

Abstract
Programmed Death Ligand 1 (PD-L1) is a critical immune checkpoint ligand whose overexpression on tumor cells provides a mechanism of escape from immune surveillance. The interaction between PD-L1 and PD-1 on T cell lymphocytes suppresses both T cell activation and effector function and is engaged by cancers to dampen anti-tumor immunity. Here, we used mRNA display to engineer an 18-residue linear peptide that binds to human PD-L1. This peptide, which we term SPAM (Signal Peptide based Affinity Maturated ligand), is non-homologous to known PD-L1 binding peptides and mAbs, with dissociation constants (KD) of 119 nM and 67 nM for unglycosylated and glycosylated human PD-L1, respectively. The SPAM peptide is highly selective for human PD-L1 and shows no significant binding to either mouse PD-L1 or human PD-L2. Competition binding assays indicate that the SPAM peptide binding site overlaps with the binding site of PD-1 as well as therapeutic anti-PD-L1 antibodies. Taken together, these results suggest that the SPAM peptide specifically binds to human PD-L1 and could potentially serve as a PD-L1 affinity agent and PD-L1/PD-1 pathway modulator.
Graphical Abstract
Introduction
The immune system constantly monitors for the formation of incipient tumor cells and eliminates nascent malignancies before they expand and metastasize (immune surveillance). Tumors, on the other hand, exploit numerous cellular and molecular mechanisms to escape removal by the immune system. Indeed, evading immune destruction is recognized as one of the hallmarks of cancer.1
Inhibitory immune checkpoint pathways are among the key mechanisms that enable tumor cells to escape recognition and lysis by T cells. Programmed Death Ligand 1 (PD-L1) is a critical immune checkpoint ligand that is expressed on the surface of tumor cells as well as their myeloid stroma and endothelium in response to inflammatory cytokines such as interferon gamma. PD-L1 engages the Programmed Cell Death Protein 1 (PD-1) receptor on T cells and suppresses immune responses by dampening their activation and effector function.2, 3 PD-L1 is also expressed on the surface of a variety of cancer cells4 and its overexpression occurs on tumor cells as a response to T cell infiltration.5 Binding of tumor PD-L1 to PD-1 on the surface of immune cells deactivates infiltrating cytotoxic T lymphocytes and Natural Killer (NK) cells, thereby attenuating anti-tumor immunity and fostering a state of tumor immune privilege.6
Immune checkpoint blockade (ICB) therapy was developed with the goal of blocking the inhibitory interaction between tumor cells and immune cells to reactivate anti-tumor immune responses. Several monoclonal antibodies (mAbs) are clinically approved to inhibit the PD-L1/PD-1 interaction. Atezolizumab,7 Avelumab,8 and Durvalumab9 bind PD-L1 whereas Pembrolizumab,10 Nivolumab,11 and Cemiplimab12 bind PD-1 and block its engagement by PD-Ligands. Despite the therapeutic success of antibodies that block the PD-1 pathway both alone and in combination with CTLA-4 blockade across a wide array of cancers, significant limitations still remain.13 Aside from their high cost and low tumor penetration,14 the majority of patients on therapeutic ICB mAbs fail to respond clinically and a subpopulation will experience potentially life-threatening immune-related adverse events.15–18 As PD-L1 testing has improved in reliability, it has become a mainstay of predicting likelihood of response to PD-1 pathway antibodies; however, all existing tests are performed on a single biopsy specimen which may or may not represent levels of PD-L1 expression across systemic disease.19 Therefore, PD-L1 ligands with antibody-like affinity, rapid tumor penetration, rapid pharmacokinetics, chemical accessibility, and low costs of production may provide a novel molecular scaffold for development of improved PD-L1-based therapeutics and affinity agents for molecular imaging and in vitro diagnostics.20
Directed evolution has previously been used to design PD-L1 binding peptides and proteins that possess a subset of these properties.21 For example, yeast display has been used to engineer a PD-L1 ligand based on the ectodomain of PD-1.14 The resulting 14 kDa inhibitor showed improved tumor penetration when compared to anti-PD-L1 antibodies and was successfully used to treat small and large tumor models as a monotherapy or in conjunction with anti-CTLA4 antibodies. In contrast, administration of an anti-PD-L1 mAb was only effective in small tumors, presumably because of attenuated penetration into large tumors due to increased molecular weight.22 In a subsequent study, a radiolabeled version of this compound was used successfully as a PET radiotracer to visualize PD-L1 expression in xenograft models.14
Small peptides that interfere with the PD-L1/PD-1 interaction are predicted to have more favorable pharmacokinetic profiles and lower immunogenicity relative to biologics.5 Peptides that disrupt the PD-L1/PD-1 interaction have been selected through phage display with dissociation constants (KD) ranging from 117 to 544 nM.5 In another study, WL12, a macrocyclic peptide developed by Bristol-Myers Squibb, showed significant binding to PD-L1 and disrupted the PD-L1/PD-1 interaction at low nanomolar concentrations.23 Although this compound showed promising results as a PET imaging agent,24, 25 its cyclic structure and multiple unnatural residues make it challenging to synthesize chemically and impossible to express in living systems.
We have used mRNA display26–29 to design novel peptides that selectively bind to human PD-L1 (hPD-L1). We designated the most promising candidate SPAM (Signal Peptide-based Affinity Matured ligand), an 18-residue linear peptide that is non-homologous to known PD-L1 binding peptides and mAbs. SPAM is highly selective for (hPD-L1) and shows essentially no binding to either mouse PD-L1 (mPD-L1; 77% homology30) or human PD-L2 (hPD-L2; more than 30% homology31, 32). The SPAM peptide binds both non-glycosylated and glycosylated forms of hPD-L1 (hPD-L1*) with a KD of 119 nM and 67 nM, respectively. Finally, the SPAM peptide binds hPD-L1 expressed on the surface of CHO cells and competes with the therapeutic antibodies Atezolizumab and Avelumab, suggesting that the SPAM binding site overlaps the PD-L1/PD-1 binding interface.
Methods
Cell Lines
Stably transfected CHO-hPD-L1 and CHO-mPD-L1 cells were developed in our laboratory from CHO-S parental cells (Thermo Fisher) and maintained in DMEM supplemented with 10% (v/v) fetal bovine serum (Sigma) and 1% (v/v) penicillin-streptomycin (Corning, Inc.). Cells were maintained at 37 °C in a humidified incubator supplemented with 5% (v/v) CO2. Expi293F cells were maintained in suspension culture with Expi293 Expression Medium (Thermo Fisher), 125 mL flasks rotated at 120 rpm in 37 °C in a humidified incubator supplemented with 7% (v/v) CO2.
Expression and Purification of Recombinant hPD-L1, mPD-L1 and mPD-1 in E. coli
gBlocks for hPD-L1, mPD-L1 and mPD-1 extracellular domains were ordered from Integrated DNA Technologies. Expression constructs for the extracellular domain of hPD-L1 were designed from A18 to T239 with a methionine added to the N-terminus and an Avi tag (GLNDIFEAQKIEWH)33 and a hexahistidine tag34 added to the C-terminus (Figure 1A). Expression constructs for the extracellular domain of mPD-1 were designed from L25 to S157 with a methionine and an alanine added to the N-terminus, and included a C83S mutation30 to prevent disulfide formation and an HA tag (YPYDVPDYA)35 and a His tag on the C-terminus (Supplementary Figure 1).
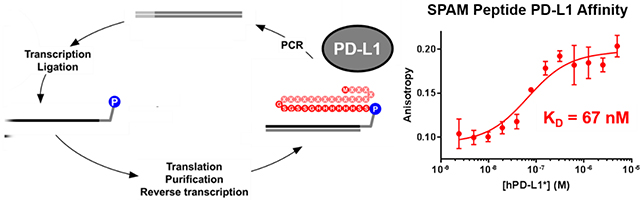
Figure 1. In vitro selection of hPD-L1 binding peptides by mRNA display.
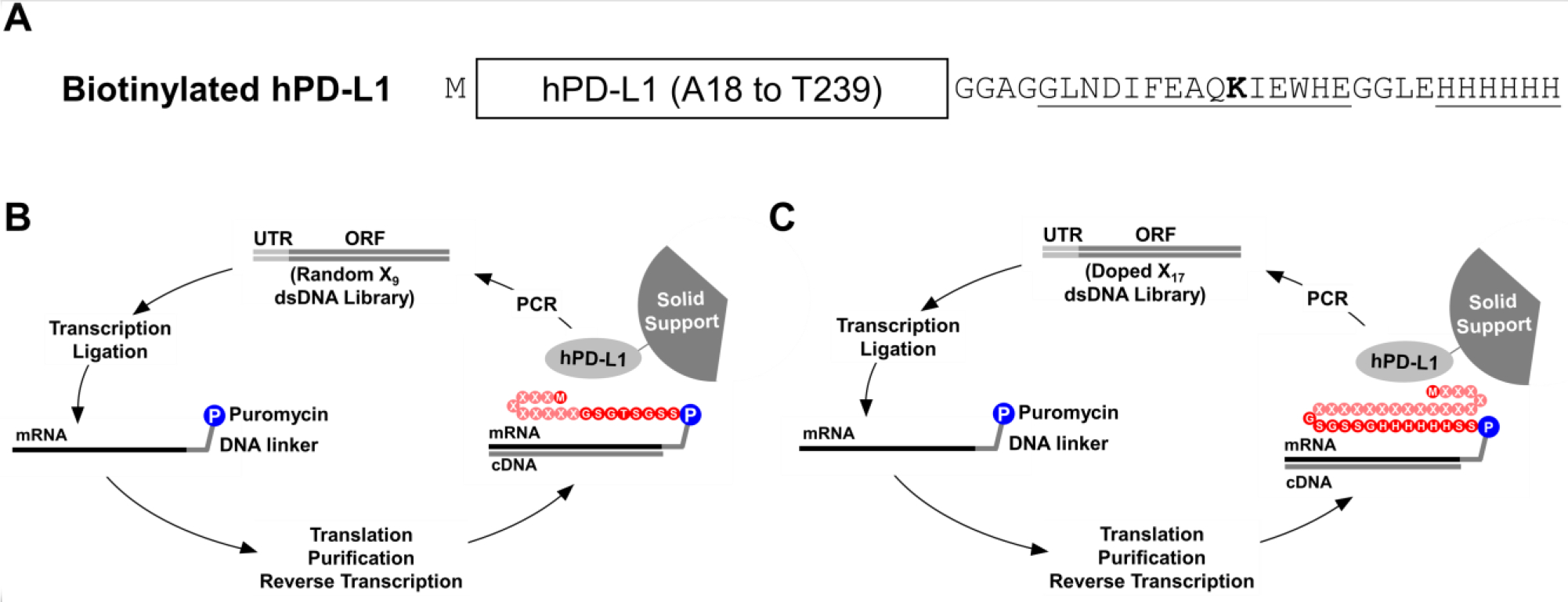
A) Biotinylated hPD-L1 target was expressed in E. coli and consisted of the extracellular domain of hPD-L1 (from A18 to T239), an N-terminal methionine, and a C-terminal hexahistidine tag and an Avi Tag, which was site-specifically biotinylated on the ε-amino group of lysine (bold) using the BirA biotin ligase. B) The initial selection was performed using a double stranded DNA library (Random X9 dsDNA Library) where the DNA was consisted of a 5’ untranslated region (UTR), a T7 promoter, a ΔTMV translation enhancer, and an open reading frame (ORF). Randomized positions are denoted with an “X” and could be any of the 20 natural amino acids. The DNA library was transcribed into RNA and ligated to a puromycin-bearing DNA linker. The template was subsequently translated, purified, and reverse transcribed to generate the mRNA-peptide fusion library, which was incubated with immobilized hPD-L1. The enriched library was amplified by PCR to generate the DNA library for the next cycle. C) A secondary selection was performed with a doped library (Doped X17 dsDNA Library) where residues denoted by an X were partially doped with the residues in hPD-L1 signal peptide.
gBlocks were PCR amplified using Taq DNA polymerase and were cloned into pET24a using NdeI and XhoI restriction enzymes, followed by Sanger sequencing. Plasmids were then transformed into BL21 (DE3) competent E. coli strains with or without pBirAcm (a pACYC184-based plasmid expressing BirA for in vivo biotinylation; Avidity LLC). Protein expression was performed in the presence of 30 mg/L kanamycin without pBirAcm, while 20 mg/L chloramphenicol was added in the presence of pBirAcm. The proteins were expressed overnight at 37 °C via auto-induction36 using ZYM-5052 and the cells were harvested using centrifugation.
Proteins were purified via denaturing affinity chromatography using an FPLC (Bio-Rad). Inclusion bodies were extracted from cell pellets using 100 mL Buffer A (100 mM NaH2PO4, 10 mM Tris-HCl, and 6 M guanidinium hydrochloride, pH 8.0) and loaded onto a Ni-NTA column. The column was washed with 100 mL Buffer B (100 mM NaH2PO4, 10 mM Tris-HCl, and 8 M Urea, pH of 8.0) and 100 mL Buffer C (100 mM NaH2PO4, 10 mM Tris-HCl, and 8 M Urea, pH of 6.3) and eluted using a linear gradient from Buffer C to Buffer D (100 mM NaH2PO4, 10 mM Tris-HCl, 8 M Urea, and 100 mM imidazole, pH of 4.5).
Fractions were analyzed using SDS-PAGE and those containing protein were combined, concentrated, and refolded in 1 M arginine, 100 mM Tris (pH=8.0), 2 mM EDTA, 0.5 mM reduced glutathione, and 0.05 mM oxidized glutathione using dilution at 4 °C overnight30 The refolded proteins were concentrated and buffer exchanged using Amicon Ultracel 10k centrifugal filters (Millipore Sigma) and then further purified using a Q Sepharose HP anion exchange column (GE Healthcare). Protein purity was analyzed using native-PAGE and SDS-PAGE and those fractions containing pure protein were combined, concentrated, and flash frozen in liquid nitrogen and stored at −80 °C.
Expression, Purification and Biotinylation of Recombinant Glycosylated Human PD-L1 (hPD-L1*) and PD-L2 (hPD-L2*) Ectodomain in Expi293F cells
pCDNA3.4 vectors encoding the hPD-L1* or hPD-L2* ectodomains were ordered from GeneArt (Thermo Fisher Scientific). The hPD-L1* vector included amino acids A18-T239, an Avi Tag and a hexahistidine tag. The hPD-L2* vector included amino acids L20-T220, an Avi Tag and a hexahistidine tag. Proteins were expressed using the Expi293F expression system per manufacturer’s instructions (Thermo Fisher Scientific) and purified at 4 °C with Ni-NTA agarose (G-Biosciences). Briefly, 293F conditioned media was 0.2 μm filtered and dialyzed 3 × 24 hours against dialysis buffer (25 mM Hepes-KOH pH 8.0, 200 mM NaCl, 5% (v/v) glycerol, 0.01% (w/v) sodium azide). Dialyzed media was mixed 1:1 with equilibration buffer (50 mM phosphate buffer pH 8.0, 300 mM NaCl, 10 mM imidazole, 0.05% (v/v) Tween-20) and 1 mL Ni-NTA resin and 20 μL 0.5 M NiSO4 were added and incubated overnight with rotation. The mixture was loaded onto a fritted column and the resin was washed once with 25 mL equilibration buffer and twice with 25 mL wash buffer (50 mM phosphate buffer pH 8.0, 300 mM NaCl and 20 mM imidazole). Protein was then eluted with 15 mL of elution buffer (50 mM phosphate buffer pH 8.0, 300 mM NaCl, 250 mM imidazole). The eluate was buffer exchanged against dialysis buffer using Amicon Ultracel 10k centrifugal filters (Millipore Sigma). Protein concentration was calculated using the Pierce BCA protein assay (Thermo Fisher Scientific) per manufacturer’s instructions. hPD-L1* yield was 2.5 mg. hPD-L2* yield was 6.3 mg protein. In vitro biotinylation of 2 mg of purified protein was performed with recombinant BirA enzyme (Avidity LLC) per manufacturer’s instructions. Biotinylation was confirmed by incubation of protein with an excess of neutravidin agarose (Thermo Fisher Scientific). The flow-through was compared with neutravidin captured protein by SDS-PAGE. Post-biotinylation, the hPD-L1* yield was 2.4 mg. Post-biotinylation the hPD-L2* yield was 0.9 mg.
Surface Plasmon Resonance (SPR) Assay
SPR analysis was performed on a Biacore T-100 at the USC Nanobiophysics core. E. coli expressed biotinylated hPD-L1 was immobilized onto a sensor chip SA (GE Healthcare Life Sciences) and different concentrations (from 1.5 × 10−7 to 2.9 × 10−5 M) of mPD-1 was flowed over the chip at a rate of 50 μL/min for 60 seconds in a buffer consisting of 50 mM HEPES-KOH pH 7.5, 150 mM NaCl, 0.05% (v/v) Tween-20 and 1 mg/mL BSA at 25 °C. The response values were fit to a one-site saturation binding model in GraphPad Prism to obtain an equilibrium binding constant.
mRNA Display Library Preparation
X9 Library:
A random DNA library encoding nine randomized positions was constructed from oligos (5’ G GGA CAA TTA CTA TTT ACA ATA ACC ATG NNK NNS NNS NNS NNS NNS NNS NNS NNK GGT AGT GGT ACG TCT GGC TCC AGC 3’) where N= A, C, G or T, S= C or G and K= G or T) and was PCR amplified together with primers 47T7Ext3 (5’ GGA TTC TAA TAC GAC TCA CTA TAG GGA CAA TTA CTA TTT ACA ATA ACC 3’) and 3Extlib3 (5’ GCT GGA GCC AGA CGT ACC ACT ACC 3’).
X17 Library:
To perform the maturation selection, a doped DNA library (X17 library) based on the wild type hPD-L1 signal peptide sequence was constructed such that the probability at each nucleotide was 70% of the wild-type nucleotide and 10% each of the other three nucleotides (corrected for the different coupling rates of the phosphoramidite monomers). X17 library template oligo (5’ G GGA CAA TTA CTA TTT ACA ATT ACA ATG 233 141 442 321 344 442 141 444 143 123 412 433 212 243 242 112 324 GGA TCC GGT TCA AGC GGT CAC 3’ where 1: 75.0% A; 7.1% G; 10.7% C; 7.1% T; 2: 10.7% A; 7.1% G; 75.0% C; 7.1% T; 3: 13.6% A; 63.6% G; 13.6% C; 9.1% T; and 4: 13.6% A; 9.1% G; 13.6% C; 63.6% T) was PCR amplified with primers SP5 (5’ GGA TTC TAA TAC GAC TCA CTA TAG GGA CAA TTA CTA TTT ACA ATT ACA 3’) and SP3 (5’ TGA ACT ATG GTG ATG ATG ATG GTG ACC GCT TGA ACC G 3’).
Both DNA libraries contained a T7 promoter sequence (5’ TAA TAC GAC TCA CTA TA 3’), a transcription start codon (5’ GGG 3’), a ΔTMV translation enhancer region derived from Tobacco Mosaic Virus (5’ ACA ATT ACT ATT TAC AAT TAC A 3’), an open reading frame and a 3’ constant sequence (for X9 library: 5’ GGT AGT GGT ACG TCT GGC TCC AGC 3’ and for X17 library: 5’ GGA TCC GGT TCA AGC GGT CAC CAT CAT CAT CAC CAT AGT TCA 3’).
To generate the mRNA library, 10 μg/mL of the PCR-amplified DNA library was incubated in transcription buffer (80 mM HEPES-KOH pH 7.5, 2 mM spermidine, 40 mM DTT, 25 mM MgCl2 and an NTP mixture consisting of 4 mM of each ATP, CTP, GTP and UTP) and briefly heated before initiating the transcription reaction using T7 RNA polymerase. The transcription reaction was incubated at 37 °C overnight and the reaction quenched by adding 0.1 volume of 0.5 M EDTA, pH 8.0. The transcribed mRNA library was gel purified using denaturing urea-PAGE, the mRNA bands excised, and the mRNA extracted using electroelution (Elutrap, Schleicher & Schuell) followed by desalting and concentrating using 0.5 mL 30k Amicon centrifugal filter (Millipore Sigma). The purified mRNA was then ligated to a puromycin-DNA linker (pF30P - 5’ 8AA AAA AAA AAA AAA AAA AAA A77 7AC C6 3’) where 6=puromycin CPG, 7=spacer phosphoramidite 9 and 8=phosphate using chemical phosphorylation reagent I (Keck Biotechnology Resource Laboratory, New Haven, CT) at the 3’ end. Splint oligos (5’ TTT TTT TTT TTT GCT GGA GCC AGA 3’ for X9 library and 5’ TTT TTT TTT TTT TGA ACT ATG GTG 3’ for X17 library) were included to facilitate the ligation. mRNA, pF30P, and splint oligo were incubated in ligation buffer and T4 DNA ligase for 90 min at room temperature. The ligated library was gel purified using denaturing urea-PAGE and electroeluted as previously described to provide the ligated mRNA library template.
mRNA Display Translation and Selection
Ligated mRNA library was translated using rabbit reticulocyte lysate (Green Hectares) in the presence of 100 mM KOAc, 0.5 mM MgOAc and 1X translation buffer consisting of 20 mM HEPES-KOH pH 7.6, 8 mM creatine phosphate, 2 mM DTT and 25 μM of each amino acid. The translation mixture was incubated at 30 °C for 1 hour followed by adding 0.5 M KCl and 50 mM MgCl2 to facilitate mRNA-peptide fusion formation. The mRNA-peptide fusion library was purified using oligo dT-agarose beads in isolation buffer (100 mM Tris-HCl, pH 8.0, 1 M NaCl and 0.2% (v/v) Triton X-100) for 1 hour at 4 °C. The beads were washed using isolation buffer and mRNA-peptide fusions were eluted in water at 65 °C. For X17 library a secondary purification was performed using Dynabeads™ His-Tag Isolation and Pulldown beads (Thermo Fisher Scientific) to selectively isolate the mRNA-peptide fusion from unfused ligated mRNA. Purified mRNA-peptide fusion library was desalted using centrisep columns (Princeton Separations) and reverse transcribed using Superscript II reverse transcriptase. The reverse transcribed library was then exposed to hPD-L1 immobilized on streptavidin or neutravidin agarose beads (Thermo Fisher scientific) or streptavidin coated magnetic beads (Dynabeads™ MyOne™ Streptavidin T1; Thermo Fisher Scientific) in blocking buffer (20 mM HEPES-KOH pH 7.5, 150 mM NaCl, 0.2% (v/v) Tween-20, 1 mg/mL BSA, 50 μg/mL tRNA and 0.02 mM biotin). For the first round, 300 pmol hPD-L1 target was used while 75 pmol was used for the subsequent rounds. Selections were performed at 4 °C except for the last two rounds of X17 library selection where the selection was performed at room temperature. After washing the beads to remove non-functional sequences, the remaining DNA was PCR amplified with library primers to generate a dsDNA library for the next round of selection. The final enriched DNA libraries were sequenced as described below.
Radiolabeled Binding Assays
[35S]-labeled peptides were translated as described above by including 2 μCi labeled methionine (Perkin Elmer) per 10 pmol of template in the translation reaction. Each peptide contained a GSGTSGSS (for X9-library derived peptides) or a GSGSSG (for X17-library derived peptides) C-terminal sequence. Binding assays using [35S]-labeled peptide-RNA fusions were similarly performed on hPD-L1 immobilized beads using 75 pmol hPD-L1 immobilized on streptavidin or neutravidin agarose beads (Thermo Fisher scientific) in blocking buffer (20 mM HEPES-KOH pH 7.5, 150 mM NaCl, 0.2% (v/v) Tween-20, 1 mg/mL BSA, 50 μg/mL tRNA and 0.02 mM biotin) at 4 °C for 1 h. The supernatant was removed and was counted. The beads were subsequently washed three times with 1 mL of blocking buffer and the washes were also counted. Finally, the beads were resuspended in 1 mL of blocking buffer and were counted separately. The ratio of the counts remaining on the beads to the total counts added to the binding reaction (the sum of the counts for supernatant, washes, and beads) was calculated to determine the percent bound.
For radioactive competition assays, hPD-L1 immobilized streptavidin agarose beads were preincubated with the desired competitor for one hour at 4 °C followed by addition of the radiolabeled peptide for 1 h. The ratio of the counts remaining on the beads to the total counts added to the binding reaction was calculated to determine the percent bound.
Serum Stability of SPAM Peptide
ROX-SPAM peptide (100 μM) was incubated in DMEM media (Thermo Fisher Scientific) supplemented with 25% (v/v) single donor human plasma (Innovative Research) at 37 °C in triplicate. At times 0, 15, 30, 120 and 300 min, 40 μL was removed and protein precipitated by the addition of 80 μL ethanol. Samples were centrifuged for 10 min at 16,000 × g and the supernatant removed for HPLC analysis. A total of 100 μL volume was injected in a HPLC gradient of 10–95% Buffer B over 15 minutes (Buffer A: dH2O with 0.1% (v/v) TFA, Buffer B: CH3CN + 0.1% (v/v) TFA). Detection was performed at 570 nm.
In a complementary test, SPAM was in vitro translated and [35S]-labeled as described above. After purification as above, the [35S]-labeled peptide-RNA fusions were incubated with DMEM media supplemented with 25% (v/v) single donor human plasma at 37 °C in triplicate for 0, 60, 120, 240 and 480 min. The binding of serum incubated peptides was then evaluated on hPD-L1 immobilized beads using 75 pmol hPD-L1 immobilized on streptavidin agarose beads (Thermo Fisher scientific) in DMEM supplemented with 25% serum buffer and 0.02 mM biotin at 4 °C for 1 h. The supernatant was removed and was counted. The beads were subsequently washed three times with 1 mL of blocking buffer and the washes were also counted. Finally, the beads were resuspended in 1 mL of blocking buffer and were counted separately. The ratio of the counts remaining on the beads to the total counts added to the binding reaction (the sum of the counts for supernatant, washes, and beads) was calculated to determine the percent bound.
Next Generation DNA Sequencing
The PCR-amplified enriched DNA libraries were uniquely barcoded and combined into a single sample and were sent for next generation Illumina DNA sequencing using a HiSeq 2500 Illumina sequencing platform at the USC Genome Core. The resulting FASTQ format file was analyzed using homemade python and bio-python scripts to translate the DNA sequences and count the frequency of the peptide sequences in each library. The number of times each sequence appeared was divided by the total number of sequences in the pool to obtain the appearance frequency.
Peptide Synthesis
All peptides were synthesized on a Prelude automated synthesis platform (Protein Technology, Inc.). Resin and amino acids were purchased from Advanced ChemTech. Rink amide AM resin (1 g, 0.47 mmol/g) was swollen in DMF for 60 min and washed twice with 6 mL DMF. The resin was deprotected with 20% (v/v) piperidine in DMF for 6 sec, 10 min and 10 min. The initial C-terminal amino acid coupling was performed with 2 equivalents (0.94 mmol) of Fmoc-propargylglycine (Pra; 313 mg), 3 equivalents of N,N-diisopropylethylamine (DIEA, 1.41 mmol, 242 μL) and 3 equivalents of N,N,N′,N′-Tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate (HBTU, 1.41 mmol, 530 mg) in 6 mL DMF for 1 hour. The resin was washed 5x with DMF and capped 2 × 30 min with 200 μL of acetic anhydride and 480 μL DIEA in DMF.
For the synthesis of peptides with a C-terminal propargylglycine residue, 100 mg Fmoc-Pra-rink amide resin was loaded onto an automated synthesizer. Synthesis was performed with 3 equivalents each of Fmoc amino acid, DIEA and HBTU in N-methyl-2-pyrrolidone (NMP) using 2 × 10 min incubations. Fmoc was deprotected with 20% (v/v) piperidine in NMP for 2.5 min. After synthesis, resin was washed 5x with NMP, 5x with DCM, and dried under vacuum for 30 min. Peptides were cleaved from the resin by addition of 3 mL of 95% (v/v) trifluoroacetic acid, 2.5 % (v/v) triisopropylsilane, and 2.5% (v/v) water for 2 hours. Peptides were precipitated in cold diethyl ether, centrifuged, washed once and dried under vacuum. The crude product was purified by reverse phase HPLC (Luna® 5 μm C18(2), LC Column 250 × 21.2 mm; Phenomenex) using gradient elution (5–95% Buffer B over 30 minutes; Buffer A: dH2O with 0.1% (v/v) TFA, Buffer B: CH3CN + 0.1% (v/v) TFA). After lyophilization, peptides were obtained as a white solid. hPD-L1-SP (MRIFAVFIFMTYWHLLNAK-Pra) yield was 19 mg (16.1%), ESI+ [M+3H]3+ expected 832.44, observed 832.89. SPAM (MPIFLDHILNKFWILHYASG-Pra) yield was 24.2 mg (20.5%), ESI+ [M+3H]3+ expected 837.10, observed 837.55. SPAM-scramble (YKLNLIHPHIFLMDWIFASG-Pra) yield was 6.6 mg (5.6%), ESI+ [M+3H]3+ expected 837.10, observed 837.55.
SPAM peptide was labeled with carboxy-X-rhodamine (ROX) by copper catalyzed click chemistry. Peptide was dissolved in DMF and added to 500 μL of click solution (16 mg/mL CuSO4·2H2O and 30 mg/mL L-ascorbic acid), 2 μL of tris-(benzyltriazolylmethyl)amine (TBTA, 100 mg/mL) and 1.5 equivalents of ROX-azide. The reaction proceeded for 1 hour at 25 °C with end-over-end rotation. The crude product was purified by reverse phase HPLC as described above. ROX-SPAM yield was 2.8 mg (64%), ESI+ [M+4H]4+ expected 782.4, observed 782.52.
ELISA/Immunoprecipitation (IP) Assays
For ELISA/IP assays, 20 μL of MyOne™ Streptavidin T1 magnetic beads Dynabeads™ (Thermo Fisher Scientific) were loaded with 100 nM biotinylated hPD-L1 in assay buffer (20 mM HEPES-KOH pH 7.5, 150 mM NaCl, 0.2% (v/v) Tween-20, 1 mg/mL BSA and 50 μg/mL tRNA) at 4 °C. The beads were subsequently washed and incubated with either DMSO or different concentrations of SPAM peptide (0.1, 1 or 10 μM) in the assay buffer for 1 h at 4 °C followed by adding HA-tagged mPD-1 (100 nM) for 1 h at 4 °C. The samples were then washed and incubated with a 1:5,000 dilution of HRP conjugated anti-HA antibody (Pierce HA Antibody, Rockford. Il, USA) for 1 h at 4 °C. The beads were washed again and developed with TMB Substrate Kit (Thermo Scientific) for 30 minutes and the reactions stopped by adding 100 μL of 2 M sulfuric acid. The absorbance at 450 nm was measured on a Biotek Synergy H1 Hybrid Multi-Mode Microplate Reader.
Fluorescence Polarization Assays
A stock solution of hPD-L1 or hPD-L1* with a 5 μM starting concentration was prepared and serially diluted to concentrations ranging from 5 μM to 2.4 nM. ROX-SPAM peptide (100 nM) was incubated with dilutions of hPD-L1 and hPD-L1* in a black 96 well polystyrene plate in the dark at room temperature with shaking. The fluorescence polarization of each sample was measured on a Synergy H4 Hybrid Multi-Mode Microplate Reader. Each measurement was carried out in triplicate and the mean value was calculated. The values were fit to a one-site saturation binding model in GraphPad Prism to obtain the corresponding equilibrium dissociation constants.
Anti-PD-L1 Antibodies
Atezolizumab and Avelumab were expressed and purified based on their reported sequences by Atum Inc. The human anti-PD-L1 antibodies 16373 and 16377 were generated by the Oncology Research for Biologics and Immunotherapy Translation (ORBIT) moonshot platform at MD Anderson Cancer Center and were also produced by Atum Inc.
Fluorescein Labeling of Antibodies
Antibodies were incubated with 8 molar equivalents of 5/6-carboxyfluorescein succinimidyl ester (Life Technologies) in the presence of 5 % (v/v) DMSO overnight at 4 °C. Dye-labeled antibodies were then purified with 7k MWCO Zeba Spin Desalting Columns (Thermo Fisher Scientific) per manufacturer’s instructions. The degree of labeling was calculated as described previously.37
Antibody Titration by Flow Cytometry
Cells were trypsinized and 200,000 cells/reaction resuspended in serum-free DMEM with a range of concentrations of Fluorescein-labeled antibodies. These samples were incubated at 4 °C for 60 min and washed three times with cold serum free DMEM. The pelleted cells were analyzed by flow cytometry with a BD FACSCalibur system using the FL1 channel. FlowJo v10.1 was used to gate to the major population and the median FL1 fluorescence reported. GraphPad Prism was used to calculate KD.
Antibody Competition by Flow Cytometry
Cells were trypsinized and 200,000 cells/reaction resuspended in serum-free DMEM with 0–20 μM SPAM, SPAM-scramble, WL-12, or BMS-202 and incubated at 4 °C for 30 min. The cells were washed once with cold serum free DMEM and incubated with 1 nM FAM-Atezolizumab, 10 nM FAM-Avelumab, 50 nM FAM-16373, or 50 nM FAM-16377 in serum free DMEM at 4 °C for 30 min. Cells were then washed three times with cold serum free DMEM. Cells were subsequently processed by flow cytometry with a BD FACSCalibur system using the FL1 channel. FlowJo v10.1 was used to gate to the major population and median FL1 fluorescence reported.
De-glycosylation and Western Blotting of hPD-L1* and CHO-hPD-L1 Proteins
CHO-hPD-L1 cells were lysed with RIPA buffer (25 mM Tris, pH 7.5, 150 mM NaCl, 0.1% (w/v) SDS, 1% (v/v) NP-40, 1% (w/v) sodium deoxycholate) and centrifuged at 16,000 × g for 10 min to pellet insoluble material. A BCA assay was used to confirm protein concentration. To remove N-linked oligosaccharides, 20 μg of the lysate was treated with 4 μL of Rapid PNGase F buffer and 1 μL of Rapid PNGase F enzyme (New England Biolabs) in a total volume of 20 μL for 2 hours at 37 °C. Recombinant hPD-L1* protein was treated in a similar manner. Control reactions without the addition of Rapid PNGase F enzyme were also performed. After incubation, 10 μL of 4x Coomassie sample buffer was added and the samples boiled at 95 °C for 5 min. Protein samples then run on a 4–15% Mini-PROTEAN TGX gel (Bio-Rad) at 120V for 1 hour and transferred to a nitrocellulose membrane at 100V for 1 hour. The membrane was then incubated in blocking buffer (TBS supplemented with 0.1% (v/v) Tween-20 [TBST] and 3% (w/v) BSA) for 1 hour at room temperature. The membrane was subsequently incubated with a 1:20,000 dilution of rabbit anti-PD-L1 antibody (E1L3N, Cell Signaling Technology) for 1 hour at room temperature, washed 3 × 5 min with TBST, then incubated in a 1:200,000 dilution of anti-rabbit-HRP antibody (AC2114, Azure Biosystems) for 1 hour at room temperature. The membrane was washed 3 × 5 min with TBST, developed with SuperSignal WestPico substrate (Thermo Fisher Scientific), and imaged with an Azure C600 gel imager (Azure Biosystems).
Results and Discussion
The goal of this project was to identify a small peptide with high affinity and high specificity for hPD-L1. In order to generate purified target for immobilization and selection, the extracellular domain of hPD-L1 (from A18 to T239) was expressed in E. coli. The protein contained an Avi tag at the C-terminus which was enzymatically biotinylated with the BirA biotin ligase.33 The biotinylated hPD-L1 was then purified using affinity chromatography and ion exchange chromatography and immobilized on neutravidin or streptavidin beads for the purpose of selection (Figure 1A).
In order to confirm that the expressed hPD-L1 was correctly folded, binding to mPD-1 was measured by SPR. The dissociation constant (KD) between recombinant hPD-L1 and PD-1 was determined to be 3.2 ± 0.3 μM using a one-site saturation binding model (Supplementary Figure 2). This value is consistent with previously reported KD values (KD ≈ 4 to 8 μM),30, 38 suggesting that the recombinant E. coli-expressed hPD-L1 is correctly folded and functional.
Primary Selection with an X9 Library
A primary selection was performed using a random X9 DNA library consisting of an open reading frame coding for nine random amino acids. The amplified DNA library was transcribed into corresponding mRNA library and ligated to a puromycin-bearing oligonucleotide. The resulting library template was translated, purified, reverse transcribed, and incubated with immobilized hPD-L1. After extensive washing, bound sequences were amplified by PCR to generate the full-length DNA library for the next selection cycle (Figure 1B). After five rounds of selection, the binding of the library to immobilized hPD-L1 increased dramatically over background (Supplementary Figure 3A), suggesting enrichment of target-binding peptide sequences. The round 6 pool was analyzed by next generation sequencing and the clones with highest frequency were identified. One of these, MRIFVIFIWA, designated as clone 1 was highly represented in the round 6 pool (Table 1). Using the Basic Local Alignment Search Tool (BLAST), we identified regions of local similarity between clone 1 and the first 10 amino acids of the hPD-L1 signal peptide (60% identity, 90% homology). This unexpected similarity prompted us to perform a maturation selection with a doped library based on the hPD-L1 signal peptide sequence.
Table 1. Selected peptides sequences.
The initial X9 library selection resulted in a clone with significant homology to the first nine residues of the hPD-L1 signal peptide. The PD-L1 signal peptide sequence was used to design the X17 library (doped at ~70% wild type at the DNA level) from which the SPAM peptide was selected. Residues that are identical between the hPD-L1 signal peptide and the selected peptides are shown in bold. The grand average of hydropathy (GRAVY Index) for the peptide sequences are calculated as the ratio of the sum of the hydropathy index of each amino acid in a sequence to the peptide length.39
| Peptide | Sequence | GRAVY Index |
|---|---|---|
| hPD-L1 signal peptide | MRIFAVFIFMTYWHLLNA | 1.25 |
| Clone 1: Selected clone from X9 library | MRIFVIFIWA | 2.16 |
| SPAM: Selected clone from X17 library | MPIFLDHILNKFWILHYA | 0.73 |
Maturation Selection and Identification of the SPAM Peptide
To enhance the affinity and selectivity of the peptide selected from the X9 library, we designed a doped library based on the wild type hPD-L1 signal peptide sequence. This library, termed X17, was constructed such that each nucleotide was doped at a 70% probability wild type frequency (and 10% of each of the other three nucleotides), which results in an average frequency of ~44% wild-type amino acid at each position in peptide sequence (Figure 1C). The hPD-L1 signal peptide itself shows some binding to streptavidin agarose beads loaded with 1.1 μM hPD-L1 (equivalent to 1,100 pmol of hPD-L1) (Supplementary Figure 4). We performed eight rounds of selection for hPD-L1 binding using the X17 library, with significant target-specific binding emerging after round 5 (Supplementary Figure 3B). Target-specific binding continued to increase in subsequent rounds even as additional selective pressure was added by increasing the binding temperature from 4 °C to room temperature. MPIFLDHILNKFWILHYA was identified as the most frequently occurring sequence by next-generation sequencing and designated as Signal Peptide-based Affinity Matured ligand (SPAM) peptide. The SPAM peptide was 33% identical to the hPD-L1 signal peptide and showed a consensus motif of MXIFπXXIXXXΩWXLXXA, where π represents an aliphatic amino acid, X represents any amino acid, and Ω represents an aromatic amino acid (Table 1).
The SPAM Peptide Binds to hPD-L1 with Mid-Nanomolar Affinity
To evaluate the affinity of SPAM for hPD-L1 and hPD-L1*, we measured the KD by fluorescence polarization. ROX-labeled SPAM (ROX-SPAM) was incubated with increasing concentrations of hPD-L1 or hPD-L1* and the fluorescence polarization of the ROX fluorophore measured at excitation and emission wavelengths of 540 nm and 620 nm, respectively. The resulting values were plotted against the concentration of hPD-L1 or hPD-L1* and fit to a one-site saturation binding model with GraphPad Prism. ROX-SPAM was found to bind hPD-L1 with a dissociation constant (KD) of 119 ± 33 nM (T= 21 °C) and hPD-L1* with a KD of 67 ± 21 nM (T= 22 °C) (Figure 2A and 2B). This affinity is significantly better than the micromolar KD between PD-1 and hPD-L130, 38 and is higher than a variety of previously reported peptides targeting hPD-L1.40, 41 (Supplementary Table 1). Although SPAM shows a weaker affinity relative to WL12,24, 25, 42, 43 it has the advantage of being a linear peptide with only natural residues, which makes it easier to chemically synthesize or express recombinantly inside cells.
Figure 2. Stability and Binding of SPAM to hPD-L1 and hPD-L1*.
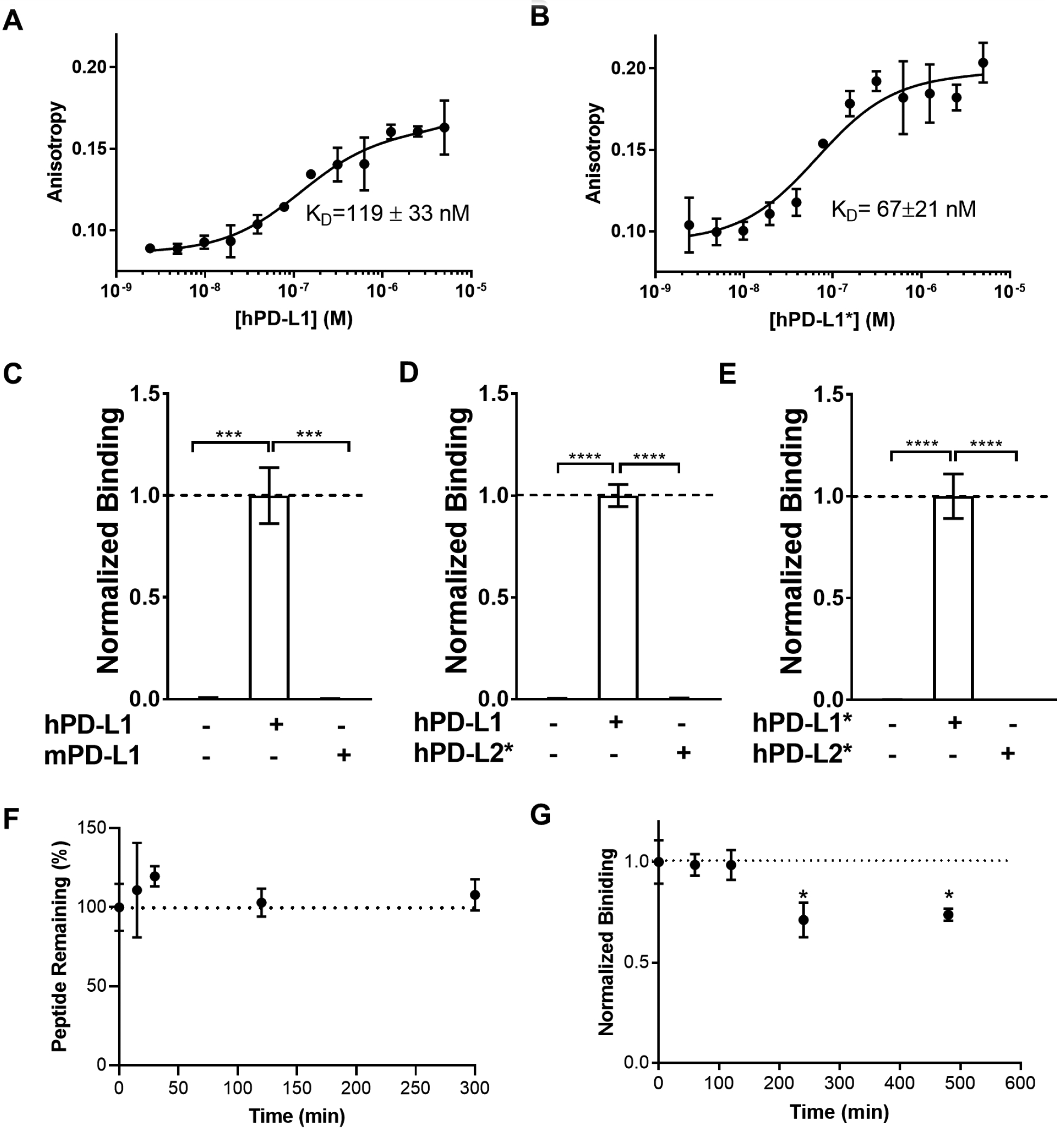
Carboxy-X-rhodamine (ROX)-labeled SPAM peptide (100 nM) was incubated with increasing concentrations (2.4 nM to 5 μM) of A) hPD-L1 and B) hPD-L1* and the anisotropy measured at 21 °C and 22 °C, respectively. Each measurement was carried out in triplicate and the mean value was reported along with the standard deviation. The values were fitted to a one-site saturation binding model to obtain the corresponding equilibrium binding constant at 21 °C and 22 °C, respectively C) [35S]-labeled SPAM binding to hPD-L1 and mPD-L1 D) [35S]-labeled SPAM binding to hPD-L1 and hPD-L2* E) [35S]-labeled SPAM binding to hPD-L1* and hPD-L2*. F) SPAM peptide is stable in human serum. ROX-SPAM peptide was incubated in 25% human serum for up to five hours and analyzed by HPLC. No change in peptide retention time was observed. G) [35S]-labeled SPAM was incubated in 25% human serum for up to eight hours and its binding to hPD-L1 was measured and normalized to the binding of [35S]-SPAM not incubated with serum (0-hour sample). Each measurement was carried out in triplicate and error bars show the standard deviation of the mean normalized binding (Students t-test; p value < 0.05 (*), < 0.01 (**), < 0.005 (***), < 0.001 (****)).
SPAM Peptide Selectively Binds to hPD-L1
SPAM was also tested for binding specificity towards mPD-L1 and hPD-L2 using radioactive binding assays. SPAM peptide mRNA was translated in vitro in the presence of [35S]-methionine to generate [35S]-labeled SPAM peptide. Radiolabeled SPAM peptide was efficiently pulled-down by hPD-L1 immobilized on streptavidin agarose but was not pulled-down by mPD-L1 (Figure 2C). [35S]-SPAM peptide was also captured by hPD-L1* (Figure 2D), although no significant binding to hPD-L2* was observed (Figure 2D and 2E). These data demonstrate that SPAM binds tightly to both glycosylated and non-glycosylated forms of hPD-L1, yet shows essentially no binding to mPD-L1 or hPD-L2 despite 77% and 34% homology to hPD-L1, respectively.30–32 SPAM therefore displays hPD-L1 specific binding, which is an important feature compared with previously reported peptides that are not hPD-L1 specific (Supplementary Table 1).
The Stability of SPAM Peptide in the Presence of Human Serum
Several previously reported PD-L1 binding peptides have used cyclization and unnatural amino acids23 or D-amino acids40 in order to increase peptide stability in serum. As SPAM is a linear peptide composed of natural amino acids, it might therefore be susceptible to degradation by proteases in human serum. We performed two complementary assays to determine the effect of serum on the SPAM/hPD-L1 interaction. We first tested SPAM peptide stability by performing an HPLC-based assay to test if SPAM is degraded by human serum. We incubated ROX-labeled SPAM with 25% human serum at 37°C, took aliquots at different time points, then analyzed the products using HPLC. We observed no degradation even after five hours of incubation nor detected any change in peptide retention time (Figure 2F), suggesting that the peptide was not proteolytically degraded in serum. We also tested to see if human serum had an effect on SPAM binding by incubating [35S]-labeled SPAM with 25% human serum at 37°C, then measuring the binding to immobilized hPD-L1. No decrease in the binding was observed after two hours of incubation, but a slight decrease in the binding was observed after four hours of incubation (Figure 2G). After eight hours of incubation, no further decrease was observed. Taken together, both assays support the conclusion that even though SPAM is a linear peptide with no unnatural amino acids, it is resistant to degradation by proteases in human serum. Previous work has shown that natural peptides can exhibit significant resistance to proteolysis44 and although SPAM was not subjected to an explicit proteolytic resistance selection, it nonetheless is resistant to proteolytic degradation by human serum. SPAM does show a small (~25%) but statistically significant loss of binding, however taken with the stability data above, this loss is unlikely to be due to proteolysis, but possibly some other modification that occurs in presence of human serum (e.g. methionine oxidation).
The C-terminal Residues of the SPAM Peptide are Critical for hPD-L1 Binding
Alanine scanning provides a framework for structure activity relationships and reveals core residues within peptides and proteins important for binding interactions.45 To determine the critical residues for the SPAM/hPD-L1 binding interaction, we translated a set of [35S]-labeled SPAM peptides where each residue was individually substituted with alanine (Figure 3A). We found that the I3A, F4A, L5A, I8A and L9A mutations had little or no significant influence on hPD-L1 binding, implying that these residues are not crucial for the SPAM affinity. P2A and Y18A variants showed decreased levels of hPD-L1 binding, although this did not reach the level of statistical significance. In contrast, the D6A, H7A, N10A, K11A, F12A, W13A, I14A, L15A, H16A, and A18 mutations completely abolished hPD-L1 binding, suggesting that these residues are critical for SPAM peptide affinity. Since the majority of these important residues are located in the C-terminal half of the peptide, these data indicate that the C-terminal portion of SPAM contributes more to hPD-L1 binding than the N-terminal portion.
Figure 3. Mutational analysis and N-terminal truncation of SPAM shows the residues critical for hPD-L1 binding.
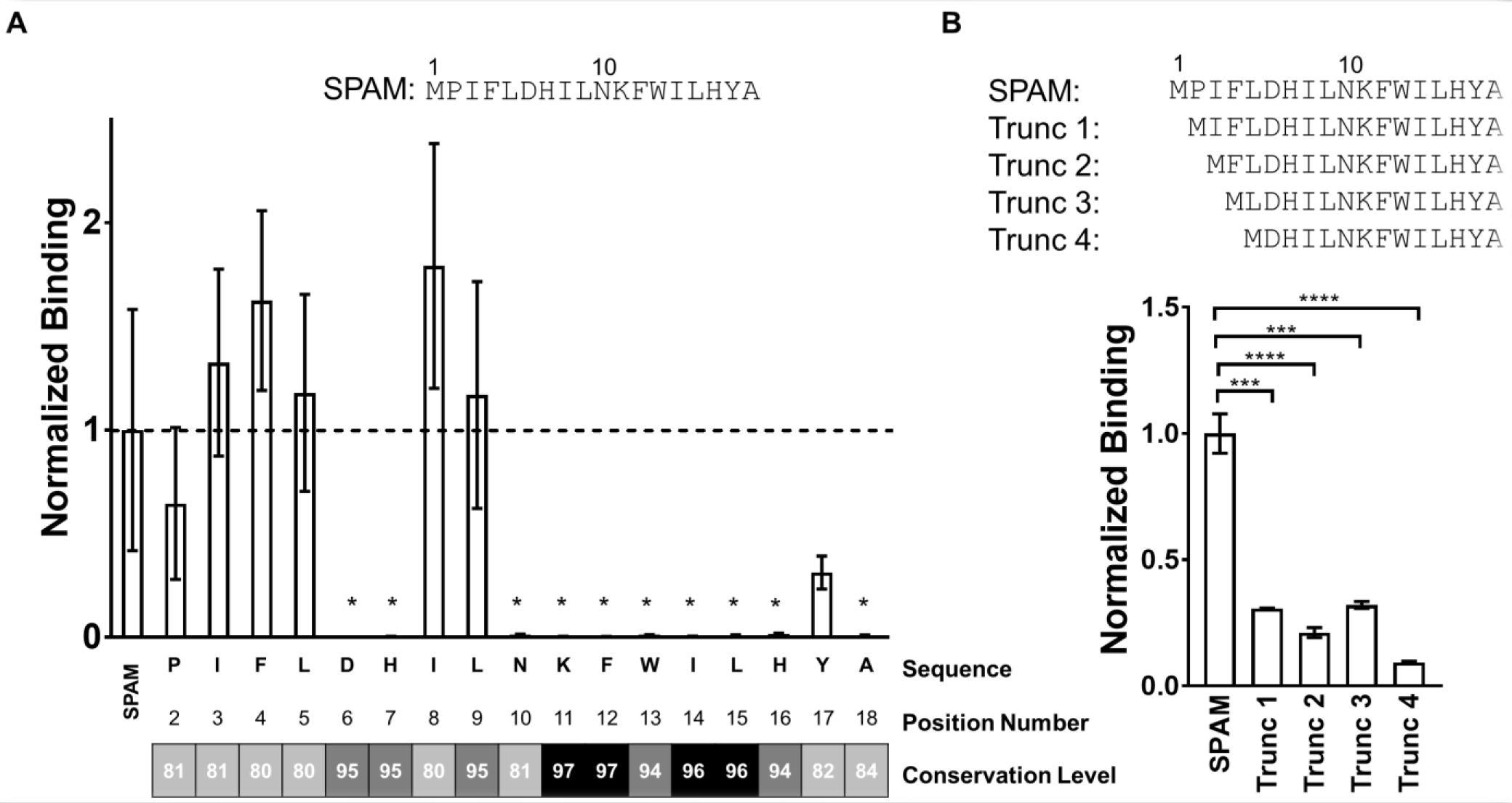
A) Mutational analysis of SPAM; [35S]-labeled SPAM variants were translated with alanine substituted at each position (2–18) and glycine substituted for alanine at position 18. hPD-L1 binding of each [35S]-SPAM alanine mutant was measured and normalized to the binding of [35S]-SPAM. The mean of the normalized binding for each SPAM variant (n=3) is shown along with the standard deviation of the mean. The conservation level for each doped residue in X17 library was obtained from next-generation sequencing data. These results were binned according to sequence family and 108 sequences from SPAM family that appeared more than 1,000 times were identified and the percent conservation relative to the SPAM peptide calculated for each position. B) [35S]-labeled N-terminally truncated versions of SPAM were translated where one (P), two (P, I), three (P, I, F) and four (P, I, F, L) residues were removed from the N-terminus of SPAM creating Trunc 1, Trunc 2, Trunc 3 and Trunc 4, respectively. hPD-L1 binding of each truncated variant was measured and normalized to the binding of [35S]-labeled SPAM. Each measurement was carried out in triplicate and error bars show the standard deviation of the mean normalized binding. (Students t-test; p value < 0.05 (*), < 0.01 (**), < 0.005 (***), < 0.001 (****)).
We analyzed the next-generation sequencing results from the round 6 pool of the X17 selection and measured the conservation of each amino acid residue relative to the SPAM peptide.46 Next generation sequencing results were first sorted into families and 108 sequences homologous to SPAM were binned and the percent identity to SPAM was determined (Supplementary Table 2). This analysis allowed residues to be sorted into three groups, highly conserved (>95%), conserved (85–95%), and modestly conserved (<85%) (Figure 3A). Most positions that are associated with abolished binding in the alanine scanning experiment showed >95% conservation in high throughput screening.
Correlation between the next-generation sequencing results and alanine scanning data is discordant for only two residues: L9 and N10. L9 is highly conserved in the final pool yet can be replaced with alanine with no significant effect on PD-L1 binding. On the other hand, N10 shows relatively low conservation in the final sequence pool, yet mutation to alanine almost completely abrogates PD-L1 binding. While these discrepancies may arise from positional differences in background mutation rate, they may also represent additional functional information. For example, mutations at position 10 are often observed in the context of multiple mutations in the N-terminal region, potentially indicating cooperativity between different parts of the SPAM peptide. Taken together, the positional scanning data and the next generation sequencing analysis data indicate that the majority of the residues in the C-terminal portion of the SPAM peptide are critical for PD-L1 binding and provide basic structure-activity information for the interaction.
These data suggest that the N-terminal residues of SPAM may therefore be dispensable, and SPAM could possibly be truncated to a shorter, yet still active peptide. To test this hypothesis and to gain a better understanding of the contribution of the N-terminal residues in the observed binding interaction, we constructed several N-terminal truncations of SPAM and compared their binding with wild type SPAM. [35S]-labeled N-terminally truncated variants of SPAM were translated where one (Trunc 1; P2 deleted), two (Trunc 2; P2, and I3 deleted), three (Trunc 3; P2, I3, and F4 deleted) and four (Trunc 4; P2, I3, F4, and L5 deleted) residues were removed from the N-terminus of SPAM. These truncated variants were tested for hPD-L1 binding and showed reduced binding (~25% or less) to hPD-L1 as compared with full-length SPAM (Figure 3B). The truncation data indicates the N-terminus does play a role in SPAM binding. Taken together with the alanine scanning data, the >75% loss of binding in these truncation variants could be due to the loss of main chain/backbone contacts between hPD-L1 and these N-terminal SPAM residues. It is also possible that the N-terminal backbone stabilizes the C-terminal portion of the SPAM peptide, facilitating its interaction with hPD-L1.
SPAM Peptide Binding is Blocked by Inhibitors of the PD-L1/PD-1 Interaction
Competition binding assays were performed to determine the functional significance of the PD-L1/SPAM peptide interaction. hPD-L1-binding ligands known to inhibit PD-1 binding (WL12, a macrocyclic hPD-L1 inhibitor peptide,23 Atezolizumab, an hPD-L1 specific monoclonal antibody,7 and mPD-1 the murine variant of PD-1) were used as competitors. All three competitor ligands significantly inhibited the SPAM/hPD-L1 interaction (Figure 4A) suggesting that SPAM peptide overlaps with at least a portion of the PD-L1 recognition site used by these ligands. As expected, control goat anti-rabbit polyclonal IgG antibody had no effect on hPD-L1 binding.
Figure 4. The SPAM: hPD-L1 interaction is disrupted by hPD-L1-binding ligands.
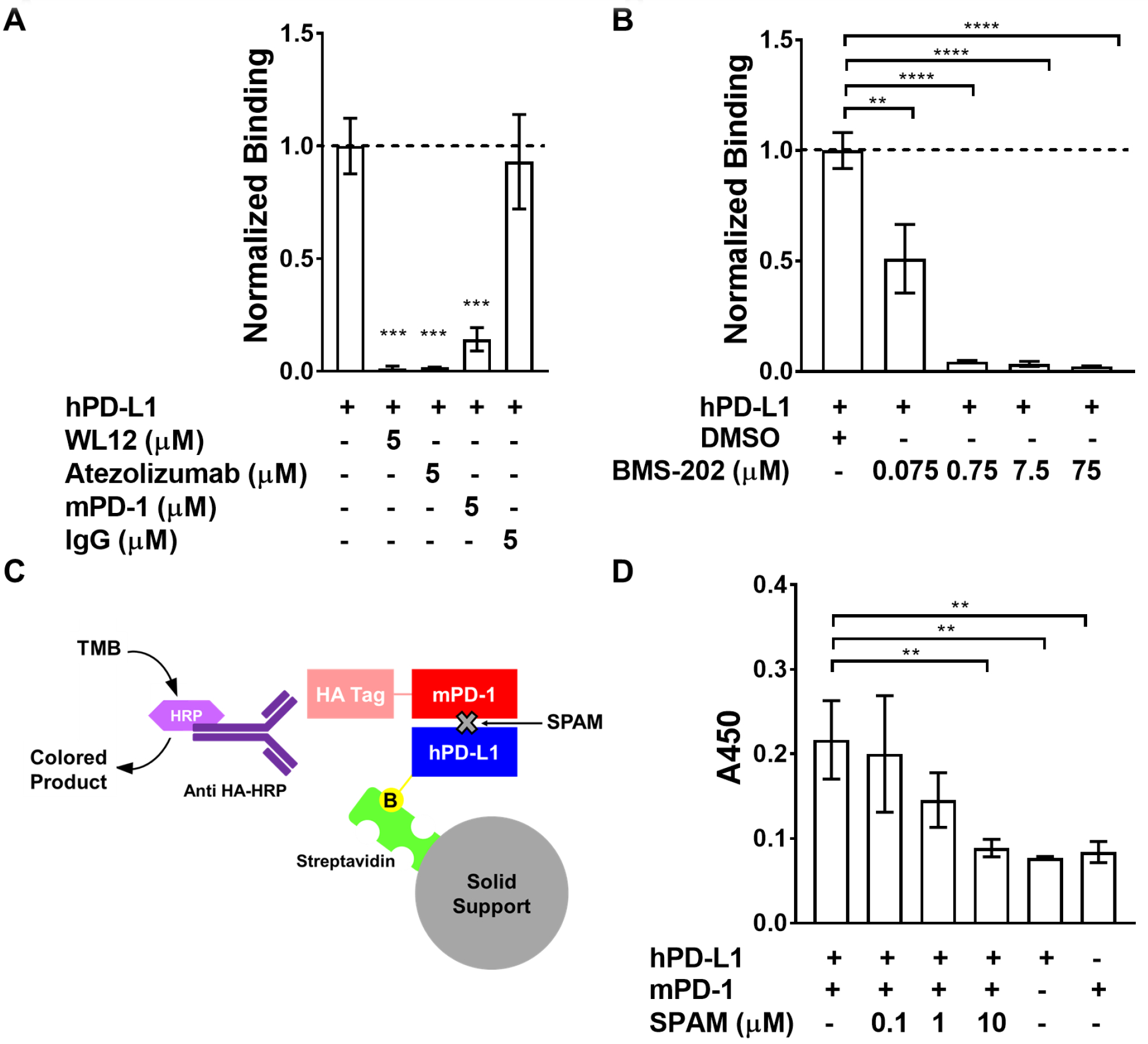
A) [35S]-labeled SPAM binding to immobilized hPD-L1 is disrupted by 5 μM WL12 (p value<0.0001), 5 μM Atezolizumab (p value<0.0001) and 5 μM mPD-1 (p value<0.0001). The presence of 5 μM IgG does not affect SPAM binding to hPD-L1 (p value > 0.05) B) [35S]-SPAM binding to hPD-L1 in the presence of varying concentrations of BMS-202. SPAM binding to hPD-L1 is almost completely inhibited by 750 nM BMS-202 (p value<0.0001). Error bars show the standard deviation of the mean for triplicate measurements. C) Schematic of the hPD-L1/mPD-1 Interaction ELISA/IP assay. D) ELISA/IP assay for hPD-L1 binding to mPD-1 in the presence and absence of different concentrations of SPAM (0.1, 1 and 10 μM). SPAM almost completely inhibits mPD-1 binding at 10 μM (p value <0.01) (Each measurement was carried out in triplicate and error bars show the standard deviation of the mean. Students t-test; p value < 0.05 (*), < 0.01 (**), < 0.005 (***), < 0.001 (****))
SPAM peptide binding was further evaluated in the presence of varying concentrations of BMS-202. BMS-202 is a small molecule that stabilizes a non-functional dimeric complex of PD-L1, blocking the PD-1 binding site and inhibiting PD-1 binding.47, 48 SPAM peptide binding is almost completely inhibited at 750 nM BMS-202 (Figure 4B), indicating that the SPAM binding site wholly or partly overlaps the binding site utilized by known PD-L1 modulating ligands.
An ELISA-based activity assay was employed to measure the potency of SPAM peptide inhibition of the hPD-L1/PD-1 interaction (Figure 4C–D). In this assay, biotinylated hPD-L1 was immobilized on streptavidin magnetic beads and incubated with various concentrations of SPAM peptide, followed by incubation with HA-tagged mPD-1 and detection with anti-HA secondary antibody. SPAM peptide almost completely disrupted PD-1 binding at 10 μM providing further evidence that SPAM can functionally inhibit the hPD-L1/mPD-1 interaction with an approximate IC50 of 1.1 μM when hPD-L1 and mPD-1 are present at a concentration of 100 nM. This observation is consistent with the fluorescence polarization data which indicates that SPAM binds to hPD-L1 with a higher affinity (KD = 60–120 nM) than the hPD-L1/mPD-1 interaction (KD ≈ 3 μM).
The competition data thus indicates that SPAM binds at the PD-1 interaction site of hPD-L1 and provides a framework for interpreting our specificity data (Figure 2). We thus compared sequence alignments and crystal structures of hPD-L1, mPD-L1 and hPD-L2 (Supplementary Figures 5 and 6) in order to determine if any structural features of the PD-1 binding site of these complexes might provide a basis for SPAM binding specificity. Despite the high level of homology between these proteins, the PD-1 interaction surface of hPD-L1 and mPD-L1 differs by a single residue (Arg113) while the PD-1 interaction surface of hPD-L1 and hPD-L2 differs by six residues. Several other residues that are not located in the PD-1 interaction surface are also different and may contribute to the specificity observed for SPAM binding (Supplementary Figure 5). We hypothesize that SPAM binding with hPD-L1 likely involves interaction with at least some of these residues, leading to the specificity of SPAM for hPD-L1.
Binding of SPAM Peptide Inhibits Binding of Anti-PD-L1 Antibodies to hPD-L1 Expressing Cells
In vitro characterization of the selected SPAM peptide revealed strong binding to the recombinant hPD-L1 ectodomain and inhibition of the PD-L1/PD-1 interaction. Although we had demonstrated that the SPAM peptide bound to hPD-L1*, we also wanted to determine if this interaction is maintained in a cellular context. To do this, we measured SPAM peptide inhibition of anti-PD-L1 antibody binding to hPD-L1 expressed on the surface of Chinese hamster ovary (CHO) cells (CHO-hPD-L1). Both Atezolizumab and Avelumab antibodies were fluorescently labeled with FAM-NHS and titrated against CHO-hPD-L1, yielding KD values of 0.2 nM and 5.1 nM, respectively (Supplementary Figure 7A). These values are in agreement with reported binding affinities.49, 50 Next, we pre-incubated CHO-hPD-L1 cells for 30 min at 4 °C with SPAM peptide, a control scrambled peptide (SPAM-scramble), or the PD-L1 inhibitors WL-1224 and BMS-20248 in serum-free media. Cells were washed and then incubated with 1 nM Fluorescein-Atezolizumab or 10 nM Fluorescein-Avelumab for 30 min at 4 °C. Cells were washed again and analyzed by flow cytometry (Figure 5A–B). At high (20 μM) concentrations of SPAM peptide, where the peptide was preincubated with cells before adding antibody, we observed a significant decrease in both Fluorescein-Atezolizumab and Fluorescein-Avelumab binding to CHO-hPD-L1. This effect was not observed from SPAM-scramble. As expected, treatment with WL-12 and BMS-202 markedly reduced antibody binding.
Figure 5. SPAM peptide reduces clinical and cross-reactive anti-PD-L1 antibody binding to CHO-hPD-L1 cells.
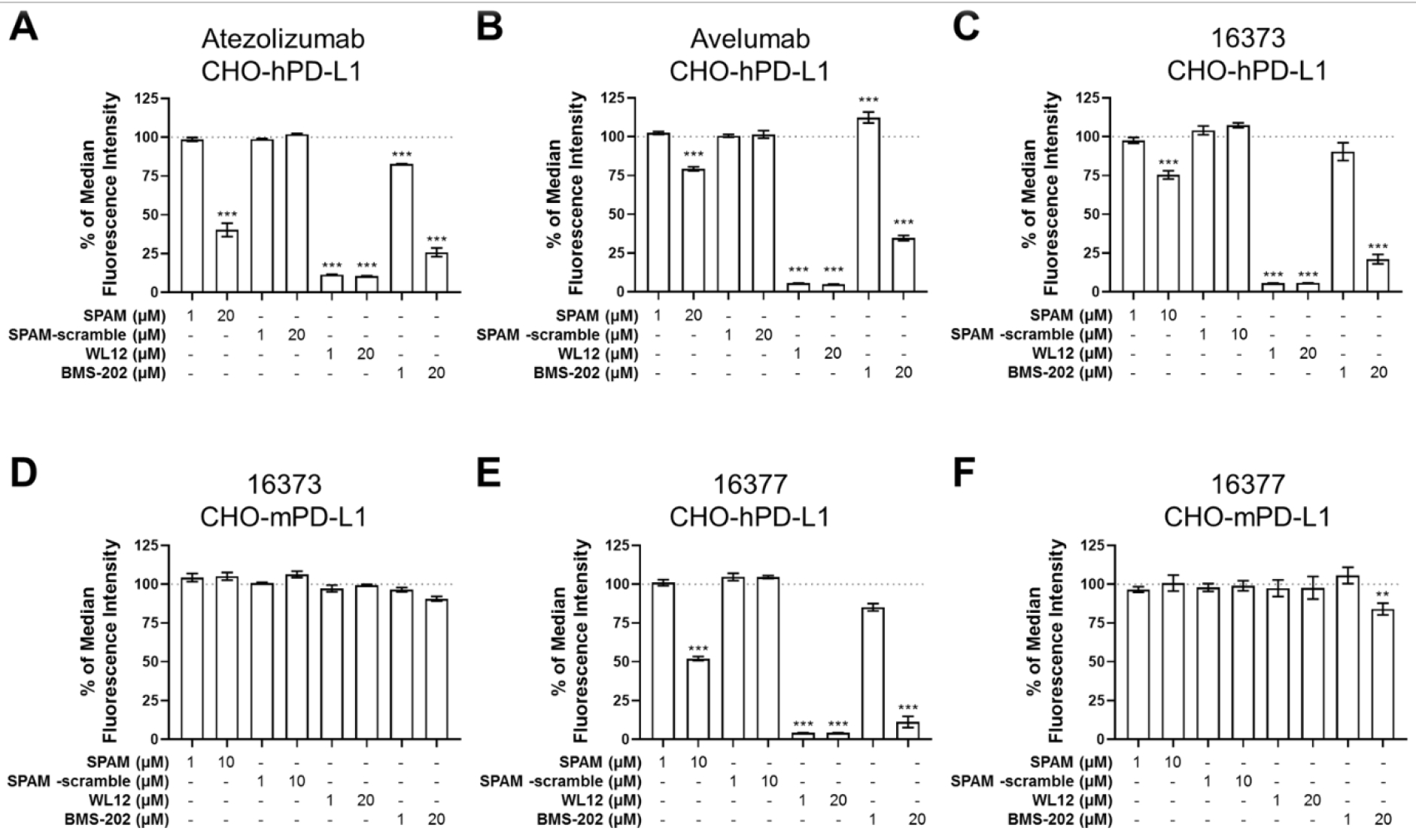
Cells were pretreated with SPAM peptide, SPAM-scramble peptide, WL-12 or BMS-202 followed by treatment with Fluorescein-labeled antibody and flow cytometry. (A) CHO-hPD-L1 cells labeled with Fluorescein-Atezolizumab. (B) CHO-hPD-L1 cells labeled with Fluorescein-Avelumab. (C) CHO-hPD-L1 cells labeled with Fluorescein-16373 antibody. (D) CHO-mPD-L1 cells labeled with Fluorescein-16373 antibody. (E) CHO-hPD-L1 cells labeled with Fluorescein-16377 antibody. (F) CHO-mPD-L1 cells labeled with Fluorescein-16373 antibody. Each measurement was carried out in triplicate and error bars show the standard deviation of the mean. One-way ANOVA with Dunnett’s multiple comparison’s test p value < 0.05 (*), < 0.01 (**), < 0.001 (***).
We performed similar experiments with CHO-mPD-L1 cells which express mPD-L1 to determine if this effect was specific to hPD-L1 expression. We have developed two cross-reactive anti-hPD-L1/mPD-L1 antibodies, 16373 and 16377. These antibodies were labeled with Fluorescein, titrated against CHO-hPD-L1 and CHO-mPD-L1 cells (Supplementary Figure 7B–C) and subjected to competition experiments as described above. Similar to the clinical antibodies, we observed blocking of Fluorescein-16373 and Fluorescein-16377 after pre-treatment with SPAM peptide, WL-12 and BMS-202 in CHO-hPD-L1 cells, however, this blocking was absent in CHO-mPD-L1 cells for all compounds tested (with the exception of 20 μM BMS-202; Figure 5C–F) confirming that hPD-L1 selectivity is maintained in cell culture.
In light of our in vitro affinity data that showed SPAM has a 67 nM affinity for hPD-L1* (Figure 2B) and the stability data that showed SPAM was not degraded by serum, the high concentrations of SPAM peptide that were necessary to effectively block antibody binding were somewhat surprising. These data suggested that the affinity of SPAM binding to hPD-L1 in a cellular context was decreased relative to our previous in vitro experiments. One possible explanation for this difference could be due to the glycosylation state of recombinant hPD-L1* protein used in vitro and hPD-L1 protein expressed on the surface of CHO-hPD-L1 cells. To compare the glycosylation state of hPD-L1* and hPD-L1 from CHO-hPD-L1 cells, we performed a western blot on hPD-L1 protein samples treated with or without PNGase F, which can remove N-linked glycans. We observed that both hPD-L1* and hPD-L1 from CHO-hPD-L1 cells show a band shift towards higher mobility, corresponding to the removal of N-linked glycans for both protein sources (Supplementary Figure 8). We do note that hPD-L1 from CHO-hPD-L1 cells shows a more drastic shift, suggesting that CHO-derived hPD-L1 is more heavily glycosylated, and could provide an explanation for the loss of affinity in SPAM binding to CHO-hPD-L1 cells; the more extensive glycosylation might interfere with the SPAM/hPD-L1 interface.
Conclusion
Dual-stage mRNA display selection for hPD-L1 ligands resulted in the identification of SPAM peptide, an 18-amino acid linear peptide with mid-nanomolar affinity and high selectivity for hPD-L1 relative to other B7 family members. The SPAM/PD-L1 interaction is significantly stronger than the PD-1/PD-L1 interaction30, 38 and is comparable or superior to previously reported linear PD-L1 binding peptides40, 41, 51–55 despite the relatively small size of the SPAM peptide. We speculate that the high affinity and selectivity of the SPAM peptide may have resulted from sequential selections with two high-diversity mRNA Display libraries (X9 and X17).
SPAM is non-homologous to known PD-L1 binding peptides and was found to competitively inhibit binding of PD-1 and ligands that interact with the PD-1 binding site. Point mutational analysis and next generation sequencing has identified numerous positions within the N-terminal region which do not significantly contribute to binding and are therefore candidates for further affinity maturation. SPAM peptide was found to inhibit the binding of anti-PD-L1 antibodies in clinical use with potencies comparable to that of the small molecule BMS-202. Although the affinity of the SPAM peptide is lower than that of the cyclic, unnatural peptide WL-12, its natural, linear architecture facilitates rapid and inexpensive synthesis or expression as a fusion protein for cell-based studies. Taken together, the results suggest that the selected linear peptide ligand selectively recognizes hPD-L1 and can potentially serve as a lead compound for further development of PD-L1 antagonists.
Supplementary Material
Acknowledgements
Funding sources for this project included: UTMDACC startup funds (SWM), a G.E. In-kind Multi-investigator Imaging (MI2) Research Award (SWM, RWR, TTT). 1-F32-EB024379-01A1 (BJE), and 1T32CA196561 (BJG). MAC and AS were partially supported by CPRIT Investigator Grant RP190218. This work acknowledges the instrumentation provided by the USC Nanobiophysics Core Facility and the USC Genome and Cytometry Core Facility as part of this work.
Footnotes
Supporting Information
The Supporting Information includes a table comparing hPD-L1 binding properties of SPAM and previously published peptides (Table S1) and a table of SPAM point mutants found in next generation sequencing data (Table S2). Figures describing expression constructs (Fig S1), the hPD-L1/mPD-1 binding interaction evaluated by Surface Plasmon Resonance (Fig S2), mRNA Display selection progress (Fig S3), hPD-L1 signal peptide-mRNA fusion binding data (Fig S4), hPD-L1 protein sequence alignment with mPD-L1 and hPD-L2 (Fig S5), the conservation of binding site residues between hPD-L1, mPD-L1, and hPD-L2 (Fig S6), the binding affinity of Fluorescein-conjugated anti-PD-L1 antibodies (Fig S7), and PNGase F treatment of PD-L1 proteins (Fig S8) are also included in the Supporting Information.
References
- 1.Hanahan D, and Weinberg RA (2011) Hallmarks of cancer: the next generation, Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- 2.Pardoll DM (2012) The blockade of immune checkpoints in cancer immunotherapy, Nat. Rev. Cancer 12, 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Riley JL (2009) PD‐1 signaling in primary T cells, Immunol. Rev 229, 114–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weber J (2010) Immune checkpoint proteins: a new therapeutic paradigm for cancer—preclinical background: CTLA-4 and PD-1 blockade, In Semin. Oncol, pp 430–439, Elsevier. [DOI] [PubMed] [Google Scholar]
- 5.Liu H, Zhao Z, Zhang L, Li Y, Jain A, Barve A, Jin W, Liu Y, Fetse J, and Cheng K (2019) Discovery of low-molecular weight anti-PD-L1 peptides for cancer immunotherapy, J. Immunother. Cancer 7, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Topalian SL, Drake CG, and Pardoll DM (2015) Immune checkpoint blockade: a common denominator approach to cancer therapy, Cancer Cell 27, 450–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ning YM, Suzman D, Maher VE, Zhang L, Tang S, Ricks T, Palmby T, Fu W, Liu Q, and Goldberg KB (2017) FDA approval summary: Atezolizumab for the treatment of patients with progressive advanced urothelial carcinoma after platinum‐containing chemotherapy, Oncologist 22, 743–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim ES (2017) Avelumab: First global approval, Drugs 77, 929–937. [DOI] [PubMed] [Google Scholar]
- 9.Syed YY (2017) Durvalumab: first global approval, Drugs 77, 1369–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hamid O, Robert C, Daud A, Hodi FS, Hwu W-J, Kefford R, Wolchok JD, Hersey P, Joseph RW, and Weber JS (2013) Safety and tumor responses with lambrolizumab (anti–PD-1) in melanoma, N. Engl. J. Med 369, 134–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, and Atkins MB (2012) Safety, activity, and immune correlates of anti–PD-1 antibody in cancer, N. Engl. J. Med 366, 2443–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Markham A, and Duggan S (2018) Cemiplimab: first global approval, Drugs 78, 1841–1846. [DOI] [PubMed] [Google Scholar]
- 13.Ingles Garces AH, Au L, Mason R, Thomas J, and Larkin J (2019) Building on the anti-PD1/PD-L1 backbone: combination immunotherapy for cancer, Expert Opin. Invest. Drugs 28, 695–708. [DOI] [PubMed] [Google Scholar]
- 14.Maute RL, Gordon SR, Mayer AT, McCracken MN, Natarajan A, Ring NG, Kimura R, Tsai JM, Manglik A, and Kruse AC (2015) Engineering high-affinity PD-1 variants for optimized immunotherapy and immuno-PET imaging, Proc. Natl. Acad. Sci. U. S. A 112, E6506–E6514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zimmer L, Goldinger SM, Hofmann L, Loquai C, Ugurel S, Thomas I, Schmidgen MI, Gutzmer R, Utikal JS, and Göppner D (2016) Neurological, respiratory, musculoskeletal, cardiac and ocular side-effects of anti-PD-1 therapy, Eur. J. Cancer 60, 210–225. [DOI] [PubMed] [Google Scholar]
- 16.Heinzerling L, and Goldinger SM (2017) A review of serious adverse effects under treatment with checkpoint inhibitors, Curr. Opin. Oncol 29, 136–144. [DOI] [PubMed] [Google Scholar]
- 17.Postow MA, Callahan MK, and Wolchok JD (2015) Immune checkpoint blockade in cancer therapy, J. Clin. Oncol 33, 1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cristescu R, Mogg R, Ayers M, Albright A, Murphy E, Yearley J, Sher X, Liu XQ, Lu H, Nebozhyn M, Zhang C, Lunceford JK, Joe A, Cheng J, Webber AL, Ibrahim N, Plimack ER, Ott PA, Seiwert TY, Ribas A, McClanahan TK, Tomassini JE, Loboda A, and Kaufman D (2018) Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy, Science 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ilie M, Hofman V, Dietel M, Soria J-C, and Hofman P (2016) Assessment of the PD-L1 status by immunohistochemistry: challenges and perspectives for therapeutic strategies in lung cancer patients, Virchows Arch 468, 511–525. [DOI] [PubMed] [Google Scholar]
- 20.Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, Patnaik A, Aggarwal C, Gubens M, and Horn L (2015) Pembrolizumab for the treatment of non–small-cell lung cancer, N. Engl. J. Med 372, 2018–2028. [DOI] [PubMed] [Google Scholar]
- 21.Sun X, Li Y, Liu T, Li Z, Zhang X, and Chen X (2017) Peptide-based imaging agents for cancer detection, Adv. Drug Delivery. Rev 110, 38–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schmidt MM, and Wittrup KD (2009) A modeling analysis of the effects of molecular size and binding affinity on tumor targeting, Mol. Cancer Ther 8, 2861–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miller MM, Mapelli C, Allen MP, Bowsher MS, Boy KM, Gillis EP, Langley DR, Mull E, Poirier MA, and Sanghvi N Macrocyclic inhibitors of the PD-1/PD-L1 and CD80 (B7–1)/PD-L1 protein/protein interactions, U.S. Patent No. 9,308,236 12 Apr. 2016. [Google Scholar]
- 24.Chatterjee S, Lesniak WG, Miller MS, Lisok A, Sikorska E, Wharram B, Kumar D, Gabrielson M, Pomper MG, and Gabelli SB (2017) Rapid PD-L1 detection in tumors with PET using a highly specific peptide, Biochem. Biophys. Res. Commun 483, 258–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kumar D, Lisok A, Dahmane E, McCoy M, Shelake S, Chatterjee S, Allaj V, Sysa-Shah P, Wharram B, and Lesniak WG (2019) Peptide-based PET quantifies target engagement of PD-L1 therapeutics, J. Clin. Invest 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roberts RW, and Szostak JW (1997) RNA-peptide fusions for the in vitro selection of peptides and proteins, Proc. Natl. Acad. Sci. U. S. A 94, 12297–12302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takahashi TT, Austin RJ, and Roberts RW (2003) mRNA display: ligand discovery, interaction analysis and beyond, Trends Biochem. Sci 28, 159–165. [DOI] [PubMed] [Google Scholar]
- 28.Takahashi TT, and Roberts RW (2009) In vitro selection of protein and peptide libraries using mRNA display, In Nucleic Acid and Peptide Aptamers, pp 293–314, Springer. [DOI] [PubMed] [Google Scholar]
- 29.Fiacco SV, Kelderhouse LE, Hardy A, Peleg Y, Hu B, Ornelas A, Yang P, Gammon ST, Howell SM, and Wang P (2016) Directed evolution of scanning unnatural‐protease‐resistant (SUPR) peptides for in vivo applications, ChemBioChem 17, 1643–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin D. Y. w., Tanaka Y, Iwasaki M, Gittis AG, Su H-P, Mikami B, Okazaki T, Honjo T, Minato N, and Garboczi DN (2008) The PD-1/PD-L1 complex resembles the antigen-binding Fv domains of antibodies and T cell receptors, Proc. Natl. Acad. Sci. U. S. A 105, 3011–3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, Iwai Y, Long AJ, Brown JA, and Nunes R (2001) PD-L2 is a second ligand for PD-1 and inhibits T cell activation, Nat. Immunol 2, 261. [DOI] [PubMed] [Google Scholar]
- 32.Lázár-Molnár E, Yan Q, Cao E, Ramagopal U, Nathenson SG, and Almo SC (2008) Crystal structure of the complex between programmed death-1 (PD-1) and its ligand PD-L2, Proc. Natl. Acad. Sci. U. S. A 105, 10483–10488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Beckett D, Kovaleva E, and Schatz PJ (1999) A minimal peptide substrate in biotin holoenzyme synthetase-catalyzed biotinylation, Protein Sci. 8, 921–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hochuli E, Bannwarth W, Döbeli H, Gentz R, and Stüber D (1988) Genetic approach to facilitate purification of recombinant proteins with a novel metal chelate adsorbent, Bio/Technology 6, 1321. [Google Scholar]
- 35.Green N, Alexander H, Olson A, Alexander S, Shinnick TM, Sutcliffe JG, and Lerner RA (1982) Immunogenic structure of the influenza virus hemagglutinin, Cell 28, 477–487. [DOI] [PubMed] [Google Scholar]
- 36.Studier FW (2005) Protein production by auto-induction in high-density shaking cultures, Protein Expr. Purif 41, 207–234. [DOI] [PubMed] [Google Scholar]
- 37.Haugland RP (1995) Coupling of monoclonal antibodies with fluorophores, In Monoclonal Antibody Protocols, pp 205–221, Springer. [DOI] [PubMed] [Google Scholar]
- 38.Zhang X, Schwartz J-CD, Guo X, Bhatia S, Cao E, Chen L, Zhang Z-Y, Edidin MA, Nathenson SG, and Almo SC (2004) Structural and functional analysis of the costimulatory receptor programmed death-1, Immunity 20, 337–347. [DOI] [PubMed] [Google Scholar]
- 39.Kyte J, and Doolittle RF (1982) A simple method for displaying the hydropathic character of a protein, J. Mol. Biol 157, 105–132. [DOI] [PubMed] [Google Scholar]
- 40.Chang HN, Liu BY, Qi YK, Zhou Y, Chen YP, Pan KM, Li WW, Zhou XM, Ma WW, and Fu CY (2015) Blocking of the PD‐1/PD‐L1 Interaction by ad‐Peptide Antagonist for Cancer Immunotherapy, Angew. Chem., Int. Ed 54, 11760–11764. [DOI] [PubMed] [Google Scholar]
- 41.Magiera‐Mularz K, Skalniak L, Zak KM, Musielak B, Rudzinska‐Szostak E, Berlicki Ł, Kocik J, Grudnik P, Sala D, and Zarganes‐Tzitzikas T (2017) Bioactive macrocyclic inhibitors of the PD‐1/PD‐L1 immune checkpoint, Angew. Chem., Int. Ed 56, 13732–13735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.De Silva RA, Kumar D, Lisok A, Chatterjee S, Wharram B, Venkateswara Rao K, Mease R, Dannals RF, Pomper MG, and Nimmagadda S (2018) Peptide-based 68Ga-PET radiotracer for imaging PD-L1 expression in cancer, Mol. Pharm 15, 3946–3952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lesniak WG, Mease RC, Chatterjee S, Kumar D, Lisok A, Wharram B, Kalagadda VR, Emens LA, Pomper MG, and Nimmagadda S (2019) Development of [18F] FPy-WL12 as a PD-L1 Specific PET Imaging Peptide, Mol. Imaging 18, 1536012119852189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Howell SM, Fiacco SV, Takahashi TT, Jalali-Yazdi F, Millward SW, Hu B, Wang P, and Roberts RW (2014) Serum stable natural peptides designed by mRNA display, Sci Rep 4, 6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Clackson T, and Wells JA (1995) A hot spot of binding energy in a hormone-receptor interface, Science 267, 383–386. [DOI] [PubMed] [Google Scholar]
- 46.Cetin M, Evenson WE, Gross GG, Jalali-Yazdi F, Krieger D, Arnold D, Takahashi TT, and Roberts RW (2017) RasIns: genetically encoded intrabodies of activated Ras proteins, J. Mol. Biol 429, 562–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chupak LS, Ding M, Martin SW, Zheng X, Hewawasam P, Connolly TP, Xu N, Yeung K-S, Zhu J, and Langley DR Compounds useful as immunomodulators, U.S. Patent No. 9,850,225 26 December 2017. [Google Scholar]
- 48.Zak KM, Grudnik P, Guzik K, Zieba BJ, Musielak B, Dömling A, Dubin G, and Holak TA (2016) Structural basis for small molecule targeting of the programmed death ligand 1 (PD-L1), Oncotarget 7, 30323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Powles T, Eder JP, Fine GD, Braiteh FS, Loriot Y, Cruz C, Bellmunt J, Burris HA, Petrylak DP, and Teng S. l. (2014) MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer, Nature 515, 558. [DOI] [PubMed] [Google Scholar]
- 50.Broos K, Lecocq Q, Xavier C, Bridoux J, Nguyen TT, Corthals J, Schoonooghe S, Lion E, Raes G, and Keyaerts M (2019) Evaluating a Single Domain Antibody Targeting Human PD-L1 as a Nuclear Imaging and Therapeutic Agent, Cancers 11, 872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kuan H, Masayuki H, Xie L, Zhang Y, Kotaro N, Hisashi S, and Zhang M-R (2019) Developing native peptide-based radiotracers for PD-L1 PET imaging and improving imaging contrast by pegylation, Chem. Commun 55, 4162–4165. [DOI] [PubMed] [Google Scholar]
- 52.Li C, Zhang N, Zhou J, Ding C, Jin Y, Cui X, Pu K, and Zhu Y (2018) Peptide blocking of PD-1/PD-L1 interaction for cancer immunotherapy, Cancer Immunol. Res 6, 178–188. [DOI] [PubMed] [Google Scholar]
- 53.Wang F, Ma J, Liu J, Jin H, and Huang D (2012) Synthetic small peptides acting on B7H1 enhance apoptosis in pancreatic cancer cells, Mol. Med. Rep 6, 553–557. [DOI] [PubMed] [Google Scholar]
- 54.Caldwell C, Johnson CE, Balaji V, Balaji GA, Hammer RD, and Kannan R (2017) Identification and validation of a PD-L1 binding peptide for determination of PDL1 expression in tumors, Sci. Rep 7, 13682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shindo Y, McDonough JS, Chang KC, Ramachandra M, Sasikumar PG, and Hotchkiss RS (2017) Anti-PD-L1 peptide improves survival in sepsis, J. Surg. Res 208, 33–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.