

Abstract
Background: Estimating single nucleotide polymorphism (SNP)-heritability (h2g) of severe malaria resistance and its distribution across the genome might shed new light in to the underlying biology. Method: We investigated h2g of severe malaria resistance from a genome-wide association study (GWAS) dataset (sample size = 11 657). We estimated the h2g and partitioned in to chromosomes, allele frequencies and annotations using the genetic relationship-matrix restricted maximum likelihood approach. We further examined non-cell type-specific and cell type-specific enrichments from GWAS-summary statistics. Results: The h2g of severe malaria resistance was estimated at 0.21 (se = 0.05, P = 2.7 × 10−5), 0.20 (se = 0.05, P = 7.5 × 10−5) and 0.17 (se = 0.05, P = 7.2 × 10−4) in Gambian, Kenyan and Malawi populations, respectively. A comparable range of h2g [0.21 (se = 0.02, P < 1 × 10−5)] was estimated from GWAS-summary statistics meta-analysed across the three populations. Partitioning analysis from raw genotype data showed significant enrichment of h2g in genic SNPs while summary statistics analysis suggests evidences of enrichment in multiple categories. Supporting the polygenic inheritance, the h2g of severe malaria resistance is distributed across the chromosomes and allelic frequency spectrum. However, the h2g is disproportionately concentrated on three chromosomes (chr 5, 11 and 20), suggesting cost-effectiveness of targeting these chromosomes in future malaria genomic sequencing studies. Conclusion: We report for the first time that the heritability of malaria resistance is largely ascribed by common SNPs and the causal variants are overrepresented in protein coding regions of the genome. Further studies with larger sample sizes are needed to better understand the underpinning genetics of severe malaria resistance.
Introduction
In spite of the global eradication efforts, malaria remains a major global public health problem with 219 million cases and 435 000 deaths in 2017 (1). Plasmodium falciparum malaria results in diverse clinical manifestations ranging from asymptomatic parasitaemia to severe malaria (2). Such a wide variation of clinical outcome is attributed to several factors including genetic factors of the host, virulence of parasite and environmental factors (2). Family studies reported that the host genetic factors (heritability) contributes ~25% of the variations observed in clinical severity of malaria in endemic populations (3). Thus, understanding the molecular basis of the natural immunity against severe malaria will speed up the development of an efficient malaria vaccine.
Driven by the wide availability of genome-wide single nucleotide polymorphism (SNP) arrays, the focus of genetic studies has been shifted from the traditional candidate gene and family-based linkage studies to GWASs. However, only a small fraction of narrow sense heritability is explained by the GWAS significant SNPs. This led to a problem commonly termed as ‘missing heritability’ (4). In effort to address this problem, several statistical methods that aim at quantifying SNP-heritability without identifying the causal variants were developed (5–7).
Packages such as the genome-wide complex trait analysis (GCTA) implement genetic relationship-matrix restricted maximum likelihood (GREML) method in which all SNPs of unrelated subjects are simultaneously analysed in a linear mixed model framework to estimate the proportion of phenotypic variations explained by genotypic variation (5). Apart from GREML approaches, Golan et al. (7) recently introduced a regression method called phenotype-correlation-genotype-correlation (PCGC) for estimation of heritability from case-control datasets. PCGC is a Haseman–Elston regression model in which a normalized phenotype is regressed on the genetic covariances of all unique pair of samples. The slope of the regression is used as an estimator of h2g. Application of these methods provided new insights in to the genetic architecture of complex diseases including autism, schizophrenia, Parkinson’s disease, type 2 diabetes, hypertension among others (8–13). Alternative contemporary statistical methods that enable estimation of h2g from publicly available GWAS-summary statistics without the need of individual genotype data are also widely available (14–16) and gained popularity due to their privacy advantages and computational costs.
Because only unrelated individuals are included, the h2g analysis offers a greater flexibility of study designs that enable us to conduct powered studies. The use of only unrelated individuals also minimizes the biases from shared environments, one of the greatest challenges in pedigree studies (5). Furthermore, h2g analytic methods allow partitioning of the cumulative heritability in to different functional categories and biological pathways and thus, provide more insights in to the underpinning biology (6, 17).
Even though a number of severe malaria GWASs have recently been implemented in endemic areas in Africa and reported few novel association variants (18–21), little is known about the h2g and its distribution across the genome. Here we present results from a comprehensive h2g study of severe malaria resistance in three African populations including Gambia, Kenya and Malawi. We estimated h2g and partitioned in to chromosomes, different minor allele frequency (MAF) bins, functional categories and cell types. We found that the h2g is disproportionately concentrated on three chromosomes (chr 5, 11 and 20) and enriched in the coding region of genome. Overall, our results suggest that malaria resistance is mainly under polygenic control.
Results
SNP-heritability estimates from genotype datasets
We estimated h2g at different quality control (QC) levels to determine the appropriate threshold (see Materials and Methods). As expected, the estimates were inflated at less stringent QC thresholds (Supplementary Material, Fig. S1). However, applying stringent QC protocols including relatedness threshold (5%), SNP differential missingness proportion (P ≤ 1 × 10−3) and SNPs missing proportion (P > 0.02) yielded a more stable ranges of h2g values that were not affected by the inclusion of additional principal components (15, 20, 50) as covariates. At the stringent QC threshold, the h2g of severe malaria resistance was 0.21 (se = 0.05, P = 2.7 × 10−5), 0.20 (se = 0.05, P = 7.5 × 10−5) and 0.17 (se = 0.05, P = 7.2 × 10−4) in Gambian, Kenyan and Malawi populations, respectively (Table 1). These estimates were approximately similar in Kenya ethnic groups such as Chonye (0.20, se = 0.07, P = 5.1 × 10−3) and Giriama (0.19, se = 0.07, P = 7.3 × 10−3). However, the estimate was slightly inflated in Mandinka ethnic group of Gambia (0.24, se = 0.06, P = 5.1 × 10−5). We did not estimate h2g for other ethnic groups because of inadequate sample sizes. Furthermore, the PCGC model yielded broadly similar results including 0.20, (se = 0.06, P = 9.7 × 10−4), 0.16 (se = 0.06, P = 8 × 10−3) and 0.23 (se = 0.07, P = 1.3 × 10−3) in Gambia, Kenya and Malawi populations, respectively.
Table 1.
h2g of severe malaria resistance determined by GCTA and PCGC methods
| Population | SNPs (n) | Samples (n) | h2 g-GCTA (%) | h2 g-PCGC (%) |
|---|---|---|---|---|
| Gambia | 1 513 822 | 4128 | 0.20(se = 0.05) | 0.20(se = 0.05) |
| Kenya | 1 579 227 | 2062 | 0.20(se = 0.05) | 0.16 (se = 0.05) |
| Malawi | 1 502 462 | 2418 | 0.17(se = 0.05) | 0.23(se = 0.06) |
| Mandinka | 1 513 822 | 1281 | 0.24 (se = 0.06) | ne* |
| Chonye | 1 579 227 | 637 | 0.20(se = 0.06) | ne* |
| Giriama | 1 579 227 | 1173 | 0.19 (se = 0.06) | ne* |
ne*: Model did not fit because of small sample size and there was no reliable estimation
SNP: single nucleotide polymorphisms, h2g-GCTA: h2g estimated using GCTA method, h2g PCGC: h2g estimated using PCGC method
Proportion of SNP-heritability attributable to GWAS loci
To quantify the effects of the known variants, we estimated the h2g without removing the severe malaria GWAS loci from the datasets (see Materials and Methods). This resulted in slight increment of the h2g estimate in Gambian [0.27 (se = 0.05, P = 1 × 10−5)] and Kenyan [0.26 (se = 0.05, P < 1 × 10−5)] populations. Repeating the GREML analysis by including rs334 as an additional covariate decreased the estimate to [0.24 (se = 0.05, P < 1 × 10−5)] and [0.23 (se = 5%, P < 1 × 10−5)] in Gambian and Kenyan populations, respectively. This suggests that the h2g attributable to the GWAS significant loci and HbS locus is approximately 0.07 and 0.03, respectively.
Partitioning SNP-heritability by chromosomes, minor allele frequencies and functional annotations
We observed no significant differences in h2g estimate obtained from separate analysis (0.24, se = 0.05, P < 1 × 10−5) and the joint analysis (0.22, se = 0.05, P = 1 × 10−5) (Supplementary Material, Fig. S2), suggesting that the population structure was adequately controlled. Moreover, we observed significant correlations between chromosomal length and h2g per chromosome (Adj r2 = 0.38, P = 0.001) (Fig. 1). However, the estimates of three chromosomes (chr 5, 11 and 20) and three other chromosomes (chr 7, 8 and 15) fell above and below the expected h2g at 95% CI, respectively. Notably, chr5 contained a considerable proportion (~0.035) of the h2g (Supplementary Material, Table S1).
Figure 1.
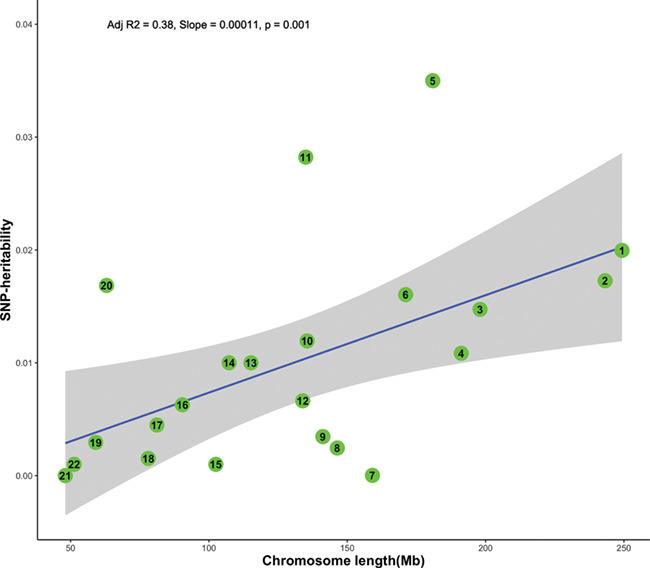
h2g per chromosome(y-axis) plotted against chromosome length (x-axis). The blue line represents the h2g estimates regressed against chromosome length. The grey shaded areas represent the 95% CI around the slope of the regression model.
We performed MAF stratified analysis to estimate the relative contribution of variants with various allele frequencies (Fig. 2). However, we did not find significant differences between the proportion of h2g attributed to different MAF bins [standards errors overlapped at 95% confidence interval (CI)]. Moreover, the total sum of our h2g estimate per bin [0.27 (se = 0.08, P = 5.3 × 10−5)] was not significantly different from the univariate estimate. We further estimated the h2g explained by genic SNPs and the intergenic SNPs at 0.165 (se = 0.05) and 0.062 (se = 0.05), respectively (Table 2). On average, a SNP residing in genic region was enriched 2.9× compared to a SNP residing in an intergenic region of the genome. This is statistically significant at 95% CI.
Figure 2.
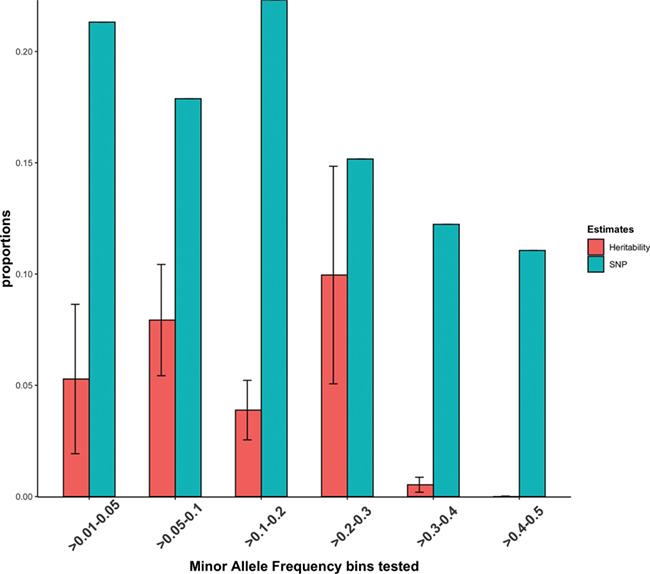
h2g partitioned in to different allele frequency spectrum. We created six MAF-bins and estimated the proportion of h2g attributed to each bin. The proportion of h2g attributed to each bin is shown in red bar and the proportion of SNPs per bin is shown by the blue bar. Error bars represent the 95% CI of the estimate.
Table 2.
h2g of severe malaria resistance partitioned in to genic and intergenic genomic regions
| SNP location | 10 kb boundary | ||
|---|---|---|---|
| SNPs(n) | h2g | h2g per SNP | |
| Genic | 727 996 | 0.165(se = 0.05) | 2.3 × 10−5 |
| Inter-genic | 785 826 | 0.062(se = 0.05) | 7.9 × 10−6 |
Functional enrichment from GWAS-summary statistics
After imputation of severe malaria GWAS-summary statistics and QC filtering, we obtained a total of 20 million high quality SNPs (see Materials and Methods). Using this dataset, we estimated the liability scale h2g at 0.21 (se = 0.02, P < 1 × 10−5). Partitioning the h2g in to 24 main genomic annotations (baseline model) showed evidences of enrichment in multiple categories including 5′UTR (11×), digital genomic footprint (DGF; 10×), enhancer (9×), coding (6×), H3K4me1 (4.9×), TSS (5×), transcription factor binding sites (TFBS; 4×) and FANTOM enhancer (4×) as shown in Figure 3. However, none of the enrichments was statistically significant after correction for multiple testing. Further cell-type specific and cell group analysis did not show significant enrichments.
Figure 3.
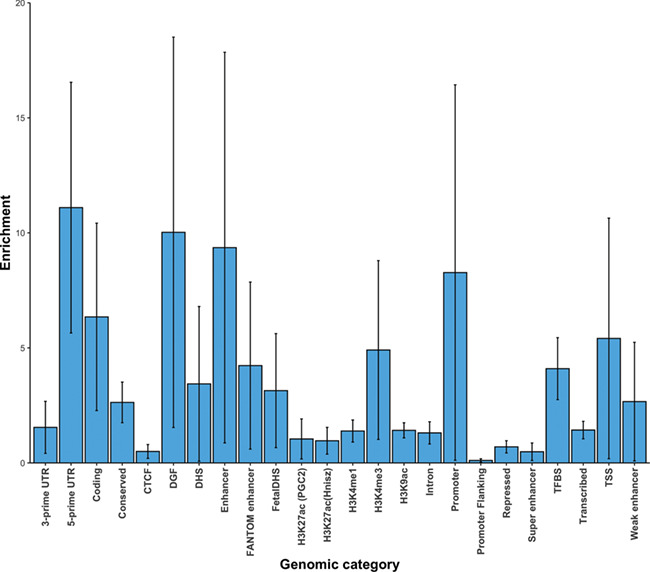
Enrichment estimates of h2g for the 24 main annotations. Error bars represent jackknife standard errors around the estimates of enrichment.
Discussions
In this study, we estimated the h2g and functional enrichment of malaria resistance in three African populations and their meta-analysis. After excluding the severe malaria resistance GWAS loci, we performed GREML analysis at different QC levels to determine the appropriate threshold; indeed, the estimates were inflated upward at less stringent QC levels and became stable at more stringent QC levels. These estimates were broadly similar across the three study populations. Except a slight inflation observed in Mandinka ethnic group which might have been underpowered because of small sample size, the estimates were also similar across the major ethnic groups. Approximately a similar range of h2g of severe malaria resistance was recently reported (22). This might suggest that substantial human genetic factors that influence malaria disease severity have been maintained across endemic populations. Consistent with our findings, a previous family-based study reported a similar range of heritability of severe malaria resistance in two different endemic populations in Kenya (4).
In contrast to the findings from other complex disease studies in which h2g is much smaller than family-based heritability values (23), our current h2g estimates were roughly close to a report from a previous family-based study (4). This might be due to the fact that the previous study underestimated the heritability estimates: First, only additive genetic effects (narrow-sense heritability) was calculated i.e. the contributions of nonadditive effects including epistasis, dominance and gene-gene interactions were not taken in to account. Second, the authors indicated that their paternity assessment was prone to misclassification, which might have underestimated the actual narrow-heritability estimate (4).
In the current study, including GWAS significant SNPs in the GREML analysis resulted in an increment of the h2g estimates by ~ 0.07 in the study populations, suggesting that the h2g attributable to the known malaria resistance GWAS loci (h2g-GWAS) is generally small. This is consistent with the hypothesis that the vast proportion of heritability of complex traits/diseases is explained by SNPs with effect sizes too small to attain the stringent genome wide significance threshold (P < 5 × 10−8) at the current sample sizes (24). Repeating the analysis by including rs334 as an additional covariate brought down these estimates by ~ 0.03, suggesting that more than a third of the h2g-GWAS is attributable to the HbS locus. This might be explained by the fact that the HbS locus has relatively larger effect sizes in the endemic populations (18).
To gain better insights in to the genetic architecture of severe malaria resistance, we partitioned the h2g in to different chromosomes, allele frequency spectrum and annotations. Separate GREML analysis and joint analysis yielded broadly similar h2g estimates, suggesting that population structure is adequately controlled. The rationale is that. However, the joint analysis in which genomic relatedness matrix (GRMs) of all chromosomes are simultaneously fitted in to a single GREML model, can effectively control the upward biases that can be created by correlated SNPs on different chromosomes (29).
Supporting the polygenic view of genetic architecture, we found a correlation between h2g per chromosome and chromosomal lengths (Adj r2 = 0.39, P = 0.001). However, the h2g is disproportionately concentrated on three chromosomes (chr 5, 11 and 20), suggesting that these chromosomes might contain loci with larger effects against the polygenic background. Thus, targeting these chromosomes using more powered studies (e.g. DNA sequencing) might be a cost-effective approach to discover new severe malaria resistance loci. Previous family-based studies reported that a region on chr5 (5q31–q33) is associated with susceptibility to mild malaria (25, 26).
MAF-stratified analysis didn’t reveal significant differences between the proportion of h2g attributed to different MAF bins. This might assert that h2g of severe malaria resistance is broadly uniform across the allele frequency spectrum and is not over-represented by rare alleles. Partitioning by annotation revealed that the h2g of severe malaria resistance is significantly enriched in SNPs residing in protein coding regions of the genome, suggesting that further studies focusing on coding regions (e.g. exome sequencing and/or exome array genotyping) might lead to the discovery novel variants.
In addition to the direct estimation of h2g from raw genotype datasets, we performed functional enrichment analysis from GWAS-summary statistics using stratified linkage disequilibrium score regression (LDSC) approach (14). To improve the performance of the analysis, we created a reference panel that is more specific to our study populations by merging the African population datasets obtained from 1000 Genomes Project and African Genome Variation Project (27). Using this panel, we created annotation files and estimated h2g of severe malaria resistance from GWAS-summary statistics meta-analysed across the study populations.
Our liability scale h2g estimate [0.21 (se = 0.02, P < 1 × 10−5)] was comparable to the direct estimates from raw genotype datasets. However, our functional enrichment analyses did not reveal significant results. One of the downsides of stratified LDSC method is that it requires very large sample sizes to detect significant enrichments (14). Of note, the coding genes and the surrounding categories were among the top annotations in our base line model. This further highlights the importance of protein coding regions of the genome in influencing the malaria disease severity.
Finally, our study had a number of caveats that might directly or indirectly affect the accuracy of estimating the true genetic heritability. First, the controls used in this study were not screened for mild malaria that might potentially bias the accuracy of h2g estimates. Second, assumptions of the models implemented for the analyses might not adequately explain the true genetic architecture of severe malaria resistance. Third, all the models implemented here do not measure the variances attributable to environmental factors. Fourth, the study is underpowered for the functional enrichment anlyses.
Conclusions
In conclusion, our study showed for the first time that heritability of severe malaria resistance is largely explained by common SNPs and is disproportionately enriched in SNPs residing in protein coding regions of the genome. Consistent with the polygenic genetic architecture, we observed that the h2g of severe malaria resistance is distributed across chromosomes and allele frequency spectrum. However, the h2g is disproportionately concentrated on three chromosomes (chr 5, 11 and 20), suggesting the cost-effectiveness of targeting these chromosomes in future malaria genomic sequencing studies. In this study, we created annotation files using population specific reference panel and showed that stratified LDSC analysis can provide reliable SNP-heritability estimates in African populations. Further studies with larger sample sizes are needed to understand the unpinning genetics and biology of severe malaria resistance trait.
Materials and Methods
Description of the study datasets
GWAS datasets of three African populations including Gambia, Kenya and Malawi were obtained from European Phenome Genome Archive (EGA) through the MalariaGen consortium standard data access protocols (28, 29). The datasets contain information about a total of 11 657 samples including 4921 samples from Gambia (2491 cases and 2430 controls), 3752 samples from Malawi (3752 cases and 3220 controls) and 2984 samples from Kenya (1506 cases and 1478 controls). Cases were obtained from children who were admitted to Hospitals and fulfilled WHO case definition for severe malaria (29) and controls were obtained from the general population (18–21). All the samples were genotyped on Illumina Omni 2.5 M array. Information about phenotypes, imputation and QC was also provided.
Quality control
The basic QC protocols including plate effects, sample relatedness, Hardy–Weinberg equilibrium, heterozygosity, missingness and differential missingness were done as described elsewhere (18, 30). Taking in to consideration that small artifacts can have substantial cumulative effects in h2g analysis (31), we applied further stringent QC filtering steps. Briefly, we aligned the quality filtered VCF files to forward stand of the human reference sequence (GRCh3) using the illumina supplied files (www.well.ox.ac.uk/~wrayner/strand) and removed all SNPs with position and strand mismatches. We further removed SNPs with MAF below 0.01, deviate from Hardy–Weinberg at P-value below 0.01 using PLINK software (32). We then implemented step-wise QC filtering based on SNPs missingness proportion, differential missingness and sample relatedness as described in (6).
Estimating heritability from genotype data
We applied GCTA (5) and PCGC (7) models to estimate the h2g of severe malaria resistance from raw genotype datasets. Briefly, we excluded the region of extended inversion (7 238 552–12 442 658) on chromosome 8p23 (33), the major histocompatibility complex (MHC) region (25 000 000–40,000,000) on chr 6 and the known severe malaria resistance loci including the ATP2B4 region on chr1:203 154 024–204 154 024, cluster of glycophorin (GYPA/B/E) region on chr4:143 000 000–146 000 000, ABO blood group region on chr9:135 630 000–136 630 000, and the sickle cell (HbS) region on chr11:2 500 000–6 500 000 to avoid potential biases from large effects.
We constructed GRMs from pruned high quality independent autosomal SNPs using GCTA software (5) and obtained list of samples with relatedness threshold >5%. We then computed GRMs using all the autosomal SNPs for each cohort and excluded one of any pair of samples with relatedness threshold >5% as recommended elsewhere (6). The final sample of unrelated individuals was 4128, 2062 and 2418 for Gambia, Kenya and Malawi, respectively. The distribution of off-diagonal element of the GRMs for each population is shown in Supplementary Material, Fig. S3.
We used population prevalence of 1% of severe malaria as previously described in (29) and included the top 10 PCs as fixed effects in the GREML analysis. We then transformed the estimates to liability scale as described in Lee et al. (34). Using the same GRMs, we estimated the h2g using PCGC model as outlined in Golan et al. (12). We also computed separate GRMs and estimated h2g for major ethnic groups in Gambia (Mandinka) and Kenya (Girimia and Chonye). Furthermore, we created GRMs in the presence of the GWAS significant SNPs and performed GREML analysis to quantify the effects of malaria resistance GWAS loci. We repeated the analysis by including rs334 as additional covariate to estimate the h2g attributable to HbS.
Partitioning SNP-heritability from genotype data
Using Gambian dataset (largest sample size), we partitioned h2g by chromosomes, MAF bins and annotations. For the partitioning analyses, we excluded the severe malaria resistance GWAS loci to minimize the potential biases from SNPs with large effects. To investigate the biases that might be created by population structure, we performed separate and joint GREML analysis using all autosomal chromosomes. We first computed GRMs for individual autosomal chromosome and estimated h2g attributed to each chromosome by separate GREML in which one chromosome is fitted to the model at a time. We then performed a joint analysis in which GRMs of all autosomal chromosomes are simultaneously fitted in to a single GREML analysis and compared the results obtained from both analyses.
In addition to this, we partitioned the h2g in to different allele frequencies and annotations. Briefly, we created five MAF bins including > 0.01–0.05, > 0.05–0.1, > 0.1–0.2, > 0.2–0.3, > 0.3–04, > 0.4–0.5, computed separate GRMs for each bin and performed joint GREML analysis. For partitioning by annotation, we mapped all the autosomal SNPs to the human reference panel hg19 in UCSC genome database (http://genome.ucsc.edu) using QCTOOLV2 (https://www.well.ox.ac.uk/~gav/qctool) and obtained a list of genic and intergenic variants. Genic variants included those SNPs mapped to genomic regions within 10 kilobases (kb) upstream and downstream of a protein coding gene. Intergenic variants included all the SNPs mapped to genomic regions outside 10 kb of a protein coding gene. We constructed separate GRMs and estimated h2g attributable to each category using the joint analysis implemented in GCTA software (5).
Functional enrichment analysis of SNP-heritability from GWAS-summary statistics
African-specific reference panel
Partitioning h2g in to cell-types and functional categories using stratified LDSC approach has recently been shedding new lights in to the genetic architecture of several complex diseases (14, 35, 36). The method is based on the fact that a given category of SNPs is enriched for h2g if SNPs with high LD to that category have higher χ2 statistics than SNPs with low LD to that category (35, 36). However, the stratified LDSC analysis require population specific reference panel and very large sample sizes to produce reliable results (14). Consequently, the current European 1000G haplotype reference panel that is used as a default in LDSC software (14, 35) might not well represent our study populations.
To address this challenge, we created a reference panel that matches with our study populations. Briefly, we merged African population datasets obtained from 1000 Genomes Project and African Genome Variation Project (27) based on overlapped variants and removed structural variants and ambiguous SNPs using plink tool (32). This resulted in a combined dataset of sample size (n = 4975). After excluding the admixed populations including Americans of African Ancestry and African Caribbean, we clustered the dataset in to East African and west African sub-regions using smart pca software (37) as shown in Supplementary Material, Fig. S4. We removed SNPs with MAF < 1%, missingness > 0.05 and HWE in controls (alpha level 0.0001), and retained a total of 22 473 268 SNPs (sample size = 2112) and 18 919 068 SNPs (sample size = 380) in east African and west African sub-regions, respectively. We finally calculated the MAF of the panel for later partitioning analysis. Owing to the fact that our study populations are comprised of both east African (Malawi and Kenya) and west Africa (Gambian) populations, we used the entire dataset as a reference panel for functional enrichment analysis.
Baseline model and functional annotations
We created baseline model and cell type specific annotations for our reference panel as described in (14). The baseline-LD model included 24 main annotations that are not cell type-specific including coding, UTRs (3′UTR and 5′UTR), promoter and intronic regions obtained from UCSC genome browser and processed by Gusev et al. (12), the histone marks (H3) such as: acetylation of histone at lysine 9 (H3K9ac), monomethylation (H3K4me1) and trimethylation (H3K4me3) of H3 at lysine 4 obtained from Trynka et al. (38), acetylation of H3 at lysine 27(H3K27ac) version one processed by Hnisz et al. (39) and version two Psychiatric Genomics Consortium, combined chromHMM and Segway predictions obtained from Hoffman et al. (40), regions that are conserved in mammals (41, 42), super enhancers (39), FANTOM5 enhancers (43), TFBS and DGF post-processed by Gusev et al (12). Around each partition, we added 500 bp windows as separate categories to prevent biases that might arise from adjacent annotations.
The 24 main annotations together with the additional windows and a category containing all SNPs yielded 53 overlapping baseline model. Next, we created 220 cell type-specific annotations for the four histone marks: H3K4me1, H3K4me3, H3K9ac and H3K27ac (14) using our reference panel and computed LD score for each annotation. We then combined the 120 cell specific annotations in to 10 cell groups including adrenal and pancreas, central nervous system, cardiovascular, connective and bone, gastrointestinal, immune and hematopoietic, kidney, liver, skeletal muscle and other as described in (14). For each of the 10 categories, we computed the corresponding LD scores.
Stratified LDSC analysis
We obtained meta-analysed GWAS-summary statistics of the three populations (n = 15 122 094 SNPs) from the previous GWAS (18). We performed imputation on this dataset using ImpG software (44). Briefly, we removed SNPs that mismatch with 1000G phase three markers, computed z-score from the association statistics and performed the imputation using ImGv.1.1 under default settings. We used all the 661 individuals labeled as ‘AFRICAN’ haplotypes in phase 1 of 1000 Genome Project version-3 calls (45). We removed all imputed SNPs with a predicted accuracy less than 0.9 and SNPs with MAF < 0.01. After QC filtering, we performed stratified LD score regression analysis using our reference panels as described in (14). Briefly, we converted the summary statistics to LDSC format, filtered SNPs with imputation accuracy greater than nine and MAF >1%, removed structural variants, ambiguous SNPs, the MHC region and significant SNPs. We then performed non-cell type- and cell type- specific analyses as described in (14).
Funding
National Research Foundation of South Africa for Funding (NRF) (grant RA171111285157/119056); The Centre for High-Performance Computing (CHPC, www.chpc.ac.za) for computing resources; The Developing Excellence in Leadership and Genetics Training for Malaria Elimination in sub-Saharan Africa (DELGEME) program (grant PD00217ML to D.D.); NIH projects (to E.C.).
Conflict of Interest statement. The authors declare no competing interests.
Authors Contributions
DD designed, performed the data analysis and drafted the manuscript, EC contributed in designing, data-analysis and revision of the manuscript and supervised the work.
Supplementary Material
Acknowledgements
We thank Kwiatkowski’s group from University of Oxford for their constructive comments and assistance. We thank Gavin Band for his advice and guidance in heritability analysis from genotype data. This work was supported through the DELTAS Africa Initiative [grant 107740/Z/15/Z]. The DELTAS Africa Initiative is an independent funding scheme of the African Academy of Sciences (AAS)‘s Alliance for Accelerating Excellence in Science in Africa (AESA) and supported by the New Partnership for Africa’s Development Planning and Coordinating Agency (NEPAD Agency) with funding from the Wellcome Trust [grant 107740/Z/15/Z] and the UK government. The views expressed in this publication are those of the author(s) and not necessarily those of AAS, NEPAD Agency, Wellcome Trust or the UK government.
References
- 1. World Health Organization (2018) World malaria report. World Health Organization, https://apps.who.int/iris/handle/10665/275867. [Google Scholar]
- 2. Kwiatkowski D.P. (2005) How malaria has affected the human genome and what human genetics can teach us about malaria. Am. J. Hum. Genet., 77, 171–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mackinnon M.J., Mwangi T.W., Snow R.W., Marsh K. and Williams T.N. (2005) Heritability of malaria in Africa. PLoS Med., 12, e340.10.1371/journal.pmed.0020340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Manolio T.A., Collins F.S., Cox N.J., Goldstein D.B., Hindorff L.A., Hunter D.J., Mccarthy M.I., Ramos E.M., Cardon L.R., Chakravarti A. et al. (2009) Finding the missing heritability of complex diseases. Nature, 461, 747–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yang J., Lee S.H., Goddard M.E. and Visscher P.M. (2011) GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet., 88, 76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Speed D., Cai N., Johnson M.R., Nejentsev S. and Balding D.J. (2017) Reevaluation of SNP heritability in complex human traits. Nat. Genet., 49, 986–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Golan D., Lander E.S. and Rosset S. (2014) Measuring missing heritability: inferring the contribution of common variants. Proc. Natl. Acad. Sci., 111, E5272–E5281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Klei L., Sanders S.J., Murtha M.T., Hus V., Lowe J.K., Willsey A.J., Moreno-de-luca D., Yu T.W., Fombonne E., Geschwind D. et al. (2012) Common genetic variants, acting additively, are a major source of risk for autism. Mol. Autism, 3(1), 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Keller M.F., Saad M., Bras J., Bettella F., Nicolaou N., Sharma M., Gibbs J.R., Simo J., Stefa H., Heutink P. et al. (2012) Using genome-wide complex trait analysis to quantify ‘missing heritability’ in Parkinson’ s disease. Hum.Mol. Genet, 21, 4996–5009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Loh P.-R., Bhatia G., Gusev A., Finucane H.K., Bulik-Sullivan B.K., Pollack S.J., Schizophrenia Working Group of Psychiatric Genomics Consortium, de Candia T.R., Lee S.H., Wray N.R. et al. (2015) Contrasting genetic architectures of schizophrenia and other complex diseases using fast variance-components analysis. Nat. Genet, 47, 1385–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee S.H., Yang J., Goddard M.E., Visscher P.M. and Wray N.R. (2012) Estimation of pleiotropy between complex diseases using single-nucleotide polymorphism-derived genomic relationships and restricted maximum likelihood. Bioinformatics, 28, 2540–2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gusev A., Lee S.H., Trynka G., Finucane H., Vilhja B.J., Xu H., Zang C., Ripke S., Bulik-sullivan B., Stahl E. et al. (2014) Partitioning heritability of regulatory and cell-type-specific variants across 11 common diseases. Am. J. Hum. Genet., 95, 535–55210.1016/j.ajhg.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. The Brian Consortium (2018) Analysis of shared heritability in common disorders of the brain. Science, 360, 8757–8769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Finucane H.K., Bulik-Sullivan B., Gusev A., Trynka G., Re-shef Y., Loh P.R., Anttila V., Xu H., Zang C., Farh K. et al. (2015) Partitioning heritability by functional annota- tion using genome-wide association summary statistics. Nat. Genet., 47, 1228–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Weissbrod O., Flint J. and Rosset S. (2018) Estimating heritability and GeneticCorrelation in case control studies directly and with summary statistics. Am. J. Hum. Genet., 103, 89–9910.1016/j.ajhg.2018.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Speed D. and Balding D.J. (2019) SumHer better estimates the SNP heritability of complex traits from summary statistics complex traits from summary statistics. Nat. Genet., 51, 277–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Visscher P.M., Macgregor S., Benyamin B., Zhu G., Gordon S., Medland S., Hill W.G., Hottenga J., Willemsen G., Boomsma D.I. et al. (2007) Genome partitioning of genetic variation for height from 11, 214 sibling pairs. Am. J. Hum. Genet., 81, 1104–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Band G., Rockett K.A., Spencer C.C.A., Kwiatkowski D.P., Band G., Si Le Q., Clarke G.M., Kivinen K., Leffler E.M., Rockett K.A. et al. (2015) A novel locus of resistance to severe malaria in a region of ancient balancing selection. Nature, 526, 253–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Timmann C., Thye T., Vens M., Evans J., May J., Ehmen C., Sievertsen J., Muntau B., Ruge G., Loag W. et al. (2012) Genome-wide association study indicates two novel resistance loci for severe malaria. Nature, 489, 443–446. [DOI] [PubMed] [Google Scholar]
- 20. Jallow M., Teo Y.Y., Small K.S., Rockett K.A., Deloukas P., Clark T.G., Kivinen K., Bojang K.A., Conway D.J., Pinder M. et al. (2009) Genome-wide and fine-resolution association analysis of malaria in West Africa. Nat Genet, 41, 657–66510.1038/ng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ravenhall M., Campino S., Sepu N., Nadjm B., Mtove G., Wangai H., Maxwell C., Olomi R., Reyburn H., Drakeley C.J. et al. (2018) Novel genetic polymorphisms associated with severe malaria and under selective pressure in North-Eastern Tanzania. PLoS Genet., 14, e1007172.https://doi.org/10.1371/journal. pgen.1007172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Malaria Genomic Epidemiology Network Consortium (2019) New insights into malaria susceptibility from the genomes of 17,000 individuals from Africa, Asia, and Oceania. bioRxiv. 10.1101/535898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mayhew A.J. and Meyre D. (2017) Assessing the heritability of complex traits in humans: methodological challenges and opportunities. Current Genom, 18, 332–34010.2174/1389202918666170307161450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Visscher P.M., Brown M.A., Mccarthy M.I. and Yang J. (2012) Five years of GWAS discovery. Am. J. Hum. Genet., 90, 7–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Flori L., Sawadogo S., Esnault C., Fre N., Fumoux F. and Rihet P. (2003) Linkage of mild malaria to the major histocompatibility complex in families living in Burkina Faso. Hum. Mol. Genet., 12, 375–378. [DOI] [PubMed] [Google Scholar]
- 26. Brisebarre A., Kumulungui B., Sawadogo S., Atkinson A., Garnier S., Fumoux F. and Rihet P. (2014) A genome scan for plasmodium falciparum malaria identifies quantitative trait loci on chromosomes 5q31, 6p21.3, 17p12, and 19p13. Malar. J., 13, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gurdasani D., Carstensen T., Tekola-Ayele F., Pagani L., Tachmazidou I., Hatzikotoulas K., Karthikeyan S., Iles L., Pollard M.O., Choudhury A. et al. (2015) The African genome variation project shapes medical genetics in Africa. Nature, 517, 327–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Parker M., Bull S.J., Vries J., Agbenyega T., Doumbo O.K. and Dominic P. (2009) Ethical data release in genome-wide association studies in developing countries. PLoSMed, 6, 11e1000143.10.1371/journal.pmed.1000143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Achidi E.A., Agbenyega T., Allen S., Amodu O., Bojang K., Conway D., Corran P., Deloukas P., Djimde A., Dolo A. et al. (2008) A global network for investigating the genomic epidemiology of malaria. Nature, 456, 732–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Band G., Le Q.S., Jostins L., Pirinen M., Kivinen K., Jallow M., Sisay-Joof F., Bojang K., Pinder M., Sirugo G. et al. (2013) Imputation-based meta-analysis of severe malaria in three African populations. PLoS Genet, 10.1371/journal.pgen.1003509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Speed D., Hemani G., Johnson M.R. and Balding D.J. (2012) Improved heritability estimation from genome-wide SNPs. Am. J. Hum. Genet., 91, 1011–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A.R., Bender D., Maller J., Sklar P., de P.I.W., Daly M.J. et al. (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet., 81, 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Antonacci F., Kidd J.M., Marques-bonet T., Ventura M., Siswara P., Jiang Z. and Eichler E.E. (2009) Characterization of six human disease-associated inversion polymorphisms. Hum. Mol. Genet., 18, 2555–256610.1093/hmg/ddp187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lee S.H., Wray N.R., Goddard M.E. and Visscher P.M. (2011) Estimating missing heritability for disease from genome-wide association studies. Am. J. Hum. Genet., 88, 294–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jiang X., Finucane H.K., Schumacher F.R., Schmit S.L., Tyrer J.P., Han Y., Michailidou K., Lesseur C., Kuchenbaecker K.B., Dennis J. et al. (2019) Shared heritability and functional enrichment across six solid cancers. Nat.Commun, 10, 431–45410.1038/s41467-018-08054-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gazal S., Finucane H.K., Furlotte N.A., Loh P. and Palamara P.F. (2017) Linkage disequilibrium dependent architecture of human complex traits shows action of negative selection. Nat Genet., 49, 1421–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Trynka G., Sandor C., Han B., Xu H., Stranger B.E., Liu X.S. and Raychaudhuri S. (2013) Chromatin marks identify critical cell types for fine mapping complex trait variants. Nat Genet., 45, 124–13010.1038/ng.2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sigova A.A., Hnisz D., Abraham B.J., Lee T.I., Lau A., Saint-andre V., Hoke H.A. and Young R.A. (2013) Resource super-enhancers in the control of cell identity and disease. Cell., 155, 934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hoffman M.M., Ernst J., Wilder S.P., Kundaje A., Harris R.S., Libbrecht M., Giardine B., Ellenbogen P.M., Bilmes J.A., Birney E. et al. (2013) Integrative annotation of chromatin elements from ENCODE data. Nuc.Ac.Res., 41, 827–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lindblad-toh K., Garber M., Zuk O., Lin M.F., Parker B.J., Washietl S., Kheradpour P., Ernst J., Jordan G., Mauceli E. et al. (2011) A high-resolution map of human evolutionary constraint using 29 mammals. Nature, 478, 476–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ward L.D. and Kellis M. (2012) Evidence of abundant purifying selection in humans for recently acquired regulatory functions. Sci., 337, 1675–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Andersson R., Gebhard C., Miguel-escalada I., Hoof I., Bornholdt J., Boyd M., Chen Y., Zhao X., Schmidl C., Suzuki T. et al. (2014) An atlas of active enhancers across human cell types and tissues. Nature, 507, 455–46110.1038/nature12787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pasaniuc B., Zaitlen N., Shi H., Bhatia G., Gusev A., Pickrell J., Hirschhorn J., Strachan D.P., Patterson N. and Price A.L. (2014) Fast and accurate imputation of summary statistics enhances evidence of functional enrichment. Bioinformatics, 30, 2906–291410.1093/bioinformatics/btu416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. The 1000 Genomes Project Consortium (2011) A map of human genome variation from population scale sequencing. Nature, 467, 1061–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.