

Abstract
Neuroendocrine and immune signaling pathways are activated following insults such as stress, injury, and infection, in a systemic response aimed at restoring homeostasis. Mitochondrial metabolism and function have been implicated in the control of immune responses. Commonly studied along with mitochondrial function, reactive oxygen species (ROS) are closely linked to cellular inflammatory responses. It is also accepted that cells experiencing mitochondrial or endoplasmic reticulum (ER) stress induce response pathways in order to cope with protein folding dysregulation, in homeostatic responses referred to as the unfolded protein responses (UPRs). Recent reports indicate that the UPRs may play an important role in immune responses. Notably, the homeostasis-regulating hormones oxytocin (OXT) and vasopressin (AVP) are also associated with the regulation of inflammatory responses and immune function. Intriguingly, OXT and AVP have been linked with ER unfolded protein responses (UPRER), and can impact ROS production and mitochondrial function. Here, we will review the evidence for interactions between these various factors and how these neuropeptides might influence mitochondrial processes.
Keywords: Unfolded protein response, microglia, NADPH oxidase, Glutathione, NF-κB
Introduction
Systemic responses to homeostatic challenges such as infection, injury, or stress impact neuroendocrine and immune signaling processes (Kotas and Medzhitov, 2015). The systemic nature of these systems likely involves a coordinated effort between hormone and inflammatory signaling processes. In response to homeostatic challenges such as brain injury or infection, immune cells respond by entering into a generally proinflammatory state characterized by the production and release of growth factors, chemokines, and cytokines designed to restore environmental homeostasis (Block et al., 2007; Kotas and Medzhitov, 2015). Similarly, disruptions in homeostasis can activate the neuroendocrine adaptations known as the stress response (Miller and O’Callaghan, 2002). While these initial responses may be useful for restoring homeostasis, prolonged dysregulation can become maladaptive (Block and Hong, 2005; Medzhitov, 2008). Many studies have begun to reveal the molecular mechanisms underlying these innate inflammatory processes. For example, mitochondria are classically thought to play a role in inflammatory cell activation through their ability to produce reactive oxygen species (ROS)(Bordt and Polster, 2014). However, more recent evidence links other aspects of mitochondrial function, i.e. mitochondrial energy production, with inflammatory processes (Mehta et al., 2017; Morris and Berk, 2015; Pearce and Pearce, 2017; van Horssen et al., 2017). Additionally, mitochondria may respond to altered homeostasis via a mechanism referred to as the mitochondrial unfolded protein response (UPRmt), a process mechanistically similar to another cellular homeostatic pathway referred to as the endoplasmic reticulum unfolded protein response (UPRER) (Celli and Tsolis, 2015; Qureshi et al., 2017). Importantly, it is becoming evident that both of these pathways are intricately linked to the immune response (Hetz, 2012; Qureshi et al., 2017).
While mitochondria appear to be intricately involved in the maintenance of cellular homeostasis, neuropeptide systems such as the oxytocin (OXT) system and the vasopressin (AVP) system are well-known as major players in tissue and overall body homeostasis (Li et al., 2016). For instance, OXT regulates energy balance, food intake, and thermogenic regulation (Chaves et al., 2013), while AVP is linked with water homeostasis, modulation of blood pressure, and maintenance of temperature homeostasis (Kozniewska and Romaniuk, 2008; Pittman et al., 1998, 1982). Similar to mitochondria, OXT and AVP have also been implicated in the regulation of inflammatory processes. Although somewhat promiscuous for each other’s receptors (see(Song and Albers, 2017)), OXT and AVP are generally thought to have opposing roles on immune function and the regulation of inflammatory responses, with OXT generally acting as an anti-inflammatory molecule and AVP acting as an inflammatory molecule (Hou and Jin, 2016; Li et al., 2016; Szmydynger-Chodobska et al., 2010). Furthermore, both OXT and AVP can modulate factors within either the UPRmt or the UPRER. Are there reasons to believe that the neuropeptides OXT and AVP may influence mitochondrial function? While literature linking OXT and AVP and mitochondria to ROS production is abundant, literature directly connecting OXT or AVP action to mitochondrial function remains somewhat limited.
Here, we briefly review evidence linking OXT and AVP to inflammatory responses. We then discuss evidence linking the UPRER and the UPRmt, and how these closely related organellar homeostatic pathways may be involved in the cellular response to immune challenges, as well as the interplay of OXT and AVP in these pathways. Finally, we review the role of mitochondria and ROS in immune regulation, and discuss the possible interactions of OXT and AVP in these diverse processes. We propose a model in which the unfolded protein responses represent a point of convergence at which mitochondria, OXT, and AVP act to regulate both systemic and cellular homeostatic responses.
Oxytocin and Vasopressin
Oxytocin (OXT) and vasopressin (AVP) are nonapeptides that differ by only 2 amino acids (Fig. 1). They are evolutionarily ancient and highly-conserved across phylogeny and regulate a wide array of physiological functions. Indeed, the separate genes for OXT and AVP likely arose from the duplication of a common ancestral gene (Acher et al., 1995). Both are primarily synthesized in the paraventricular and supraoptic nuclei of the hypothalamus (PVN and SON, respectively) (Brownstein et al., 1980; Young and Gainer, 2003). Magnocellular neurons within the PVN and SON then project to the posterior pituitary gland, from which both OXT and AVP are released into the periphery where they act as hormones to regulate hydromineral balance, cardiovascular tone and smooth muscle function (Kelly et al., 2014). OXT is primarily involved in the stimulation of uterine contractions during parturition, as well as the milk ejection reflex (Belin et al., 1984; Fuchs and Poblete, 1970). In contrast, AVP (also known as antidiuretic hormone; ADH) acts in the kidney to regulate water retention and blood pressure (Silva et al., 1969). Parvocellular neurons within the PVN also release AVP into the anterior pituitary where it augments the neuroendocrine stress response by stimulating adrenocorticotropic hormone release, leading to the release of glucocorticoids from the adrenal cortex (Gillies et al., 1982). Conversely, OXT released by PVN parvocellular neurons into the anterior pituitary dampens the activity of the hypothalamic-pituitary-adrenal axis in response to stress (Kelly et al., 2014). Interestingly, the PVN neurons that respond to body fluid homeostatic challenges appear to be the same PVN neurons that modulate behavioral and physiological responses to psychological stress (Smith et al., 2016), allowing for a well-coordinated reaction to perceived changes in homeostatic processes. Within the brain, both OXT and AVP can be released from axons, dendrites, and somas of neurons (Landgraf and Neumann, 2004; Ludwig and Leng, 2006), acting as neuropeptides to modulate a wide array of neural systems and functions, including social behavior, anxiety, learning and memory, circadian rhythms, appetite, temperature regulation, and immune function, often in sex-specific ways (for review see;(Dumais and Veenema, 2016; Goodson, 2008; Stoop, 2012)).
Figure 1. Oxytocin and vasopressin.

Chemical structures of oxytocin (OXT) and vasopressin (AVP). Highlighted in red are the two amino acids in which OXT and AVP differ. Highlighted in blue are the disulfide bonds present in both OXT and AVP.
OXT and AVP can signal via several G protein-coupled receptors - the oxytocin receptor (OTR) and the vasopressin receptors V1a, V1b, and V2 (V1aR, V1bR, and V2R, respectively), with distinct forms of the V2R (V2aR, mammals; V2bR, birds) present in different vertebrate species (Ocampo Daza et al., 2012). Importantly, both OXT and AVP display some degree of binding affinity for each other’s receptors, leaving open the possibility for crosstalk between these two systems (see (Song and Albers, 2017) for review). Depending on which G protein their receptors are coupled to, OXT and AVP can activate multiple downstream signaling pathways. For example, in vagal neurons, OXT has been shown to activate two distinct signaling pathways. The first is cAMP-dependent, but PKA-independent, while the other is PLC-β and cAMP-independent (Alberi et al., 1997).
The role of oxytocin and vasopressin in inflammation and immune activation
Dysregulation of the innate immune system has been implicated in numerous pathologies, from autism spectrum disorders (Masi et al., 2015) and traumatic brain injury (Villapol et al., 2017) to cardiac disease (Frantz and Nahrendorf, 2014) and obesity (Castoldi et al., 2015). In several instances, OXT and AVP have been shown to have opposing roles on immune function and the regulation of inflammatory responses (Hou and Jin, 2016; Li et al., 2016; Szmydynger-Chodobska et al., 2010). For instance, OXT promotes the growth of rat thymic glands in culture while AVP inhibits growth (Ficek, 1983). Moreover, OXT reduces stress-induced corticosterone (CORT) release and anxiety-like states, and promotes social bonding to decrease stress-induced illnesses (Gobrogge and Wang, 2015). Conversely, AVP, acting together with corticotropin releasing hormone (CRH), facilitates CORT release and enhances autonomic reactivity to stress, thereby increasing vulnerability to psychological and physical illnesses (Gobrogge and Wang, 2015).
The opposing roles of OXT and AVP appear to extend to immune function in microglia, with OXT dampening inflammatory pathways and AVP amplifying them. The bacterial endotoxin lipopolysaccharide (LPS) and the cytokine interferon-γ (IFN-γ) have been shown to activate innate immune cells, inducing a state that can be broadly described as proinflammatory, i.e. characterized by the production of nitric oxide (NO) and cytokines such as tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β) (Mehta et al., 2017). In response to LPS stimulation, OXT inhibits brain inflammatory processes mediated by microglia. For instance, pretreatment with OXT i) dampens the LPS-induced upregulation of Iba1 (a microglial marker) in both BV2 cells (a microglial cell line) and primary microglia and ii) inhibits the LPS-stimulated production of TNF-α, IL-1β, inducible nitric oxide synthase (iNOS) and cyclooxygenase 2 (COX2) in these microglial cells (Yuan et al., 2016). Additionally, OXT intranasal pretreatment significantly reduces in vivo expression of TNF-α and IL-1β in the prefrontal cortex of BALB/C mice injected intraperitoneally with LPS (Yuan et al., 2016). Intriguingly, this OXT pretreatment regimen decreases LPS-stimulated phosphorylation of ERK and p38 but not c-Jun N-terminal kinase (JNK) MAPK and fails to suppress phosphorylation of the p65 subunit of NF-κB (Yuan et al., 2016). Thus, these data suggest that OXT’s inhibition of proinflammatory markers following LPS exposure is not mediated through the blockade of NF-κB activation but may be mediated through microglial OTRs, which are significantly increased following an in vitro LPS challenge (Yuan et al., 2016). Along these lines, Iba1 staining in the medial amygdala and lateral septum of OTR knockout (KO) mice was ~2x as strong as compared to wild type mice, and revealed enlarged microglial cell bodies and increased cell branches, possibly indicative of increased microglial activation in the absence of OTRs (Miyazaki et al., 2016). These OTR knockout mice also showed reduced levels of the postsynaptic scaffolding protein PSD95, a reduction that was blocked when mice were treated with minocycline, a drug known to inhibit microglial activation (Miyazaki et al., 2016). In another assay, combined medial hypothalamic lesions that significantly reduced OXT mRNA levels led to increased NF-κB pathway activation, and increased mRNA expression of TNF-α and IL-1β (Roth et al., 2016). Furthermore, following cerebral ischemia, male mice that had been either pretreated with exogenous OXT or socially housed showed a reduction in infarct size, neuroinflammation, and oxidative stress as compared to mice socially housed but administered an oxytocin receptor antagonist (OTA) (Karelina et al., 2011). When microglial cultures were prepared from the brains of socially isolated mice, OXT treatment dampened microglial activity following LPS treatment (Karelina et al., 2011). Taken together, the above studies point to a critical role of OXT and OTRs in limiting inflammatory processes mediated by microglia (Karelina et al., 2011). It should be noted that neonatal OXT treatment during the course of normal development (and in the absence of an immune challenge) increases the expression of the astrocytic marker GFAP, but not the microglial marker CD68 (Havránek et al., 2017), suggesting that OXT may not always be directly linked with microglial function. Interestingly, OXT pretreatment significantly reduces Iba1/TNFα co-labeling but not GFAP/TNFα co-labeling following LPS challenge (Yuan et al., 2016), suggesting that OXT’s effects on glial subtypes may depend on the presence or absence of inflammation.
Unlike OXT, AVP promotes inflammatory processes in the brain in response to various forms of brain injury, including ischemic stroke. By virtue of its central role as a regulator of tissue water homeostasis, AVP exacerbates brain capillary water permeability, edema, blood-brain barrier (BBB) disruption and neuronal cell loss within various models of traumatic brain injury (Dickinson and Betz, 1992; Bemana and Nagao, 1999; Trabold et al., 2008; Vakili et al., 2005). In an attempt to gain insight into the cellular mechanisms underlying AVP’s pathophysiological role, Szmydynger-Chodobska et al. (2010) studied AVP-deficient (Avpdi/di) Brattleboro rats in a controlled cortical impact model of traumatic brain injury. Compared to Avpdi/di rats that lacked AVP, wild-type rats had a significantly greater influx of neutrophil and monocyte chemoattractants (i.e. CXC and CC chemokines) in response to cortical injury. Furthermore, AVP amplified the TNF-α-dependent production of CXC and CC chemokines through activation of the c-Jun N-terminal kinase (Szmydynger-Chodobska et al., 2010). Importantly, increases in CXC and CC chemokines have been shown to facilitate the recruitment of inflammatory cells, thereby disrupting the BBB and potentiating ischemic damage (Chen et al., 2003). Interestingly, the increased levels of AVP that have been reported in the CSF of patients following traumatic brain injury and ischemic stroke (Barreca et al., 2001; Sørensen et al., 1985; Xu et al., 2007) may be derived from activated microglia. For instance, in their model of a controlled cortical impact traumatic brain injury, Szmydynger-Chodobska et al. observed both an increase in AVP synthesis in the hypothalamus and cerebral cortex near the lesion site and that the majority of this AVP was produced by microglia in close contact with cortical microvessels and/or perivascular macrophages (Szmydynger-Chodobska et al., 2011). Furthermore, following the induction of a proinflammatory phenotype using LPS or IFNγ in primary cultures of microglia from neonatal and adult C57Bl/6 mice, microglia showed significantly increased sensitivity to AVP, as compared to control cultures not exposed to LPS or IFNγ (Pannell et al., 2014). The above studies highlight a role for AVP in potentiating brain inflammatory processes and suggest that microglia are a source of AVP in response to brain injury.
Several disparate studies also point to a critical role for OXT and AVP in microglial or macrophage-related inflammation following LPS immune challenge or injury to organ systems other than the brain. Within LPS-stimulated THP-1 macrophages, OXT attenuates the secretion of the proinflammatory cytokine IL-6 (Szeto et al., 2008). Furthermore, LPS-treatment of human THP-1, mouse RAW 264.7 macrophage cells, or primary human monocyte-derived macrophages resulted in upregulation of OTR mRNA 10–250 fold, and treatment with OXT again dampened LPS-induced production of IL-6 (Szeto et al., 2017). This upregulation of OTR appears to be NF-κB dependent, as the NF-κB inhibitor CAPE prevented the LPS-induced upregulation of OTR mRNA, as well as the LPS-induced increase in IL-6 (Szeto et al., 2017). Thus, in response to LPS challenge, OTRs are upregulated in both microglia and macrophages. OXT dampens proinflammatory pathways in both cell types, but through an NF-κB-independent mechanism in microglia (Yuan et al., 2016) and a NF-κB-dependent mechanism in macrophages (Szeto et a., 2017) (Fig. 2). In contrast to OXT signaling, LPS induces a ~65% decrease in AVP receptor expression in THP-1 macrophages and AVP treatment does not alter LPS-induced IL-6 secretion from these cells (Szeto et al., 2017).
Figure 2. Disparate effects of oxytocin on microglial and macrophage lipopolysaccharide responses.

Lipopolysaccharide (LPS) induces an upregulation of oxytocin receptor (OTR) in both microglia (left) and macrophages (right). In microglia, oxytocin (OXT) dampens inflammatory factor transcription by preventing the phosphorylation of p38 and ERK in the MAPK pathway. In macrophages, OXT dampens inflammatory factor transcription by preventing phosphorylation of the p65 subunit of NF-κB. LPS downregulates vasopressin receptor 2 (VR2) expression in macrophages, while the effect on vasopressin receptors (AVPR; subtype not yet specified) in microglia is not yet known
Following myocardial infarction, rats infused with OXT showed diminished cellular apoptosis in the myocardium and a suppression of inflammation via a reduction in neutrophils, macrophages, and T lymphocytes and decreased TNF-α and IL-6 expression (Jankowski et al., 2010). Importantly, knockdown of the microglial receptor P2X7R by either P2X7-siRNA or by pharmacological inhibition via the antagonist Brilliant Blue-G reduced IL-1β and TNF-α mRNA and protein in the PVN of Sprague-Dawley rats following acute myocardial infarction (Du et al., 2015). As OXT has been demonstrated to regulate cardiovascular responses following myocardial infarction, the authors then examined the effect of P2X7R knockdown on OXT and AVP neurons within the hypothalamus. Knockdown of P2X7R by siRNA following acute myocardial infarction decreased the number of both AVP-positive and OXT-positive neurons in the PVN (Du et al., 2015), suggesting that activation of P2X7R within the PVN may produce proinflammatory cytokines that activate AVP and OXT neurons, which then modulate sympathetic nerve activity (Du et al., 2015). Within a model of early-stage cecal ligation puncture sepsis, OXT treatment also attenuates the release of nitrite, IL-1β, and TNF-α from isolated macrophages (Oliveira-Pelegrin et al., 2013).
It is important to note that in addition to its opposing roles to OXT, AVP has also been described as i) having a much more limited role in immune system regulation compared with OXT ii) a neuropeptide that is supplemental to OXT for neuroendocrine-immune regulation and iii) a hormone that acts together with OXT to sequentially stimulate the maturation of the immune system (Li et al., 2016).
The unfolded protein response - from endoplasmic reticulum to mitochondria
The endoplasmic reticulum (ER) is the first step for proteins in the secretory pathway, where these proteins are properly folded and undergo post-translational modifications (Schröder and Kaufman, 2005a). Disturbances in any of these protein-folding or trafficking processes can result in ER stress (Navid and Colbert, 2017). It is well characterized that cells experiencing ER stress activate a series of mechanisms to cope with alterations in protein folding in order to restore cellular homeostasis in a response termed the endoplasmic reticulum unfolded protein response (UPRER) (Hetz, 2012). The UPRER notifies the cytosol and nucleus about the protein-folding status in the ER in order to reduce the unfolded protein load (Hetz et al., 2011; Schröder and Kaufman, 2005b), while also removing irreversibly damaged cells by apoptosis (Hetz, 2012). The UPRER has also been implicated in the regulation of various cellular processes, from energy homeostasis and lipid/cholesterol metabolism to cell differentiation and inflammation (Rutkowski and Hegde, 2010), indicating crosstalk between metabolic and stress pathways (Hetz, 2012).
The UPRER is activated through several major stress sensors: activating transcription factors 4–6 (ATF4–6), inositol-requiring protein 1 (IRE1), and protein kinase RNA-like ER kinase (PERK) (Ron and Walter, 2007)(Fig. 3a). Two distinct cascades of events occur in cells experiencing ER stress. First, PERK activation leads to the inhibition of protein translation through phosphorylation of eukaryotic translation initiator factor 2α (eIF2α) (Harding et al., 2000). PERK phosphorylates eIF2α, inducing translation of ATF4 mRNA, which is then responsible for controlling pro-survival genes relating to protein folding, autophagy, redox homeostasis, and amino acid metabolism (Ameri and Harris, 2008; Schröder and Kaufman, 2005b). A large-scale degradation pathway referred to as macroautophagy is also activated once the UPRER is brought online (Kroemer et al., 2010), and the translocation of targeted proteins into the ER is then prevented through quality control (Kang et al., 2006) and co-translational degradation mechanisms (Oyadomari et al., 2006). The second cascade utilizes three distinct transcription factors to induce a large-scale gene response: ATF4, XBP1, and ATF6 (Hetz, 2012)(Fig. 3a). Each of these stress sensors then promote the activation of different transcription factors, resulting in upregulation of UPRER target genes (Ron and Walter, 2007). For instance, under ER stress conditions the kinase IRE1α dimerizes and autotransphosphorylates, leading to the activation of a cytosolic RNase domain (Walter and Ron, 2011). This activated IRE1α then excises an intron in the mRNA encoding the transcription factor X box-binding protein 1 (XBP1), shifting the mRNA reading frame, resulting in a stable transcription factor called spliced XBP1 (XBP1s) (Lee et al., 2002; Yoshida et al., 2001). XBP1s upregulates the expression of genes responsible for the ER-associated degradation (ERAD) pathway, phospholipid synthesis, protein folding, and ER protein entry (Asada et al., 2011; Hetz et al., 2011; Lee et al., 2003). Activating transcription factor 6 (ATF6) translocates to the Golgi under ER stress conditions, from which a cleaved cytosolic fragment (ATF6f) that regulates ERAD and XBP1 genes is released (Haze et al., 1999; Lee et al., 2002; Yamamoto et al., 2007). In response to proteotoxic stress induced by proteasome inhibition, ATF4 and CHOP induce the expression of ATF5. Knockdown of ATF5 mitigates apoptosis, suggesting that the transcriptional activation of the UPRER promotes apoptotic cell death (Teske et al., 2013). Taken together, these studies suggest that XBP1, ATF4–6, and CHOP regulate a large network of overlapping genes that modulate cellular adaptation to ER stress.
Figure 3. Unfolded protein responses.

(a) Major players in the endoplasmic reticulum unfolded protein response (UPRER). Inositol-requiring protein 1α = IRE1α. X box-binding protein 1 = XBP1. Spliced XBP1 = XBP1s. Protein kinase RNA-like ER kinase = PERK. Eukaryotic translation initiator factor 2α = eIF2α. Activating transcription factors 4–6 (ATF4–6). CCAAT-enhancer-binding protein homologous protein = CHOP. (b) Major players in the mammalian mitochondrial unfolded protein response (UPRmt). Note that ATF5 may be regulated by mitochondrial import efficiency in addition to eIF2α, ATF4, and CHOP due to the presence of a mitochondrial targeting sequence. General control non-derepressible-2 = GCN2. Eukaryotic translation initiator factor 2α = eIF2α. Activating transcription factors 4–5 (ATF4–5). CCAAT-enhancer-binding protein homologous protein = CHOP. Mitochondrial targeting sequence = MTS.
Similar to the ER predisposition to accumulate misfolded proteins and respond to ER stress, mitochondria are prone to dysregulation of protein import and various mitochondrial stressors and possess the ability to respond to these stressors by their own stress signaling pathway referred to as the mitochondrial unfolded protein response (UPRmt)(Qureshi et al., 2017) (Fig. 2b). Although the UPRmt was discovered in mammalian cells, much of our current knowledge comes from discoveries made in Caenorhabditis elegans (C. elegans). For a thorough comparison of the UPRmt in C. elegans vs. mammals, see (Qureshi et al., 2017). The UPRmt is induced by conditions such as mitochondrial DNA (mtDNA) depletion, alterations to mitochondrial oxidative phosphorylation, and inhibition of mitochondrial protein quality control machinery (Nargund et al., 2012; Yoneda et al., 2004). Additionally, mitochondrially-generated reactive oxygen species (ROS) can alter mitochondrial protein folding, disrupting the proper stoichiometry of the protein complexes that make up the mitochondrial electron transport chain (ETC), intensifying the protein misfolding environment and activating the UPRmt (Martinus et al., 1996; Runkel et al., 2013; Wang et al., 2017).
The pivotal regulator of the UPRmt is ATFS-1 in C. elegans (Nargund et al., 2012), thought to be somewhat comparable to ATF4, ATF5, and ATF6 in the UPRER. ATFS-1 contains both a nuclear localization sequence (NLS) and a mitochondria targeting sequence (MTS) (Nargund et al., 2015, 2012). Under resting conditions, ATFS-1 is imported into the mitochondria and degraded by the Lon protease (Nargund et al., 2012). However, mitochondrial stress reduces mitochondrial import efficiency (Wrobel et al., 2015), resulting in ATFS-1 accumulation in the cytosol, and eventual targeting to the nucleus (Qureshi et al., 2017). Importantly, following import into the mitochondrial matrix, ATFS-1 is degraded, indicating that the efficiency of mitochondrial import is a negative regulator of the UPRmt (Melber and Haynes, 2018). The bZIP transcription factor ATF5 in mammals is regulated by the efficiency of mitochondrial import, similar to ATFS-1 in C. elegans (Fiorese et al., 2016). Similar to ATFS-1, ATF5 contains a MTS (Fig. 3B), possibly allowing it to respond to impaired mitochondrial import efficiency (Melber and Haynes, 2018). However, recent studies have also identified ATF4 as a key regulator of UPRmt in mammals (Quirós et al., 2017) further highlighting the need for more research into the direct comparison of ATFS-1 in C. elegans and ATF4/ATF5 in the mammalian UPRmt. In a process similar to the UPRER, this nuclear localization of ATFS-1 in C. elegans induces a transcriptional response to restore proper mitochondrial functioning (Nargund et al., 2012). Activation of the mammalian UPRmt is also thought to involve two other bZIP transcription factors, ATF4 and CHOP (Michel et al., 2015; Quirós et al., 2017). Mitochondrial stress-induced phosphorylation of eIF2α upregulates translation of all three of the transcription factors ATF5, ATF4, and CHOP (Melber and Haynes, 2018). The phosphorylation and activation of eIF2α is primarily mediated by the kinase general control non-derepressible-2 (GCN2)(Baker et al., 2012; Melber and Haynes 2018), which is itself activated by mitochondrial stress, reactive oxygen species, and amino acid depletion (Melber and Haynes, 2018). In C. elegans lacking GCN2, worm development was significantly delayed in response to the mitochondrial toxin rotenone, and GCN2 deletion significantly impaired worms’ ability to accommodate an increased unfolded protein load, indicating the importance of GCN2 in the protection of mitochondrial function during mitochondrial stressors (Baker et al. 2012). Although it has not yet been shown in the context of mitochondrial stress, ATF4 and CHOP are thought to also regulate ATF5 transcription (Teske et al., 2013; Zhou et al., 2008), highlighting the likely interacting signaling mechanisms between the UPRmt and UPRER. It should be noted that reduction of mitochondrial protein import through disruption of the inner mitochondrial membrane channel Tim23 activated CHOP without activation of ATF5, suggesting that in some cases ATF5 and CHOP are activated independently (Oliveira and Hood 2018). Intriguingly, the UPRmt was recently demonstrated to be influenced by the neuropeptide FLP-2 in C. elegans (Shao et al., 2016), highlighting the possibility that neuropeptides such as AVP and OXT may also regulate unfolded protein responses in mammals.
How does the UPRmt attenuate stress? Surprisingly, in addition to the strong nuclear localization of ATFS-1 induced following episodes of mitochondrial stress, a limited quantity of ATFS-1 still accumulates in the mitochondria during cellular stress (Nargund et al., 2015, 2012). ATFS-1 downregulates oxidative phosphorylation genes encoded by mtDNA and negatively regulates genes responsible for oxidative phosphorylation encoded in the nucleus (Nargund et al., 2015). It is thought to regulate these nuclear-encoded genes through both indirect and direct binding of their promoters (Nargund et al., 2015). It is proposed that ATFS-1 limits oxidative phosphorylation gene transcription until mitochondria recover their protein folding capacity as a mechanism to promote mitochondrial recovery (Qureshi et al., 2017). ATFS-1 also upregulates the expression of genes in the glycolysis pathway (Nargund et al., 2015, 2012), thereby ensuring that cells possess enough ATP to promote mitochondrial repair and survival when there is improper oxidative phosphorylation functioning (Qureshi et al., 2017). As observed with chronic inflammation, prolonged activation of the UPRmt may predispose cells to disease. For example, a metabolic switch to glycolysis via ATFS-1 could lead to the Warburg effect (see (Liberti and Locasale, 2016)) for review of Warburg effect) and may facilitate conditions which could promote tumorigenesis. Although they are distinct processes, it is notable that the UPRER and UPRmt share many similar modulatory processes (Fig. 3), indicating crosstalk and the possibility of collaboration between these cellular stress response pathways (Qureshi et al., 2017). Of note, mitochondrial respiration is induced by ER stress, and knockdown of the UPRER sensor protein IRE1 impaired mitochondrial respiratory activation (Knupp et al. 2018). Uncoupling the mitochondrial electron transport chain to increase mitochondrial oxygen consumption was able to rescue cell survival following ER stress (Knupp et al. 2018), indicative of a direct link between mitochondrial function (and possibly the UPRmt) and the UPRER.
Interactions of oxytocin and vasopressin with the UPRER and immune responses
An increasing number of reports indicate that the UPRER plays an important role in inflammatory responses through activation of the transcription factors ATF4, ATF5, ATF6, IRE1, and PERK. For example, PERK signaling has been found to activate NF-κB and Nrf2, two transcription factors known to regulate redox metabolism and inflammation (Schröder and Kaufman, 2005b). Furthermore, siRNA inhibition of the UPRER transcription factors ATF4 (downstream of PERK) and XBP1 (downstream of IRE1) demonstrated their necessity in the production of the proinflammatory factors IL-8, IL-6, and MCP1 by human aortic endothelial cells (Gargalovic et al., 2006). Moreover, TLR4 stimulation (i.e. LPS) induces upregulation of XBP1 precursor mRNA(Roach et al., 2007), while XBP1 deficiency significantly dampens macrophage production of IL-6 (Martinon et al., 2010). LPS treatment also triggers splicing of XBP1 mRNA, thereby increasing XBP1s action on UPRER target genes, and enhancing transcription of proinflammatory cytokines(Martinon et al., 2010). Utilizing a mouse model of multiple sclerosis (experimental autoimmune encephalomyelitis (EAE)), Ta et al observed a marked increase in the expression of the ATF6-target chaperones GRP94 and GRP78 (Ta et al., 2016). EAE in ATF6α−/− mice significantly dampened microglia/macrophage iNOS expression and lowered levels of the NF-κB subunit p65 compared to EAE in ATF6α+/+ mice (Ta et al., 2016), suggesting a vital role for key elements of the UPRER in the induction of inflammatory responses.
Given the important roles that both OXT/AVP and the UPRER play in inflammatory responses, it stands to reason that these hormones may interact with the UPRER to modulate inflammatory signaling and disease states. OXT has been shown to modulate both the Akt/PI3K pathways and the Akt/mammalian target of rapamycin complex I (mTORC1) pathways that regulate ribosomal protein and translation factor activity (Kelleher and Bear, 2008; Klein et al., 2013). Interestingly, upregulation of the mTORC1/eukaryotic translation factor 4E (eIF4E) pathway is associated with Autism-related gene mutations in TSC1/2, NF1, PTEN and fragile X mental retardation protein 1 FMRP1 (Kelleher and Bear, 2008). Furthermore, aberrant protein translation has been linked with Autism Spectrum Disorder (ASD) through mutations in FMRP1 and eIF4E (Gkogkas et al., 2013; Kelleher and Bear, 2008; Neves-Pereira et al., 2009; Wang and Doering, 2013). Given these findings, work by Klein et al explored the possibility that OXT regulates protein translation in the gut via the UPRER(Klein et al., 2014). Klein et al. found that OXT significantly increased phosphorylation of eIF2α and its upstream activator PERK in cultured Caco2BB enterocyte cells, indicative of activation of the UPRER. However, it must be noted that this response was partially insensitive to an OTA (Klein et al., 2014). OXT (7.8–62.5 nM) also increased the XBP1 splice variant, XBP1s, in an OTA-dependent manner (Klein et al., 2014). The ER-resident chaperone BiP is responsive to XBP1 during ER stress and is involved in the clearance of misfolded proteins (Lee et al., 2003). As expected, BiP levels in Caco2BB cells were increased by OXT treatment, demonstrating that OXT can activate the UPRER stress pathway in enterocytes and that this interaction may be protective during a postnatal critical period of gut development (Klein et al., 2014).
In a subsequent study, a protective role of OXT for developing enterocytes was further examined in a model utilizing LPS exposure (Klein et al., 2016). Caco2BB enterocytes stimulated with LPS demonstrate classical markers of NF-κB pathway activation, with an increase in the pIκB/IκB ratio and lower overall IκB levels consistent with dissociation of IκB’s inhibitory association with NF-κB (Klein et al., 2016). OXT treatment (7.8 nM) prevented the LPS-induced increase in pIκB/IκB ratio, and increased IkB levels by itself, thereby enhancing NF-κB inhibition (Klein et al., 2016). LPS treatment mildly reduced phosphorylation of the UPRER factor eIF2α. As phosphorylation of eIF2α inactivates its protein translation abilities, LPS treatment appears to be stimulating eIF2α-induced protein translation (Klein et al., 2016). However, OXT + LPS significantly increased phospho-eIF2α levels in comparison to LPS alone, suggesting that OXT reduces protein translation during cellular stress in response to LPS treatment, as a possible protective mechanism (Klein et al., 2016). LPS decreased levels of the important UPRER transcription factor XBP1s, an effect that was reversed with OXT treatment (Klein et al., 2016). These findings suggest that OXT can modulate the UPRER in response to LPS immune challenge. While OXT + LPS treatment significantly upregulated markers of the UPRER in comparison to LPS treatment, LPS treatment alone did not significantly downregulate phosphorylation of PERK, phosphorylation or total levels of IRE1α, or BiP levels (Klein et al., 2016). Finally, 40 min of OXT exposure upregulated ATF4 expression in primary osteoblasts (Di Benedetto et al., 2014), providing further evidence linking OXT action and the UPRER. Although limited, adequate evidence exists linking OXT and the UPRER to merit further studies into this intriguing relationship and its hypothesized protective role for developing enterocytes (Klein et al., 2016).
AVP, also referred to as antidiuretic hormone, assists in the regulation of water homeostasis in the periphery (Bourque, 2008) and is also linked to the UPRER. AVP secretion, which occurs in response to dehydration and salt loading, is induced by osmosensitive organs that project to magnocellular neurons of the supraoptic nucleus (SON) and paraventricular nucleus (PVN) (McKinley et al., 2003). This secretion of AVP serves to stimulate reabsorption of water in the kidney (Boone and Deen, 2008). Intriguingly, mRNA for the ER-resident chaperone BiP is expressed in AVP neurons in the SON and PVN under basal conditions and is upregulated following dehydration (Hagiwara et al., 2012), suggesting that BiP may be involved in ER homeostasis in SON and PVN neurons following dehydration. A study by Azuma et al. examined the role of the UPRER transcription factor ATF6 in the function of the AVP water homeostasis system (Azuma et al., 2014). Using ATF6α−/− mice, they found that urine volumes were increased in ATF6α−/− but not ATF6α+/+ mice undergoing water deprivation. As expected, BiP mRNA expression was upregulated in ATF6α+/+ but not in ATF6α−/− animals following water deprivation (Azuma et al., 2014). Mice possessing a mutation inducing familial neurohypophysial diabetes insipidus (FNDI) demonstrate marked reduction in AVP-positive neurons in the SON following dehydration (Hagiwara et al., 2014). FNDI/ATF6α−/− mice subjected to water deprivation had fewer AVP-positive neurons than FNDI mice alone following 12 weeks of water deprivation (Azuma et al., 2014), indicating that ATF6 and the UPRER are necessary for the AVP system to maintain water homeostasis in response to dehydration. CREB3L1 has been described as a transducer of ER stress that shares a high sequence similarity to ATF6, and functions in the modulation of the ER response to stress (Honma et al., 1999; Kondo et al., 2011; Murakami et al., 2009). Interestingly, CREB3L1 can induce AVP gene expression through direct binding to the AVP promoter (Greenwood et al., 2014). mRNA expression of the UPRER factor genes BiP, Atf4, Chop, and Creb3l1 were significantly upregulated in the SON following either 3 days of water deprivation or 7 days of salt loading (M. Greenwood et al., 2015). BiP, Atf4, and Creb3l1 expression were also increased in the PVN, whereas Chop expression remained stagnant (M. Greenwood et al., 2015), suggesting an important role of the UPRER in the response of the AVP system to stressors. Additionally, studies profiling the transcriptome of the SON following salt loading demonstrated upregulation of the UPRER genes ATF4, ATF6, and XBP1, as well as the UPRmt gene ATF5, as well as many genes comprising the mitochondrial electron transport chain (M. P. Greenwood et al., 2015). Finally, evidence linking AVP and the UPRER in the periphery was demonstrated by Crambert et al: AVP has previously shown to increase extracellular osmolality, and an increase in extracellular osmolality was demonstrated to stimulate the ATF6 and PERK arms of the UPRER (Crambert et al., 2014).
Interactions of oxytocin and vasopressin with the UPRmt and immune response
Although less well-studied than the UPRER, the UPRmt is thought to play an important role in inflammatory processes, possibly by supporting the ability of a cell to withstand damage from harmful pathogens by modulating mitochondrial recovery (Qureshi et al., 2017). Disrupting mitochondrial function in C. elegans via inhibition of the mitochondrial protease SPG-7 induced expression of the mitochondrial chaperones hsp-6 and hsp-60, as well as genes involved in the innate immune response, such as abf-2, lys-2, and irg-1 (Liu et al., 2014; Pellegrino et al., 2014; Wang et al., 2017). Microbes are able to activate the UPRmt through the production of mitochondrial toxins such as cyanide (Liu et al., 2014; Pellegrino et al., 2014), reminiscent of mitochondrial toxins activating NLRP3 inflammasome-induced inflammatory processes (Zhou et al., 2011). siRNA-mediated knockout of the UPRmt transcription factor ATFS-1 significantly reduced survival of C. elegans exposed to the gram-negative bacteria P. aeruginosa (Pellegrino et al., 2014). Additionally, the mitochondrial chaperone HSP-60, whose transcription is upregulated during UPRmt activation (Haynes et al., 2010; Nargund et al., 2015, 2012), increases the MAPK innate immunity response by binding to the SEK-1/MAPK kinase 3 (Jeong et al., 2017). Although indirect, knock-out of the adipocyte fatty acid binding protein (FABP4/aP2) in macrophages attenuated the UPRmt and reduced NLRP3 activation (Steen et al., 2017), suggesting a putative link between the UPRmt and inflammation. Further studies into the relationship between both the UPRER and the UPRmt and inflammatory responses are vital.
While direct links between OXT and/or AVP and the UPRmt have not yet been found, an indirect link between these neurohormones and mitochondrial proteostasis may come from localization of the mitochondrial uncoupling protein 2 (UCP2). Uncoupling proteins (UCPs) are anion carriers on the inner mitochondrial membrane capable of ‘uncoupling’ the mitochondrial proton gradient from ATP production (Echtay, 2007). An early study by Horvath et al. to localize UCP2 expression in the brain found that it was primarily localized in the perikaryon of the supraoptic, paraventricular, suprachiasmatic, and arcuate nuclei of the hypothalamus in both rodents and non-human primates (Diano et al., 2000; Horvath et al., 1999). Neurons expressing UCP2 were also found to primarily express corticotropin-releasing hormone, AVP, and OXT (Horvath et al., 1999). Due to the perikaryal localization of UCP2 in OXT- and AVP-expressing neurons, the authors suggested that activated UCP2 may be able to modulate the actions of these homeostatic circuits (Horvath et al., 1999). Intriguingly, mitochondrial UCP2 may also be connected to the FABP4/aP2 regulation of the UPRmt and inflammation previously described, wherein FABP4/aP2 knockout diminished the UPRmt as well as NLRP3 inflammasome activation (see(Steen et al., 2017)). Another study from this group demonstrated that FABP4/aP2 inhibition upregulated expression of UCP2, and that blocking this upregulation of UCP2 mRNA prevented the effects seen by FABP4/aP2 inhibition (Xu et al., 2015). The above literature highlights the need for studies examining the direct role of OXT and/or AVP on mitochondrial protein expression as it relates to unfolded protein responses or inflammatory processes.
Together, these studies suggest at least an indirect connection between the OXT and AVP systems and both the UPRER and UPRmt. To our knowledge, no studies have treated animals with OXT and/or AVP and followed with RNA sequencing or PCR analyses to determine if there are changes in expression of UPRER or UPRmt genes. Such studies would significantly improve our understanding of any links between these complex signaling pathways. In summary, there is compelling evidence linking OXT and AVP signaling to the UPRER, yet future studies are necessary to further elucidate the effects of OXT and AVP signaling on the UPRER, and to begin to investigate any interactions between OXT and/or AVP and the closely-related UPRmt. While direct links between OXT/AVP action and the UPRmt have not yet been elucidated, a possible link is suggested by the interactions of these neurohormones with other mitochondrial functions, which will be explored in subsequent sections.
Mitochondria and the innate immune system
Recent studies indicate that mitochondrial metabolism may play an important role in controlling the homeostatic function of immune cells (Mehta et al., 2017). While mitochondrial metabolism has been implicated in controlling aspects of many types of immune cells, we will focus on two cells of the innate immune system, macrophages and microglia. Impairments in the mitochondrial ETC have been observed in proinflammatory-stimulated macrophages (Haschemi et al., 2012; Vats et al., 2006) and microglia (Jaber et al., 2017; Orihuela et al., 2016). At the same time, an increase in glycolytic flux was found to be necessary for macrophages to display an inflammatory state (Palsson-McDermott et al., 2015; Tannahill et al., 2013). Interestingly, inflammatory macrophages have a tricarboxylic acid (TCA) cycle that is segmented into three distinct processes as opposed to the normal single functioning cycle (Jha et al., 2015). This leads to buildup of the metabolite itaconate (Jha et al., 2015; Michelucci et al., 2013), which is a weak inhibitor of Complex II of the mitochondrial ETC (Lampropoulou et al., 2016). This TCA segmentation can eventually lead to reverse electron transfer (RET) reactive oxygen species (ROS) production, and subsequent IL-1β production (Mills et al., 2016). In fact, preventing RET ROS was demonstrated to upregulate expression of the anti-inflammatory cytokine interleukin-10 (IL-10) (Mills et al., 2016).
While mitochondrial function is affected by activation of the innate immune system, mitochondria may themselves be immunogenic (Krysko et al., 2011; Rongvaux, 2017). The innate immune response can be activated upon cellular injury by release of endogenous damage-associated molecular patterns (DAMPs) (Matzinger, 1994). Since mitochondria are thought to be of bacterial origin (endosymbiotic theory;(Sagan, 1967)), they may also possess molecular motifs similar to bacteria(Zhang et al., 2010), potentially allowing them to act as DAMPs themselves. For example, a study by Zhang and colleagues found that mitochondrial DNA (mtDNA) was increased in plasma from major trauma patients (Zhang et al., 2010). Using these so-called mitochondrial DAMPs (mtDAMPs), the authors demonstrated that treatment of human polymorphonuclear neutrophils (PMNs) with mtDAMPs (mtDNA and formyl peptides) to mimic bacterial challenge activated MAP kinases through TLR9 and formyl peptide receptor-1 (Zhang et al., 2010). DAMPs can activate the NLRP3 inflammasome and induce the innate immune system to produce proinflammatory cytokines such as IL-1β (Schroder and Tschopp, 2010). Compounds known to inhibit mitochondrial function (rotenone, TTFA, or antimycin A) have also been shown to trigger activation of the NLRP3 inflammasome (Zhou et al., 2011). mtDNA, particularly mtDNA containing oxidatively damaged nucleotides, can induce inflammation, whereas nuclear DNA was unable to induce inflammatory responses (Collins et al., 2004; Nakahira et al., 2011; Shimada et al., 2012), further suggesting a role for mitochondria in the onset of inflammatory processes. Additionally, cardiolipin, a mitochondrial inner membrane lipid, directly binds to and is necessary for the activation of NLRP3 (Iyer et al., 2013). Another piece of evidence linking mitochondria to innate immune activation stems from studies showing that necrotic dendritic or melanoma cells induce NLRP3 activation by releasing mitochondrial molecules such as ATP that can trigger the purinergic P2X7 receptor (Ghiringhelli et al., 2009; Iyer et al., 2009).
Oxytocin, vasopressin, and mitochondria - is there a direct link?
While direct evidence connecting OXT or AVP action to mitochondrial processes remains limited, several recent studies suggest that there may be links between these hormones and mitochondrial function. Oxygen-glucose deprivation (OGD) paradigms model ischemia at birth. During OGD, neurons progress to anoxic depolarization, a process characterized by a dramatic fall in ATP, the failure of ATP-dependent ionic transports, a build-up of intracellular Ca2+, and the subsequent production of free radicals and cellular death of neurons (Khazipov et al, 2008). This process is accelerated by the administration of OTA’s at birth and attenuated by exogenous OXT (Tyzio et al., 2006; Ceanga et al., 2010), and certainly impairs mitochondrial function (Lipton, 1999), but direct effects of OXT on mitochondria during inflammatory signaling have rarely been assessed. A study by Gravina et al. demonstrated that OXT treatment induced an acute depolarization of the mitochondrial membrane potential in isolated myometrial cells (Gravina et al., 2011). This effect was likely due to the OXT-mediated increase in the intracellular calcium concentration and subsequent stimulation of the ATP synthase (Gravina et al., 2011). Additional evidence linking OXT to mitochondrial function stems from a recent report by Kaneko et al. utilizing oxygen glucose deprivation (OGD) to model ischemia at birth (Kaneko et al., 2016). The authors showed that pretreatment of embryonic day 18 primary rat neural cells with OXT increased cell viability, preserved ‘mitochondrial activity’ as measured by MTT assay and provided protection against OGD-induced ROS via a reduction in GSSG and high mobility group box 1 (HMGB1) (Kaneko et al., 2016). More recently, Amini-Khoei et al. (2017) have shown that within a model of early life stress (i.e. maternal separation; MS), OTX administration in mice mitigates the effects of MS on mitochondrial function by decreasing ROS formation and NO levels and increasing ATP and GSH levels. Furthermore, OT-treated mice subjected to MS showed reduced expression of neuroinflammatory genes, such as IL-1B, Myd88, TNF-α, Tlr4 and Nlrp3, compared to saline-injected controls (Amini-Khoei et al., 2017).
Early studies by Lehninger and Neubert demonstrated that both OXT and AVP were capable of stimulating mitochondrial swelling through water uptake (Lehninger and Neubert, 1961). GSH, which alone does not induce mitochondrial swelling, significantly potentiates the ability of OXT and AVP to swell isolated mitochondria (Lehninger and Neubert, 1961). The addition of ATP reversed liver mitochondrial swelling triggered by either OXT alone or OXT in combination with GSH, while the effects of ATP on AVP actions were unfortunately not reported (Lehninger and Neubert, 1961). These findings were in agreement with earlier findings showing the potentiation of mitochondrial swelling by combining disulfides and thiols (Neubert and Lehninger, 1962). As both AVP and OXT contain disulfides (see blue bonds in Fig. 1), the authors suggested that the active group of both hormones may be their disulfide groups (Lehninger and Neubert, 1961). Studies about the protective effect of OXT against ischemia-reperfusion injury support the idea that the disulfide group of OXT may be important in its actions. Following ischemia-reperfusion in rabbits, OXT significantly reduced myocardial infarct size and reduced the number of arrhythmias, an effect that was abolished by treatment with the mitochondrial KATP channel blocker 5-HD, but not by the iNOS inhibitor 1400W (Das and Sarkar, 2012). The finding that blocking the mitochondrial KATP channel prevented OXT-mediated protection directly implicates OXT action in mitochondrial function. However, the fact that iNOS inhibition prevented ischemic preconditioning-mediated protection but not OXT-mediated preconditioning protection (Das and Sarkar, 2012) suggests that OXT treatment may be acting similarly to nitric oxide. As nitric oxide can protect against oxidative damage through reversible s-nitrosylation of cysteines (Kohr et al., 2011; Sun et al., 2006), it may be that the disulfide bonds on OXT similarly protect cysteines from oxidation. Elucidating the ability of OXT to affect cysteine oxidation is a critical next step in understanding the protective actions of this fascinating molecule. This could also explain the ability of GSH to potentiate the ability of OXT and AVP to mediate mitochondrial swelling (Lehninger and Neubert, 1961). GSH can reduce disulfide bonds, forming mixed disulfide bonds, and thereby freeing up a sulfur for subsequent attack (Chakravarthi et al., 2006). GSH may break the disulfide bonds in OXT or AVP, allowing them to subsequently attack any cysteine residues in mitochondria. Protein thiols can undergo reversible modifications resulting in nitrosylation or disulfide bonds. However, cysteines can also undergo less reversible and more toxic modifications such as oxidation (Fig. 4)(Zimmet and Hare, 2006). It may be that OXT and/or AVP can form disulfide bonds with vulnerable protein thiols that can later be reversed, thereby protecting them from harmful cysteine oxidation. The observation that an OTA partially failed to reverse OXT-induced eIF2α phosphorylation and PERK activation (Klein et al., 2014) is suggestive of an alternative mechanism of OXT action. While it may be that OXT was acting off-target at the AVP receptor (see(Kelly and Goodson, 2014) for discussion of OXT and AVP receptor promiscuity), other mechanisms such as the ability of OXT to modulate thiol modifications should also be explored.
Figure 4. Oxytocin, vasopressin, and thiol modifications.
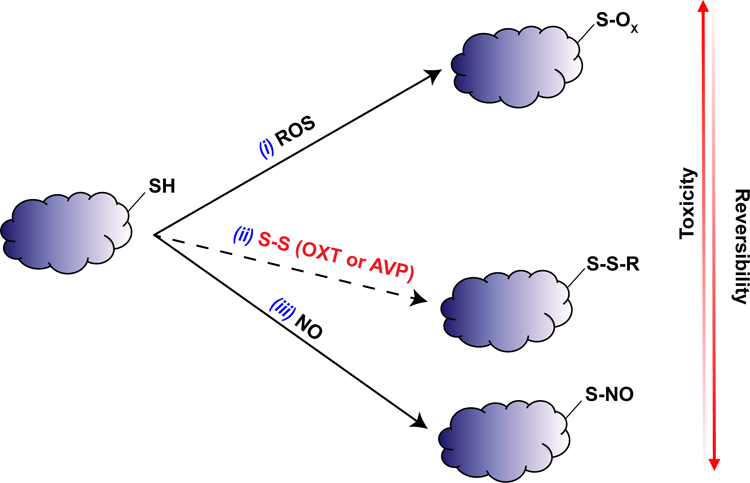
Diagram of proposal that OXT and AVP may be protective against oxidative mitochondrial damage through reversible thiol modifications. (i) ROS can oxidize protein thiols to form sulfenic, sulfinic, or sulfonic acids (S-OX) which are highly toxic and difficult to reverse. (ii) The disulfide bond present in OXT or AVP may react with protein thiols to form disulfide bonds (S-S-R) themselves, which are less toxic and more reversible than oxidized thiols. (iii) Nitric oxide (NO) can react with protein thiols to form S-nitrosothiols (S-NO), which are readily reversible and much less toxic than oxidized thiols.
Additional evidence linking AVP action and mitochondrial function stems from a recent study by Sims et al. In this study, male rats were exposed to a decompensated hemorrhagic shock model, which strongly decreased mean arterial blood pressure, pituitary and serum AVP levels, as well as inhibited mitochondrial Complex I and Complex II-linked respiration and Complex I-III linked enzyme activities in isolated kidney mitochondria (Sims et al., 2017). Resuscitation with AVP rescued all of the above measures, indicating that AVP can protect against hemorrhagic shock, potentially through the restoration of mitochondrial function (Sims et al., 2017).
Recent evidence suggests that some G-protein coupled receptors (GPCRs), such as the type-1 cannabinoid receptor (CB1), can be localized to the outer mitochondrial membrane itself, where they are responsible for regulation of cellular respiration (Bénard et al., 2012; Harkany and Horvath, 2017; Hebert-Chatelain et al., 2016). Additionally, nuclear receptors such as the estrogen receptor, thyroid hormone receptor, and peroxisome proliferators-activated receptor gamma 2 have been found localized in mitochondria (Casas et al., 2000; Chen et al., 2004; Lee et al., 2008; Wrutniak et al., 1995), suggesting that non-classical mitochondrial receptors may localize to and function at the mitochondrial membrane. OXT and AVP primarily function through the oxytocin receptor and vasopressin receptors, all of which are GPCRs (Ocampo Daza et al., 2012). Future studies are necessary exploring whether the OTRs or AVP receptors can localize to mitochondrial membranes, and if so, in what functions they may be involved. While promising, literature exploring direct actions of OXT and/or AVP on mitochondrial function (Table 1) remain vastly underdeveloped and may prove vital to understanding the complex interplay of these neurohormones for mitochondrial function and homeostatic immune responses.
Table 1.
Effects of oxytocin and vasopressin on mitochondria.
| Oxytocin | Vasopressin | |
|---|---|---|
| Induce mitochondrial swelling | (Lehninger and Neubert 1961) | (Lehninger and Neubert 1961) |
| Depolarize mitochondrial membrane potential | (Gravina et al 2011) | - |
| Protect ‘mitochondrial activity’ against oxygen-glucose deprivation | (Kaneko et al 2016) | - |
| Protect ‘mitochondrial activity’ against effects of maternal separation | (Amini-Khoei et al 2017) | - |
| Interact with mitochondrial KATP channel | (Das and Sarkar 2012) | - |
| Restore CI, CII, and CI-III ETC activity following hemorrhagic shock | - | (Sims et al 2017) |
Reactive Oxygen Species
A common signaling pathway implicated in the innate immune response is the activation of toll-like receptors and cytokine receptors (Block et al., 2007). Subsequent to receptor binding, there is well-characterized evidence of an increased production of reactive oxygen species (ROS) such as nitric oxide, peroxynitrite, superoxide, and hydrogen peroxide (Block et al., 2007; Palmer and Paulson, 1997). ROS have been proposed to be second messengers in cytokine responses (Palmer and Paulson, 1997), and can induce NF-κB nuclear translocation (Haddad and Land, 2002; Martindale and Holbrook, 2002), a key step in proinflammatory factor production (Zeng et al., 2012; Zheng et al., 2012). ROS are thought to promote or enhance inflammatory processes through the activation of MAPK and downstream NF-κB signaling pathways (Bordt and Polster, 2014; Martindale and Holbrook, 2002). While NADPH oxidase gp91 subunit knockout mice established NADPH oxidase as the main source of extracellular superoxide production during innate immune activation (Qin et al., 2004), mitochondria may produce further ROS important for inflammatory processes (Bordt and Polster, 2014; Qin et al., 2004). While the source of ROS and the extent to which they regulate the inflammatory response remain contentious, that oxidative stress and inflammation are intricately linked seems certain.
As OXT, AVP, and ROS are shown to regulate inflammatory processes, what then is the effect of OXT and AVP on the regulation of ROS? As OXT has been described as possessing anti-inflammatory properties, it is reasonable to predict that it may also affect oxidative signaling. Indeed, work by Bikyikli et al utilizing an E. coli injection model in male rats demonstrated that OXT treatment reversed E. coli-induced increases in creatinine, blood urea, and TNF-α (Biyikli et al., 2006). They also found that OXT reversed E. coli-triggered increases in renal tissue malondialdehyde (MDA), myeloperoxidase (MPO), and decreased levels of glutathione (GSH), all markers of oxidative damage (Biyikli et al., 2006), indicating that OXT may possess the ability to negatively regulate oxidative damage. Within two different models of experimentally induced colitis, OXT treatment similarly reduced MDA and MPO levels, increased GSH levels, and decreased inflammatory cell infiltration and submucosal edema (Cetinel et al., 2010; Işeri et al., 2005) while the administration of the OTA atosiban reversed these protective effects (Cetinel et al., 2010). Incubation of THP-1 monocytes and macrophages with OXT dramatically decreased NADPH oxidase-dependent superoxide production and attenuated production of the inflammatory cytokine IL-6 (Szeto et al., 2008), suggestive of an intricate link between the actions of this neurohormone and the ROS/inflammatory balance. Newborn rats from mothers treated with the OTA atosiban showed increased markers of oxidative stress in their plasma and heart tissue, suggesting that OTR activation may protect against oxidative stress. Interestingly, no differences were found in the oxidative status in brains of these newborn rats (Simsek et al., 2012). However, within a model of cerebral ischemia, OTX pretreatment increases brain antioxidant levels via an elevation of glutathione peroxidase and decreases oxidative stress exposure via an increase in GSH/GSSG, effects that are blocked by an OTA (Karelina et al., 2011). Furthermore, in a model of early life stress, maternal separation decreased GSH and increased ROS formation in the hippocampus of mice, effects that were prevented with OTX i.c.v injections and reversed by the OTA atosiban (Amini-Khoei et al., 2017). Finally, OTX significantly reduced Cisplatin-induced neurotoxicity in mice by decreasing plasma levels of MDA and TNF-α and increasing GSH levels (Akman et al., 2015). All together, these studies provide support that OTX signaling protects tissues from oxidative damage.
While OXT appears to act as an antioxidant in many cases, at least one study suggests that the opposite may be true, or alternatively that there may be concentration-dependent effects of OXT. Levels of MDA and GSH in plasma and red blood cells were studied in human spontaneous or OXT-augmented births (Schneid-Kofman et al., 2009). Although no differences were found in MDA levels, indicating no change in lipid peroxidation between groups, GSH was significantly decreased in cord blood from OXT-induced births, suggestive of an increase in oxidative stress with exogenous OXT treatment as opposed to endogenous OXT release (Schneid-Kofman et al., 2009). While the lower GSH levels found in this study may indicate an increase in oxidative stress, the GSH/GSSG ratio is generally thought to be a more accurate marker of oxidative damage (Zitka et al., 2012) and would be a welcome addition to future studies. Further studies into the relationship between OXT treatment and oxidative stress following birth are necessary, especially in light of the fact that 23–50% of labors are induced using synthetic OXT (Declercq et al., 2014; Hamilton et al., 2015). Importantly, one study provides evidence that the neuroprotective effects of OXT at birth for fetal brain tissue might be concentration dependent (Ceanga et al., 2010). Within a model of oxygen-glucose deprivation (OGD) model of hypoxia-ischemia, Ceanga et al. showed that OXT is maximally neuroprotective at 1 µM but this protection significantly decreases at concentrations above or below 1 µM (Ceanga et al., 2010).
Much less is currently known regarding the role of AVP in the regulation of ROS. Intriguingly, AVP reduced hemorrhagic shock-triggered ROS production as measured by dichlorofluorescein fluorescence in isolated kidney mitochondria, as well as reducing levels of the lipid peroxidation and protein nitration markers 4-hydroxynonenal and 3-nitrotyrosine (Sims et al., 2017). While ROS are often thought of only in terms of their ability to damage, they are often also essential in the body as signaling molecules. In fact, ROS produced by hyperosmolarity in the supraoptic nucleus (SON) of the hypothalamus are necessary for noradrenergic-mediated increases in AVP levels (St-Louis et al., 2014, 2012). Given the well-defined links between OXT/AVP & inflammation as well as ROS & inflammation (Table 2), it is clear that further studies into the effects of OXT and AVP on ROS production are necessary.
Table 2.
Effects of oxytocin and vasopressin on reactive oxygen species.
| Oxytocin | Vasopressin | |
|---|---|---|
| Decrease malondialdehyde (MDA) and myeloperoxidase (MPO) | (Biyikli et al 2016) (Işeri et al 2005) (Cetinel et al 2010) |
- |
| Increase glutathione (GSH) | (Biyikli et al 2016) (Işeri et al 2005) (Cetinel et al 2010) |
- |
| Decrease glutathione (GSH) | (Schneid-Kofman et al 2009) | - |
| Reduce hemorrhagic shock-induced dichlorofluorescein fluorescence | - | (Sims et al 2017) |
Conclusion
While there is increasing interest in the roles of the UPRER and UPRmt, mitochondrial function, as well as OXT and AVP signaling in inflammatory responses, to date the intersection of these seemingly disparate pathways in the regulation of inflammatory responses remains understudied. While this review does not attempt to claim a definitive role for OXT and AVP in mitochondrial function or unfolded protein responses, we have discussed possible convergence points in the hope that these ideas spur future research. We have discussed the generally inflammatory role of AVP and anti-inflammatory role of OXT on the innate immune response, as well as evidence linking OXT and AVP to the regulation of the cellular homeostatic responses known as the unfolded protein responses in both the endoplasmic reticulum (UPRER) and the mitochondria (UPRmt). Mitochondrial function has been increasingly implicated in regulation of immune responses. While studies examining the effects of OXT and/or AVP on mitochondrial function remain limited, a growing literature suggests that these neurohormones may directly interact with mitochondria, and that these interactions could have vast effects on mitochondrial bioenergetic functioning, signaling, interaction with the unfolded protein response, and production of reactive oxygen species. These multitudes of neurohormone-mitochondrial interactions may play a significant role in regulating immune responses to a variety of homeostatic challenges, and are a vital area of future research.
References
- Acher R, Chauvet J, Chauvet MT (1995) Man and the chimaera. Selective versus neutral oxytocin evolution. Adv Exp Med Biol 395:615–627 [PubMed] [Google Scholar]
- Akman T, Akman L, Erbas O, et al. (2015) The preventive effect of oxytocin to Cisplatin-induced neurotoxicity: an experimental rat model. Biomed Res Int 2015:167235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberi S, Dreifuss JJ, Raggenbass M (1997) The oxytocin-induced inward current in vagal neurons of the rat is mediated by G protein activation but not by an increase in the intracellular calcium concentration. Eur J Neurosci 9:2605–2612 [DOI] [PubMed] [Google Scholar]
- Ameri K, Harris AL (2008) Activating transcription factor 4. Int J Biochem Cell Biol 40:14–21 [DOI] [PubMed] [Google Scholar]
- Amini-Khoei H, Mohammadi-Asl A, Amiri S, et al. (2017) Oxytocin mitigated the depressive-like behaviors of maternal separation stress through modulating mitochondrial function and neuroinflammation. Prog Neuropsychopharmacol Biol Psychiatry 76:169–178 [DOI] [PubMed] [Google Scholar]
- Asada R, Kanemoto S, Kondo S, et al. (2011) The signalling from endoplasmic reticulum-resident bZIP transcription factors involved in diverse cellular physiology. J Biochem 149:507–518 [DOI] [PubMed] [Google Scholar]
- Azuma Y, Hagiwara D, Lu W, et al. (2014) Activating transcription factor 6α is required for the vasopressin neuron system to maintain water balance under dehydration in male mice. Endocrinology 155:4905–4914 [DOI] [PubMed] [Google Scholar]
- Baker BM, Nargund AM, Sun T, Haynes CM (2012) Protective coupling of mitochondrial function and protein synthesis via the eIF2α kinase GCN-2. PLoS Genet 8:e1002760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreca T, Gandolfo C, Corsini G, et al. (2001) Evaluation of the secretory pattern of plasma arginine vasopressin in stroke patients. Cerebrovasc Dis 11:113–118 [DOI] [PubMed] [Google Scholar]
- Belin V, Moos F, Richard P (1984) Synchronization of oxytocin cells in the hypothalamic paraventricular and supraoptic nuclei in suckled rats: direct proof with paired extracellular recordings. Exp Brain Res 57:201–203 [DOI] [PubMed] [Google Scholar]
- Bénard G, Massa F, Puente N, et al. (2012) Mitochondrial CB₁ receptors regulate neuronal energy metabolism. Nat Neurosci 15:558–564 [DOI] [PubMed] [Google Scholar]
- Biyikli NK, Tuğtepe H, Sener G, et al. (2006) Oxytocin alleviates oxidative renal injury in pyelonephritic rats via a neutrophil-dependent mechanism. Peptides 27:2249–2257 [DOI] [PubMed] [Google Scholar]
- Block ML, Hong J-S (2005) Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog Neurobiol 76:77–98 [DOI] [PubMed] [Google Scholar]
- Block ML, Zecca L, Hong J-S (2007) Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci 8:57–69 [DOI] [PubMed] [Google Scholar]
- Boone M, Deen PMT (2008) Physiology and pathophysiology of the vasopressin-regulated renal water reabsorption. Pflugers Arch 456:1005–1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordt EA, Polster BM (2014) NADPH oxidase- and mitochondria-derived reactive oxygen species in proinflammatory microglial activation: a bipartisan affair? Free Radic Biol Med 76:34–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourque CW (2008) Central mechanisms of osmosensation and systemic osmoregulation. Nat Rev Neurosci 9:519–531 [DOI] [PubMed] [Google Scholar]
- Brownstein MJ, Russell JT, Gainer H (1980) Synthesis, transport, and release of posterior pituitary hormones. Science 207:373–378 [DOI] [PubMed] [Google Scholar]
- Casas F, Domenjoud L, Rochard P, et al. (2000) A 45 kDa protein related to PPARgamma2, induced by peroxisome proliferators, is located in the mitochondrial matrix. FEBS Lett 478:4–8 [DOI] [PubMed] [Google Scholar]
- Castoldi A, Naffah de Souza C, Câmara NOS, Moraes-Vieira PM (2015) The Macrophage Switch in Obesity Development. Front Immunol 6:637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceanga M, Spataru A, Zagrean A-M (2010) Oxytocin is neuroprotective against oxygen-glucose deprivation and reoxygenation in immature hippocampal cultures. Neurosci Lett 477:15–18 [DOI] [PubMed] [Google Scholar]
- Celli J, Tsolis RM (2015) Bacteria, the endoplasmic reticulum and the unfolded protein response: friends or foes? Nat Rev Microbiol 13:71–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cetinel S, Hancioğlu S, Sener E, et al. (2010) Oxytocin treatment alleviates stress-aggravated colitis by a receptor-dependent mechanism. Regul Pept 160:146–152 [DOI] [PubMed] [Google Scholar]
- Chakravarthi S, Jessop CE, Bulleid NJ (2006) The role of glutathione in disulphide bond formation and endoplasmic-reticulum-generated oxidative stress. EMBO Rep 7:271–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaves VE, Tilelli CQ, Brito NA, Brito MN (2013) Role of oxytocin in energy metabolism. Peptides 45:9–14 [DOI] [PubMed] [Google Scholar]
- Chen JQ, Delannoy M, Cooke C, Yager JD (2004) Mitochondrial localization of ERalpha and ERbeta in human MCF7 cells. Am J Physiol Endocrinol Metab 286:E1011–22 [DOI] [PubMed] [Google Scholar]
- Chen Y, Hallenbeck JM, Ruetzler C, et al. (2003) Overexpression of monocyte chemoattractant protein 1 in the brain exacerbates ischemic brain injury and is associated with recruitment of inflammatory cells. J Cereb Blood Flow Metab 23:748–755 [DOI] [PubMed] [Google Scholar]
- Collins LV, Hajizadeh S, Holme E, et al. (2004) Endogenously oxidized mitochondrial DNA induces in vivo and in vitro inflammatory responses. J Leukoc Biol 75:995–1000 [DOI] [PubMed] [Google Scholar]
- Crambert G, Ernandez T, Lamouroux C, et al. (2014) Epithelial sodium channel abundance is decreased by an unfolded protein response induced by hyperosmolality. Physiol Rep 2: 10.14814/phy2.12169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das B, Sarkar C (2012) Is preconditioning by oxytocin administration mediated by iNOS and/or mitochondrial K(ATP) channel activation in the in vivo anesthetized rabbit heart? Life Sci 90:763–769 [DOI] [PubMed] [Google Scholar]
- Declercq ER, Sakala C, Corry MP, et al. (2014) Major Survey Findings of Listening to Mothers(SM) III: Pregnancy and Birth: Report of the Third National U.S. Survey of Women’s Childbearing Experiences. J Perinat Educ 23:9–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diano S, Urbanski HF, Horvath B, et al. (2000) Mitochondrial uncoupling protein 2 (UCP2) in the nonhuman primate brain and pituitary. Endocrinology 141:4226–4238 [DOI] [PubMed] [Google Scholar]
- Di Benedetto A, Sun L, Zambonin CG, et al. (2014) Osteoblast regulation via ligand-activated nuclear trafficking of the oxytocin receptor. Proc Natl Acad Sci U S A 111:16502–16507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du D, Jiang M, Liu M, et al. (2015) Microglial P2X₇ receptor in the hypothalamic paraventricular nuclei contributes to sympathoexcitatory responses in acute myocardial infarction rat. Neurosci Lett 587:22–28 [DOI] [PubMed] [Google Scholar]
- Dumais KM, Veenema AH (2016) Vasopressin and oxytocin receptor systems in the brain: Sex differences and sex-specific regulation of social behavior. Front Neuroendocrinol 40:1–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echtay KS (2007) Mitochondrial uncoupling proteins--what is their physiological role? Free Radic Biol Med 43:1351–1371 [DOI] [PubMed] [Google Scholar]
- Ficek W (1983) Physiological dependency between the hypothalamus and the thymus of Wistar rats. IV. Organotypic culture of the thymus in the presence of hypophyseal hormones, vasopressin, and oxytocin. Gegenbaurs Morphol Jahrb 129:445–458 [PubMed] [Google Scholar]
- Fiorese CJ, Schulz AM, Lin Y-F, et al. (2016) The Transcription Factor ATF5 Mediates a Mammalian Mitochondrial UPR. Curr Biol 26:2037–2043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frantz S, Nahrendorf M (2014) Cardiac macrophages and their role in ischaemic heart disease. Cardiovasc Res 102:240–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs AR, Poblete VF Jr (1970) Oxytocin and uterine function in pregnant and parturient rats. Biol Reprod 2:387–400 [DOI] [PubMed] [Google Scholar]
- Gargalovic PS, Gharavi NM, Clark MJ, et al. (2006) The unfolded protein response is an important regulator of inflammatory genes in endothelial cells. Arterioscler Thromb Vasc Biol 26:2490–2496 [DOI] [PubMed] [Google Scholar]
- Ghiringhelli F, Apetoh L, Tesniere A, et al. (2009) Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med 15:1170–1178 [DOI] [PubMed] [Google Scholar]
- Gillies GE, Linton EA, Lowry PJ (1982) Corticotropin releasing activity of the new CRF is potentiated several times by vasopressin. Nature 299:355–357 [DOI] [PubMed] [Google Scholar]
- Gkogkas CG, Khoutorsky A, Ran I, et al. (2013) Autism-related deficits via dysregulated eIF4E-dependent translational control. Nature 493:371–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobrogge K, Wang Z (2015) Neuropeptidergic regulation of pair-bonding and stress buffering: Lessons from voles. Horm Behav 76:91–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodson JL (2008) Nonapeptides and the evolutionary patterning of sociality. Prog Brain Res 170:3–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravina FS, Jobling P, Kerr KP, et al. (2011) Oxytocin depolarizes mitochondria in isolated myometrial cells. Exp Physiol 96:949–956 [DOI] [PubMed] [Google Scholar]
- Greenwood M, Bordieri L, Greenwood MP, et al. (2014) Transcription factor CREB3L1 regulates vasopressin gene expression in the rat hypothalamus. J Neurosci 34:3810–3820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwood M, Greenwood MP, Paton JFR, Murphy D (2015a) Transcription Factor CREB3L1 Regulates Endoplasmic Reticulum Stress Response Genes in the Osmotically Challenged Rat Hypothalamus. PLoS One 10:e0124956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwood MP, Mecawi AS, Hoe SZ, et al. (2015b) A comparison of physiological and transcriptome responses to water deprivation and salt loading in the rat supraoptic nucleus. Am J Physiol Regul Integr Comp Physiol 308:R559–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddad JJ, Land SC (2002) Redox/ROS regulation of lipopolysaccharide-induced mitogen-activated protein kinase (MAPK) activation and MAPK-mediated TNF-alpha biosynthesis. Br J Pharmacol 135:520–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagiwara D, Arima H, Morishita Y, et al. (2012) BiP mRNA expression is upregulated by dehydration in vasopressin neurons in the hypothalamus in mice. Peptides 33:346–350 [DOI] [PubMed] [Google Scholar]
- Hagiwara D, Arima H, Morishita Y, et al. (2014) Arginine vasopressin neuronal loss results from autophagy-associated cell death in a mouse model for familial neurohypophysial diabetes insipidus. Cell Death Dis 5:e1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton BE, Martin JA, Osterman MJK, et al. (2015) Births: Final Data for 2014. Natl Vital Stat Rep 64:1–64 [PubMed] [Google Scholar]
- Harding HP, Novoa I, Zhang Y, et al. (2000) Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell 6:1099–1108 [DOI] [PubMed] [Google Scholar]
- Harkany T, Horvath TL (2017) (S)Pot on Mitochondria: Cannabinoids Disrupt Cellular Respiration to Limit Neuronal Activity. Cell Metab 25:8–10 [DOI] [PubMed] [Google Scholar]
- Haschemi A, Kosma P, Gille L, et al. (2012) The sedoheptulose kinase CARKL directs macrophage polarization through control of glucose metabolism. Cell Metab 15:813–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havránek T, Lešťanová Z, Mravec B, et al. (2017) Oxytocin Modulates Expression of Neuron and Glial Markers in the Rat Hippocampus. Folia Biol 63:91–97 [PubMed] [Google Scholar]
- Haynes CM, Yang Y, Blais SP, et al. (2010) The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Mol Cell 37:529–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haze K, Yoshida H, Yanagi H, et al. (1999) Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell 10:3787–3799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert-Chatelain E, Desprez T, Serrat R, et al. (2016) A cannabinoid link between mitochondria and memory. Nature 539:555–559 [DOI] [PubMed] [Google Scholar]
- Hetz C (2012) The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol 13:89–102 [DOI] [PubMed] [Google Scholar]
- Hetz C, Martinon F, Rodriguez D, Glimcher LH (2011) The unfolded protein response: integrating stress signals through the stress sensor IRE1α. Physiol Rev 91:1219–1243 [DOI] [PubMed] [Google Scholar]
- Honma Y, Kanazawa K, Mori T, et al. (1999) Identification of a novel gene, OASIS, which encodes for a putative CREB/ATF family transcription factor in the long-term cultured astrocytes and gliotic tissue. Brain Res Mol Brain Res 69:93–103 [DOI] [PubMed] [Google Scholar]
- Horvath TL, Warden CH, Hajos M, et al. (1999) Brain uncoupling protein 2: uncoupled neuronal mitochondria predict thermal synapses in homeostatic centers. J Neurosci 19:10417–10427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou D, Jin F (2016) Model Roles of the Hypothalamo-Neurohypophysial System in Neuroscience Study. Biochem Pharmacol 5: 10.4172/2167-0501.1000211 [DOI] [Google Scholar]
- Işeri SO, Sener G, Sağlam B, et al. (2005) Oxytocin ameliorates oxidative colonic inflammation by a neutrophil-dependent mechanism. Peptides 26:483–491 [DOI] [PubMed] [Google Scholar]
- Iyer SS, He Q, Janczy JR, et al. (2013) Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 39:311–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer SS, Pulskens WP, Sadler JJ, et al. (2009) Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc Natl Acad Sci U S A 106:20388–20393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaber SM, Bordt EA, Bhatt NM, et al. (2017) Sex differences in the mitochondrial bioenergetics of astrocytes but not microglia at a physiologically relevant brain oxygen tension. Neurochem Int. 10.1016/j.neuint.2017.09.003 [DOI] [PMC free article] [PubMed]
- Jankowski M, Bissonauth V, Gao L, et al. (2010) Anti-inflammatory effect of oxytocin in rat myocardial infarction. Basic Res Cardiol 105:205–218 [DOI] [PubMed] [Google Scholar]
- Jeong D-E, Lee D, Hwang S-Y, et al. (2017) Mitochondrial chaperone HSP-60 regulates anti-bacterial immunity via p38 MAP kinase signaling. EMBO J 36:1046–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha AK, Huang SC-C, Sergushichev A, et al. (2015) Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 42:419–430 [DOI] [PubMed] [Google Scholar]
- Kaneko Y, Pappas C, Tajiri N, Borlongan CV (2016) Oxytocin modulates GABAAR subunits to confer neuroprotection in stroke in vitro. Sci Rep 6:35659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang S-W, Rane NS, Kim SJ, et al. (2006) Substrate-specific translocational attenuation during ER stress defines a pre-emptive quality control pathway. Cell 127:999–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karelina K, Stuller KA, Jarrett B, et al. (2011) Oxytocin mediates social neuroprotection after cerebral ischemia. Stroke 42:3606–3611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelleher RJ 3rd, Bear MF (2008) The autistic neuron: troubled translation? Cell 135:401–406 [DOI] [PubMed] [Google Scholar]
- Kelly AM, Goodson JL (2014) Social functions of individual vasopressin-oxytocin cell groups in vertebrates: what do we really know? Front Neuroendocrinol 35:512–529 [DOI] [PubMed] [Google Scholar]
- Klein BY, Tamir H, Hirschberg DL, et al. (2013) Oxytocin modulates mTORC1 pathway in the gut. Biochem Biophys Res Commun 432:466–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein BY, Tamir H, Hirschberg DL, et al. (2014) Oxytocin modulates markers of the unfolded protein response in Caco2BB gut cells. Cell Stress Chaperones 19:465–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein BY, Tamir H, Hirschberg DL, et al. (2016) Oxytocin opposes effects of bacterial endotoxin on ER-stress signaling in Caco2BB gut cells. Biochim Biophys Acta 1860:402–411 [DOI] [PubMed] [Google Scholar]
- Knupp J, Arvan P, Chang A (2018) Increased mitochondrial respiration promotes survival from endoplasmic reticulum stress. Cell Death Differ. 10.1038/s41418-018-0133-4 [DOI] [PMC free article] [PubMed]
- Kohr MJ, Sun J, Aponte A, et al. (2011) Simultaneous measurement of protein oxidation and S-nitrosylation during preconditioning and ischemia/reperfusion injury with resin-assisted capture. Circ Res 108:418–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo S, Saito A, Asada R, et al. (2011) Physiological unfolded protein response regulated by OASIS family members, transmembrane bZIP transcription factors. IUBMB Life 63:233–239 [DOI] [PubMed] [Google Scholar]
- Kotas ME, Medzhitov R (2015) Homeostasis, inflammation, and disease susceptibility. Cell 160:816–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozniewska E, Romaniuk K (2008) Vasopressin in vascular regulation and water homeostasis in the brain. J Physiol Pharmacol 59 Suppl 8:109–116 [PubMed] [Google Scholar]
- Kroemer G, Mariño G, Levine B (2010) Autophagy and the integrated stress response. Mol Cell 40:280–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krysko DV, Agostinis P, Krysko O, et al. (2011) Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol 32:157–164 [DOI] [PubMed] [Google Scholar]
- Lampropoulou V, Sergushichev A, Bambouskova M, et al. (2016) Itaconate Links Inhibition of Succinate Dehydrogenase with Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab 24:158–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landgraf R, Neumann ID (2004) Vasopressin and oxytocin release within the brain: a dynamic concept of multiple and variable modes of neuropeptide communication. Front Neuroendocrinol 25:150–176 [DOI] [PubMed] [Google Scholar]
- Lee A-H, Iwakoshi NN, Glimcher LH (2003) XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol 23:7448–7459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Sharma S, Kim J, et al. (2008) Mitochondrial nuclear receptors and transcription factors: who’s minding the cell? J Neurosci Res 86:961–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Tirasophon W, Shen X, et al. (2002) IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev 16:452–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehninger AL, Neubert D (1961) Effect of oxytocin, vasopressin, and other disulfide hormones on uptake and extrusion of water by mitochondria. Proc Natl Acad Sci U S A 47:1929–1936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberti MV, Locasale JW (2016) The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem Sci 41:211–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Wang P, Wang SC, Wang Y-F (2016) Approaches Mediating Oxytocin Regulation of the Immune System. Front Immunol 7:693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Samuel BS, Breen PC, Ruvkun G (2014) Caenorhabditis elegans pathways that surveil and defend mitochondria. Nature 508:406–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig M, Leng G (2006) Dendritic peptide release and peptide-dependent behaviours. Nat Rev Neurosci 7:126–136 [DOI] [PubMed] [Google Scholar]
- Martindale JL, Holbrook NJ (2002) Cellular response to oxidative stress: signaling for suicide and survival. J Cell Physiol 192:1–15 [DOI] [PubMed] [Google Scholar]
- Martinon F, Chen X, Lee A-H, Glimcher LH (2010) TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat Immunol 11:411–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinus RD, Garth GP, Webster TL, et al. (1996) Selective induction of mitochondrial chaperones in response to loss of the mitochondrial genome. Eur J Biochem 240:98–103 [DOI] [PubMed] [Google Scholar]
- Masi A, Quintana DS, Glozier N, et al. (2015) Cytokine aberrations in autism spectrum disorder: a systematic review and meta-analysis. Mol Psychiatry 20:440–446 [DOI] [PubMed] [Google Scholar]
- Matzinger P (1994) Tolerance, danger, and the extended family. Annu Rev Immunol 12:991–1045 [DOI] [PubMed] [Google Scholar]
- McKinley MJ, McAllen RM, Davern P, et al. (2003) The sensory circumventricular organs of the mammalian brain. Adv Anat Embryol Cell Biol 172:III–XII, 1–122, back cover [DOI] [PubMed] [Google Scholar]
- Medzhitov R (2008) Origin and physiological roles of inflammation. Nature 454:428–435 [DOI] [PubMed] [Google Scholar]
- Mehta MM, Weinberg SE, Chandel NS (2017) Mitochondrial control of immunity: beyond ATP. Nat Rev Immunol. 10.1038/nri.2017.66 [DOI] [PubMed]
- Melber A, Haynes CM (2018) UPRmtregulation and output: a stress response mediated by mitochondrial-nuclear communication. Cell Res 28:281–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel S, Canonne M, Arnould T, Renard P (2015) Inhibition of mitochondrial genome expression triggers the activation of CHOP-10 by a cell signaling dependent on the integrated stress response but not the mitochondrial unfolded protein response. Mitochondrion 21:58–68 [DOI] [PubMed] [Google Scholar]
- Michelucci A, Cordes T, Ghelfi J, et al. (2013) Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc Natl Acad Sci U S A 110:7820–7825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DB, O’Callaghan JP (2002) Neuroendocrine aspects of the response to stress. Metabolism 5:5–10 [DOI] [PubMed] [Google Scholar]
- Mills EL, Kelly B, Logan A, et al. (2016) Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell 167:457–470.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki S, Hiraoka Y, Hidema S, Nishimori K (2016) Prenatal minocycline treatment alters synaptic protein expression, and rescues reduced mother call rate in oxytocin receptor-knockout mice. Biochem Biophys Res Commun 472:319–323 [DOI] [PubMed] [Google Scholar]
- Morris G, Berk M (2015) The many roads to mitochondrial dysfunction in neuroimmune and neuropsychiatric disorders. BMC Med 13:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami T, Saito A, Hino S-I, et al. (2009) Signalling mediated by the endoplasmic reticulum stress transducer OASIS is involved in bone formation. Nat Cell Biol 11:1205–1211 [DOI] [PubMed] [Google Scholar]
- Nakahira K, Haspel JA, Rathinam VAK, et al. (2011) Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 12:222–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nargund AM, Fiorese CJ, Pellegrino MW, et al. (2015) Mitochondrial and nuclear accumulation of the transcription factor ATFS-1 promotes OXPHOS recovery during the UPR(mt). Mol Cell 58:123–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nargund AM, Pellegrino MW, Fiorese CJ, et al. (2012) Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science 337:587–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navid F, Colbert RA (2017) Causes and consequences of endoplasmic reticulum stress in rheumatic disease. Nat Rev Rheumatol 13:25–40 [DOI] [PubMed] [Google Scholar]
- Neubert D, Lehninger AL (1962) The effect of thiols and disulfides on water uptake and extrusion by rat liver mitochondria. J Biol Chem 237:952–958 [PubMed] [Google Scholar]
- Neves-Pereira M, Müller B, Massie D, et al. (2009) Deregulation of EIF4E: a novel mechanism for autism. J Med Genet 46:759–765 [DOI] [PubMed] [Google Scholar]
- Ocampo Daza D, Lewicka M, Larhammar D (2012) The oxytocin/vasopressin receptor family has at least five members in the gnathostome lineage, inclucing two distinct V2 subtypes. Gen Comp Endocrinol 175:135–143 [DOI] [PubMed] [Google Scholar]
- Oliveira AN, Hood DA (2018) Effect of Tim23 Knockdown in vivo on Mitochondrial Protein Import and Retrograde Signaling to the UPRmt in Muscle. Am J Physiol Cell Physiol. 10.1152/ajpcell.00275.2017 [DOI] [PMC free article] [PubMed]
- Oliveira-Pelegrin GR, Saia RS, Cárnio EC, Rocha MJA (2013) Oxytocin affects nitric oxide and cytokine production by sepsis-sensitized macrophages. Neuroimmunomodulation 20:65–71 [DOI] [PubMed] [Google Scholar]
- Orihuela R, McPherson CA, Harry GJ (2016) Microglial M1/M2 polarization and metabolic states. Br J Pharmacol 173:649–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyadomari S, Yun C, Fisher EA, et al. (2006) Cotranslocational degradation protects the stressed endoplasmic reticulum from protein overload. Cell 126:727–739 [DOI] [PubMed] [Google Scholar]
- Palmer HJ, Paulson KE (1997) Reactive oxygen species and antioxidants in signal transduction and gene expression. Nutr Rev 55:353–361 [DOI] [PubMed] [Google Scholar]
- Palsson-McDermott EM, Curtis AM, Goel G, et al. (2015) Pyruvate kinase M2 regulates Hif-1α activity and IL-1β induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab 21:65–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce EJ, Pearce EL (2017) Immunometabolism in 2017: Driving immunity: all roads lead to metabolism. Nat Rev Immunol. 10.1038/nri.2017.139 [DOI] [PMC free article] [PubMed]
- Pellegrino MW, Nargund AM, Kirienko NV, et al. (2014) Mitochondrial UPR-regulated innate immunity provides resistance to pathogen infection. Nature 516:414–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittman QJ, Chen X, Mouihate A, et al. (1998) Arginine vasopressin, fever and temperature regulation. Prog Brain Res 119:383–392 [DOI] [PubMed] [Google Scholar]
- Pittman QJ, Lawrence D, McLean L (1982) Central effects of arginine vasopressin on blood pressure in rats. Endocrinology 110:1058–1060 [DOI] [PubMed] [Google Scholar]
- Qin L, Liu Y, Wang T, et al. (2004) NADPH oxidase mediates lipopolysaccharide-induced neurotoxicity and proinflammatory gene expression in activated microglia. J Biol Chem 279:1415–1421 [DOI] [PubMed] [Google Scholar]
- Quirós PM, Prado MA, Zamboni N, et al. (2017) Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J Cell Biol 216:2027–2045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qureshi MA, Haynes CM, Pellegrino MW (2017) The mitochondrial unfolded protein response: Signaling from the powerhouse. J Biol Chem 292:13500–13506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roach JC, Smith KD, Strobe KL, et al. (2007) Transcription factor expression in lipopolysaccharide-activated peripheral-blood-derived mononuclear cells. Proc Natl Acad Sci U S A 104:16245–16250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D, Walter P (2007) Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 8:519–529 [DOI] [PubMed] [Google Scholar]
- Rongvaux A (2017) Innate immunity and tolerance toward mitochondria. Mitochondrion. 10.1016/j.mito.2017.10.007 [DOI] [PubMed]
- Roth CL, D’Ambrosio G, Elfers C (2016) Activation of nuclear factor kappa B pathway and reduction of hypothalamic oxytocin following hypothalamic lesions. J Syst Integr Neurosci 2:79–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runkel ED, Liu S, Baumeister R, Schulze E (2013) Surveillance-activated defenses block the ROS-induced mitochondrial unfolded protein response. PLoS Genet 9:e1003346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutkowski DT, Hegde RS (2010) Regulation of basal cellular physiology by the homeostatic unfolded protein response. J Cell Biol 189:783–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagan L (1967) On the origin of mitosing cells. J Theor Biol 14:255–274 [DOI] [PubMed] [Google Scholar]
- Schneid-Kofman N, Silberstein T, Saphier O, et al. (2009) Labor augmentation with oxytocin decreases glutathione level. Obstet Gynecol Int 2009:807659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder K, Tschopp J (2010) The inflammasomes. Cell 140:821–832 [DOI] [PubMed] [Google Scholar]
- Schröder M, Kaufman RJ (2005a) ER stress and the unfolded protein response. Mutat Res 569:29–63 [DOI] [PubMed] [Google Scholar]
- Schröder M, Kaufman RJ (2005b) The mammalian unfolded protein response. Annu Rev Biochem 74:739–789 [DOI] [PubMed] [Google Scholar]
- Shao L-W, Niu R, Liu Y (2016) Neuropeptide signals cell non-autonomous mitochondrial unfolded protein response. Cell Res 26:1182–1196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada K, Crother TR, Karlin J, et al. (2012) Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36:401–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva YJ, Moffat RC, Walt AJ (1969) Vasopressin effect on portal and systemic hemodynamics. Studies in intact, unanesthetized humans. JAMA 210:1065–1068 [PubMed] [Google Scholar]
- Sims CA, Yuxia G, Singh K, et al. (2017) Supplemental arginine vasopressin during the resuscitation of severe hemorrhagic shock preserves renal mitochondrial function. PLoS One 12:e0186339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simsek Y, Celik O, Karaer A, et al. (2012) Elevated cardiac oxidative stress in newborn rats from mothers treated with atosiban. Arch Gynecol Obstet 285:655–661 [DOI] [PubMed] [Google Scholar]
- Smith AS, Tabbaa M, Lei K, et al. (2016) Local oxytocin tempers anxiety by activating GABAA receptors in the hypothalamic paraventricular nucleus. Psychoneuroendocrinology 63:50–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Z, Albers HE (2017) Cross-talk among oxytocin and arginine-vasopressin receptors: Relevance for basic and clinical studies of the brain and periphery. Front Neuroendocrinol. 10.1016/j.yfrne.2017.10.004 [DOI] [PMC free article] [PubMed]
- Sørensen PS, Gjerris A, Hammer M (1985) Cerebrospinal fluid vasopressin in neurological and psychiatric disorders. J Neurol Neurosurg Psychiatry 48:50–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steen KA, Xu H, Bernlohr DA (2017) FABP4/aP2 Regulates Macrophage Redox Signaling and Inflammasome Activation via Control of UCP2. Mol Cell Biol 37: 10.1128/MCB.00282-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- St-Louis R, Parmentier C, Grange-Messent V, et al. (2014) Reactive oxygen species are physiological mediators of the noradrenergic signaling pathway in the mouse supraoptic nucleus. Free Radic Biol Med 71:231–239 [DOI] [PubMed] [Google Scholar]
- St-Louis R, Parmentier C, Raison D, et al. (2012) Reactive oxygen species are required for the hypothalamic osmoregulatory response. Endocrinology 153:1317–1329 [DOI] [PubMed] [Google Scholar]
- Stoop R (2012) Neuromodulation by oxytocin and vasopressin. Neuron 76:142–159 [DOI] [PubMed] [Google Scholar]
- Sun J, Steenbergen C, Murphy E (2006) S-nitrosylation: NO-related redox signaling to protect against oxidative stress. Antioxid Redox Signal 8:1693–1705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szeto A, Nation DA, Mendez AJ, et al. (2008) Oxytocin attenuates NADPH-dependent superoxide activity and IL-6 secretion in macrophages and vascular cells. Am J Physiol Endocrinol Metab 295:E1495–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szeto A, Sun-Suslow N, Mendez AJ, et al. (2017) Regulation of the macrophage oxytocin receptor in response to inflammation. Am J Physiol Endocrinol Metab 312:E183–E189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szmydynger-Chodobska J, Fox LM, Lynch KM, et al. (2010) Vasopressin amplifies the production of proinflammatory mediators in traumatic brain injury. J Neurotrauma 27:1449–1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szmydynger-Chodobska J, Zink BJ, Chodobski A (2011) Multiple sites of vasopressin synthesis in the injured brain. J Cereb Blood Flow Metab 31:47–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ta HM, Le TM, Ishii H, et al. (2016) Atf6α deficiency suppresses microglial activation and ameliorates pathology of experimental autoimmune encephalomyelitis. J Neurochem 139:1124–1137 [DOI] [PubMed] [Google Scholar]
- Tannahill GM, Curtis AM, Adamik J, et al. (2013) Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 496:238–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teske BF, Fusakio ME, Zhou D, et al. (2013) CHOP induces activating transcription factor 5 (ATF5) to trigger apoptosis in response to perturbations in protein homeostasis. Mol Biol Cell 24:2477–2490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Horssen J, van Schaik P, Witte M (2017) Inflammation and mitochondrial dysfunction: A vicious circle in neurodegenerative disorders? Neurosci Lett. 10.1016/j.neulet.2017.06.050 [DOI] [PubMed]
- Vats D, Mukundan L, Odegaard JI, et al. (2006) Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab 4:13–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villapol S, Loane DJ, Burns MP (2017) Sexual dimorphism in the inflammatory response to traumatic brain injury. Glia 65:1423–1438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P, Ron D (2011) The unfolded protein response: from stress pathway to homeostatic regulation. Science 334:1081–1086 [DOI] [PubMed] [Google Scholar]
- Wang H, Doering LC (2013) Reversing autism by targeting downstream mTOR signaling. Front Cell Neurosci 7:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Gao K, Liu Y (2017) UPRmtcoordinates immunity to maintain mitochondrial homeostasis and animal fitness. Mitochondrion. 10.1016/j.mito.2017.11.004 [DOI] [PubMed]
- Wrobel L, Topf U, Bragoszewski P, et al. (2015) Mistargeted mitochondrial proteins activate a proteostatic response in the cytosol. Nature 524:485–488 [DOI] [PubMed] [Google Scholar]
- Wrutniak C, Cassar-Malek I, Marchal S, et al. (1995) A 43-kDa protein related to c-Erb A alpha 1 is located in the mitochondrial matrix of rat liver. J Biol Chem 270:16347–16354 [DOI] [PubMed] [Google Scholar]
- Xu H, Hertzel AV, Steen KA, et al. (2015) Uncoupling lipid metabolism from inflammation through fatty acid binding protein-dependent expression of UCP2. Mol Cell Biol 35:1055–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Su W, Huang W-D, et al. (2007) Effect of AVP on brain edema following traumatic brain injury. Chin J Traumatol 10:90–93 [PubMed] [Google Scholar]
- Yamamoto K, Sato T, Matsui T, et al. (2007) Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev Cell 13:365–376 [DOI] [PubMed] [Google Scholar]
- Yoneda T, Benedetti C, Urano F, et al. (2004) Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. J Cell Sci 117:4055–4066 [DOI] [PubMed] [Google Scholar]
- Yoshida H, Matsui T, Yamamoto A, et al. (2001) XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107:881–891 [DOI] [PubMed] [Google Scholar]
- Young WS 3rd, Gainer H (2003) Transgenesis and the study of expression, cellular targeting and function of oxytocin, vasopressin and their receptors. Neuroendocrinology 78:185–203 [DOI] [PubMed] [Google Scholar]
- Yuan L, Liu S, Bai X, et al. (2016) Oxytocin inhibits lipopolysaccharide-induced inflammation in microglial cells and attenuates microglial activation in lipopolysaccharide-treated mice. J Neuroinflammation 13:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng K-W, Zhang T, Fu H, et al. (2012) Schisandrin B exerts anti-neuroinflammatory activity by inhibiting the Toll-like receptor 4-dependent MyD88/IKK/NF-κB signaling pathway in lipopolysaccharide-induced microglia. Eur J Pharmacol 692:29–37 [DOI] [PubMed] [Google Scholar]
- Zhang Q, Raoof M, Chen Y, et al. (2010) Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464:104–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W, Zheng X, Liu S, et al. (2012) TNFα and IL-1β are mediated by both TLR4 and Nod1 pathways in the cultured HAPI cells stimulated by LPS. Biochem Biophys Res Commun 420:762–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D, Palam LR, Jiang L, et al. (2008) Phosphorylation of eIF2 directs ATF5 translational control in response to diverse stress conditions. J Biol Chem 283:7064–7073 [DOI] [PubMed] [Google Scholar]
- Zhou R, Yazdi AS, Menu P, Tschopp J (2011) A role for mitochondria in NLRP3 inflammasome activation. Nature 469:221–225 [DOI] [PubMed] [Google Scholar]
- Zimmet JM, Hare JM (2006) Nitroso-redox interactions in the cardiovascular system. Circulation 114:1531–1544 [DOI] [PubMed] [Google Scholar]
- Zitka O, Skalickova S, Gumulec J, et al. (2012) Redox status expressed as GSH:GSSG ratio as a marker for oxidative stress in paediatric tumour patients. Oncol Lett 4:1247–1253 [DOI] [PMC free article] [PubMed] [Google Scholar]