to sequencing errors, and thus also were filtered out. Mutations mapping to the background SNP reference were filtered out. For each read, we checked if there are T > C mutations for sense strand or A > G mutations for antisense strand, and labeled these mutated reads as newly synthesized.
Each cell was characterized by two digital gene expression matrixes from the full sequencing data and newly synthesized RNA data as described above. Genes with expression in equal or less than 5 cells were filtered out. Cells with fewer than 2,000 UMIs or more than 80,000 UMIs were discarded. Cells with doublet score > 0.2 by doublet analysis pipeline Scrublet/0.274 were removed.
The dimensionality of the data was first reduced with PCA (after selecting the top 2,000 genes with highest variance) on digital gene expression matrix on either full gene expression data or the newly synthesized gene expression data by Monocle 3/alpha2,75. The top 10 PCs were selected for dimensionality reduction analysis with uniform manifold approximation and projection (UMAP/0.3.2), a recently proposed algorithm based on Riemannian geometry and algebraic topology to perform dimension reduction and data visualization33. For joint analysis, we combined top 10 PCs calculated on the whole transcriptome and top 10 PCs on the newly synthesized transcriptome for each single cell before dimension reduction with UMAP. Cell clusters were done via densityPeak algorithm implemented in Monocle 3/alpha2,75. We first performed UMAP analysis on joint information of all processed cells, and identified an outlier cluster (724 out of 7,404 cells). These cells were marked by high level expression of GATA3, a marker of differentiated cells45, and were filtered out before downstream analysis.
Linking transcription factors (TFs) to their regulated genes
We sought to identify links between TFs and their regulated genes based on expression covariance. Cells with more than 10,000 UMIs detected, and genes (including TFs) with newly synthesis reads detected in more than 10% of all cells were selected. On average, these TFs are detected as expressed in ~58% of cells. Of note, a small number of TFs with high expression overall but low expression within newly synthesized reads were filtered out at this step (14 TFs with expression in >50% of cells; 75 of TFs with expression in >20% of cells, filtered consequent to this). The full gene expression and newly synthesized gene count per cell were normalized by cell-specific library size factors computed on the full gene expression matrix by estimateSizeFactors in Monocle 3/alpha2,75, log transformed, centered, then scaled by scale() function in R. For each gene detected, a LASSO regression model was constructed with package glmnet/v.2.076 to predict the normalized expression levels, based on the normalized expression of 853 TFs annotated in the “motifAnnotations_hgnc” data from package RcisTarget/v1.2.138, by fitting the following model:
where Gi is the adjusted gene expression value for gene i. It is calculated by the newly synthesized mRNA count for each cell, normalized by cell specific size factor (SGi) estimate by estimateSizeFactors in Monocle 3/alpha2,75 on the full expression matrix of each cell, and log transformed:
To simplify downstream comparison between genes, we standardize the response Gi prior to fitting the model for each gene i with the scale() function in R.
Similar with Gi, Ti is the adjusted TF expression value for each cell. It is calculated by the full TF expression count for each cell, normalized by cell specific size factor (SGi) estimate by estimateSizeFactors in Monocle 3/alpha2,75 on the full expression matrix of each cell, and log transformed:
Prior to fitting, Ti is standardized with the scale() function in R.
Although negative correlations between a TF’s expression and a gene’s new synthesis rate could reflect the activity of a transcriptional repressor, we felt that the more likely explanation for negative links reported by glmnet was mutually exclusive patterns of cell-state specific expression and TF activity. Thus during prediction, we excluded TFs with negatively correlated expression with a potential target gene’s synthesis rate, and also low regression coefficient (<= 0.03) links. We identified a total of 6,103 links between TFs and regulated genes. A modified strategy without filtering negatively correlated TF-gene synthesis rate pairs identified 47 additional repressive TF-gene links between 9 TFs and 46 genes (Supplementary Table 2).
Our approach aims to identify TFs that may regulate each gene, by finding the subset that can be used to predict its expression in a regression model. However, a TF with expression correlated with a gene’s expression does not definitively mean that it is directly regulating that gene. To identify putatively direct targets within this set, we intersected the links with TFs profiled in ENCODE ChIP-seq experiments37. Out of the 6,103 links between TFs and genes by LASSO regression, 1,086 links have TFs characterized in ENCODE. 807 of these 1,086 links were validated by target TF binding sites near gene promoters from ENCODE77, a 4.3-fold enrichment relative to background expectation (odds ratio for validation = 2.89 for links identified in LASSO regression vs. 0.67 for background, p-value < 2.2e-16, two-sided Fisher’s Exact test). Only gene sets with significant enrichment of the correct TF ChIP-seq binding sites were retained (two-sided Fisher’s Exact test, FDR of 5%), and further pruned to remove indirect target genes without TF binding data support. Ultimately, 591 links were retained by this approach.
To expand the set of validated TF-gene links, we further applied package SCENIC38, a pipeline to construct gene regulatory networks based on the enrichment of target TF motifs in the 10 kb window around genes’ promoters. Each co-expression module identified by LASSO regression was analyzed using cis-regulatory motif analysis using RcisTarget/v1.2.138. Only modules with significant motif enrichment of the correct TF regulator were retained, and pruned to remove indirect target genes without motif support. We filtered the TF-gene links by three correlation coefficient thresholds (0.3, 0.4 and 0.5), and combined all links validated by RcisTarget38. In total, there were 509 links validated this motif-based approach.
Combining both approaches, we identified a total of 986 TF-gene regulatory links by the covariance between TF expression and gene synthesis rate, validated by DNA binding data or motif analysis. To evaluate the possibility that the links were artifacts of regularized regression, we permuted the order of single cell TF expression (Ti) and performed the same analyses. No links were identified after this permutation.
Applying a similar strategy to all mRNA (rather than only newly synthesized mRNA) revealed 2,108 TF-gene links, with 532 identified by both approaches. The 448 TF-gene links uniquely identified by analysis of newly synthesized mRNA exhibited higher correlations between TF expression and newly synthesized mRNA than all mRNA (mean Spearman’s correlation of 0.19 vs. 0.16, respectively; p-value = 5.3e-8, two-sided Wilcoxon rank sum test). The TF-gene links identified exclusively by analysis of all mRNAs corresponded lower mRNA synthesis rates for linked genes (mean UMI count, normalized by size factor for newly synthesised mRNA: 1.20 for genes with links by newly synthesized mRNAs vs 0.97 for genes with links identified solely through analysis of all mRNAs; p-value < 2.2e-16, two-sided Wilcoxon rank sum test).
Ordering cells based on the activity of functional TF modules
To calculate TF “activity” in each cell, newly synthesized UMI counts for genes linked to each of the 27 TFs were scaled by library size, log-transformed, aggregated and then mapped to Z-scores. As TFs with highly correlated or anti-correlated activity suggest they may function in linked biological processes, we calculated the absolute Pearson’s correlation coefficient between each pair of TF activity, and based on this we clustered TFs by ward.d2 clustering method in package pheatmap/1.0.1278. Five functional TF modules were identified and annotated based on their functions.
To characterize the dynamics of cells in relation to potentially independent cellular processes, cells were ordered by the activity of cell cycle related TFs (TF module 1) or GR activity related TFs (TF module 3) with UMAP33(metric = “cosine”, n_neighbors = 30, min_dist = 0.01). The cell cycle progression trajectory were validated by cell cycle gene markers in Seurat/2.3.435. Three cell cycle phases were identified by densityPeak algorithm implemented in Monocle 3/alpha2,75, on the UMAP coordinates ordered by cell cycle TF modules. As each main cell cycle phase still showed variable TF activity and cell cycle marker expression, we segmented each phase to early/middle/late states by k-means clustering (k = 3), and recovered a total of nine cell cycle states. Three GR response states were identified by densityPeak algorithm implemented in Monocle 3/alpha2,75.
Past transcriptome state recovery from sci-fate
To infer the past transcriptome state (i.e. the cell state before 4sU labeling commenced), we assume mRNA half-lives are consistent across different DEX treatment durations. This assumption is further validated by self-consistency check later. Under this assumption, the partly degraded bulk transcriptome before the 2 hour 4sU labeling should be the same between no DEX and 2 hour DEX treated cells. Thus, for any given gene, differences in whole transcriptomes (bulk) between these time points should be equal to differences in the newly synthesized transcriptomes (bulk), corrected by technique’s detection rate:
A0h is the aggregated UMI count for all cells in no DEX treatment group; S0h is the library size (total UMI count of cells) at no DEX treatment; N0h is the aggregated newly synthesized UMI count for all cells in no DEX treatment group; A2h is the aggregated UMI count for all cells in 2 hour DEX treatment group; S2h is the library size (total UMI count of cells) in 2 hour DEX treatment group; N2h is the aggregated newly synthesized UMI count for all cells in 2 hour DEX treatment group; α is the detection rate for each gene in sci-fate. As cells from different time points were profiled in the same experiment and the UMI counts detected per cell were similar across conditions (Supplementary Fig. 2ab), we assume the same overall RNA amount in the 0h and 2h samples, and normalize the aggregated gene count reads by total counts of each time point. For experiments where this assumption may not stand, spike-in standards could be used to control for differences in the overall amount of mRNA between conditions. In theory, one detection rate can be calculated for each gene. However, for genes with minor differences of newly synthesis rate between two conditions, the estimated α is dominated by noise. We thus selected genes showing higher differences in normalized newly synthesis rate between two conditions: we first tested a series of threshold for gene filtering and calculated the α for each gene. We then plotted the relationship between threshold and the ratio of genes with out-range α values (< 0 or > 1). We selected the threshold that was at the knee point of the plot, resulting in 186 genes selected (Supplementary Fig. 5a). The differences in newly synthesized mRNA of these genes highly correlates with the differences in mRNA expression level (Pearson’s r = 0.93, Supplementary Fig. 5b), suggesting the new RNA detection rate is stable across genes (Supplementary Fig. 5c). Here we use the median detection rate across 186 selected genes in order to estimateα.
We next computed the mRNA degradation rate across each 2 hour interval. As the A549 cell population can be regarded stable without external perturbation, for 2 hour DEX treated cells, its past state (before 2 hour 4sU labeling) should be the same as the 0 hour DEX treated cells. Expanding on this logic, the past state (before 4sU labeling) for T = 0/2/4/6/8/10 hour DEX treated cells should be similar to the profiled T = 0/0/2/4/6/8 hour cells, respectively:
At1 is the aggregated UMI count for all cells in t1; St1 is is the library size (the total UMI count of cells) at t1; Nt1 is the aggregated newly synthesized UMI count for all cells at t1; α is the estimated detection rate of sci-fate; At0 is the aggregated UMI count for all cells in t0; St0 is the library size (the total UMI count of cells) at t0; β is 1 - gene specific degradation rate between t0 and t1, and is related with the mRNA half life γ by:
The β was calculated for each of 14,587 genes across each 2 hour interval of DEX treatment. As the self-consistency check mentioned above, the gene degradation rates are highly correlated across different DEX treatment times (Supplementary Fig. 5g). We therefore used the average degradation rate for each gene for downstream analysis.
With the overall sci-fate detection rate as well as per-gene degradation rates estimated, the past transcriptome state of each cell can be estimated by:
αt1 is the single cell UMI count in t1; nt1 is the single cell newly synthesized UMI count at t1; α is the detection rate for each gene in sci-fate. Here we use the median detection rate across 186 selected genes as its estimates; β is 1 - gene specific degradation rate between t0 and t1. at0 is the estimated single cell transcriptome in a past time point t0, with all negative values (15.6% of values on average) converted to 0.
The detection rate (α) of the newly synthesised transcriptome is experiment-specific and mainly depends on the 4sU labeling concentration. Generally, a lower 4sU concentration will lead to a lower 4sU incorporation rate to newly synthesised mRNA, which will reduce the detection rate of newly synthesised transcripts. Additionally, the length of sequencing reads may affect the detection rate, as discussed above. In the case of our experiments and as noted above, we used relatively long reads (on average, 75 bp of transcript-derived sequence obtained per read) to potentially increase detection rate. We also designed the experiments such that all treatment conditions shares the same 4sU treatment concentration and incubation time in the same cell type. Furthermore, all cells were sequenced in the same sequencing run, such that all conditions are expected to share a similar α. For sci-fate experiments with different labeling conditions or sequencing settings, the αwould need to be re-estimated for each part of the experiment. Of note, our simplifying assumption that gene-specific degradation rates are constant across our DEX time course might not be a good choice in other systems. Specifically, in systems where the gene-specific degradation rates are expected or observed to substantially vary over time, these should be estimated for each time interval separately.
Linkage analysis to build single cell state trajectory
The goal of what we call here “linkage analysis” is to associate each cell with parent and child cells at different time points, i.e. single cell state trajectories. Our approach is based on a fact that the past transcriptome states (before 2 hour 4sU labeling) of cells at t1 should share the same cell population distribution with the profiled transcriptome states of cells at t0 (2 hours earlier than t1), assuming there is no cell apoptosis. We thus applied a published manifold alignment strategy to identify common cell states between two data sets, based on common sources of variation35. As a result, whole transcriptomes from t0 cells and recovered past transcriptomes from t1 cells are aligned in the same UMAP space. This analysis is based on an assumption that for intermediate time points, we are oversampling the space of physiologically distinct states in this time course. Violation of this and other assumptions can be detected by outliers during alignment of the two data sets. For each cell A from t1, we selected its nearest neighbor in t0 as its parent state in the alignment UMAP space. Similarly, for each cell from t0, we selected its nearest neighbor in t1 as its child cell state. Of note, the link is not necessary to be bi-directional: the parent state of one cell may be linked to a different child cell. After the parent and child states were identified for each cell (except cells at the start and end time points), we then extend each cell trajectory by searching for the linked parent cell of each cell's parent, and similarly the linked child cell of each cell's child. Thus each single cell can be characterized by a single cell state transition path across all six time points spanning 10 hours. As multiple cells (>50) are profiled for each of the 27 defined cell state, stochastic cell state transition processes can also potentially be captured.
Dimensionality reduction and clustering analysis
For dimensionality reduction on single cell transcriptomes, the top 5 PCs for full transcriptomes and top 5 PCs for newly synthesized transcriptomes were selected for each state, and combined in temporal order along single cell state trajectory for UMAP analysis. Main cell trajectory types were identified by density peak clustering algorithm79.
With cell state proportion at the beginning time point (0 hour treatment) and cell state transition probabilities estimated from the data, we first predicted the cell state distribution after 2 hours, assuming the cell state transitions in DEX treatment are cell-autonomous, time-independent, Markovian processes. Similarly, the cell state distribution at later time points can be predicted from the cell state distribution 2 hours before.
For RNA velocity analysis of these same data, single cell spliced/unspliced expression matrices were generated using the command line interface of velocyto/v.0.1732 with the default run_smartseq2 mode on single cell bam files. Cell transition direction inference were performed with an optimized scalable RNA velocity analysis toolkit scVelo/v.0.1.17 and scanpy/v.1.4.1 with default settings32,80. For integrating treatment time information into the RNA velocity analysis of the 0h and 2h timepoints, we prohibited transitions of cells within 0h, and instead selected the future state as the cell at the 2h time point with the highest transition probability.
Cell state instability and cell state distance calculations
We defined cell state instability as the proportion of cells in a given state ‘moving’ to any other state at the next time point. To calculate cell state distances, we first sampled equal number (n = 50) of cells from each state, and separately aggregated the full transcriptome and newly synthesized transcriptome of sampled cells of that state (i.e. in this ‘joint transcriptome’, each gene is represented by two columns, one for the whole transcriptome and one for the newly synthesized transcriptome). The cell state distance is calculated as the Pearson's correlation coefficient between the joint transcriptomes of two different states.
Data Availability
The data generated by this study can be downloaded in raw and processed forms from the NCBI Gene Expression Omnibus (GSE131351).
Code Availability
Scripts for processing sci-fate sequencing were written in python and R with code available at https://github.com/JunyueC/sci-fate_analysis.
Supplementary Material
Supplementary Table 1: Differentially expressed (DE) genes between 0 hour and 2 hour DEX time points (Cell number n = 1,054 for 0h and 1,049 for 2h). The “qval” is the false detection rate for the gene differential expression test (one-sided likelihood ratio test) across different DEX treatment points.
Supplementary Table 2: Links between 27 TFs and 532 putatively regulated genes by LASSO regression analysis with 6,680 cells (with group name: activating links by newly synthesized mRNA, link number n = 986). The table also includes repressive TF-gene links (group name: repressive links by newly synthesized mRNA, link number n = 47) as well as links obtained by considering full mRNA expression (group name: links by full mRNA, link number n = 2,108).
Supplementary Table 3: Beta value (β, 1 - Gene degradation rate) in each 2 hour time window, estimated via sci-fate.
Acknowledgements
We thank the members of the Shendure labs for helpful discussions, particularly X. Huang, R. Blecher, B. Martin, F. Chardon and R. Qiu; J. Rose, D. Maly, L. VandenBosch, and T. Reh lab for sharing the NIH/3T3 cell line; J. McFaline-Figueroa for sharing the A549 cell line. This work was funded by the Paul G. Allen Frontiers Foundation (Allen Discovery Center grant to JS and CT), grants from the NIH (DP1HG007811 and R01HG006283 to JS; DP2 HD088158 to CT), the W. M. Keck Foundation (to CT and JS), the Dale. F. Frey Award for Breakthrough Scientists (to CT), the Alfred P. Sloan Foundation Research Fellowship (to CT), and the Brotman Baty Institute for Precision Medicine. JS is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Competing Financial Interests Statement
F.J.S. declares competing financial interests in the form of stock ownership and paid employment by Illumina, Inc. One or more embodiments of one or more patents and patent applications filed by Illumina may encompass the methods, reagents, and data disclosed in this manuscript.
References
- 1.Trapnell C et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol 32, 381–386 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Qiu X et al. Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 14, 979–982 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wolf FA et al. PAGA: graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells. Genome Biol. 20, 59 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haghverdi L, Büttner M, Wolf FA, Buettner F & Theis FJ Diffusion pseudotime robustly reconstructs lineage branching. Nat. Methods 13, 845–848 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Setty M et al. Wishbone identifies bifurcating developmental trajectories from single-cell data. Nat. Biotechnol 34, 637–645 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Street K et al. Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genomics 19, 477 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moris N, Pina C & Arias AM Transition states and cell fate decisions in epigenetic landscapes. Nat. Rev. Genet 17, 693–703 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Herzog VA et al. Thiol-linked alkylation of RNA to assess expression dynamics. Nat. Methods 14, 1198–1204 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schofield JA, Duffy EE, Kiefer L, Sullivan MC & Simon MD TimeLapse-seq: adding a temporal dimension to RNA sequencing through nucleoside recoding. Nat. Methods 15, 221–225 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cao J et al. Comprehensive single-cell transcriptional profiling of a multicellular organism. Science 357, 661–667 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cleary MD, Meiering CD, Jan E, Guymon R & Boothroyd JC Biosynthetic labeling of RNA with uracil phosphoribosyltransferase allows cell-specific microarray analysis of mRNA synthesis and decay. Nat. Biotechnol 23, 232–237 (2005). [DOI] [PubMed] [Google Scholar]
- 12.Dolken L et al. High-resolution gene expression profiling for simultaneous kinetic parameter analysis of RNA synthesis and decay. RNA 14, 1959–1972 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miller C et al. Dynamic transcriptome analysis measures rates of mRNA synthesis and decay in yeast. Mol. Syst. Biol 7, 458–458 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Duffy EE et al. Tracking Distinct RNA Populations Using Efficient and Reversible Covalent Chemistry. Mol. Cell 59, 858–866 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schwalb B et al. TT-seq maps the human transient transcriptome. Science 352, 1225–1228 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Rabani M et al. Metabolic labeling of RNA uncovers principles of RNA production and degradation dynamics in mammalian cells. Nat. Biotechnol 29, 436–442 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miller MR, Robinson KJ, Cleary MD & Doe CQ TU-tagging: cell type–specific RNA isolation from intact complex tissues. Nat. Methods 6, 439–441 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cusanovich DA et al. Multiplex single cell profiling of chromatin accessibility by combinatorial cellular indexing. Science 348, 910–914 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cao J et al. Comprehensive single-cell transcriptional profiling of a multicellular organism. Science 357, 661–667 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cao J et al. Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science 361, 1380–1385 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ramani V et al. Massively multiplex single-cell Hi-C. (2016) doi: 10.1101/065052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mulqueen RM et al. Highly scalable generation of DNA methylation profiles in single cells. Nat. Biotechnol 36, 428–431 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vitak SA et al. Sequencing thousands of single-cell genomes with combinatorial indexing. Nat. Methods 14, 302–308 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rosenberg AB et al. Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science 360, 176–182 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yin Y et al. High-Throughput Single-Cell Sequencing with Linear Amplification. Mol. Cell 76, 676–690.e10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cao J et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Buckingham JC Glucocorticoids: exemplars of multi-tasking. Br. J. Pharmacol 147, S258 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reddy TE et al. Genomic determination of the glucocorticoid response reveals unexpected mechanisms of gene regulation. Genome Res. 19, 2163–2171 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.John S et al. Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nat. Genet 43, 264–268 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reddy TE, Gertz J, Crawford GE, Garabedian MJ & Myers RM The Hypersensitive Glucocorticoid Response Specifically Regulates Period 1 and Expression of Circadian Genes. Mol. Cell. Biol 32, 3756–3767 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vockley CM et al. Direct GR Binding Sites Potentiate Clusters of TF Binding across the Human Genome. Cell 166, 1269–1281.e19 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.La Manno G et al. RNA velocity of single cells. Nature 560, 494 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McInnes L, Healy J, Saul N & Großberger L UMAP: Uniform Manifold Approximation and Projection. Journal of Open Source Software 3, 861 (2018). [Google Scholar]
- 34.Binder EB The role of FKBP5, a co-chaperone of the glucocorticoid receptor in the pathogenesis and therapy of affective and anxiety disorders. Psychoneuroendocrinology vol. 34 S186–S195 (2009). [DOI] [PubMed] [Google Scholar]
- 35.Butler A, Hoffman P, Smibert P, Papalexi E & Satija R Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dataset - ENCODE Transcription Factor Binding Site Profiles. http://amp.pharm.mssm.edu/Harmonizome/dataset/ENCODE+Transcription+Factor+Binding+Site+Profiles.
- 37.The ENCODE Project Consortium. The ENCODE (ENCyclopedia Of DNA Elements) Project. Science 306, 636–640 (2004). [DOI] [PubMed] [Google Scholar]
- 38.Aibar S et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods 14, 1083–1086 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Han H et al. TRRUST v2: an expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 46, D380–D386 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kuleshov MV et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Research vol. 44 W90–W97 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boruk M, Savory JGA & Haché RJG AF-2-Dependent Potentiation of CCAAT Enhancer Binding Proteinβ -Mediated Transcriptional Activation by Glucocorticoid Receptor. Mol. Endocrinol 12, 1749–1763 (1998). [DOI] [PubMed] [Google Scholar]
- 42.Qin W et al. Identification of functional glucocorticoid response elements in the mouse FoxO1 promoter. Biochem. Biophys. Res. Commun 450, 979–983 (2014). [DOI] [PubMed] [Google Scholar]
- 43.Sheela Rani CS, Elango N, Wang S-S, Kobayashi K & Strong R Identification of an Activator Protein-1-Like Sequence as the Glucocorticoid Response Element in the Rat Tyrosine Hydroxylase Gene. Mol. Pharmacol 75, 589 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fischer M & Müller GA Cell cycle transcription control: DREAM/MuvB and RB-E2F complexes. Crit. Rev. Biochem. Mol. Biol 52, 638–662 (2017). [DOI] [PubMed] [Google Scholar]
- 45.Chou J, Provot S & Werb Z GATA3 in development and cancer differentiation: cells GATA have it! J. Cell. Physiol 222, 42–49 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Madhurima Biswas JYC Role of Nrf1 in antioxidant response element-mediated gene expression and beyond. Toxicol. Appl. Pharmacol 244, 16 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ryoo I-G & Kwak M-K Regulatory crosstalk between the oxidative stress-related transcription factor Nfe2l2/Nrf2 and mitochondria. Toxicol. Appl. Pharmacol 359, 24–33 (2018). [DOI] [PubMed] [Google Scholar]
- 48.Heer R, Robson CN, Shenton BK & Leung HY The role of androgen in determining differentiation and regulation of androgen receptor expression in the human prostatic epithelium transient amplifying population. J. Cell. Physiol 212, 572–578 (2007). [DOI] [PubMed] [Google Scholar]
- 49.Meixner A, Karreth F, Kenner L, Penninger JM & Wagner EF Jun and JunD-dependent functions in cell proliferation and stress response. Cell Death Differ. 17, 1409–1419 (2010). [DOI] [PubMed] [Google Scholar]
- 50.Li M et al. Krüppel-Like Factor 5 Promotes Epithelial Proliferation and DNA Damage Repair in the Intestine of Irradiated Mice. Int. J. Biol. Sci 11, 1458–1468 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Eberlé D, Hegarty B, Bossard P, Ferré P & Foufelle F SREBP transcription factors: master regulators of lipid homeostasis. Biochimie 86, 839–848 (2004). [DOI] [PubMed] [Google Scholar]
- 52.Shermoen AW & O’Farrell PH Progression of the cell cycle through mitosis leads to abortion of nascent transcripts. Cell 67, 303–310 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Palozola KC et al. Mitotic transcription and waves of gene reactivation during mitotic exit. Science 358, 119–122 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Parsons GG & Spencer CA Mitotic repression of RNA polymerase II transcription is accompanied by release of transcription elongation complexes. Mol. Cell. Biol 17, 5791–5802 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sanchez-Alvarez M, Zhang Q, Finger F, Wakelam MJO & Bakal C Cell cycle progression is an essential regulatory component of phospholipid metabolism and membrane homeostasis. Open Biol. 5, 150093 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Harmon JM, Norman MR, Fowlkes BJ & Thompson EB Dexamethasone induces irreversible G1 arrest and death of a human lymphoid cell line. J. Cell. Physiol 98, 267–278 (1979). [DOI] [PubMed] [Google Scholar]
- 57.Greenberg AK et al. Glucocorticoids inhibit lung cancer cell growth through both the extracellular signal-related kinase pathway and cell cycle regulators. Am. J. Respir. Cell Mol. Biol 27, 320–328 (2002). [DOI] [PubMed] [Google Scholar]
- 58.Erhard F et al. scSLAM-seq reveals core features of transcription dynamics in single cells. Nature 571, 419–423 (2019). [DOI] [PubMed] [Google Scholar]
- 59.Hendriks G-J et al. NASC-seq monitors RNA synthesis in single cells. Nat. Commun 10, 3138 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Baran-Gale J, Chandra T & Kirschner K Experimental design for single-cell RNA sequencing. Briefings in Functional Genomics vol. 17 233–239 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen W et al. UMI-count modeling and differential expression analysis for single-cell RNA sequencing. Genome Biol. 19, 70 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Matsushima W et al. SLAM-ITseq: sequencing cell type-specific transcriptomes without cell sorting. Development 145, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sharma U et al. Small RNAs are trafficked from the epididymis to developing mammalian sperm. (2017) doi: 10.1101/194522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gay L et al. Mouse TU tagging: a chemical/genetic intersectional method for purifying cell type-specific nascent RNA. Genes Dev. 27, 98–115 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hastie T & Stuetzle W Principal Curves. Journal of the American Statistical Association vol. 84 502 (1989). [Google Scholar]
- 66.Wickham H ggplot2: Elegant Graphics for Data Analysis. (Springer, 2016). [Google Scholar]
Methods-only References
- 67.Muhar M et al. SLAM-seq defines direct gene-regulatory functions of the BRD4-MYC axis. Science 360, 800–805 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lindenbaum P JVarkit: java-based utilities for Bioinformatics. figshare (2015). [Google Scholar]
- 70.FelixKrueger. FelixKrueger/TrimGalore. GitHub https://github.com/FelixKrueger/TrimGalore. [Google Scholar]
- 71.Li H et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Picard Tools - By Broad Institute. http://broadinstitute.github.io/picard/.
- 73.Koboldt DC et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568–576 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wolock SL, Lopez R & Klein AM Scrublet: computational identification of cell doublets in single-cell transcriptomic data. (2018) doi: 10.1101/357368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.cole-trapnell-lab. cole-trapnell-lab/monocle-release. GitHub https://github.com/cole-trapnell-lab/monocle-release. [Google Scholar]
- 76.Friedman J, Hastie T & Tibshirani R Regularization Paths for Generalized Linear Models via Coordinate Descent. J. Stat. Softw 33, (2010). [PMC free article] [PubMed] [Google Scholar]
- 77.Dataset - ENCODE Transcription Factor Binding Site Profiles. http://amp.pharm.mssm.edu/Harmonizome/dataset/ENCODE+Transcription+Factor+Binding+Site+Profiles.
- 78.raivokolde. raivokolde/pheatmap. GitHub https://github.com/raivokolde/pheatmap. [Google Scholar]
- 79.Rodriguez A & Laio A Clustering by fast search and find of density peaks. Science 344, 1492–1496 (2014). [DOI] [PubMed] [Google Scholar]
- 80.Wolf FA, Angerer P & Theis FJ SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1: Differentially expressed (DE) genes between 0 hour and 2 hour DEX time points (Cell number n = 1,054 for 0h and 1,049 for 2h). The “qval” is the false detection rate for the gene differential expression test (one-sided likelihood ratio test) across different DEX treatment points.
Supplementary Table 2: Links between 27 TFs and 532 putatively regulated genes by LASSO regression analysis with 6,680 cells (with group name: activating links by newly synthesized mRNA, link number n = 986). The table also includes repressive TF-gene links (group name: repressive links by newly synthesized mRNA, link number n = 47) as well as links obtained by considering full mRNA expression (group name: links by full mRNA, link number n = 2,108).
Supplementary Table 3: Beta value (β, 1 - Gene degradation rate) in each 2 hour time window, estimated via sci-fate.
Data Availability Statement
The data generated by this study can be downloaded in raw and processed forms from the NCBI Gene Expression Omnibus (GSE131351).