

Abstract
Cellular interactions in the tumor microenvironment (TME) significantly govern cancer progression and drug response. The efficacy of clinical immunotherapies has fostered an exponential interest in the tumor immune microenvironment, which in turn has engendered a pressing need for robust experimental systems modeling patient-specific tumor-immune interactions. Traditional 2D in vitro tumor immunotherapy models have reconstituted immortalized cancer cell lines with immune components, often from peripheral blood. However, newly developed 3D in vitro organoid culture methods now allow the routine culture of primary human tumor biopsies and increasingly incorporate immune components. Here, we present a viewpoint on recent advances, and propose translational applications of tumor organoids for immuno-oncology research, immunotherapy modeling, and precision medicine.
Organoid Culture Systems for Modeling the Tumor Immune Microenvironment
Tumors comprise both neoplastic cells and a diversity of non-neoplastic host components, termed the tumor microenvironment (TME; see Glossary), which fosters carcinogenesis, tumor progression, and metastases of malignant cells. The complex TME includes mesenchymal-derived cells (pericytes and fibroblasts), resident or infiltrating vascular structure (endothelium), and an immune cellular network (innate and adaptive immune cells). These immune cells, including lymphocytes (T and B cells), natural killer (NK) cells, macrophages, dendritic cells (DCs), eosinophils, mast cells, and myeloid-derived suppressor cells (MDSCs), populate the tumor and can be derived from local tissue-resident populations, or infiltrate from secondary lymphoid organs (draining lymph nodes, spleen, Peyer’s patches, and mucosal tissues). The immune TME is regulated by the balance between cellular and humoral components and diverse inflammatory responses to support the growth of neoplasms into an advanced tumor biomass [1–3].
The crucial functions of immunity during tumorigenesis have been historically illustrated by well-established cancer predisposition during inflammatory and immunodeficiency states [4,5]. Recently, the potential of cancer immunomodulation was unequivocally demonstrated by the transformative anticancer efficacies of both cellular and pharmacological immunotherapies. Such immunotherapies systemically augment the immune surveillance of the body and/or locally modulate the tumor immune microenvironment. These immunotherapies include tumor antigen-targeted monoclonal antibodies, vaccines, pattern recognition receptor (PRR)-targeted therapies, and other nonspecific small molecules [interleukins (ILs), interferons, and colony-stimulating factors], which have all been used in clinical settings [6,7]. Breakthrough immunotherapy approaches that have revolutionized conventional cancer therapy are: (i) immune checkpoint inhibitors (ICIs) [8–10], such as therapeutic monoclonal antibodies against programmed cell death-1 (PD-1)/PD-L1 (programmed cell death-1 ligand) and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) to unleash cytotoxic T cell effector functions; and (ii) adoptive T cell therapies (ACT), including chimeric antigen receptor (CAR)- and T cell receptor (TCR)-T cells, as well as bulk tumor-infiltrating lymphocyte (TIL) therapy [11–13].
For decades, cancer research has utilized in vitro 2D cell cultures, and in vivo xenografts or genetically engineered animal models. The former enables viral transduction, pharmacological intervention, and multiplexed drug screening, while the latter provides the dynamic context of tumor tissue structure and vasculature. However, both conventional in vitro and in vivo models insufficiently model the complex immunobiology of native human tumors. 2D cultures can be co-cultured with different types of exogenously added heterogeneous cells to simulate cell-cell communication in tumors [14–16]. Addition of peripheral blood mononuclear cells (PBMCs) can be used for exploration of immunotherapeutic agents [17]. Furthermore, such reconstituted cells are typically not from the endogenous intratumoral stroma, and adherent monolayer cancer cells do not replicate 3D morphological structures. Furthermore, oncogene and tumor suppressor biology may be less accurate in 2D versus 3D culture [14,18]. Humanized immuno-oncology models are generated by the engraftment of patient-derived xenografts (PDXs) into immunodeficient mice bearing human immune cells, but cost, time, throughput, and complete immunocompatibility, remain challenges [19,20].
The recent advent of in vitro human organoid culture embodies a new approach to studying tumor immunobiology. As originally defined, organoids are 3D in vitro cultures of normal tissues with multiple cell lineages, including stem cells and differentiated cells, and tissue architecture in vitro [21–23]. However, organoid technology has been rapidly adapted to cancer modeling [24]. On the one hand, a forward genetic strategy can be used in which organoids from wild-type tissues or induced pluripotent stem cells (iPSCs) are engineered to bear oncogene or tumor suppressor mutations [25–29]. On the other hand, organoid methods can now robustly propagate human tumor biopsies in vitro (patient-derived organoids, PDOs). The large-scale application of 3D PDO culture has transformed in vitro cancer biology, allowing the establishment of large tumor biobanks capturing the histological and mutational diversity of human cancers [30–32]. Furthermore, current PDOs represent relatively early passage material, as opposed to long-passaged 2D cancer cell lines that, through continued genomic instability, may no longer represent the genetics of their original tumors [33,34]. Here, we discuss various organoid culture strategies in which tumor cells are grown with native or reconstituted TME immune components (Figure 1, Key Figure and Table 1). We also propose applications of tumor organoids recapitulating the immune TME for: (i) investigating cancer immunobiology; (ii) testing cancer immunotherapeutics; and (iii) developing novel approaches for personalized medicine (Figure 2).
Figure 1.
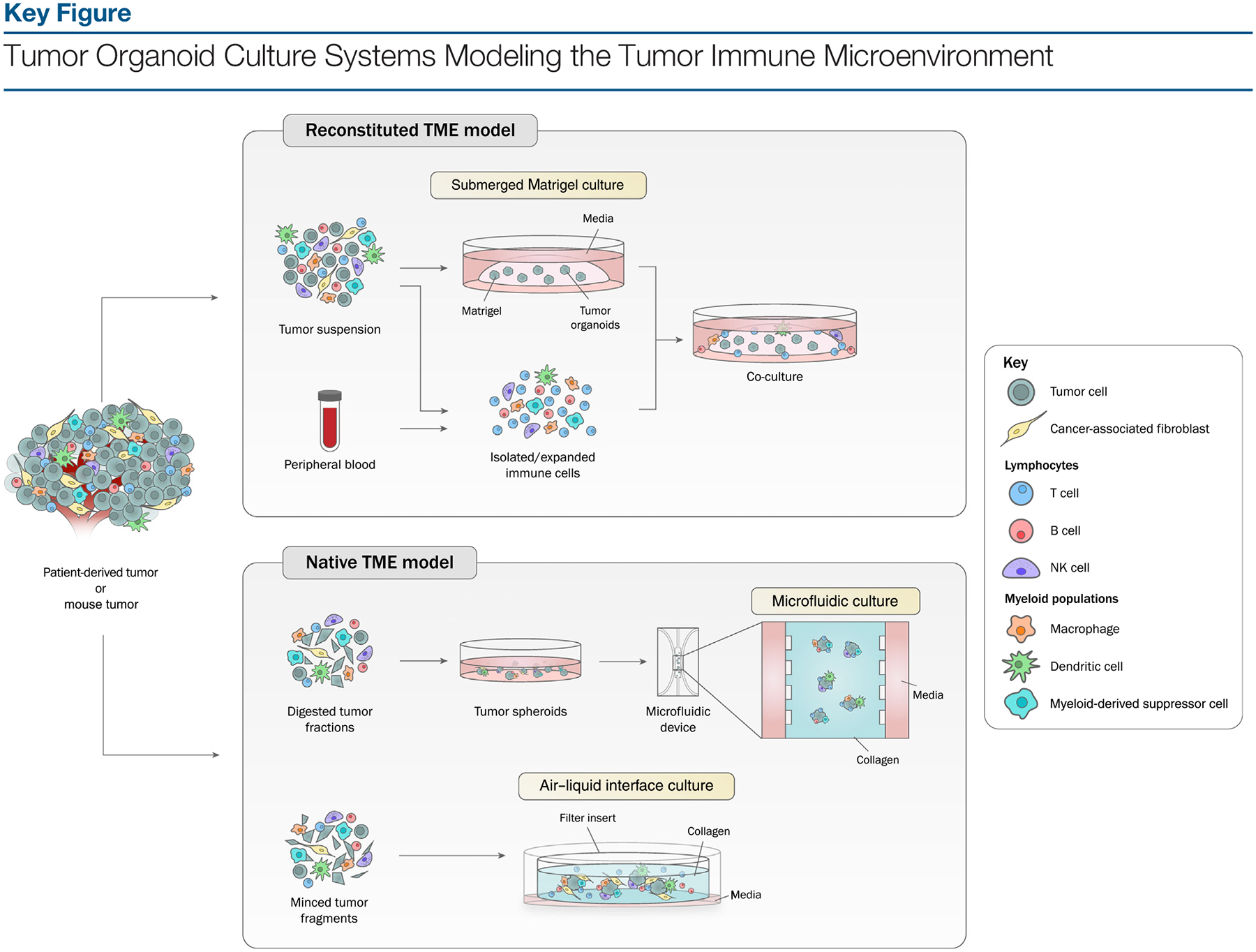
The tumor immune microenvironment can be generated in organoids by two types of approaches. In reconstituted models, organoids containing exclusively tumor cells, often from physically and enzymatically dissociated tissues, are cultured in extracellular matrix domes (e.g., Matrigel or BME-2) and submerged beneath tissue culture medium. Exogenous immune cells, such as those from autologous peripheral blood or tumor, are isolated and subsequently co-cultured with grown organoids. In holistic native TME models, the intrinsic immune microenvironment of tumor specimens is preserved along with tumor cells without reconstitution. Tumor spheroids from digested tumor tissues can be mixed with collagen and injected into microfluidic culture devices. Alternatively, in air-liquid interface (ALI) culture, minced primary tissue fragments containing both tumor cells and immune components are embedded in collagen gels within an inner transwell dish. The top of the collagen gel is exposed to air, allowing cells access to a sufficient oxygen supply. Abbreviation: NK, natural killer.
Table 1.
Overview of Ex Vivo Tumor Organoid Culture Systems Modeling the Tumor Immune Microenvironment
| Feature | Method | ||
|---|---|---|---|
| Submerged Matrigel culture | Microfluidic 3D culture | ALI culture | |
| Source | Patient-derived and mouse-derived tumor specimens | ||
| Tissue processing before culture | Tissues are dissociated physically and enzymatically (e.g., collagenase, dispase, and trypsin) | Tissues are dissociated physically and enzymatically (collagenase); samples are passed over filters to collect 40–100 μm-sized spheroid fractions, subsequently maintained in ultra-low-attachment plates | Tissues are physically minced into fragments |
| Matrix | Matrigel | Collagen | Collagen |
| Culture instrument | Any size of plate or dish | 3D microfluidic culture device | Diverse cell culture inserts and dishes or plates, including multiwells |
| Plating procedure | Cell-Matrigel mixture is plated; medium is added over Matrigel | Spheroid-collagen mixture is injected into central gel region of device; medium is added into media channels on both sides | Minced tumor tissue fragments are embedded in collagen and plated on bottom collagen layer; medium is added into an outer dish; top of collagen layer is exposed to air |
| Cell types of components retained in culture | Tumor cells exclusively; difficult to maintain stromal components long-term | Tumor cells, tumor-infiltrating lymphoid and myeloid cells, including DCs, MDSCs, and TAMs; determined by flow cytometry | Tumor cells, native immune cells (T and B cells, myeloid cells, macrophages, and NK cells) and stromal fibroblasts; determined by flow cytometry, single cell RNA-seq, and immunofluorescence |
| Culture period | Long-term culture to maintain and expand tumor organoids; short-term reconstitutive co-culture with different types of immune cell | Short-term culture; long-term culture is not reported | Tumor cells can propagate long-term; immune cells and fibroblasts in both human and mouse organoids decline over a 1–2-month period |
| Advantages | Easy to enrich and expand tumor organoids; can recapitulate genetic and morphological alterations of original tumor; potential recapitulation of clinical responses to chemotherapy and/or radiation | Requires small number of cells and small amount of medium and reagents to test; preserves multiple different types of cell in TME; enables study of tumor-immune interactions | Recapitulates genetic and morphological alterations of original tumor; preserves diverse immune cells and fibroblasts in TME; enables study of tumor–immune interactions |
| Limitations | Lack of native immune and stromal components; exogenously added TME only | Size limitation; requires specialized equipment; restricted to native tumor-infiltrating immune cells; does not reflect recruitment of circulating immune cells into tumor | Creation of uniformly sized organoids; restricted to native tumor-infiltrating immune cells; does not reflect recruitment of circulating immune cells into tumor |
| Co-culture system to reconstitute immune TME | Organoids can be co-cultured with PBMCs, primary leukocytes, TAMs, and DCs that are added to medium | Immune cells (Jurkat cells) can be added in medium to assess T cell infiltration into organotypic tumor spheroids; immune TME of primary tissue is faithfully reconstituted | Immune TME of primary tissue is faithfully reconstituted |
| Potential of immune cells in culture | Co-culture of autologous PDOs and PBMCs enriches tumor-reactive T cells, which can be used to assess efficiency of T-cell mediated cytotoxicity; enables assessment of tumor organoid killing by co-culture with TILs and CAR cells | Recapitulates response to anti-PD-1 antibody; useful culture system to test therapeutic combinations to enhance response to PD-1 response; secreted cytokine profiling | Recapitulate functional T cell activation and tumor-killing responses to anti-PD-1 and anti-PD-L1 antibodies; TCR repertoire highly conserved between TILs of original tumor and ALI PDOs |
| Refs | [37–40,50,52,53,72,92] | [43,44,55] | [36,46] |
Figure 2.
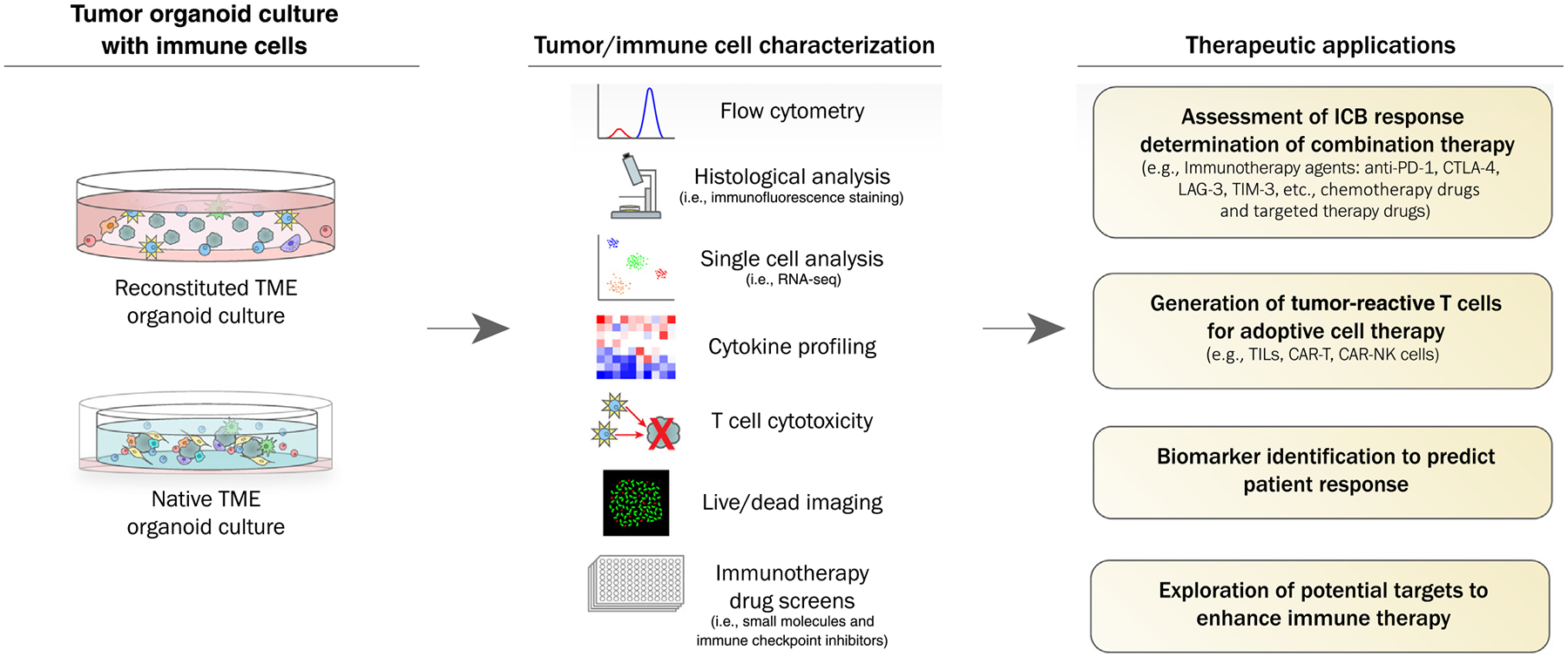
Patient-Derived Organoids (PDOs) for Personalized Cancer Immunotherapy. The native immune tumor microenvironment (TME) can be modeled using PDO air-liquid interface (ALI) organoids or PDO tumor (PDOT) microfluidic devices; alternatively, the TME can be reconstituted by adding purified immune populations to submerged tumor epithelial organoids. Multiple downstream applications include defining interactions between tumor cells and immune cells, development of immunotherapies, biomarker research, and prediction of individualized patient responses. Abbreviations: CAR, chimeric antigen receptor; CTLA-4, cytotoxic T-lymphocyte-associated protein 4; ICB, immune checkpoint blockade; NK, natural killer; PD-1, programmed cell death-1; TIL, tumor-infiltrating lymphocyte.
Submerged Matrigel Culture
A commonly used organoid method is to culture dissociated tumor cells in a dome or flat gel of 3D Matrigel, underneath tissue culture medium. In this ‘submerged Matrigel’ procedure, various growth factors and/or pathway inhibitors are supplemented depending on tissue type [27,34,35]. Exact culture conditions are customized for specific tumor histologies, but often include additives, such as Wnt3a, R-spondin, epidermal growth factor (EGF), and bone morphogenetic (BMP) inhibitor Noggin, which allow the stem cells to undergo self-renewal and differentiation (e.g., in intestinal organoids) [21]. These supplemented culture media have also been used for air-liquid interface (ALI) culture (see later) [36]. Submerged Matrigel PDOs facilitate cancer disease modeling and drug screening by recapitulating not only the genetic and phenotypic diversity of original tumors, but also by potentially modeling functional patient responses to clinical treatment [30,31,37–41]. Of note, typical submerged Matrigel PDOs exclusively enrich epithelial cancer cells but fail to retain stromal components [21]. Thus, TME modeling in this culture system requires a combinational co-culture of exogenously added immune cell types, as described later.
Microfluidic 3D Culture
Spheroid-based organotypic cultures within collagen gels in 3D microfluidic culture devices have been adapted to culture murine- or patient-derived tumors [42]. Tumor spheroids are allowed to grow in a 3D gel in the center region of the device with media from the media channels running parallel to, and located on either side of, the central region. Murine- and patient-derived organotypic tumor spheroids (MDOTS/PDOTS) from syngeneic immunocompetent murine models and patient tumor specimens, such as melanoma and Merkel cell carcinoma, can be cultured and evaluated for 1–2 weeks [43–45]. Flow cytometric immune cell profiling showed that MDOTS and PDOTS retain autologous lymphocytes (B cell and T cell) and myeloid populations [monocyte, DC, MDSC, and tumor-associated macrophages (TAM)] as well as tumor cells [43].
Air-Liquid Interface Culture
In this method, tumor organoids grown from minced primary tissue fragments are embedded in a collagen gel in an inner transwell dish. Culture medium in an outer dish diffuses via the permeable transwell into the inner dish and the top of collagen layer is exposed to air via an ALI, allowing cells access to a sufficient oxygen supply [23,25,46]. The culture of tissue fragments allows PDO generation as cancer cells en bloc alongside endogenous native stromal and immune components without reconstitution, which is distinct from submerged Matrigel culture.
Initially, ALI organoids from diverse normal tissues, including small intestine, colon, stomach, and pancreas, were shown to incorporate both epithelial and mesenchymal components [23,25]. Subsequently, the ALI organoid method was developed to culture PDOs from human biopsies, such as melanoma, renal cell carcinoma (RCC), and non-small cell lung cancer (NSCLC), and mouse tumors in syngeneic immunocompetent mice [36]. ALI PDOs preserve not only the genetic alterations of the original tumor, but also the complex cellular composition and architecture of the TME. Indeed, both tumor parenchyma and stroma are retained, including fibroblasts and a variety of endogenous infiltrating immune cell populations [36].
Reconstitution Approaches for Studying the Immune TME in Organoids
Since conventional submerged Matrigel organoids contain epithelial cells exclusively, any investigation of the TME in this system necessitates addition of exogenous stromal cell types. Such reconstitution has been utilized to supplement PDO cultures with cancer-associated fibroblasts (CAFs) [35,47–49]. Accordingly, human ductal adenocarcinoma (PDAC) organoids co-cultured with CAFs revealed that CAF-secreted Wnt drives organoid growth in Wnt-nonproducing PDAC subtypes [35]. In addition, co-culture of pancreatic stellate cells (PSCs) with PDAC organoids produces desmoplastic stroma and reveals two distinct CAF subtypes from PSCs: IL-6-expressing inflammatory CAFs activated by paracrine secreted factors from tumor cells, and high α-smooth muscle actin (αSMA)-expressing myofibroblast-like CAFs that interact with tumor cells [48]. The co-culture of organoids and CAFs has identified tumor-secreted ligands, IL-1 and transforming growth factor (TGF)β, which promote distinct inflammatory and myofibroblast CAF subtypes, respectively [49].
Diverse immune cell reconstitution of submerged Matrigel organoids has also been performed. Addition of mouse mammary adenocarcinoma-derived CD4+ T cells with and without associated macrophages (TAMs) to mammary epithelial organoids from the MMTV-PyMT mouse model demonstrated that CD4+ T lymphocytes enhanced organoid disruption and invasive behavior by directly activating a protumorigenic TAM phenotype via CD4+ T cell-secreted IL-4 [50]. Co-culture of patient-matched CAFs and peripheral blood lymphocytes (PBLs) with PDAC organoids demonstrated myofibroblast-like CAF activation and lymphocyte infiltration into Matrigel, migrating toward the tumor organoids [47]. A more complex setup involved the co-culture of cytotoxic T lymphocytes (CTLs) with DCs primed by tumor antigen released from mouse gastric tumor organoids; here, stimulated-CTLs killed gastric tumor organoids in the presence of anti-PD-L1 antibody, suggesting that the reconstitution of multiple immune cells would allow the study of tumor-immune and immune-immune cell interactions [51]. Additional reconstitution models of tumor organoid co-cultures with autologous PBLs to generate tumor-reactive T cells [52] or DCs in a Helicobacter pylori infection model [53,54] are discussed later in detail.
Holistic Approaches for Studying the Immune TME in Organoids
In contrast to adding exogenous immune components to epithelial-only organoids, holistic approaches culture tumor epithelium together with stroma endogenous immune cells as a cohesive unit without reconstitution. On the one hand, in a microfluidic culture, organotypic tumor spheroids (spheroids are 40–100 μm in diameter) are cultured as MDOTS/PDOTS with collagen in microfluidic devices for 5–9 days, preserving tumor cells and endogenous immune cells, such as lymphocyte and myeloid populations, without reconstitution [43]. This method allows the study of endogenous immune-tumor interactions or analysis of T cell infiltration into tumor spheroids by adding T cells, such as Jurkat cells, into the media [55]. On the other hand, in ALI organoid culture, large regions of tumors can be grown in their native state and thus also faithfully preserve a diversity of endogenous immune cells, including T cells [T helper (Th), cytotoxic (Tc), regulatory (Treg), and exhausted (Tex)], B cells, NK cells, and macrophages, as identified by single cell sequencing [36]. Notably, ALI PDOs can also preserve the TCR repertoire of the original fresh tumor, and expanded T cell clonotypes can correspond to ‘exhausted’ T cell phenotypes, similar to what has been observed in human fresh tumors, such as clear cell RCC [36].
ALI PDO cultures can be grown from a diversity of tumor sites, including colon, lung, pancreas, colon, and kidney, and, at least over the short-term (30 days), can accurately recapitulate the histology and mutational burden of the original malignancies [36]. By contrast to the tumor epithelium, which can be serially passaged and cryopreserved, the immune components of ALI PDOs decline over time and, despite IL-2 supplementation, do not persist beyond ~2 months [36]. Furthermore, preservation of the vasculature that carries immune cells could improve modeling of the physiological immune cell circulation, but perfusion would likely remain a challenge (see Outstanding Questions). Nevertheless, the ALI organoid approach affords a holistic strategy to in vitro immune TME modeling that can explore complex crosstalk between multiple distinct cellular populations.
Outstanding Questions.
How can one apply organoids to understanding basic mechanisms of action of immunotherapies?
How can increased immune cellular complexity be both incorporated into, and maintained in, organoid cultures?
Can culture duration be extended, to the degree of establishing organoid biobanks containing tumor cells and immune components for model standardization across laboratories?
How can organoids be best deployed for the development of current and novel immunotherapies?
Can organoids accurately assess clinical responsiveness to pharmacological or cellular immunotherapies, and can this be performed in real-time?
Applying Organoids to Cancer Immunotherapy Research
Recapitulating the Immune Checkpoint Inhibitor Response
ICIs targeting PD1/PD-L1 and CTLA-4 have demonstrated clinical responses in diverse advanced cancers, including melanoma [9,56,57], cutaneous squamous cell cancer [58], NSCLC [8,59], RCC [60], head and neck cancer [61], and mismatch repair-deficient tumors, regardless of histology [62]. Organoid biobanks have been established from multiple types of cancer, including colon [30,34,40,63,64], rectum, stomach [31,65], pancreas [66], liver [67,68], breast [32], ovary [69,70], and prostate [71], with distribution through entities such as the Human Cancer Models Initiative (HCMI)i. While these epithelial-only PDOs are widely available, their lack of immune components hinders immunotherapy assessment, such as the response to ICIs.
As one solution, immune checkpoint treatment has been performed on epithelial-only submerged Matrigel organoids reconstituted with exogenous immune components [72,73]. Notably, co-culture of patient derived-tumor-only organoids with autologous TILs after expansion of organoids and TILs separately, enabled TIL migration toward PDOs through Matrigel, and tumor cell cytotoxicity, suggesting the use of this co-culture system to assess cytotoxic TIL function [72]. The potential application for immunotherapy screening was suggested by co-culture of patient-derived tumor spheroids with autologous TILs treated with immunomodulatory antibodies targeting MICA/B and NKG2A antigens in colorectal cancer (CRC) [73]. We posit that, on the one hand, such reconstitution approaches can enable reproducible investigation by long-term expansion of the epithelial component; however, the addition of single immune cell types may not fully recapitulate the complex interplay between different immune populations following immunomodulatory drug treatment, either singly or in combination.
On the other hand, holistic culture systems including microfluidic and ALI strategies can be used to functionally model ICI. In 3D microfluidic cultures, MDOTS/PDOTS generated from mouse and human tumors can recapitulate the therapeutic sensitivity and resistance in vivo to PD-1 blockade for short-term cultures through the assessment of TIL cytotoxicity against tumor cells by tumor live/dead staining (e.g., PD1-sensitive MC38 and GL261 tumors, PD1-intermediate-sensitive CT26, PD-1- resistant B16F10, human melanoma, and Merkel cell carcinoma) [43]. In ALI cultures, ALI organoids grown from mouse tumors inoculated into syngeneic immunocompetent mice (B16-SIY, MC38, and A20) manifest CD8+ CTL activation and tumor killing in response to anti-PD-1/PD-L1 antibodies, accompanied by antigen-specific clonal CD8+ T cell expansion, where T cell functions were assessed by flow cytometry-based immune phenotyping and tumor live/dead staining [74]. Similarly, PDOs from diverse human cancer biopsies, such as NSCLC, melanoma, and RCC, exhibited CD8+ T cell expansion, activation, and subsequent tumor cell killing after 1 week of anti-PD-1 antibody treatment [74].
Extending the Therapeutic Reach of Immunotherapy
The numerous promising immunotherapy clinical outcomes to date have been counterbalanced by both intrinsic and acquired resistance [75,76]. Furthermore, several tumor histologies appear refractory to checkpoint inhibition. Organoids represent a potential in vitro approach to: (i) optimizing the efficacy of existing immunotherapies; and (ii) functionally assessing novel approaches.
Clinical immunotherapy trials increasingly explore combinatorial treatments, leveraging the distinct biology of multiple immune checkpoints (e.g., PD-1, CTLA-4, LAG-3, TIM-3, and VISTA) [10] and immunosuppressive cellular populations, such as Tregs [77] and MDSCs [78]. Moreover, immunotherapy is further added to modalities such as chemotherapy [79], radiotherapy [80], or antiangiogenic therapy [81]. Addition of molecularly targeted therapy to ICI is possible, as with MEK or BRAF inhibitors in melanoma [82]. Combining ICI with blockade of locally acting paracrine pathways within the TME, such as TGFβ [83], or chemokine receptors, such as CCR4 [84], and CXCR4 [85], is underway. Other potential targets for immunotherapy combinations include tumor cell activation of JAK-STAT [86], MAPK [87], Wnt/β-catenin [88], and NF-κB pathways [89], which can promote the release of immunomodulatory cytokines and chemokines.
In vitro culture systems are further being deployed to explore novel mechanisms, therapeutic combinations, and putative biomarkers relevant to ICI response and resistance. For example, cytokine profiling of MDOTS identified particularly high concentrations of Ccl2 in a mouse CT26 tumor of partial sensitivity to PD1 blockade [43]. Blockade of TBK1/TKKε innate immune signaling kinases inhibited immunosuppressive cytokine production by CT26 tumor cells and enhanced cytotoxicity of the tumor in combination with PD-1 blockade [43]. Similar cytokine profiling of PDOTS from anti-PD1-responsive human cancers, such as melanoma, thyroid cancer, and Merkel cell carcinoma, identified acute production of cytokines and chemokines, such as CC-chemokine ligand 19 (CCL19) and CXV chemokine ligand 13 (CXCL13), after anti-PD1 treatment; induction of CCL19/CXCL13 was consistent with response in paired clinical samples with melanoma after PD-1 inhibition [43]. In another study, small-molecule screening identified cyclin-dependent kinase (CDK) 4/6 inhibitors as compounds enhancing T cell activation in PD-1-overexpressing Jurkat T cells. Combination CDK4/6 inhibition and PD-1 blockade significantly induced tumor cell death in vitro in MC38 murine-derived MDOTS, as evidenced from tumor live/dead staining as well as from T cell-mediated tumor growth inhibition in vivo in syngeneic MC38 and CT26 mouse models [45]. Such studies highlight the potential of mechanistic in vitro immunotherapy drug studies, and could be extended to immune reconstitution of submerged Matrigel organoids or to holistic ALI organoids.
Application of PDOs for Adoptive Cell Immunotherapy
ACT immunotherapy represents a viable alternative to ICI and utilizes genetically engineered T cells with CARs or high-affinity TCRs recognizing tumor-enriched antigens, or alternatively, bulk autologous TILs. These strategies utilize ex vivo expansion of antitumor lymphocytes followed by reinfusion into patients [12]. Organoids are increasingly finding application for ACT research, such as in CAR-T cell therapy development. While CAR-T cells targeting CD19 exhibit impressive activity in hematological malignancies, such as B cell lymphoma [90] and acute lymphoblastic leukemia [91], solid tumor efficacy has been elusive [12]. PDOs have now been utilized to model tumor antigen-specific cytotoxicity of CAR-NK92 targeting EGFRvIII or FRIZZLED on CRC organoids [92]. This organoid-immune cell co-culture system could be used to assess CAR-mediated tumor-specific cytotoxicity in normal and tumor organoids [92]. Epithelial-only submerged Matrigel organoids, while lacking immune components, can serve as an antigen source enabling the selection of tumor-reactive lymphocytes. For instance, the generation of tumor-reactive CD8+ T cells by co-cultures of CRC or NSCLC PDOs with autologous PBMCs, in medium supplemented with IL-2, anti-CD28, and anti-PD1, can expand such lymphocytes with MHC-dependent cytotoxicity against autologous tumors, but not normal cells [52]. More complex organoid systems incorporating immune elements such as microfluidic or ALI methods might be similarly used for mechanistic or translational ACT studies, although further robust investigations are warranted.
Precision Medicine and Predicting the Immunotherapy Response
Conventional Matrigel PDOs can represent a promising platform for evaluating the functional responses of patients with cancer to chemotherapeutic drugs [37,39,93] and combined chemoradiation [37,39]. However, significant caveats include the need for reproduction in validation cohorts, the effects of tumor heterogeneity, the need for rapid real-time analysis, and, ultimately, the correlation against patient response and overall survival. Nevertheless, the application of PDOs to predict individualized responses to conventional treatment modalities is an active area of investigation.
The identification of biomarkers portending successful patient responses to immunotherapy has been problematic. Several biomarkers aid stratification of patient responses to anti-PD-1/PD-L1 checkpoint inhibition, including PD-L1 expression [94], CD8+ TIL density [95], mismatch repair deficiency (MMR), high microsatellite instability (MSI-H) [62], tumor mutation burden (TMB) [62], tumor neoantigen expression [96], and TCR clonality [97], as well as immune gene signatures [98]. However, the selection of appropriate patient populations to enrich clinical checkpoint inhibitor response rates remains challenging.
Organoid modeling of patient-specific immunotherapy responses suffers from the same limitations as the prediction of chemotherapy and radiotherapy, but with the additional challenges of requiring both tumor and immune cell compartments. Here, holistic systems may find particular utility because they contain the immune TME. The evaluation of cytokine profiling and T cell cytotoxicity against tumor cells for 1 week in microfluidic culture PDOTS-retaining tumor cells and autologous immune components might provide the potential to predict or assess a patient’s response to ICI treatment [43]. Alternatively, ALI organoids co-preserving tumor epithelium alongside diverse endogenous immune elements might model responses to immune checkpoint inhibition by evaluating T cell functions through flow cytometry, fluorescence staining, and tumor killing [72]. These and other predictive approaches will require prospective validation and correlation with clinical outcomes, but represent a substantial opportunity for clinical translation via identification of cohorts optimally responsive to immunotherapies.
Modeling Cancer-Associated Inflammation in Tumor Organoids
Chronic inflammation, accounting for 15–20% of cancer deaths, can promote both tumorigenesis and treatment resistance [4]. Most commonly, these inflammatory diseases can include inflammatory bowel disease (IBD) in CRC, hepatitis B or C virus (HBV/HCV) infection in hepatocellular carcinoma (HCC), H. pylori-induced gastritis, and Barrett’s esophagus [99]. Oncogenic viruses, such as Epstein-Barr Virus (EBV) and human papilloma virus (HPV), not only encode transforming proteins, but can also elicit inflammation [99,100]. The substantial TME reciprocal crosstalk between neoplastic and inflammatory cells incorporates dynamic components of the adaptive, humoral, and innate immune systems, to cooperatively regulate cancer-associated inflammation and tumorigenesis [4,101]. Thus, organoid models might facilitate fundamental insights into such cancer-associated inflammation, providing opportunities for disease prevention and treatment.
Numerous infection models relevant to cancer utilize organoids, such as HBV [102,103] and HCV [104] infection of liver organoids and H. pylori infection of gastric organoids [105–107]. Microinjection of H. pylori into gastric organoids induced strong inflammatory responses, including the release of IL-8 [105]. The reciprocal interactions between epithelial cells and carcinogenic pathogens is illustrated by gastric organoids producing urease, a chemoattractant to H. pylori, after which the bacteria then deliver the bacterially encoded CagA transforming protein to the gastric epithelium [107]. Additionally, the prolonged exposure of carcinogenic Escherichia coli carrying the pathogenicity island pks to human intestinal organoids can elicit oncogenic mutations [108].
Such infection systems can be extended to organoid co-cultures with immune cells. Upon co-culture of human gastric organoids and DCs isolated from PBMC, H. pylori microinjection into gastric organoids increased DC recruitment into gastric organoids in a gastric epithelium secreted chemokine-dependent manner, followed by DC phagocytosis of H. pylori applied to the organoid lumen; this was evidenced from differential fluorescence expression, labeling each cell and H. pylori, allowing the visualization of their interactions [53]. H. pylori also induced PD-L1 expression on gastric epithelium mediated by sonic hedgehog (Shh) signaling, and the inhibition of PD-L1-induced organoid cell death in a co-culture of CTLs, DCs, and human gastric organoids. This suggested that H. pylori-induced PD-L1 expression is protective against cytotoxic activity [54]. In addition to immune reconstitutive approaches, holistic microfluidic or ALI organoid strategies might also find application. Such organoid models containing triads of immune cells, tumor cells, and pathogens have the potential to elaborate a carcinogenic interplay.
Concluding Remarks
Recent advances in 3D organoid cultures of human malignant cells, engineered either from primary wild-type tissues or directly from tumor biopsies, are rapidly transforming the landscape of in vitro cancer experimentation and clinical translation. Intrinsic to the application of organoids to the study of tumor immunology is the need to incorporate immune components alongside tumor epithelium, as currently addressed by reconstitutive and holistic approaches. Optimally, the immune TME within organoids would contain an entire diversity of immune cells, including T, B, NK, and myeloid cells and other innate immune cells, and might conceivably incorporate both tumor-infiltrating and peripheral immune cell populations. The additional overlay of infectious and carcinogenic pathogens certainly adds further complexity (see Outstanding Questions). Co-culture of tumor organoids with peripheral immune cells from PBMCs or lymph nodes might also allow modeling of the cancer-immunity cycle, including T cell priming/activation, trafficking/infiltration of T cells into tumors, and recognition/killing of tumor cells by T cells. Introducing pathogens or commensal microbiota into tumor organoids with immune cells could ostensibly recapitulate cancer-associated inflammation and carcinogenesis through pathogen, epithelial-immune cell interactions, enabling an assessment of immunomodulatory outcomes and immunotherapeutic responses. Furthermore, these culture systems may enable investigations of innate immunity in promoting T cell effector function through inclusion of relevant cell types, such as DCs, monocytes, macrophages, mast cells, and NK cells, and further allow interrogation of toll-like receptor (TLR), NOD-like receptor (NLR), and stimulator of interferon genes (STING) agonists.
By analogy to biobanks of epithelial-only organoids, long-term cryopreservable PDO cultures containing immune components might allow experimental standardization of models, which otherwise are ‘n = 1’ models that differ between laboratories. Substantial challenges remain in the long-term culture and preservation of immune cells within organoids; alternatively, matched tumor epithelium and immune cell components could be banked in parallel and then reconstituted. Additional supplements, such as IL-2, anti-CD3, and anti-CD28 antibodies, might facilitate the long-term maintenance of immune cells, but recapitulation of antitumor responses would certainly require robust validation. Primary organoid cultures of peripheral immune organoids, such as lymph nodes, might offer opportunities for investigating the crosstalk between immune cells, tumor cells, and local TME-specific events (see Outstanding Questions).
An important translational challenge for immune organoid research is represented by modeling sensitivity and resistance to immunotherapies, spanning checkpoint inhibition, novel pathways, and ACT strategies. In addition to elucidating underlying resistance pathways, organoids might facilitate therapeutic efforts, such as in vitro screening and optimizing pharmacological and cellular immunotherapies; and might also allow real-time determination of patient sensitivity to single or combinatorial treatments, if short-term responses in culture can indeed accurately reflect clinical responses and long-term outcomes (see Outstanding Questions). Ultimately, one can anticipate that current and future organoid methodologies will substantially further immuno-oncology basic science and translation, forging a path towards realizing the already considerable promise of human tumor immunotherapy.
Highlights.
Diverse in vitro culture methods allow modeling of tumor immunity.
‘Tumor-only’ 3D patient-derived organoid (PDO) systems can be reconstituted with exogenously added immune components.
Holistic air-liquid interface (ALI) tumor organoid cultures and microfluidic cultures can recapitulate the tumor microenvironment (TME) by preserving endogenous stromal components including diverse immune cells (B, T, and natural killer cells, and macrophages) without reconstitution.
Tumor-infiltrating lymphocytes in the native TME of ALI PDOs or microfluidic PDO tumor spheroids can model programmed cell death-1 (PD-1)-dependent immune checkpoint inhibition and tumor cytotoxicity.
Regardless of methodology, in vitro culture models of the immune TME allow exploration of tumor immunology and novel immunotherapeutic targets.
Acknowledgments
The authors gratefully acknowledge Amanda Mah for figure preparation, Yuan-Hung Lo and Kasper Karlsson for helpful discussion, and support from the Emerson Collective, Ludwig Cancer Research, National Institutes of Health (NIH) CA217851, CA224081, AI116484, and the Human Cancer Models Initiative.
Glossary
- Air-liquid interface (ALI)
culture method by which cells are grown in a collagen matrix where the top of gel is exposed to the air
- B16F10
murine melanoma cell line
- Chimeric antigen receptor (CAR)
genetically engineered T cell receptors that recognize tumor antigens
- CT26
murine colon carcinoma cell line
- Cytotoxic T lymphocyte (CTL)
effector T lymphocytes that kill antigen-expressing target cells
- Exhausted T cell
dysfunctional T cells characterized by loss of effector function, expression of multiple inhibitory receptors, and altered transcriptional network due to chronic antigen exposure, such as viral infection and cancer
- GL261
murine glioma cell line
- High microsatellite instability (MSI-H)
high frequency of mutations within microsatellites that are short, repeated sequences of DNA
- Immune checkpoint inhibitor (ICI)
inhibitors, often monoclonal antibody based, that neutralize inhibitory immune checkpoints, allowing immune cells to recognize and attack cancer
- MC38
murine colon adenocarcinoma cell line
- Patient-derived organoids (PDOs)
3D cultures of patient’s tumor cells grown in matrix that retain the architecture, genetic, and phenotypic changes of the primary tumor
- Pattern recognition receptors (PRRs)
receptors capable of recognizing molecules such as pathogens; have crucial roles in innate immunity
- Peripheral blood lymphocytes (PBLs)
blood cells comprising T, B, NK, and NKT cells. Depending on the purpose, specific lymphocytes, such as CD3+ T cells, are isolated and used as PBLs
- Peripheral blood mononuclear cells (PBMCs)
unfractionated circulating blood cells comprising lymphocytes (T, B, NK, and NKT cells), monocytes, and DCs
- Tumor-infiltrating lymphocytes (TILs)
lymphocytes that infiltrate tumors
- Tumor microenvironment (TME)
complex cellular milieu surrounding tumor epithelium, including diverse supporting cell types, such as activated fibroblasts, blood vessels, infiltrating immune cells, and extracellular matrix
- Tumor neoantigen
tumor-specific antigens generated by somatic mutations
- α-Smooth muscle actin (αSMA)
often used as a myofibroblast marker
Resources
References
- 1.Palucka AK and Coussens LM (2016) The basis of oncoimmunology. Cell 164, 1233–1247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Quail DF and Joyce JA (2013) Microenvironmental regulation of tumor progression and metastasis. Nat. Med 19, 1423–1437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gajewski TF et al. (2013) Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol 14, 1014–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mantovani A et al. (2008) Cancer-related inflammation. Nature 454, 436–444 [DOI] [PubMed] [Google Scholar]
- 5.Kitamura T et al. (2015) Immune cell promotion of metastasis. Nat. Rev. Immunol 15, 73–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Houot R et al. (2011) Targeting immune effector cells to promote antibody-induced cytotoxicity in cancer immunotherapy. Trends Immunol 32, 510–516 [DOI] [PubMed] [Google Scholar]
- 7.Demaria O et al. (2019) Harnessing innate immunity in cancer therapy. Nature 574, 45–56 [DOI] [PubMed] [Google Scholar]
- 8.Socinski MA et al. (2018) Atezolizumab for first-line treatment of metastatic nonsquamous NSCLC. N. Engl. J. Med 378, 2288–2301 [DOI] [PubMed] [Google Scholar]
- 9.Larkin J et al. (2019) Five-year survival with combined nivolumab and ipilimumab in advanced melanoma. N. Engl. J. Med 381, 1535–1546 [DOI] [PubMed] [Google Scholar]
- 10.Pardoll DM (2012) The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 12, 252–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hammerl D et al. (2018) Adoptive T cell therapy: new avenues leading to safe targets and powerful allies. Trends Immunol 39, 921–936 [DOI] [PubMed] [Google Scholar]
- 12.Rosenberg SA and Restifo NP (2015) Adoptive cell transfer as personalized immunotherapy for human cancer. Science (New York, N.Y.) 348, 62–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tran E et al. (2016) T-cell transfer therapy targeting mutant KRAS in cancer. N. Engl. J. Med 375, 2255–2262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Riedl A et al. (2017) Comparison of cancer cells in 2D vs 3D culture reveals differences in AKT-mTOR-S6K signaling and drug responses. J. Cell Sci 130, 203–218 [DOI] [PubMed] [Google Scholar]
- 15.Sung PJ et al. (2019) Cancer-associated fibroblasts produce Netrin-1 to control cancer cell plasticity. Cancer Res 79, 3651–3661 [DOI] [PubMed] [Google Scholar]
- 16.Faget J et al. (2011) Early detection of tumor cells by innate immune cells leads to T(reg) recruitment through CCL22 production by tumor cells. Cancer Res 71, 6143–6152 [DOI] [PubMed] [Google Scholar]
- 17.Mo X et al. (2019) HTiP: high-throughput immunomodulator phenotypic screening platform to reveal IAP antagonists as anti-cancer immune enhancers. Cell Chem. Biol 26, 331–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Han K et al. (2020) CRISPR screens in cancer spheroids identify 3D growth-specific vulnerabilities. Nature 580, 136–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jespersen H et al. (2017) Clinical responses to adoptive T-cell transfer can be modeled in an autologous immune-humanized mouse model. Nat. Commun 8, 707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao Y et al. (2018) Development of a new patient-derived xenograft humanised mouse model to study human-specific tumour microenvironment and immunotherapy. Gut 67, 1845–1854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sato T et al. (2009) Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459, 262–265 [DOI] [PubMed] [Google Scholar]
- 22.Fujii M et al. (2018) Human intestinal organoids maintain self-renewal capacity and cellular diversity in niche-inspired culture condition. Cell Stem Cell 23, 787–793 [DOI] [PubMed] [Google Scholar]
- 23.Ootani A et al. (2009) Sustained in vitro intestinal epithelial culture within a Wnt-dependent stem cell niche. Nat. Med 15, 701–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Drost J and Clevers H (2018) Organoids in cancer research. Nat. Rev. Cancer 18, 407–418 [DOI] [PubMed] [Google Scholar]
- 25.Li X et al. (2014) Oncogenic transformation of diverse gastrointestinal tissues in primary organoid culture. Nat. Med 20, 769–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matano M et al. (2015) Modeling colorectal cancer using CRISPR-Cas9-mediated engineering of human intestinal organoids. Nat. Med 21, 256–262 [DOI] [PubMed] [Google Scholar]
- 27.Drost J et al. (2015) Sequential cancer mutations in cultured human intestinal stem cells. Nature 521, 43–47 [DOI] [PubMed] [Google Scholar]
- 28.Crespo M et al. (2017) Colonic organoids derived from human induced pluripotent stem cells for modeling colorectal cancer and drug testing. Nat. Med 23, 878–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Drost J et al. (2017) Use of CRISPR-modified human stem cell organoids to study the origin of mutational signatures in cancer. Science (New York, N.Y.) 358, 234–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van de Wetering M et al. (2015) Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 161, 933–945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yan HHN et al. (2018) A comprehensive human gastric cancer organoid biobank captures tumor subtype heterogeneity and enables therapeutic screening. Cell Stem Cell 23, 882–897 [DOI] [PubMed] [Google Scholar]
- 32.Sachs N et al. (2018) A living biobank of breast cancer organoids captures disease heterogeneity. Cell 172, 373–386 [DOI] [PubMed] [Google Scholar]
- 33.Lee SH et al. (2018) Tumor evolution and drug response in patient-derived organoid models of bladder cancer. Cell 173, 515–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fujii M et al. (2016) A colorectal tumor organoid library demonstrates progressive loss of niche factor requirements during tumorigenesis. Cell Stem Cell 18, 827–838 [DOI] [PubMed] [Google Scholar]
- 35.Seino T et al. (2018) Human pancreatic tumor organoids reveal loss of stem cell niche factor dependence during disease progression. Cell Stem Cell 22, 454–467 [DOI] [PubMed] [Google Scholar]
- 36.Neal JT et al. (2018) Organoid modeling of the tumor immune microenvironment. Cell 175, 1972–1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ganesh K et al. (2019) A rectal cancer organoid platform to study individual responses to chemoradiation. Nat. Med 25, 1607–1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Driehuis E et al. (2019) Oral mucosal organoids as a potential platform for personalized cancer therapy. Cancer Discov 9, 852–871 [DOI] [PubMed] [Google Scholar]
- 39.Pasch CA et al. (2019) Patient-derived cancer organoid cultures to predict sensitivity to chemotherapy and radiation. Clin. Cancer Res 25, 5376–5387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vlachogiannis G et al. (2018) Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 359, 920–926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roerink SF et al. (2018) Intra-tumour diversification in colorectal cancer at the single-cell level. Nature 556, 457–462 [DOI] [PubMed] [Google Scholar]
- 42.Sontheimer-Phelps A et al. (2019) Modelling cancer in microfluidic human organs-on-chips. Nat. Rev. Cancer 19, 65–81 [DOI] [PubMed] [Google Scholar]
- 43.Jenkins RW et al. (2018) Ex vivo profiling of PD-1 blockade using organotypic tumor spheroids. Cancer Discov 8, 196–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aref AR et al. (2018) 3D microfluidic ex vivo culture of organotypic tumor spheroids to model immune checkpoint blockade. Lab Chip 18, 3129–3143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Deng J et al. (2018) CDK4/6 inhibition augments antitumor immunity by enhancing T-cell activation. Cancer Discov 8, 216–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li X et al. (2016) An air-liquid interface culture system for 3D organoid culture of diverse primary gastrointestinal tissues. Methods Mol. Biol 1422, 33–40 [DOI] [PubMed] [Google Scholar]
- 47.Tsai S et al. (2018) Development of primary human pancreatic cancer organoids, matched stromal and immune cells and 3D tumor microenvironment models. BMC Cancer 18, 335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ohlund D et al. (2017) Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med 214, 579–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Biffi G et al. (2019) IL1-Induced JAK/STAT signaling is antagonized by TGFbeta to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov 9, 282–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.DeNardo DG et al. (2009) CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell 16, 91–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chakrabarti J et al. (2018) Hedgehog signaling induces PD-L1 expression and tumor cell proliferation in gastric cancer. Oncotarget 9, 37439–37457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dijkstra KK et al. (2018) Generation of tumor-reactive T cells by co-culture of peripheral blood lymphocytes and tumor organoids. Cell 174, 1586–1598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sebrell TA et al. (2019) A novel gastric spheroid co-culture model reveals chemokine-dependent recruitment of human dendritic cells to the gastric epithelium. Cell Mol. Gastroenterol. Hepatol 8, 157–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Holokai L et al. (2019) Increased programmed death-ligand 1 is an early epithelial cell response to Helicobacter pylori infection. PLoS Pathog 15, e1007468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kitajima S et al. (2019) Suppression of STING associated with LKB1 loss in KRAS-driven lung cancer. Cancer Discov 9, 34–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hodi FS et al. (2010) Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med 363, 711–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Robert C et al. (2011) Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N. Engl. J. Med 364, 2517–2526 [DOI] [PubMed] [Google Scholar]
- 58.Migden MR et al. (2018) PD-1 Blockade with cemiplimab in advanced cutaneous squamous-cell carcinoma. N. Engl. J. Med 379, 341–351 [DOI] [PubMed] [Google Scholar]
- 59.Garon EB et al. (2015) Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med 372, 2018–2028 [DOI] [PubMed] [Google Scholar]
- 60.Motzer RJ et al. (2015) Nivolumab versus everolimus in advanced renal-cell carcinoma. N. Engl. J. Med 373, 1803–1813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ferris R and Gillison ML (2017) Nivolumab for squamous-cell cancer of head and neck. N. Engl. J. Med 376, 596. [DOI] [PubMed] [Google Scholar]
- 62.Le DT et al. (2015) PD-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med 372, 2509–2520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Weeber F et al. (2015) Preserved genetic diversity in organoids cultured from biopsies of human colorectal cancer metastases. Proc. Natl. Acad. Sci. U. S. A 112, 13308–13311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yao Y et al. (2020) Patient-derived organoids predict chemoradiation responses of locally advanced rectal cancer. Cell Stem Cell 26, 17–26.e6 [DOI] [PubMed] [Google Scholar]
- 65.Seidlitz T et al. (2019) Human gastric cancer modelling using organoids. Gut 68, 207–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Huang L et al. (2015) Ductal pancreatic cancer modeling and drug screening using human pluripotent stem cell- and patient-derived tumor organoids. Nat. Med 21, 1364–1371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Broutier L et al. (2017) Human primary liver cancer-derived organoid cultures for disease modeling and drug screening. Nat. Med 23, 1424–1435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nuciforo S et al. (2018) Organoid models of human liver cancers derived from tumor needle biopsies. Cell Rep 24, 1363–1376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kopper O et al. (2019) An organoid platform for ovarian cancer captures intra- and interpatient heterogeneity. Nat. Med 25, 838–849 [DOI] [PubMed] [Google Scholar]
- 70.Hill SJ et al. (2018) Prediction of DNA repair inhibitor response in short-term patient-derived ovarian cancer organoids. Cancer Discov 8, 1404–1421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Beshiri ML et al. (2018) A PDX/organoid biobank of advanced prostate cancers captures genomic and phenotypic heterogeneity for disease modeling and therapeutic screening. Clin. Cancer Res 24, 4332–4345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kong JCH et al. (2018) Tumor-infiltrating lymphocyte function predicts response to neoadjuvant chemoradiotherapy in locally advanced rectal cancer. JCO Precis. Oncol 2, 1–15 [DOI] [PubMed] [Google Scholar]
- 73.Courau T et al. (2019) Cocultures of human colorectal tumor spheroids with immune cells reveal the therapeutic potential of MICA/B and NKG2A targeting for cancer treatment. J. Immunother. Cancer 7, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Neal JT and Kuo CJ (2016) Organoids as models for neoplastic transformation. Annu. Rev. Pathol 11, 199–220 [DOI] [PubMed] [Google Scholar]
- 75.Sharma P et al. (2017) Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 168, 707–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hugo W et al. (2016) Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell 165, 35–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sakaguchi S et al. (2008) Regulatory T cells and immune tolerance. Cell 133, 775–787 [DOI] [PubMed] [Google Scholar]
- 78.Ostrand-Rosenberg S and Fenselau C (2018) Myeloid-derived suppressor cells: immune-suppressive cells that impair antitumor immunity and are sculpted by their environment. J. immunol. (Baltimore, Md.: 1950) 200, 422–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pfirschke C et al. (2016) Immunogenic chemotherapy sensitizes tumors to checkpoint blockade therapy. Immunity 44, 343–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sharabi AB et al. (2015) Radiation and checkpoint blockade immunotherapy: radiosensitisation and potential mechanisms of synergy. Lancet Oncol 16, e498–e509 [DOI] [PubMed] [Google Scholar]
- 81.Wallin JJ et al. (2016) Atezolizumab in combination with bevacizumab enhances antigen-specific T-cell migration in metastatic renal cell carcinoma. Nat. Commun 7, 12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ribas A et al. (2019) Combined BRAF and MEK inhibition with PD-1 blockade immunotherapy in BRAF-mutant melanoma. Nat. Med 25, 936–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mariathasan S et al. (2018) TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 554, 544–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Doi T et al. (2019) A Phase I study of the anti-CC chemokine receptor 4 antibody, mogamulizumab, in combination with nivolumab in patients with advanced or metastatic solid tumors. Clin. Cancer Res 25, 6614–6622 [DOI] [PubMed] [Google Scholar]
- 85.D’Alterio C et al. (2019) Targeting CXCR4 potentiates anti-PD-1 efficacy modifying the tumor microenvironment and inhibiting neoplastic PD-1. J. Exp. Clin. Cancer Res 38, 432–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zaretsky JM et al. (2016) Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med 375, 819–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ebert PJR et al. (2016) MAP kinase inhibition promotes T cell and anti-tumor activity in combination with PD-L1 checkpoint blockade. Immunity 44, 609–621 [DOI] [PubMed] [Google Scholar]
- 88.Luke JJ et al. (2019) WNT/β-catenin pathway activation correlates with immune exclusion across human cancers. Clin. Cancer Res 25, 3074–3083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lim S-O et al. (2016) Deubiquitination and stabilization of PD-L1 by CSN5. Cancer Cell 30, 925–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Schuster SJ et al. (2017) Chimeric antigen receptor T cells in refractory B-cell lymphomas. N. Engl. J. Med 377, 2545–2554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Park JH et al. (2018) Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N. Engl. J. Med 378, 449–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Schnalzger TE et al. (2019) 3D model for CAR-mediated cytotoxicity using patient-derived colorectal cancer organoids. EMBO J 38, e100928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tiriac H et al. (2018) Organoid profiling identifies common responders to chemotherapy in pancreatic cancer. Cancer Discov 8, 1112–1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Topalian SL et al. (2012) Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med 366, 2443–2454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chen PL et al. (2016) Analysis of immune signatures in longitudinal tumor samples yields insight into biomarkers of response and mechanisms of resistance to immune checkpoint blockade. Cancer Discov 6, 827–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.McGranahan N et al. (2016) Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 351, 1463–1469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tumeh PC et al. (2014) PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Thorsson V et al. (2018) The immune landscape of cancer. Immunity 48, 812–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Coussens LM and Werb Z (2002) Inflammation and cancer. Nature 420, 860–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ferber MJ et al. (2003) Integrations of the hepatitis B virus (HBV) and human papillomavirus (HPV) into the human telomerase reverse transcriptase (hTERT) gene in liver and cervical cancers. Oncogene 22, 3813–3820 [DOI] [PubMed] [Google Scholar]
- 101.Diakos CI et al. (2014) Cancer-related inflammation and treatment effectiveness. Lancet Oncol 15, e493–e503 [DOI] [PubMed] [Google Scholar]
- 102.Crignis ED et al. (2019) Human liver organoids; a patient-derived primary model for HBV infection and related hepatocellular carcinoma. bioRxiv Published online March 5, 2019. 10.1101/568147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Nie YZ et al. (2018) Recapitulation of hepatitis B virus-host interactions in liver organoids from human induced pluripotent stem cells. EBioMedicine 35, 114–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Baktash Y et al. (2018) Single particle imaging of polarized hepatoma organoids upon hepatitis C virus infection reveals an ordered and sequential entry process. Cell Host Microbe 23, 382–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bartfeld S et al. (2015) In vitro expansion of human gastric epithelial stem cells and their responses to bacterial infection. Gastroenterology 148, 126–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Shibata W et al. (2017) Helicobacter-induced gastric inflammation alters the properties of gastric tissue stem/progenitor cells. BMC Gastroenterol 17, 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Huang JY et al. (2015) Chemodetection and destruction of host urea allows Helicobacter pylori to locate the epithelium. Cell Host Microbe 18, 147–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Pleguezuelos-Manzano C et al. (2020) Mutational signature in colorectal cancer caused by genotoxic pks(+) E. coli. Nature 580, 269–273 [DOI] [PMC free article] [PubMed] [Google Scholar]