

Abstract
Protein docking is essential for structural characterization of protein interactions. Besides providing the structure of protein complexes, modeling of proteins and their complexes is important for understanding the fundamental principles and specific aspects of protein interactions. The accuracy of protein modeling, in general, is still less than that of the experimental approaches. Thus, it is important to investigate the applicability of docking techniques to modeled proteins. We present new comprehensive benchmark sets of protein models for the development and validation of protein docking, as well as a systematic assessment of free and template-based docking techniques on these sets. As opposed to previous studies, the benchmark sets reflect the real case modeling/docking scenario where the accuracy of the models is assessed by the modeling procedure, without reference to the native structure (which would be unknown in practical applications). We also expanded the analysis to include docking of protein pairs where proteins have different structural accuracy. The results show that, in general, the template-based docking is less sensitive to the structural inaccuracies of the models than the free docking. The near-native docking poses generated by the template-based approach, typically, also have higher ranks than those produces by the free docking (although the free docking is indispensable in modeling the multiplicity of protein interactions in a crowded cellular environment). The results show that docking techniques are applicable to protein models in a broad range of modeling accuracy. The study provides clear guidelines for practical applications of docking to protein models.
Keywords: protein interactions, structure prediction, protein modeling, benchmarking, interactome
INTRODUCTION
Protein-protein interactions are essential for life processes. Despite significant advances, the experimental techniques for structure determination are capable of solving only a fraction of protein structures, leaving the rest to computational modeling. The role of modeling is even greater in characterizing protein-protein interactions, given the multiplicity of protein partners and binding modes.1–4 Methods for predicting the structure of protein-protein complexes (protein docking) fall into two broad categories: (1) free docking where sampling of the binding modes, based on structural and physicochemical complementarity of the proteins, is performed with no knowledge of similar experimentally determined complexes, and (2) comparative (or homology, template-based) docking, which relies on the structures of similar complexes (templates).5–16 Both docking protocols typically consist of a scan stage where a global search is performed for an approximate structure of the complex,17 followed by post-processing of the putative matches by scoring (re-ranking by more accurate and often computationally expensive functions).18 It has been shown that integration of the scan and the scoring (typically, by using the scoring function to bias the sampling) generally leads to better docking results than running independent sampling and scoring procedures.19 Structural refinement by various minimization protocols improves the accuracy of the predictions.4,17,20–24
In terms of the intermolecular energy, the scan in the free docking methodology typically means the intermolecular energy landscape-wide search for the binding funnel. The free docking success depends heavily on a number of factors (the force field, characteristics of the intermolecular energy landscape, and such). For example, enzyme-inhibitor complexes typically can be modeled with a reasonable accuracy, whereas antigen-antibody complexes are less predictable. A substantial conformational change upon binding makes the free docking particularly difficult.6
Comparative docking uses similarity to known structures of protein-protein complexes to determine the plausible mutual arrangements of the target proteins. Thus, the comparative docking scan stage is just global or partial alignment of the sequence or the structure (or both) of the target proteins to the pool of templates (co-crystallized protein-protein complexes).6 Along with the robustness of the alignment protocol, the success of comparative docking is determined by the size and diversity of the template library. It has been shown that templates for docking of structurally characterized individual proteins are widely available.25 Consequently, comparative docking has been successful in quaternary structure prediction.26 Combining free and comparative docking methods yields more successful predictions than either method alone.27,28 Since the comparative docking methods are based on proteins alignments, as opposed to the free docking which is based on surface complementarity, they are generally less sensitive to the conformational changes upon binding.29
Because of limitations of the experimental techniques, docking often has to rely on the models of proteins instead of the higher-resolution experimentally resolved structures.4,30,31 A thorough investigation of low-resolution free docking on a large set of protein models of different accuracy showed that such docking determines the gross structural features of the complex for a significant portion of the protein models, including highly inaccurate ones.32 More recently, high-resolution docking benchmarking on a diverse set of protein models revealed that meaningful predictions can be obtained even for models with significant distortions, and that the comparative docking is much less sensitive to the inaccuracies of the protein models compared to the free docking.33 That study was based on the benchmark set of protein models with accuracy assessed by RMSD from the protein native structure, and was limited to protein pairs of similar accuracy.
To test the ability of docking procedures on predicting the structure of protein model-model complexes in the real case scenario, we built docking benchmark sets of protein models generated by the Phyre modeling pipeline,34 with accuracy assessed by the Phyre ranks, for 171 and 963 binary protein complexes from the Dockground docking benchmark set 435 and GWIDD database,36,37 respectively. The sets are different from previously generated ones,38,39 in which arrays of protein models were generated according RMSD to the native structure. The current benchmark sets rather reflect the real case modeling/docking scenario where the accuracy of the models is assessed by the modeling procedure, with no reference to the native structure (which would be unknown in practical applications). The sets are available at http://dockground.compbio.ku.eduand http://www.gwyre.org.
Free and template-based docking was thoroughly benchmarked on the sets. The accuracy of the docking was assessed according to the CAPRI criteria.40 The docking success rate was correlated with the Phyre modeling parameters for individual proteins, rather than the native structure of the proteins. For the first time, the exhaustive benchmarking was done for protein pairs in which proteins are modeled at different levels of accuracy (e.g. low accuracy “receptor” and high accuracy “ligand”), again reflecting the real-case scenario of practical applications.
METHODS
Datasets
The initial sets of experimentally determined bound protein structures consisted of 445 proteins from 223 binary complexes in the Dockground docking benchmark set 4 (http://dockground.compbio.ku.edu),35 and 2821 proteins from 1818 co-crystallized human protein-protein complexes in the GWIDD database (http://gwidd.compbio.ku.edu).36,37 The modeling pipeline (see below) produced five models for each of 342 proteins (171 complexes) from the Dockground set, and 1120 proteins (963 complexes) from the GWIDD. The PDB codes and chain IDs of corresponding experimentally determined complexes are in Supplemental Tables S1 and S2. All complexes in the Dockground set were hetero-dimeric, whereas in the GWIDD set ~80% of complexes consisted of proteins identical by structure and sequence (Fig. S1).
Protein modeling
The models of individual proteins were generated using the batch processing facility of the Phyre2 web server. Full details of the methodology are available in Ref34. Briefly, a protein sequence to be modeled is first scanned against a large sequence database to construct a profile hidden Markov model (HMM). This HMM is then scanned against a similarly constructed library of template HMMs of known structure using the program HHsearch.41 The template library was constructed from a non-redundant subset of proteins of known structure taken from the PDB such that no two proteins share > 80% sequence identity. The result of this template detection step is a list of proteins and alignments ranked by the confidence score of HHsearch, reflecting the probability that the aligned pair is homologous. The top 20 such template alignments were then used to construct crude backbone models, often with missing insertions or deletions. The latter were modeled using a loop modeling procedure based on a search of a library of protein structure fragments. Candidate loops were fitted to the backbone models using cyclic coordinate descent and ranked by a range of empirical energy functions. Finally, full-atom models with side chains were constructed by program SCWRL,42 which uses a backbone-dependent library of side chain rotamers with associated probabilities derived from frequency of observation in the PDB. SCWRL attempts to fit the highest probability rotamers for each amino acid while avoiding clashes. The end result of the Phyre2 processing was a maximum of 20 full atom models with associated confidence scores. Conventionally, the models with a confidence score < 0.9 were considered inaccurate. Another important aspect in choice of the models was the necessity to avoid self-hits. Thus, the selection of monomers models was governed by conjunction of two criteria: (i) a model must have confidence > 0.9, and (ii) a model must have sequence identity < 95% to avoid self-hits. The relatively high threshold for self-hits in modeling of monomers was used because this study was not aimed at benchmarking the modeling of monomers (where that would be a critical issue), but at determination of the relative success rates of docking of the modeled monomers. Application of the above two conditions to sets of 20 models for each monomer of a target protein-protein complex resulted in discrimination of some models that have high rank. Thus, there were situations where models with rank 1 were removed, while models of the same monomer having rank 2 remained in the set. In such cases, we re-ranked the models to preserve their continuous numeration in the order of decreasing accuracy. Such filtering and re-ranking lead to different numbers of available models for different monomers. In order to make the benchmark sets consistent and homogeneous we narrowed the corresponding sets of models for each monomer to top five. This resulted in 171 complexes from the Dockground set, and 963 complexes from the GWIDD set (the discarded complexes had < 5 models for at least one of the monomers). The larger protein in the complex was designated as “receptor” and the smaller one as “ligand.” Most proteins had > 0.9 sequence coverage (in the Dockground set 77% of receptors and 88% of ligands, and in the GWIDD set 77% of receptors and 79% of ligands). Due to the above strict requirements for choosing models for further docking, the confidence scores produced by Phyre2 could not be used as a measure of model quality, because all selected modes had high values of that score.
Protein docking
We performed free and template-based docking of protein models corresponding to the experimentally determined protein structures in the datasets of complexes. For each heterodimeric complex, we had 25 distinct combinations of 5 models of the receptor and 5 models of the ligand. Homodimeric complexes had 15 distinct combinations (above-diagonal part of the symmetric 5x5 matrix plus the diagonal). We performed both the “diagonal” docking (receptor and ligand of same modeling rank) and “cross” docking (receptor and ligand of different modeling rank) and analyzed these two groups of results separately. In the earlier studies,32,33 only the diagonal docking was performed. Docking of a protein pair was defined as successful if at least one correct prediction (acceptable, medium of high quality according to the CAPRI criteria40) was present in the top N predictions. Docking success rate was calculated as a fraction of successful docking predictions for all protein pairs (171 for the Dockground set and 963 for the GWIDD set) with the same combination of Phyre2 modeling ranks. The reference complex structures used for calculations of the CAPRI criteria - ligand RMSD (L-RMSD), interface RMSD (i-RMSD) and fraction of predicted native contacts (fnat), were obtained by structural alignment of the protein models onto the corresponding bound structures.
Docking protocols
Free docking was performed by our FFT (Fast Fourier Transform) program GRAMM43 with 3.5 Å grid step and 10° angular intervals. The top 300,000 matches initially ranked by the shape complementarity, were re-scored by the semi-empirical atom-atom contact potential AACE18.44 Template based docking was performed by the full-structure alignment protocol.45 The individual proteins were structurally aligned by TM-align46 to the monomers of the Dockground template library47 consisting of 4,950 co-crystallized binary complexes. Scoring of the initial models was performed by the combined scoring function.48
RESULTS AND DISCUSSION
Rank of protein models
In practical applications, known characteristics of protein models are not be based on the protein native structure (which is unknown). Such characteristics can be the rank of the protein model evaluated by an energy function, sequence identity between target and template proteins, etc. Thus, in this study, we investigated how the free and comparative docking success rates depend on the protein model rank produced by the Phyre2 modeling pipeline (see Methods). This rank is correlated with the e-values of the sequence alignments estimated by HHsearch.41 Thus, the same rank for different proteins (even within the same complex) may comprise wide ranges of sequence identity to the templates and deviation from the native bound structures (Fig. 1 and Fig. S2 for protein models in the Dockground and GWIDD datasets, respectively). The vast majority (~ 95 % and ~ 87 % of rank 1 models in the Dockground and GWIDD, respectively) reproduce well the fold of the native structures (TM-score > 0.6, gray areas in Fig. 1 and Fig. S2). This fraction slightly decreases (by ~10%) with the decrease of the model rank. The share of these models with the low target/template sequence identity (< 40%, dark gray areas in Figs. 1 and S2) increases from ~45% and 48% for the rank 1 models, to ~72% and 63% for the rank 5 models of the Dockground receptors and ligands, respectively. Corresponding increases for the receptor, ligand and homodimers in the GWIDD datasets are from ~40%, 28%, and 32% for rank 1, to ~67%, 60%, and 63% for rank 5. The rank 1 models tend to have higher values of both target/template sequence identity and target/native structure similarity compared to models of lower ranks (Figs. S3 and S4 for models in the Dockground and GWIDD sets, respectively). Characteristics of the models generated for homodimers in the GWIDD set are qualitatively similar to those generated for the GWIDD heterodimers (Figs. S2 and S4).
Figure 1. Correlation of model/template sequence identity and model/native structure similarity for protein models.
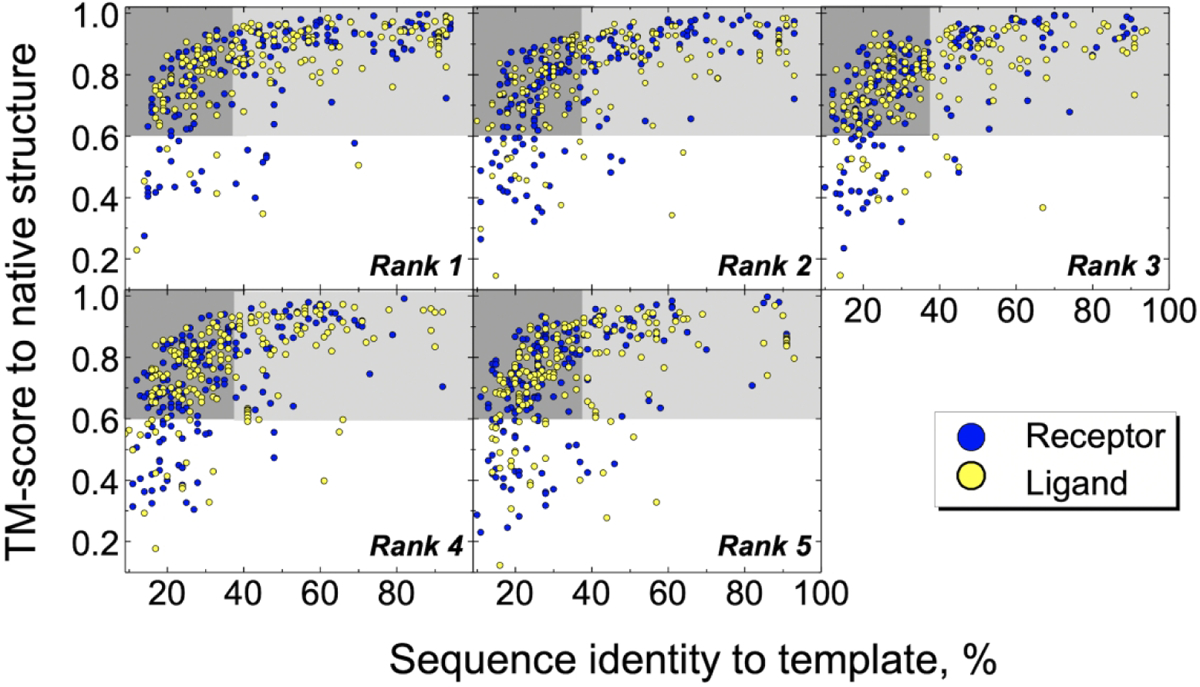
The models were generated for proteins in the Dockground benchmark set. Gray areas represent models which reproduce the fold of the native structure. The dark-gray areas contain models with low target/template sequence identity.
In this study, we chose not to use independent structure quality assessment tools as a measure of model accuracy. These tools evolve rapidly,49 and an assessment based on them would be soon out of date. Thus, the simpler sequence identity metric will be useful for longer. We used comparison with the native structure as the true measure of the accuracy. For the real case modeling scenario, when the native structure is unknown, we opted for simplicity and greater clarity to the broad user community, by correlating outputs of representative modeling/docking procedures, which could be typical choices in real-life applications, and which at the same time reflect the core accuracy-based trends in free and template-based docking paradigms.
Docking protein models of the same rank
We tested free and template-based docking on all possible combinations of protein models ranks. However, for quantitative comparison with the earlier work,33 we also used results of the “diagonal” docking (see Methods), where protein models have the same rank (Fig. 2). The diagonal docking also provides most meaningful results for the homo-dimeric complexes in the GWIDD set, as in such case both models used in the docking have the same accuracy.
Figure 2. Success rates in top-N predictions.
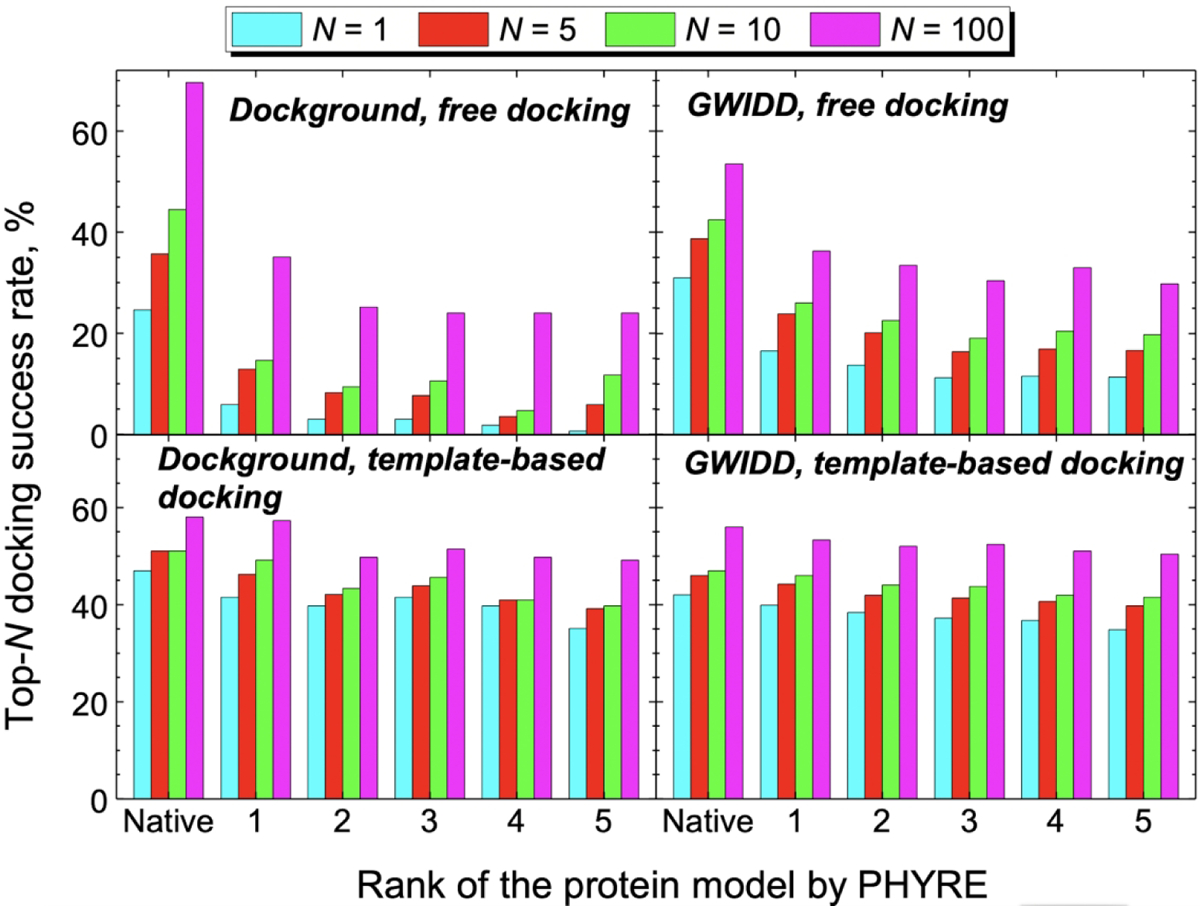
The data is for the “diagonal” docking (docking of the same rank models). The docking success rate is defined as a fraction of complexes in the set, for which at least one near-native match is in the top-N predictions (see Methods). The total number of complexes for each model rank is 171 for the Dockground set and 963 for the GWIDD set.
For both Dockground and GWIDD sets, the most significant drop in the free docking success rates for all top N docking poses (N = 1, 5, 10, and 100) was observed when the native structures of the interacting proteins are substituted by the models of rank 1. This drop constitutes ~30 – 80% of the native-structure success rate depending on the dataset and N (Fig. 2, top panels). Further decrease in the free docking success rate with the decrease of the model rank is not so significant, adding at most ~15% to the initial drop. When the model quality was estimated in terms of model-to-native RMSD, the free docking success rate decreased monotonically with the deterioration of the model quality.33 This indicates that models generated by Phyre have, on average, similar values (~ 2 – 3 Å) model-to-native RMSDs for all model ranks (Figs S3 and S4). The free docking relies on the shape complementarity of the interacting proteins. Thus, even a small distortion of the models’ surface may lead to a significantly different pool of docking poses, pushing the near-native predictions down, and sometime out of the list of the predictions (Fig. S5).
As models rank decreases, the template-based docking success rate (Fig. 2, bottom panels) drops much less than in the free docking. The maximum drop from the native structure to rank 5 models is ~ 30% for the top-1 success in the Dockground set (compared to ~95% drop for the corresponding free docking success rate). This indicates low sensitivity of the comparative docking to the models’ inaccuracies. The comparative docking relies on the structural similarity of the entire folds of the target and the template proteins. Thus, as long as the model preserves the native fold, good templates (i.e. yielding near-native docking poses) identified for the native structures, are detected for the models as well. Consequently, there should be no significant change in the docking success rate.33
The observed small fluctuations in the free and comparative docking success rates for models of ranks 2 to 5 in both datasets are related to the degree of arbitrariness in ranking of many Phyre models as their ranking parameters (e.g., e-values output by the HHsearch) are practically undistinguishable.
The differences in the free docking success rates between the Dockground and the GWIDD sets reflect different ways these sets were generated: nonredundant (Dockground) vs. redundant (GWIDD), and heterodimeric (Dockground) vs. primarily homodimeric (GWIDD). Homodimeric GWIDD complexes have consistently lower success rates than the heterodimeric complexes for both docking protocols and all top-N docking success criteria (Figs. S5 and S6), This is related to generally smaller interface areas of the homodimers in the GWIDD dataset (Fig. S7) as well as to likely presence of non-biological homodimeric assemblies.
The near-native docking predictions in the template-based protocol tend to be at the top of the list for both the native proteins and models in both sets (Fig. S8, bottom panels). In the free docking protocol, a significant part of the near-native poses is low-ranked (Fig. S8, top panels). This correlates well with the observations in our earlier work.33 Free and comparative docking protocols have different origins of sensitivity to the models’ inaccuracy. The free docking is inherently based on protein surface geometric and physicochemical complementarity, and thus is significantly influenced by the local structural distortions in the protein models. The comparative docking requires just the similarity of the protein folds, which leads to much greater tolerance to the models’ quality. This illustrated by an example in Figure 3. The structural inaccuracy introduced by modeling destroys the cluster of the free docking predictions in the correct place. At the same time, the correct prediction by the comparative docking, based on a good template, holds for the whole range of modeling accuracy levels.
Figure 3. Example of docking predictions by free and template-based docking.
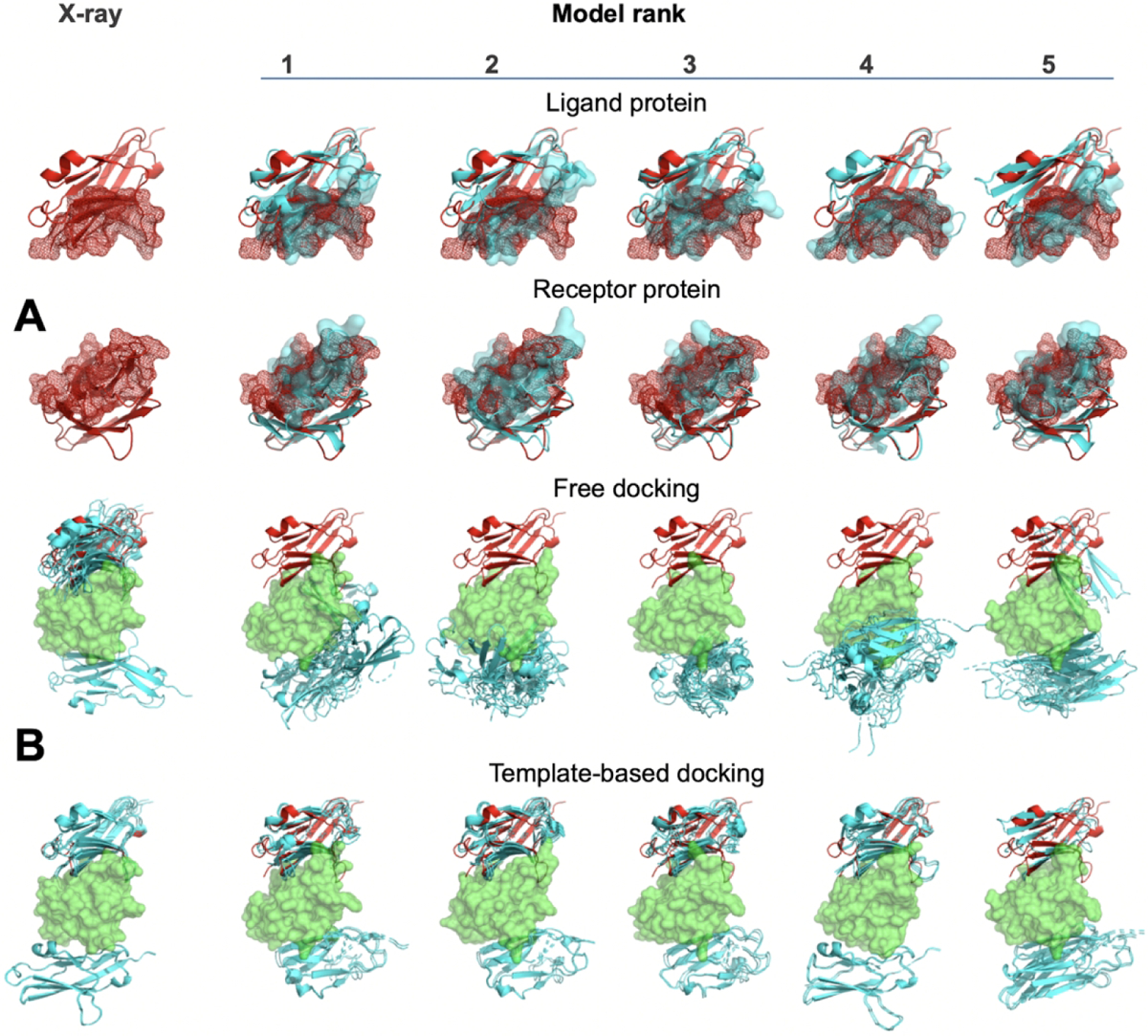
The docking was performed on co-crystallized chains of 3rnk and their models. (A) The models (cyan) are superimposed onto the native structures (red). The interface is shown by surface. (B) The receptor is in green, and the ligand in the experimentally determined position is in red. The top five predictions of the ligand pose in each docking are in cyan. The models’ inaccuracy interferes with the free docking clustering at the near-native pose, whereas the correct comparative docking prediction remains in place.
Docking protein models of different rank
In the real-case scenario, protein models to be docked may have different structural accuracy. For simplicity of the analysis, the previous studies only addressed the cases of the same model accuracy (“diagonal” docking, see Methods).32,33 In this study, we address this issue head-on. We expressed the model accuracy in terms of the rank given by the modeling software (Phyre). Thus, we performed free and template-based docking of models with all possible combinations of the rank values, from 1 to 5 (“cross” docking, see Methods). The dependence of the docking success rates on model rank in the cross docking is qualitatively similar to the one described above for the diagonal docking. The docking success rates, generally, decrease with lower rank of the protein models (Figs. 4 and 5 for the free and template-based docking, respectively). Compared to the diagonal dockings, such decrease tends to be smaller for the docking with only one protein of decreasing rank. Also, the drop in the success rate tends to be smaller for docking of a fixed-rank receptor with the decreasing-rank ligand (columns in Figs. 4 and 5), compared to the opposite situation where the decreasing-rank receptors are docked to ligands of the same rank (rows in Figs. 4 and 5). The interface of the heterodimeric protein-protein complexes is generally asymmetric - the larger protein’s binding site is typically concave.50 Thus, for consistency, in our studies, the larger protein is assigned as receptor, which is likely the cause of this observed difference. The difference in such decrease tends to get smaller with more relaxed definitions of the docking success (larger N in the near-native match in top N predictions criteria). For example, the free docking top-1 and top-100 success rates decrease by 59% and 19%, respectively, for docking receptor rank 1 with ligand decreasing rank 1 to 5. In the diagonal docking, such drop is 91% and 31%, respectively.
Figure 4. Success rates of free cross-docking of models.
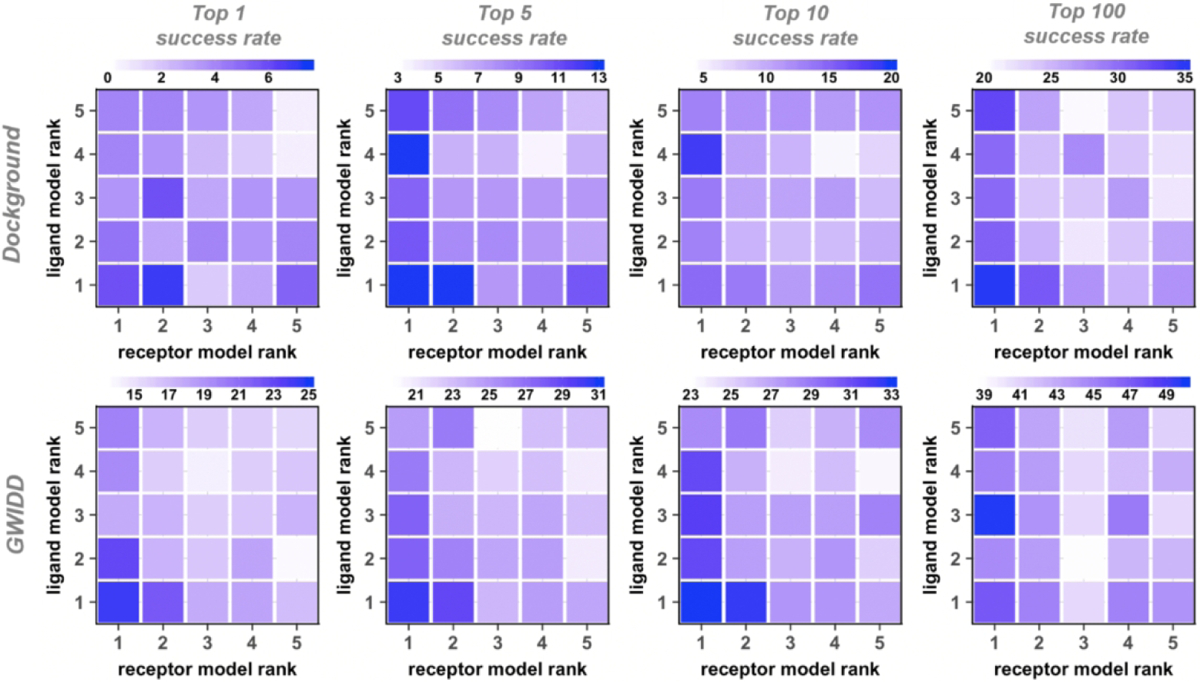
Each panel has its own color scale. The success rate is defined as in Figure 2. For the GWIDD set, results are shown only for heterodimers.
Figure 5. Success rates of template-based cross-docking of models.
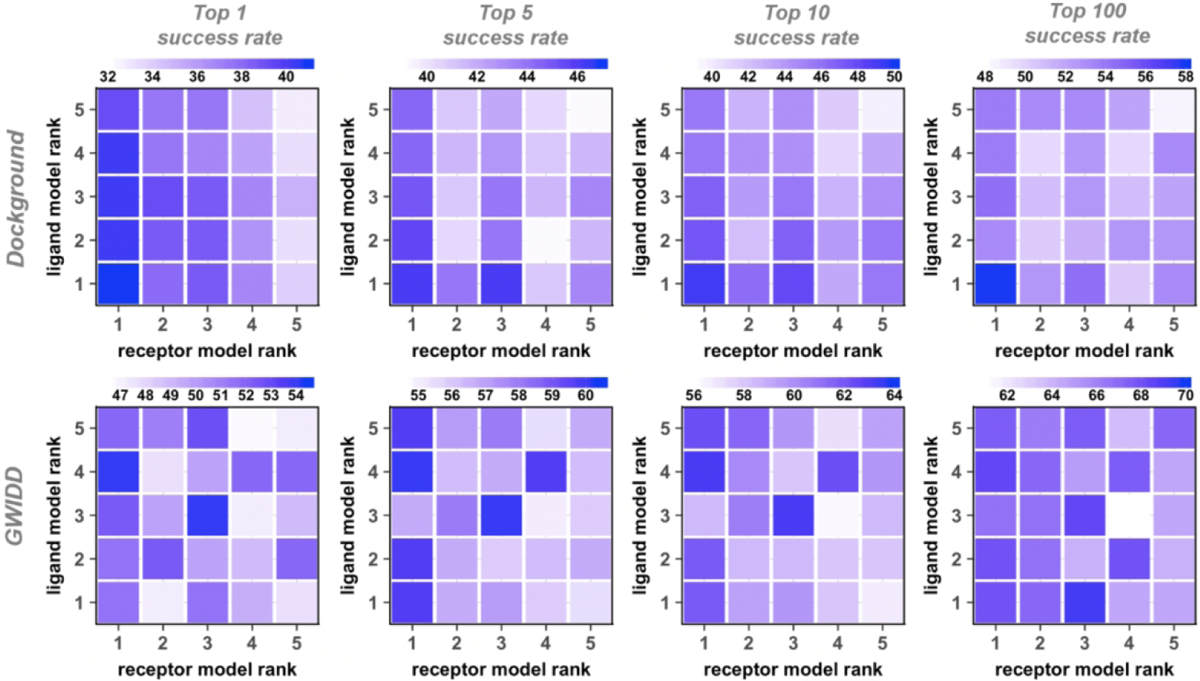
The success rate is defined as in Figure 2. For the GWIDD set, results are shown only for heterodimers.
The rank of a protein model is software specific. Thus, we mapped the docking success rates to the model’s sequence identity to the template (Figs. S9 – S11 and Fig. 6 for top-1 top-5 top-10 and top-100, respectively), and to model’s structure similarity to the native protein (Figs. S12 – S14 and Fig. 7). Again, there is a general trend of decreasing free and comparative docking success rates with the decrease of a model’s similarity to its template and its native structure. However, it was difficult to quantify these drops due to insufficient number of models in some ranges of the sequence identity and structure similarity (Figs. S3 and S4 for the Dockground and GWIDD sets, correspondingly), which lead to fluctuations in the docking success rates. When the docking success rates are remapped to structural similarity between the protein models and the docking templates, the template-based docking success rate monotonically decreases with the decrease of the TM-scores (Fig. S15).
Figure 6. Cross-docking success rates remapped on model/template sequence identity.
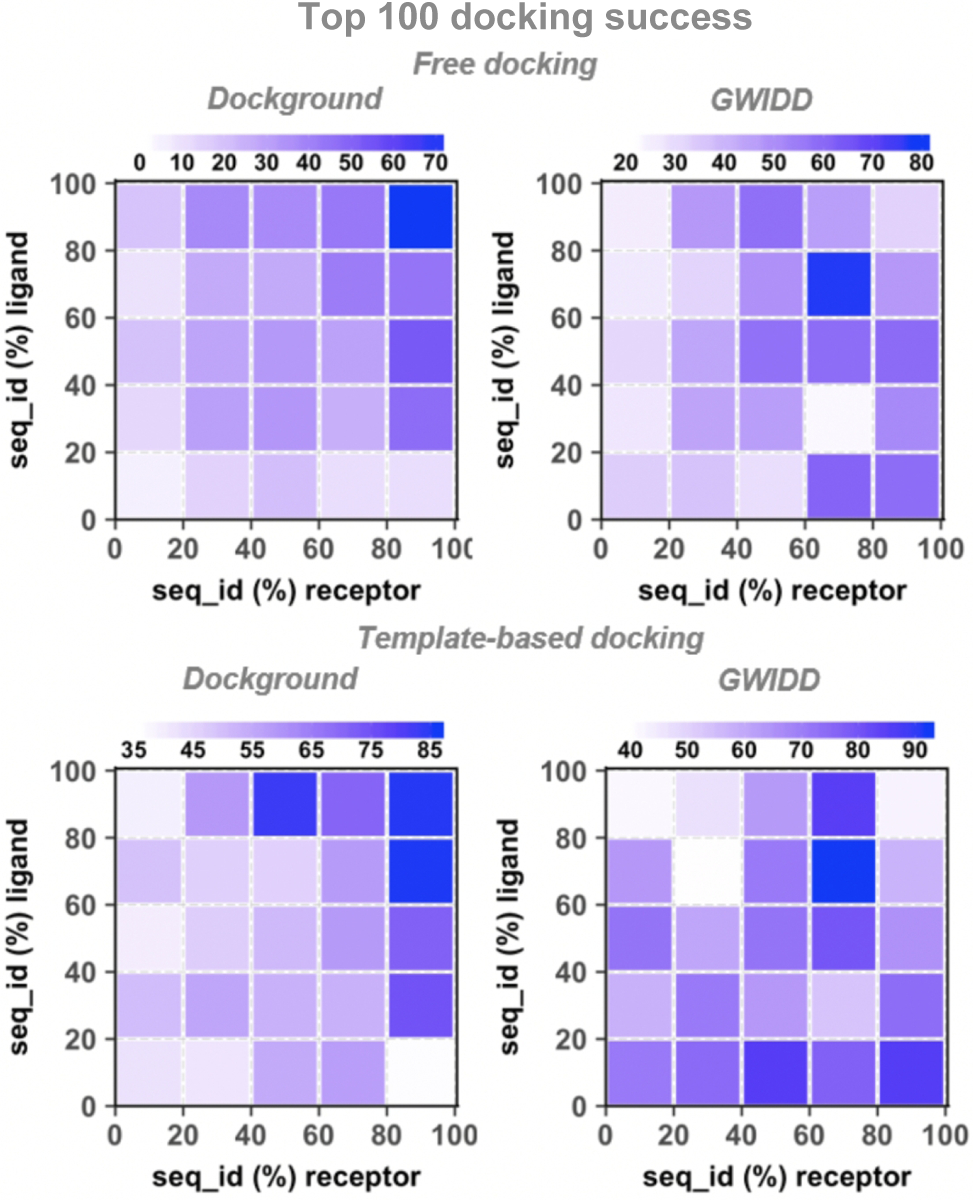
The success rate is defined as in Figure 2. For the GWIDD set, results are shown only for heterodimers.
Figure 7. Cross-docking success rates remapped on model/native structure similarity.
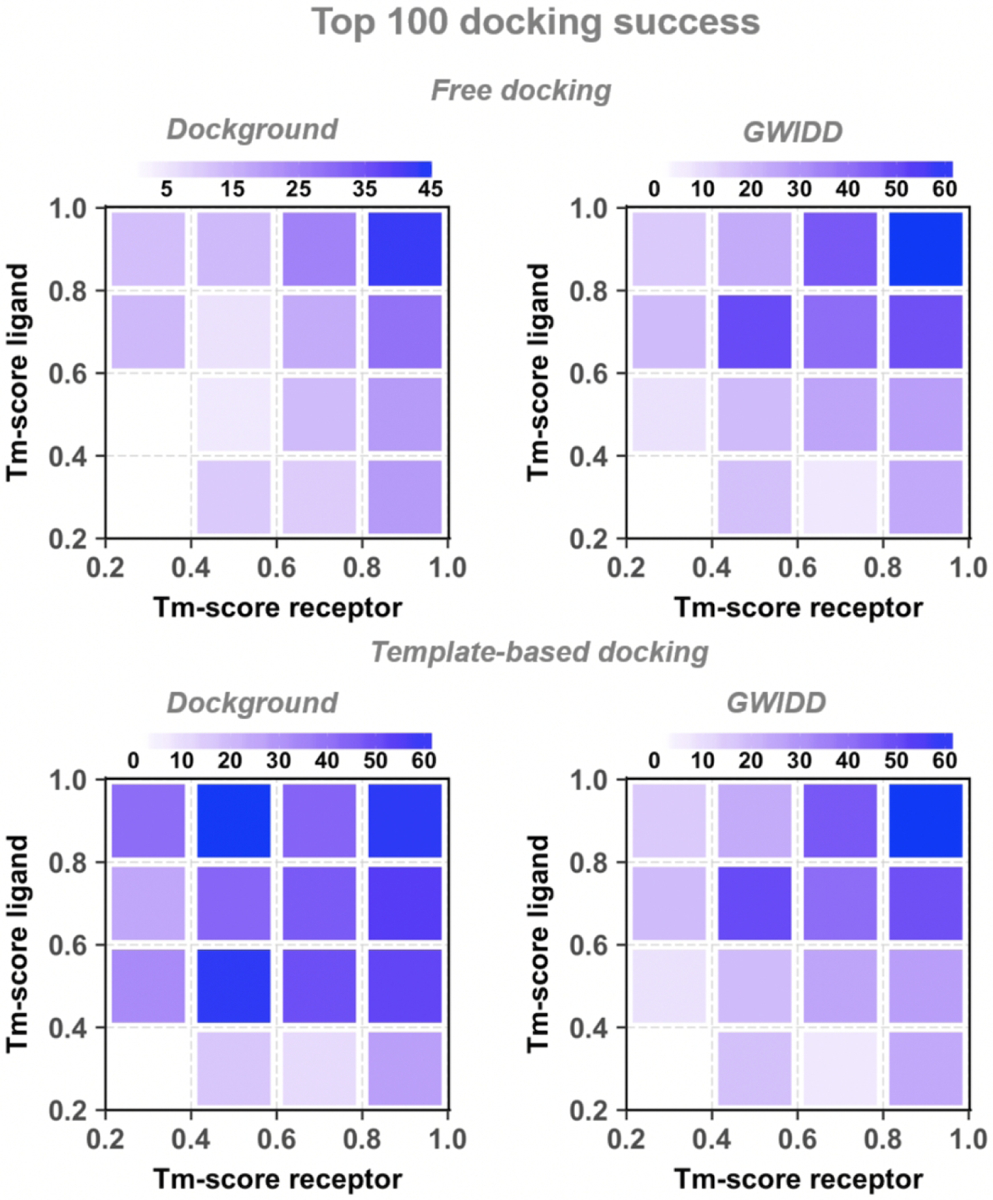
The success rate is defined as in Figure 2. For the GWIDD set, results are shown only for heterodimers.
CONCLUSIONS AND FUTURE DIRECTIONS
For development and validation of docking methodologies to predict complexes of modeled proteins, we generated two benchmark sets of protein models totaling 1134 binary complexes. To reflect the real-case modeling scenario, the models were ranked without the use of corresponding native protein structures, which would be unknown in practical applications. The modeling rank generated by Phyre2 was used as a parameter describing the model’s accuracy. We performed a comprehensive benchmarking of template-based and free protein-protein docking techniques on the sets. The docking protocols were applied to protein pairs of different modeling rank (cross docking). The results showed that the free docking success rate for the experimentally determined structures is significantly higher than that for the corresponding protein models. At the same time, the template-based docking was equally effective for docking of the protein models and docking of the experimentally determined structures. The near-native predictions by template-based docking also, generally, had higher ranks than those by the free docking. Recent CAPRI experiments also showed that the template-based approach is the best strategy for modeling protein-protein complexes when a reliable template is available.28,51 The results of this study were generated by widely utilized docking protocols based on surface complementarity (free docking) and structure similarity (template-based docking), which are typical paradigms in the docking field.6 Thus, whereas the absolute values of the docking success rates depend on the specific search/scoring method, the relative changes of these rates with respect to the structural accuracy of the proteins (the goal of this study) should be indicative of the docking in general. The results show that docking methods are applicable to protein models, including less accurate ones. The study also provides clear guidelines for using docking techniques in the structural modeling of interactomes where most protein structures will be models of limited accuracy. In the future, we will expand the scope of the study by systematic application of other protein modeling pipelines and docking approaches, such as template-based docking by structural alignments of the interfaces.8,9
Supplementary Material
ACKNOWLEDGMENTS
The authors wish to acknowledge Lawrence Kelly of the Imperial College London for generating models of proteins in the benchmark sets, and Matthew Copeland of the University of Kansas for assistance with the online resource. This study was supported by NIH grant R01GM074255 and NSF grant DBI1565107.
REFERENCES
- 1.Keskin O, Tuncbag N, Gursoy A. Predicting protein-protein interactions from the molecular to the proteome level. Chem Rev. 2016;116:4884–4909. [DOI] [PubMed] [Google Scholar]
- 2.Mosca R, Pons T, Ceol A, Valencia A, Aloy P. Towards a detailed atlas of protein–protein interactions. Curr Opin Struct Biol. 2013;23:929–940. [DOI] [PubMed] [Google Scholar]
- 3.Zhang QC, Petrey D, Deng L, et al. Structure-based prediction of protein-protein interactions on a genome-wide scale. Nature. 2012;490:556–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vakser IA. Low-resolution structural modeling of protein interactome. Curr Opin Struct Biol. 2013;23:198–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Szilagyi A, Zhang Y. Template-based structure modeling of protein–protein interactions. Curr Opin Struct Biol. 2014;24:10–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vakser IA. Protein-protein docking: From interaction to interactome. Biophys J. 2014;107:1785–1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xue LC, Rodrigues JPGLM, Dobbs D, Honavar V, Bonvin AMJJ. Template-based protein-protein docking exploiting pairwise interfacial residue restraints. Brief Bioinform. 2017;18:458–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muratcioglu S, Guven-Maiorov G, Keskin O, Gursoy A. Advances in template-based protein docking by utilizing interfaces towards completing structural interactome. Curr Opin Struct Biol. 2015;35:87–92. [DOI] [PubMed] [Google Scholar]
- 9.Estrin M, Wolfson HJ. SnapDock-template-based docking by Geometric Hashing. Bioinformatics. 2017;33:i30–i36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mirabello C, Wallner B. InterPred: A pipeline to identify and model protein-protein interactions. Proteins. 2017;85:1159–1170. [DOI] [PubMed] [Google Scholar]
- 11.Lua RC, Marciano DC, Katsonis P, Adikesavan AK, Wilkins AD, Lichtarge O. Prediction and redesign of protein-protein interactions. Prog Biophys Mol Biol. 2014;116:194–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dapkunas J, Olechnovic K, Venclovas C. Structural modeling of protein complexes: Current capabilities and challenges. Proteins. 2019;87:1222–1232. [DOI] [PubMed] [Google Scholar]
- 13.Baek M, Park T, Woo H, Seok C. Prediction of protein oligomer structures using GALAXY in CASP13. Proteins. 2019;87:1233–1240. [DOI] [PubMed] [Google Scholar]
- 14.Waterhouse A, Bertoni M, Bienert S, et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucl Acids Res. 2018;46:W296–W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Negroni J, Mosca R, Aloy P. Assessing the applicability of template-based protein docking in the twilight zone. Structure. 2014;22:1356–1362. [DOI] [PubMed] [Google Scholar]
- 16.Porter KA, Padhorny D, Desta I, et al. Template-based modeling by ClusPro in CASP13 and the potential for using co-evolutionary information in docking. Proteins. 2019;87:1241–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Park H, Lee H, Seok C. High-resolution protein–protein docking by global optimization: Recent advances and future challenges. Curr Opin Struct Biol. 2015;35:24–31. [DOI] [PubMed] [Google Scholar]
- 18.Moal IH, Moretti R, Baker D, Fernandez-Recio J. Scoring functions for protein–protein interactions. Curr Opin Struct Biol. 2013;23:862–867. [DOI] [PubMed] [Google Scholar]
- 19.Vajda S, Hall DR, Kozakov D. Sampling and scoring: A marriage made in heaven. Proteins. 2013;81:1874–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roy Burman SS, Yovanno RA, Gray JJ. Flexible backbone assembly and refinement of symmetrical homomeric complexes. Structure. 2019;27:1041–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pfeiffenberger E, Bates PA. Refinement of protein-protein complexes in contact map space with metadynamics simulations. Proteins. 2019;87:12–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mashiach E, Nussinov R, Wolfson HJ. FiberDock: Flexible induced-fit backbone refinement in molecular docking. Proteins. 2010;78:1503–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Viswanath S, Ravikant DVS, Elber R. Improving ranking of models for protein complexes with side chain modeling and atomic potentials. Proteins. 2013;81:592–606. [DOI] [PubMed] [Google Scholar]
- 24.Schindler CEM, de Vries SJ, Zacharias M. iATTRACT: Simultaneous global and local interface optimization for protein–protein docking refinement. Proteins. 2015;83:248–258. [DOI] [PubMed] [Google Scholar]
- 25.Kundrotas PJ, Zhu Z, Janin J, Vakser IA. Templates are available to model nearly all complexes of structurally characterized proteins. Proc Natl Acad Sci U S A. 2012;109:9438–9441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lensink MF, Velankar S, Baek M, Heo L, Seok C, Wodak SJ. The challenge of modeling protein assemblies: The CASP12-CAPRI experiment. Proteins. 2018;86:257–273. [DOI] [PubMed] [Google Scholar]
- 27.Vangaveti S, Vreven T, Zhang Y, Weng Z. Integrating ab initio and template-based algorithms for protein-protein complex structure prediction. Bioinformatics. 2020;doi: 10.1093/bioinformatics/btz623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Porter KA, Desta I, Kozakov D, Vajda S. What method to use for protein–protein docking? Curr Opin Struct Biol. 2019;55:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Szilagyi A, Zhang Y. Template-based structure modeling of protein-protein interactions. Curr Opin Struct Biol. 2014;24:10–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Im W, Liang J, Olson A, Zhou HX, Vajda S, Vakser IA. Challenges in structural approaches to cell modeling. J Mol Biol. 2016;428:2943–2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Keskin O, Tuncbag N, Gursoy A. Predicting protein-protein interactions from the molecular to the proteome level. Chemical Rev. 2016;116:4884–4909. [DOI] [PubMed] [Google Scholar]
- 32.Tovchigrechko A, Wells CA, Vakser IA. Docking of protein models. Protein Sci. 2002;11:1888–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Anishchenko I, Kundrotas PJ, Vakser IA. Modeling complexes of modeled proteins. Proteins. 2017;85:470–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kelley LA, Sternberg MJ. Protein structure prediction on the Web: A case study using the Phyre server. Nat Protoc. 2009;4:363–371. [DOI] [PubMed] [Google Scholar]
- 35.Kundrotas PJ, Anishchenko I, Dauzhenka T, et al. Dockground: A comprehensive data resource for modeling of protein complexes. Protein Sci. 2018;27:172–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kundrotas PJ, Zhu Z, Vakser IA. GWIDD: Genome-wide protein docking database. Nucleic Acids Res. 2010;38:D513–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kundrotas PJ, Zhu Z, Vakser IA. GWIDD: a comprehensive resource for genome-wide structural modeling of protein-protein interactions. Hum Genomics. 2012;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Anishchenko I, Kundrotas PJ, Tuzikov AV, Vakser IA. Protein models: The Grand Challenge of protein docking. Proteins. 2014;82:278–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Anishchenko I, Kundrotas PJ, Tuzikov AV, Vakser IA. Protein models docking benchmark 2. Proteins. 2015;83:891–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lensink MF, Wodak SJ. Docking, scoring, and affinity prediction in CAPRI. Proteins. 2013;81:2082–2095. [DOI] [PubMed] [Google Scholar]
- 41.Soding J Protein homology detection by HMM-HMM comparison. Bioinformatics. 2005;21:951–960. [DOI] [PubMed] [Google Scholar]
- 42.Krivov GG, Shapovalov MV, Dunbrack RL. Improved prediction of protein side-chain conformations with SCWRL4. Proteins. 2009;77:778–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vakser IA. Protein docking for low-resolution structures. Protein Eng. 1995;8:371–377. [DOI] [PubMed] [Google Scholar]
- 44.Anishchenko I, Kundrotas PJ, Vakser IA. Contact potential for structure prediction of proteins and protein complexes from Potts model. Biophys J. 2018;115:809–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sinha R, Kundrotas PJ, Vakser IA. Docking by structural similarity at protein-protein interfaces. Proteins. 2010;78:3235–3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang Y, Skolnick J. TM-align: A protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005;33:2302–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Anishchenko I, Kundrotas PJ, Tuzikov AV, Vakser IA. Structural templates for comparative protein docking. Proteins. 2015;83:1563–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kundrotas PJ, Anishchenko I, Badal VD, Das M, Dauzhenka T, Vakser IA. Modeling CAPRI targets 110–120 by template-based and free docking using contact potential and combined scoring function. Proteins. 2018;86 Suppl 1:302–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Won J, Baek M, Monastyrskyy B, Kryshtafovych A, Seok C. Assessment of protein model structure accuracy estimation in CASP13: Challenges in the era of deep learning. Proteins. 2019;87:1351–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nicola G, Vakser IA. A simple shape characteristic of protein-protein recognition. Bioinformatics. 2007;23:789–792. [DOI] [PubMed] [Google Scholar]
- 51.Lensink MF, Brysbaert G, Nadzirin N, et al. Blind prediction of homo- and hetero-protein complexes: The CASP13-CAPRI experiment. Proteins. 2019;87:1200–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.