

Abstract
Introduction
Dementia pathogenesis begins years before clinical symptom onset, necessitating the understanding of premorbid risk mechanisms. Here we investigated potential pathogenic mechanisms by assessing DNA methylation associations with dementia risk factors in Alzheimer's disease (AD)–free participants.
Methods
Associations between dementia risk measures (family history, AD genetic risk score [GRS], and dementia risk scores [combining lifestyle, demographic, and genetic factors]) and whole‐blood DNA methylation were assessed in discovery and replication samples (n = ~400 to ~5000) from Generation Scotland.
Results
AD genetic risk and two dementia risk scores were associated with differential methylation. The GRS associated predominantly with methylation differences in cis but also identified a genomic region implicated in Parkinson disease. Loci associated with dementia risk scores were enriched for those previously associated with body mass index and alcohol consumption.
Discussion
Dementia risk measures show widespread association with blood‐based methylation, generating several hypotheses for assessment by future studies.
Keywords: alcohol, Alzheimer's disease, dementia, DNA methylation, genetic risk score, risk factors
1. BACKGROUND
The pathophysiology of dementia begins many years, possibly decades, before the emergence of clinical symptoms. 1 This long prodromal phase highlights the need for preventative strategies prior to the development of irreversible brain damage. As such, understanding premorbid risk mechanisms is critical. Several approaches to identify individuals at risk of developing dementia have been devised, including the summation of genetic risk, in the form of genetic risk scores (GRSs), the consideration of family history, and the calculation of risk scores, which incorporate multiple lifestyle, demographic, and genetic risk factors. 2 , 3 , 4
DNA methylation is an epigenetic modification which, in some contexts, is associated with gene expression variation. Altered gene expression has been identified in the blood and post‐mortem brains of AD patients, 5 , 6 and post‐mortem brain‐based studies have identified associations between DNA methylation and AD and its neuropathological hallmarks. 7 , 8 , 9 Blood‐based studies, while limited by small sample sizes, have also found evidence for AD‐associated methylation differences. 10 , 11 It is not, however, possible to determine from these studies whether methylation differences precede AD onset, making them potentially etiologically informative, or whether they reflect ongoing pathology, compensatory mechanisms and/or treatment effects. Studies that have identified associations between variation in blood‐based DNA methylation and risk factors for dementia (eg, carrying the apolipoprotein E (APOE) ε4 haplotype, 12 , 13 aging, 14 and obesity 15 ) suggest that the assessment of methylation in this tissue may yield insights into the pathways and processes that lead to dementia.
In this study, by assessing associations between multiple measures of dementia risk and blood‐based DNA methylation in AD‐free participants, we aim to further understand the mechanisms conferring dementia risk and characterize the role of methylation in these processes.
2. METHODS
2.1. Participants
Participants were drawn from the Generation Scotland: Scottish Family Health Study (GS:SFHS). 16 , 17 The cohort comprises ≈24,000 participants ≥18 years of age at recruitment. At a baseline clinical appointment, participants were phenotyped for a range of health, demographic, and lifestyle factors, and provided physical measurements and samples for DNA extraction. GS:SFHS has been granted ethical approval from the NHS Tayside Committee on Medical Research Ethics, on behalf of the National Health Service (05/S1401/89), and has Research Tissue Bank Status (15/ES/0040). GS:SFHS participants provided broad and enduring written informed consent for biomedical research.
RESEARCH IN CONTEXT
Systematic review: Several studies have investigated associations between DNA methylation and individual dementia risk factors (eg, aging and obesity) but none has compared multiple risk measures. We compared the methylation signatures of multifactorial dementia risk scores, an Alzheimer's disease (AD) genetic risk score (GRS), and dementia family history in the two largest single‐cohort blood‐based methylation samples.
Interpretation: In AD‐free participants, we identified methylation associations with an AD GRS and two midlife dementia risk scores, with no overlap between the GRS‐ and risk score–associated loci. The GRS analysis identified loci in cis to significant genome‐wide association study (GWAS) regions and a new putative AD‐risk locus, previously implicated in Parkinson disease. Loci associated with a midlife dementia risk overlapped with those associated with alcohol consumption.
Future directions: Longitudinal analyses should be performed to assess the pathogenic role of the identified loci. Analyses to assess the role of the putative novel dementia‐risk locus are warranted.
2.2. Calculation of dementia risk scores
Four dementia risk scores, henceforth referred to as CAIDE1, CAIDE2, 2 Li, 3 and Reitz, 4 were calculated using data that were collected at GS:SFHS enrollment or obtained through record linkage (see Figure 1, Supplementary Methods and Table S1 for information on the contributing variables). To generate each risk score, the contributing variables were scaled and weighted according to the original studies, and summed. The Reitz score 4 was calculated using weightings devised when considering participants with both a “possible” and “probable” diagnosis of Alzheimer's disease (AD). Each score was calculated for participants within the appropriate age‐range (CAIDE1/2: 39‐64 years 2 ; Li: ≥60 years 3 ; Reitz: ≥65 years). 4
FIGURE 1.
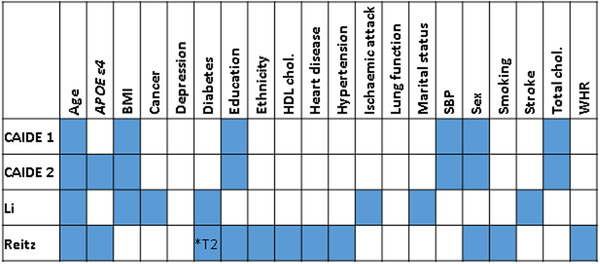
The variables contributing to each dementia risk score are indicated by filled blue boxes. Abbreviations: APOE ε4, apolipoprotein E ε4; BMI, body mass index; HDL, high‐density lipoprotein; chol, cholesterol; SBP, systolic blood pressure; T2, type 2; WHR, waist‐hip ratio
2.3. Genotyping and calculation of Alzheimer's disease genetic risk score
GS:SFHS genotyping has been described previously 18 , 19 (Supplementary Methods). The AD GRS was calculated using the lead single‐nucleotide polymorphism (SNP) from each of the 26 genome‐wide significant loci identified through a meta‐analysis of parental AD and AD 20 (Table S2). Each participant's score was generated by summing their dosage of each risk allele, weighted by the corresponding GWAS effect estimate.
2.4. DNA methylation profiling
Whole blood DNA methylation was profiled using the Infinium MethylationEPIC BeadChip (Illumina Inc.) in two sets of GS:SFHS participants at two separate times, leading to a natural discovery (n = 5190) and replication (n = 4583) design, as described previously 21 , 22 , 23 (Supplementary Methods). Discovery and replication sample normalization was performed separately and the data were converted to M‐values. Participants in the replication sample were unrelated (SNP‐based relatedness <0.05) to each other and/or those in the discovery sample. A correction for relatedness was applied to the discovery sample (Supplementary Methods).
Prior to analyses, poor‐performing probes (Supplementary Methods), sex chromosome probes, participants with unreliable self‐report data, suspected XXY genotype, or self‐reported AD (n = 5) were excluded. The final discovery data set comprised 777,193 loci in 5087 participants; the replication data set comprised 773,860 loci in 4450 participants. Subsequent analyses of the methylation data were carried out using R versions 3.6.0. or 3.6.1. 24
2.5. Epigenome‐wide association studies
Epigenome‐wide association studies (EWASs) were performed using linear regression modeling, implemented in limma. 25 CpG sites (M‐values) were modeled as the dependent variable and the dementia risk measure was the predictor‐of‐interest. Additional covariates included in the standard models are detailed below:
2.5.1. Discovery sample
CpG site (M‐values pre‐corrected for relatedness, estimated cell count proportions, and processing batch) ∼ dementia risk measure + age + sex + smoking status + pack years + 20 methylation principal components
2.5.2. Replication sample
CpG site (M‐values) ∼ dementia risk measure+ age + sex + smoking status + pack years + estimated cell count proportions (granulocytes, natural killer cells, B lymphocytes, CD4+ T lymphocytes, and CD8+ T lymphocytes) + processing batch + 20 methylation principal components
The variables “smoking status,” “pack years,” and the methylation principal components are explained in the Supplementary Methods.
A number of sensitivity analyses for the CAIDE1 score were performed in which additional covariates were included one‐by‐one, using the same thresholds for categorizing continuous variables as implemented in the risk score. These were body mass index (BMI; ≤30 kg/m2 or >30 kg/m2); systolic blood pressure (SBP; ≤140 mm Hg or >140 mm Hg); total cholesterol (TC; ≤6.5 mmol/L or > 6.5 mmol/L); years of education (≥10, >6 and <10, or ≤6); self‐reported alcohol consumption (log10‐transformed (+1) units of alcohol/week), and a DNA methylation alcohol consumption score derived using the R package dnamalci. 26 , 27
Limma was used to calculate empirical Bayes moderated t‐statistics from which P values were obtained. The significance threshold in the discovery sample was P ≤ 3.6 × 10−8. 28 Sites attaining significance in the discovery sample were assessed in the replication sample using a Bonferroni‐adjusted threshold of 0.05/no. sites assessed.
2.6. EWAS meta‐analysis
Inverse standard error‐weighted fixed effects meta‐analyses of the discovery and replication EWAS results were performed using METAL. 29 Sites attaining a meta‐analysis P ≤ 3.6 × 10−8 were considered significant.
2.7. Identification of differentially methylated regions
Differentially methylated regions (DMRs) were identified using the dmrff.meta function in dmrff. 30 DMRs were defined as regions containing 2 to 30 sites with consistent direction of effect and EWAS meta‐analysis P values ≤.05 separated by ≤500 bp. DMRs with Bonferroni‐adjusted P values ≤.05 were declared significant.
2.8. EWAS and GWAS catalog look‐ups
The GWAS Catalog v1.0.2. was downloaded from https://www.ebi.ac.uk/gwas/docs/file-downloads (December 16, 2019) 34 and queried using gene names annotated to probes containing differentially methylated positions (DMPs) identified in the meta‐analysis (meta‐DMPs) for the phenotype‐of‐interest. GWAS traits enriched for association (P ≤ 1 × 10−5) with genes containing meta‐DMPs were identified using Fisher's exact test. Enrichment was declared significant when P ≤ 1.26 × 10−5 (0.05/3980 traits assessed).
The EWAS Catalog was downloaded from http://www.ewascatalog.org/ (03/07/19) 35 and queried using the significant DMPs probe IDs. EWAS traits enriched for association (P ≤ 1 × 10−5) with meta‐DMPs were identified using Fisher's exact test. Enrichment was declared significant when P ≤ 3.31 × 10−4 (0.05/151 traits assessed).
2.9. Gene ontology/KEGG pathway analyses
Gene ontology (GO) and KEGG pathway analyses were implemented using a modified version of the missMethyl gometh function 31 (Supplementary Methods). The target list comprised probes showing suggestive association with the phenotype‐of‐interest (P ≤ 1 × 10−5) in the EWAS or DMR analysis, and the gene universe included all probes in the analyses. Enrichment was assessed using a hypergeometric test, accounting for the bias arising from the variation in the number of probes per gene. Significance thresholds of P ≤ 2.20 × 10−6 and P ≤ 1.48 × 10−4 were applied to allow for a Bonferroni‐correction for the 22,750 GO terms and 337 KEGG pathways assessed, respectively.
2.10. Identification of meQTLs
Methylation quantitative trait loci (meQTLs) for the AD GRS‐associated DMPs were identified using the discovery sample. The quality control, normalization, and pre‐correction of the data prior to the meQTL analyses have been described previously 32 (Supplementary Methods). The resulting residuals were inverse‐rank normal transformed and entered as the dependent variable in simple linear model GWASs to identify meQTLs. GWASs were implemented using REGSCAN v0.5. 33 SNPs that were associated with a DMP with P ≤ 5 × 10−8/49 (Bonferroni correction for the 49 DMPs for which meQTL results were available), an info score ≥0.8, and had a MAF > 0.01 were declared to be meQTLs.
Where a meta‐DMP associated with AD GRS harbored a SNP at the CpG site, linkage disequilibrium between the CpG SNP and the nearest SNP contributing to the GRS was assessed using the LDpair Tool using data from the British in England and Scotland (GBR) population (https://ldlink.nci.nih.gov/?tab=ldpair; June 09, 2020). 36
3. RESULTS
3.1. Epigenome‐wide asssociation study sample demographics
Participant numbers and sample demographic information are shown in Table S3.
3.2. Genetic risk for Alzheimer's disease
3.2.1. Identification of differentially methylated positions
An epigenome‐wide association study (EWAS) of the Alzheimer's disease (AD) genetic risk score (GRS) identified 32 differentially methylated positions (DMPs) in the discovery sample (1.06 × 10−30 ≤P≤ 2.22 × 10−8; Table S4). Of these, 31 showed replicated association (1.07 × 10−30 ≤ P ≤ 8.33 × 10−4; Table S5). Meta‐analysis of the discovery and replication samples identified 68 DMPs (6.15 × 10−48 ≤ P ≤ 3.45 × 10−8; Table 1; Table S6; Figure 2).
TABLE 1.
Top 20 DMPs associated with the AD GRS in a meta‐analysis of the discovery and replication samples
| ID | Chr. | BP a | Gene symbol | Effect | SE | P |
|---|---|---|---|---|---|---|
| cg10757760 | 2 | 127893054 | 0.0568 | 0.0039 | 6.15 × 10−48 | |
| cg04441687 | 11 | 85869322 | 0.0586 | 0.0041 | 1.12 × 10−46 | |
| cg26631131 | 19 | 45240591 | 0.0385 | 0.0029 | 1.56 × 10−39 | |
| cg02887598 | 2 | 127841945 | BIN1 | −0.0986 | 0.0085 | 2.51 × 10−31 |
| cg19116668 | 7 | 99932089 | PMS2L1 | 0.049 | 0.0051 | 1.54 × 10−21 |
| cg18959616 | 11 | 85814918 | 0.0656 | 0.0072 | 5.85 × 10−20 | |
| cg02521229 | 11 | 60019236 | 0.0658 | 0.0073 | 1.61 × 10−19 | |
| cg03579757 | 7 | 100091793 | NYAP1 | 0.0342 | 0.0038 | 2.33 × 10−19 |
| cg16618979 | 7 | 143108841 | AC092214.10 | 0.0974 | 0.0109 | 5.11 × 10−19 |
| cg03526776 | 6 | 41159608 | TREML2 | −0.0402 | 0.0045 | 7.06 × 10−19 |
| cg11461311 | 2 | 127782614 | RP11‐521O16.1;RP11‐521O16.2 | 0.0321 | 0.0036 | 1.31 × 10−18 |
| cg00436254 | 2 | 127862614 | BIN1 | 0.0256 | 0.003 | 6.76 × 10−18 |
| cg06750524 | 19 | 45409955 | APOE | 0.05 | 0.0059 | 2.18 × 10−17 |
| cg23423086 | 11 | 85856245 | −0.0365 | 0.0043 | 2.37 × 10−17 | |
| cg22906224 | 7 | 99728672 | AC073842.19 | −0.0392 | 0.0047 | 3.55 × 10−17 |
| cg05908241 | 7 | 143109367 | AC092214.10 | 0.0283 | 0.0035 | 6.62 × 10−16 |
| cg17830204 | 7 | 99819110 | GATS;PVRIG;STAG3;AC005071.1 | 0.0308 | 0.0039 | 4.78 × 10−15 |
| cg19590598 | 2 | 127782813 | RP11‐521O16.1;RP11‐521O16.2 | 0.0282 | 0.0036 | 5.22 × 10−15 |
| cg08871934 | 10 | 11720283 | −0.0343 | 0.0044 | 9.59 × 10−15 | |
| cg09555818 | 19 | 45449301 | APOC2;APOC4 | −0.0498 | 0.0065 | 1.24 × 10−14 |
Abbreviations: BP, base position; Chr., chromosome; SE, standard error, DMP; differentially methylated position; GRS, genetic risk score.
Base position in genome assembly hg19/GRCh37.
FIGURE 2.
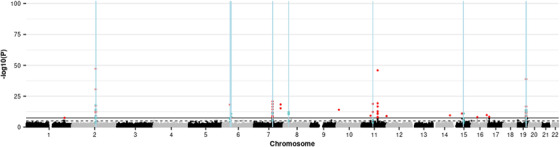
Manhattan plot showing the results of the epigenome‐wide association study (EWAS) meta‐analysis of the Alzheimer's disease (AD) genetic risk score (GRS), and the positions of differentially methylated regions (DMRs) identified in a meta‐DMR analysis. Each point represents one of the 772,453 loci included in the EWAS meta‐analysis, with the point's position being determined by genomic position (x‐axis) and significance in the EWAS meta‐analysis (–log10 P value; y‐axis). Sites attaining genome‐wide significance (P ≤ 3.6 × 10−8) are indicated in red and those that are involved in a significant DMR (Bonferroni‐correct P ≤ .05) are indicated in blue. The locations of DMRs are further indicated by vertical blue lines. The solid horizontal line is the threshold for genome‐wide significance (P ≤ 3.6 × 10−8) and the dashed line indicates a suggestive significance threshold (P ≤ 1 × 10−5)
Sixty‐one of the 68 meta‐DMPs were located within 18 of the 26 genome‐wide association study (GWAS) loci used to produce the GRS, 20 with six of the remaining seven being located within 30 kb of one. Four of the associated CpGs have a single nucleotide polymorphism (SNP) at the CpG site. Density plots of the signal at these sites indicated a potential SNP effect on methylation at three of the CpGs (Figure S1). All three of these SNPs are in high linkage disequilibrium with the nearest SNP included in the GRS (D’ = 1 for cg02887598 and cg16618979, and D’ = 0.64 for cg12568536). Formal methylation quantitative trait loci (meQTL) analysis could be performed for 49/68 DMPs: of these, 48 were associated in cis but not trans, with genetic variants located in the GWAS loci. 20 Methylation at the remaining DMP, cg14354618, which is not located within or in proximity (<30 kb) to a GWAS locus, was associated in trans with genetic variation on chromosome 19 (chr19: 868083‐1188756; hg19/GRCh37), which overlaps a GWAS locus. 20
There was no overlap between the meta‐DMPs identified as being associated with the CAIDE1 score and the GRS. To explore the reason for this lack of overlap, the Pearson correlation coefficient was calculated between the two scores. This was small and non‐significant (r = −.018, P = .326).
Querying the GWAS catalog 34 with the 34 gene names annotated to the 68 meta‐DMPs unsurprisingly identified many terms related to AD and its neuropathological hallmarks (Table S7), the most significant being “AD or family history of AD” (P = 1.77 × 10−27). No significant enrichment was identified when querying the EWAS catalog; however, this catalogue comprises results from studies using the 450K array on which only 30/68 meta‐DMPs were measured.
3.2.2. Identification of differentially methylated regions
The differentially methylated region (DMR) meta‐analysis identified 18 significant DMRs comprising 41 CpGs, of which 20 were identified by the meta‐EWAS (Table S8; Figure 2). Seventeen of the DMRs overlap with loci that were in the GWAS, 20 and the 18th is located <7 kb from the nearest GWAS locus. The longest DMR spans a 302 bp region ≈13.7 kb upstream of BIN1, whereas the most significant spans a 199 bp region ≈22.8 kb downstream of BIN1. Both show increased methylation with increased GRS.
Gene ontology (GO) analysis using the combined DMP and DMR results identified 18 terms, the most significant of which was “amyloid‐beta formation” (P = 3.68 × 10−10; Table 2). No significant KEGG pathways were identified.
TABLE 2.
GO terms showing significant enrichment for probes where methylation is associated the AD GRS
| Ontology | Term | Proportiona | P |
|---|---|---|---|
| BP | Amyloid beta formation | 6/34 | 3.68 × 10‐10 |
| BP | Negative regulation of amyloid precursor protein catabolic process | 5/16 | 4.62 × 10‐10 |
| BP | Amyloid precursor protein catabolic process | 6/44 | 1.89 × 10‐9 |
| BP | Amyloid‐beta metabolic process | 6/47 | 2.88 × 10‐9 |
| BP | Regulation of amyloid‐beta formation | 5/28 | 1.12 × 10‐8 |
| BP | Amyloid precursor protein metabolic process | 6/63 | 1.66 × 10‐8 |
| CC | Protein‐lipid complex | 6/39 | 2.53 × 10‐8 |
| BP | Regulation of amyloid precursor protein catabolic process | 5/35 | 3.52 × 10‐8 |
| BP | Negative regulation of amyloid‐beta formation | 4/13 | 3.79 × 10‐8 |
| BP | Reverse cholesterol transport | 4/20 | 8.38 × 10‐8 |
| BP | Protein‐lipid complex subunit organization | 6/49 | 1.38 × 10‐7 |
| CC | High‐density lipoprotein particle | 5/26 | 2.42 × 10‐7 |
| BP | Chylomicron remnant clearance | 3/9 | 1.12 × 10‐6 |
| BP | Triglyceride‐rich lipoprotein particle clearance | 3/9 | 1.12 × 10‐6 |
| BP | Protein‐lipid complex assembly | 6/32 | 1.13 × 10‐6 |
| CC | Plasma lipoprotein particle | 6/37 | 1.34 × 10‐6 |
| CC | Lipoprotein particle | 5/37 | 1.34 × 10‐6 |
| BP | Very‐low‐density lipoprotein particle remodeling | 3/12 | 2.10 × 10‐6 |
Abbreviations: BP, biological process; CC, cellular component; GRS, genetic risk score; GO, gene ontology.
Number of significant target list‐associated Entrez IDs associated with the gene ontology term/total number of Entrez IDs associated with the GO term. The target list comprised probes that met a nominal threshold for association with the AD GRS of P ≤ 1 × 10−5
3.3. Mid‐life dementia risk scores
The CAIDE1 and CAIDE2 risk scores assess the risk of developing dementia in 20 years' time in individuals 39 to 64 years of age. 2 CAIDE2 takes into account the same risk factors as CAIDE1 (with different weightings) and also considers apolipoprotein E (APOE) ε4 carrier status.
3.3.1. Identification of differentially methylated positions
An EWAS of the CAIDE1 score in the discovery sample identified 76 DMPs (3.29 × 10−20 ≤P≤ 3.49 × 10−8; Table S9), of which 65 replicated (7.76 × 10−18 ≤ P ≤ 6.48 × 10−4; Table S10). Meta‐analysis of the discovery and replication samples identified 227 DMPs (1.20 × 10−29 ≤ P ≤ 3.58 × 10−8; Figure 3; Table 3; Table S11).
FIGURE 3.
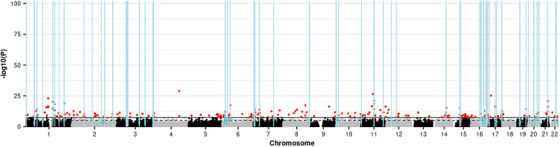
Manhattan plot showing the results of the epigenome‐wide association study (EWAS) meta‐analysis of the CAIDE1 dementia risk score and the positions of differentially methylated regions (DMRs) identified in a meta‐DMR analysis. Each point represents one of the 772,453 loci included in the EWAS meta‐analysis, with the point's position being determined by genomic position (x‐axis) and significance in the EWAS meta‐analysis (–log10 P value; y‐axis). Sites attaining genome‐wide significance (P ≤ 3.6 × 10−8) are indicated in red and those that are involved in a significant DMR (Bonferroni‐correct P ≤ .05) are indicated in blue. The locations of DMRs are further indicated by vertical blue lines. The solid horizontal line is the threshold for genome‐wide significance (P ≤ 3.6 × 10−8) and the dashed line indicates a suggestive significance threshold (P ≤ 1 × 10−5)
TABLE 3.
Top 20 DMPs associated with the CAIDE1 risk score in a meta‐analysis of the discovery and replication samples
| ID | Chr. | BP | Gene symbol | Direction | effect | SE | P |
|---|---|---|---|---|---|---|---|
| cg06690548 | 4 | 139162808 | SLC7A11 | – | −0.0766 | 0.0068 | 1.20 × 10‐29 |
| cg19758958 | 11 | 62319222 | AHNAK | – | −0.0256 | 0.0024 | 4.22 × 10‐27 |
| cg11024682 | 17 | 17730094 | SREBF1 | ++ | 0.026 | 0.0025 | 5.31 × 10‐26 |
| cg14476101 | 1 | 120255992 | PHGDH | – | −0.0391 | 0.0039 | 1.14 × 10‐23 |
| cg00574958 | 11 | 68607622 | CPT1A | – | −0.0413 | 0.0043 | 8.30 × 10‐22 |
| cg06500161 | 21 | 43656587 | ABCG1 | ++ | 0.0225 | 0.0024 | 2.10 × 10‐21 |
| cg19693031 | 1 | 145441552 | TXNIP | – | −0.0369 | 0.004 | 8.74 × 10‐21 |
| cg22699725 | 1 | 207242586 | PFKFB2 | ++ | 0.0275 | 0.003 | 6.89 × 10‐20 |
| cg00683922 | 1 | 207242569 | PFKFB2 | ++ | 0.0272 | 0.003 | 2.70 × 10‐19 |
| cg05325763 | 11 | 68607719 | CPT1A | – | −0.0394 | 0.0044 | 2.90 × 10‐19 |
| cg22976567 | 1 | 156074182 | LMNA | – | −0.026 | 0.0029 | 7.06 × 10‐19 |
| cg02715788 | 8 | 119974400 | – | −0.0225 | 0.0026 | 4.44 × 10‐18 | |
| cg18120259 | 6 | 43894639 | LOC100132354 | – | −0.0209 | 0.0024 | 4.49 × 10‐18 |
| cg11376147 | 11 | 57261198 | SLC43A1 | – | −0.0236 | 0.0028 | 2.98 × 10‐17 |
| cg00163198 | 11 | 130767760 | SNX19 | ++ | 0.0237 | 0.0028 | 4.89 × 10‐17 |
| cg16246545 | 1 | 120255941 | PHGDH | – | −0.0255 | 0.003 | 5.16 × 10‐17 |
| cg01270753 | 9 | 101944336 | RP11‐96L7.2 | – | −0.034 | 0.0041 | 5.27 × 10‐17 |
| cg08857797 | 17 | 40927699 | VPS25 | ++ | 0.0236 | 0.0028 | 5.86 × 10‐17 |
| cg16740586 | 21 | 43655919 | ABCG1 | ++ | 0.0243 | 0.0029 | 7.52 × 10‐17 |
| cg26457483 | 1 | 120256112 | PHGDH | – | −0.0308 | 0.0037 | 1.76 × 10‐16 |
Abbreviations: BP, base position; Chr., chromosome; SE, standard error; DMP, differentially methylated position.
aBase position in genome assembly hg19/GRCh37.
An EWAS of the CAIDE2 score in the discovery sample identified 18 DMPs (1.96 × 10−17 ≤P≤ 3.24 × 10−8; Table S12), of which 17 replicated (4.91 × 10−12 ≤ P ≤ 2.01 × 10−3; Table S13). Meta‐analysis of the discovery and replication samples identified 59 DMPs (3.56 × 10−22 ≤ P ≤ 3.21 × 10−8; Table S14). Fifty‐four of the CAIDE2 meta‐DMPs were also identified by the CAIDE1 meta‐analysis; given this overlap, subsequent analyses focus on CAIDE1.
The CAIDE1‐associated meta‐DMPs were explored using the EWAS and GWAS catalogs. 34 Significant enrichment was identified for 16 EWAS traits/conditions, with “Body mass index [BMI]” being the most significantly enriched (P = 2.96 × 10−118; Table S15). Two alcohol‐related traits: “alcohol consumption per day” (P = 9.70 × 10−29) and “gamma‐glutamyl transferase” (P = 8.55 × 10−25) also showed enrichment. GWAS catalog enrichment analysis identified only one significant term, “Eosinophil counts” (P = 4.97 × 10−8).
Sensitivity analyses
The extent to which the BMI component of the CAIDE1 score drives the observed CAIDE1 associations was assessed by performing an EWAS meta‐analysis in which BMI was included as an additional covariate. Co‐varying for BMI resulted in only 11 of the original 227 meta‐DMPs remaining significant (Table S11), with the correlation between the effect estimates for all sites between the two analyses being r = 0.745 (95% confidence interval (CI) = 0.744 to 0.746). Compared with BMI, co‐varying for other components of the CAIDE1 score resulted in a larger numbers of CAIDE1 meta‐DMPs remaining significant (SBP: 94/227; TC: 190/227; education: 221/227; Table S11); correlations with the effect estimates of the original analysis were higher (SBP: r = 0.815, 95% CI = 0.815 to 0.816; TC: r = 0.938, 95% CI = 0.937 to 0.938); education: r = 0.990, 95% CI = 0.990 to 0.990).
Assessment of the involvement of alcohol consumption in the CAIDE1 EWAS results
Twenty‐four of the 88 meta‐DMPs that are represented on the 450K array, including the most significant DMP, cg06690548, have previously been associated with alcohol consumption (P ≤ 1 × 10−5). 26 Similarly, an EWAS meta‐analysis of self‐reported alcohol consumption in our methylation sample identified 5599 DMPs at a suggestive threshold of P ≤ 1 × 10−5 (unpublished data, Clarke et al.); of these, 49 show a significant association with CAIDE1 with a consistent direction of effect. This overlap is highly significant (P < 2 × 10−16).
Because alcohol consumption showed a small but significant correlation with CAIDE1 score (r = 0.091; 95% CI = 0.065 to 0.118; P = 2.60 × 10−11), the potential for alcohol consumption to drive the observed associations between CAIDE1 and DNA methylation was assessed by including alcohol consumption measured by (1) self‐report or (2) a polyepigenetic risk score 26 , 27 as an additional covariate in the CAIDE1 EWAS. Neither measure of alcohol consumption resulted in a substantial change in effect estimates (self‐reported: r = 0.948, 95% CI = 0.948 to 0.948; DNA methylation score: r = 0.993, 95% CI = 0.993 to 0.993), with 166 and 191 of the 227 CAIDE1 meta‐DMPs remaining significant after the inclusion of the self‐reported and DNA methylation score coefficients, respectively (Table S11).
3.3.2. Identification of differentially methylated regions
The DMR meta‐analysis of the discovery and replication samples identified 57 CAIDE1‐associated DMRs (all Bonferroni‐adjusted P < 0.044; Table S16), each comprising two to seven CpGs. In total, the 57 DMRs involve 179 sites, of which 35 were significant in the EWAS meta‐analysis. The most significant DMR (Bonferroni‐adjusted P = 1.67 × 10−20) comprises four hypomethylated CpGs spanning a 115 bp intronic region of CPT1A (chr11: 68607622‐68607737; hg19/GRCh37). The longest DMR spans a 1.1 kb intronic region of JARID2 (chr6: 15504844‐15505949; hg19/GRCh37).
GO and KEGG pathway analyses found no enrichment for biological processes or pathways among the CAIDE1 DMP or DMR CpG sites (min. P GO = 8.46 × 10−4; minutes. PKEGG = 3.51 × 10−3).
3.4. Other measures of dementia risk
The other measures of dementia risk assessed were (1) dementia family history (FH) and (2) two late‐life dementia risk scores that predict the risk of developing dementia in those older than 60 or 65 years of age. 3 , 4 EWASs of the discovery sample (minimum (min). P FH = 8.47 × 10−7; min. P Li = 3.91 × 10−8; min P Reitz = 1.58 × 10−6), meta‐EWASs of the discovery and replication samples (min. P FH = 1.15 × 10−6; min. P Li = 1.80 × 10−6; min. P Reitz = 8.99 × 10−7), and DMR analyses (min. Bonferroni‐adjusted P FH = 0.439; min. Bonferroni‐adjusted P Li = 0.208; min. Bonferroni‐adjusted P Reitz = 1) failed to identify any significant associations.
4. DISCUSSION
We have assessed DNA methylation associations with a range of dementia risk measures in large discovery and replication samples comprising Alzheimer's disease (AD)‐free participants, and we report multiple loci as being associated with AD genetic risk and two multifactorial mid‐life risk scores for dementia.
All but one of the loci associated with the AD genetic risk score (GRS) were located within 30 kb of the genome‐wide association study (GWAS) loci used to derive the GRS, 20 with methylation quantitative trait loci (meQTL) analysis supporting involvement of cis meQTLs. Only one differentially methylated position (DMP), cg14354618 on chromosome 11, was an exception to this pattern, being associated with trans meQTLs in a GWAS risk locus on chromosome 19. cg14354618 is located in a CpG island in AP001979.1. Genetic variation annotated to AP001979.1 has been associated with Parkinson's disease, 37 , 38 body fat percentage, 39 and sugar consumption 40 but has not been associated with AD in large‐scale GWASs. 41 , 42 There is a degree of overlap between the clinical features and pathologies associated with AD and Parkinson's disease, with certain genetic variants being associated with both. 43 , 44 Moreover, obesity and hyperglycemia have been implicated as dementia risk factors. 45 Taken together, these findings suggest this locus to be a plausible AD‐risk locus, which warrants further investigation.
Considering both the meta‐DMP and differentially methylated region (DMR) results, two regions harbor a large number of AD GRS‐associated sites. These regions contain (1) BIN1 and (2) PVRL2, APOE, APOC4, and APOC2 (henceforth referred to as the APOE locus). The APOE locus has not been identified previously by brain‐based epigenome‐wide association studies (EWASs) of AD neuropathological hallmarks 7 , 46 or a blood‐based AD case‐control EWAS 44 ; larger samples might be required to detect association between methylation at this locus and AD.
In contrast, several studies have identified altered methylation of BIN1 in AD patients or in association with AD neuropathological hallmarks. 7 , 47 , 48 These findings are of particular interest, as altered BIN1 brain expression has been reported in AD 49 , 50 , 51 and DNA methylation has been suggested to regulate BIN1’s expression. 52 We identified a mixture of hyper‐ and hypomethylation in the upstream region and gene body and hypermethylation in the downstream region. Although none of the identified sites directly replicated those identified by previous studies, it is noteworthy that one of our hypermethylated meta‐DMPs (cg18813565) is located only 31 bp from a site (that failed our quality control) at which increased methylation in the dorsolateral prefrontal cortex has been associated with neuritic plaque burden and AD diagnosis. 47 Moreover, this site contributes to a hypermethylated DMR that spans a 199 bp region located ≈23 kb downstream of BIN1. This region overlaps with non‐coding RNAs, RP11‐521O16.1, and RP11‐521O16.2, suggesting the possibility that altered methylation of these non‐coding RNAs might alter BIN1 expression. This hypothesis should be assessed by future studies.
The CAIDE1 risk score is a composite score formed by the weighted summation of age, sex, body mass index (BMI), years in education, systolic blood pressure, and total cholesterol. 2 It is designed for the prediction of dementia in 20 years' time in individuals 39 to 64 years of age, which it does with an area under the curve of 0.77 (95% confidernce interval 0.71 to 0.83). Because age and sex were covariates in our analytical models, the differential methylation observed in this study reflects the modifiable “lifestyle” components of the score. Our analyses revealed BMI to be the primary driver of the CAIDE1‐associated methylation differences. We identified significant overlap between the sites associated with CAIDE1 and those that have been associated previously with alcohol consumption. Strikingly, the most significant CpG in our analysis of CAIDE1 was also the most significant CpG in a recent Generation Scotland: Scottish Family Health Study (GS:SFHS) alcohol consumption EWAS (unpublished data, Clarke et al.). Although alcohol consumption was significantly correlated with CAIDE1 score, it could not account for the CAIDE1‐associated differences in methylation. This finding is of interest in light of the observed associations between excessive alcohol consumption and dementia risk. 53 Our findings suggest that the risk factors contributing to the CAIDE1 score and alcohol consumption might confer risk for dementia via independent effects on common pathways.
The loci implicated by our analyses of the AD GRS and the CAIDE1 score did not overlap, and they did not implicate common genes. In keeping with this, the correlation between the scores was small and non‐significant. This lack of concordance might be attributable to differences in the methodology used to create the scores: although the CAIDE1 score was trained using a sample comprising mixed dementia cases (of which ≈75% were diagnosed with AD), 2 the AD GRS was devised using a sample comprising AD and proxy AD cases. 20 Moreover, the CAIDE 1 score predominantly comprises cardiovascular risk factors for dementia, meaning that it is likely to identify a sub‐population of those at risk for dementia.
We did not observe any DNA methylation associations with AD family history (FH) or two late‐life dementia risk scores. The lack of associations with AD FH is somewhat surprising, as this has been shown previously to be a good AD proxy‐phenotype. 20 Our failure to observe significant associations for these traits may reflect a lack of statistical power, particularly as the samples available for the late‐life dementia risk scores were relatively small.
It is important to note some additional strengths and limitations to our study. Although it would clearly be desirable to study DNA methylation in brain tissue, growing evidence highlights the contribution of systemic factors to dementia pathogenesis. 54 Thus methylation studies in the blood are necessary to provide a holistic characterization of the processes that contribute to dementia development. Moreover, profiling blood methylation permits both longitudinal analyses to characterize the dynamic processes underlying dementia pathogenesis and biomarker identification.
An important limitation of our study is that the use of a cross‐sectional design means that causal inferences cannot be drawn. A corollary of this is that it is difficult to determine whether the methylation differences assessed play a causal role in the development of dementia. In some cases, causal inference analyses to assess relationships with important intermediary variables such as cognitive ability and cognitive decline together with Mendelian randomization may help delineate likely causality; future studies should assess this possibility.
Ultimately, the longitudinal assessment of cognitive decline and the development of dementia will also be necessary to address questions about causation. Moreover, the availability of longitudinal data would also permit the development of an epigenetic (and potentially a multifactorial genetic, epigenetic, and lifestyle factors) predictor of dementia. An important conceptual issue that must be considered when attempting to determine causality from longitudinal data is that that the pathogenesis of dementia is itself a gradual process involving quantitative changes in multiple biological systems, which eventually result in the binary diagnosis of dementia. As such, it might not be possible to strictly delineate the temporal relationship between risk factors, their biological correlates and the onset of dementia. Instead, the identification of co‐occurring processes might yield experimentally testable hypotheses. An additional factor to consider is that the non‐genetic risk factors that contribute to the scores assessed are themselves only associated with the development of dementia and do not necessarily play a causal role. Future studies that aim to delineate the causal contribution of these factors to dementia will facilitate the development of risk scores whose primary purpose is for use in the investigation of pathogenic mechanisms.
Other limitations concern the quality of the variables used in the dementia risk scores: self‐reporting may have resulted in errors, and the blood samples used for cholesterol measurements were not taken at a consistent time of day or after a consistent fasting length. Furthermore, we considered only a sub‐set of putative dementia risk factors. The demographic and lifestyle risk factors considered in this study were selected due to their involvement in validated composite risk scores; 2 , 3 , 4 however, it would be of interest to examine DNA methylation associations of other well‐supported dementia risk factors in future studies.
Here we present the first comprehensive characterization of associations between blood DNA methylation and dementia risk, performed in the largest single‐cohort methylation samples collected to date. We identify several CpGs where methylation is associated with dementia risk measures and identify a putative novel AD risk locus. Our findings suggest a number of hypotheses for assessment by future studies, which should include longitudinal assessments of the causal nature of methylation in dementia pathogenesis.
CONFLICTS OF INTEREST
Andrew M. McIntosh has received grant support from Pfizer, Eli Lilly, Janssen, and The Sackler Trust. These sources are not connected to the current investigation. Andrew M. McIntosh has also received speaker's fees from Janssen and Illumina. The remaining authors report no conflicts of interest.
Supporting information
Supplementary Information
Supplementary Information
Supplementary Information
Supplementary Information
Supplementary Information
Supplementary Information
Supplementary Information
Supplementary Information
Supplementary Information
Supplementary Information
Supplementary Information
Supplementary Information
Supplementary Information
Supplementary Information
Supplementary Information
Supplementary Information
Supplementary Information
Supplementary Information
ACKNOWLEDGMENTS
This work was supported by a Wellcome Strategic Award “STratifying Resilience and Depression Longitudinally” (STRADL; 104036/Z/14/Z) to AMM, KLE, CSH, DJP, and others, and an MRC Mental Health Data Pathfinder Grant (MC_PC_17209) to AMM and DJP. REM is supported by an Alzheimer's Research UK major project grant (ARUK‐PG2017B‐10). KV is funded by the Wellcome Translational Neuroscience PhD Programme at the University of Edinburgh (108890/Z/15/Z). ADB would like to acknowledge funding from the Wellcome PhD training fellowship for clinicians (204979/Z/16/Z), the Edinburgh Clinical Academic Track (ECAT) program. Generation Scotland received core support from the Chief Scientist Office of the Scottish Government Health Directorates (CZD/16/6) and the Scottish Funding Council (HR03006). Genotyping of the GS:SFHS samples was carried out by the Genetics Core Laboratory at the Clinical Research Facility, Edinburgh, Scotland and was funded by the UK's Medical Research Council and the Wellcome (104036/Z/14/Z). DNA methylation profiling of the GS:SFHS samples was funded by the Wellcome Strategic Award (10436/Z/14/Z), with additional funding from a 2018 NARSAD Young Investigator Grant from the Brain & Behavior Research Foundation (27404). We are grateful to all the families who took part, the general practitioners and the Scottish School of Primary Care for their help in recruiting them, and the entire Generation Scotland team, which includes interviewers, computer and laboratory technicians, clerical workers, research scientists, volunteers, managers, receptionists, healthcare assistants, and nurses.
Walker RM, Bermingham ML, Vaher K, et al. Epigenome‐wide analyses identify DNA methylation signatures of dementia risk. Alzheimer's Dement. 2020;12:e12078 10.1002/dad2.12078
Contributor Information
Rosie M. Walker, Email: rwalke13@staffmail.ed.ac.uk.
Kathryn L. Evans, Email: kathy.evans@igmm.ed.ac.uk.
REFERENCES
- 1. Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimer's Dementia:. 2011;7:280‐292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kivipelto M, Ngandu T, Laatikainen T, Winblad B, Soininen H, Tuomilehto J. Risk score for the prediction of dementia risk in 20 years among middle aged people: a longitudinal, population‐based study. Lancet Neurol. 2006;5:735‐741. [DOI] [PubMed] [Google Scholar]
- 3. Li J, Ogrodnik M, Devine S, Auerbach S, Wolf PA, Au R. Practical risk score for 5‐, 10‐, and 20‐year prediction of dementia in elderly persons: Framingham Heart Study. Alzheimer's Dementia. 2018;14:35‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Reitz C, Tang MX, Schupf N, Manly JJ, Mayeux R, Luchsinger JA. A summary risk score for the prediction of Alzheimer disease in elderly persons. Arch Neurol. 2010;67:835‐841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Xu M, Zhang DF, Luo R, et al. A systematic integrated analysis of brain expression profiles reveals YAP1 and other prioritized hub genes as important upstream regulators in Alzheimer's disease. Alzheimer's Dementia. 2018;14:215‐229. [DOI] [PubMed] [Google Scholar]
- 6. Hampel H, Vergallo A, Afshar M, et al. Blood‐based systems biology biomarkers for next‐generation clinical trials in Alzheimer's disease. Dialogues Clin Neurosci. 2019;21:177‐191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. De Jager PL, Srivastava G, Lunnon K, et al. Alzheimer's disease: early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat Neurosci. 2014;17:1156‐1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lunnon K, Smith R, Hannon E, et al. Methylomic profiling implicates cortical deregulation of ANK1 in Alzheimer's disease. Nat Neurosci. 2014;17:1164‐1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gasparoni G, Bultmann S, Lutsik P, et al. DNA methylation analysis on purified neurons and glia dissects age and Alzheimer's disease‐specific changes in the human cortex. Epigenetics Chromatin. 2018;11:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fransquet PD, Lacaze P, Saffery R, McNeil J, Woods R, Ryan J. Blood DNA methylation as a potential biomarker of dementia: a systematic review. Alzheimer's Dementia. 2018;14:81‐103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Madrid A, Hogan KJ, Papale LA, Clark LR, Asthana S, Johnson SC, et al. DNA Hypomethylation in blood links B3GALT4 and ZADH2 to Alzheimer's disease. J Alzheimer's Dis. 2018;66:927‐934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Foraker J, Millard SP, Leong L, et al. The APOE gene is differentially methylated in Alzheimer's disease. J Alzheimer's dis. 2015;48:745‐755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ma Y, Smith CE, Lai CQ, et al. Genetic variants modify the effect of age on APOE methylation in the genetics of lipid lowering drugs and diet network study. Aging Cell. 2015;14:49‐59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Slieker RC, Relton CL, Gaunt TR, Slagboom PE, Heijmans BT. Age‐related DNA methylation changes are tissue‐specific with ELOVL2 promoter methylation as exception. Epigenetics Chromatin. 2018;11:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wahl S, Drong A, Lehne B, Loh M, Scott WR, Kunze S, et al. Epigenome‐wide association study of body mass index, and the adverse outcomes of adiposity. Nature. 2017;541:81‐86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Smith BH, Campbell A, Linksted P, et al. Cohort profile: generation Scotland: Scottish Family Health Study (GS:sFHS). The study, its participants and their potential for genetic research on health and illness. IntJ Epidemiol. 2013;42:689‐700. [DOI] [PubMed] [Google Scholar]
- 17. Smith BH, Campbell H, Blackwood D, et al. Generation Scotland: the Scottish Family Health Study; a new resource for researching genes and heritability. BMC Med Genetics. 2006;7:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kerr SM, Campbell A, Murphy L, et al. Pedigree and genotyping quality analyses of over 10,000 DNA samples from the Generation Scotland: Scottish Family Health Study. BMC Med Genetics. 2013;14:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nagy R, Boutin TS, Marten J, Huffman JE, Kerr SM, Campbell A, et al. Exploration of haplotype research consortium imputation for genome‐wide association studies in 20,032 Generation Scotland participants. Genome Med. 2017;9:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Marioni RE, Harris SE, Zhang Q, et al. GWAS on family history of Alzheimer's disease. Transl Psychiatry. 2018;8:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Barbu MC, Walker RM, Howard DM, et al. Epigenetic prediction of major depressive disorder. medRxiv. 2019:19001123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Madden RA, McCartney DL, Walker RM, et al. Birth weight predicts psychiatric and physical health, cognitive function, and DNA methylation differences in an adult population. bioRxiv. 2019:664045. [Google Scholar]
- 23. Bermingham ML, Walker RM, Marioni RE, et al. Identification of novel differentially methylated sites with potential as clinical predictors of impaired respiratory function and COPD. EBioMedicine. 2019;43:576‐586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. R Core Team. A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2019. [Google Scholar]
- 25. Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA‐sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liu C, Marioni RE, Hedman AK, et al. A DNA methylation biomarker of alcohol consumption. Mol Psychiatry. 2018;23:422‐433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yousefi PD, Richmond R, Langdon R, et al. Validation and characterization of a DNA methylation alcohol biomarker across the life course. bioRxiv. 2019:591404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Saffari A, Silver MJ, Zavattari P, et al. Estimation of a significance threshold for epigenome‐wide association studies. Genet Epidemiol. 2018;42:20‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta‐analysis of genomewide association scans. Bioinformatics. 2010;26:2190‐2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Suderman M, Staley JR, French R, Arathimos R, Simpkin A, Tilling K. dmrff: identifying differentially methylated regions efficiently with power and control. bioRxiv. 2018:508556. [Google Scholar]
- 31. Phipson B, Maksimovic J, Oshlack A. missMethyl: an R package for analyzing data from Illumina's HumanMethylation450 platform. Bioinformatics. 2016;32:286‐288. [DOI] [PubMed] [Google Scholar]
- 32. Zeng Y, Amador C, Xia C, et al. Parent of origin genetic effects on methylation in humans are common and influence complex trait variation. Nat Commun. 2019;10:1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Haller T, Kals M, Esko T, Magi R, Fischer K. RegScan: a GWAS tool for quick estimation of allele effects on continuous traits and their combinations. Brief Bioinform. 2015;16:39‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Buniello A, MacArthur JAL, Cerezo M, et al. The NHGRI‐EBI GWAS catalog of published genome‐wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019;47:D1005‐D12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. MRC‐IEU. EWAS Catalog. Available at: http://www.ewascatalog.org/. Accessed July 3, 2019
- 36. Machiela MJ, LDlink Chanock SJ. a web‐based application for exploring population‐specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics. 2015;31:3555‐3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chang D, Nalls MA, Hallgrimsdottir IB, et al. A meta‐analysis of genome‐wide association studies identifies 17 new Parkinson's disease risk loci. Nat Genet. 2017;49:1511‐1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nalls MA, Pankratz N, Lill CM, et al. Large‐scale meta‐analysis of genome‐wide association data identifies six new risk loci for Parkinson's disease. Nat Genet. 2014;46:989‐993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hubel C, Gaspar HA, Coleman JRI, et al. Genomics of body fat percentage may contribute to sex bias in anorexia nervosa. Am J Med Genet B Neuropsychiatr Genet. 2019;180:428‐438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hwang LD, Lin C, Gharahkhani P, et al. New insight into human sweet taste: a genome‐wide association study of the perception and intake of sweet substances. Am J Clin Nutr. 2019;109:1724‐1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kunkle BW, Grenier‐Boley B, Sims R, et al. Genetic meta‐analysis of diagnosed Alzheimer's disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat Genet. 2019;51:414‐430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lambert JC, Ibrahim‐Verbaas CA, Harold D, et al. Meta‐analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet. 2013;45:1452‐1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Desikan RS, Schork AJ, Wang Y, et al. Genetic overlap between Alzheimer's disease and Parkinson's disease at the MAPT locus. Mol Psychiatry. 2015;20:1588‐1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nuytemans K, Maldonado L, Ali A, et al. Overlap between Parkinson disease and Alzheimer disease in ABCA7 functional variants. Neurol Genet. 2016;2:e44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Alford S, Patel D, Perakakis N, Mantzoros CS. Obesity as a risk factor for Alzheimer's disease: weighing the evidence. Obes Rev. 2018;19:269‐280. [DOI] [PubMed] [Google Scholar]
- 46. Lunnon K, Smith RG, Cooper I, Greenbaum L, Mill J, Beeri MS. Blood methylomic signatures of presymptomatic dementia in elderly subjects with type 2 diabetes mellitus. Neurobiol Aging. 2015;36:e1‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chibnik LB, Yu L, Eaton ML, et al. Alzheimer's loci: epigenetic associations and interaction with genetic factors. Ann Clin Transl Neurol. 2015;2:636‐647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Salcedo‐Tacuma D, Melgarejo JD, Mahecha MF, et al. Differential methylation levels in CpGs of the BIN1 gene in individuals with Alzheimer disease. Alzheimer Dis Assoc Disord. 2019;33:321‐326. [DOI] [PubMed] [Google Scholar]
- 49. Chapuis J, Hansmannel F, Gistelinck M, et al. Increased expression of BIN1 mediates Alzheimer genetic risk by modulating tau pathology. Mol Psychiatry. 2013;18:1225‐1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. De Rossi P, Buggia‐Prevot V, Clayton BL, et al. Predominant expression of Alzheimer's disease‐associated BIN1 in mature oligodendrocytes and localization to white matter tracts. Mol Neurodegener. 2016;11:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Glennon EB, Whitehouse IJ, Miners JS, et al. BIN1 is decreased in sporadic but not familial Alzheimer's disease or in aging. PLoS One. 2013;8:e78806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wechsler‐Reya R, Sakamuro D, Zhang J, Duhadaway J, Prendergast GC. Structural analysis of the human BIN1 gene. Evidence for tissue‐specific transcriptional regulation and alternate RNA splicing. J Biol Chem. 1997;272:31453‐31458. [DOI] [PubMed] [Google Scholar]
- 53. Xu W, Wang H, Wan Y, Tan C, Li J, Tan L, et al. Alcohol consumption and dementia risk: a dose‐response meta‐analysis of prospective studies. Eur J Epidemiol. 2017;32:31‐42. [DOI] [PubMed] [Google Scholar]
- 54. Pluvinage JV, Wyss‐Coray T. Systemic factors as mediators of brain homeostasis, ageing and neurodegeneration. Nat Rev Neurosci. 2020;21(2):93‐102. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information
Supplementary Information
Supplementary Information
Supplementary Information
Supplementary Information
Supplementary Information
Supplementary Information
Supplementary Information
Supplementary Information
Supplementary Information
Supplementary Information
Supplementary Information
Supplementary Information
Supplementary Information
Supplementary Information
Supplementary Information
Supplementary Information
Supplementary Information