

Abstract
Mutations in sonic hedgehog (SHH) signaling promote aberrant proliferation and tumor growth. SHH-medulloblastoma (MB) is among the most frequent brain tumor among children less than three years of age. While key components of the SHH-pathway are well known, we hypothesized that new candidate drivers of SHH)-driven medulloblastoma might be identified from databases and systems analysis. Using a data-driven systems biology approach a medulloblastoma-specific interactome was built. We discovered ABCC4, an ATP-binding cassette transporter, as a candidate driver of SHH-medulloblastoma. Accordingly, increased ABCC4 expression was related to the poor overall survival of SHH-MB patients. Further, knockdown of ABCC4 expression markedly blunted the constitutive activation of the SHH pathway secondary to Ptch1 or Sufu insufficiency. In a clinically relevant murine SHH medulloblastoma model, targeted ablation of Abcc4 in primary tumors significantly reduced tumor burden and extended the lifespan of tumor-bearing mice. Our studies reveal ABCC4 as a potent SHH pathway regulator and a new candidate target with the potential to improve SHH-medulloblastoma.
Significance:
ABCC4 is a novel regulator of the SHH pathway, that when knocked down, markedly improves survival in a mouse model of SHH-medulloblastoma. ABCC4 appears to be new potential target to treat SHH-medulloblastoma.
INTRODUCTION
Medulloblastoma (MB) is the most common malignant pediatric brain tumor for which standard of care therapy includes tumor resection followed by radiation and chemotherapy (Ramaswamy and Taylor, 2017). However, in children, the use of craniospinal irradiation has been deemed unacceptable due to the developmental side-effects and chemotherapeutic regimens have been prioritized. In the Sonic hedgehog (SHH) MB, patients with TP53 mutations and MYCN amplification exhibit poor clinical outcome likely due to de-novo resistance, representing the highest risk form of SHH-MB (Zhukova et al., 2013). While initially promising, therapy targeted at the key SHH activator, Smoothened (SMO), was shown ineffective as SHH-MB acquired therapy-induced SMO mutations, producing drug resistance and ultimately relapse (Atwood et al., 2015; Yauch et al., 2009). An additional liability that limited the broad implementation of the SMO inhibitor, vismodegib, was that children exposed to prolonged treatment developed irreversible growth plate fusions (Robinson et al., 2017). As SHH cancers frequently harbor tumorgenic SHH pathway mutations downstream of SMO (e.g., SUFU (Taylor et al., 2002)) new targets amenable to manipulation are needed. To identify candidate new regulators that restrain or block the SHH pathway we screened publicly available data sets to identify new candidate drivers of SHH-MB. The studies described herein identify a previously unknown regulator of the Sonic Hedgehog pathway, the ABC transporter, ABCC4.
RESULTS
ABCC4 is highly expressed in SHH-driven medulloblastoma
Human MB has been stratified, based on molecular signature, into four primary subgroups, WNT (Wingless), SHH (Sonic hedgehog), Group 3, and Group 4. Interrogating publicly available datasets on MBs, we found ABCC4 was the only ABC transporter with specific high expression in the SHH subgroup (Supplementary Figure 1A,1A, S1B). The DNA copy number for the ABCC4 gene was unchanged (Supplementary Figure 1C). Gene expression analysis of human MB revealed that ABCC4, unlike other ABC transporters found in the brain, was uniquely co-expressed with SHH pathway genes such as GLI1–3 (Figure 1B). Using a large human MB gene expression dataset (Cavalli et al., 2017), we evaluated the link between ABCC4 expression and survival in the primary MB subgroups. High ABCC4 expression was significantly correlated with reduced overall survival of SHH-MB patients (P=0.0025). Other MB subgroups did not show this relationship (Figure 1C, S1D).
Figure 1. ABCC4 is highly expressed in SHH-MB.
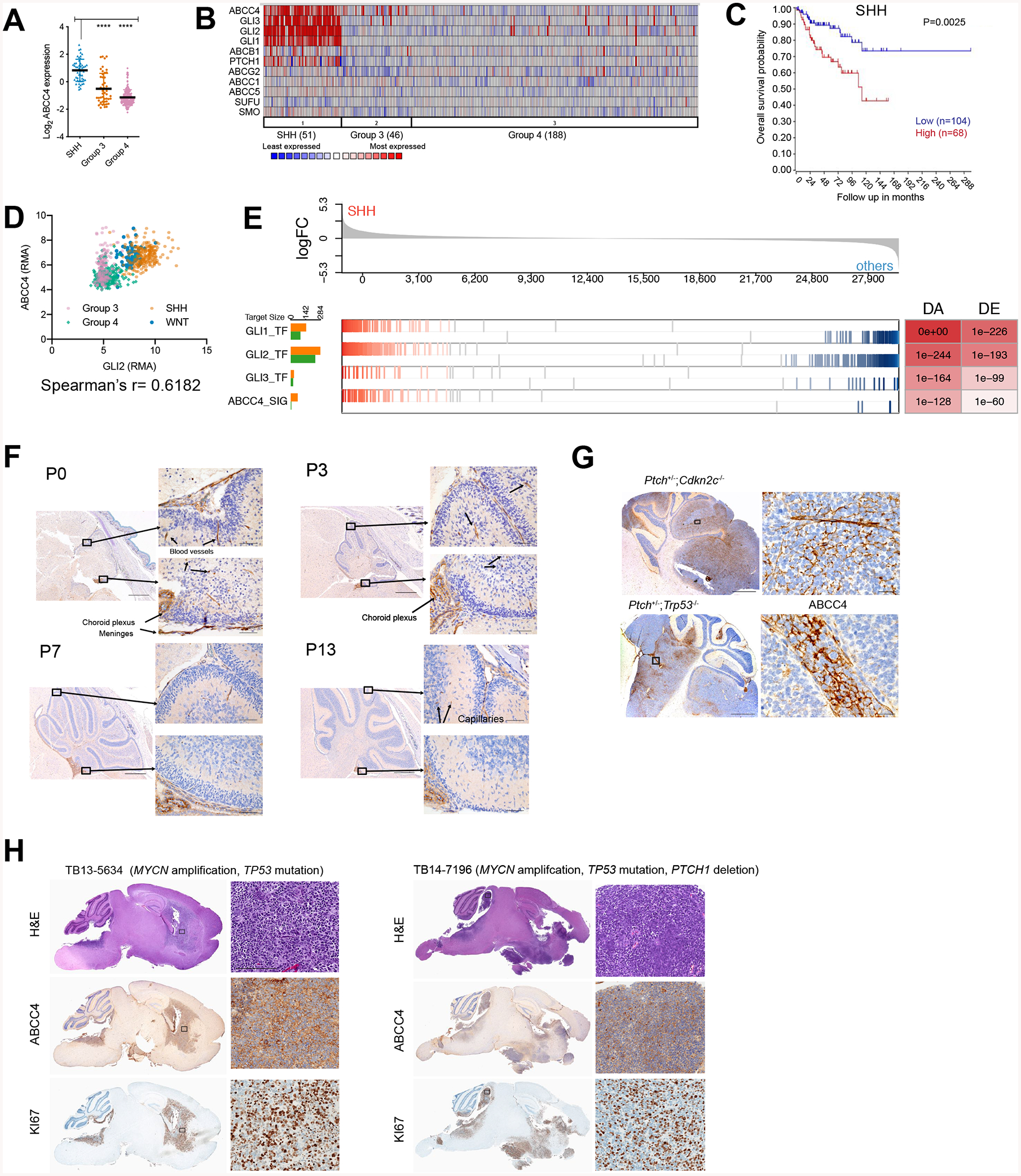
(A) Abcc4 expression in MB cohort from GSE37385, n= 1087 (Northcott et al., 2012b). * P < 0.05, ** P < 0.01, **** P < 0.0001, one-way ANOVA.
(B) Data from (A) where ABCC4 expression and SHH signaling genes are compared in a heatmap.
(C) SHH-MB patients with available survival and gene expression information from GSE85217, n = 763 (Cavalli et al., 2017). The relationship between ABCC4 expression and survival time was analyzed using the Kaplan-Meier method with log-rank statistics.
(D) Analysis of correlation between GLI2 and ABCC4 expression levels from GSE85217 data, n =763. Spearman’s correlation r was 0.6182, P<0.0001.
(E) Enrichment of predicted GLI1, GLI2, GLI3 transcriptional regulons and ABCC4 signaling regulon in differentially expressed genes between SHH and other MB subgroups from GSE85217, n - 763 (Cavalli et al., 2017). P values of regulon enrichment and differential expression of the hub gene are indicated in the DA DE table.
F) Wild type mouse brains were fixed at indicated post-natal days (P0, P3, P7, and P10) and immunostained with antibodies to mouse ABCC4 (anti-ABCC4). Scale bars = 5 mm and 50 μm
(G) Sagittal sections of SHH-MB mouse models (Ptch+/−;Cdkn2c−/−, and Ptch+/−;Trp53−/−) immunostained with anti-ABCC4. Scale bar = 5 mm and 25 μm.
(H) Sagittal sections of SHH-MB patient-derived xenografts. Sections were stained with H&E and with anti-KI67 and anti-ABCC4. Scale bar = 5 mm and 200 μm.
The strong upregulation of ABCC4 mRNA in SHH-MB led us to investigate if the GLI transcription factors targeted ABCC4. Publicly available datasets (GSE85217) revealed a positive correlation between GLI2 and ABCC4 mRNA levels (Figure 1D). Notably, the highest expression of ABCC4 was found in the SHH subgroup where the level of GLI2 was also the highest (Figure 1D). In parallel with the expression correlation analyses, we used NetBID (Du et al., 2018), a data-driven systems biology approach to reconstruct a medulloblastoma-specific interactome (MBi) using microarray gene expression profiles of 383 medulloblastoma patients collected from published datasets (Jones et al., 2012; Kool et al., 2014; Northcott et al., 2014; Robinson et al., 2012) with the same platform ([HG-U133_Plus_2]). MBi identified ABCC4 as a driver for SHH-MB, with the predicted GLI and ABCC4 regulons being significantly enriched in SHH subgroup from Cavalli’s MB dataset (Figure 1E). In the same dataset, GLIs and ABCC4 MBi inferred activities are significantly higher in SHH-MB (Supplementary Figure 1E) with a strong positive correlation for both inferred activities and mRNA level (Supplementary Figure 1F, 1D).
We further identified conserved GLI2 binding sites within the ABCC4 gene. These sites coincide with active enhancer marks (H3K27ac peaks) in MB patients, providing further support for GLI2 contribution to ABCC4 regulation. (Supplementary Figure 1G). The contribution of GLI2 to ABCC4 expression was investigated in Ptch−/− cells to bypass the impact of SHH ligand mediated activation. Ptch−/− MEFs were treated with GANT61, an SHH pathway inhibitor that blocks GLI-mediated transcription by preventing GLI binding to DNA (Lauth et al., 2007). GANT61 treatment dose-dependently suppressed GLI1 with ABCC4 showing a similar dose-dependent suppression, thus supporting the idea that ABCC4 is upregulated by constitutive SHH activation (Supplementary Figure 1H).
Because activation of the SHH pathway is critical for granule neuron progenitors (GNPs) proliferation and migration during cerebellar development (Roussel and Hatten, 2011), we interrogated the murine cerebellum during postnatal development (P0, P3, P7, and P13) for ABCC4 protein expression using immunohistochemistry (IHC). At the peak SHH activity in the developing cerebellum, postnatal day 7 (P7), immunochemical ABCC4 was barely detected, but as expected it was expressed in meninges and in the choroid plexus (Leggas et al., 2004) (Figure 1F). ABCC4 expression in the cerebellum was generally very low at the start of SHH signaling (P0) all the way through the termination of signaling (P13) (Figure 1F). In contrast, in two different mouse models of SHH-MB (Ptch+/−;Cdkn2c−/− and Ptch+/−;Trp53−/−) (Uziel et al., 2005), ABCC4 was robustly expressed at the plasma membrane (Figure 1G). Furthermore, strong ABCC4 immunoreactivity was observed in patient-derived orthotopic xenografts (PDOXs) of SHH-MB (Smith et al., 2019) (Figure 1H). These results showing high expression of ABCC4 in SHH-MB are consistent with regulation by GLI transcription factors.
ABCC4 is a positive regulator of SHH signaling
To determine if the amount of ABCC4 affects the canonical ligand-activated SHH signaling, we overexpressed human ABCC4 in NIH3T3 cells, an SHH-responsive model. Enforced expression of ABCC4 alone is not sufficient to relieve PTCH repression of SMO to activate the pathway (Figure 2A). In contrast, with an agonist that binds SMO (SAG), ABCC4 potentiates GLI1 expression in a dose-dependent manner (Figure 2A).
Figure 2. Abcc4 is a positive regulator of SHH signaling.
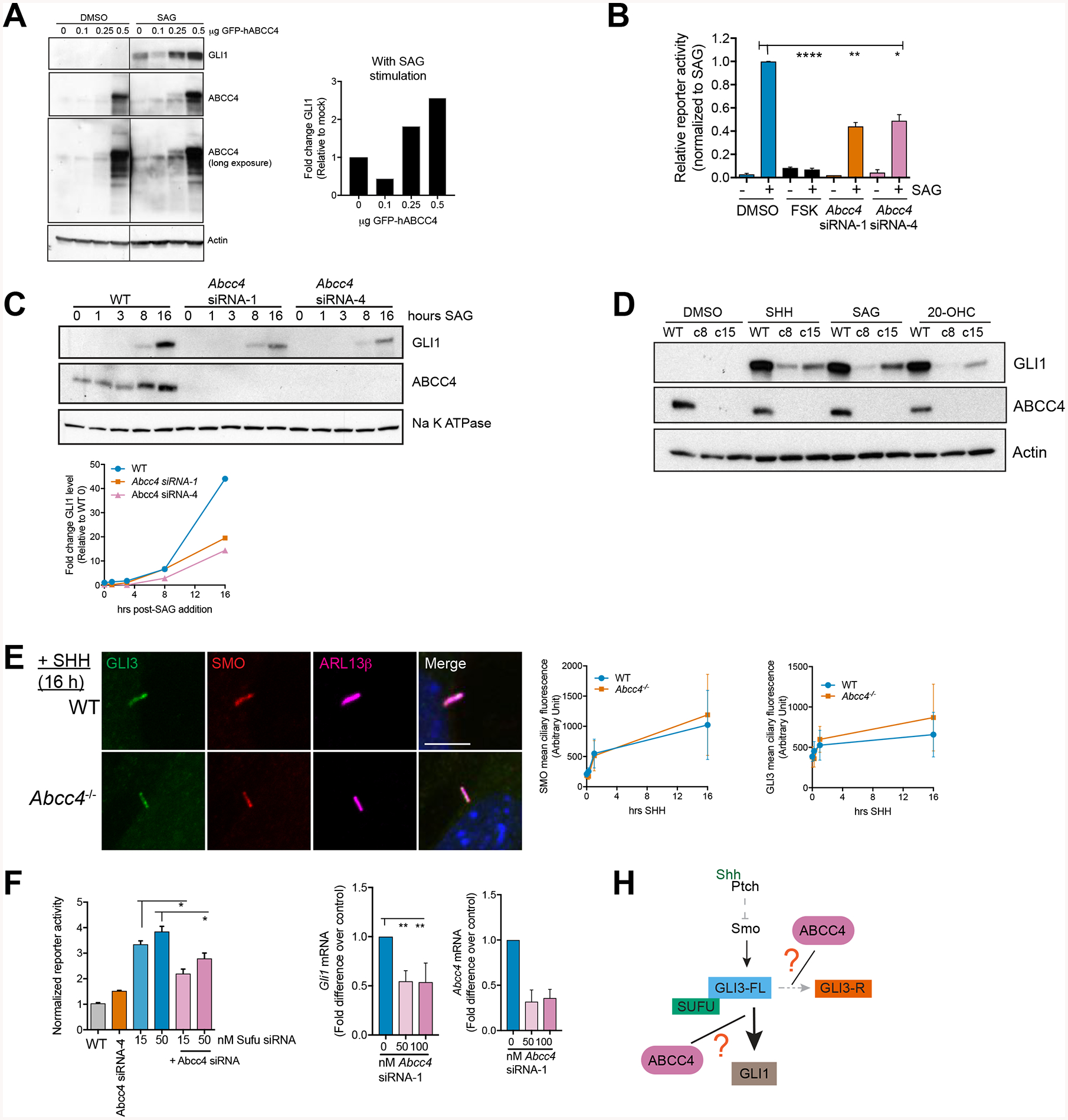
(A) NIH3T3 cells were transfected with plasmid encoding human ABCC4 and treated with SAG (200 nM) for 16 hours. GLI1 protein level was quantified by densitometry and normalized to the actin loading control (bar graph). Blot is of the same exposure time where irrelevant lanes were removed and marked with black line.
(B) Light2 cells expressing Gli1 reporter construct were transfected with Abcc4 siRNA (15 nM), forskolin/FSK (50 μM), or DMSO control for 24 hours prior to addition of SAG (200 nM). Bars are mean (± SEM) of three independent experiments. * P < 0.05, ** P < 0.01, **** P < 0.0001. One-way ANOVA.
(C) NIH3T3 cells were transfected with Abcc4 siRNAs (15 nM). Cells were harvested at indicated time points post-SAG addition (200 nM). Represenative blot of at least three independent experiments. Levels of indicated proteins were measured by immunoblotting. Na K ATPase was used as a loading control. Lines represent band intensities of blots shown. GLI1 was normalized to DMSO-treated control of WT cells.
(D) WT or clonal Abcc4−/− NIH3T3 were treated with SHH, SAG (200 nM), or 20-OHC (10 uM) for 16 hours. Levels of indicated proteins were measured by immunoblotting. All blots are representative of at least three independent experiments.
(E) WT or Abcc4−/− NIH3T3 cells were treated SAG (200 nM) for 16 hours. Cells were fixed, permeabilized, and stained with SMO, GLI3, and ciliary GTPase, Arl13b. Representative images and quantification are shown. Bars are mean (± SD) of n=40–50 cells/group. Scale bar = 5 μm.
(F) Light2 Gli reporter cells were transfected with indicated Abcc4 and Sufu siRNAs for 24 hours prior reporter activity measurement. Bars are mean (± SD) of a representative experiment. * P < 0.05. One-way ANOVA.
(G) SufuKD cells were transfected with control or Abcc4 siRNA. RNA was isolated 16 hours post-serum starvation. Levels of indicated transcripts were measured by qRT-PCR. Means (± SEM) of two independent experiments are shown. ** P < 0.01. One-way ANOVA.
(H) Schematic of ABCC4 locus in the SHH pathway. ABCC4 functions downstream of SMO where it may affect GLI3-FL to GLI-R conversion or it may act downstream of SUFU.
As ABCC4 overexpression enhances SHH output, we tested if the converse was true: Does Abcc4 absence impair SHH transcription? Abcc4 knockdown by siRNA decreased Gli1 transcription as determined with a cell line expressing Gli1 reporter construct (Light2 cells) (Figure 2B). Furthermore, using various agonists, we investigated where Abcc4 affects the SHH pathway. Wildtype (WT), Abcc4 siRNA-transfected NIH3T3, or Abcc4-null cells were treated with SHH-conditioned media to activate the pathway at the level of PTCH. SAG and 20-OHC act downstream of PTCH by directly binding to SMO. Irrespective of the agonist’s mechanism of SHH pathway activation, ABCC4 absence impaired signaling output (GLI1) at both the mRNA and protein level (Figure 2C,D, Supplementary Figure 2). Furthermore, level of other downstream SHH target genes, including Ptch1 and Ccnd1, were also suppressed by Abcc4-deficiency (Supplementary Figure 2). Together, these results suggest ABCC4 is required for optimal SHH pathway activation and that SMO activation is a necessary prerequisite for ABCC4 to elicit its positive effect on the pathway.
Binding of SHH to PTCH triggers translocation and enrichment of SMO and GLI3 in primary cilia, which promotes formation of activated GLI3 (Pak et al., 2016). Blockade of SMO translocation to primary cilia has been shown to disrupt GLI activation (Wu et al., 2012). Additionally, translocation and enrichment of SMO and GLI3 at primary cilia has been reported to occur within minutes of ligand addition (Wen et al., 2010). We sought to determine if ABCC4 affected pathway activation at this step. The rate of GLI3 and SMO translocation in Abcc4-null cells was almost identical to WT (Figure 2E), suggesting the ciliary translocation and enrichment step, a prerequisite for canonical ligand-dependent pathway activation, does not depend on ABCC4.
SUFU associates with GLI3 to restrain its nuclear entry thereby blocking its ability to activate transcription (Humke et al., 2010). To determine if ABCC4 genetically interacts with SUFU to influence SHH signaling, we performed knockdown of Sufu in a NIH3T3 cell line harboring a stable Gli transcriptional reporter gene. The resulting high luciferase activity was consistent with the expected Gli1-mediated transcriptional activation that was promoted by the absence of SUFU (Figure 2F). Next, Abcc4 knockdown was superimposed on the Sufu knockdown (SufuKD) cells. Gli1 reporter activity was further reduced by knockdown of Abcc4 (Figure 2F). Knockdown of Abcc4 in SufuKD cells also reduced the expression of the endogenous Gli1 mRNA indicating Abcc4 knockdown is not simply affecting the Gli reporter construct (Figure 2G). In total, these results indicate that ABCC4 modulates the SHH pathway post-ciliary translocation of SMO and GLI3 and suggest it might modulate either GLI3-FL conversion to repressor (GLI3-R) or act downstream of SUFU (Figure 2H).
ABCC4 is required to maintain nuclear GLI3-FL level
Upon pathway activation, GLI3-A, a labile species, enters the nucleus, acting as a transcriptional activator (Humke et al., 2010; Wen et al., 2010). An investigation into whether ABCC4 affects either the kinetics and/or the distribution of nuclear and cytoplasmic GLI3 was performed by subcellular fractionation. Enrichment of the nuclear fraction was confirmed by the nuclear marker, Lamin A/C. As expected, the shorter, repressor form of GLI3, GLI3-R was mostly found in the nucleus (Figure 3A). Notably, in the absence of Abcc4, at 16 hours post SAG treatment, GLI3-FL was almost undetectable in the nucleus (requiring a long exposure for detection) unlike WT cells (Figure 3A). Nuclear GLI3-FL appeared to accumulate time-dependently in WT cells after SAG treatment whereas in Abcc4-deficient cells it only appeared transiently at 2 hours post-agonist addition (Figure 3A, B). GLI3-R distribution was comparable between WT and siRNA-treated cells throughout the time course (Figure 3B). This result indicates that GLI3-FL nuclear translocation is not defective in Abcc4-deficient cells. Importantly, Gli3 mRNA level is similar to WT cells at 16 hours, suggestive of a post-translational effect on GLI3-FL (Supplementary Figure 2). Notably, compared to WT, ABCC4 only alters the cytosolic and nuclear distribution of only GLI3-FL with little impact on the distribution of GLI3-R.
Figure 3. ABCC4 absence alters nuclear GLI3-FL level and stability.
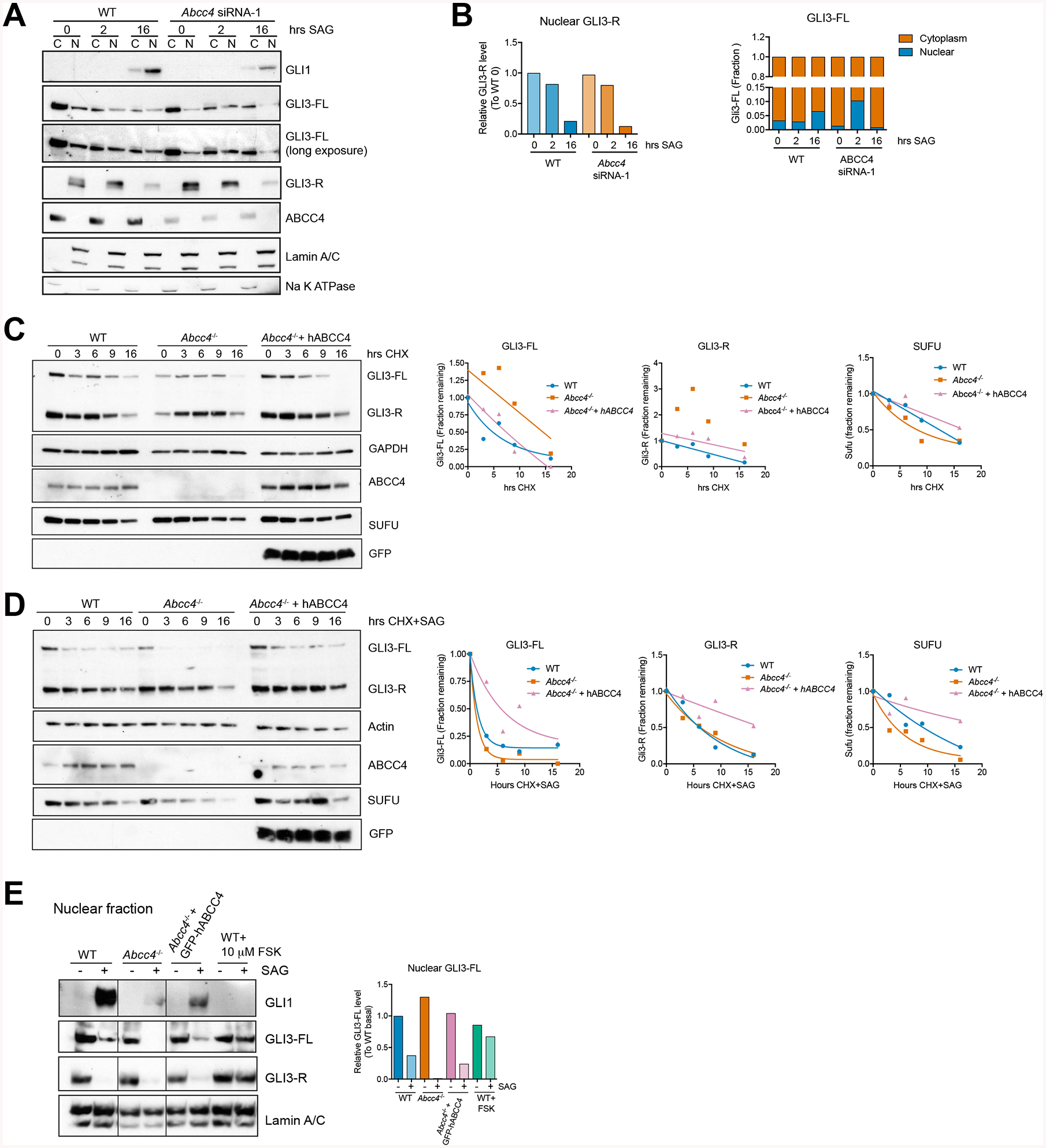
(A) Cytoplasmic (C) and nuclear (N) distribution of full-length (FL) and short (R) forms of GLI3 in NIH3T3 cells. Cells were transfected with control or siRNA targeting Abcc4 and treated with SAG for durations indicated. Lysates were fractionated into nuclear and cytosolic fractions.
(B) Densitometry analyses of (A). Lamin A/C and Na K ATPase were used as nuclear and cytoplasmic loading controls, respectively. Bars represent relative abundance of indicated GLI3 species in the cytosol (C) and nuclear (N) fractions.
(C,D) WT, Abcc4−/−, or Abcc4−/− cells stably expressing human ABCC4 were treated with CHX (100 μg/mL) with or without SAG (200 nM) for durations indicated. GLI3 and SUFU levels were represented as fractions relative to the respective genotypes. Data points were fitted to best-fit single exponential decay curves.
(E) WT, Abcc4−/−, or Abcc4−/− cells transiently transfected with GFP-tagged human ABCC4 were treated with SAG (200 nM), Forskolin (10 μM), or DMSO control for 16 hours and fractionated as in (A). Bars represent relative abundance of nuclear full-length GLI3 (GLI3-FL) to WT DMSO control. Irrelevant lanes were removed and marked with a black line. All blots in this figure is a representative of at least two independent experiments.
Previous study showed that nuclear GLI3-FL is labile with a short half-life (Humke et al., 2010). Under basal conditions, ABCC4 absence did not alter the half-lives of both Gli3-FL and Gli3-R species compared to WT cells (Figure 3C). However, upon SHH-activation, ABCC4 loss dramatically reduced the stability of GLI3-FL (Figure 3D). Re-expression of human ABCC4 to levels similar to wildtype in Abcc4−/− cells restored GLI3-FL stability to levels comparable to that of WT (Figure 3D). SUFU, a known regulator of GLI3-FL stability (Humke et al., 2010; Wang et al., 2010), also had a shorter half-life in the absence of ABCC4, which was also rescued by re-expression of ABCC4. These experiments indicate that ABCC4 modulates the stability of GLI3-FL.
Next, we expressed human ABCC4 in Abcc4−/− cells to determine if ABCC4 re-expression affects nuclear GLI3-FL and GLI1. ABCC4 expression restored the nuclear levels of both GLI1 and Gli3-FL (Figure 3E). Because ABCC4 has the ability to export cAMP (Copsel et al., 2011), we evaluated whether elevation of intracellular cAMP generally reduces nuclear GLI3-FL and GLI1 levels. WT cells were treated with forskolin (FSK), which by adenylyl cyclase activation elevates intracellular cAMP concentration. Like absence of ABCC4, FSK treatment blocked GLI1 expression, however in contrast with ABCC4, FSK treatment increased GLI3-R without affecting nuclear GLI3-FL level (Figure 3E, last lane). Thus, ABCC4 deficiency promotes nuclear GLI3-FL loss, whereas an overall rise in intracellular cAMP elevation by FSK, did not, revealing a distinct ABCC4-mediated regulatory mechanism.
ABCC4 alters membrane-specific cAMP and downstream PKA activity to modulate SHH signaling
Since ABCC4 has the ability to export cAMP (Copsel et al., 2014; Li et al., 2007; Sassi et al., 2012), we next sought to determine if ABCC4 modulates SHH signaling by modulating cAMP level. NetBID predicted that the PKA regulon was significantly lower in SHH-MB, which shows the opposite enrichment pattern compared to the GLI transcription factors and ABCC4 regulons (Figure 4A). We confirmed that ABCC4 is capable of exporting cAMP in our model system, NIH3T3 cells. Following acute treatment with FSK and IBMX, reagents that dramatically elevate cAMP level, cAMP effluxed from the cells was measured in culture supernatant. Abcc4-deficient cells exported about two-fold less cAMP to the media (Figure 4B). Addition of exogenous human ABCC4 to Abcc4−/− cells restored cAMP export (Figure 4B). We did not observe a significant change in bulk intracellular cAMP level when SHH pathway was active (Supplementary Figure 3A) and the amount of PKA was comparable between WT and Abcc4−/− cells (Supplementary Figure 3B).
Figure 4. ABCC4 modulates cAMP level and PKA activity to regulate SHH signaling.
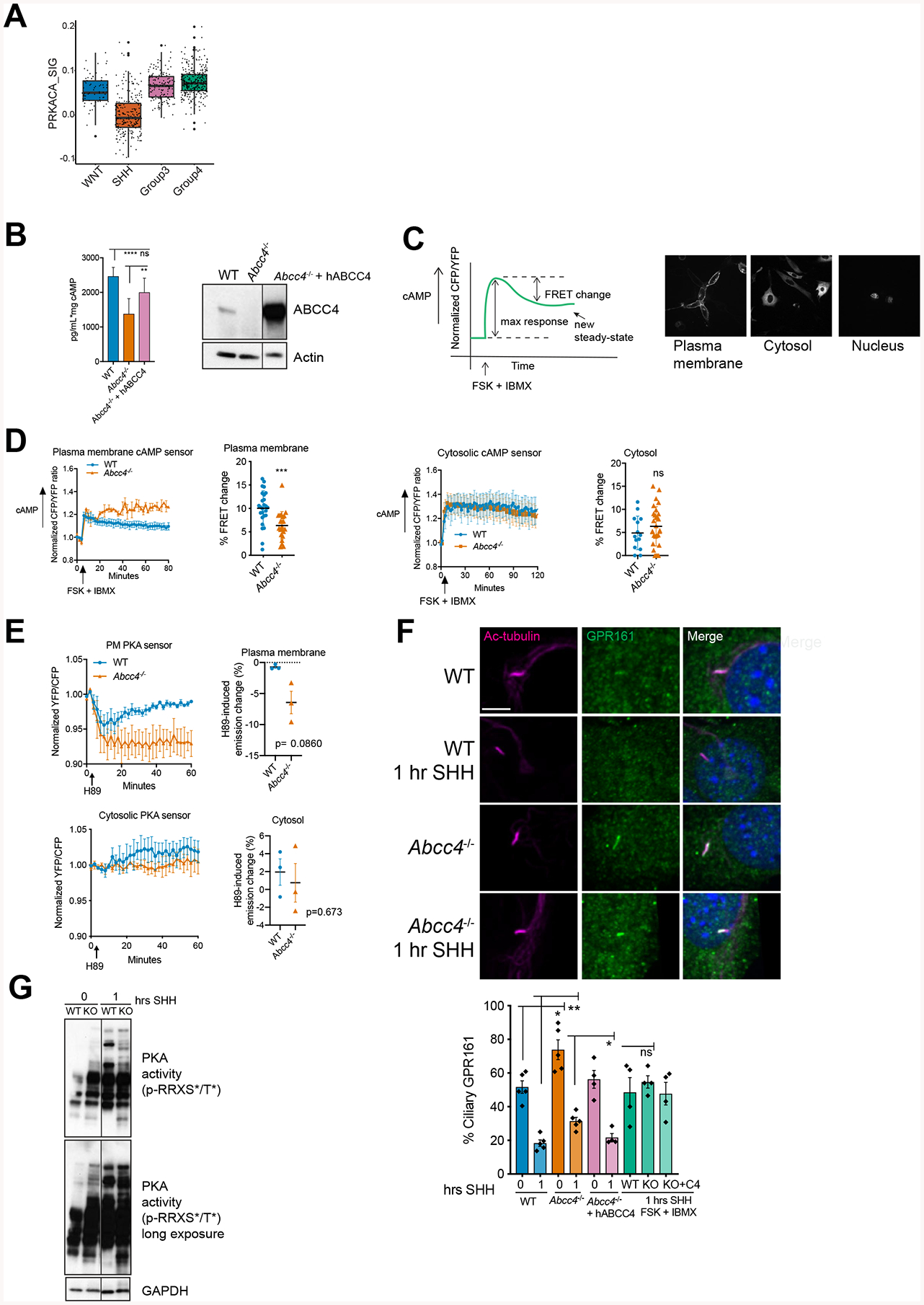
(A) Enrichment of predicted PRKACA signaling regulon in differentially expressed genes between SHH and other MB subgroups from GSE85217 (Cavalli et al., 2017). P values of regulon enrichment and differential expression of the hub gene are indicated in DA DE table.
(B) WT, Abcc4−/−, Abcc4−/− cells transiently transfected with GFP-tagged human ABCC4 were treated with forskolin (50 μM) and IBMX (100 μM) for 1 hour. cAMP level in the media was measured by ELISA. Bars represent means (± SD) of two independent experiments. ** P < 0.01, **** P <0.0001, ns = not significant, one-way ANOVA. Level of ABCC4 was measured by immunoblotting. Irrelevant lanes were removed and marked with black line.
(C) Schematic of live-cell imaging FRET experiment. Arrow indicates addition of forskolin (50 μM) and IBMX (100 μM). Representative images of plasma membrane, cytosolic, and nuclear-targeted cAMP sensors.
(D) Representative live-cell imaging analyses of WT or Abcc4−/− NIH3T3 expressing plasma membrane or cytosolic cAMP FRET sensors. Percentage of FRET change is represented as means (± SD) of three independent experiments. * P < 0.05, ** P < 0.01, *** P <0.001, Mann-Whitney test.
(E) Live-cell imaging analyses of WT or Abcc4−/− NIH3T3 expressing plasma membrane or cytosolic PKA FRET sensors. Emission ratio time course (means ± SD) of three independent experiments are shown. H89-induced emission change was calculated by subtracting the emission ratio at the start with the end point emission ratio at 60 minutes and represented as a percent emission change. Mann-Whitney test.
(F) Indicated cells were treated SHH-conditioned media. At indicated time points, cells were fixed, permeabilized, and stained with GPR161 and acetylated-a-tubulin to label cilia. Bars are mean (± SEM) of four or five independent experiments with n=40–50 cells/experiment/group. * P < 0.05, ** P < 0.01, ns = not significant, student’s t-test.
(G) WT or Abcc4−/− NIH3T3 cells were treated SHH for 30 minutes or 1 hour. PKA activity was measured by probing for phosphorylation of PKA substrates at consensus PKA motif, RRXS*/T*. Irrelevant lanes were removed and marked with black line.
cAMP signaling elicits specific responses to various cellular stimuli. Such a specific response is achieved through compartmentalization of cAMP signaling (Steinberg and Brunton, 2001). The idea of compartmentalized cAMP signaling led us to hypothesize that ABCC4 might affect the SHH pathway by regulating cAMP specifically at the plasma membrane. To interrogate cAMP changes at distinct subcellular compartments, we utilized genetically encoded cAMP biosensors (ICUE3 sensors) targeted to the plasma membrane, cytosol, and nucleus (DiPilato et al., 2004; Sample et al., 2012). Cells expressing these biosensors were treated with FSK to activate adenylyl cyclase and IBMX to inhibit phosphodiesterases (PDEs) (Figure 4C). These treatments allowed us to concentrate on cAMP changes predominantly mediated by ABCC4 efflux and not degradation by PDEs. Upon treatment, the cyan to yellow emission ratio was increased, indicative of elevated cAMP levels (Figure 4C,D). Addition of FSK and IBMX rapidly increased cAMP level in both WT and Abcc4−/− cells in all subcellular compartments monitored (Figure 4D, Supplementary Figure 3C). No significant difference was observed in either the amplitude of maximum response or the time to reach maximum response between the genotypes (Supplementary Figure 3C,C). As expected, the cAMP response at the plasma membrane was faster than the cytoplasm and nucleus (Supplementary Figure 3C,D).
Following the maximum cAMP response, cells re-establish a new, lower steady-state cAMP level (Figure 4C). Abcc4 deficiency significantly impaired the ability of cells to create a lower cAMP steady-state at the plasma membrane (which we refer to as “FRET change”) (Figure 4D). This contrasts with the cytosol and nucleus, where the cells’ ability to establish a new steady-state level was unimpiared by Abcc4 absence (Figure 4D, S3C). Together, these results highlight that ABCC4 is capable of determining the duration of sustained cAMP concentration at the plasma membrane.
We next investigated how ABCC4 absence impacts the direct downstream target of cAMP, protein kinase A (PKA). PKA activity at the plasma membrane and cytosol was monitored using PKA activity FRET biosensors (AKAR4 sensors). The AKAR4 sensor harbors an FHA1 phosphoamino acid binding domain and a PA substrate peptide. When PKA activity is high, the substrate peptide is phosphorylated, the FHA domain then binds to the phosphorylated substrate thus inducing the FRET event (Depry et al., 2011; Sample et al., 2012). A high ratio of YFP over CFP emission signifies strong PKA activity. We hypothesized that in Abcc4−/− cells, the majority of the sensors exist in phosphorylated form in the resting state. As such, inhibition of PKA by H89 will decrease the FRET ratio as a consequence of sensor dephosphorylation by endogenous phosphatases. When WT and Abcc4−/− cells expressing membrane PKA sensors were treated with H89, there was a much greater decrease in the FRET ratio for cells lacking ABCC4 (Figure 4E). This indicated that the amount of phosphorylated sensor was much greater at the resting state in Abcc4−/− cells compared to WT, suggesting a greater PKA activity. This difference between the genotypes was not observed for the cytosolic PKA sensors and indicates that ABCC4 specifically influences plasma membrane PKA activity (Figure 4E). In total, these results show that cAMP and PKA activity are regulated by plasma membrane ABCC4.
As GPR161 is implicated in PKA-dependent basal repression of SHH signaling (Mukhopadhyay et al., 2013; Pal and Mukhopadhyay, 2014), we investigated if GPR161 might be an intermediary in the altered response to the SHH agonist in Abcc4−/− cells. Notably Abcc4−/− cells had more GPR161 positive cilia (Figure 4F). Exit of GPR161 is required for strong SHH pathway activation. After SHH pathway activation, the rate of GPR161 is much slower from Abcc4−/− cells(Figure 4F). Importantly, restoration of ABCC4 to levels similar to WT in Abcc4−/− cells both reduced the proportion of GPR161 positive cilia and enhanced its rate of export to levels comparable to WT cells. We show that the egress of GPR161 from cilia in response to SHH can be suppressed by strongly in WT, KO and KO with restored ABCC6, by elevating cAMP with forskolin and IBX (Figure 4F).
Next, we utilized a phospho-specific antibody that recognizes phosphorylation of PKA substrates at the consensus PKA motif, RRXS*/T* as an orthogonal approach to assay PKA activity. The magnitude of PKA substrate phosphorylation in cells lacking Abcc4 under basal conditions was greater than WT cells (Figure 4G). Extending this, one hour after signaling initiation, the number of phosphorylated substrates appears more extensive in Abcc4−/− cells (Figure 4G), which may in part be related to retention of GPR161 at primary cilia. These results further support ABCC4 role in modulating plasma membrane cAMP level and PKA activity during initiation of SHH signaling.
Increased PKA activity can alter the overall phosphorylation status of PKA targets including GLI2/3 and SUFU, subsequently altering GLI signaling output (Chen et al., 2011; Niewiadomski et al., 2014). Consistent with this, we observed, by quantitative phosphoproteomics, differential phosphorylation of SUFU, GLI2, and GLI3 in Abcc4−/− cells compared to WT (Supplementary Figure 3D).
Perturbing ABCC4 as a strategy to impair aberrant SHH signaling
Aberrant, disease-like ligand-independent SHH signaling might also be modulated by ABCC4. Ptch1 and Sufu restrain SHH signaling by different mechanisms and genetic alterations in each have been linked to SHH-MB (Brugier̀es et al., 2012; Northcott et al., 2012a; Taylor et al., 2002). Loss of Ptch1 relieves the repression of SMO, allowing it to ultimately activate the GLI transcription factors. In contrast, Sufu mutations can produce a truncated, inactive protein, causig the constitutive presence of GLI proteins in the nucleus (Taylor et al., 2002). In Ptch−/− mouse embryonic fibroblast (MEF) and Sufu knockdown NIH3T3 cells (SufuKD), ABCC4 was upregulated at both the protein and mRNA level compared to wildtype (WT) NIH3T3 cells (Figure 5A). Importantly, suppression of Abcc4 in both Ptch−/− and SufuKD cells resulted in a dose-dependent reduction in the activity of the SHH pathway as measurable by GLI1 protein and mRNA levels (Figure 5B–D).
Figure 5. Targeting Abcc4 is a viable strategy to impair aberrant SHH signaling in vitro and in SHH-MB.
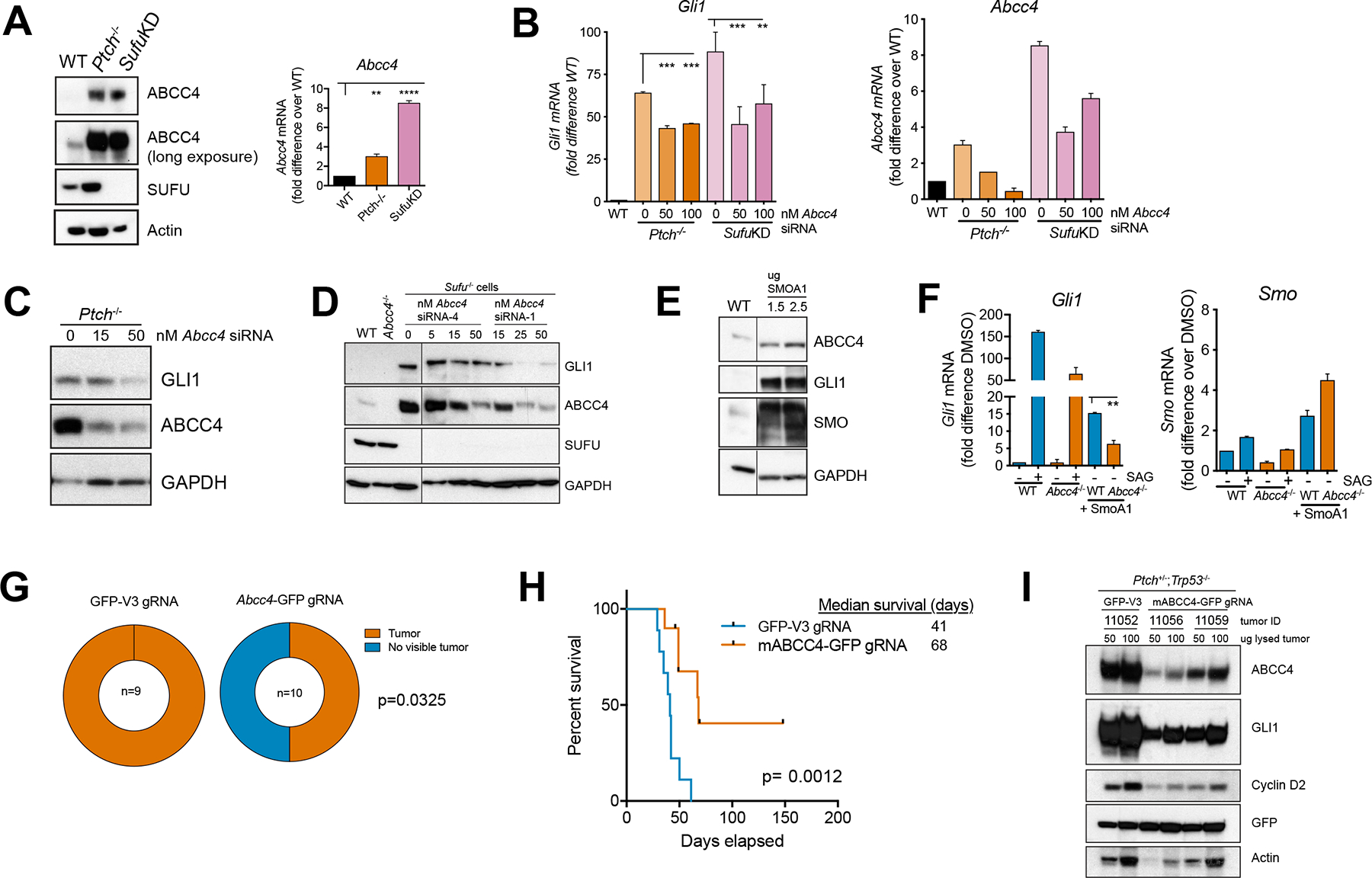
(A) NIH3T3, Ptch−/− and Sufu knockdown (SufuKD) cells were harvested 16 hours post-serum starvation. Levels of indicated proteins were determined by immunoblotting. RNA was isolated from Ptch−/−, SufuKD, and WT NIH3T3 cells. Levels of indicated transcripts were measured by qRT-PCR. Bars represent mean (± SD) cumulative from two independent experiments. * P < 0.05, ** P < 0.01, *** P <0.001. One-way ANOVA.
(B) Ptch−/− and SufuKD cells were transfected with Abcc4 siRNA. RNA was isolated and levels of indicated transcripts were measured by qRT-PCR. Bars represent mean (± SD). ** P < 0.01 *** P < 0.001, one-way ANOVA.
(C,D) as in (B) except proteins were harvested. Levels of indicated proteins were determined by immunoblotting. Irrelevant lanes were removed and marked with black line. Representive blot from two independent experiments.
(E) NIH3T3 cells were transfected with exogenous SmoA1. Levels of indicated proteins were determined by immunoblotting. Irrelevant lanes were removed and marked with a black line. Bars represent abundance of indicated proteins relative to untreated control. Representive blot from two independent experiments.
(F) WT or Abcc4−/− NIH3T3 transfected with SMOA1. RNA was isolated and levels of indicated transcripts were measured by qRT-PCR. Representive of two independent experiments. Bar represents mean (± SD) . ** P < 0.01 *** P < 0.001, one-way ANOVA.
(G) Tumor incidence from between control gRNA mice recipients compared to Abcc4 gRNA mice recipients. *p < 0.05, Fisher’s exact test.
(H) Kaplan-Meier with log-rank statistics survival curve of CD-1 nude mice bearing Ptch−/−;Trp53−/− SHH-MB tumor infected with either control gRNA or gRNA targeting exon 4 of mouse Abcc4 (n=9–10/group. Two independent experiments).
(I) Representative tumor chunks obtained from (D) were lysed and levels of indicated proteins were measured by immunoblotting.
Acquired resistance to SMO inhibitors re-activates the hedgehog pathway, secondary to mutations within SMO such as W535L (SMOA1 mutant) (Atwood et al., 2015; Dijkgraaf et al., 2011; Espstein et al., 1998). The oncogenic SMO mutant resulted in a ligand-independent expression of GLI and increased ABCC4 expression (Figure 5E). SMOA1 expression i increased Gli1 mRNA level even in the absence of ligand, indicative of aberrant signaling, which was strongly reduced in Abcc4-null cells (Figure 5F). These results show that aberrant Hedgehog can be markedly reduced by suppression of ABCC4.
We next investigated if suppression of Abcc4 alters SHH-MB pathogenesis. SHH-MB arising from Ptch+/−;Trp53−/− tumors were lentivirally-transduced with an all-in-one CRISPR-Cas9 plasmid encoding GFP and Cas9, with or without Abcc4 gRNA (targeting exon 4 of mouse Abcc4). Multiple gRNAs were designed and tested but gRNA targeting exon 4 reduced ABCC4 to the greatest extent (data not shown). These tumor cells were then implanted into the cortices of naïve recipients and disease progression was monitored. In two independent experiments, among a total of 10 mice that were implanted with Abcc4 gRNA-infected tumor cells, only 50% of the mice developed pathologically discernible MB, whereas in the control group, all of the mice readily developed MB (Figure 5G). Furthermore, transduction of tumors with Abcc4 gRNA delayed tumor progression in mice (p=0.0012) (Figure 5H). When moribund mice were sacrificed, tumor cells were isolated and analyzed by western blotting to evaluate SHH signaling. All GFP levels in tumors were similar indicating comparable transduction. As we demonstrated in NIH3T3 cells with knockdown of Abcc4, SHH target genes including GLI1 and Cyclin D2 were decreased in tumors where ABCC4 level was reduced (Figure 5I). Notably, among the Abcc4 gRNA cohort that developed tumor, ABCC4 levels were much lower than the control gRNA cohort (Figure 5I). These results show that ABCC4 is required for maximal tumor expansion and SHH pathway action in vivo.
To further explore the molecular connections between ABCC4 and GLIs, we extracted a subnetwork from MB interactome. We found GLIs share mutual downstream targets, not only within GLI family members, but also with ABCC4. Interestingly, only GLI2 showed a direct regulatory relationship with ABCC4. Additionally, PKA coregulated multiple target genes with GLI family members too (Figure 6). In total, these results integrate ABCC4 into a regulatory network important in SHH-MB pathogenesis.
Figure 6. Interconnectivity of ABCC4, PKA, and GLI transcription factors inferred by NetBID analyses.
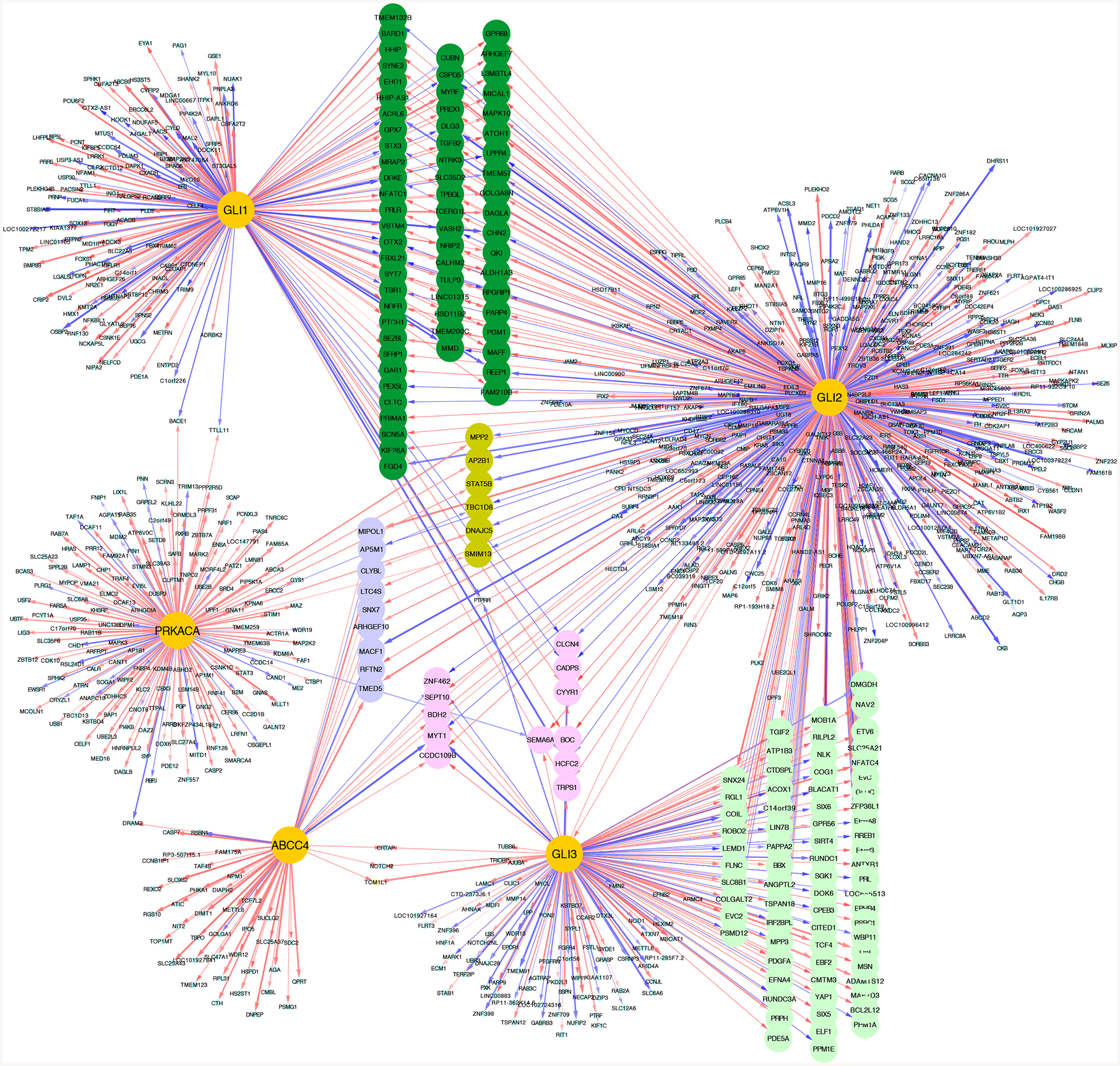
The hub genes are highlighted in golden. Red and blue lines indicate positive and negative regulation effect between hub gene and its targets, respectively. Line thickness indicate the strength of the effect. Mutual downstream targets are highlighted in different colors.
DISCUSSION
In this study, through bioinformatic data mining and a systems biology approach, we discovered ABCC4 as a candidate driver of SHH-MB. We further validated that ABCC4 level contributes to SHH-MB pathogenesis, a finding supporting the strong relationship between poor prognosis in SHH-MB and high ABCC4 expression. Importantly, suppression of ABCC4 was especially salutary when applied to a murine model of SHH-MB as lifespan was extended significantly and associated with a strongly reduced tumor burden. While ABCC4 overexpression alone does not spontaneously activate the SHH pathway, when the pathway is activated, ABCC4 function is crucial to elicit maximal pathway activation. These findings highlight how upregulation of ABCC4 expression is intimately linked to the activation of the SHH pathway, promoting a heretofore unknown “feedforward” mechanism, which suggests that reduction in ABCC4 function might be a general strategy to yield overall improvements in SHH-MB outcomes.
We propose the following model for ABCC4 modulation of SHH signaling (Figure 7). The enrichment of cilia with GLI2/3 after SHH activation is unimpaired by ABCC4 deficiency. However, sustained plasma membrane cAMP in ABCC4 absence, delays ciliary exit of GPR161, which is closely related to PKA hyperactivation. Increased PKA activity enhances phosphorylation of PKA targets including GLI3 which correlates with its reduced stability and is likely related to an alteration in its phosphorylation. Importantly, re-expression of ABCC4 rescues the stability of GLI3 and restores GPR161 exit from cilia.
Figure 7. Model of ABCC4 function in SHH signaling.
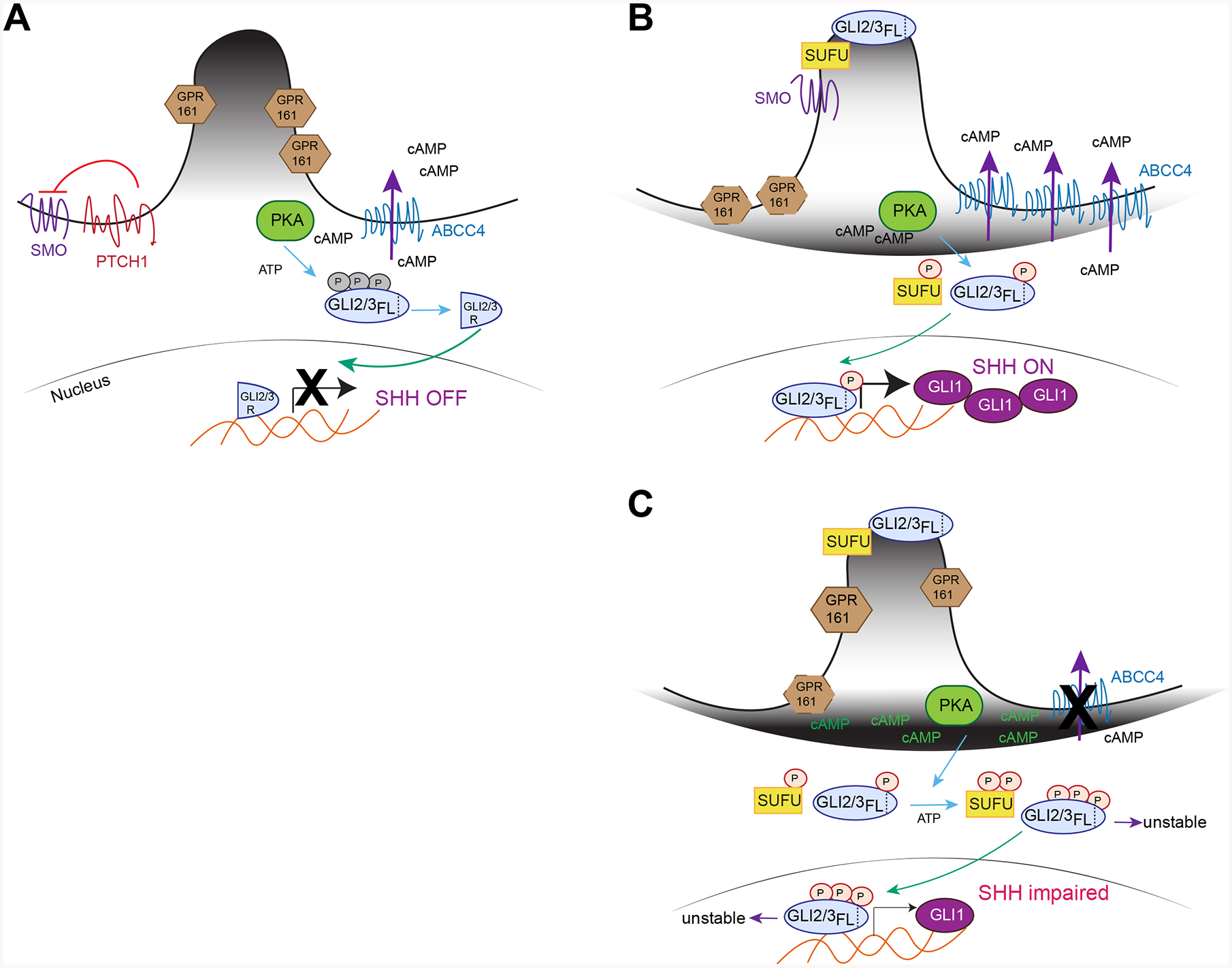
(A) In the absence of SHH agonist, PTCH prevents SMO activation and GPR161 is retained at primary cilia where it regulates cAMP level. GLI2/3 is phosphorylated by PKA at inhibitory sites and proteolytically cleaved to act as a transcriptional repressor. ABCC4 is a plasma membrane, ATP-dependent cAMP efflux transporter.
(B) When SHH binds to PTCH, SMO is activated where it enters primary cilia and induces GPR161 exit. SUFU and GLI2/3 also translocate to primary cilia, a step necessary for GLI2/3 conversion to transcriptional activator. It is unknown whether PKA activating phosphorylation on GLI2/3 occurs prior or after ciliary enrichment. When SHH pathway is active, ABCC4 is upregulated where it regulates membrane cAMP pool and PKA activity. ABCC4 acts as a restrain to allow for just enough PKA to be active to phosphorylate key SHH proteins such as GLI2/3 at specific sites required to ensure proper propagation of signaling. These PKA sites are distinct from that of (A).
(C) In the absence of ABCC4, cAMP accumulates at the plasma membrane, GPR161 ciliary exit is delayed and PKA becomes over-activated. These results in GLI3-FL destabilization, leading the reduced GLI1 level. Over-activated PKA can also alter the phosphorylation codes in GLI2/3 and SUFU that are distinct from (B).
ABCC4’s role in SHH-MB disease progression is important in the following context: 1) High ABCC4 expression is prognostic of poor overall survival in human SHH-MB, 2) ABCC4 expression is correlated to GLI2, which is amplified in 29% of human SHH-MB cases (Cavalli et al., 2017), 3) Loss of Abcc4 suppressed aberrant SHH signaling caused by Ptch1 and Sufu inactivation, genetic lesions common to MB, 4) A medulloblastoma-specific interactome revealed ABCC4, an ATP-binding cassette transporter, as a potential driver of SHH-MB. To that end, we employed a mouse model of SHH-MB harboring either one Ptch1 allele with Trp53 loss (Uziel et al., 2005), genetic alterations typically found in human SHH-MB (Northcott et al., 2017). Importantly, ABCC4 is highly expressed in these tumors (Figure 1G,H). Targeted Abcc4 ablation decreased tumor incidence, extended survival of tumor-bearing mice, and reduced SHH signaling even in tumors without complete Abcc4 ablation. Collectively, these results show ABCC4 regulates SHH-MB progression.
How ABCC4 accomplishes this could be evaluated by placing these tumors in in vitro cultures. However, the traditional cell culture media conditions used for primary murine MB (e.g., Group 3 MB (Morfouace et al., 2015)) does not retain SHH signaling (Sasai et al., 2006) and established cell lines appear to not faithfully execute SHH signaling. A less attractive, alternative might be to investigate, in model cell line systems (with constitutive hedgehog pathway activation and high ABCC4 expression) whether suppression of ABCC4 impacts either cell growth or survival. Furthermore, understanding the crosstalk between the GLI transcription factors and ABCC4 may provide future insights into targeting ABCC4-mediated positive regulation of SHH signaling in MB (Figure 6).
Inhibition of ABCC4 as a strategy to improve survival of SHH-MB is advantageous because ABCC4 promotes the SHH pathway downstream from SMO activation where most of the MB driver mutations (e.g. SUFU) or amplifications occur in the most aggressive cases of MB (Cavalli et al., 2017). There are important additional reasons for the appeal of this approach, not the least of which has been described herein, but also because current frontline MB chemotherapy includes the camptothecins, topotecan and irinotecan, well-known ABCC4 substrates (Leggas et al., 2004; Murray et al., 2017). Thus, an effective ABCC4 inhibitor would be envisioned as both restraining MB tumor cell growth and decreasing tumor cell survival by affecting ABCC4 function, while concomitantly blocking camptothecin export, thereby further increasing the intratumoral concentration and tumorocidal activity.
MATERIALS AND METHODS
Cell lines
WT and Abcc4−/− NIH3T3 cells were cultured in Dulbecco’s modified eagle medium (DMEM), containing 10% bovine calf serum, 0.05 mg/ml penicillin and streptomycin, 2 mM glutamine, 1 mM sodium pyruvate, and 0.1 mM non-essential amino acids. Light2 cells were cultured in the same media with the addition of 0.4 mg/mL G418 and 0.15 mg/mL zeocin. To generate Abcc4−/− NIH 3T3 cells, 3 μg CRISPR-Cas9 plasmid containing Abcc4 gRNA targeting exon 1 was transfected to 200,000 cells/well in 6 wells plate using lipofectamine 2000 at 1:2 DNA:lipid ratio. Media was replaced the next day and cells were harvested, sorted for GFP+ population, and plated at single cell density/well in 96 wells plates. Multiple single-cell derived clones were analyzed by western blotting using at least 100 ug lysate and extended film exposure to ensure complete ablation of Abcc4. Clones that showed complete depletion by western blot were expanded and gene deletion was verified and mapped by sequencing. Ptch−/− and SufuKD mouse embryonic fibroblast (MEF) cells were cultured in DMEM containing 10% fetal bovine serum, 0.05 mg/mL penicillin and streptomycin, and supplemented with 2 mM glutamine.
Mice
Female CD-1 nu/nu mice (6–12 weeks old) were used as cranial transplant recipient. All animal experiments were performed in accordance to and approved by St. Jude Children’s Research Hospital Animal Care and Use Committee. After transplant, animals were monitored daily for symptoms including doming of the head, ataxia, and reduced activity. Moribund mice were humanely sacrificed, and tumors were isolated for downstream analyses such as RNA and protein extraction.
METHOD DETAILS
Bioinformatics analyses
Gene expression and heatmaps from Figure 1A, Supplementary Figure 1A–C were extracted from Oncomine (www.oncomine.org). The relationship between ABCC4 and survival data of MB patients (GSE85217 dataset) were obtained from R2 genomics analysis and visualization platform (http://r2.amc.nl).
NetBID analysis to identify drivers in SHH-MB
We applied the network-based integrative NetBID (Du et al., 2018). algorithm to identify “hidden” drivers in SHH subgroup using gene expression profiles from medulloblastoma patients. We first reverse-engineered a medulloblastoma-specific interactome (MBi) using published microarray data (Jones et al., 2012; Kool et al., 2014; Northcott et al., 2014; Robinson et al., 2012) from platform HG-U133_Plus_2 (total 383 samples) against 1,621 transcription factors and 5,791 signaling proteins. We removed the batch effects from different datasets by using the “removeBatchEffect” function in limma (Ritchie et al., 2015). The data-driven MBi resulted in 1,567,381 edges and 26,126 nodes. We then applied NetBID (signed=TRUE) on comparing hub gene inferred activity between SHH versus all the other subgroups from the cohort GSE85217 (Cavalli et al., 2017). Finally, we subtracted the network of our hub genes of interest GLI1/2/3, ABCC4 and PRKACA from MBi and visualized their interconnections by Cytoscape. The NetBID package can be found online at: https://github.com/jyyulab/NetBID. An updated version NetBID2 will be released soon.
Gene Set Enrichment Analysis
We applied NetBID to do gene set enrichment assay: We used MBi network predicted regulons as gene sets and summarized differential gene expression profile between SHH versus other subgroups from cohort GSE85217. Differential activity (DA) p values were used as an enrichment score of each regulon. We also verified GSEA result by other two packages: JavaGSEA and fGSEA, with 1000 permutations and parameters as the default setting.
siRNA and plasmid transfections
NIH3T3 cells were seeded on a well in 6 wells plate at 150,000 cells/well. The next day, cells were transfected with siRNA at indicated final concentration using 5 μL lipofectamine 2000/siRNA. Unless otherwise indicated, the final concentrations of siRNAs are: 5 nM Abcc4 siRNA-4; 15 nM Abcc4 siRNA-1; 5 nM for non-targeting control; 50 nM Sufu siRNA.
For cDNA transfection, cells were seeded at 150,000 cells/well in 6 wells plate. The next day, cells were transfected using lipofectamine 2000 at 1:2 dna:lipid ratio. For example, 2 ug of plasmid was preincubated with 4 uL lipofectamine 2000 for 10 mins in Opti-MEM at room temperature before dropwise addition to the cells.
Cell culture condition to assay for SHH signaling
For each treatment, WT or Abcc4−/− NIH3T3 cells were seeded in a well of 6 wells plate at a density of 150,000 cells/well. The next day, cells were transfected as described above. 24 hours post-transfection, media was replaced with media containing 0.5% serum. Pathway activation was initiated using 200 nM SAG, SHH-conditioned media, or 10 μM 20-OHC for 16 hours unless indicated otherwise. Post-treatment, cells were harvested by trypsinization, and lysed as described below. For cyclohexamide treatment, cells were treated post-starvation in 0.5% serum media at 100 ug/mL cyclohexamide.
Western blot
Cells were lysed in modified RIPA buffer (50 mM Tris-Cl pH 7.4, 300 mM NaCl, 2% NP-40, 0.25% deoxycholate) on ice for 30 minutes supplemented with 10 mM N-etyl-malemaide (NEM), 1 mM phenylmethylsulfonyl fluoride (PMSF), 1x EDTA-free protease inhibitor cocktail, 1x phosphatase inhibitor, and 10 μM MG132. Lysates were vortexed every 10 minutes, and then cleared by centrifugation after 30 minutes incubation on ice. Proteins were measured by BCA and equal amounts were loaded to 4–15% SDS gel. Gels were transferred to PVDF membrane and blocked with 5% milk in TBST (Tris-buffered saline with 0.1% tween-20). Primary antibodies were incubated overnight at 4°C in blocking buffer or 5% BSA in TBST (for GLI1 and phospho antibodies). Horseradish peroxidase (HRP)-conjugated secondary antibodies incubation were performed at room temperature. Signal was detected by ECL detection and blots were exposed on film.
Quantitative real-time PCR (qRT-PCR)
RNA was isolated using RNAeasy kit. cDNA was synthesized using iscript reverse transfection reagent using 750 ng – 1 μg RNA and used for template in SYBR green ER reaction. Actin was used as the normalization control. Relative gene expression was calculated using the 2−ΔΔCt method.
Subcellular fractionation
Separation of nuclear fraction from cytosolic fraction (also contains heavy membrane fraction), was performed using NE-PER nuclear and cytosolic extraction kit. Proteins were measured by BCA assay and equal amounts were loaded on SDS-PAGE and analyzed by western blot with specific antibodies. If the fractions were dilute, samples were concentrated by adding tricholoroacetic acid (TCA) to a final concentration of 10%. Samples were incubated on ice for 30 minutes and recovered by centrifugation. Pellets were then resuspended by vigorous vortexing in 2x SDS-sample buffer containing fresh 20 mM DTT.
Gli1 luciferase reporter assay
Light2 cells are NIH3T3 stably expressing 8x Gli1-binding sites fused to a luciferase(Taipale et al., 2000). Cells were seed at 100,000 cells/well in 24 wells plate. Indicated siRNAs were transfected at the time of plating (reverse transfection). After 24 hours, medium was replaced with 0.5% serum DMEM and pathway stimulation was initiated using 200 nM SAG, 100 uL/mls SHH-N-CM, or 10 uM 20-OHC for 48 hours. Gli1 luciferase activity was measured using the dual-luciferase assay system.
Immunofluorescence
Cells were seeded on #1.5 glass coverslips in 12 wells plate. The next day, cells were transfected with indicated siRNAs or plasmids. 24 hours post-transfection, medium was replaced with 0.5% serum DMEM. SHH pathway was then initiated by 200 nM SAG or SHH. At indicated time points, medium was removed, and cells were washed quickly with PBS. Cells were then fixed in 4% methanol-free formaldehyde for 15 minutes at room temperature. Cells were permeabilized and blocked in 0.2% triton x-100 with 10% normal goat serum in PBS for 1 hour, and incubated overnight with primary antibodies in 1% normal goat serum in PBS at 4°C. After 3, 5 minutes PBS washes, cells were incubated in fluorochrome-conjugated secondary antibodies for 1 hour at room temperature, washed with PBS thrice, and mounted on ProLong gold diamond with DAPI.
FRET acquisition and analysis
For FRET live-cell imaging using cAMP ((Lyn-ICUE3, ICUE3 and NLS-ICUE3) or PKA sensors WT or Abcc4−/− NIH3T3 cells were seeded onto 2 wells Lab-Tek glass dish. Cells were transfected as described under siRNA and plasmid transfections section. The next day, medium was replaced with 0.5% serum DMEM without phenol red. 48 hours post-transfection, 10 μM FSK and 100 μM IBMX were added to stimulate cAMP elevation in the cells. Dual emission ratio imaging was performed using a Marianas spinning disk confocal (SDC) imaging system on a Zeiss Axio Observer inverted microscope platform using a Zeiss Plan-Apochromat 63X (1.4 NA) oil objective. Images were taken every 60 s with an exposure time of 100–200 ms. Data were normalized by setting the emission ratio prior to small molecule addition to 1. % FRET change = (Rmax-Rnewsteadystate)/Rmax * 100 where R is emission ratio.
For Live-cell imaging using PKA sensors AKAR4 (Lyn-AKAR4, AKAR-4 NES) sensors, 10 μM H89 was added instead of FSK and IBMX. Area under the curve (AUC) of cells expressing plasma membrane PKA sensor was measured by integrating the net decrease in FRET over time from 30–60 min using 1 as the baseline.
Lentiviral transduction of SHH-MB tumor cells
For vector construction, oligonucleotides coding for gRNA targeting exon 3 of mouse Abcc4 were cloned into a modified lentiCRISPRv2 plasmid (carrying GFP marker instead of puromycin). Lentiviral supernatant containing lentiCRISPRv2 with mABCC4 gRNA was produced in HEK293T cells.
For transduction, frozen SHH-MB tumor cells were thawed and resuspended in neurobasal media. High-titer lentiviral supernatant was added to resuspended tumor, mixed every 10 minutes for 30 minutes, and cells were harvested by centrifugation 5 minutes after transduction. Cells were then resuspended in matrigel for transplant.
Animal husbandry and Orthotropic transplants
Tumor cells were implanted into the cortex of 6–8 weeks old, female CD-1 nu/nu cranial transplant recipient. All animal experiments were performed in accordance to and approved by St. Jude Children’s Research Hospital Animal Care and Use Committee. After transplant, animals were monitored daily for symptoms including doming of the head, ataxia, and reduced activity. Moribund mice were humanely sacrificed, and tumors were isolated for downstream analyses such as RNA and protein extraction.
Quantitative analysis of proteome and phosphoproteome by 11-plex TMT-LC/LC-MS/MS
The proteome and phosphoproteome profiling was performed essentially according to systematically optimized protocols (Bai et al., 2017; Niu et al., 2017)
Cell harvest and protein extraction, digestion, and TMT labeling
The wild type and Abcc4−/− NIH3T3 cells treated with Shh for durations indicated. Cells were harvested and cell pellets were washed twice with ice cold PBS and resuspended in fresh lysis buffer (50 mM HEPES, pH 8.5, 8 M urea, 0.5% sodium deoxycholate and phosphatase inhibitor cocktail (PhosphoSTOP, Roche) with a buffer to cell pellet ratio of 10:1 (v/v). After addition of glass beads (0.5 mm in diameter, Sigma), the cells were lysed with Bullet Blender (Next Advance). Protein concentration was quantified by the BCA protein assay and confirmed on a short Coomassie-stained SDS gel with titrated BSA as a standard(Xu et al., 2009). Proteins (about 1 mg) from each sample were digested with Lys-C (Wako, 1:100 w/w) at room temperature for 2 h, diluted 4 times with 50 mM HEPES, pH 8.5, to reduce urea to 2 M, and further digested with trypsin (Promega, 1:50 w/w) overnight at room temperature. The digestion was terminated with 1% trifluoroacetic acid, followed by desalting with Sep-Pak C18 cartridge (Waters), and drying by speedvac. Each sample was then resuspended in 50 mM HEPES, pH 8.5, and individually labeled with 11-plex TMT reagents following the manufacturer’s instruction. Finally, the labeled samples were equally mixed, desalted again and dried by speedvac.
LC/LC-MS/MS analysis of TMT labeled peptides and phosphopeptides
The analysis was performed essentially based on our optimized platform(Wang et al., 2015). The mixed TMT labeled peptides were solubilized in buffer A (10 mM ammonium formate, pH 8) and divided for the analyses of whole proteome (10%) and phosphoproteome (90%). The 10% portion was separated by basic pH RPLC (an XBridge C18 column, 4.6 mm × 25 cm, 3.5 μm resin, Waters) a gradient from 15% to 65% buffer B (95% acetonitrile plus buffer A, flow rate of 0.4 ml/min). 40 fractions were then analyzed by acidic pH RPLC (75 μm × 15 cm, 1.9 μm C18 resin, Dr. Maisch GmbH, Germany) on an Orbitrap Fusion MS (Thermo Fisher Scientific). Peptides were eluted by 20–45% buffer B gradient in ~2 h (buffer A: 0.2% formic acid, 5% DMSO; buffer B: buffer A plus 65% acetonitrile, flow rate of 0.25 μl/min). Mass spectrometer was operated in data-dependent mode with a survey with one MS scan (60K resolution, 1 × 106 AGC target and 50 ms maximal ion time) and 20 MS/MS high resolution scans (60K resolution, 1 × 105 AGC target, 105 ms maximal ion time, HCD, 35 normalized collision energy).
The remaining 90% peptides were fractionated basic pH RPLC on the same column but with a shallow gradient (15% to 45% buffer B), and ~40 fractions were subjected to optimized phosphopeptide enrichment by TiO2 beads (GL sciences). The enriched phosphopeptides analyzed by acidic pH RPLC on a similar nanoscale column but with a small ID of 50 μm to increase sensitivity, and a modified gradient (9–35% buffer B, flow rate of 0.15 μl/min). The enriched phosphopeptides analyzed by acidic pH RPLC on a similar nanoscale column but with a small ID of 50 μm to increase sensitivity, and a modified gradient (9–35% buffer B, flow rate of 0.15 μl/min).
The acquired MS/MS data were search against Uniprot human database (88,717 protein entries) by our recently developed JUMP program to improve the sensitivity and specificity (Wang et al., 2015). Searching parameters included precursor ion and product ion mass tolerance (± 10 ppm), static mass shift for TMT-tagged N-terminus and lysine (+229.16293), dynamic modification for phosphorylation (+79.96633) of serine, threonine and tyrosine and oxidation (+15.99492) of methionine, three maximal modification sites, fully tryptic restriction (with two maximal missed cleavages), and the assignment of a, b, and y ions. Putative peptide spectra matches were filtered by MS mass accuracy and matchiworng scores (Jscore and ΔJn) to reach a protein FDR lower than 1%. If one peptide was matched to multiple proteins, the peptide was represented by the protein with the highest peptide-spectrum matches (PSM) according to the rule of parsimony. In addition, phosphosite reliability was re-analyzed by the localization score (Lscore) in the JUMP software suite (Tan et al., 2017).
TMT-based protein and phosphopeptide quantification by the JUMP software suite
The analysis was performed as previously reported with following modifications(Niu et al., 2017): (i) exacting TMT reporter ion intensities of each PSM; (ii) correcting the raw intensities based on isotopic distribution of each labeling reagent (e.g. TMT126 generates 91.8%, 7.9% and 0.3% of 126, 127, 128 m/z ions, respectively); (iii) excluding PSMs of very low intensities (e.g. minimum intensity of 1,000 and median intensity of 5,000); (iv) removing sample loading bias by normalization with the trimmed median intensity of all PSMs; (v) calculating the mean-centered intensities across samples (e.g. relative intensities between each sample and the mean), which has the merits as follows: (1) each scan is fairly treated regardless of its intensity level to avoid bias toward highly or lowly abundant PSMs; (2) outliers can be readily detected by statistical tests (e.g. Dixon’s Q-test or generalized extreme Studentized deviate test); (vi) summarizing protein or phosphopeptide relative intensities by averaging related PSMs. To generate a combined quantification table for multiple batches, two common samples were included in each batch as an internal standard, by which MS intensities were normalized for batch effect correction as reported (Stewart et al., 2018).
Immunohistochemistry
Brains, tumors, and spines were formalin-fixed, paraffin-embedded, and sectioned. 5 μM thick sections were stained with H&E and indicated antibodies.
Western blot quantification
Films were scanned as 600 dpi grayscale TIFF images. Images were analyzed on ImageStudioLite software. Band intensities were measured and normalized to loading controls such actin, gapdh, lamin A/C or histone (for nuclear fraction). Data were transferred to Prism for further analysis and figure representation.
Imaging analysis
For quantitative intensity measurements, images were acquired and compared with identical gain, laser power, and exposure time. Mean ciliary intensities were measured on Imaris.
Quantitation and Statistical methods
Statistical parameters are reported within the figure legends. All statistical analyses, which include student t-tests, one- or two-way ANOVAs, Mann-Whitney, Mantel-Cox test were calculated using GraphPad Prism 8.
Supplementary Material
Supplemental Figure Legends
Supplementary Figure 1 (related to Figure 1) (A) Cyclic nucleotide transporters (ABCC4, ABCC11, ABCC5, and ABCG2) expression in MB cohort compared in a heatmap from GSE37385 (Northcott et al., 2012b).
(B) ABCC4 expression in pediatric MB cohort from (Cavalli et al., 2017; Kool et al., 2008; Robinson et al., 2012). * P < 0.05, ** P < 0.01, **** P < 0.0001. One-way ANOVA.
(C) ABCC4 DNA copy number profile across MB subgroups (Northcott et al., 2012b). * P < 0.05, ** P < 0.01, **** P < 0.0001. One-way ANOVA.
(D) MB patients with available survival and gene expression information from GSE85217 (Cavalli et al., 2017). The relationship between ABCC4 mRNA expression and survival time across all MB subgroup was analyzed using the Kaplan-Meier method with log-rank statistics.
(E) ABCC4, GLI1 and GLI2 NetBID inferred activity in pediatric MB cohort from GSE85217(Cavalli et al., 2017; Kool et al., 2008; Robinson et al., 2012). **** P < 0.0001. One-way ANOVA. (All the P values <0.0001)
(F) Analysis of correlation between ABCC4 and GLI2 NetBID inferred activity from GSE85217(Cavalli et al., 2017). Spearman’s correlation r was 0.8182, P<0.0001.
(G) GLI2 ChIP-Seq and H3K27ac tracks indicating conserved enhancer element found in within ABCC4. GLI2 ChIP-Seq data was obtained from ENCODE (accession: ENCSR978EQY or GSE105977). H3K27ac data was from MB cohorts Two potential conserved binding sites within the first intron were identified and marked in purple and orange. GLI2 motifs loci in the bindings sites were marked with darker line. One HEK293-specific site was marked in teal.
(H) Ptch−/− MEF cells were treated with indicated concentrations of GANT61 for 48 hours. Levels of indicated proteins were measured by western blotting.
Supplementary Figure 2 (related to Figure 2) (A) NIH3T3 cells were transfected with indicated Abcc4 siRNAs (15 nM). Cells were harvested at indicated time points post-SAG (200 nM). RNA was isolated at indicated time points. Levels of indicated transcripts were measured by qRT-PCR. Representative data of two independent experiments. Bars represent mean (± SD). * P < 0.05, ** P < 0.01, *** P <0.001. Two-way ANOVA.
Supplementary Figure 3 (related to Figure 4) (A) Intracellular cAMP level in WT or Abcc4−/− cells were measured by ELISA 16 hours post SAG (200 nM) treatment or DMSO control. Bars represent cAMP level relative to WT, untreated control.
(B) WT, Abcc4−/− Abcc4−/−expressing exogenous human ABCC4 NIH3T3 cells were treated with SHH for duration indicated. Levels of indicated proteins were measured by western blotting.
(C) WT or Abcc4−/− NIH3T3 expressing nuclear cAMP FRET sensors. Representative live-cell imaging analysis. Percentage of FRET change is represented as means (± SD) of three independent experiments. * P < 0.05, ** P < 0.01, *** P <0.001, Mann-Whitney test.
(D) The maximum change in FRET upon FSK and IBMX addition relative to basal. Comparison of kinetics of maximum FRET change upon FSK and IBMX addition.
(E) Abundance of indicated phosphopeptides in WT and Abcc4−/− cells treated with SHH measured by tandem-mass-tag (TMT) mass spectrometry.
Acknowledgements
This study was supported by P30 CA021765 Cancer Center Support grant (J.D.S), R01AG053987 (J.P.), and ALSAC. We thank Drs. Young-Goo Han and Stacy Ogden, Frederique Zindy, and members of the Schuetz lab for helpful discussions. Microscopy images were acquired at Cell & Tissue Imaging Center, which is supported by SJCRH and NCI (P30 CA021765). We thank Drs. Victoria Frohlich, Aaron Pitre, Jennifer Peters, and Sharon King for technical help with microscopy data acquisition and analyses. We thank the staff of Flow Cytometry and Cell Sorting Shared Resource (NCI grant P30 CA021765). Dr. Young-Goo Han for SufuKD cell line, and Dr. Philip Beachy for Ptch−/−, Light2, and HEK293-SHH producing cell lines.
Footnotes
Conflict of Interest:
The authors declare no potential conflicts of interest.
References
- Atwood SX, Sarin KY, Whitson RJ, Li JR, Kim G, Rezaee M, Ally MS, Kim J, Yao C, Chang ALS, et al. (2015). Smoothened Variants Explain the Majority of Drug Resistance in Basal Cell Carcinoma. Cancer Cell 27, 342–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann VA, Mayrhofer JE, Ilouz R, Tschaikner P, Raffeiner P, Röck R, Courcelles M, Apelt F, Lu T-W, Baillie GS, et al. (2016). Gpr161 anchoring of PKA consolidates GPCR and cAMP signaling. Proc. Natl. Acad. Sci. U. S. A 113, 7786–7791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai B, Tan H, Pagala VR, High AA, Ichhaporia VP, Hendershot L, and Peng J (2017). Deep Profiling of Proteome and Phosphoproteome by Isobaric Labeling, Extensive Liquid Chromatography, and Mass Spectrometry. Methods Enzymol. 585, 377–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brugier̀es L, Remenieras A, Pierron G, Varlet P, Forget S, Byrde V, Bombled J, Puget S, Caron O, Dufour C, et al. (2012). High frequency of germline SUFU mutations in children with desmoplastic/nodular medulloblastoma younger than 3 years of age. J. Clin. Oncol 30, 2087–2093. [DOI] [PubMed] [Google Scholar]
- Cavalli FMG, Remke M, Rampasek L, Peacock J, Shih DJH, Luu B, Garzia L, Torchia J, Nor C, Morrissy AS, et al. (2017). Intertumoral Heterogeneity within Medulloblastoma Subgroups. Cancer Cell 31, 737–754.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Yue S, Xie L, Pu XH, Jin T, and Cheng SY (2011). Dual phosphorylation of suppressor of fused (Sufu) by PKA and GSK3?? regulates its stability and localization in the primary cilium. J. Biol. Chem 286, 13502–13511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copsel S, Garcia C, Diez F, Vermeulem M, Baldi A, Bianciotti LG, Russel FGM, Shayo C, and Davio C (2011). Multidrug resistance protein 4 (MRP4/ABCC4) regulates cAMP cellular levels and controls human leukemia cell proliferation and differentiation. J. Biol. Chem 286, 6979–6988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copsel S, Bruzzone A, May M, Beyrath J, Wargon V, Cany J, Russel FGM, Shayo C, and Davio C (2014). Multidrug resistance protein 4/ ATP binding cassette transporter 4: a new potential therapeutic target for acute myeloid leukemia. Oncotarget 5, 9308–9321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depry C, Allen MD, and Zhang J (2011). Visualization of PKA activity in plasma membrane microdomains. Mol. BioSyst 7, 52–58. [DOI] [PubMed] [Google Scholar]
- Dijkgraaf GJP, Alicke B, Weinmann L, Januario T, West K, Modrusan Z, Burdick D, Goldsmith R, Robarge K, Sutherlin D, et al. (2011). Small molecule inhibition of GDC-0449 refractory smoothened mutants and downstream mechanisms of drug resistance. Cancer Res. 71, 435–444. [DOI] [PubMed] [Google Scholar]
- DiPilato LM, Cheng X, and Zhang J (2004). Fluorescent indicators of cAMP and Epac activation reveal differential dynamics of cAMP signaling within discrete subcellular compartments. Proc. Natl. Acad. Sci 101, 16513–16518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du X, Wen J, Wang Y, Karmaus PWF, Khatamian A, Tan H, Li Y, Guy C, Nguyen T-LM, Dhungana Y, et al. (2018). Hippo/Mst signalling couples metabolic state and immune function of CD8α+ dendritic cells. Nature 558, 141–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espstein EH, de Sauvage FJ, Xie J, Murone M, Luoh S-M, Ryan A, Gu Q, Zhang C, Bonifas JM, Lam C-W, et al. (1998). Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature 391, 90–92. [DOI] [PubMed] [Google Scholar]
- Huang Y, Roelink H, and McKnight GS (2002). Protein kinase A deficiency causes axially localized neural tube defects in mice. J. Biol. Chem 277, 19889–19896. [DOI] [PubMed] [Google Scholar]
- Humke EW, Dorn KV, Milenkovic L, Scott MP, and Rohatgi R (2010). The output of Hedgehog signaling is controlled by the dynamic association between Suppressor of Fused and the Gli proteins. Genes Dev. 24, 670–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DTW, Jäger N, Kool M, Zichner T, Hutter B, Sultan M, Cho Y-J, Pugh TJ, Hovestadt V, Stütz AM, et al. (2012). Dissecting the genomic complexity underlying medulloblastoma. Nature 488, 100–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kool M, Koster J, Bunt J, Hasselt NE, Lakeman A, van Sluis P, Troost D, Meeteren NS, Caron HN, Cloos J, et al. (2008). Integrated Genomics Identifies Five Medulloblastoma Subtypes with Distinct Genetic Profiles, Pathway Signatures and Clinicopathological Features. PLoS One 3, e3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kool M, Jones DTDTWDTWDTW, Jäger N, Northcott PAPAA, Pugh TJTJTJ, Hovestadt V, Piro RMM, Esparza LAA, Markant SLL, Remke M, et al. (2014). Genome Sequencing of SHH Medulloblastoma Predicts Genotype-Related Response to Smoothened Inhibition. Cancer Cell 25, 393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauth M, Bergström A, Shimokawa T, and Toftgård R (2007). Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc. Natl. Acad. Sci. U. S. A 104, 8455–8460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leggas M, Adachi M, Scheffer GL, Sun D, Wielinga P, Du G, Mercer KE, Zhuang Y, Panetta JC, Johnston B, et al. (2004). Mrp4 confers resistance to topotecan and protects the brain from chemotherapy. Mol. Cell. Biol 24, 7612–7621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Krishnamurthy PC, Penmatsa H, Marrs KL, Wang XQ, Zaccolo M, Jalink K, Li M, Nelson DJ, Schuetz JD, et al. (2007). Spatiotemporal coupling of cAMP transporter to CFTR chloride channel function in the gut epithelia. Cell 131, 940–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfouace M, Cheepala S, Jackson S, Fukuda Y, Patel YT, Fatima S, Kawauchi D, Shelat AA, Stewart CF, Sorrentino BP, et al. (2015). ABCG2 Transporter Expression Impacts Group 3 Medulloblastoma Response to Chemotherapy. Cancer Res. 75, 3879–3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay S, Wen X, Ratti N, Loktev A, Rangell L, Scales SJ, and Jackson PK (2013). The Ciliary G-Protein-Coupled Receptor Gpr161 Negatively Regulates the Sonic Hedgehog Pathway via cAMP Signaling. Cell 152, 210–223. [DOI] [PubMed] [Google Scholar]
- Murray J, Valli E, Yu DMT, Truong AM, Gifford AJ, Eden GL, Gamble LD, Hanssen KM, Flemming CL, Tan A, et al. (2017). Suppression of the ATP-binding cassette transporter ABCC4 impairs neuroblastoma tumour growth and sensitises to irinotecan in vivo. Eur. J. Cancer 83, 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niewiadomski P, Kong J, Ahrends R, Ma Y, Humke E, Khan S, Teruel M, Novitch B, and Rohatgi R (2014). Gli protein activity is controlled by multisite phosphorylation in vertebrate hedgehog signaling. Cell Rep. 6, 168–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu M, Cho J-H, Kodali K, Pagala V, High AA, Wang H, Wu Z, Li Y, Bi W, Zhang H, et al. (2017). Extensive Peptide Fractionation and y 1 Ion-Based Interference Detection Method for Enabling Accurate Quantification by Isobaric Labeling and Mass Spectrometry. Anal. Chem 89, 2956–2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northcott PA, Jones DTW, Kool M, Robinson GW, Gilbertson RJ, Cho Y-J, Pomeroy SL, Korshunov A, Lichter P, Taylor MD, et al. (2012a). Medulloblastomics: the end of the beginning. Nat. Rev. Cancer 12, 818–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northcott PA, Shih DJH, Peacock J, Garzia L, Sorana Morrissy A, Zichner T, Stütz AM, Korshunov A, Reimand J, Schumacher SE, et al. (2012b). Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature 488, 49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northcott PA, Lee C, Zichner T, Stütz AM, Erkek S, Kawauchi D, Shih DJH, Hovestadt V, Zapatka M, Sturm D, et al. (2014). Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature 511, 428–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northcott PA, Buchhalter I, Morrissy AS, Hovestadt V, Weischenfeldt J, Ehrenberger T, Gröbner S, Segura-Wang M, Zichner T, Rudneva VA, et al. (2017). The whole-genome landscape of medulloblastoma subtypes. Nature 547, 311–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pak E, Segal RA, Ahlfeld J, Favaro R, Pagella P, Kretzschmar HA, Nicolis S, Schuller U, Amakye D, Jagani Z, et al. (2016). Hedgehog Signal Transduction: Key Players, Oncogenic Drivers, and Cancer Therapy. Dev. Cell 38, 333–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal K, and Mukhopadhyay S (2014). Primary cilium and sonic hedgehog signaling during neural tube patterning: role of GPCRs and second messengers. Dev. Neurobiol 75, n/a–n/a. [DOI] [PubMed] [Google Scholar]
- Pitre A, Ge Y, Lin W, Wang Y, Fukuda Y, Temirov J, Phillips AH, Peters JL, Fan Y, Ma J, et al. (2017). An unexpected protein interaction promotes drug resistance in leukemia. Nat. Commun 8, 1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswamy V, and Taylor MD (2017). Medulloblastoma : From Myth to Molecular. 1–10. [DOI] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47–e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson G, Parker M, Kranenburg TA, Lu C, Chen X, Ding L, Phoenix TN, Hedlund E, Wei L, Zhu X, et al. (2012). Novel mutations target distinct subgroups of medulloblastoma. Nature 488, 43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson GW, Kaste SC, Chemaitilly W, Bowers DC, Laughton S, Smith A, Gottardo N, Partap S, Bendel A, Wright KD, et al. (2017). Irreversible growth plate fusions in children with medulloblastoma treated with a targeted hedgehog pathway inhibitor. Oncotarget 8, 69295–69302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roussel MF, and Hatten ME (2011). Cerebellum development and medulloblastoma. Curr. Top. Dev. Biol 94, 235–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sample V, DiPilato LM, Yang JH, Ni Q, Saucerman JJ, and Zhang J (2012). Regulation of nuclear PKA revealed by spatiotemporal manipulation of cyclic AMP. Nat. Chem. Biol 8, 375–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasai K, Romer JT, Lee Y, Finkelstein D, Fuller C, McKinnon PJ, and Curran T (2006). Shh pathway activity is down-regulated in cultured medulloblastoma cells: implications for preclinical studies. Cancer Res. 66, 4215–4222. [DOI] [PubMed] [Google Scholar]
- Sassi Y, Abi-Gerges A, Fauconnier J, Mougenot N, Reiken S, Haghighi K, Kranias EG, Marks AR, Lacampagne A, Engelhardt S, et al. (2012). Regulation of cAMP homeostasis by the efflux protein MRP4 in cardiac myocytes. FASEB J. 26, 1009–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, and Ideker T (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SMC, Bianski BM, Orr BA, Harknett G, Onar-Thomas A, Gilbertson RJ, Merchant TE, Roussel MF, and Tinkle CL (2019). Preclinical Modeling of Image-Guided Craniospinal Irradiation for Very-High-Risk Medulloblastoma. Int. J. Radiat. Oncol 103, 728–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg SF, and Brunton LL (2001). Compartmentation of G protein-coupled signaling pathways in cardiac myocytes. Annu. Rev. Pharmacol. Toxicol 41, 751–773. [DOI] [PubMed] [Google Scholar]
- Stewart E, McEvoy J, Wang H, Chen X, Honnell V, Ocarz M, Gordon B, Dapper J, Blankenship K, Yang Y, et al. (2018). Identification of Therapeutic Targets in Rhabdomyosarcoma through Integrated Genomic, Epigenomic, and Proteomic Analyses. Cancer Cell 34, 411–426.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taipale J, Chen JK, Cooper MK, Wang B, Mann RK, Milenkovic L, Scott MP, and Beachy PA (2000). Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature 406, 1005–1009. [DOI] [PubMed] [Google Scholar]
- Tan H, Yang K, Li Y, Shaw TI, Wang Y, Blanco DB, Wang X, Cho J-H, Wang H, Rankin S, et al. (2017). Integrative Proteomics and Phosphoproteomics Profiling Reveals Dynamic Signaling Networks and Bioenergetics Pathways Underlying T Cell Activation. Immunity 46, 488–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MD, Liu L, Raffel C, Hui C, Mainprize TG, Zhang X, Agatep R, Chiappa S, Gao L, Lowrance A, et al. (2002). Mutations in SUFU predispose to medulloblastoma. Nat. Genet 31, 306–310. [DOI] [PubMed] [Google Scholar]
- Tiecke E, Turner R, Sanz-Ezquerro JJ, Warner A, and Tickle C (2007). Manipulations of PKA in chick limb development reveal roles in digit patterning including a positive role in Sonic Hedgehog signaling. Dev. Biol 305, 312–324. [DOI] [PubMed] [Google Scholar]
- Tuson M, He MM, and Anderson KV (2011). Protein kinase A acts at the basal body of the primary cilium to prevent Gli2 activation and ventralization of the mouse neural tube. Development 138, 4921–4930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uziel T, Zindy F, Xie S, Lee Y, Forget A, Magdaleno S, Rehg JE, Calabrese C, Solecki D, Eberhart CG, et al. (2005). The tumor suppressors Ink4c and p53 collaborate independently with Patched to suppress medulloblastoma formation. Genes Dev. 19, 2656–2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C. bing, Pan Y, and Wang B (2010). Supressor of fused and Spop regulate the stability, processing, and function of Gli2 and Gli3 full-length activators but not their repressors. Development 137, 2001–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Yang Y, Li Y, Bai B, Wang X, Tan H, Liu T, Beach TG, Peng J, and Wu Z (2015). Systematic Optimization of Long Gradient Chromatography Mass Spectrometry for Deep Analysis of Brain Proteome. J. Proteome Res 14, 829–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen X, Lai CK, Evangelista M, Hongo J-A, de Sauvage FJ, and Scales SJ (2010). Kinetics of hedgehog-dependent full-length Gli3 accumulation in primary cilia and subsequent degradation. Mol. Cell. Biol 30, 1910–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C-C, Hou S, Orr BA, Kuo BR, Youn YH, Ong T, Roth F, Eberhart CG, Robinson GW, Solecki DJ, et al. (2017). mTORC1-Mediated Inhibition of 4EBP1 Is Essential for Hedgehog Signaling-Driven Translation and Medulloblastoma. Dev. Cell 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu VM, Chen SC, Arkin MR, and Reiter JF (2012). Small molecule inhibitors of Smoothened ciliary localization and ciliogenesis. Proc. Natl. Acad. Sci 109, 13644–13649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P, Duong DM, and Peng J (2009). Systematical Optimization of Reverse-Phase Chromatography for Shotgun Proteomics. J. Proteome Res 8, 3944–3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yauch RL, Dijkgraaf GJP, Alicke B, Januario T, Ahn CP, Bazan JF, Kan Z, Seshagiri S, and Hann CL (2009). Smoothened mutation confers resistance to a hedgehog pathway inhibitor in medulloblastoma. 23231, 572–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Zhuang T, Lin Q, Yang B, Xu X, Xin G, Zhu S, Wang G, Yu B, Zhang T, et al. (2018). Patched1–ArhGAP36–PKA–Inversin axis determines the ciliary translocation of Smoothened for Sonic Hedgehog pathway activation. Proc. Natl. Acad. Sci 201804042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhukova N, Ramaswamy V, Remke M, Pfaff E, Shih DJH, Martin DC, Castelo-Branco P, Baskin B, Ray PN, Bouffet E, et al. (2013). Subgroup-specific prognostic implications of TP53 mutation in medulloblastoma. J. Clin. Oncol 31, 2927–2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure Legends
Supplementary Figure 1 (related to Figure 1) (A) Cyclic nucleotide transporters (ABCC4, ABCC11, ABCC5, and ABCG2) expression in MB cohort compared in a heatmap from GSE37385 (Northcott et al., 2012b).
(B) ABCC4 expression in pediatric MB cohort from (Cavalli et al., 2017; Kool et al., 2008; Robinson et al., 2012). * P < 0.05, ** P < 0.01, **** P < 0.0001. One-way ANOVA.
(C) ABCC4 DNA copy number profile across MB subgroups (Northcott et al., 2012b). * P < 0.05, ** P < 0.01, **** P < 0.0001. One-way ANOVA.
(D) MB patients with available survival and gene expression information from GSE85217 (Cavalli et al., 2017). The relationship between ABCC4 mRNA expression and survival time across all MB subgroup was analyzed using the Kaplan-Meier method with log-rank statistics.
(E) ABCC4, GLI1 and GLI2 NetBID inferred activity in pediatric MB cohort from GSE85217(Cavalli et al., 2017; Kool et al., 2008; Robinson et al., 2012). **** P < 0.0001. One-way ANOVA. (All the P values <0.0001)
(F) Analysis of correlation between ABCC4 and GLI2 NetBID inferred activity from GSE85217(Cavalli et al., 2017). Spearman’s correlation r was 0.8182, P<0.0001.
(G) GLI2 ChIP-Seq and H3K27ac tracks indicating conserved enhancer element found in within ABCC4. GLI2 ChIP-Seq data was obtained from ENCODE (accession: ENCSR978EQY or GSE105977). H3K27ac data was from MB cohorts Two potential conserved binding sites within the first intron were identified and marked in purple and orange. GLI2 motifs loci in the bindings sites were marked with darker line. One HEK293-specific site was marked in teal.
(H) Ptch−/− MEF cells were treated with indicated concentrations of GANT61 for 48 hours. Levels of indicated proteins were measured by western blotting.
Supplementary Figure 2 (related to Figure 2) (A) NIH3T3 cells were transfected with indicated Abcc4 siRNAs (15 nM). Cells were harvested at indicated time points post-SAG (200 nM). RNA was isolated at indicated time points. Levels of indicated transcripts were measured by qRT-PCR. Representative data of two independent experiments. Bars represent mean (± SD). * P < 0.05, ** P < 0.01, *** P <0.001. Two-way ANOVA.
Supplementary Figure 3 (related to Figure 4) (A) Intracellular cAMP level in WT or Abcc4−/− cells were measured by ELISA 16 hours post SAG (200 nM) treatment or DMSO control. Bars represent cAMP level relative to WT, untreated control.
(B) WT, Abcc4−/− Abcc4−/−expressing exogenous human ABCC4 NIH3T3 cells were treated with SHH for duration indicated. Levels of indicated proteins were measured by western blotting.
(C) WT or Abcc4−/− NIH3T3 expressing nuclear cAMP FRET sensors. Representative live-cell imaging analysis. Percentage of FRET change is represented as means (± SD) of three independent experiments. * P < 0.05, ** P < 0.01, *** P <0.001, Mann-Whitney test.
(D) The maximum change in FRET upon FSK and IBMX addition relative to basal. Comparison of kinetics of maximum FRET change upon FSK and IBMX addition.
(E) Abundance of indicated phosphopeptides in WT and Abcc4−/− cells treated with SHH measured by tandem-mass-tag (TMT) mass spectrometry.